

Abstract
Engineering T cells and natural killer (NK) cells with anti-HIV chimeric antigen receptors (CAR) has emerged as a promising strategy to eradicate HIV-infected cells. However, current anti-HIV CARs are limited by targeting a single epitope of the HIV envelope glycoprotein gp160, which cannot counter the enormous diversity and mutability of viruses. Here, we report the development of a universal CAR-NK cell, which recognizes 2,4-dinitrophenyl (DNP) and can subsequently be redirected to target various epitopes of gp160 using DNP-conjugated antibodies as adaptor molecules. We show that this CAR-NK cell can recognize and kill mimic HIV-infected cell lines expressing subtypes B and C gp160. We additionally find that anti-gp160 antibodies targeting membrane-distal epitopes (including V1/V2, V3, and CD4bs) are more likely to activate universal CAR-NK cells against gp160+ target cells, compared with those targeting membrane-proximal epitopes located in the gp41 MPER. Finally, we confirm that HIV-infected primary human CD4+ T cells can be effectively killed using the same approach. Given that numerous anti-gp160 antibodies with different antigen specificities are readily available, this modular universal CAR-NK cell platform can potentially overcome HIV diversity, thus providing a promising strategy to eradicate HIV-infected cells.
Graphical Abstract
INTRODUCTION
Despite three decades of research, human immunodeficiency virus (HIV) infection has remained largely incurable, with more than 36 million people worldwide currently living with the virus. The limitation of current antiretroviral therapy (ART) is that it can effectively suppress HIV replication but not eradicate infected cells, which become latent HIV reservoirs that can induce viral rebound upon treatment interruptions. Developing a method to eradicate HIV-infected cells is thus of critical importance to finding a universal cure for HIV infection.1
A promising approach is to use chimeric antigen receptor (CAR)-engineered T cells and natural killer (NK) cells.2, 3 CARs are hybrid receptors consisting of an extracellular antigen-recognition domain, typically a single-chain variable fragment (scFv) antibody, and an intracellular signaling domain, such as a fusion of CD3ζ and CD28 or 4-1BB fragments that are responsible for T-cell activation. CAR expression can thus enable T cells and NK cells to recognize and kill target cells expressing the antigen of interest. This approach has been primarily used to treat cancer and achieved remarkable success; for example, the US FDA has approved the use of anti-CD19 CAR-T cells to treat highly refractory B-cell leukemia and lymphoma.4 It is known that HIV-infected cells express the viral envelope glycoprotein gp160 (a complex between gp120 and gp41) at the cell surface via viral budding.5 As such, there has been a growing interest in developing anti-gp160 CAR-modified immune cells to eradicate HIV-infected cells.6–10 For example, Ali et al. showed that CD8+ T cells modified with anti-gp160 CARs could induce lysis of HIV type 1 (HIV-1) infected cells.7 Additionally, Zhen et al. reported that hematopoietic stem cells engineered with anti-HIV CARs could differentiate into T cells and NK cells and suppress HIV replication in humanized mice.9
However, a unique challenge of targeting HIV-1 is that the virus is enormously diverse and highly mutable.11 HIV-1 is classified into four genetically distinct groups (M, N, O, and P), and the M group alone, which is the most common, contains at least nine subtypes (A–D, F-H, and J-K). The genetic variation of HIV strains can be up to 35% between subtypes and 20% within the same subtype, leading to the expression of extensively diversified gp160 proteins.12 Since current anti-HIV CARs are mostly designed based on individual broadly neutralizing antibodies (bNAbs) recognizing a single epitope of HIV-1 gp160, they cannot cover all HIV strains. Bi- and tri-specific anti-HIV CARs have recently been developed, which demonstrate enhanced anti-HIV breadth and potency.13–15 However, a combination of two or three epitopes is still not sufficient to cover all HIV variants, as indicated by previous studies on multispecific bNAbs.16, 17 Furthermore, HIV can rapidly mutate under pressure, which may lead to the selection or generation of escape mutants.18, 19 Therefore, the ability to further expand the epitope coverage of anti-HIV CARs is highly desirable.
Here, we report the development of a chemically-directed universal CAR-NK cell capable of recognizing various epitopes of gp160 from different HIV-1 subtypes. Instead of targeting gp160 directly, this CAR recognizes 2,4-dinitrophenyl (DNP), a small-molecule ligand, and its antigen specificity can subsequently be redirected to target different epitopes of HIV-1 gp160 variants using DNP-conjugated antibodies as adaptor molecules (Figure 1). As a proof of concept, we show that in the presence of adaptor molecules, anti-DNP CAR-NK cells can specifically target and kill mimic HIV-infected cell lines expressing gp160 of subtypes B and C. We additionally find that adaptor molecules targeting membrane-distal epitopes, which have higher accessibility, are more likely to activate anti-DNP CAR-NK cells against gp160+ target cells compared with those targeting membrane-proximal epitopes. Finally, we verify that the above approach is effective in killing HIV-infected primary human T cells. This modular universal CAR-NK cell platform, combined with numerous readily-available anti-gp160 antibodies, can potentially be used to overcome HIV-1 diversity and enhance the eradication of infected cells, which is essential to ultimately finding a universal cure for HIV.
Figure 1.
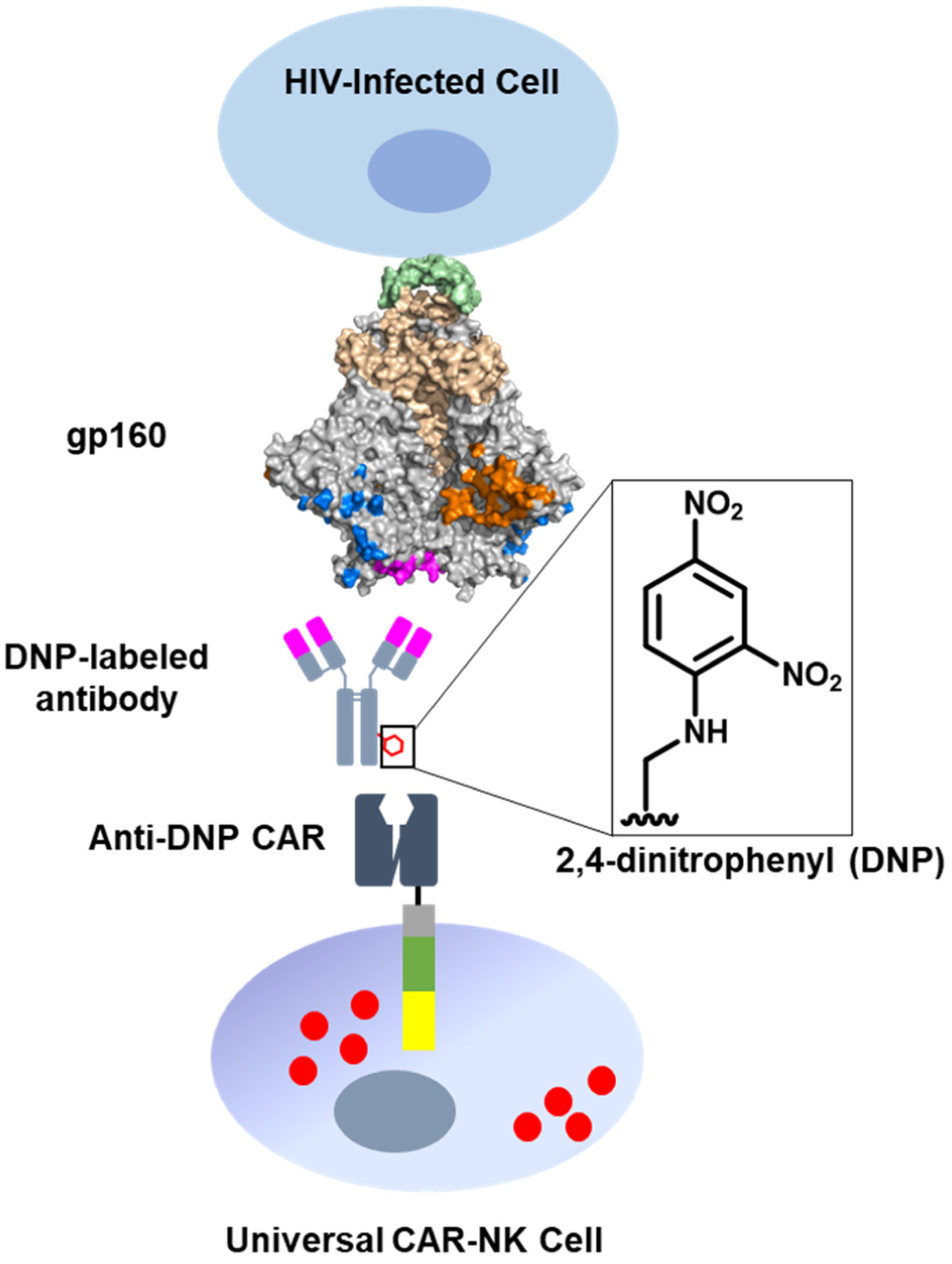
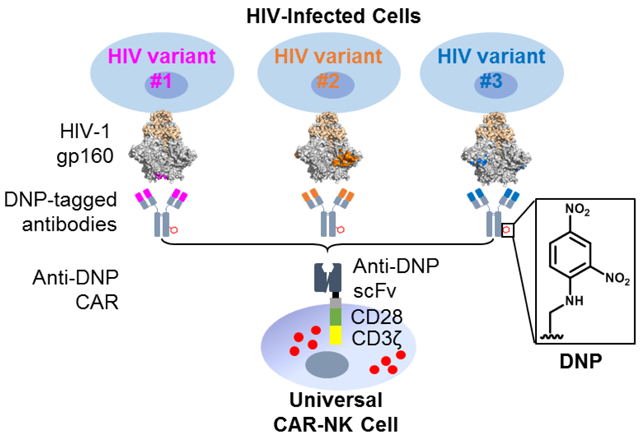
A schematic illustration of using a universal CAR-NK cell to target an HIV-infected cell. The NK cell is engineered to express an anti-DNP CAR, which can be redirected by DNP-labeled antibodies to target HIV-1 gp160 expressed on the infected-cell surface.
RESULTS
Genetic engineering of NK cells to express an anti-DNP CAR
We started by designing a DNP-mediated universal CAR. We chose DNP as the target of the CAR for three reasons. First, the sequences of many anti-DNP antibodies are readily available for CAR design. Second, DNP-conjugated anti-gp160 antibodies can be conveniently generated by chemical conjugation. Third, DNP is biocompatible as it has previously been used in the design of other therapeutic molecules, such as antibody-recruiting molecules.20, 21 As depicted in Figure 2A, our anti-DNP CAR consists of an N-terminal HA-tag for the detection of CAR expression, an extracellular anti-DNP scFv, a CD28 transmembrane domain (CD28TM), and intracellular CD28/CD3ζ signaling domains. The anti-DNP scFv was designed based on a high-affinity mouse anti-DNP antibody IgG2a-2, in the format of VL-(GGGGS)3-VH.22, 23 The CD28/CD3ζ signaling domains have been broadly used in the design of second-generation CARs.24 The gene of the anti-DNP CAR was assembled by overlap extension PCR and inserted to the pFUW lentiviral vector.25 This construct was then used to generate lentiviral particles in HEK293T cells.
Figure 2.
