

Abstract
Disclosed herein is a photochemical Heck-type arylation of vinylphenols with non-activated aryl and heteroaryl halides under visible light irradiation. Preliminary mechanistic studies suggested that the colored vinylphenolate anions acted as a strong reducing photoactivator to directly activate (hetero)aryl halides without the need for any sacrificial reductants. The photochemically generated aryl radicals coupled with another molecule of vinylphenol to afford the Heck-type arylation product in a regiospecific and stereoselective manner. The developed photochemical arylation protocol showed exceptional functional group tolerance and was successfully applied in the challenging late-stage modification of natural products without any protection–deprotection procedures.
A regioselective and stereoselective Heck-type arylation of vinylphenols with non-activated (hetero)aryl halides was realized under visible light irradiation. The vinylphenolate anions acted as strong reducing photoactivators to activate (hetero)aryl halides.
Introduction
Introducing aryl and heteroaryl units into molecular scaffolds is one of the fundamental chemical transformations in the synthesis of natural products, pharmaceuticals, and functional materials.1 Over the past few decades, transition-metal-mediated arylation reactions have been well established, although there are still some problems associated with these methods (i.e. expensive transition metal catalysts, specific and air-sensitive ligands, and elevated temperatures).2 New, cheap, efficient, and environmentally safe methodologies to realize arylation are always of great interest. In recent years, visible-light-induced radical arylation methods have been developed as attractive alternatives under mild conditions.3 However, various photo-redox processes for arylation reactions require the use of electron-poor aryl diazonium salts4 or diaryliodonium salts5 as radical precursors (Scheme 1a). The reductive cleavage of non-activated (hetero)aryl halides, which are the most readily available and the cheapest arylation reagents, is limited by the redox properties of typical photocatalysts and the energy of the visible photon due to the high negative reduction potentials of (hetero)aryl halides.6
Scheme 1. (a) Typical aryl radical precursors in visible-light-mediated arylation reactions. (b) Reported strategies of visible-light-induced aryl halide activation. (c) Redox-neutral photochemical Heck-type arylation of vinylphenols with (hetero)aryl halides.

In this context, development of powerful reducing photo-reductants is an important synthetic goal. As depicted in Scheme 1b, recent examples of successful activations of (hetero)aryl halides with visible light involve three mechanistic scenarios: (i) direct single electron transfer (SET) from strongly reducing iridium,6a,7 copper,8N-phenylphenothiazine (PTH),9 or 9-anthrolate anion10 photocatalysts; (ii) indirect two-photon processes with organic dyes, such as perylene bisimides (PBIs),6b rhodamine 6G (Rh-6G),11 1,8-dihydroxyanthraquinone (Aq-OH),12 and 9,10-dicyanoanthracene (DCA),13 by consecutive photo-induced SET cascades or with butane-2,3-dione (BD, sensitizer) and 2,5-di-phenyloxazole (PPO, annihilator) by triplet–triplet annihilation;14 (iii) two-center photo-redox catalysis in which Ru(bpy)3Cl2 absorbs visible light and transfers the energy to pyrene which enables the redox reactions.15 Although these photo-catalytic methods are highly innovative, most of them require a large excess of sacrificial reductants such as tertiary amines, sodium formate, or Hantzsch ester (HEH), resulting in a fast competing hydrogen atom abstraction by the reactive aryl radicals to give undesired side products. Consequently, the reactivity of the resulting aryl radical has been confined to hydrodehalogenation,6,7d,12,14 C–H arylation of electron-rich heteroarenes,6b,10–13,15 and hydroarylation of alkenes.7a–c,8,9 However, the redox-neutral photochemical Heck-type arylation of alkenes under visible light is still a challenge and the only known example was reported by Chu and co-workers for the pyridylation of alkenes.16 Evidently, development of new powerful photo-reductants that operate using mild visible light and do not require sacrificial reductants is highly warranted to alleviate side reactions and expand the synthetic applicability of aryl radicals from (hetero)aryl halides. Phenolate anions have been used as an attractive model system for a photoinduced electron ejection mechanism study;17 nevertheless, the photochemical reactivities of excited phenolate anions have received little attention. In 2015, Melchiorre and co-workers reported a redox-neutral photochemical strategy for the direct aromatic perfluoroalkylation of phenols.18 In this reaction, excited phenolate anions directly acted as photoreductants to activate perfluoroalkyl iodides. Inspired by these studies, we questioned whether phenolate anions could be employed as photoreductants to activate (hetero)aryl halides using a similar mechanistic model. Herein, we demonstrated that the colored vinylphenolate anions, in situ generated by deprotonation of vinylphenols, reached an electronically excited state upon visible-light irradiation and then acted as effective photo-reductants for the reductive activation of non-activated (hetero)aryl halides (Scheme 1c). This photochemical reaction overcame the need for sacrificial reductants and afforded Heck-type arylation products in a regiospecific and stereoselective manner under redox-neutral conditions.
