

Abstract
Intrinsically disordered proteins (IDPs) are an important class of proteins that do not fold to a well-defined three-dimensional shape but rather adopt an ensemble of inter-converting conformations. This feature makes their experimental characterization challenging and invites a theoretical and computational approach to complement experimental studies. In this review, we highlight the recent progress in developing new computational and theoretical approaches to study the structure and dynamics of monomeric and order higher assemblies of IDPs, with a particular emphasis on their phase separation into protein-rich condensates.
Introduction
Intrinsically disordered proteins (IDPs) - those which do not adopt a folded structure in isolation - are now recognized to play key roles in cellular signaling and transcription.[1] An important feature of IDPs is their ability to self-assemble into solid fibrillar structures (a process often linked to pathological outcomes for the cell) as well as liquid assemblies formed through the process of liquid-liquid phase separation. Several recent reviews have extensively covered the process of fibril formation, and we focus here on IDP monomers and liquid-liquid phase separated assemblies [2,3].
Computational and theoretical methods have proven extremely valuable in understanding the structure and function of intrinsically disordered proteins: firstly, they help to resolve details that would be difficult to tease out from experiment alone, owing to the disordered nature of these proteins, and secondly they can provide a framework for understanding the properties of IDPs and their relation to protein sequence. Building upon fundamental work on IDP structure and function using a variety of computational and theoretical tools, rapid progress has been made in the last few years spurred by the role of IDPs in the formation of membraneless organelles (MLOs) via liquid-liquid phase separation (LLPS) [4,5]. Transferable physics-based models, which have been parameterized or/and tested against experimental data, can not only help interpret experimental data but can also provide detailed predictive information on important scientific questions [6,7]. Most importantly, because they are defined by the protein sequence, such models do not require any modifications to the potential parameters based on available knowledge from experiment, before being applied to a specific protein system of interest. We note that this does not preclude subsequent integration of experimental data to improve the accuracy of the generated ensemble [8–10]. Here, we restrict our review primarily to transferable physics-based models.
We start by reviewing recent developments in simulation models for IDPs, ranging from coarse-grained to all-atom simulation methods. We then describe recent methodological developments enabling the determination of phase equilibria for liquid-liquid phase separation directly from molecular simulations. We conclude by describing newly developed theoretical approaches for single chain IDPs and formation of IDP complexes.
All-atom simulations
All-atom explicit solvent simulations have the in principle the highest resolution and accuracy, considering recent improvements in protein force fields [11] [12–14] and can capture highly specific interactions that would be averaged out in coarse graining. IDPs have served as an essential benchmark for improving the accuracy and transferability of such models to study conformational and dynamical properties of proteins. As discussed in recent work [13,15], all-atom models can now provide a rather accurate atomic view of global and local properties of IDPs, although there remain deficiencies such as their low cooperativity relative to experiment [13]. The extensive application of some of these models (Amber03ws/Amber99SBws, Amber99SB-disp) to IDPs has highlighted the remarkable accuracy with which experimental observables based on nuclear magnetic resonance (NMR)[16], small-angle X-ray scattering (SAXS)[17,18], and single-molecular Förster resonance energy transfer (smFRET)[18] can be computed from the simulated ensembles [19]. The physics-based models provide a natural path to study the dynamical relaxation properties of IDPs as opposed to the ensemble refinement methods, which are most easily applied to average structural/thermodynamic properties. There is emerging evidence that the helical propensity for IDPs (especially low-complexity prion-like domains) is not entirely consistent with NMR-derived data, which may be related to the presence of specific residues in large proportion within these sequences thereby requiring residue-level fine-tuning of parameters [14]. One should note that most algorithms used to compute experimental observables (such as chemical shifts) from the simulated ensemble were parameterized using data for folded proteins [20]. More work is needed to test and refine these empirical methods to derive experimental measurables.
One approach to reducing the computational cost of all-atom protein simulations is the use of implicit solvent. By integrating out only solvent degrees of freedom, the atomistic chemical detail is retained, but a large fraction of the computational cost is saved. Although many implicit solvent models are not appropriate for IDPs, being too collapsed, the one that has shown the most promise at the time of writing is the ABSINTH model[21], which has recently been updated to improve backbone conformational preferences [22].
However, even with implicit solvent, it is challenging to use atomistic models for studying LLPS [23–25], as system sizes and sampling requirements to study phase coexistence are computationally prohibitive with current state-of-the-art computer hardware. But such methods can be used in conjunction with coarse-grained (CG) models to obtain essential insights into the atomic-level details of inter-residue interactions [26,27]. Currently, CG models for IDPs provide the most direct path to understanding sequence-determinants of LLPS and to identifying molecular interactions responsible for self-assembly.
Coarse-grained models
Coarse-grained models have been utilized with great success in understanding protein folding, protein aggregation, and macromolecular crowding, as well as many applications in polymer physics [28,29]. There is significant flexibility in the level of detail included in a CG model; it can range from one-bead-per-protein to several-beads-per-residue. Several CG IDP models have been proposed recently that differ in terms of how bonded and nonbonded interactions are represented, which is ultimately dictated by the computational efficiency and the intended application of the model. The models used to study monomer properties are more complex and are meant to capture additional molecular details accurately [30]. On the other hand, the models intended to explore the large-scale assembly of IDPs are much simpler and mostly rely on modeling proteins as bead-spring polymers with a particular emphasis on parameterizing nonbonded interactions between CG beads meant to represent different amino acids.