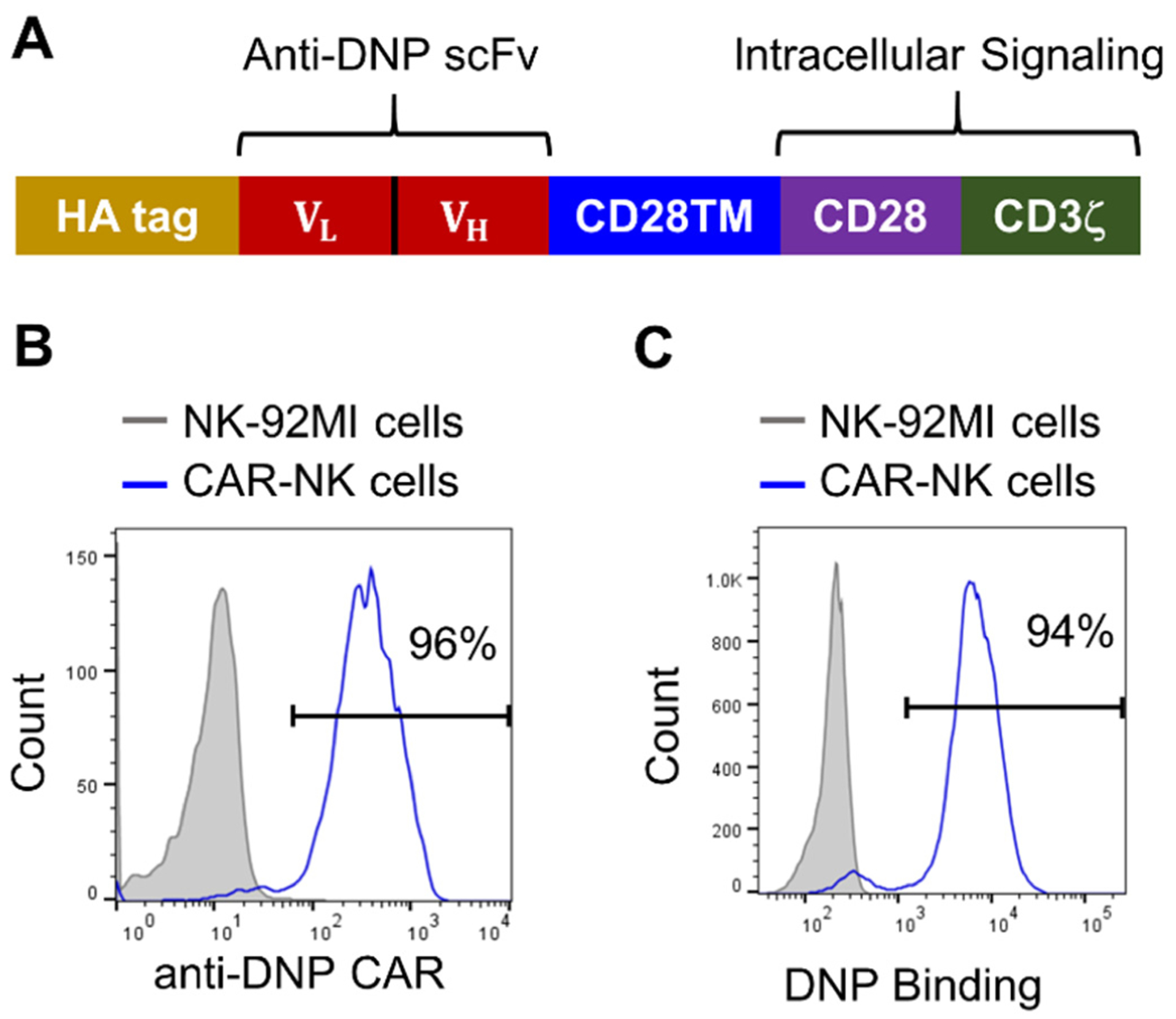
Generation and characterization of anti-DNP CAR-NK cells. (A) Schematic representation of the anti-DNP CAR construct. CD28TM: CD28 transmembrane domain. (B) Verification of anti-DNP CAR expression on NK-92MI cells after lentiviral transduction and magnetic cell sorting. CAR-NK cells were stained with an anti-HA-tag antibody and a PE-conjugated secondary antibody and then analyzed by flow cytometry. (C) Verification of the DNP-binding ability of CAR-NK cells by flow cytometry. Cells were stained with DNP-conjugated PE.
Next, we engineered NK-92 cells, a highly cytotoxic human NK cell line, to express anti-DNP CARs. Compared with allogeneic T cells, NK cells have the advantage of having little or no risk of inducing graft-versus-host diseases (GVHD). There is thus an increasing interest in engineering CAR-NK cells, including both primary cells and cell lines, as potential off-the-shelf cellular therapeutics.26–29 We chose NK-92 as the host cell because it can be continuously expanded to provide an unlimited number of homogenous cells, and CAR-engineered NK-92 cells have been safely utilized in clinical trials against cancer.30–32 Since the growth of NK-92 cells is dependent on interleukin 2 (IL-2), we used an IL-2-secreting NK-92-derived cell line (NK-92MI) as the host cell for CAR engineering.33 NK-92MI cells were transduced with lentiviral particles generated above to express anti-DNP CARs. The CAR expression was confirmed by staining with an anti-HA-tag antibody and a phycoerythrin (PE)-conjugated secondary antibody, followed by flow cytometric analysis. The initial lentiviral transduction led to approximately 3% CAR-expressing cells, which were subsequently enriched to 96% purity by magnetic cell sorting (Figure 2B). To verify whether the extracellular anti-DNP scFv was expressed in the functional conformation, CAR-NK cells were stained with a homemade DNP-conjugated fluorescent protein PE. Flow cytometric analysis showed that approximately 94% of cells were stained positively, confirming that they were able to recognize DNP (Figure 2C).
Redirection of anti-DNP CAR-NK cells to target subtype B gp160+ cells using a DNP-conjugated antibody
Because HIV-infected cells express the viral envelope glycoprotein gp160 at the cell surface, we developed a gp160-expressing cell line as a mimic of HIV-infected cells to be targeted by CAR-NK cells. The cell line was generated by transfecting HEK293 cells with the vector pConBgp160-opt, which encodes a full-length subtype B consensus gp160.34 The expression of gp160 was confirmed by cell surface staining using a human anti-gp120 antibody VRC01 and a PE-conjugated anti-human IgG polyclonal antibody. After transfection using Lipofectamine 2000 (a lipid-based transfection reagent), approximately 15–20% of cells expressed gp160, which were selected by G418 (Geneticin) and then further enriched to a purity of 99% by fluorescence-activated cell sorting (FACS) (Figure 3A).
Figure 3.
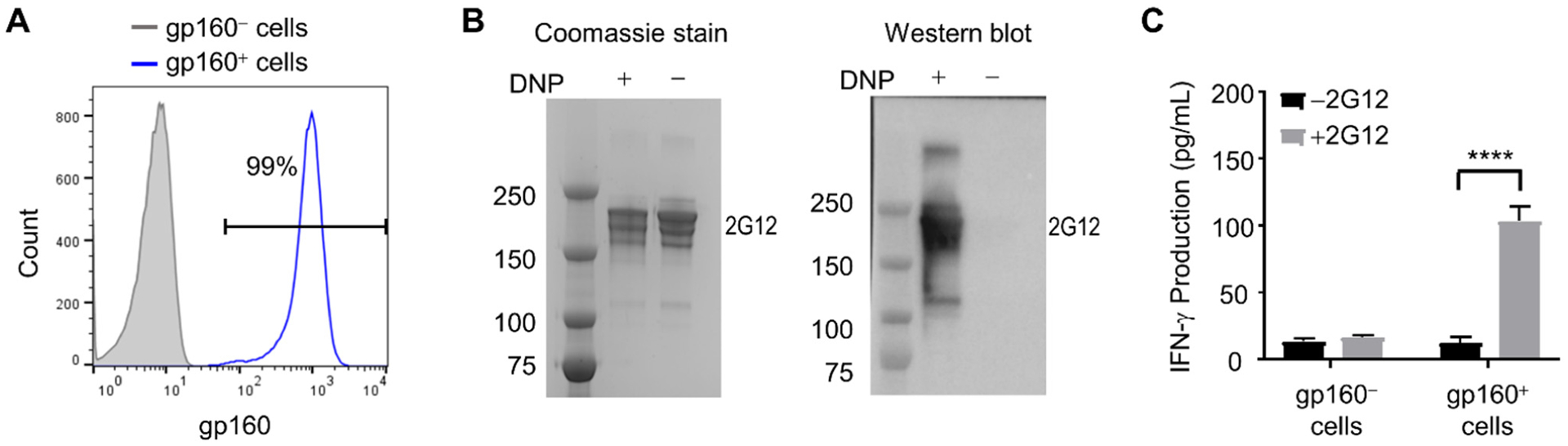
Redirection of anti-DNP CAR-NK cells to target subtype B gp160-expressing cells by the DNP-conjugated antibody 2G12. (A) Flow cytometry of the subtype B gp160-expressing HEK293 cell line. Expression of gp160 was verified by staining with the anti-gp160 antibody VRC01, followed by a PE-conjugated anti-human IgG antibody. (B) SDS-PAGE of the DNP-modified and unmodified antibody 2G12, followed by coomassie blue staining and western blot analysis using a goat polyclonal anti-DNP antibody. (C) IFN-γ production by anti-DNP CAR-NK cells in response to gp160-positive or negative target cells, with or without DNP-conjugated antibody 2G12 (10 nM). The concentrations of IFN-γ in the culture supernatant were determined by ELISA. Data are presented as the mean ± SD of triplicate samples. Statistical significance is calculated by two-way ANOVA and Tukey’s post-hoc analysis. **** p<0.0001.
Next, we generated a DNP-conjugated anti-gp160 antibody as the adaptor molecule. We chose 2G12, a bNAb that recognizes the high-mannose glycan epitope proximal to the variable region V3 of gp160.35, 36 DNP conjugation was conducted using N-(2,4-dinitrophenyl)-6-aminocaproic acid N-succinimidyl ester (DNP-NHS ester). The attachment of DNP to the antibody was verified by SDS-PAGE, followed by western blot analysis (Figure 3B). The conjugate contained approximately two DNP moieties per antibody, as determined by measuring the absorbance at 280 nm (for the antibody) and 360 nm (for DNP) (see Supporting Information for detailed methods).
To test the feasibility of using DNP-conjugated 2G12 to redirect anti-DNP CAR-NK cells against gp160+ cells, we used an in vitro co-culture assay. Specifically, anti-DNP CAR-NK cells were co-cultured overnight with subtype B gp160+ HEK293 cells at an effector-to-target (E:T) ratio of 1:1, with or without the DNP-conjugated antibody 2G12 (10 nM). In the negative control, CAR-NK cells were co-cultured with plain, gp160-negative HEK293 cells. The activation of CAR-NK cells was assessed by measuring the secretion of interferon-γ (IFN-γ) using an enzyme-linked immunosorbent assay (ELISA). As expected, gp160-negative cells could not activate anti-DNP CAR-NK cells, regardless of whether DNP-conjugated 2G12 was present or not. Subtype B gp160+ cells alone could not activate CAR-NK cells either, but they became stimulatory in the presence of DNP-conjugated 2G12, inducing CAR-NK cells to secrete a significant amount of IFN-γ (Figure 3C). Together, these results indicate that anti-DNP CAR-NK cells can be redirected to target gp160+ cells.
To determine the optimal concentration of adaptor molecules, we cultured CAR-NK cells and gp160+ target cells with DNP-conjugated 2G12 at different concentrations (0, 0.4, 2, 10, and 50 nM). We found that IFN-γ production reached a plateau when the 2G12 adaptor molecule was used at 2 nM or above (Supporting Information, Figure S1). Based on this result, we used 2G12 and other bNAb-derived adaptor molecules at 2 nM (unless otherwise specified) in all subsequent experiments.