Results and discussion
Optimization of reaction conditions
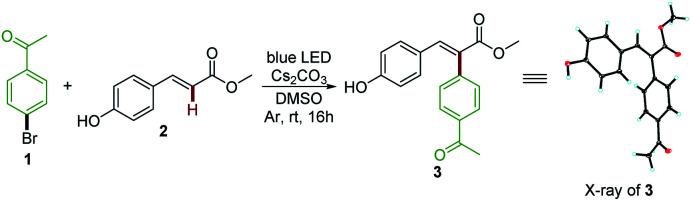
Vinylphenol is an essential motif in a wide range of biologically active natural products and pharmaceuticals that accomplish a series of important functions.19 Our initial explorations focused on methyl 4-hydroxycinnamate 2, a key chromophore of the photoactive yellow protein (PYP).19e,20 After investigating a range of reaction parameters, we discovered that upon irradiating a mixture of 4′-bromoacetophenone 1 and 2 in DMSO with an 18 W blue light emitting diode (LED) lamp at room temperature for 16 hours with Cs2CO3 as the base, Heck arylation product 3 was formed in 87% yield (Table 1, entry 1). The structure of the desired product 3 was unambiguously assigned by X-ray crystallography. Interestingly, this photochemical process exhibited a complete opposite regioselectivity compared with traditional palladium catalyzed Heck arylation reactions,21 and afforded tri-substituted alkenes in a stereoselective manner (E/Z > 19 : 1). Controlled reactions established that this reaction did not occur in the dark even after heating to 80 °C, thus demonstrating the photochemical nature of the reaction (entries 2 and 3). Irradiating with a 23 W compact fluorescent light (CFL) bulb or a 30 W white LED lamp resulted in a drop in yields (entries 4 and 5). The in situ formed phenolate anion of 2 was essential for this transformation because the reaction was completely inhibited without the base Cs2CO3 (entry 6). Spectroscopic investigations confirmed that 2 was completely deprotonated by Cs2CO3 (see ESI Note 1†). Finally, a survey of bases including inorganic and organic bases (KHCO3, Na2CO3, K2CO3, K3PO4, DBU, TMG, and Et3N) and other solvents (DMF, acetone, CH3CN, and DCM) found that they were not comparable to the combination of Cs2CO3 and DMSO (entries 7–17).
The model reaction and the reaction parameters evaluated.
| ||
|---|---|---|
| Entrya | Change from standard conditions | Yieldb (%) |
| 1 | None | 87 |
| 2 | No light | 0 |
| 3 | No light and heated to 80 °C | 0 |
| 4 | A 23 W CFL, instead of a blue LED | 56 |
| 5 | A 30 W white LED, instead of a blue LED | 50 |
| 6 | No base | 0 |
| 7 | KHCO3, instead of Cs2CO3 | 53 |
| 8 | Na2CO3, instead of Cs2CO3 | 40 |
| 9 | K2CO3, instead of Cs2CO3 | 73 |
| 10 | K3PO4, instead of Cs2CO3 | 43 |
| 11 | DBU, instead of Cs2CO3 | 51 |
| 12 | TMG, instead of Cs2CO3 | 54 |
| 13 | Et3N, instead of Cs2CO3 | 15 |
| 14 | DMF, instead of DMSO | 72 |
| 15 | CH3CN, instead of DMSO | 50 |
| 16 | Acetone, instead of DMSO | 66 |
| 17 | DCM, instead of DMSO | 18 |
Conditions: 1.0 equiv. of 4′-bromoacetophenone 1, 2.0 equiv. of methyl 4-hydroxycinnamate 2, 3.0 equiv. of base [0.10 M]; all solvents were rigorously degassed by freeze/pump/thaw cycles.
