

Abstract
The conversion of unactivated carbon-hydrogen (C–H) bonds to carbon–nitrogen (C-N) bonds is a highly valued transformation. Existing strategies typically accomplish such reactions at only a single C–H site because the first derivatization diminishes the reactivity of surrounding C–H bonds. Here, we show that alkylated arenes can undergo vicinal C–H diamination reactions to form 1,2-diamine derivatives via an electrophotocatalytic strategy, using acetonitrile as both solvent and nitrogen source. The reaction is catalyzed by a trisaminocyclopropenium (TAC) ion, which undergoes anodic oxidation to furnish a stable radical dication while the cathodic reaction reduces protons to molecular hydrogen. Irradiation of the TAC radical dication (wavelength of maximum absorption of 450 to 550 nanometers) with a white-light compact fluorescent light generates a strongly oxidizing photoexcited intermediate. Depending on the electrolyte used, either 3,4-dihydroimidazole or 2-oxazoline products are obtained.
Chemical reactions that convert ubiquitous but relatively inert carbon-hydrogen (C–H) bonds to valuable (C–N) bonds can greatly accelerate the construction of complex molecules, particularly those relevant to the biomedical enterprise (1). Accordingly, a variety of C–H amination reactions have been devised, ranging from classic transformations such as the Hofmann-Löffler-Freytag reaction to modern methods that involve transition metal or photoredox catalysis (2, 3). Despite the power and scope of these advances, the vast majority of such methods result in the transformation of only single C–H bonds, whereas many synthetic campaigns require the installation of numerous C–N linkages. Thus, the development of chemical reactions that effect multiple C–H bond activation events concurrently promise great value to the field of chemical synthesis. However, one of the major challenges to developing such reactions is that the installation of heterofunctionality tends to deactivate surrounding bonds towards the typical mechanistic modes of C–H activation. Accordingly, few reaction technologies have been reported that accomplish this type of multipotent functionalization on proximal C–H bonds (4).
We recently reported a strategy for potent oxidation chemistry that combined the energy of light and electricity within a single catalyst, a process termed electrophotocatalysis (EPC) (5–7). This strategy involved electrochemical oxidation of the trisaminocyclopropenium (TAC) ion 1 under a relatively mild electrochemical potential and concomitant visible light irradiation to excite the resulting radical dication intermediate 2 (Fig. 1A). The photoexcited radical dication 3 is an extremely potent oxidant, which we demonstrated could promote challenging reactions such as the oxidative functionalization of benzene and other electron-poor arenes or the regioselective C–H functionalization of ethers. We speculated that the oxidizing power of 1 might also enable other C–H bond activation manifolds. In particular, we hypothesized that under the right conditions, the electrophotocatalytic (8–13) approach might lead to the generation of carbocation intermediates, which would enable the Ritter-type functionalization of C–H bonds without the need for an external chemical oxidant. Ritter-type reactions involve the generation of a carbocation 5 with subsequent trapping by a nitrile (usually as solvent), which leads to the formation of nitrilium ion intermediates and, after hydrolysis, amide products 6 (Fig. 1B) (14, 15). If the carbocation is generated from the corresponding C-H bond, Ritter-type processes provide an avenue to achieve C-H amination, and several methods have been developed that accomplish this type of transformation (16–18). We speculated that the strongly oxidizing yet selective conditions offered by TAC EPC might enable a sequence of multiple Ritter-type C-H functionalization reactions, in which the initially formed acetamide group facilitated a second amination reaction at an adjacent (vicinal) position. If feasible, such a process could enable the regioselective amination of two C-H bonds by using simply visible light, a mild electrochemical potential, and a common solvent (acetonitrile) as the nitrogen source rather than potentially explosive nitrene precursors. Here we report the realization of this electrophotocatalytic diamination of vicinal C-H bonds to furnish either dihydroimidazoles 8 or 2-oxazolines 9, depending on the electrolyte used (Fig. 1C).
