

Abstract
The ROS1 fusion kinase is an attractive antitumor target. Though with significant clinical efficacy, the well-known first-generation ROS1 inhibitor (ROS1i) crizotinib inevitably developed acquired resistance due to secondary point mutations in the ROS1 kinase. Novel ROS1is effective against mutations conferring secondary crizotinib resistance, especially G2032R, are urgently needed. In the present study, we evaluated the antitumor efficacy of SAF-189s, the new-generation ROS1/ALK inhibitor, against ROS1 fusion wild-type and crizotinib-resistant mutants. We showed that SAF-189s potently inhibited ROS1 kinase and its known acquired clinically resistant mutants, including the highly resistant G2032R mutant. SAF-189s displayed subnanomolar to nanomolar IC50 values against ROS1 wild-type and mutant kinase activity and a selectivity vs. other 288 protein kinases tested. SAF-189s blocked cellular ROS1 signaling, and in turn potently inhibited the cell proliferation in HCC78 cells and BaF3 cells expressing ROS1 fusion wild-type and resistance mutants. In nude mice bearing BaF3/CD74–ROS1 or BaF3/CD74–ROS1G2032R xenografts, oral administration of SAF-189s dose dependently suppressed the growth of both ROS1 wild-type- and G2032R mutant-driven tumors. In a patient-derived xenograft model of SDC4–ROS1 fusion NSCLC, oral administration of SAF-189s (20 mg/kg every day) induced tumor regression and exhibited notable prolonged and durable efficacy. In addition, SAF-189s was more potent than crizotinib and comparable to lorlatinib, the most advanced ROS1i known against the ROS1G2032R. Collectively, these results suggest the promising potential of SAF-189s for the treatment of patients with the ROS1 fusion G2032R mutation who relapse on crizotinib. It is now recruiting both crizotinib-relapsed and naive ROS1-positive NSCLC patients in a multicenter phase II trial (ClinicalTrials.gov Identifier: NCT04237805).
Keywords: ROS1 kinase, ROS1 inhibitor, SAF-189s, lorlatinib, crizotinib resistance, G2032R mutant, NSCLC
Introduction
Oncogenic c-ros oncogene (ROS1) fusion kinases have been found in various cancers, including non-small cell lung cancer (NSCLC), and are attractive antitumor targets [1–3]. Crizotinib is a well-known first-generation inhibitor of ALK and ROS1 and has shown significant clinical efficacy in ROS1 fusion-positive NSCLC [4, 5]. However, similar to patients treated with other kinase inhibitors [6, 7], patients treated with crizotinib also develop acquired resistance via secondary point mutations in ROS1 kinase. Some mutations associated with acquired resistance, such as G2032R, S1986Y, S1986F, D2033N, L2026M, and L1951R, have been identified in patients who relapse on crizotinib [8]. ROS1G2032R is the most frequent secondary mutation [9]. Novel ROS1is active against known resistance-associated mutants, especially the ROS1G2032R mutant, are needed.
SAF-189s is a novel new-generation ALK inhibitor with central nervous system (CNS)-penetrating properties that was codeveloped by Fochon Pharmaceuticals Ltd and the Shanghai Institute of Materia Medica. In the multicenter dose-finding portion phase I/II trial of SAF-189s in ALK-positive NSCLC, notable antitumor activity was observed in both crizotinib-naive and crizotinib-relapsed patients, particularly in patients with CNS metastasis, and SAF-189s was very tolerable [10]. In parallel, in the present study, SAF-189s was found to be effective against ROS1 kinase and a series of clinically resistant mutants, including the highly resistant G2032R mutant. Moreover, SAF-189s displayed substantial efficacy against both ROS1 wild-type (wt)- and G2032R mutant-driven tumors. Based on these impressive preclinical and clinical data, both crizotinib-relapsed and naive ROS1-positive NSCLC patients are being recruited for the multicenter phase II trial (ClinicalTrials.gov identifier: NCT04237805).
Here, for the first time, we explored the preclinical antitumor activity of SAF-189s against tumor cell lines and subcutaneous (s.c.) xenograft models derived from ROS1 fusion wt and crizotinib-resistant tumors.
Materials and methods
Compound
SAF-189s was designed and synthesized by Fochon Pharmaceuticals Ltd (WO 2014071832, WO 2017133663). Crizotinib, ceritinib, and lorlatinib were purchased from Selleck Chemicals (Houston, USA).