Anti-gp160 antibodies targeting membrane-distal epitopes are more likely to activate CAR-NK cells compared with those targeting membrane-proximal epitopes
Numerous bNAbs recognizing different epitopes of HIV-1 gp160 have previously been identified.37 We asked whether the epitope location on gp160 could affect the ability of a bNAb to redirect anti-DNP CAR-NK cells against gp160+ cells. To address this question, we selected 12 well-characterized bNAbs recognizing four different regions of gp160, including: 1) PG9, PGT145, and PG16, which recognize glycan epitopes in the V1/V2 region of gp120; 2) 2G12, PGT128, and 10-1074, which recognize glycan epitopes in or proximal to the V3 region of gp120; 3) VRC01, b12, and 3BNC117, which recognize the CD4-binding site (CD4bs) on gp120; and 4) 10E8, 4E10, and 2F5, which recognize the gp41 membrane-proximal external region (MPER) (Figure 4A).35, 36 We conjugated these antibodies with DNP, and then compared their ability to redirect anti-DNP CAR-NK cells against subtype B gp160+ target cells using in vitro co-culture assays.
Figure 4.
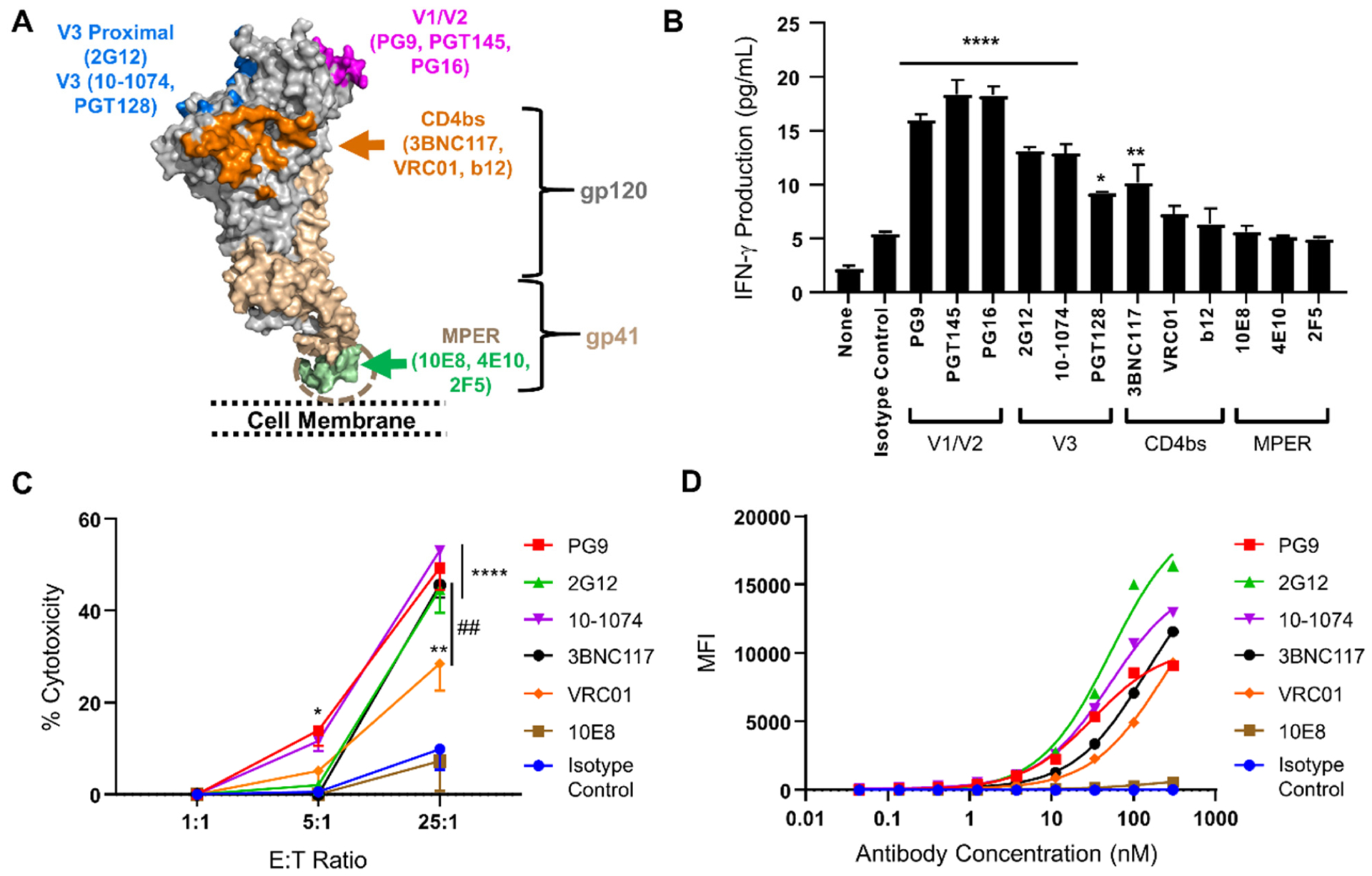
Epitope locations of anti-gp160 antibodies affect their abilities to redirect anti-DNP CAR-NK cells against subtype B gp160+ cells. (A) Illustration of the epitope locations of 12 anti-gp160 antibodies tested in this study. The image was generated based on the structures of HIV-1 BG505 SOSIP.664 Env trimer (PDB ID: 5T3Z) and MPER region (PDB ID: 6E8W).42, 43 (B) IFN-γ production by anti-DNP CAR-NK cells against subtype B gp160-expressing cells in the presence of different DNP-conjugated bNAbs (2 nM). IFN-γ concentrations were determined by ELISA. Data are presented as mean ± SD. Statistical significance is calculated by one-way ANOVA and Dunnett’s post-hoc test compared with the isotype control. * p<0.05, ** p<0.01, **** p<0.0001. (C) Cytotoxicity of anti-DNP CAR-NK cells against subtype B gp160+ cells at multiple E:T ratios and with different DNP-conjugated bNAbs (2 nM). Cells were stained with a viability dye and analyzed by flow cytometry. The percentage of cytotoxicity was calculated as [(A-B)/Ax100], in which A and B were the numbers of viable gp160+ cells after the cell co-culture was incubated without and with the addition of DNP-conjugated antibodies, respectively. Data are presented as the mean ± SD of triplicate samples. Statistical significance is calculated by two-way ANOVA and Tukey’s post-hoc analysis. * p<0.05,** p<0.01,**** p<0.0001 vs. the isotype control. ## p<0.01 comparing 3BNC117 to VRC01 at the 25:1 E:T ratio. (D) Analysis of the binding potencies of bNAbs against subtype B gp160+ cells. Cells were stained by bNAbs or the isotype control at a series of three-fold dilutions (from 0.045 nM to 300 nM) followed by a PE-conjugated anti-human IgG. The average fluorescence intensity per cell was measured by flow cytometry. Data are presented as the mean ± SD of triplicate samples. Nonlinear regression is used to fit saturation binding curves.
We first assessed the levels of CAR-NK cell activation using IFN-γ production assays. CAR-NK cells and gp160+ cells at a 1:1 ratio were incubated with DNP-conjugated bNAbs (2 nM). As negative controls, cells were either incubated with DNP-conjugated human IgG (isotype control) or without any antibodies. After four hours, the concentration of IFN-γ in the culture supernatant was measured by ELISA. We found that all three V1/V2-targeting antibodies—PG9, PGT145, and PG16—were able to strongly activate CAR-NK cells against gp160+ target cells, leading to a three- to four-fold increase in IFN-γ production as compared with cells incubated with the isotype control (Figure 4B). The three V3-targeting antibodies—2G12, PGT128, and 10-1074—also significantly activated CAR-NK cells, leading to a two- to threefold increase in IFN-γ production. Among the three CD4bs antibodies, 3BNC117 activated CAR-NK cells to produce IFN-γ about two-fold more than the isotype control, but VRC01 and b12 did not significantly activate CAR-NK cells. Strikingly, none of the three MPER-binding antibodies—10E8, 4E10, and 2F5—could activate CAR-NK cells against subtype B gp160+ target cells.
Next, we assessed CAR-NK cell-mediated killing of gp160+ cells using a flow cytometry-based cytotoxicity assay.38 For ease of detection, subtype B gp160+ cells were pre-labeled with fluorescein using carboxyfluorescein succinimidyl ester (CFSE). These cells were mixed with non-labeled gp160-negative cells at a 1:1 ratio, and then co-cultured with anti-DNP CAR-NK cells at varying E:T ratios (1:1, 5:1, and 25:1), with or without DNP-conjugated bNAbs (2 nM). Eight hours later, cells were stained with a viability dye and subjected to flow cytometric analysis. The levels of cytotoxicity were determined based on the percentage of gp160+ cells killed by CAR-NK cells. We selected six bNAbs to be tested in the cytotoxicity assay, including 1) PG9, 2G12, 10-1074, and 3BNC117, which were effective in the IFN-γ assay, and 2) VRC01 and 10E8, which were ineffective in the IFN-γ assay. We found that the first four bNAbs were also more potent in the cytotoxicity assay, activating CAR-NK cells to kill approximately 50% of gp160+ cells after an eight-hour incubation at the 25:1 E:T ratio (Figure 4C). VRC01 was less potent, inducing CAR-NK cells to kill 28.5% of gp160+ cells. The MPER-targeting bNAb 10E8, however, did not induce any significant killing as compared with the isotype control, which was consistent with its lack of activity in the IFN-γ assay. At lower E:T ratios, the cytotoxicity of CAR-NK cells was significantly reduced. Indeed, after an eight-hour incubation, only PG9 and 10-1074 were able to activate anti-DNP CAR-NK cells to kill approximately 10–15% of gp160+ cells at the 5:1 E:T ratio, and no cytolysis was detectable at the 1:1 E:T ratio. Importantly, CAR-NK cells did not kill gp160-negative HEK293 cells under any conditions, indicating that the targeting was gp160-specific (Supporting Information, Figure S2A).
The above results suggest that the V1/V2, V3, and CD4bs-targeting antibodies have a greater likelihood of activating anti-DNP CAR-NK cells against gp160+ target cells than do MPER-targeting antibodies. A possible reason is that MPER-targeting bNAbs have poor epitope accessibility due to local steric hindrance and thus cannot efficiently bind to gp160 on the target cell surface. To compare the binding potency of bNAbs, we conducted a cell surface staining assay. Specifically, subtype B gp160+ cells were stained with bNAbs at a three-fold serial dilution (from 300 to 0.045 nM), followed with a PE-conjugated secondary antibody against human IgG. Cells were then subjected to flow cytometric analysis, and the average fluorescence intensity per cell was plotted against the concentration of bNAb. As shown in Figure 4D, the V1/V2 and V3-targeting antibodies PG9, 10-1074, and 2G12 strongly bound to subtype B gp160. Interestingly, the saturated staining with PG9 was weaker than that of 2G12 and 10-1074. This is probably because gp160 proteins are expressed as trimers on the cell surface, and the three epitopes of PG9 within the same trimer are located close to each other, preventing the simultaneous binding of three PG9 antibodies.39 The two CD4bs antibodies 3BNC117 and VRC01 also positively bound to gp160+ cells but did not reach binding saturation within the tested concentration range (0.045–300 nM). However, the MPER-targeting antibody 10E8, even at 300 nM, could not stain gp160+ cells to any significant extent as compared with the isotype control, suggesting that it has poor epitope accessibility. Overall, we found that the gp160-binding potency of antibody was roughly consistent with its ability to redirect anti-DNP CAR-NK cells against gp160+ cells (Figures 4B–4D). It is possible that other factors, such as the binding orientation of CARs over gp160 and the intermembrane distance between effector and target cells, can also affect the potency of CAR-NK cells,40, 41 which remain to be determined in future studies.