Fig. 1. Electrophotocatalytic amination of C-H Bonds:
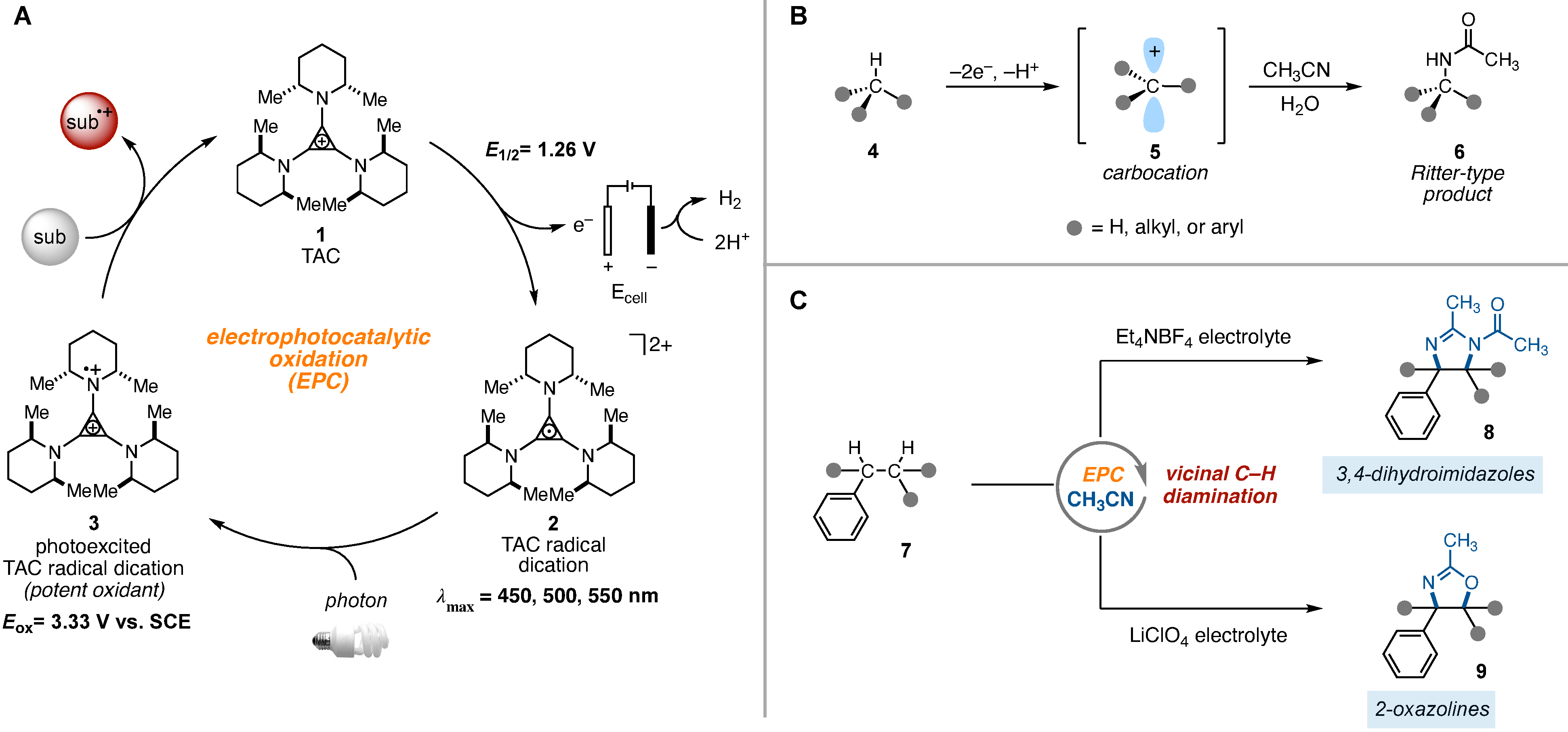
(A) Generic electrophotocatalytic cycle with trisaminocyclopropenium (TAC) 1. (B) Ritter-type C-H amination reaction. (C) Electrophotocatalytic vicinal C-H diamination or oxyamination reactions reported in this work. Sub, substrate; subox, oxidized substrate; Me, methyl; Et, ethyl; Ac, acetyl; Eox, oxidation potential; λmax, wavelength of maximum absorption.