Cell lines
The HCC78 and BaF3 cell lines were obtained from Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH. HighFive cells were obtained from the American Type Culture Collection. All cell lines used in this study were acquired between August 2014 and March 2016 and were maintained in the medium suggested by the suppliers. Cells were confirmed to be mycoplasma free and were passaged less than 25–30 times after resuscitation. Cell lines were characterized using short tandem repeat markers by Genesky Biopharma Technology (last tested in 2017).
Expression and purification of the human ROS1 kinase domain
The sequence of the GST-tagged fusion human ROS1 kinase domain was synthesized by Generay Biotech and cloned into the pFastBacHTB insect cell expression vector (Life Technologies). Baculovirus stock was generated using the Bac-to-Bac baculovirus expression system. HighFive cells were harvested 48 h after baculovirus infection. Protein purification was performed using Glutathione Sepharose™ 4B Media (Thermo Fisher).
Kinase inhibition assay
Active ALK proteins were purchased from Eurofins (Fremont, USA), and the kinase activity of ROS1 and ALK was assessed using ELISA, as previously reported [11]. IC50 values were calculated by dose–response curve fitting using GraphPad Prism. The broad kinase selectivity profile of SAF-189s (0.001, 0.01, 0.1, and 1 μM) was determined by Eurofins (UK) by screening the compound against 299 human recombinant kinases.
ATP competition assay
An ELISA-based ATP competition assay was performed by introducing increasing concentrations of ATP. The inhibition of ROS1 kinase activity at different ATP concentrations (5, 25, 125, or 625 μM) was assessed with a series of fixed concentrations of SAF-189s. Inhibition rate–concentration curves were used to distinguish competitive and noncompetitive inhibitors.
Cell proliferation and viability assays
Cells were seeded into 96-well plates overnight, incubated with a series of concentrations of the compounds or control solvent for 72 h, and then subjected to a sulforhodamine B or cell counting kit-8 assay. IC50 values were calculated by dose–response curve fitting with a SoftMax Pro-based four-parameter method.
Western blot analysis
After the indicated treatment, cells were lysed in 1× sodium dodecyl sulfate (SDS). Cell lysates were subsequently separated by SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. Proteins were incubated with the appropriate primary antibodies (against phospho-ROS1, phospho-SHP2, phospho-ERK1/2, phospho-AKT, phospho-STAT3, ROS1, SHP2, ERK1/2, AKT, STAT3, GAPDH, and actin) (Cell Signaling Technology, Beverly, MA, USA) followed by horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG. Protein bands were detected using enhanced chemiluminescence detection reagent (Thermo Fisher Scientific, Rockford, IL, USA).
In vivo antitumor activity assay
Cell line-derived xenograft (CDX) model
Female nude mice (4–6 weeks old) were housed and maintained under specific pathogen-free conditions. Tumor cells (5 × 106 in 200 μL) were grown as s.c. tumors in nude mice. Treatment experiments were started when the tumor volume (TV) reached 100–150 mm3, and mice were randomized to the vehicle control or compound-treated group (oral, qd) and treated for 14 days. The number of mice per group in the vehicle- and compound-treated groups was 12 and 6, respectively. The tumor size was measured twice weekly. The CDX model studies were approved by the Institutional Animal Care and Use Committee (IACUC) at Shanghai Institute of Materia Medica.
Patient-derived xenograft (PDX) model
Animal studies using the LU-01-0414 NSCLC PDX model were performed by WuXi AppTec (Shanghai, China) (n = 6/group). The mice were orally administered vehicle control, SAF-189s, or crizotinib at the indicated dose once daily for 35 days. In addition, 20 mg/kg SAF-189s was administered for another 25 days, and administration was then stopped for an additional 20 days until we ended the experiment. The PDX model study was approved by the IACUCs of WuXi AppTec.
We calculated the TVs with the following formula: TV = (width2 × length)/2. Tumor growth inhibition (TGI) (%) values in the compound-treated and vehicle-treated mice were calculated on the final day of the study with the following formula: 100 × {1 − [(VTreated Final Day − V Treated Day 0)/(VControl Final Day − VControl Day 0)]}.
Immunohistochemistry (IHC)
Tumor specimens were fixed in 4% paraformaldehyde and embedded in paraffin. IHC was performed by Shanghai ZuoChengBio Co., Ltd. using a Vectastain ABC Kit (Vector Laboratories). The Ki67 antibody was purchased from Shanghai ZuoChengBio Co., Ltd.
Statistical analysis
Statistical analysis was performed by one-way ANOVA using GraphPad Prism 8. P < 0.05 was considered statistically significant.