A recent CG framework in this spirit has emerged as a useful computational tool to study sequence-specific changes in the phase behavior of disordered proteins [31]. Due to the simplicity of the model, it can be easily extended to represent small structural motifs or large folded domains [19], post-translational modifications [32], thermoresponsive behavior [33], and even interactions with nucleic acids [34,35] that are quite relevant for the function of MLOs. It is useful to note that most current CG models either lack or have not been tested well for partial secondary structure propensities in IDPs. The prevalence of secondary structure elements in the heterogeneous ensemble of IDPs is considered functionally important for their binding mechanism as well as the LLPS propensity. We believe this to be an important future direction of research in the physics-based CG modeling of IDPs [36]. An additional direction for improvement may be the use of more than a single bead per residue, thus allowing backbone and side-chain interactions to be distinguished [37].
Despite the simplicity of a CG model, it is almost impossible to sample the thermodynamic phase diagram of IDPs undergoing LLPS using standard sampling techniques employed in computational biophysics research such as parallel tempering. Dignon et al. proposed to use a coexistence sampling method that was previously applied successfully to sample phase behavior of short homopolymers by Howard et al. [38], and which has now become the de facto standard for computing the LLPS phase diagrams of IDPs [39]. We note that other methods that rely on the protein density differences between the two phases are also being used, most often in the context of highly simplified patchy particle models of proteins or for on-lattice systems [40,41]. We believe that this is an area of opportunity to identify suitable existing methodologies in soft matter and polymer simulations or develop new techniques that are computationally more efficient. This is especially important in the case of multicomponent assemblies of disordered proteins due to the expansion of the parameter space in terms of the relative composition of different components [42].
We note that there is an inherent limitation with the use of simplified models concerning the mapping of simulation temperature and timescales to laboratory conditions. Therefore, appropriate additional validation steps should be taken before applying these models to study issues that are typically outside the scope of the initial parameterization.
Theoretical approaches
Theoretical approaches based on polymer physics principles have provided essential insights into the behavior of monomeric IDPs. An important contribution of such methods has been the identification of low-dimensional order parameters based on relevant sequence descriptors. While earlier mean field theories of protein collapse[43] and phase separation[44] characterized proteins in terms of average sequence properties, effects of sequence order are now being included. For example, sequence charge decoration (SCD) [45] or κ [46], which are highly similar parameters to describe sequence patterning effects[47], can faithfully describe global conformational properties (such as Rg) of IDPs with predominantly polyelectrolytic or polyampholytic sequences nearly