After extensive screening of reaction conditions-including the cell potential, electrolyte, acid additive, and reaction time-we identified reaction conditions that enabled the efficient conversion of a variety of benzylic hydrocarbons to the corresponding N-acyl-4,5-dihydroimidazole adducts (Fig. 2; optimization with phenylcyclopentane is provided in table S1). The reaction set-up involved visible light irradiation with a white compact fluorescent light (CFL) of a solution of the substrate, 8 mol% TAC 1, and tetraethylammonium tetrafluoroborate (Et4NBF4) in 5:1 acetonitrile:trifluoroacetic acid (TFA) solution within a divided electrolytic cell (carbon felt anode and platinum plate cathode) under controlled potential [2.4 V, anode potential (Eanode) = 1.3 V vs. Ag/AgCl], The TAC catalyst and the substrate were contained within the anodic chamber, which was where the C-H diamination chemistry occurred. The electrochemical reaction was balanced by the cathodic reduction of protons to molecular hydrogen, providing an effectively traceless redox by-product. Using these conditions, a variety of benzylic hydrocarbons were found to undergo vicinal C–H diamination. No more than trace amounts of such products could be detected without the catalyst 1 or in the absence of irradiation. In addition, no traditional oxidants were found to effect this transformation, including K2S2O8, oxone, tert-butyl hydrogen peroxide (TBHP), MnO2, Mn(OAc)3, HClO4, phenyliodine(III) diacetate (PIDA), ceric ammonium nitrate (CAN), or O2.
Fig. 2. Substrate scope of electrophotocatalytic vicinal C–H diamination.

All yields are of isolated products. Products were obtained as racemic mixtures; wedge and dash depictions indicate relative stereochemical relationships. (A) Diamination of secondary alkylbenzenes. (B) Diamination of primary alkylbenzenes. Experimental details are provided in the supplementary materials. An asterisk indicates run at 2.2 V; a dagger symbol (†) indicates work-up with NaHCO3 (aq) and CH3OH; and a double dagger symbol (‡) indicates nBu4NPF6 instead of Et4NBF4. SM, starting materials. Compound 36 was deacylated upon workup.
Using this procedure, isopropylbenzene (cumene) was converted to adduct 10 in 72% yield (Fig. 2). Halogen substitution on the aryl ring was also tolerated (11 to 14), as was a protected anilino nitrogen (15) and a bromomethyl substituent (16). On the other hand, 4-isopropylbiphenyl did not lead to any of the diaminated product 17; instead, the reaction mixture turned black, and an unidentified mixture of products was generated. Despite the strongly oxidizing conditions, a benzylic trifluoroacetamide group was reasonably well tolerated (18). When 4-methylisopropylbenzene (p-cymene) was used, a 51% yield of the diaminated product 19 was generated, along with an 11% yield of the product arising from Ritter reaction of the benzylic methyl group. This alternative process was even more competitive with 4-ethylisopropylbenzene, with which a 41% yield of the diaminated adduct 20 was accompanied by 41% of the ethyl-functionalized product. By contrast, 1,4-diisopropylbenzene underwent efficient conversion to adduct 21 in which only one of the isopropyl groups was diaminated. The isomeric 1,3-diisopropylbenzene also furnished adduct 22, although the efficiency was notably lower in this case. In the reactions to form 21 and 22, we only observed products resulting from functionalization of one of the benzylic carbons. Similarly, methyl 2,4,6-triisopropylbenzoate and 2,4,6-triisopropylbromobenzene were functionalized exclusively on the 4-isopropyl group to furnish 23 and 24, respectively, in good to high yields.
We also examined the site selectivity for amination of the nonbenzylic carbon (25 to 32). In all cases, functionalization of methylene carbons occurred in preference to methyl carbons, even when the presence of a sterically demanding group (29) or electron-withdrawing groups (30 and 31) might have modified the outcome. By contrast, a substrate probing the competition between a methyl and a methine carbon led exclusively to adduct 32 in which the methyl was functionalized preferentially.
Because α,α-diaryl amines are a valuable substructure in biomedically relevant compounds, we also investigated this transformation on gem-diaryl substrates. We found that 1,1-diphenylethane reacted efficiently to furnish compound 33 in 80% yield. Fluorine substituents on one (34) or both (35) rings was tolerated, albeit with a slight decrease in yield. Meanwhile, the compatibility of alcohol, ester, alkyl fluoride, and amide substituents enabled the synthesis of the more highly functionalized adducts 36 to 39.