Results
SAF-189s is highly potent against ROS1 wt and G2032R mutant kinases
SAF-189s significantly inhibited ROS1 kinase activity in biochemical assays (IC50 = 0.5 nM). In addition, as shown in Fig. 1a–d and Table 1, SAF-189s was more potent than crizotinib and ceritinib and comparable to lorlatinib, the most advanced ROS1i known to be active against ROS1G2032R. Moreover, SAF-189s effectively inhibited the kinase activity of a series of reported crizotinib-resistant ROS1 mutants, especially the most frequently identified mutant, ROS1G2032R (Fig. 1b–d and Table 1). The efficacy of SAF-189s was equivalent to that of lorlatinib, although the inhibitory effect of these two compounds against some ROS1 mutants was slightly weaker than that against wt ROS1. However, ceritinib, another next-generation ROS1i, could not overcome the ROS1G2032R mutant, consistent with previously reported data [12]. In addition, SAF-189s is an ATP-competitive ROS1i according to kinetic tests (Fig. 1e). Therefore, these findings indicate that in addition to being an ALK inhibitor, SAF-189s is also a highly potent, ATP-competitive ROS1i with notable potency against a series of crizotinib-resistant mutants, including ROS1G2032R. Furthermore, the broad kinase selectivity of SAF-189s was investigated in a panel of 299 other protein kinases (Table S1). In contrast to its high potency against ROS1 and the ALK family (with an IC50 of ~1 nM), SAF-189s had no obvious inhibitory effect against the other 288 (96.3%) tested kinases, with more than 200-fold (214 kinases) or 20-fold (74 kinases) selectivity over ROS1 (IC50 = 0.5 nM), demonstrating the selectivity of SAF-189s for ALK and ROS1. In the present study, we focused on ROS1.
Fig. 1. SAF-189s is highly potent against ROS1 wild-type and G2032R mutant kinases.

Kinase activity inhibition curve of SAF-189s against ROS1 (a), ROS1G2032R (b), and ROS1L2026M (c). d IC50 values of SAF-189s, lorlatinib, ceritinib, and crizotinib against the kinase activity of ROS1 and a series of crizotinib-resistant mutants. e SAF-189s functions in an ATP-competitive manner to inhibit ROS1 kinases. The values shown in Fig.1 represent the mean ± SD.
Table 1.
Biochemical activities (IC50, Mean ± SD, nM) of ROS1 inhibitors and SAF-189s.
| Kinase | SAF-189s | Crizotinib | Ceritinib | Lorlatinib |
|---|---|---|---|---|
| ROS1 | 0.5 ± 0.1 | 1.5 ± 0.2 | 2.7 ± 0.8 | 0.3 ± 0.2 |
| ROS1G2032R | 12.4 ± 5.4 | 222.5 ± 55.2 | 271.4 ± 82.8 | 17.3 ± 3.7 |
| ROS1L2026M | 0.2 ± 0.2 | 9.8 ± 1.3 | 2.6 ± 0.2 | 2.4 ± 0.1 |
| ROS1S1986F | 0.7 ± 0.4 | 3.1 ± 0.9 | 3.7 ± 1.1 | 0.4 ± 0.2 |
| ROS1S1986Y | 0.3 ± 0.3 | 2.9 ± 0.7 | 5.7 ± 1.4 | 0.4 ± 0.3 |
| ROS1D2033N | 2.9 ± 0.9 | 20.2 ± 3.3 | 24.0 ± 2.5 | 2.2 ± 0.3 |
We further evaluated the cellular activity of SAF-189s on targeting ROS1 and its potential to overcome resistance mutants. ROS1 fusions preferentially signal through the autophosphorylation of ROS1 and the phosphorylation of SHP2, MEK/ERK, PI3K/AKT, and STAT3 [13]. Therefore, we examined the impact of SAF-189s on these pathways. As shown in Fig. 2a, in the HCC78 NSCLC cell line harboring a ROS1 gene translocation (SLC34A2–ROS1), the phosphorylation of ROS1, SHP2, AKT, and ERK was significantly inhibited upon treatment with SAF-189s or other ROS1is, whereas the p-STAT3 level remained unchanged, consistent with the previous observation of ROS1i treatment in HCC78 cells [14]. Moreover, 50 nM SAF-189s almost completely blocked ROS1 signaling activation (Fig. 2a). The activity of SAF-189s was much more potent than that of crizotinib and ceritinib; indeed, the potency of SAF-189s at 50 nM was almost comparable to that of crizotinib and ceritinib at 500 nM (Fig. 2a).