Redirection of anti-DNP CAR-NK cells to target subtype C gp160+ cells
To evaluate the generality of the universal CAR-NK cell approach, we additionally used it to target HIV-1 subtype C gp160. HEK293 cells were modified to express a subtype C consensus gp160 protein using the vector pConCgp160-opt as previously reported.34, 44 Subtype C gp160+ cells were then incubated with anti-DNP CAR-NK cells in the presence of different DNP-conjugated bNAbs (2 nM). Similar to our findings for subtype B gp160+ cells, we found that a majority of the nine V1/V2, V3, and CD4bs-targeting antibodies were able to strongly activate CAR-NK cells to produce IFN-γ in response to subtype C gp160+ cells, while none of the three MPER-targeting antibodies could significantly activate CAR-NK cells compared with the isotype control (Figure 5A). Surprisingly, 2G12, one of the V3-targeting antibodies that were highly effective against subtype B gp160 (Figure 4B), completely lost its ability to activate CAR-NK cells to produce IFN-γ against subtype C gp160 (Figure 5A). The cytotoxicity assay also showed that 2G12 could not redirect CAR-NK cells to kill subtype C gp160+ cells, neither could the MPER-targeting antibody 10E8 and the isotype control (Figure 5B, and Supporting Information, Figure S2B). For comparison, PG9 and 10-1074 redirected CAR-NK cells to kill about 30% of subtype C gp160+ cells after an eight-hour incubation at a 25:1 E:T ratio, and 3BNC117 and VRC01 led to lysis of about 20% of target cells. We then used these antibodies to stain subtype C gp160+ cells. The result showed that PG9 and 10-1074 had the highest binding potency, followed by 3BNC117 and VRC01, while 2G12, 10E8, and the isotype control exhibited little or no binding ability within the tested concentration range (0.045–300 nM) (Figure 5C).
Figure 5.
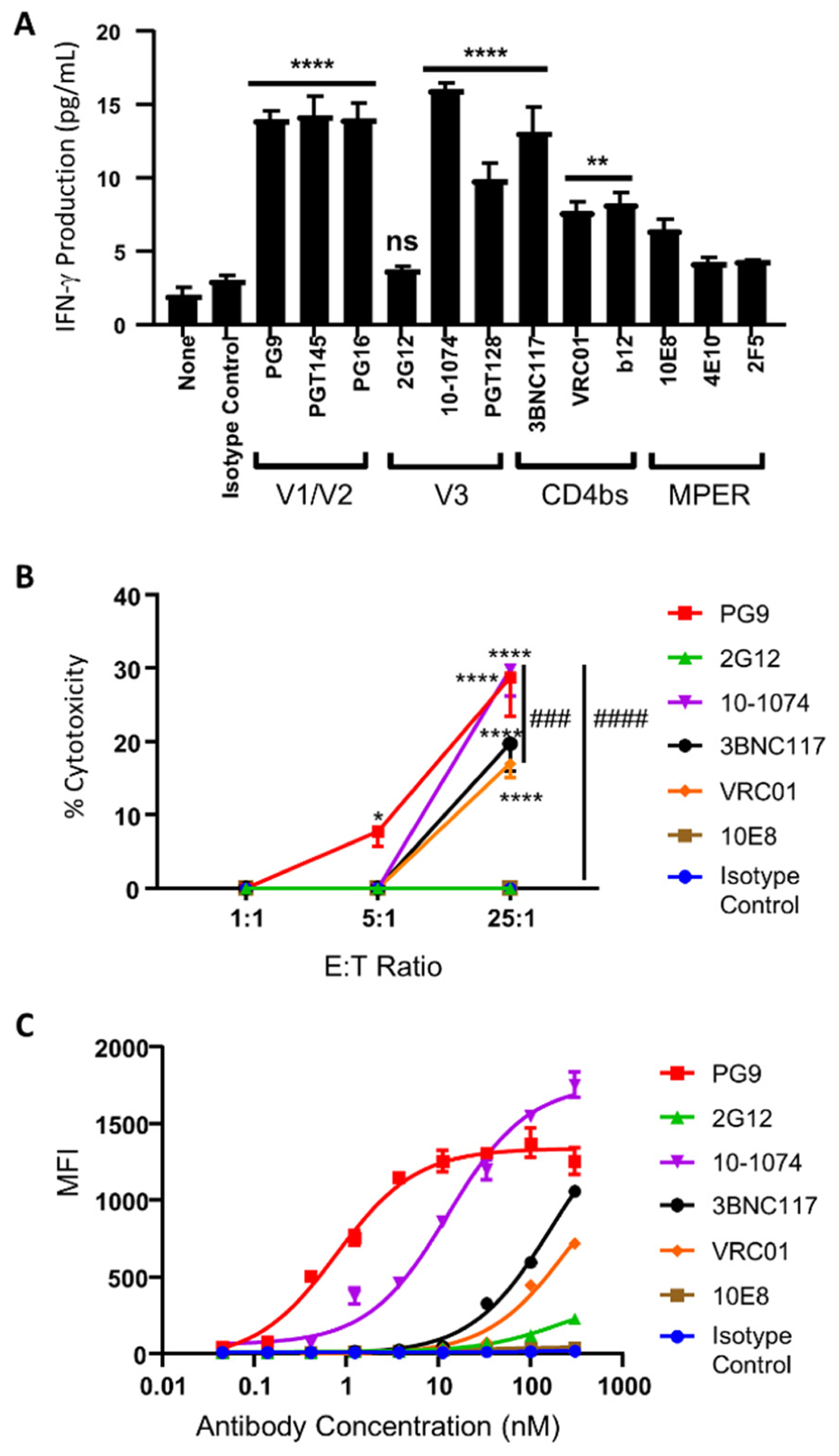
Targeting subtype C gp160+ cells by universal CAR-NK cells. (A) IFN-γ production by anti-DNP CAR-NK cells against subtype C gp160+ cells in the presence of DNP-conjugated bNAbs (2 nM). The concentrations of IFN-γ were determined by ELISA. Data are presented as the mean ± SD of triplicate samples. Statistical significance is calculated by one-way ANOVA and Dunnett’s post-hoc test compared with the isotype control. ns: not significant, ** p<0.01, **** p<0.0001. (B) Cytotoxicity of anti-DNP CAR-NK cells against subtype C gp160+ cells at multiple E:T ratios and with different DNP-conjugated bNAbs (2 nM). Data are presented as the mean ± SD of triplicate samples. Statistical significance is calculated by two-way ANOVA and Tukey’s post-hoc analysis. * p<0.05, **** p<0.0001 vs. the isotype control. ### p<0.001, #### p<0.0001 comparing % cytotoxicity of 10-1074 to VRC01 and 2G12, respectively, at the 25:1 E:T ratio. (C) Flow cytometric analysis of the binding potency of each bNAb against subtype C gp160-expressing cells. Data are presented as the mean ± SD of triplicate samples.
Therefore, as with the experiments on subtype B gp160, the relative binding potency of antibodies for subtype C gp160 was largely consistent with their ability to induce IFN-γ production and cytotoxicity of anti-DNP CAR-NK cells in response to subtype C gp160+ cells. Importantly, 2G12 demonstrates a dramatically different ability to activate CAR-NK cells against subtypes B and C gp160+ target cells, highlighting the advantage of using a universal CAR whose antigen specificity is not limited by a single antibody. We also noted that the universal CAR-NK cells were significantly more cytotoxic to subtype B than to subtype C gp160+ cells (Figures 4C and 5B). For example, with the 10-1074-derived adaptor molecule, the universal CAR-NK cells were able to kill about 53.1% of subtype B gp160+ cells but only 29.8% of subtype C gp160+ cells. Likewise, the 3BNC117-derived adaptor molecule induced CAR-NK cells to kill 45.5% of subtype B gp160+ cells but only 19.7% of subtype C gp160+ cells. The preferential targeting of subtype B over subtype C gp160 is likely because most bNAbs have been developed against subtype B HIV-1 variants.45 To enhance the cytotoxicity against subtype C gp160+ cells, it would be necessary to use other bNAbs that are more specialized for subtype C gp160.
Next, we examined whether different bNAbs could be used in combination to further enhance the efficacy of universal CAR-NK cells against a mixture of different subtypes of gp160-expressing cells. To this end, CAR-NK cells and subtypes B and C gp160+ cells were co-cultured at a 25:1:1 ratio, in the presence of either 10-1074 (5 nM), 3BNC117 (5 nM), or both. After an 8-hour incubation at 37°C, cells were stained with a viability dye (7-AAD), and the numbers of viable gp160+ cells were measured by flow cytometry. The percentages of gp160+ cells killed were found as follows: 41% with 10-1074, 31% with 3BNC117, and 60% with both of them (Supporting Information, Figure S3). Hence, a combination of 10-1074 and 3BNC117 was more potent than either alone in eliciting CAR-NK cell cytotoxicity against subtypes B and C gp160+ cells. This result highlights the potentiality of using universal CAR-NK cells supplemented with a cocktail of bNAbs to target diverse gp160+ cells, which warrants further investigation.
Comparison of universal and conventional CAR-NK cells
The major advantage of the universal CAR-NK cells developed above is that they can be coupled with various adaptor molecules to expand epitope coverage. However, it is unclear whether the potency of universal CAR-NK cells is comparable to that of conventional bNAb-based single-specificity CAR-NK cells. To address this question, we designed a 2G12-based anti-gp160 CAR (2G12 CAR) and generated CAR-NK cells using the same approach as described above. We then compared the cytotoxicity of universal CAR-NK cells (in combination with DNP-conjugated 2G12) and 2G12 CAR-NK cells against subtype B gp160+ target cells. We found that at an E:T ratio of 25:1, the universal and the conventional CAR-NK cells killed about 50% and 60% of gp160+ cells, respectively (Figure 6). At the 5:1 and 1:1 E:T ratios, neither of them was effective. These results suggest that despite having a greater epitope breadth, the universal CAR-NK cells are only slightly less potent than the conventional single-specificity CAR-NK cells.
Figure 6.