We also investigated the capacity of this reaction to functionalize ring systems. Reaction of phenylcyclopentane led to the bicyclic compound in 85% yield, formed as a 5:1 mixture of N-acyl isomers (40 and 40’). Similarly, the six- and seven-membered ring products 41 and 42, respectively, were also produced as regioisomeric mixtures, whereas eight-membered and 12-membered ring products 43 and 44, respectively, were produced as single isomers. Some of the yields for cyclic substrates were improved by the use of tetrabutylammonium phosphate (TBAPF6) as the electrolyte. The spiro compound 45 was also accessible, demonstrating functionalization of a C–H bond in a strained ring. Furthermore, in addition to acetonitrile, other nitriles could also be employed in this reaction, giving rise to diaminated products 46 to 48 derived from propionitrile, butyl nitrite, or benzonitrile as the nitrogen source.
We also evaluated the diamination process with unbranched benzylic substrates. Both n-propylbenzene and halogenated n-propylbenzenes reacted with CH3CN to deliver products 49 to 51 in moderate yields. For n-propylbenzene, two isomers (49 and 49’) were obtained in a 3:1 ratio; the major product had the opposite acyl group regioselectivity to that observed with the branched substrates. Similar regioselectivity was also observed when longer alkyl chain substrates such as n-butylbenzene (52 and 52’) and n-dodecylbenzene (55 and 55’) were employed. Additionally, cyclic indane could be functionalized to furnish adduct 56 in 32% yield.
During the course of our studies, we discovered that changing the electrolyte from Et4NBF4 to LiClO4 resulted in an alternative vicinal C–H difunctionalization product: 2-oxazolines (Fig. 3). Except for the cases noted below, the isomers with the nitrogen atom in the benzylic position were produced. Thus, cumene and halogenated derivatives gave rise to oxazolines 57 to 59 in low to modest yields, along with nearly equal yields of the diaminated products. Other oxidants we tried did not deliver any of the oxazoline or diamination products (tables S3 and S4). With substrates bearing a pendant carboxyl substituent, the oxazoline products 60 and 61 were the major ones. Product yields were somewhat higher with gem-diaryl substrates (62 to 64). Remarkably, products 62 and 64 were generated as the alternative oxazoline isomers, with the oxygen atom in the benzylic position. Similarly, reaction of 3-phenylpentane led to the formation of the O-benzylic oxazoline regioisomer 65 in modest yield as a 1:1 mixture of diastereomers. In general, substrates bearing electron-rich aryl rings did not participate well in this process; however, we were able to prepare adducts 66 to 68 in modest yields. The reason for the formation of oxazolines when changing the electrolyte to a perchlorate salt is not obvious, but presumably, the counterions (supplied by the electrolyte) impact the stability of key cationic intermediates and their susceptibility to nucleophilic attack by acetonitrile or oxygen nucleophiles.
Fig. 3. Electrophotocatalytic vicinal C–H oxyamination.