Fig. 2. SAF-189s significantly inhibits the downstream signaling pathway of ROS1 and resistance mutants.

a SAF-189s, crizotinib and ceritinib suppressed ROS1 pathway activation in HCC78 cells. HCC78 cells were lysed after 1h of treatment with the corresponding compound and subjected to Western blot analysis. Effects of SAF-189s, crizotinib, ceritinib, and lorlatinib on ROS1 phosphorylation and downstream signaling in BaF3/CD74–ROS1 (b), BaF3/CD74–ROS1G2032R (c), and BaF3/CD74–ROS1L2026M (d) cells. The corresponding cells were lysed after 0.5 h of treatment with the indicated compound and subjected to Western blot analysis.
Furthermore, we generated a “ROS1-dependent” context by introducing a CD74–ROS1 fusion using a mouse pro-B BaF3 cell line that is recognized to be dependent on IL-3 for survival, and the introduction of oncogenic kinases allows cell growth to be independent of IL-3 [15, 16]. The levels of p-ROS1 and p-SHP2, as well as those of p-AKT and p-ERK, were similarly reduced in BaF3/CD74–ROS1 cells (Fig. 2b).
Next, to investigate the potential of SAF-189s to overcome resistance mutants, BaF3 cells engineered to express crizotinib-resistant mutants, including the G2032R and L2026M mutants, were used. As expected, SAF-189s showed potent inhibitory activity against ROS1 signaling in ROS1G2032R mutant cells. Moreover, the potency of SAF-189s against signaling through the G2032R mutant and L2026M mutant was comparable to that of lorlatinib, as observed in the CD74–ROS1 wt context (Fig. 2c, d). In contrast, crizotinib and ceritinib had only a marginal effect on ROS1 signaling in the above BaF3 cell lines harboring resistance mutations (Fig. 2c, d). These data suggest that SAF-189s significantly inhibits the kinase activity and related downstream signaling of both wt ROS1 and resistance mutants.
SAF-189s potently inhibits cell proliferation driven by ROS1 fusion wt and resistance mutants
We then investigated the antiproliferative effect of SAF-189s in cells expressing ROS1 fusion wt and resistance mutants. The antiproliferative potency of SAF-189s, crizotinib, ceritinib, and lorlatinib was compared. As shown in Table 2, SAF-189s significantly inhibited the proliferation of HCC78 and BaF3/CD74–ROS1 cells, both of which harbor the wt ROS1 fusion. The respective IC50 values were 49.4 and 1.1 nM. Moreover, SAF-189s was 20-fold more potent than crizotinib or ceritinib and comparable to lorlatinib (Table 2). Next, we extended our investigation to the context of cells expressing a resistance mutant. The BaF3 cell line expressing the CD74–ROS1 fusion protein with the G2032R mutation was resistant to both crizotinib and ceritinib (Table 2), consistent with reported data. In contrast, SAF-189s and lorlatinib showed more potent growth inhibitory effects on BaF3 cells expressing the G2032R and L2026M-mutated ROS1 fusion protein than crizotinib (Table 2), further confirming that SAF-189s can block downstream signaling in resistant ROS1G2032R and ROS1L2026M mutant cells. Similarly, SAF-189s significantly inhibited the proliferation of BaF3 cells expressing other resistance mutants, including the S1986F, S1986Y, and D2033N mutants (Table 2). Taken together, these in vitro results suggest that SAF-189s potently inhibits the wt ROS1 fusion kinase and overcomes crizotinib resistance mutations, including G2032R.
Table 2.
Cellular activities (IC50, Mean ± SD, nM) of ROS1 inhibitors and SAF-189s.