Isolated yields by chromatography.
Substrate scope
We then evaluated the synthetic potential of this photochemical Heck-type arylation strategy. As detailed in Fig. 1, various (hetero)aryl halides including iodides, bromides and chlorides were successfully coupled to 2. In general, aryl iodides displayed reactivity higher than that of aryl bromides and chlorides with shorter reaction time (3–8). It is noteworthy that the activation of aryl chlorides by a transition-metal complex is full of challenges and is normally conducted at high temperature due to the inertness of the C–Cl bond.22 We observed that the aryl chlorides also afforded the corresponding products in good yields under mild photochemical Heck-type arylation conditions (3–8). Moreover, the site of the substitution on the aromatic ring had no detrimental effects on this photochemical transformation, and o-, m- and p-substituted aryl halides were found to be good substrates (6–8). For electron-neutral and electron-rich aryl iodides, longer reaction times were needed and relatively lower yields were obtained, which were attributed to the associated slow electron transfer processes (9–16). However, the corresponding bromides or chlorides did not afford products under standard reaction conditions, which might be due to their much lower reduction potentials.7d,13,23 Overall, this photochemical Heck-type arylation method was compatible with aryl halides containing a wide range of functional groups, including –COCH3 (3), –CHO (4), –CO2Me (5), –CN (6–8), –NHAc (12), –NH2 (13), –OH (14 and 15), –COOH (16 and 17), and –NO2 (18), providing a useful handle for subsequent functionalization and diversification of the corresponding products. We next evaluated the activation of polycyclic aromatic hydrocarbon halides, and found that naphthalene (19 and 20), fluorene (21), anthracene (22), and phenanthrene (23) were efficient coupling partners. In addition, this newly developed system was also extended to various biologically important heterocyclic aryl halides, which are ubiquitous among pharmaceutical products. Heterocycles such as pyridine (24 and 25), isoquinoline (26 and 27), quinoline (28 and 29), pyrazine (30), thiophene (31) and carbazole (32) were readily used as radical precursors and provided the desired products with good yields.
Fig. 1. Substrate scope of (hetero)aryl halides. General conditions: (hetero)aryl halide (0.10 mmol), 2 (0.20 mmol), Cs2CO3 (0.30 mmol), and DMSO (1.0 mL). Isolated yields were reported. The E/Z ratio was measured by 1H NMR.

Next, we extended the substrate scope to other vinylphenols (Fig. 2). Under the standard conditions, the reaction of vinylphenols with different electron-withdrawing substituents on the double bond, including thioester (33), amide (34), and ketone moieties (35–37), proceeded smoothly and gave the Heck-type arylation products in good to excellent yields. Notably, this transformation was not only limited to electron-withdrawing groups; non- (38), alkyl- (39) and aryl- (40) substituted vinylphenols were also found to be compatible with this reaction system. This approach even enabled the regiospecific and stereoselective synthesis of the congested tetra-substituted olefin 41. In addition, a series of substituents on the aromatic ring were well tolerated (42–45), and this photo-induced process was also effective for substrates with the phenolic hydroxyl positioned meta (46) or ortho (47) to the double bond. In order to demonstrate the practicability of this transformation, we further evaluated the method for late-stage functionalization applications of natural products. The methods for arylation on the C ring of flavonoids are limited and multiple step procedures are involved including Pd-catalyzed Suzuki coupling with aromatic boronic acids24 or the Pd-catalyzed Negishi coupling by a Lewis acid-triggered zincation.25 We found that coumarin (48) and flavonoid (49–52) derivatives were easily arylated via the photochemical protocol in one step and the products were afforded with good yields. The natural products with additional carbohydrate appendices, such as the pharmaceutical ingredients chlorogenic acid (53), esculin (54), and scutellarin (55), were also directly arylated under standard conditions without any protection procedures. The aryl chlorides were also tested as the aryl reagents, and the corresponding arylated products were obtained in moderate yields (48–53).