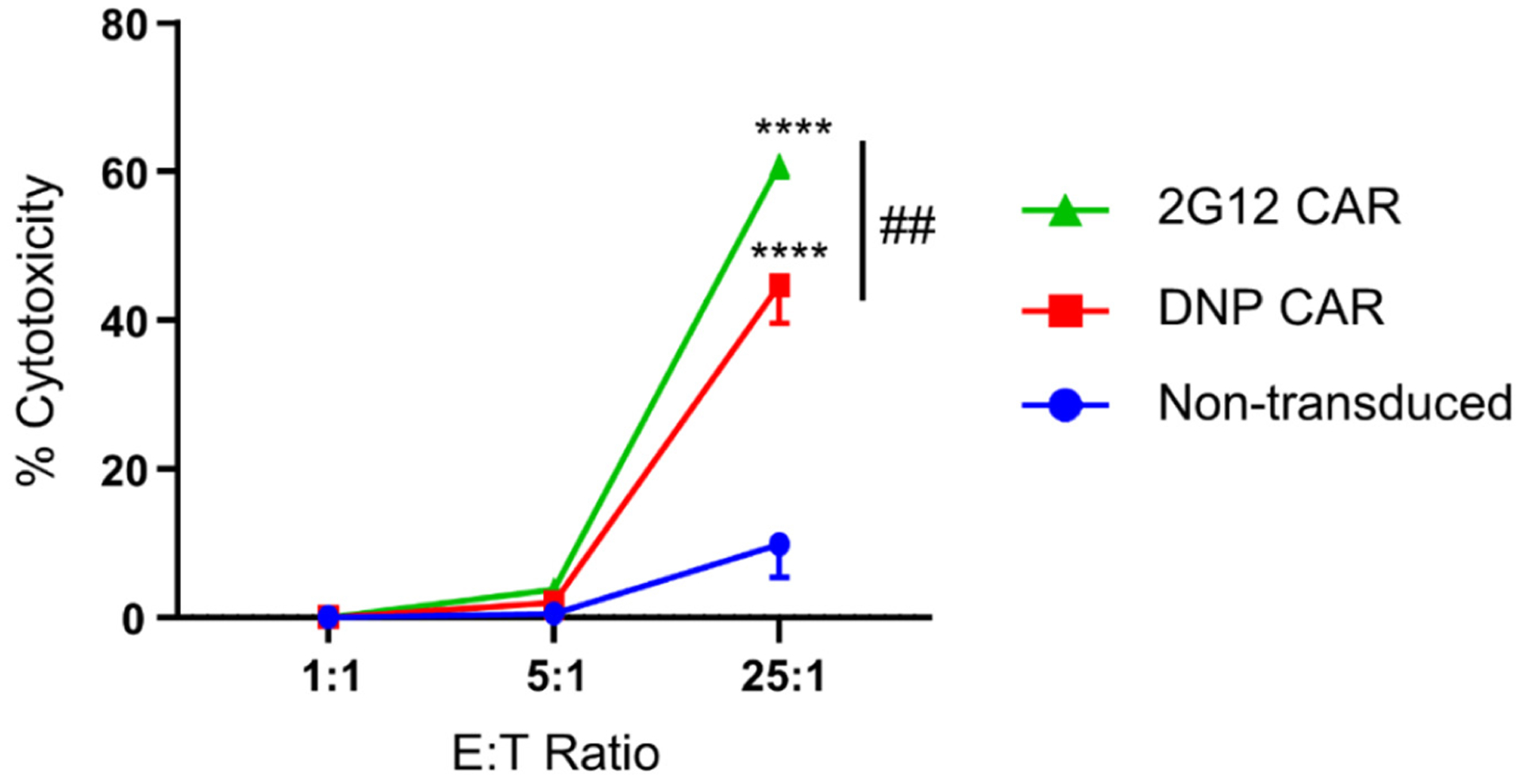
Comparison of the universal and the conventional 2G12-based CAR-NK cells in killing subtype B gp160-expressing cells. Cells were co-cultured eight hours at 1:1, 5:1, or 25:1 E:T ratios. The cytotoxicity was determined based on the percentage of gp160-positive cells killed by CAR-NK cells. Data are presented as the mean ± SD of triplicate samples. Statistical significance is calculated by two-way ANOVA and Tukey’s post-hoc analysis. **** p<0.0001 vs. non-transduced NK cells. ## p<0.01 comparing the percentages of cytotoxicity of 2G12-CAR-NK cells to that of DNP-CAR-NK cells at the 25:1 E:T ratio.
Killing of HIV-infected primary human CD4+ T cells
Next, we examined whether universal CAR-NK cells could kill HIV-infected T cells. To this end, human primary CD4+ T cells from healthy donors were stimulated with anti-CD3/CD28 beads for two days and were infected with HIV-1 NL4-3 (a commonly used X4-tropic subtype B HIV-1 virus) for 10 days. Infected CD4+ T cells were pre-stained with CellTrace Far Red, incubated with 10 nM of DNP-conjugated bNAbs for 20 minutes, and then co-cultured with anti-DNP CAR-NK cells at a 20:1 E:T ratio for 16 hours. Afterward, cells were stained with Zombie Violet (a fixable viability dye), and live HIV-infected cells were identified by intracellular staining for HIV-1 Gag. We observed an effective killing of HIV Gag-expressing CD4+ T cells by CAR-NK cells with various DNP-modified bNAbs as compared with the no-antibody control (Figure 7). In particular, we found that PG9, PG16, 2G12, 10-1074, and 3BNC117 (which target membrane-distal epitopes) were more effective than 10E8 (which targets a membrane-proximal epitope), similar to what we found with the gp160+ cell line (Figure 4B).
Figure 7.
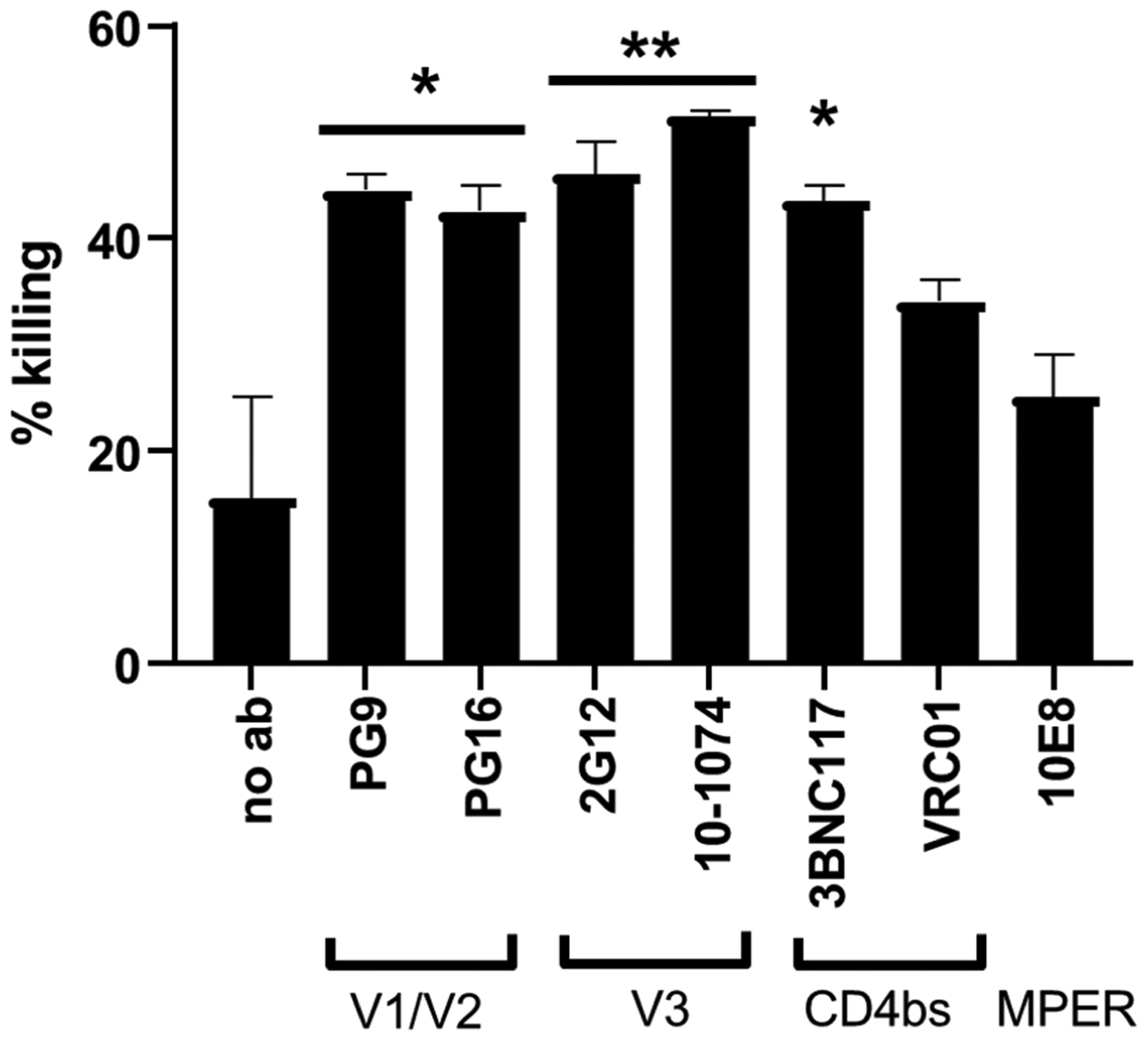
Universal CAR-NK cells mediate an effective killing of HIV-infected CD4+ T cells. CAR-NK cells and HIV-infected CD4+ T cells were co-cultured at a 20:1 E:T ratio with or without DNP-modified bNAbs (10 nM). After overnight incubation, the cytotoxicity of CAR-NK cells was assessed by flow cytometry. % killing was calculated as [(A-B)/Ax100], in which A and B were the percentages of live Gag-expressing CD4+ T cells without and with the treatment of CAR-NK cells, respectively. Data are presented as the mean ± SD of duplicate samples. Statistical significance is calculated by a one-way ANOVA and Tukey’s post-hoc analysis. * p<0.05, ** p<0.01, vs. the negative control in which no antibody was added.
Redirection of anti-DNP CAR-NK cells to target malignant B cells
HIV-infected patients have an approximately 60–200 fold higher risk of developing non-Hodgkin’s lymphoma (NHL) when compared with the general population, presumably due to their compromised immune system.46 We speculated that the universal CAR-NK cell could be used to eradicate HIV-associated lymphoma as well. As a proof of concept, we conjugated DNP to the antibody FMC63 recognizing CD19, a biomarker overexpressed in most B-cell malignancies. We then used the conjugate to redirect anti-DNP CAR-NK cells against REH, a malignant B cell line. The flow cytometry-based cytotoxicity assay showed that at an E:T ratio of 25:1, approximately 41% of REH cells remained alive after overnight incubation, that is, 59% of cells were killed by CAR-NK cells (Supporting Information, Figure S4). Our universal DNP-directed CAR-NK cell platform hence provides a potential solution to eradicate both infected and malignant cells.
The universal CAR-NK cell-mediated killing of gp160+ cells is not due to ADCC
It should be noted that primary NK cells express CD16 (FcγRIII) to recognize antibodies and kill antibody-coated cells using the mechanism of antibody-dependent cellular cytotoxicity (ADCC). Since NK-92 cells do not express CD16,47, 48 the CAR-NK cell-mediated killing of gp160+ cells observed above is not due to ADCC. This notion is consistent with our finding that DNP-conjugated PG9 can effectively activate anti-DNP CAR-NK to target subtype B gp160+ cells, but the unconjugated PG9 cannot (Supporting Information, Figure S5).
Engineering anti-DNP CAR-T cells to target gp160+ cells
Finally, we examined whether the same technique could be used to expand the specificity of CAR-T cells. Primary human T cells were activated with anti-CD3/CD28 beads and recombinant IL-2 for three days and then transduced with the lentiviral vector encoding anti-DNP CAR. Flow cytometric analysis showed that about 36% of T cells expressed anti-DNP CARs. CAR-T cells were first blocked with an anti-human CD4 antibody (clone: RPA-T4) to prevent syncytia formation between CD4+ T cells and gp160+ cells,49 and then co-cultured with subtype B gp160+ HEK293 cells at a 1:1 E:T ratio in the presence of 10 nM of DNP-modified bNAbs or isotype control. After a 48-hour incubation at 37°C, cell viability was assessed by flow cytometry. The result showed that DNP-conjugated 2G12 and 10-1074 enabled anti-DNP CAR-T cells to kill about 34% and 29% of gp160+ cells, respectively (Supporting Information, Figure S6). It is thus clear that the technique described here applies to both T cells and NK cells.
DISCUSSION
In this proof-of-concept study, we developed a novel approach that can potentially enable CAR-NK cells to target and eradicate multiple variants of HIV-infected cells. Our strategy involves generating an anti-DNP CAR-NK cell that can be redirected to target various epitopes of HIV-1 gp160 using DNP-conjugated antibodies as adaptor molecules. We showed that the universal CAR-NK cells could be redirected to recognize and kill both subtypes B and C gp160+ cell lines and HIV-infected human primary T cells. Given that numerous anti-gp160 antibodies with different specificities are readily available, this approach can significantly expand the epitope coverage of anti-HIV CAR-NK cells, thus providing a promising strategy to overcome viral diversity.