| Cell lines | SAF-189s | Crizotinib | Ceritinib | Lorlatinib |
|---|---|---|---|---|
| HCC78 | 49.4 ± 21.8 | 739.3 ± 65.1 | 1712.0 ± 556.8 | 33.0 ± 11.0 |
| BaF3/CD74–ROS1 | 1.1 ± 0.6 | 32.2 ± 7.6 | 102.1 ± 11.5 | 1.2 ± 0 |
| BaF3/CD74–ROS1G2032R | 141.0 ± 16.8 | 629.2 ± 131.3 | 1256.8 ± 67.8 | 262.0 ± 11.1 |
| BaF3/CD74–ROS1L2026M | 3.1 ± 0.5 | 162.8 ± 35.0 | 209.4 ± 53.8 | 9.0 ± 1.3 |
| BaF3/CD74–ROS1D2033N | 15.6 ± 4.8 | 177.2 ± 14.5 | 686.8 ± 25.6 | 8.6 ± 1.4 |
| BaF3/CD74–ROS1S1986F | 0.8 ± 0.2 | 51.5 ± 4.2 | 163.2 ± 9.5 | 2.9 ± 0.8 |
| BaF3/CD74–ROS1S1986Y | 1.2 ± 0.7 | 52.9 ± 2.2 | 74.7 ± 10.6 | 1.8 ± 0.2 |
| BaF3 + IL-3 | 1490.3 ± 223.1 | 1317.4 ± 28.2 | 1787.0 ± 342.5 | >10000 |
SAF-189s shows potent antitumor efficacy against ROS1 wt- and G2032R mutant-driven tumors
The in vivo antitumor efficacy of SAF-189s was further assessed in established xenograft models derived from BaF3 cells expressing native CD74–ROS1 and the CD74–ROS1G2032R mutant. The oral administration of SAF-189s displayed dramatic antitumor activity in CD74–ROS1 xenografts. The TGI rates of SAF-189s were 97.3%, 106.7%, and 107.1% at doses of 5, 10, and 20 mg/kg, respectively (Fig. S1a). Moreover, all of the mice in the group receiving 10 and 20 mg/kg exhibited complete tumor regression (Fig. 3a).
Fig. 3. SAF-189s shows potent antitumor efficacy against ROS1 wild-type- and G2032R mutant-driven tumors.

Nude mice bearing BaF3/CD74–ROS1 (a) or BaF3/CD74–ROS1G2032R (b) subcutaneous xenografts were administered vehicle or the compounds once daily for 14 days. The values shown in a and b represent the mean ± SEM. c, d Tumor samples were collected at the end of treatment, and intratumoral Ki67 expression was assessed by immunohistochemical (IHC) analysis. BaF3/CD74–ROS1 (c) or BaF3/CD74–ROS1G2032R (d). Nude mice bearing BaF3/CD74–ROS1 (e) or BaF3/CD74–ROS1G2032R (f) subcutaneous xenografts were administered vehicle or compounds once daily for 14 days. Body weights were measured twice a week and are shown as the mean ± SEM. The values shown in a and b represent the mean ± SEM. ***P < 0.001; by one-way ANOVA.
In contrast to its potent antitumor activity in the BaF3/CD74–ROS1 model (TGI, 103.5%) (Fig. S1a), 50 mg/kg crizotinib exhibited only marginal effects in the BaF3/CD74–ROS1G2032R mutant model (TGI, 22.8%) (Fig. S1b), indicating crizotinib resistance in the CD74–ROS1G2032R model. However, SAF-189s treatment led to marked TGI in the BaF3/CD74–ROS1G2032R mutant model (Fig. 3b). Notably, the effect of SAF-189s at 30 mg/kg was comparable to that of lorlatinib at 30 mg/kg (data not shown). The intratumoral Ki67 proliferation index was then analyzed in the above models, and we found a significant decrease in the Ki67 level in the groups treated with the optimal dose of SAF-189s (Fig. 3c, d). Moreover, SAF-189s was well tolerated at all tested doses (Fig. 3e, f).
Collectively, these findings indicate that SAF-189s, a potent new-generation ROS1i, is effective against reported clinically resistant mutants, including the highly resistant G2032R mutant.
SAF-189s shows potent antitumor efficacy against the SDC4–ROS1 fusion in the PDX model
Compared with the traditional CDX model, which lacks molecular and cellular heterogeneity, the PDX model is expected to serve as an improved disease model by increasing the diversity of molecular damage and retaining 3D tumor-stromal cell components and interactions. We thus investigated the in vivo efficacy of SAF-189s in the ROS1 fusion-positive NSCLC PDX model (LU-01-0414, harboring the SDC4–ROS1 fusion).
We treated the PDX model with SAF-189s (1.25, 5, and 20 mg/kg) for 35 days. In addition, SAF-189s (20 mg/kg) was administered for further 25 days and then stopped for an additional 20 days to observe whether the tumor would rebound until we ended the experiment (Fig. 4a). As shown in Fig. 4b, c, SAF-189s inhibited tumor growth in a dose-dependent manner during the 35-day treatment period. Moreover, 20 mg/kg SAF-189s caused tumor regression, with a TV reduction of greater than 97.9%. After SAF-189s treatment (20 mg/kg), one of the six tumor-bearing mice experienced complete tumor regression.
Fig. 4. SAF-189s shows potent antitumor efficacy against the SDC4–ROS1 fusion in the patient-derived xenograft model.