quantitatively. Recently, it was shown that one could complement SCD with an additional order parameter based on sequence hydropathy decoration (SHD), which can account for the presence of uncharged residues, to describe the conformational properties of a large number of IDPs simulated using a simple CG model [48] (Figure 1A). It has also been shown that using a similar theoretical approach one can describe the average inter-residue distances within an IDP, giving qualitative agreement with all-atom simulation results (Figure 1B) [49]. Chan and co-workers have also recently developed a parameter similar to SCD to describe association of oppositely charged chains [50].
Figure 1.
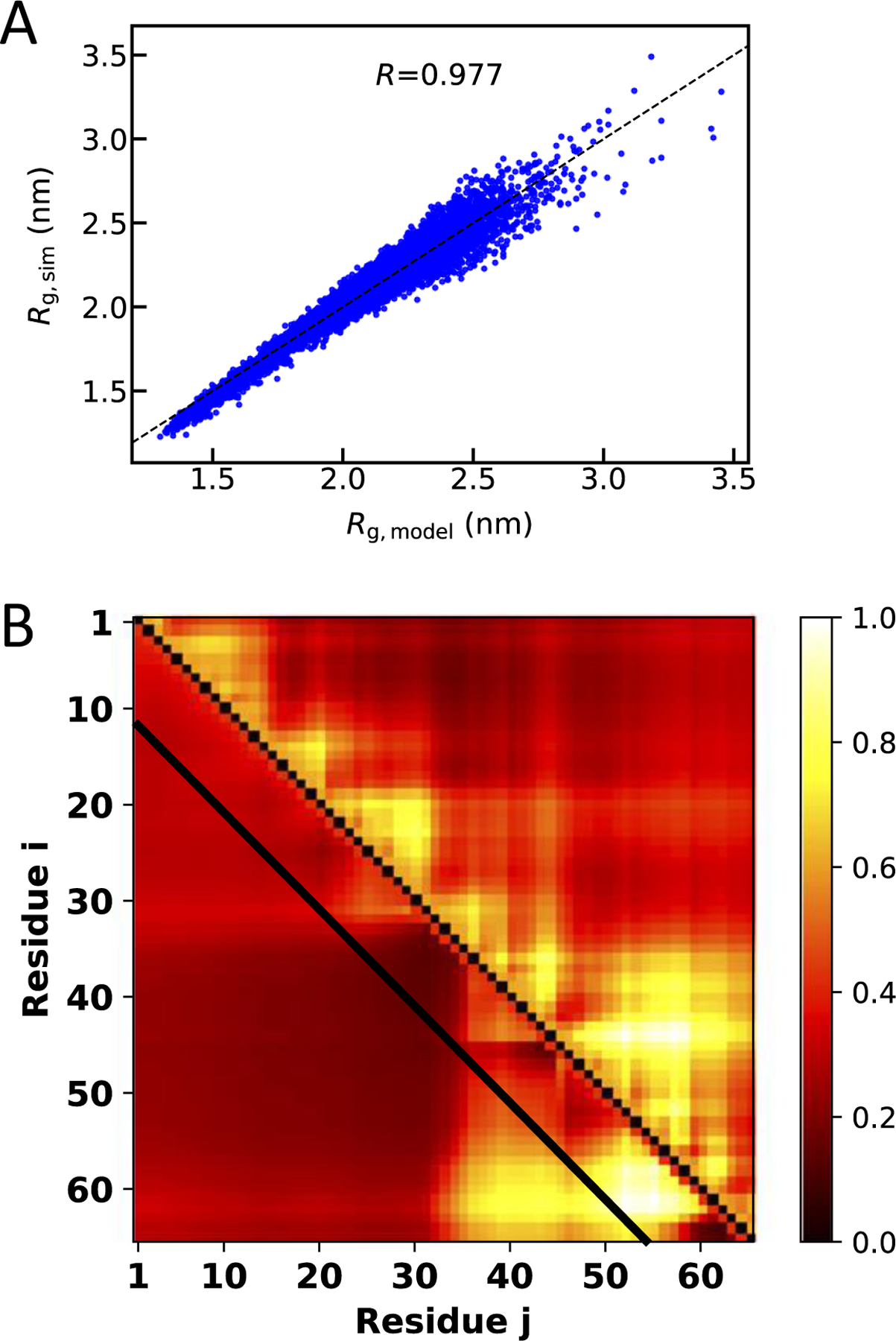
Comparison of analytical prediction of single-chain properties with simulation. (A) Radius of gyration computed from a combination of hydrophobic and charge patterning [48] compared with coarse-grained simulations. (B) Mean residue-residue distances (normalized to a [0,1] scale) from heteropolymer theory (lower left) compared with all-atom simulations [49].
Going beyond single chain properties and the association of pairs of chains, it is clearly of interest to obtain predictive theoretical models to describe phase separation. Towards this end, it was found in a recent study that the correlation between single-chain collapse transition temperature (Tθ) and critical temperature of LLPS (Tc) expected for homopolymers of infinite length could also apply to the behavior of heteropolymeric IDPs of the order of 100 residues in length (Figure 2) [51]. It is important to note that these results were preceded by Lin and Chan’s study on the dependence of protein Rg and Tc for IDPs of fixed chain lengths[52]. These theories and empirical models can be used to infer properties of IDPs in a high-throughput manner, which can potentially be used for the design of synthetic IDPs with tunable properties and associated assembly behavior, see e.g., recent work on predicting the thermoresponsive behavior of IDPs (Dignon, 2019, 10.1021/acscentsci.9b00102).
Figure 2.
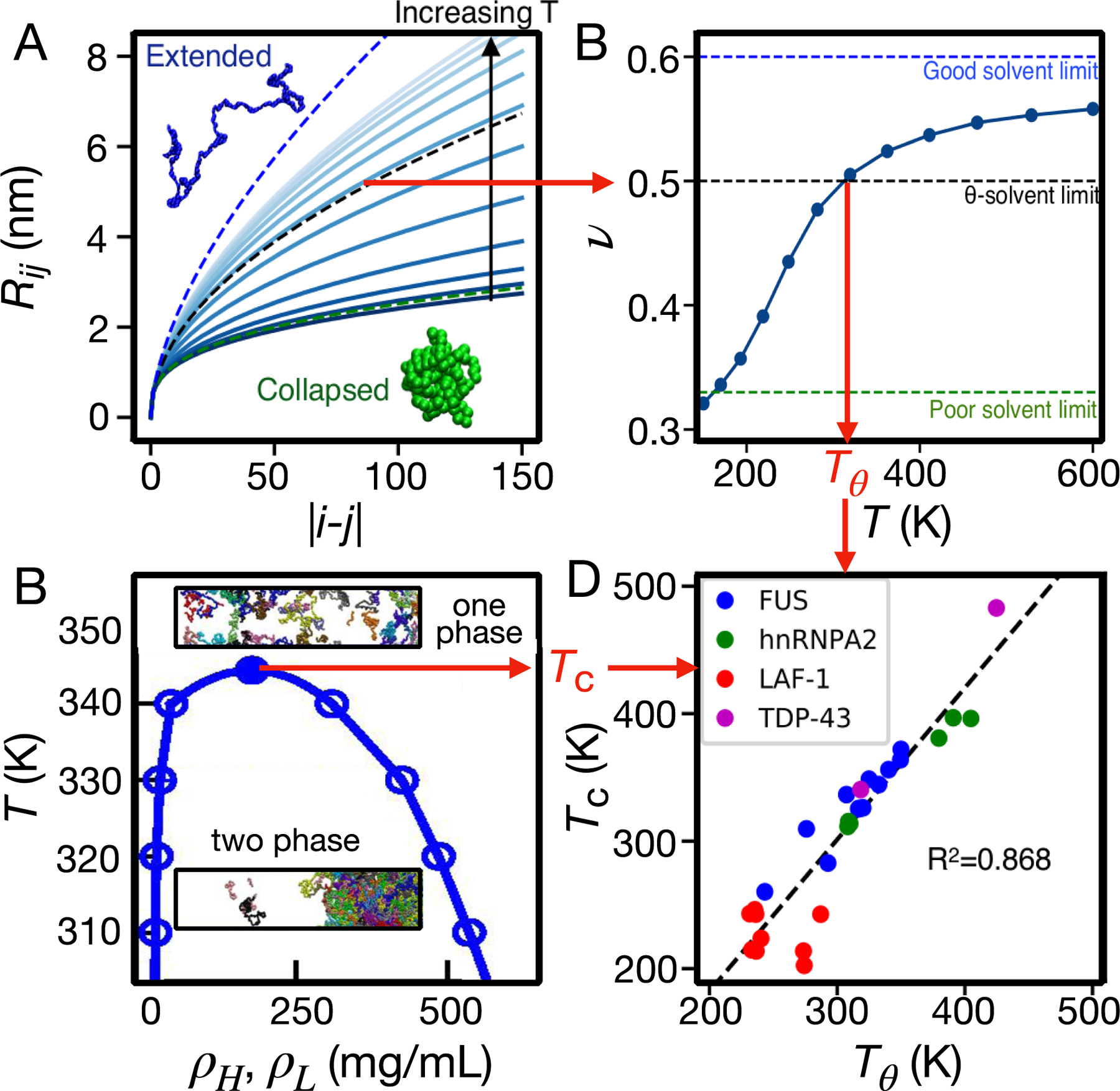
Relating single chain properties to phase separation. (A) Internal distance scaling for a single protein chain yields an effective scaling exponent ν as a function of temperature (B) – Tθ is the temperature at which v = 1/2. (C) Slab simulations (shown as insets) as a function of temperature establish the one-phase and two-phase regions of the phase diagram and an estimate of the critical temperature Tc. (D) Tc is remarkably well correlated with Tθ for a range of different sequences [51].
An important contribution of theory-based approaches has been to study the LLPS behavior of IDPs at much lower computational cost than molecular simulations of LLPS to provide important biophysical insights. Proto-models, including field theory, scaling theory, and ion release models, described in a recent review by Sing [53] in the context of complex coacervation of polyampholytes and polyelectrolytes, and that themselves build on earlier work in the context of polymer melts [54,55], are prime for extension and application to IDPs. We focus here on analytical and computational implementations of the polymer field theory model, as this is the most developed of the proto-models to date for IDP LLPS. In particular, field