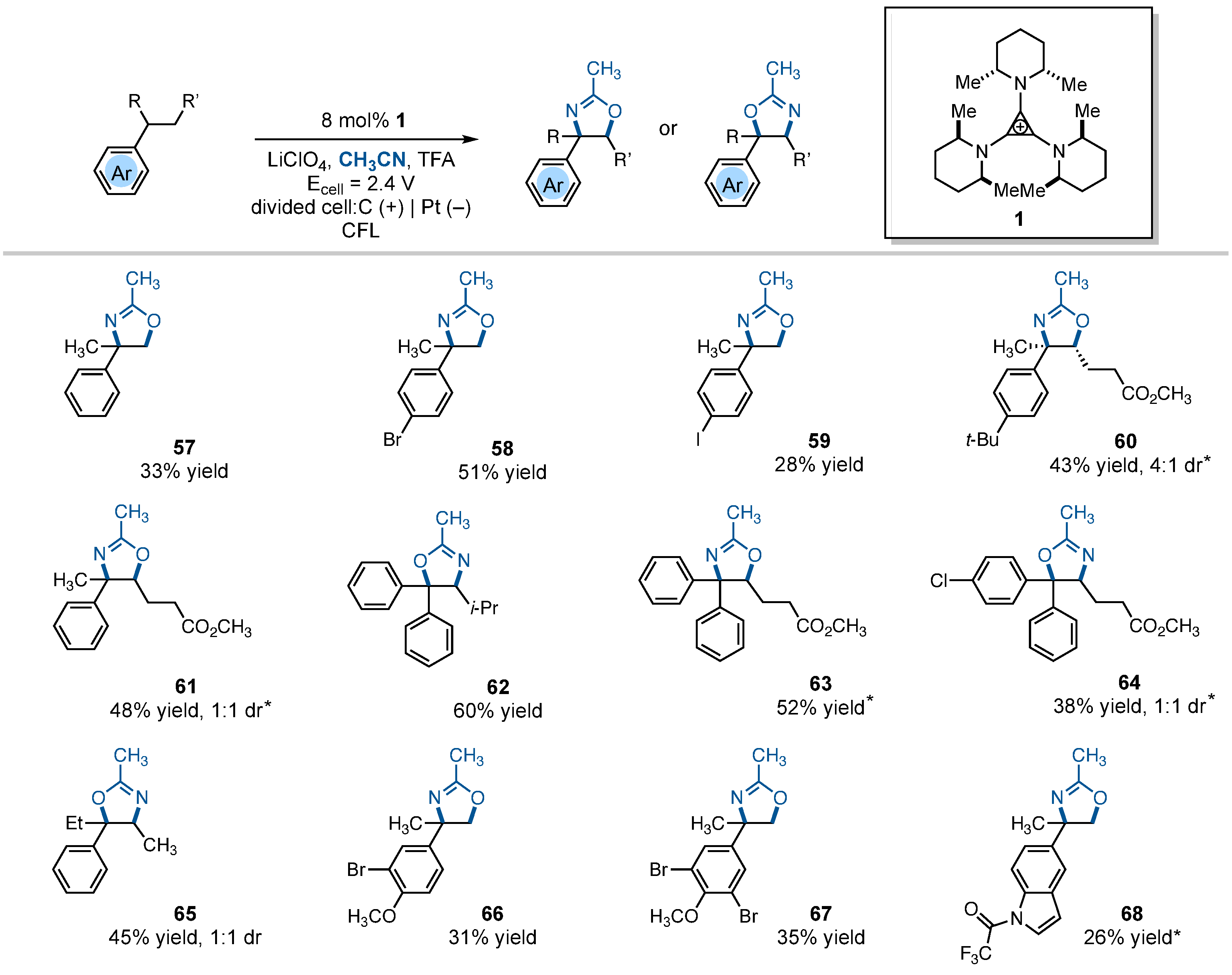
Detailed reaction conditions for each substrate are provided in the supplementary materials. Products were obtained as racemic mixtures; wedge and dash depictions indicate relative stereochemical relationships. An asterisk indictes run at 2.2 V. i-Pr, isopropyl.
Because late stage C–H functionalization processes offer powerful tools for the diversification of medicinal compound libraries, we decided to test this difunctionalization chemistry on several molecules that are close analogues of known biologically active molecules (Fig. 4A). Thus, we found that a dibromoisatin derivative could be diaminated to produce compound 69 in 42% yield. Isatin derivatives have been investigated for a number of medicinal properties, including antitumor and antiviral activities (19). We also found that the celecoxib (Celebrex) analogue 70 could be produced in 56% yield under standard conditions. FKGK11 is a group VIA calcium-independent phospholipase A2 inhibitor (20), and we found that the methylated derivative analogue 71 could be accessed in 38% yield. Meanwhile, an analogue of thalidomide 5HPP-33, which has shown antiproliferative activity against nine cancer cell lines in vitro (21), was converted to 72 in 50% yield after protection of the phenolic hydroxyl group as a triflate group. We also found that a retinoic acid receptor agonist and a CYP11B1 inhibitor (22, 23), both of which possessed a biaryl moiety, were amenable to the diamination procedure to furnish analogues 73 and 74, respectively. Successful synthesis of the insertraline analogue 75 proceeded in 68% yield despite the presence of a potentially sensitive benzylic C–N group. In addition to the diamination products, we also found that the oxyamination procedure was operable in a more complex setting, allowing direct access to products 76 and 77.