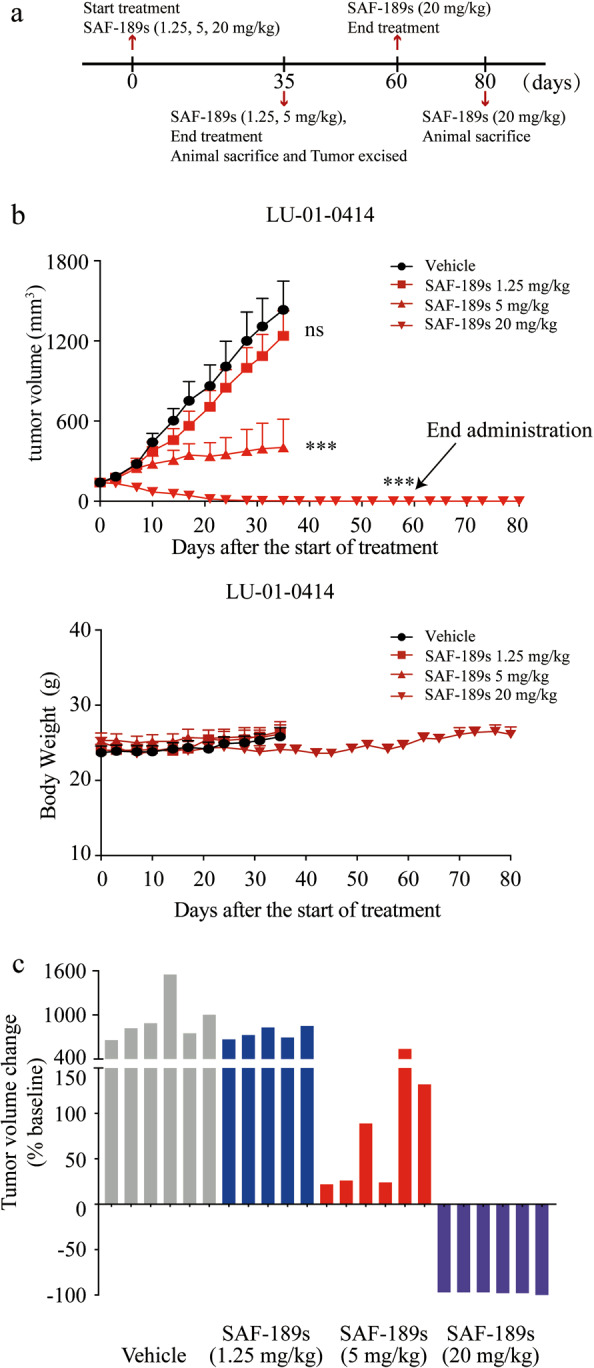
a Schematic of the SAF-189s treatment schedule. Tumor growth and survival were monitored until the experimental endpoints. b, c Nude mice bearing the PDX model (LU-01-0414) with the ROS1 fusion were administered vehicle control or SAF-189s once daily. Body weight is shown as the mean ± SEM in the bottom of b. The changes in tumor volumes in all treated groups at the end of the 35-day treatment period are shown in c. Body weights were measured twice a week, and there was no significant body weight loss in nude mice bearing tumors. The values shown in b represent the mean ± SEM. ***P < 0.001; by one-way ANOVA.
After treatment with 20 mg/kg SAF-189s for another 25 days, five of the six mice exhibited complete tumor regression. Notably, SAF-189s resulted in prolonged and durable posttreatment efficacy, with no tumor regrowth for 20 days post treatment until we ended the experiment (Fig. 4a). In addition, SAF-189s was well tolerated at the tested dose (Fig. 4b). These data indicate that SAF-189s can effectively inhibit tumor growth in the PDX model. Importantly, the tumor will not rebound following treatment with SAF-189s at a dose of 20 mg/kg after ending administration.
Discussion
ROS1 is a clinically validated therapeutic target in NSCLC. Novel ROS1is that can overcome the secondary mutations conferring resistance to the first-generation inhibitor crizotinib, especially the frequently identified G2032R mutation, are urgently needed.