Fig. 2. Substrate scope of vinylphenol derivatives. General conditions: 1 (0.10 mmol), vinylphenol (0.20 mmol), Cs2CO3 (0.30 mmol), and DMSO (1.0 mL). Isolated yields were reported. The E/Z ratio was measured by 1H NMR.

Mechanistic investigation
Previously, we reported a visible light-induced α-arylation of oxindole through the formation of an electron-donor–acceptor (EDA) complex between oxindole enolate and (hetero)aryl halides.26 The formation of an EDA complex was unambiguously confirmed by a significant bathochromic shift in the UV-vis spectra when the solution of oxindole enolate was mixed with (hetero)aryl halides. Therefore, we originally proposed that a similar photon-absorbing EDA complex between the phenolate anion of 2 and aryl halide 1 was formed to enable the arylation. However, we found that the colorless solution of vinylphenol 2(orange line in Fig. 3a) immediately turned to a bright yellow color upon addition of Cs2CO3 in the absence of aryl halide 1 (blue line in Fig. 3a). Moreover, we did not observe any color change after aryl halide 1 was added to the solution of the phenolate anion of 2 (red line in Fig. 3a, perfectly overlapped with the absorption of the phenolate anion of 2). These results excluded the formation of a ground-state EDA complex association with the phenolate anion of 2 and aryl halide 1,18,27 but indicated that the photon-absorbing ability of the phenolate anion of 2 in the visible spectral region was responsible for triggering the aryl radical from its halide 1.
Fig. 3. (a) UV/Vis absorption spectra of DMSO solutions (0.10 M) of 1, 2, a mixture of 2 and Cs2CO3, and a mixture of 1, 2 and Cs2CO3. (b) Quenching of emission from the phenolate of 2 (5 × 10−5 M in DMSO) in the presence of increasing amounts of 1. (c) Proposed mechanistic pathway.

To better understand this process, we investigated the photo-physical behaviors of the phenolate anion of 2. The redox potential of the excited phenolate anion of 2 was estimated to be −2.48 V vs. SCE on the basis of electrochemical and spectroscopic measurements (see ESI Note 3†). Therefore, the reduction of 4′-bromoacetophenone 1 (−1.83 V vs. SCE in DMSO, see ESI Fig. 7†) by the excited phenolate anion of 2 is thermodynamically feasible. Furthermore, a series of Stern–Volmer quenching studies revealed that the excited state of the phenolate anion of 2 was effectively quenched by aryl halide 1 (Fig. 3b). The addition of radical inhibitor (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) or electron-transfer scavenger 1,4-dinitrobenzene strongly inhibited the photochemical Heck-type arylation reaction, which indicated that a radical pathway proceeding through a single-electron transfer (SET) process was highly possible in this reaction.
On the basis of these observations, we proposed a possible mechanism as depicted in Fig. 3c. Methyl 4-hydroxycinnamate 2 was deprotonated by Cs2CO3 to generate the colored phenolate anion A, which reached an electronically excited state (A*) under blue-light irradiation. Then a SET process from the excited phenolate anion A* to 4′-bromoacetophenone 1 occurred to produce a phenoxyl radical (B) and an aryl radical (C). The latter was confirmed by the isolation of compound 56 during the TEMPO trapping experiments (see ESI Note 5†). There are two possible pathways to generate arylation product 3. The first one is the direct coupling between the delocalized phenoxyl radical B and aryl radical C, and the second one is the addition of radical C to another molecule of phenolate anion A to afford the radical anion intermediate D. In the TEMPO trapping experiments, we also isolated compound 57, which was generated by trapping intermediate D with TEMPO, and thus confirmed the second pathway (see ESI Note 5†). The quantum yield for the model reaction was determined to be Φ = 0.58 (see ESI Note 6†), indicating that the reported arylation reaction was a non-chain process28 or an inefficient chain process.29 Therefore, the subsequent pathways can be diverted into two types. In path a, the radical anion intermediate D might transfer a hydrogen to radical B to afford arylation product 3 and recycle methyl 4-hydroxycinnamate 2. Path b includes a SET process between the radical anion intermediate D and 4′-bromoacetophenone 1 to give 3 and regenerate aryl radical C through a chain mechanism.