Fig. 4. Synthetic applications of electrophotocatalytic vicinal C–H diamination.

(A) Bioactive compound analogs prepared by means of electrophotocatalytic vicinal C–H diamination or oxyamination. (B) 1,2-Diamine synthesis. (C) Dihydroimidazole synthesis. (D) Bioactive compound synthesis. Detailed reaction conditions are provided in the supplementary materials. Products were obtained as racemic mixtures; wedge and dash depictions indicate relative stereochemical relationships. Products 80 and 81 isolated as bis tosylate salts. Ph, phenyl; Tf, trifluoromethanesulfonate.
Because of the importance of 1,2-diamines for pharmaceutical synthesis, ligands for catalysis, and other applications, it would be highly appealing to synthesize these structures directly from abundant sources. We found that a small modification to our electrophotocatalytic procedure led to the isolation of free 1,2-diamines in good yields (Fig. 4B). Following the diamination procedure described above, treating the crude reaction mixture with KOH, ethanol, and ethylene glycol and heating to reflux furnished the diamine products 78 to 84 (80 and 81 were isolated as their bis-toluenesulfonic acid salts). Cumene (99, <$0.03 per milliliter), a petroleum-derived feedstock of the Hock reaction for industrial phenol production, could be efficiently converted into valuable 2-phenylpropane-1,2-diamine 78 ($569 per gram) via this route (24). Alternatively, by using similar hydrolysis conditions but at room temperature, the free dihydroimidazole adducts 85 to 90 could be obtained (Fig. 4C). To further demonstrate the synthetic potential of this method for the generation of valuable compounds, we executed several short synthetic sequences (Fig. 4D). The Y5 receptor antagonists 93 and 94 were prepared in high yield by the electrophotocatalytic diamination of diarylethanes 91 and 92, followed by hydrolysis and amidine formation (25). Alternatively, diamination and hydrolysis of 91, conversion to piperazine 97, and acylation afforded the A2 adenosine receptor inhibitor 98 (26, 27). Meanwhile, trifluoromethyl compound 95 was engaged in a similar sequence to synthesize the vasopressin agonist 96 in short order (28). Lastly, cumene (99) could also be directly transformed into the important β-amino alcohol 100 ($357 per gram) (24) via a simple two-step process of oxyamination and hydrolysis.
Regarding the mechanism, we believe the reaction begins with Ritter-type amination of the substrate’s benzylic C–H bond in a process that accords with known electrochemical Ritter-type reactions (Fig. 5) (29, 30). In this case, the TAC photoexcited radical dication 3 effects single-electron oxidation of the substrate 99 [half-peak potential (Ep/2) = 2.12 V versus saturated calomel electrode (SCE) in 5:1 CH3CN:TFA] to produce radical cation 101, after which deprotonation and a second oxidation (presumably by TAC radical dication 2) reveals the cation 102. Solvolysis of 102 then furnishes the Ritter adduct 103. Notably, acetamide 103 converts to the dihydroimidazole product 10 when subjected to the standard reaction conditions. The pathway for the second C–H bond amination is less certain. One likely possibility is that the initially formed Ritter product 103 undergoes a reversible, acid-catalyzed elimination reaction to produce α-methylstyrene 104 (31). Single-electron oxidation of α-methylstyrene 104 (Ep/2 = 1.72 V vs. SCE in 5:1 CH3CN:TFA) (32) with subsequent solvent trapping (33) and oxidation events would then lead to the dihydroimidazole product 10 or the oxazoline 57, depending on the electrolyte. We have been able to detect trace amounts of styrenic products in these reactions (fig. S12), and we have indirectly implicated the formation of 104 under related conditions (supplementary materials). However, α-methylstyrene 104 did not furnish any diaminated products when subjected to the standard conditions and instead led to only polymeric material. We speculate that only small steady-state quantities of this intermediate are generated under the reaction conditions, thereby allowing the diamination to occur without the dimerization or oligomerization self-reactions to which styrenes are prone. If so, the conditions are remarkable in that they are potent enough to enable the oxidative functionalization of unactivated C–H bonds while still allowing for the selective reaction of a notoriously sensitive intermediate. A more in-depth mechanistic discussion can be found in the supplementary materials.