The next-generation ALK inhibitor SAF-189s is currently in a multicenter phase II trial (ClinicalTrials.gov identifier: NCT04237805). Intriguingly, in addition to targeting ALK, SAF-189s also potently inhibits ROS1 and is effective against reported clinically resistant mutants, showing subnanomolar to nanomolar biochemical potency against wt ROS1 kinase and the G2032R, L2026M, S1986F, S1986Y, and D2033N mutants in the present study. Similarly, using a cancer cell line and a series of BaF3 model cell lines, we found that SAF-189s inhibits cell proliferation mediated by ROS1 fusion wt and resistance mutants. Moreover, SAF-189s was more potent than crizotinib and ceritinib and comparable to lorlatinib, the most advanced ROS1i known, against the ROS1G2032R mutant. In both native CD74–ROS1- and CD74–ROS1G2032R mutant-driven CDX models, SAF-189s significantly suppressed tumor growth. Indeed, in the G2032R-resistant mutant model, the efficacy of 30 mg/kg SAF-189s was comparable to that of 30 mg/kg lorlatinib. In the NSCLC PDX model harboring a ROS1 fusion, 20 mg/kg SAF-189s induced tumor regression, and five of the six mice exhibited complete tumor regression. Notably, SAF-189s resulted in prolonged and durable posttreatment efficacy, as indicated by the absence of tumor regrowth for another 20 days after the 60-day treatment period until we ended the experiment.
New-generation ROS1is are active against crizotinib-resistant mutants, such as the L2026M gatekeeper and L1951R, S1986Y/F, and D2033N mutants. However, only a few have been reported to exhibit activity against the most common solvent front mutant, G2032R, including lorlatinib, repotrectinib, and DS-6051b [12, 17, 18]. Interestingly, it could not be ignored that, in preclinical studies, these inhibitors achieved strong antitumor efficacy only at much higher doses in the ROS1 fusion G2032R xenograft model than in the wt ROS1 fusion model. Similar observations were observed at the cellular level [12, 17, 18]. Likewise, this result was similar to our observations after treatment with SAF-189s and lorlatinib both in vitro and in vivo. However, repotrectinib was recently reported to show promising clinical activity in patients with the ROS1 fusion G2032R mutation [17]. In addition, the results from the dose-finding portion phase I/II study of SAF-189s showed that it is very tolerable [10]. These results together suggest the promising potential of SAF-189s for the treatment of patients with the ROS1 fusion G2032R mutation who relapse on crizotinib.
In conclusion, our data demonstrate that SAF-189s, a potent new-generation ROS1 kinase inhibitor, is able to overcome a series of crizotinib-resistant mutants, including the high-frequency G2032R mutant. Overall, these results support its clinical development in crizotinib-relapsed and naive ROS1-positive NSCLC patients.
Supplementary information
Acknowledgements
This research was supported by grants from the “Personalized Medicines–Molecular Signature-based Drug Discovery and Development” Strategic Priority Research Program of the Chinese Academy of Sciences (Nos XDA12020000 for MYG and XDA12020103 for JA), the Natural Science Foundation of China for Innovation Research Group (No. 81821005 for MYG), the Natural Science Foundation of China (Nos 81773762 and 81473243 for JA), the National Science & Technology Major Project “Key New Drug Creation and Manufacturing Program” of China (No. 2018ZX09711002-011-013 for JA), the Collaborative Innovation Cluster Project of Shanghai Municipal Commission of Health and Family Planning (Grant 2019CXJQ02), and the Collaborative Innovation Cluster Project of Shanghai Municipal Commission of Health and Family Planning (2020CXJQ02). We thank Heng-lei Lu for the support in assessing the IHC staining, Dr. Zuo-quan Xie for assistance with the English polishing of the manuscript.
Author contributions
MYG, JD, and JA, conceived the project; JA, supervised the project; ZJX, YCJ, DQS, XP, YLG, and YFF, performed the research; ZJX, YCJ, DQS, XP, and JA, analyzed the data; XDZ, and WBW, contributed to SAF-189s design and synthesis; and ZJX, DQS, and JA, wrote the paper.
Competing interests
WBW is a co-founder and shareholder of Fochon Pharmaceuticals, a company developing the SAF-189s; and XDZ is a full-time employee of Fochon Pharmaceuticals. The other authors declare that they have no competing interest.
Supplementary information
The online version of this article (10.1038/s41401-020-00513-3) contains supplementary material, which is available to authorized users.