Importantly, we find that bNAbs targeting membrane-distal regions of gp160 (including V1/V2, V3, and CD4bs) have a much higher likelihood of activating universal CAR-NK cells against gp160+ cells than those targeting membrane-proximal epitopes located in the gp41 MPER. Our results are in contrast to previous studies on anti-cancer CAR-T cells and bispecific T cell engagers (BiTE), which report that targeting membrane-proximal epitopes confers more effective T-cell activation than targeting membrane-distal epitopes.50–53 For example, CAR-T cells have been actively developed to target different regions of CD22, a B-cell leukemia antigen containing seven extracellular immunoglobulin (Ig)-like domains (numbered 1–7, from distal to proximal). James et al. reported that targeting the most distal Ig domains 1 and 2 (by HD39-derived CAR) was less effective than targeting Ig domain 3 (by RFB4-derived CAR),51 and Haso et al. further found that the latter was less effective than targeting the most proximal Ig domains 5–7 (by m971-derived CAR).53
The discrepancy between our findings and others may stem from the poor accessibility of membrane-proximal epitopes in our study. These epitopes, located in the HIV-1 gp41 MPER, are incredibly close to the cell membrane, e.g., within 10 Å for the 10E8 epitope (Supporting Information, Figure S7). As such, they may become hardly accessible due to local steric hindrance, consistent with our observation that there is little or no staining of gp160+ cells by 10E8 (Figures 4D and 5C). The membrane-proximal epitopes in previous cancer studies are not nearly as proximal as HIV-1 MPER epitopes. For example, the m971 and RFB4 epitopes of CD22 are up to 114 Å and 152–190 Å from the cell membrane, respectively. They are similar to—or even farther away than—the membrane-distal epitopes of HIV-1 gp160, which are within 110 Å from the cell membrane (Figure S7). There is thus a significant variation in actual membrane proximity among different membrane-proximal epitopes, which may explain the contradictory results from our study and previous studies. Taken together, we speculate that there is an optimal distance between the epitope and the cell membrane to activate CAR-T or CAR-NK cells. If the epitope is too membrane-distal, it will allow the protein phosphatase CD45, which has a large extracellular domain, to diffuse into the immunological synapse to inhibit lymphocyte signaling, as suggested by the kinetic segregation model.54 However, if the epitope is too membrane-proximal, its accessibility will deteriorate, countering the positive effects of CD45 exclusion on lymphocyte activation. The optimal epitope distance may also be adjustable by using CARs with different hinge lengths.55
Another possible cause of the discrepancy is that many HIV-1 MPER-targeting bNAbs preferably bind to the fusion-intermediate state over the pre-fusion conformation of gp41.56–58 In this study, we expect that gp160 is in the pre-fusion state because it lacks binding to its receptor CD4 and co-receptor CCR5 or CXCR4.59 This factor may have contributed to the reduced binding efficacy between MPER-targeting bNAbs and gp160+ cells, which in turn weakens the cytotoxicity of CAR-NK cells.
While universal CAR-T cells mediated by biotin, fluorescein isothiocyanate (FITC), peptide neoepitopes, and leucine zipper have previously been developed to target cancer,60–65 this study is the first to engineer NK cells with a universal CAR and to apply it for targeting HIV-1 epitopes. It is important to note that allogeneic T cells from healthy donors carry a high risk of inducing GVHD due to the expression of highly diverse T cell receptors, while allogeneic NK cells have little or no such risk. Therefore, the universal CAR-NK cell can potentially be developed as an off-the-shelf cellular therapeutic for all patients.
Although our universal CAR-NK cell approach is somewhat similar to ADCC, it has several important potential advantages. First, the universal CAR is more versatile because it is compatible with all types of antibodies, including IgA which cannot effectively induce NK cell-mediated ADCC. Second, it is likely more specific and safer, because anti-DNP CAR-NK cell-mediated killing is strictly dependent on DNP-conjugated antibodies and thus will not be induced by serum antibodies. Third, the potency of universal CAR-NK cells can be further enhanced by incorporating multiple signaling domains. Indeed, it was recently reported that CARs containing the transmembrane and intracellular signaling domains of NKG2D, 2B4, DAP10, and CD3ζ could enable NK cells to more effectively kill ovarian cancer cells, as compared to a CAR containing the CD16 intracellular signaling domain.28 Finally, the universal CAR-NK cell approach and ADCC are not mutually exclusive because the DNP-conjugated bNAbs can still recruit primary NK cells to respond to gp160+ cells.
A potential limitation of the DNP-directed universal CAR-NK cell approach is that up to 1% of naturally-generated antibodies in humans can recognize DNP,66, 67 which can compete with the DNP-conjugated adaptor molecules in binding and stimulating anti-DNP CAR-NK cells. This can potentially be addressed by designing higher-affinity anti-DNP CARs to strengthen universal CAR-NK cell recognition of DNP-conjugated adaptor molecules. It is also necessary to examine the potential toxicity of DNP-conjugated antibodies, which will be investigated in animal studies in the future.
Future research will focus on the continued enhancement of the potency of the universal CAR-NK cells. One promising approach is to use NK cell-specific signaling domains, such as NKG2D, 2B4, and CD16, which have led to enhanced anti-cancer CAR-NK cells.28 Another approach is to optimize adaptor molecules, for example, by incorporating DNP into anti-gp160 antibodies at an optimal site.68, 69 Future research should also validate the optimal universal CAR-NK cells using human primary CD4+ T cells infected with various HIV-1 strains and humanized mouse models of HIV-1 infection.
CONCLUSION
In summary, our proof-of-concept study demonstrates that a single universal CAR-NK cell can be redirected to target various epitopes of HIV-1 envelope glycoprotein gp160. Given that numerous anti-gp160 antibodies are readily available, this modular approach can significantly expand the epitope coverage of CAR-NK cells, making it possible to overcome the extraordinary diversity and mutability of HIV-1. Recently, Herzig et al. reported the development of a universal CAR-T cell platform, which is mediated by an orthogonal receptor-ligand pair and demonstrates a greater breadth and control for attacking the latent HIV reservoir.70 Their work thus robustly corroborates the feasibility of using a universal CAR to target diverse HIV-1 variants. However, as noted in their paper, an important hurdle of using universal CAR-T cells in the clinic is the high cost to isolate, expand, and transduce autologous T cells from each patient, and a potential solution is to create allogeneic T cells that do not induce GVHD, e.g., by disabling T cell receptors. Unlike T cells, allogeneic NK cells have little or no risk of inducing GVHD. Therefore, an attractive potential of our universal CAR-NK cell platform is that it can potentially be developed as a low-cost, off-the-shelf cellular therapeutic for eradicating HIV infection in many patients.
METHODS
Construction of the anti-DNP CAR lentiviral vector.
The human CD8 α signal peptide was used to translocate the anti-DNP CAR to the cellular membrane. The gene fragment consisting of the CD8 α signal peptide, HA-tag, and anti-DNP scFv was synthesized by Integrated DNA Technologies (IDT, San Diego, CA). It was then amplified by PCR using primers CD8 signal-BamH I-F and αDNP-R. The gene fragment consisting of the CD8 α chain hinge domain, the CD28 transmembrane domain, the CD28 intracellular domain, and the CD3ζ intracellular domain was amplified from a parental lentiviral plasmid pFUW-anti-CD19-CD28-CD3ζ encoding an anti-CD19 CAR (a gift from Prof. Pin Wang at USC), using primers CD8 hinge-F and CD28-CD3ζ-EcoR I-R. The two gene fragments were linked together by overlap PCR, digested with BamH I and EcoR I, and ligated into the pFUW linear vector. The ligation product was transformed into DH5α cells. Single colonies grown on LB-agar plates containing 100 μg/ml ampicillin were picked and grown in the LB liquid medium. The plasmid was purified, sequence verified, and then used to generate lentiviral particles by calcium-phosphate transfection of HEK293T cells.
Generation of anti-DNP CAR-NK cells.
To generate the lentivirus, HEK293T cells (~600,000 cells/mL) were plated in a 100 mm dish and transfected the next day with 13 μg of the pFUW-aDNP CAR and 6 μg of lentiviral packaging plasmids (pVSVG, pRRE, and pREV) using calcium phosphate transfection. After 4 hours of incubation at 37°C, the supernatant was changed to fresh complete DMEM media with 10% FBS and antibiotics and incubated at 37°C for lentivirus production. After three days of incubation, the lentivirus supernatant was filtered using a 0.45 μm Supor membrane filter and concentrated ~10X using an Amicron 100kDa MWCO concentrator. Anti-DNP CAR-NK cells were generated by lentiviral transduction using the retronectin-based technique. Non-treated 24-well plates were coated with 10μg/mL retronectin at room temperature for 2 hours. Plates were then blocked using a 1X phosphate buffered saline (PBS) supplemented with 2% bovine serum albumin (BSA) solution at room temperature for 30 minutes. After blocking, the plate was washed two times using sterile 1X PBS before lentiviral transduction. To initiate the transduction, about ~400,000 cells/mL of NK-92MI cells in RPMI complete culture medium supplemented with 20% FBS were plated per well and mixed with an equal volume of CAR lentivirus in the retronectin-coated plate. Then the plate was centrifuged at 1,200 g for 90 minutes followed by overnight incubation at 37°C. The next day, the transduced NK-92MI cells were washed to remove the excess lentivirus and cultured in RPMI complete culture medium with 20% FBS. After 48 hours, the transduction efficiency was verified by flow cytometry using the LSRII (BD Biosciences, San Jose, CA). CAR-NK cells were enriched two times using the MagniSort streptavidin positive selection beads. The 2G12 CAR-NK cells were also generated using this method.
Generation of gp160-expressing cell lines.
HEK293 cells were transfected with the plasmid pConBgp160-opt (for subtype B gp160) or pConCgp160-opt (for subtype C gp160) (45 μg/each) and Lipofectamine 2000 (144 μL) in a 15 cm cell culture dish. After incubation at 37°C for 6 hours, the excess Lipofectamine and plasmid were removed by media changes. After an additional two-day incubation, G418 (400 μg/mL for Subtype B, 500 μg/mL for Subtype C) was added to the cell culture to select for cells expressing these genes. All cell colonies after selection were collected. A small aliquot was stained with VRC01 and PE-conjugated secondary anti-human IgG polyclonal antibodies and analyzed by flow cytometry to confirm gp160 expression. The rest of the cells were further purified by FACS using a BD FACSAria II sorter. After further growth, cells were stained and analyzed by flow cytometry using the LSRII flow cytometer (BD Biosciences, San Jose, CA) to further verify the purity and the expression efficiency of gp160.
IFN-γ production assays.
The gp160-expressing HEK293 cells were incubated with DNP-conjugated bNAb or isotype control at the specified concentration(s) for 20 minutes at room temperature. Next, antibody-treated gp160-expressing cells and DNP-CAR NK cells were co-cultured at a 1:1 E:T ratio in a U-bottom 96-well plate (50,000 cells each per well, in triplicate). Cells were incubated in an incubator maintained at 37oC and with 5% CO2. After 4 hours of incubation, cells were centrifuged. Supernatant from each well (100 μL) was collected, and the concentration of IFN-γ was analyzed using an ELISA kit (Thermo Fisher Scientific) by following the manufacturer’s instructions.
Flow cytometry-based cytotoxicity assays.
The subtype B or C gp160-expressing HEK293 cells were labeled with CFSE (2.5 μM) in 1X PBS (supplemented with 2% FBS) for 5 min at room temperature, and washed with RPMI 1640 complete medium (supplemented with 10% FBS) for three times. The CFSE-labeled gp160-expressing cells and the non-labeled gp160-negative cells (50,000/each) were mixed at a 1:1 ratio and then incubated with one of the seven DNP-conjugated antibodies (VRC01, 3BNC117, 2G12, 10-1074, PG9, 10E8, and isotype control) at a concentration of 2 nM for 20 min at room temperature. Then anti-DNP CAR-NK cells were added to the mixture of gp160-positive and negative cells at either 1:1, 5:1, or 25:1 E:T ratios (with respect to DNP-CAR NK cells and gp160+ cells) in triplicate. After 8 hours of incubation at 37°C, cells were collected and stained with APC-labeled human anti-HLA-A2 antibody for separating HEK293 cells (HLA-A2+) from CAR-NK cells (HLA-A2−) and aqua live/dead cell stain reagent. Cells were analyzed by flow cytometry using the LSRII (BD Biosciences, San Jose, CA), and the cytotoxic activity was assessed based on the number of remaining live gp160+ cells, calculated using the FlowJo software (Ashland, OR).