theoretic simulations with Complex Langevin sampling (FTS-CL) offers a computational efficient protocol as it considers a single IDP in the field of other IDPs rather than embedding many-body interactions. FTS begin with a particle-based model, and the particle-based canonical partition function is converted through a Hubbard-Stratonovich transformation into a field-theoretic partition function, in which the excluded volume and electrostatic interactions are decoupled through auxiliary fields [56]. The Hamiltonian of the system, expressed in terms of fluctuating chemical (ω) and electrostatic (φ) potential fields and the partition function Q[ω,φ] of a single chain in the complex-valued conjugate fields, is then sampled numerically using complex Langevin Dynamics. Phase diagrams for IDP LLPS can be readily obtained from FTS-CL, as detailed in references [57,58]. An alternate approach to FTS to obtain phase diagrams is to turn to analytical approaches. One of the most successful approaches has been the Random Phase Approximation (RPA), an theory initially developed by polymer physicists De Gennes [59] and Erukhimovich [60]in the context of polyelectrolytes, and recently applied to IDP coacervation [61]. This method considers Gaussian fluctuations of the chemical and electrostatic fields (as opposed to full compositions fluctuations as was the case with FTS). This approach rivals FTS simulations at a fraction of the computational cost at high densities, but the Gaussian approximation renders this analytical approach less accurate than FTS at low densities where charge and density fluctuations are magnified. Both RPA and FTS models have been able to shed insight into how sequence patterning affects the ability of peptides to LLPS. In particular, a study of peptides consisting of the amino acids E and K revealed that a blocky sequences phase separate with greater ease than scrambled sequences. Coupling the RPA/FTS results with coarse-grained simulations indicate that in this model, the interactions that drive LLPS are the same interactions that drive the collapse of an individual protein [58]. Of note is a recent improvement in the RPA formalism developed by Chan and Ghosh that combines the traditional RPA model with a renormalized Gaussian-chain formulation [62]. This combination (termed rG-RPA) gives a better representation of the conformational heterogeneity that arises when multiple chains interact and it seeks to account for pH, salt, and sequence effects. Both rG-RPA and FTS-CL are emerging as leading methodologies for efficient generation of phase diagrams for IDP LLPS, but as with all theoretical models, they will need further validation against experimental data. FTS applied to IDP is still in its infancy and restricted to date to implicit solvent representations of the polymer chain, with a limited amino acid library. Nonetheless, this approach has been successful in modeling the LLPS behavior of the Tau protein in the presence of RNA, predicting phase diagrams and identifying which segments of Tau are responsible for phase separation (Figure 3) [63,64]. On-going research involves building in explicit solvent into the FTS model and including additional sequence-specific details, and importantly extending the studies to a larger set of IDP proteins and validating results using experiment.