Conclusions
In summary, this study established the ability of vinylphenolate anions to function as a strong reducing photoactivator to activate a broad range of aryl and heteroaryl halides, including iodides, bromides, and chlorides, when irradiated by visible light at room temperature. The resulting aryl radical participated in a redox-neutral photochemical Heck-type arylation reaction that could not be realized by previously known visible light-induced photoredox processes. This method allows for the installation of aryl and heteroaryl units on vinylphenols with high regiospecificity and stereoselectivity and tolerates a wide range of functional groups. Furthermore, the potential utility of this transformation has been demonstrated by late-stage functionalization of complex natural products.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was financially supported by the Natural Science Foundation of Yunnan Province (2018FY001015), China Postdoctoral Science Foundation (2018M643540), the National Natural Science Foundation of China (21871228), and the Program for Changjiang Scholars and Innovative Research Team in University (IRT_17R94). We acknowledge the Advanced Analysis and Measurement Center of Yunnan University for help with NMR experiments.
Electronic supplementary information (ESI) available. CCDC 1917425. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc06184c
Notes and references
- (a) Dounay A. B. Overman L. E. Chem. Rev. 2003;103:2945. doi: 10.1021/cr020039h. [DOI] [PubMed] [Google Scholar]; (b) Nicolaou K. C. Bulger P. G. Sarlah D. Angew. Chem., Int. Ed. 2005;44:4442. doi: 10.1002/anie.200500368. [DOI] [PubMed] [Google Scholar]; (c) Okamoto K. Zhang J. Housekeeper J. B. Marder S. R. Luscombe C. K. Macromolecules. 2013;46:8059. doi: 10.1021/ma401190r. [DOI] [Google Scholar]
- Ackermann L., Modern Arylation Methods, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2009 [Google Scholar]
- (a) Wang C.-S. Dixneuf P. H. Soulé J.-F. Chem. Rev. 2018;118:7532. doi: 10.1021/acs.chemrev.8b00077. [DOI] [PubMed] [Google Scholar]; (b) Ghosh I. Marzo L. Das A. Shaikh R. König B. Acc. Chem. Res. 2016;49:1566. doi: 10.1021/acs.accounts.6b00229. [DOI] [PubMed] [Google Scholar]; (c) Qiu G. Li Y. Wu J. Org. Chem. Front. 2016;3:1011. doi: 10.1039/C6QO00103C. [DOI] [Google Scholar]; (d) Twilton J. Le C. Zhang P. Shaw M. H. Evans R. W. MacMillan D. W. C. Nat. Rev. Chem. 2017;1:0052. doi: 10.1038/s41570-017-0052. [DOI] [Google Scholar]
- (a) Hari D. P. König B. Angew. Chem., Int. Ed. 2013;52:4734. doi: 10.1002/anie.201210276. [DOI] [PubMed] [Google Scholar]; (b) Kalyani D. McMurtrey K. B. Neufeldt S. R. Sanford M. S. J. Am. Chem. Soc. 2011;133:18566. doi: 10.1021/ja208068w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hari D. P. Schroll P. König B. J. Am. Chem. Soc. 2012;134:2958. doi: 10.1021/ja212099r. [DOI] [PubMed] [Google Scholar]; (d) Guo W. Lu L.-Q. Wang Y. Wang Y.-N. Chen J.-R. Xiao W.-J. Angew. Chem., Int. Ed. 2015;54:2265. doi: 10.1002/anie.201408837. [DOI] [PubMed] [Google Scholar]; (e) Zoller J. Fabry D. C. Rueping M. ACS Catal. 2015;5:3900. doi: 10.1021/acscatal.5b00668. [DOI] [Google Scholar]