Fig. 5. Mechanistic rationale for electrophotocatalytic vicinal C–H diamination.
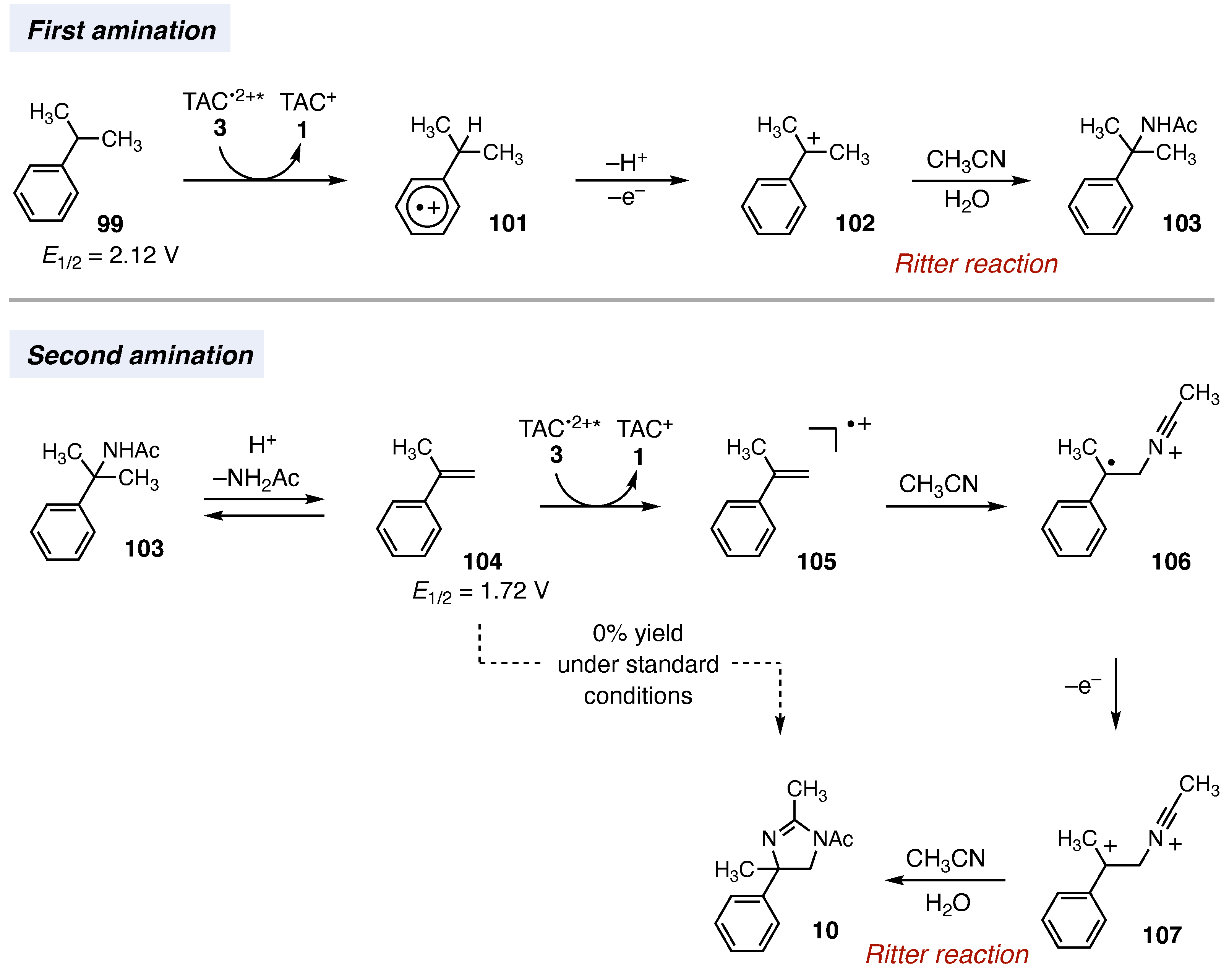
Voltages were measured in a 5:1 mixture of CH3CN and TFA to mimic the reaction conditions and are relative to SCE.