Figure 3.
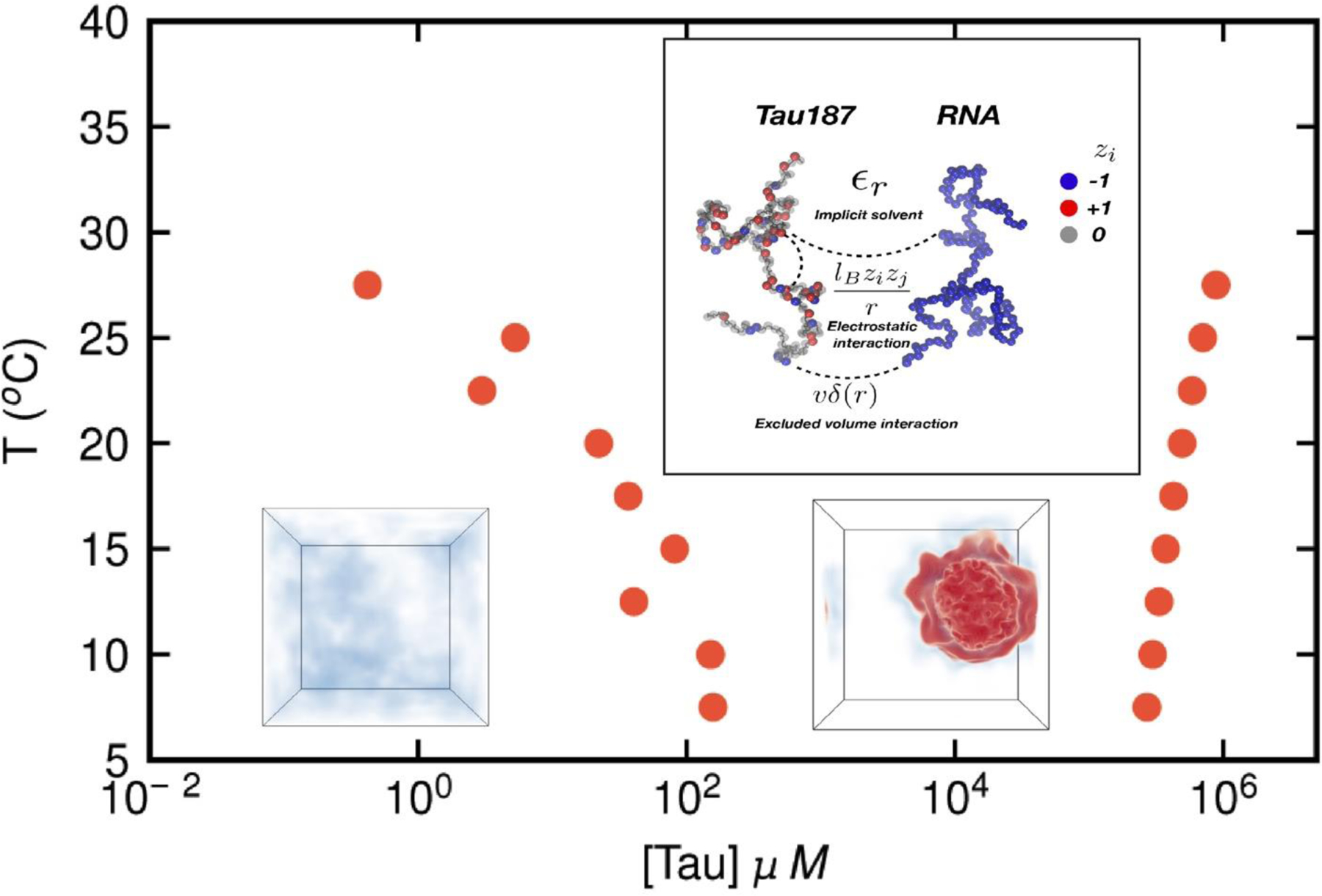
Field Theoretic Simulations of the LLPS of the Tau protein. The upper inset panel shows the coarse-grained models of Tau and RNA that serve as the input for the FTS-CL simulations. The red circles correspond to the boundaries of the phase diagram. The inner envelope corresponds to the phase separated state (shown by the red density in the FTS-CL snapshot) [63].
Conclusions
IDPs are involved in crucial biological functions and their experimental characterization is singularly challenging. Computer simulations have become an indispensable complement to experiment, as they are uniquely positioned to provide molecular mechanistic insights into the behavior of individual IDPs as well as IDP complexes. The last two years have seen tremendous progress in the development of accurate atomistic force fields, a novel framework for coarse-grained simulations of IDPs and LLPS, and powerful analytical and field theoretic models for LLPS. We are at a particularly exciting juncture in terms of the development of novel computational and theoretical tools for IDPs, and we anticipate that simulation and theory will play an essential part in coming years in unraveling the many mysteries that still surround the unique biopolymers that are IDPs.