Cytotoxicity assays against HIV-infected primary CD4+ T cells
Primary CD4+ T cells were purified from healthy PBMCs with CD4 microbeads (Miltenyi # 130-045-101) and stimulated with plate-bound anti-CD3 and soluble anti-CD28 antibodies for 2 days. Then cells were infected with HIV-1 NL4-3 (500 ng of p24 per million cells) and cultured in 5 ng/ml IL-2 (Peprotech human IL-2 #200-02-1MG) for 10 days. Infected CD4 cells were pre-stained with Celltrace Farred (ThermoFisher #C34564), incubated with or without DNP-conjugated antibodies (10 nM) for 20 minutes, and then co-cultured with anti-DNP CAR-NK cells at a 20:1 E:T ratio for 16 hours. For the negative control, infected CD4 T cells were cultured without anti-DNP CAR-NK cells. After 16 hours of co-incubation, cells were stained with zombie violet fixable viability dye (Biolegend #423113), fixed, and stained intracellularly using BD Cytofix/Cytoperm kit (BD #554714) for HIV core antigen (Clone KC57, Beckman Coulter, #6604667). Cells were analyzed by flow cytometry using a MACSQuant analyzer 10 (Miltenyi) and flowjo (BD).
Generation of anti-DNP CAR-T cells
Cryopreserved human PBMCs (10 × 106 cells) were thawed, washed, and resuspended in 8 mL complete RPMI 1640 medium supplemented with 10% FBS, and then stimulated with 50 IU/mL of recombinant IL-2 and 75μL of Human T-Activator CD3/CD28 Dynabeads (ThermoFisher #11131D) for three days. The stimulated PBMCs (1 × 106 cells) were transferred to a non-treated 24-well plate precoated with retronectin (10 μg/mL). Anti-DNP CAR lentiviral particles were generated by calcium phosphate transfection of HEK293T cells and were subsequently concentrated using Lenti-X Concentrator solution (Takara Biosciences #631232). Then the lentivirus supernatant was added to PBMCs, followed by spinoculation with 100 IU/mL IL-2 at 1,200Xg for 90 minutes at room temperature. After transduction, PBMCs were washed three times and then cultured for a week in complete RPMI medium supplemented with 50 IU/mL IL-2 and Human T-Activator Dynabeads. CAR expression was verified by flow cytometry, and CAR-T cells were used directly in the cytotoxicity assay against gp160+ cells.
Safety Statement.
Lentivirus vectors were used in this study to engineer NK cells with chimeric antigen receptors. These vectors are classified as level 2 biohazard material. HIV-1 NL4-3 was used to infect human CD4+ T cells. This virus is classified as a level 2+ biohazard material.
Supplementary Material
ACKNOWLEDGMENTS
We thank P. Wang (University of Southern California) for the gift of the parental anti-CD19 CAR lentiviral vector. All anti-gp160 antibodies and gp160-expressing plasmids were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH.
Funding Sources
This study is supported by a startup fund from the School of Pharmacy at the University of Southern California (J.X.), a Pre-Doctoral Fellowship in Pharmaceutical Sciences from the American Foundation for Pharmaceutical Education (R.M.L.), and two grants from the National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health (NIH) under grant numbers R21AI155117 (J.X. and A.Z.) and R21AI140866 (A.Z.).
Footnotes
The authors declare no competing financial interest.
REFERENCES
- 1.Churchill MJ, Deeks SG, Margolis DM, Siliciano RF, and Swanstrom R (2016) HIV reservoirs: what, where and how to target them, Nat Rev Microbiol 14, 55–60. [DOI] [PubMed] [Google Scholar]
- 2.Kuhlmann AS, Peterson CW, and Kiem HP (2018) Chimeric antigen receptor T-cell approaches to HIV cure, Current opinion in HIV and AIDS 13, 446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maldini CR, Ellis GI, and Riley JL (2018) CAR T cells for infection, autoimmunity and allotransplantation, Nat Rev Immunol 18, 605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fesnak AD, June CH, and Levine BL (2016) Engineered T cells: the promise and challenges of cancer immunotherapy, Nat Rev Cancer 16, 566–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miranda LR, Schaefer BC, Kupfer A, Hu Z, and Franzusoff A (2002) Cell surface expression of the HIV-1 envelope glycoproteins is directed from intracellular CTLA-4-containing regulated secretory granules, Proc Natl Acad Sci U S A 99, 8031–8036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu B, Zou F, Lu L, Chen C, He D, Zhang X, Tang X, Liu C, Li L, and Zhang H (2016) Chimeric Antigen Receptor T Cells Guided by the Single-Chain Fv of a Broadly Neutralizing Antibody Specifically and Effectively Eradicate Virus Reactivated from Latency in CD4+ T Lymphocytes Isolated from HIV-1-Infected Individuals Receiving Suppressive Combined Antiretroviral Therapy, J Virol 90, 9712–9724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ali A, Kitchen SG, Chen ISY, Ng HL, Zack JA, and Yang OO (2016) HIV-1-Specific Chimeric Antigen Receptors Based on Broadly Neutralizing Antibodies, J Virol 90, 6999–7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hale M, Mesojednik T, Romano Ibarra GS, Sahni J, Bernard A, Sommer K, Scharenberg AM, Rawlings DJ, and Wagner TA (2017) Engineering HIV-Resistant, Anti-HIV Chimeric Antigen Receptor T Cells, Molecular therapy : the journal of the American Society of Gene Therapy 25, 570–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhen A, Kamata M, Rezek V, Rick J, Levin B, Kasparian S, Chen IS, Yang OO, Zack JA, and Kitchen SG (2015) HIV-specific Immunity Derived From Chimeric Antigen Receptor-engineered Stem Cells, Molecular therapy : the journal of the American Society of Gene Therapy 23, 1358–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sahu GK, Sango K, Selliah N, Ma Q, Skowron G, and Junghans RP (2013) Anti-HIV designer T cells progressively eradicate a latently infected cell line by sequentially inducing HIV reactivation then killing the newly gp120-positive cells, Virology 446, 268–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taylor BS, Sobieszczyk ME, McCutchan FE, and Hammer SM (2008) The challenge of HIV-1 subtype diversity, N Engl J Med 358, 1590–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lynch RM, Shen T, Gnanakaran S, and Derdeyn CA (2009) Appreciating HIV type 1 diversity: subtype differences in Env, AIDS Res Hum Retroviruses 25, 237–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu L, Patel B, Ghanem MH, Bundoc V, Zheng Z, Morgan RA, Rosenberg SA, Dey B, and Berger EA (2015) Novel CD4-Based Bispecific Chimeric Antigen Receptor Designed for Enhanced Anti-HIV Potency and Absence of HIV Entry Receptor Activity, J Virol 89, 6685–6694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghanem MH, Bolivar-Wagers S, Dey B, Hajduczki A, Vargas-Inchaustegui DA, Danielson DT, Bundoc V, Liu L, and Berger EA (2018) Bispecific chimeric antigen receptors targeting the CD4 binding site and high-mannose Glycans of gp120 optimized for anti-human immunodeficiency virus potency and breadth with minimal immunogenicity, Cytotherapy 20, 407–419. [DOI] [PubMed] [Google Scholar]
- 15.Anthony-Gonda K, Bardhi A, Ray A, Flerin N, Li M, Chen W, Ochsenbauer C, Kappes JC, Krueger W, Worden A, Schneider D, Zhu Z, Orentas R, Dimitrov DS, Goldstein H, and Dropulic B (2019) Multispecific anti-HIV duoCAR-T cells display broad in vitro antiviral activity and potent in vivo elimination of HIV-infected cells in a humanized mouse model, Science translational medicine 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steinhardt JJ, Guenaga J, Turner HL, McKee K, Louder MK, O’Dell S, Chiang CI, Lei L, Galkin A, Andrianov AK, N, A. D.-R., Bailer RT, Ward AB, Mascola JR, and Li Y (2018) Rational design of a trispecific antibody targeting the HIV-1 Env with elevated antiviral activity, Nature communications 9, 877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bournazos S, Gazumyan A, Seaman MS, Nussenzweig MC, and Ravetch JV (2016) Bispecific Anti-HIV-1 Antibodies with Enhanced Breadth and Potency, Cell 165, 1609–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deng K, Pertea M, Rongvaux A, Wang L, Durand CM, Ghiaur G, Lai J, McHugh HL, Hao H, Zhang H, Margolick JB, Gurer C, Murphy AJ, Valenzuela DM, Yancopoulos GD, Deeks SG, Strowig T, Kumar P, Siliciano JD, Salzberg SL, Flavell RA, Shan L, and Siliciano RF (2015) Broad CTL response is required to clear latent HIV-1 due to dominance of escape mutations, Nature 517, 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei X, Decker JM, Wang S, Hui H, Kappes JC, Wu X, Salazar-Gonzalez JF, Salazar MG, Kilby JM, Saag MS, Komarova NL, Nowak MA, Hahn BH, Kwong PD, and Shaw GM (2003) Antibody neutralization and escape by HIV-1, Nature 422, 307–312. [DOI] [PubMed] [Google Scholar]
- 20.Parker CG, Domaoal RA, Anderson KS, and Spiegel DA (2009) An antibody-recruiting small molecule that targets HIV gp120, J Am Chem Soc 131, 16392–16394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McEnaney PJ, Parker CG, Zhang AX, and Spiegel DA (2012) Antibody-recruiting molecules: an emerging paradigm for engaging immune function in treating human disease, ACS Chem Biol 7, 1139–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gonzalez ML, and Waxman FJ (2001) Relationship between complement activation and renal deposition of immune complexes made with IgG2a monoclonal antibodies, Clin Immunol 100, 362–371. [DOI] [PubMed] [Google Scholar]
- 23.White KD, Frank MB, Foundling S, and Waxman FJ (1996) Effect of immunoglobulin variable region structure on C3b and C4b deposition, Mol Immunol 33, 759–768. [DOI] [PubMed] [Google Scholar]
- 24.Maher J, Brentjens RJ, Gunset G, Riviere I, and Sadelain M (2002) Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor, Nat Biotechnol 20, 70–75. [DOI] [PubMed] [Google Scholar]
- 25.Lois C, Hong EJ, Pease S, Brown EJ, and Baltimore D (2002) Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors, Science 295, 868–872. [DOI] [PubMed] [Google Scholar]
- 26.Glienke W, Esser R, Priesner C, Suerth JD, Schambach A, Wels WS, Grez M, Kloess S, Arseniev L, and Koehl U (2015) Advantages and applications of CAR-expressing natural killer cells, Frontiers in pharmacology 6, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Daher M, and Rezvani K (2018) Next generation natural killer cells for cancer immunotherapy: the promise of genetic engineering, Curr Opin Immunol 51, 146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Y, Hermanson DL, Moriarity BS, and Kaufman DS (2018) Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity, Cell stem cell 23, 181–192 e185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Siegler EL, Kim YJ, Chen X, Siriwon N, Mac J, Rohrs JA, Bryson PD, and Wang P (2017) Combination Cancer Therapy Using Chimeric Antigen Receptor-Engineered Natural Killer Cells as Drug Carriers, Molecular therapy : the journal of the American Society of Gene Therapy 25, 2607–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tang X, Yang L, Li Z, Nalin AP, Dai H, Xu T, Yin J, You F, Zhu M, Shen W, Chen G, Zhu X, Wu D, and Yu J (2018) First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia, American journal of cancer research 8, 1083–1089. [PMC free article] [PubMed] [Google Scholar]