The compatibility of the diamination with a reasonable diversity of functionality lends some degree of optimism that this reaction could be of practical utility. Meanwhile, the power of combining light and electrical energy within the operation of a single catalyst has been further shown to hold value for advancing synthetic capabilities.
Supplementary Material
Data and materials availability:
Data are available in the supplementary materials.
Acknowledgments:
Funding:
Research reported in this publication was supported by the National Institutes of Health under R35 GM127135.
Footnotes
References and Notes:
- 1.Richter MF et al. , Nature 545, 299–304 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davies HM, Manning JR, Nature 451, 417–424 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park Y, Kim Y, Chang S, Chem. Rev. 117, 9247–9301 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Prusinowski AF, Twumasi RK, Wappes EA, Nagib DA, J. Am. Chem. Soc. 142, 5429–5438 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang H et al. , Angew. Chem. Int. Ed. 58, 13318–13322 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang H, Lambert TH, Angew. Chem. Int. Ed. 59, 658–662 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang H, Strater ZM, Lambert TH, J. Am. Chem. Soc. 142, 1698–1703 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moutet JC, Reverdy G, J. Chem. Soc. Chem. Comm 654–655 (1982). [Google Scholar]
- 9.Scheffold R, Orlinski R, J. Am. Chem. Soc. 105, 7200–7202 (1983). [Google Scholar]
- 10.Wang F, Stahl SS, Angew. Chem. Int. Ed. 58, 6385–6390 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yan H, Hou ZW, Xu HC, Angew. Chem. Int. Ed. 58, 4592–4595 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Zhang L et al. , Nat. Catal. 2, 366–373 (2019). [Google Scholar]
- 13.Zhang W, Carpenter KL, Lin S, Angew. Chem. Int. Ed. 59, 409–417 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ritter JJ, Minieri PP, J. Am. Chem. Soc. 70, 4045–4048 (1948). [DOI] [PubMed] [Google Scholar]
- 15.Ritter JJ, Kalish J, J. Am. Chem. Soc. 70, 4048–4050 (1948). [DOI] [PubMed] [Google Scholar]
- 16.Michaudel Q, Thevenet D, Baran PS, J. Am. Chem. Soc. 134, 2547–2550 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kiyokawa K, Takemoto K, Minakata S, Chem. Commun. 52, 13082–13085 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Duhamel T, Martinez MD, Sideri IK, Muniz K, ACS Catalysis 9, 7741–7745 (2019). [Google Scholar]
- 19.Krishnegowda G et al. , Bioorg. Med. Chem. 19, 6006–6014 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baskakis C et al. , J. Med. Chem. 51, 8027–8037 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hashimoto Y, Bioorg. Med. Chem. 10, 461–479 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Lund BW et al. , J. Med. Chem. 48, 7517–7519 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Hille UE, Zimmer C, Vock CA, Hartmann RW, ACS. Med. Chem. Lett. 2, 2–6 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Prices are from the following vendors: cumene (Sigma-Aldrich, 1 liter); 2-phenylpropane-1,2-diamine (SKU, 1 g); and 2-Amino-2-phenyl-propan-1-ol (SKU, 1 g).
- 25.Sato N et al. , Bioorg. Med. Chem. Lett. 19, 1670–1674 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Guandalini L et al. , Bioorg. Med. Chem. Lett. 25, 1700–1704 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Vidal Juan B et al. , PCT. Int. Appl 2003000694 (2003). [Google Scholar]
- 28.Fuerstner C et al. , PCT. Int. Appl 2012028644 (2012). [Google Scholar]
- 29.Becker JY, Byrd LR, Miller LL, So Y-H, J. Am. Chem. Soc. 97, 853–856 (1975). [Google Scholar]
- 30.Becker JY, Byrd LR, Miller LL, J. Am. Chem. Soc. 96, 4718–4719 (1974). [Google Scholar]
- 31.Metallinos C, Nerdinger S, Snieckus V. Org. Lett, 1, 1183–1186 (1999). [Google Scholar]
- 32.Roth HG, Romero NA, Nicewicz DA, Synlett 27, 714–723 (2016). [Google Scholar]
- 33.Lijser HJP, Arnold DR, J. Org. Chem. 62, 8432–8438 (1997). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available in the supplementary materials.