Acknowledgement
This work was supported in part by the National Institutes of Health R01NS116176 (J.M.), R01GM120537 (J.M.), R01AG05605 (J.-E. S), National Science Foundation grants DMR2004796 (J.M), MCB-1716956 (J.-E.S.), TG-MCA05S027 (J.-E.S.), CNS-1725797 (J.-E. S) and DMR 1720256 (J.-E.S). R.B. was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health.
References
- 1.Babu MM: The contribution of intrinsically disordered regions to protein function, cellular complexity, and human disease. Biochemical Society Transactions 2016, 44:1185–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ilie IM, Caflisch A: Simulation Studies of Amyloidogenic Polypeptides and Their Aggregates. Chemical Reviews 2019, 119:6956–6993. [DOI] [PubMed] [Google Scholar]
- 3.Carballo-Pacheco M, Strodel B: Advances in the Simulation of Protein Aggregation at the Atomistic Scale. Journal of Physical Chemistry B 2016, 120:2991–2999. [DOI] [PubMed] [Google Scholar]
- 4.Choi JM, Holehouse AS, Pappu RV: Physical Principles Underlying the Complex Biology of Intracellular Phase Transitions. Annual Review of Biophysics, Vol 49, 2020 2020, 49:107–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dignon GL, Zheng WW, Mittal J: Simulation methods for liquid-liquid phase separation of disordered proteins. Current Opinion in Chemical Engineering 2019, 23:92–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levine ZA, Shea JE: Simulations disordered proteins and systems with conformational heterogeneity. Current Opinion in Structural Biology 2017, 43:95–103. [DOI] [PubMed] [Google Scholar]
- 7.Ruff KM, Pappu RV, Holehouse AS: Conformational preferences and phase behavior of intrinsically disordered low complexity sequences: insights from multiscale simulations. Current Opinion in Structural Biology 2019, 56:1–10. [DOI] [PubMed] [Google Scholar]
- 8.Bottaro S, Lindorff-Larsen K: Biophysical experiments and biomolecular simulations: A perfect match? Science 2018, 361:355–360. [DOI] [PubMed] [Google Scholar]
- 9.Pietrek LM, Stelzl LS, Hummer G: Hierarchical Ensembles of Intrinsically Disordered Proteins at Atomic Resolution in Molecular Dynamics Simulations. Journal of Chemical Theory and Computation 2020, 16:725–737. [DOI] [PubMed] [Google Scholar]
- 10.Holmstrom ED, Holla A, Zheng W, Nettels D, Best RB, Schuler B: Accurate Transfer Efficiencies, Distance Distributions, and Ensembles of Unfolded and Intrinsically Disordered Proteins From Single-Molecule FRET. Methods Enzymol 2018, 611:287–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Best RB: Computational and theoretical advances in studies of intrinsically disordered proteins. Current Opinion in Structural Biology 2017, 42:147–154. [DOI] [PubMed] [Google Scholar]
- 12.Best RB, Zheng WW, Mittal J: Balanced Protein Water Interactions Improve Properties of Disordered Proteins and Non-Specific Protein Association (vol 10, pg 5113, 2014). Journal of Chemical Theory and Computation 2015, 11:1978–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robustelli P, Piana S, Shaw DE: Developing a molecular dynamics force field for both folded and disordered protein states. Proceedings of the National Academy of Sciences of the United States of America 2018, 115:E4758–E4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang WS, Fawzi NL, Mittal J: Refining All-Atom Protein Force Fields for Polar-Rich, Prion-like, Low-Complexity Intrinsically Disordered Proteins. Journal of Physical Chemistry B 2020, 124:9505–9512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zerze GH, Zheng WW, Best RB, Mittal J: Evolution of All-Atom Protein Force Fields to Improve Local and Global Properties. Journal of Physical Chemistry Letters 2019, 10:2227–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bellaiche MMJ, Best RB: Molecular Determinants of Abeta42 Adsorption to Amyloid Fibril Surfaces. J Phys Chem Lett 2018, 9:6437–6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shrestha UR, Juneja P, Zhang Q, Gurumoorthy V, Borreguero JM, Urban V, Cheng X, Pingali SV, Smith JC, O’Neill HM, et al. : Generation of the configurational ensemble of an intrinsically disordered protein from unbiased molecular dynamics simulation. Proc Natl Acad Sci U S A 2019, 116:20446–20452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zheng W, Borgia A, Buholzer K, Grishaev A, Schuler B, Best RB: Probing the Action of Chemical Denaturant on an Intrinsically Disordered Protein by Simulation and Experiment. J Am Chem Soc 2016, 138:11702–11713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Conicella AE, Dignon GL, Zerze GH, Schmidt HB, D’Ordine AM, Kim YC, Rohatgi R, Ayala YM, Mittal J, Fawzi NL: TDP-43 alpha-helical structure tunes liquid-liquid phase separation and function. Proc Natl Acad Sci U S A 2020, 117:5883–5894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lincoff J, Haghighatlari M, Krzeminski M, Teixeira JMC, Gomes GNW, Gradinaru CC, Forman-Kay