- (a) Baralle A. Fensterbank L. Goddard J.-P. Ollivier C. Chem.–Eur. J. 2013;19:10809. doi: 10.1002/chem.201301449. [DOI] [PubMed] [Google Scholar]; (b) Liu Y.-X. Xue D. Wang J.-D. Zhao C.-J. Zou Q.-Z. Wang C. Xiao J. Synlett. 2013;24:507. doi: 10.1055/s-0032-1318155. [DOI] [Google Scholar]; (c) Tobisu M. Furukawa T. Chatani N. Chem. Lett. 2013;42:1203. doi: 10.1246/cl.130547. [DOI] [Google Scholar]; (d) Fumagalli G. Boyd S. Greaney M. F. Org. Lett. 2013;15:4398. doi: 10.1021/ol401940c. [DOI] [PubMed] [Google Scholar]; (e) Neufeldt S. R. Sanford M. S. Adv. Synth. Catal. 2012;354:3517. doi: 10.1002/adsc.201200738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Nguyen J. D. D'Amato E. M. Narayanam J. M. R. Stephenson C. R. J. Nat. Chem. 2012;4:854. doi: 10.1038/nchem.1452. [DOI] [PubMed] [Google Scholar]; (b) Ghosh I. Ghosh T. Bardagi J. I. König B. Science. 2014;346:725. doi: 10.1126/science.1258232. [DOI] [PubMed] [Google Scholar]
- (a) Kim H. Lee C. Angew. Chem., Int. Ed. 2012;51:12303. doi: 10.1002/anie.201203599. [DOI] [PubMed] [Google Scholar]; (b) Boyington A. J. Riu M.-L. Y. Jui N. T. J. Am. Chem. Soc. 2017;139:6582. doi: 10.1021/jacs.7b03262. [DOI] [PubMed] [Google Scholar]; (c) Aycock R. A. Wang H. Jui N. T. Chem. Sci. 2017;8:3121. doi: 10.1039/C7SC00243B. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Devery J. J. Nguyen J. D. Dai C. Stephenson C. R. J. ACS Catal. 2016;6:5962. doi: 10.1021/acscatal.6b01914. [DOI] [Google Scholar]
- Michelet B. Deldaele C. Kajouj S. Moucheron C. Evano G. Org. Lett. 2017;19:3576. doi: 10.1021/acs.orglett.7b01518. [DOI] [PubMed] [Google Scholar]
- Boyington A. J. Seath C. P. Zearfoss A. M. Xu Z. Jui N. T. J. Am. Chem. Soc. 2019;141:4147. doi: 10.1021/jacs.9b01077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmalzbauer M. Ghosh I. König B. Faraday Discuss. 2019;215:364. doi: 10.1039/C8FD00176F. [DOI] [PubMed] [Google Scholar]
- Ghosh I. König B. Angew. Chem., Int. Ed. 2016;55:7676. doi: 10.1002/anie.201602349. [DOI] [PubMed] [Google Scholar]
- Bardagi J. I. Ghosh I. Schmalzbauer M. Ghosh T. König B. Eur. J. Org. Chem. 2018;2018:34. doi: 10.1002/ejoc.201701461. [DOI] [Google Scholar]
- Neumeier M. Sampedro D. Májek M. de la Peña O'Shea V. A. Jacobi von Wangelin A. Pérez-Ruiz R. Chem.–Eur. J. 2018;24:105. doi: 10.1002/chem.201705326. [DOI] [PubMed] [Google Scholar]
- Majek M. Faltermeier U. Dick B. Pérez-Ruiz R. Jacobi von Wangelin A. Chem.–Eur. J. 2015;21:15496. doi: 10.1002/chem.201502698. [DOI] [PubMed] [Google Scholar]
- Ghosh I. Shaikh R. S. König B. Angew. Chem., Int. Ed. 2017;56:8544. doi: 10.1002/anie.201703004. [DOI] [PubMed] [Google Scholar]
- Zhu S. Qin J. Wang F. Li H. Chu L. Nat. Commun. 2019;10:749. doi: 10.1038/s41467-019-08669-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Chen X. Larsen D. S. Bradforth S. E. van Stokkum I. H. M. J. Phys. Chem. A. 2011;115:3807. doi: 10.1021/jp107935f. [DOI] [PubMed] [Google Scholar]; (b) Ichino T. Fessenden R. W. J. Phys. Chem. A. 2003;107:9257. doi: 10.1021/jp030153j. [DOI] [PubMed] [Google Scholar]; (c) Fessenden R. W. Verma N. C. J. Am. Chem. Soc. 1976;98:243. doi: 10.1021/ja00417a044. [DOI] [Google Scholar]; (d) Lewis G. N. Lipkin D. J. Am. Chem. Soc. 1942;64:2801. doi: 10.1021/ja01264a025. [DOI] [Google Scholar]