- 31.Tonn T, Schwabe D, Klingemann HG, Becker S, Esser R, Koehl U, Suttorp M, Seifried E, Ottmann OG, and Bug G (2013) Treatment of patients with advanced cancer with the natural killer cell line NK-92, Cytotherapy 15, 1563–1570. [DOI] [PubMed] [Google Scholar]
- 32.Zhang C, Oberoi P, Oelsner S, Waldmann A, Lindner A, Tonn T, and Wels WS (2017) Chimeric Antigen Receptor-Engineered NK-92 Cells: An Off-the-Shelf Cellular Therapeutic for Targeted Elimination of Cancer Cells and Induction of Protective Antitumor Immunity, Front Immunol 8, 533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tam YK, Maki G, Miyagawa B, Hennemann B, Tonn T, and Klingemann HG (1999) Characterization of genetically altered, interleukin 2-independent natural killer cell lines suitable for adoptive cellular immunotherapy, Human gene therapy 10, 1359–1373. [DOI] [PubMed] [Google Scholar]
- 34.Andre S, Seed B, Eberle J, Schraut W, Bultmann A, and Haas J (1998) Increased immune response elicited by DNA vaccination with a synthetic gp120 sequence with optimized codon usage, J Virol 72, 1497–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sievers SA, Scharf L, West AP Jr., and Bjorkman PJ (2015) Antibody engineering for increased potency, breadth and half-life, Current opinion in HIV and AIDS 10, 151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kwong PD, and Mascola JR (2012) Human antibodies that neutralize HIV-1: identification, structures, and B cell ontogenies, Immunity 37, 412–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Corti D, and Lanzavecchia A (2013) Broadly neutralizing antiviral antibodies, Annu Rev Immunol 31, 705–742. [DOI] [PubMed] [Google Scholar]
- 38.Jedema I, van der Werff NM, Barge RM, Willemze R, and Falkenburg JH (2004) New CFSE-based assay to determine susceptibility to lysis by cytotoxic T cells of leukemic precursor cells within a heterogeneous target cell population, Blood 103, 2677–2682. [DOI] [PubMed] [Google Scholar]
- 39.Yasmeen A, Ringe R, Derking R, Cupo A, Julien JP, Burton DR, Ward AB, Wilson IA, Sanders RW, Moore JP, and Klasse PJ (2014) Differential binding of neutralizing and non-neutralizing antibodies to native-like soluble HIV-1 Env trimers, uncleaved Env proteins, and monomeric subunits, Retrovirology 11, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adams JJ, Narayanan S, Liu B, Birnbaum ME, Kruse AC, Bowerman NA, Chen W, Levin AM, Connolly JM, Zhu C, Kranz DM, and Garcia KC (2011) T cell receptor signaling is limited by docking geometry to peptide-major histocompatibility complex, Immunity 35, 681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Srivastava S, and Riddell SR (2015) Engineering CAR-T cells: Design concepts, Trends Immunol 36, 494–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gristick HB, von Boehmer L, West AP Jr., Schamber M, Gazumyan A, Golijanin J, Seaman MS, Fatkenheuer G, Klein F, Nussenzweig MC, and Bjorkman PJ (2016) Natively glycosylated HIV-1 Env structure reveals new mode for antibody recognition of the CD4-binding site, Nat Struct Mol Biol 23, 906–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fu Q, Shaik MM, Cai Y, Ghantous F, Piai A, Peng H, Rits-Volloch S, Liu Z, Harrison SC, Seaman MS, Chen B, and Chou JJ (2018) Structure of the membrane proximal external region of HIV-1 envelope glycoprotein, Proc Natl Acad Sci U S A 115, E8892–E8899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kothe DL, Li Y, Decker JM, Bibollet-Ruche F, Zammit KP, Salazar MG, Chen Y, Weng Z, Weaver EA, Gao F, Haynes BF, Shaw GM, Korber BT, and Hahn BH (2006) Ancestral and consensus envelope immunogens for HIV-1 subtype C, Virology 352, 438–449. [DOI] [PubMed] [Google Scholar]
- 45.Li M, Gao F, Mascola JR, Stamatatos L, Polonis VR, Koutsoukos M, Voss G, Goepfert P, Gilbert P, Greene KM, Bilska M, Kothe DL, Salazar-Gonzalez JF, Wei X, Decker JM, Hahn BH, and Montefiori DC (2005) Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies, J Virol 79, 10108–10125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grogg KL, Miller RF, and Dogan A (2007) HIV infection and lymphoma, Journal of clinical pathology 60, 1365–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kudo K, Imai C, Lorenzini P, Kamiya T, Kono K, Davidoff AM, Chng WJ, and Campana D (2014) T lymphocytes expressing a CD16 signaling receptor exert antibody-dependent cancer cell killing, Cancer Res 74, 93–103. [DOI] [PubMed] [Google Scholar]
- 48.Klingemann H, Boissel L, and Toneguzzo F (2016) Natural Killer Cells for Immunotherapy - Advantages of the NK-92 Cell Line over Blood NK Cells, Front Immunol 7, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chowdhury MI, Koyanagi Y, Suzuki M, Kobayashi S, Yamaguchi K, and Yamamoto N (1992) Increased production of human immunodeficiency virus (HIV) in HIV-induced syncytia formation: an efficient infection process, Virus Genes 6, 63–78. [DOI] [PubMed] [Google Scholar]
- 50.Li J, Stagg NJ, Johnston J, Harris MJ, Menzies SA, DiCara D, Clark V, Hristopoulos M, Cook R, Slaga D, Nakamura R, McCarty L, Sukumaran S, Luis E, Ye Z, Wu TD, Sumiyoshi T, Danilenko D, Lee GY, Totpal K, Ellerman D, Hotzel I, James JR, and Junttila TT (2017) Membrane-Proximal Epitope Facilitates Efficient T Cell Synapse Formation by Anti-FcRH5/CD3 and Is a Requirement for Myeloma Cell Killing, Cancer Cell 31, 383–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.James SE, Greenberg PD, Jensen MC, Lin Y, Wang J, Till BG, Raubitschek AA, Forman SJ, and Press OW (2008) Antigen sensitivity of CD22-specific chimeric TCR is modulated by target epitope distance from the cell membrane, J Immunol 180, 7028–7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bluemel C, Hausmann S, Fluhr P, Sriskandarajah M, Stallcup WB, Baeuerle PA, and Kufer P (2010) Epitope distance to the target cell membrane and antigen size determine the potency of T cell-mediated lysis by BiTE antibodies specific for a large melanoma surface antigen, Cancer Immunol Immunother 59, 1197–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haso W, Lee DW, Shah NN, Stetler-Stevenson M, Yuan CM, Pastan IH, Dimitrov DS, Morgan RA, FitzGerald DJ, Barrett DM, Wayne AS, Mackall CL, and Orentas RJ (2013) Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia, Blood 121, 1165–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Davis SJ, and van der Merwe PA (2006) The kinetic-segregation model: TCR triggering and beyond, Nat Immunol 7, 803–809. [DOI] [PubMed] [Google Scholar]
- 55.Sharma P, and Kranz DM (2016) Recent advances in T-cell engineering for use in immunotherapy, F1000Research 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen J, Frey G, Peng H, Rits-Volloch S, Garrity J, Seaman MS, and Chen B (2014) Mechanism of HIV-1 neutralization by antibodies targeting a membrane-proximal region of gp41, J Virol 88, 1249–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Frey G, Peng H, Rits-Volloch S, Morelli M, Cheng Y, and Chen B (2008) A fusion-intermediate state of HIV-1 gp41 targeted by broadly neutralizing antibodies, Proc Natl Acad Sci U S A 105, 3739–3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alam SM, Morelli M, Dennison SM, Liao HX, Zhang R, Xia SM, Rits-Volloch S, Sun L, Harrison SC, Haynes BF, and Chen B (2009) Role of HIV membrane in neutralization by two broadly neutralizing antibodies, Proc Natl Acad Sci U S A 106, 20234–20239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pancera M, Zhou T, Druz A, Georgiev IS, Soto C, Gorman J, Huang J, Acharya P, Chuang GY, Ofek G, Stewart-Jones GB, Stuckey J, Bailer RT, Joyce MG, Louder MK, Tumba N, Yang Y, Zhang B, Cohen MS, Haynes BF, Mascola JR, Morris L, Munro JB, Blanchard SC, Mothes W, Connors M, and Kwong PD (2014) Structure and immune recognition of trimeric pre-fusion HIV-1 Env, Nature 514, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Urbanska K, Lanitis E, Poussin M, Lynn RC, Gavin BP, Kelderman S, Yu J, Scholler N, and Powell DJ Jr. (2012) A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor, Cancer Res 72, 1844–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lohmueller JJ, Ham JD, Kvorjak M, and Finn OJ (2017) mSA2 affinity-enhanced biotin-binding CAR T cells for universal tumor targeting, Oncoimmunology 7, e1368604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ma JS, Kim JY, Kazane SA, Choi SH, Yun HY, Kim MS, Rodgers DT, Pugh HM, Singer O, Sun SB, Fonslow BR, Kochenderfer JN, Wright TM, Schultz PG, Young TS, Kim CH, and Cao Y (2016) Versatile strategy for controlling the specificity and activity of engineered T cells, Proc Natl Acad Sci U S A 113, E450–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tamada K, Geng D, Sakoda Y, Bansal N, Srivastava R, Li Z, and Davila E (2012) Redirecting gene-modified T cells toward various cancer types using tagged antibodies, Clin Cancer Res 18, 6436–6445. [DOI] [PubMed] [Google Scholar]
- 64.Cao Y, Rodgers DT, Du J, Ahmad I, Hampton EN, Ma JS, Mazagova M, Choi SH, Yun HY, Xiao H, Yang P, Luo X, Lim RK, Pugh HM, Wang F, Kazane SA, Wright TM, Kim CH, Schultz PG, and Young TS (2016) Design of Switchable Chimeric Antigen Receptor T Cells Targeting Breast Cancer, Angew Chem Int Ed Engl 55, 7520–7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cho JH, Collins JJ, and Wong WW (2018) Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses, Cell 173, 1426–1438 e1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Farah FS (1973) Natural antibodies specific to the 2,4-dinitrophenyl group, Immunology 25, 217–226. [PMC free article] [PubMed] [Google Scholar]
- 67.Karjalainen K, and Makela O (1976) Concentrations of three hapten-binding immunoglobulins in pooled normal human serum, Eur J Immunol 6, 88–93. [DOI] [PubMed] [Google Scholar]
- 68.Axup JY, Bajjuri KM, Ritland M, Hutchins BM, Kim CH, Kazane SA, Halder R, Forsyth JS, Santidrian AF, Stafin K, Lu Y, Tran H, Seller AJ, Biroc SL, Szydlik A, Pinkstaff JK, Tian F, Sinha SC, Felding-Habermann B, Smider VV, and Schultz PG (2012) Synthesis of site-specific antibody-drug conjugates using unnatural amino acids, Proc Natl Acad Sci U S A 109, 16101–16106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ren W, Ji A, Wang MX, and Ai HW (2015) Expanding the Genetic Code for a Dinitrophenyl Hapten, Chembiochem 16, 2007–2010. [DOI] [PubMed] [Google Scholar]
- 70.Herzig E, Chan K, Packard TA, Vardi N, Schwarzer R, Gramatica A, Deeks SG, Williams SR, Landgraf K, Killeen N, Martin DW, Weinberger LS, and Greene WC (2019) Attacking Latent HIV with convertibleCAR-T Cells, a Highly Adaptable Killing Platform, Cell 179, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.