JD, Head-Gordon T: Extended experimental inferential structure determination method in determining the structural ensembles of disordered protein states. Communications Chemistry 2020, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vitalis A, Pappu RV: ABSINTH: a new continuum solvation model for simulations of polypeptides in aqueous solutions. J Comput Chem 2009, 30:673–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choi JM, Pappu RV: Improvements to the ABSINTH Force Field for Proteins Based on Experimentally Derived Amino Acid Specific Backbone Conformational Statistics. J Chem Theory Comput 2019, 15:1367–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng WW, Dignon GL, Xu X, Regy RM, Fawzi NL, Kim YC, Best RB, Mittal J: Molecular details of protein condensates probed by microsecond-long atomistic simulations. bioRxiv 2020.08.05.237008 2020. [DOI] [PMC free article] [PubMed]
- 24.Paloni M, Bailly R, Ciandrini L, Barducci A: Unraveling Molecular Interactions in Liquid-Liquid Phase Separation of Disordered Proteins by Atomistic Simulations. J Phys Chem B 2020, 124:9009–9016. [DOI] [PubMed] [Google Scholar]
- 25.Welsh TJ, Krainer G, Espinosa JR, Joseph JA, Sridhar A, Jahnel M, Arter WE, Saar K, Alberti S, Collepardo-Guevara R, et al. : Single particle zeta-potential measurements reveal the role of electrostatics in protein condensate stability 2020.
- 26.Murthy AC, Dignon GL, Kan Y, Zerze GH, Parekh SH, Mittal J, Fawzi NL: Molecular interactions underlying liquid-liquid phase separation of the FUS low-complexity domain. Nature Structural & Molecular Biology 2019, 26:637–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schuster BS, Dignon GL, Tang WS, Kelley FM, Ranganath AK, Jahnke CN, Simpkins AG, Regy RM, Hammer DA, Good MC, et al. : Identifying sequence perturbations to an intrinsically disordered protein that determine its phase-separation behavior. Proceedings of the National Academy of Sciences of the United States of America 2020, 117:11421–11431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kmiecik S, Gront D, Kolinski M, Wieteska L, Dawid AE, Kolinski A: Coarse-Grained Protein Models and Their Applications. Chemical Reviews 2016, 116:7898–7936. [DOI] [PubMed] [Google Scholar]
- 29.Noid WG: Perspective: Coarse-grained models for biomolecular systems. Journal of Chemical Physics 2013, 139. [DOI] [PubMed] [Google Scholar]
- 30.Wu H, Wolynes PG, Papoian GA: AWSEM-IDP: A Coarse-Grained Force Field for Intrinsically Disordered Proteins. Journal of Physical Chemistry B 2018, 122:11115–11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dignon GL, Zheng WW, Kim YC, Best RB, Mittal J: Sequence determinants of protein phase behavior from a coarse-grained model. Plos Computational Biology 2018, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perdikari TM, Jovic N, Dignon GL, Kim YC, Fawzi NL, Mittal J: A predictive coarse-grained model for position-specific effects of post-translational modifications on disordered protein phase separation. bioRxiv 2020.06.12.148650 2020. [DOI] [PMC free article] [PubMed]
- 33.Dignon GL, Zheng WW, Kim YC, Mittal J: Temperature-Controlled Liquid-Liquid Phase Separation of Disordered Proteins. Acs Central Science 2019, 5:821–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Regy RM, Dignon GL, Zheng W, Kim YC, Mittal J: Sequence dependent co-phase separation of RNA-protein mixtures elucidated using molecular simulations. bioRxiv 2020:2020.2007.2007.192047. [DOI] [PMC free article] [PubMed]
- 35.Alshareedah I, Moosa MM, Raju M, Potoyan DA, Banerjee PR: Phase transition of RNA−protein complexes into ordered hollow condensates. Proceedings of the National Academy of Sciences 2020, 117:15650–15658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao YN, Cortes-Huerto R, Kremer K, Rudzinski JF: Investigating the Conformational Ensembles of Intrinsically Disordered Proteins with a Simple Physics-Based Model. Journal of Physical Chemistry B 2020, 124:4097–4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baul U, Chakraborty D, Mugnai ML, Straub JE, Thirumalai D: Sequence Effects on Size, Shape, and Structural Heterogeneity in Intrinsically Disordered Proteins. J Phys Chem B 2019, 123:3462–3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Silmore KS, Howard MP, Panagiotopoulos AZ: Vapour-liquid phase equilibrium and surface tension of fully flexible Lennard-Jones chains. Molecular Physics 2017, 115:320–327. [Google Scholar]