- Filippini G. Nappi M. Melchiorre P. Tetrahedron. 2015;71:4535. doi: 10.1016/j.tet.2015.02.034. [DOI] [Google Scholar]
- (a) Huang M.-T. Smart R. C. Wong C.-Q. Conney A. H. Cancer Res. 1988;48:5941. [PubMed] [Google Scholar]; (b) Sato Y. Itagaki S. Kurokawa T. Ogura J. Kobayashi M. Hirano T. Sugawara M. Iseki K. Int. J. Pharm. 2011;403:136. doi: 10.1016/j.ijpharm.2010.09.035. [DOI] [PubMed] [Google Scholar]; (c) Lin L.-L. Liu A.-J. Liu J.-G. Yu X.-H. Qin L.-P. Su D.-F. J. Cardiovasc. Pharmacol. 2007;50:327. doi: 10.1097/FJC.0b013e3180cbd0e7. [DOI] [PubMed] [Google Scholar]; (d) Fraser J. A. Sperber W. H. J. Food Prot. 1988;51:762. doi: 10.4315/0362-028X-51.10.762. [DOI] [PubMed] [Google Scholar]; (e) Baca M. Borgstahl G. E. O. Boissinot M. Burke P. M. Williams D. R. Slater K. A. Getzoff E. D. Biochemistry. 1994;33:14369. doi: 10.1021/bi00252a001. [DOI] [PubMed] [Google Scholar]
- van der Horst M. A. Arents J. C. Kort R. Hellingwerf K. J. Photochem. Photobiol. Sci. 2007;6:571. doi: 10.1039/B701072A. [DOI] [PubMed] [Google Scholar]
- (a) Caló V. Nacci A. Monopoli A. Laera S. Cioffi N. J. Org. Chem. 2003;68:2929. doi: 10.1021/jo026877t. [DOI] [PubMed] [Google Scholar]; (b) Moreno-Mañas M. Pérez M. Pleixats R. Tetrahedron Lett. 1996;37:7449. doi: 10.1016/0040-4039(96)01608-5. [DOI] [Google Scholar]; (c) Sharma D. Kumar S. Shil A. K. Guha N. R. Bandna Das P. Tetrahedron Lett. 2012;53:7044. doi: 10.1016/j.tetlet.2012.10.062. [DOI] [Google Scholar]
- Grushin V. V. Alper H. Chem. Rev. 1994;94:1047. doi: 10.1021/cr00028a008. [DOI] [Google Scholar]
- Yin H. Jin Y. Hertzog J. E. Mullane K. C. Carroll P. J. Manor B. C. Anna J. M. Schelter E. J. J. Am. Chem. Soc. 2016;138:16266. doi: 10.1021/jacs.6b05712. [DOI] [PubMed] [Google Scholar]
- (a) Hyup Joo Y. Kwan Kim J. Kang S.-H. Noh M.-S. Ha J.-Y. Kyu Choi J. Min Lim K. Hoon Lee C. Chung S. Bioorg. Med. Chem. Lett. 2003;13:413. doi: 10.1016/S0960-894X(02)00952-6. [DOI] [PubMed] [Google Scholar]; (b) Quintin J. Roullier C. Thoret S. Lewin G. Tetrahedron. 2006;62:4038. doi: 10.1016/j.tet.2006.02.024. [DOI] [Google Scholar]
- Klier L. Bresser T. Nigst T. A. Karaghiosoff K. Knochel P. J. Am. Chem. Soc. 2012;134:13584. doi: 10.1021/ja306178q. [DOI] [PubMed] [Google Scholar]
- Liang K. Li N. Zhang Y. Li T. Xia C. Chem. Sci. 2019;10:3049. doi: 10.1039/C8SC05170D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Silvi M. Arceo E. Jurberg I. D. Cassani C. Melchiorre P. J. Am. Chem. Soc. 2015;137:6120. doi: 10.1021/jacs.5b01662. [DOI] [PubMed] [Google Scholar]; (b) Bahamonde A. Melchiorre P. J. Am. Chem. Soc. 2016;138:8019. doi: 10.1021/jacs.6b04871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cismesia M. A. Yoon T. P. Chem. Sci. 2015;6:5426. doi: 10.1039/C5SC02185E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argüello J. E. Peñéñory A. B. Rossi R. A. J. Org. Chem. 2000;65:7175. doi: 10.1021/jo001141m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.