References
- 1.Lin JJ, Shaw AT. Recent advances in targeting ROS1 in lung cancer. J Thorac Oncol. 2017;12:1611–25. doi: 10.1016/j.jtho.2017.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schram AM, Chang MT, Jonsson P, Drilon A. Fusions in solid tumours: diagnostic strategies, targeted therapy, and acquired resistance. Nat Rev Clin Oncol. 2017;14:735–48. doi: 10.1038/nrclinonc.2017.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Uguen A, De Braekeleer M. ROS1 fusions in cancer: a review. Future Oncol. 2016;12:1911–28. doi: 10.2217/fon-2016-0050. [DOI] [PubMed] [Google Scholar]
- 4.Bergethon K, Shaw AT, Ou SH, Katayama R, Lovly CM, McDonald NT, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. 2012;30:863–70. doi: 10.1200/JCO.2011.35.6345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shaw AT, Ou SH, Bang YJ, Camidge DR, Solomon BJ, Salgia R, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med. 2014;371:1963–71. doi: 10.1056/NEJMoa1406766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi YL, Soda M, Yamashita Y, Ueno T, Mano H. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363:1734–9. doi: 10.1056/NEJMoa1007478. [DOI] [PubMed] [Google Scholar]
- 7.Kobayashi S, Boggon TJ, Dayaram T, Jänne PA, Halmos B. EGFR mutation and resistance of non–small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–92. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 8.Rotow J, Bivona TG. Understanding and targeting resistance mechanisms in NSCLC. Nat Rev Cancer. 2017;17:637–58. doi: 10.1038/nrc.2017.84. [DOI] [PubMed] [Google Scholar]
- 9.Awad MM, Katayama R, McTigue M, Liu W, Deng YL, Brooun A, et al. Acquired resistance to crizotinib from a mutation in CD74-ROS1. N Engl J Med. 2013;368:2395–401. doi: 10.1056/NEJMoa1215530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang JJ, Zhou JY, Yang N, Wu ZL, Sun J, Hui AM, et al. SAF-189s in previously treated patients with advanced ALK-rearranged non-small cell lung cancer (NSCLC): results from the dose-finding portion in a single arm, first-in-human phase 1/2 study. ASCO Annual Meeting. 2020;e21689.
- 11.Yan W, Wang X, Dai Y, Zhao B, Yang X, Fan J, et al. Discovery of 3-(5’-Substituted)-benzimidazole-5-(1-(3,5-dichloropyridin-4-yl)ethoxy)-1H-indazo les as potent fibroblast growth factor receptor inhibitors: design, synthesis, and biological evaluation. J Med Chem. 2016;59:6690–708. doi: 10.1021/acs.jmedchem.6b00056. [DOI] [PubMed] [Google Scholar]
- 12.Zou HY, Li Q, Engstrom LD, West M, Appleman V, Wong KA, et al. PF-06463922 is a potent and selective next-generation ROS1/ALK inhibitor capable of blocking crizotinib-resistant ROS1 mutations. Proc Natl Acad Sci U S A. 2015;112:3493–8. doi: 10.1073/pnas.1420785112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ou SHI, Tan J, Yen Y, Soo RA. ROS1 as a ‘druggable’ receptor tyrosine kinase: lessons learned from inhibiting the ALK pathway. Expert Rev Anticancer Ther. 2012;12:447–56. doi: 10.1586/era.12.17. [DOI] [PubMed] [Google Scholar]
- 14.Davies KD, Le AT, Theodoro MF, Skokan MC, Aisner DL, Berge EM, et al. Identifying and targeting ROS1 gene fusions in non-small cell lung cancer. Clin Cancer Res. 2012;18:4570–9. doi: 10.1158/1078-0432.CCR-12-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Melnick JS, Janes J, Kim S, Chang JY, Sipes DG, Gunderson D, et al. An efficient rapid system for profiling the cellular activities of molecular libraries. Proc Natl Acad Sci U S A. 2006;103:3153–8. doi: 10.1073/pnas.0511292103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Warmuth M, Kim S, Gu XJ, Xia G, Adrian F. Ba/F3 cells and their use in kinase drug discovery. Curr Opin Oncol. 2007;19:55–60. doi: 10.1097/CCO.0b013e328011a25f. [DOI] [PubMed] [Google Scholar]
- 17.Drilon A, Ou SI, Cho BC, Kim DW, Lee J, Lin JJ, et al. Repotrectinib (TPX-0005) is a next-generation ROS1/TRK/ALK inhibitor that potently inhibits ROS1/TRK/ALK solvent-front mutations. Cancer Discov. 2018;8:1227–36. doi: 10.1158/2159-8290.CD-18-0484. [DOI] [PubMed] [Google Scholar]
- 18.Katayama R, Gong B, Togashi N, Miyamoto M, Kiga M, Iwasaki S, et al. The new-generation selective ROS1/NTRK inhibitor DS-6051b overcomes crizotinib resistant ROS1-G2032R mutation in preclinical models. Nat Commun. 2019;10:3604. doi: 10.1038/s41467-019-11496-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.