- 39.Espinosa JR, Joseph JA, Sanchez-Burgos I, Garaizar A, Frenkel D, Collepardo-Guevara R: Liquid network connectivity regulates the stability and composition of biomolecular condensates with many components. Proc Natl Acad Sci U S A 2020, 117:13238–13247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ghosh A, Mazarakos K, Zhou HX: Three archetypical classes of macromolecular regulators of protein liquid-liquid phase separation. Proceedings of the National Academy of Sciences of the United States of America 2019, 116:19474–19483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Choi JM, Dar F, Pappu RV: LASSI: A lattice model for simulating phase transitions of multivalent proteins. Plos Computational Biology 2019, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mao S, Chakraverti-Wuerthwein MS, Gaudio H, Košmrlj A: Designing the Morphology of Separated Phases in Multicomponent Liquid Mixtures. Physical Review Letters 2020, 125. [DOI] [PubMed] [Google Scholar]
- 43.Alonso DO, Dill KA: Solvent denaturation and stabilization of globular proteins. Biochemistry 1991, 30:5974–5985. [DOI] [PubMed] [Google Scholar]
- 44.Fields GB, Alonso DO, Stigter D, Dill KA: Theory for the aggregation of proteins and copolymers. J. Phys. Chem 1992, 96:3974–3981. [Google Scholar]
- 45.Sawle L, Ghosh K: A theoretical method to compute sequence dependent configurational properties in charged polymers and proteins. J Chem Phys 2015, 143:085101. [DOI] [PubMed] [Google Scholar]
- 46.Das RK, Pappu RV: Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proc Natl Acad Sci U S A 2013, 110:13392–13397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ashbaugh HS: Tuning the globular assembly of hydrophobic/hydrophilic heteropolymer sequences. J Phys Chem B 2009, 113:14043–14046. [DOI] [PubMed] [Google Scholar]
- 48.Zheng WW, Dignon G, Brown M, Kim YC, Mittal J: Hydropathy Patterning Complements Charge Patterning to Describe Conformational Preferences of Disordered Proteins. Journal of Physical Chemistry Letters 2020, 11:3408–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huihui J, Ghosh K: An analytical theory to describe sequence-specific inter-residue distance profiles for polyampholytes and intrinsically disordered proteins. J Chem Phys 2020, 152:161102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Amin AN, Lin YH, Das S, Chan HS: Analytical Theory for Sequence-Specific Binary Fuzzy Complexes of Charged Intrinsically Disordered Proteins. J Phys Chem B 2020, 124:6709–6720. [DOI] [PubMed] [Google Scholar]
- 51.Dignon GL, Zheng W, Best RB, Kim YC, Mittal J: Relation between single-molecule properties and phase behavior of intrinsically disordered proteins. Proc Natl Acad Sci U S A 2018, 115:9929–9934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin YH, Chan HS: Phase Separation and Single-Chain Compactness of Charged Disordered Proteins Are Strongly Correlated. Biophys J 2017, 112:2043–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sing CE, Perry SL: Recent progress in the science of complex coacervation. Soft Matter 2020, 16:2885–2914. [DOI] [PubMed] [Google Scholar]
- 54.Kyrylyuk AV, Fraaije JGEM: Structure formation in films of weakly charged block polyelectrolyte solutions. Journal of Chemical Physics 2004, 121:9166–9171. [DOI] [PubMed] [Google Scholar]
- 55.Muthukumar M: 50th Anniversary Perspective: A Perspective on Polyelectrolyte Solutions. Macromolecules 2017, 50:9528–9560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Delaney KT, Fredrickson GH: Recent Developments in Fully Fluctuating Field-Theoretic Simulations of Polymer Melts and Solutions. Journal of Physical Chemistry B 2016, 120:7615–7634. [DOI] [PubMed] [Google Scholar]
- 57.Delaney KT, Fredrickson GH: Theory of polyelectrolyte complexation-Complex coacervates are self-coacervates. Journal of Chemical Physics 2017, 146. [DOI] [PubMed] [Google Scholar]
- 58.McCarty J, Delaney KT, Danielsen SPO, Fredrickson GH, Shea JE: Complete Phase Diagram for Liquid-Liquid Phase Separation of Intrinsically Disordered Proteins. Journal of Physical Chemistry Letters 2019, 10:1644–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gennes PGd: Scaling concepts in polymer physics Ithaca, N.Y.: Cornell University Press; 1979. [Google Scholar]
- 60.Borue VY, Erukhimovich IY: A Statistical-Theory of Weakly Charged Poly-Electrolytes - Fluctuations, Equation of State, and Microphase Separation. Macromolecules 1988, 21:3240–3249. [Google Scholar]
- 61.Lin YH, Chan HS: Phase Separation and Single-Chain Compactness of Charged Disordered Proteins Are Strongly Correlated. Biophysical Journal 2017, 112:2043–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lin YH, Brady JP, Chan HS, Ghosh K: A unified analytical theory of heteropolymers for sequence-specific phase behaviors of polyelectrolytes and polyampholytes. Journal of Chemical Physics 2020, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lin YX, McCarty J, Rauch JN, Delaney KT, Kosik KS, Fredrickson GH, Shea JE, Han S: Narrow equilibrium window for complex coacervation of tau and RNA under cellular conditions. Elife 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang X, Vigers M, McCarty J, Rauch JN, Fredrickson GH, Wilson MZ, Shea JE, Han S, Kosik KS: The proline-rich domain promotes Tau liquid-liquid phase separation in cells. J Cell Biol 2020, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]