

Dear Editor,
Male infertility is defined as the inability to achieve pregnancy in a fertile female partner even after 12 months of unprotected intercourse. In humans, it accounts for 40%–50% of cases of infertility,1 and it occurs most often due to problems with sperm production or sperm delivery. Male infertility is commonly due to deficiencies in semen, and semen quality is used as a surrogate measure of male fecundity. Primary ciliary dyskinesia (PCD) is a rare cause of male infertility.2,3 It is an autosomal recessive genetic condition that causes abnormal function of cilia. Cilia are microscopic finger-like projections on the surface of the cells. They are found in the linings of the airway, the reproductive system, and other organs and tissues. Similar to cilia, flagella are tail-like structures that are present on the sperm and enable sperm motility by a propulsion mechanism, which is a rare cause of male infertility. Kartagener's syndrome is a type of PCD associated with a mirror-image reversal of the heart and other internal organs (situs inversus).4,5 In this paper, we used whole-genome sequencing to identify a novel deletion mutation of the PIH1 domain containing 3 (PIH1D3) gene in infertile siblings from one family of four generations.
Based on computer-assisted sperm assignment, the proband (III-2) was diagnosed as having nonmotile sperm. During the patient's consultation for infertility, we were informed that he had normal karyotype and the Y chromosome azoospermia factor (YqAZF) microdeletion was not found. We also acquired his history and he had a history of bronchiectasis, bronchopneumonia, and ejaculatory duct cyst. Upon evaluating family history, we were informed that the patient's uncle has no children for 40 years since marriage (no medical records of infertility), and his 16-year-old cousin (III-3; Figure 1a and 1b) also has bronchiectasis. Furthermore, his cousin has dextrocardia (Figure 1b). We obtained peripheral whole blood samples from the patient and from his parents, cousin, aunt, uncle, and grandparents. DNA was extracted from the samples using a DNA mini kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Blood-extracted genomic DNA samples were then subjected to whole-exome sequencing, and data analysis was performed by the Yueer Gene Technology Company (Shanghai, China). The proband provided a sperm sample through masturbation (III-2). This study was approved by the Ethics Committee of the Shanghai General Hospital (Shanghai, China), and all participants had provided written informed consents (No. 2016KY131).
Figure 1.
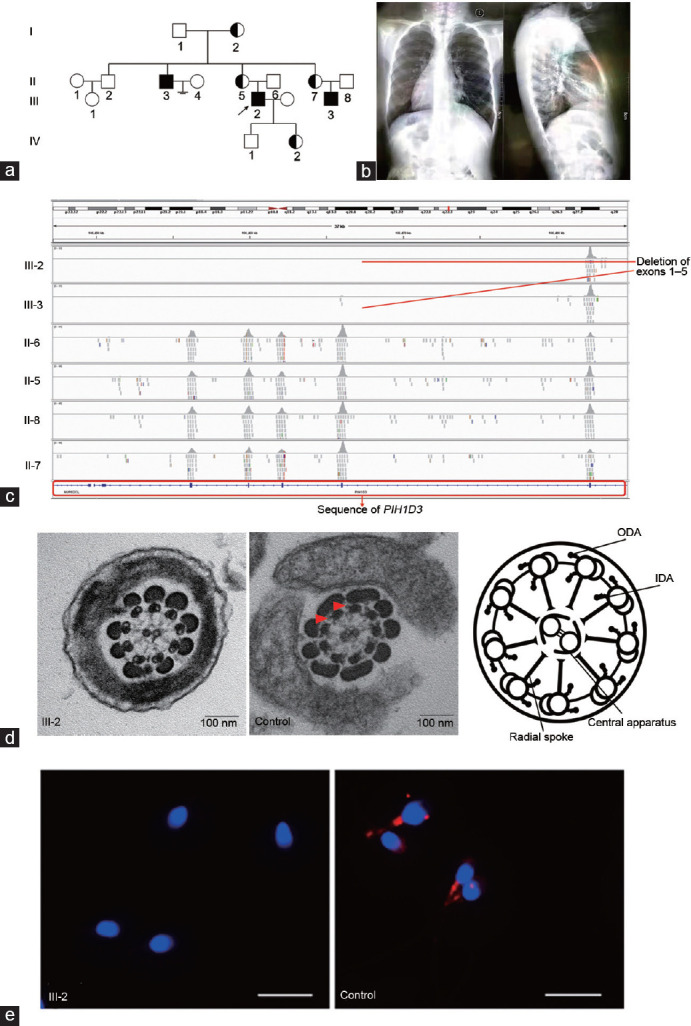
Results of the mutational analyses in PIH1D3. Pedigrees of the PCD-affected family and clinical features. (a) Hemizygous loss-of-function mutations in PIH1D3 located on the X chromosome were identified in one family. Pedigree of the PCD-affected family. PCD-affected siblings are indicated in black, and the unaffected siblings are indicated in white. (b) Chest X-ray shows situs inversus totalis, chronic airway disease with bronchiectasis in the middle lobe, and situs inversus totalis in III-3. (c) All blood samples were detected with whole-exome sequencing. The proband and his cousin had deletion of the PIH1D3 gene exons 1–5. (d) Analysis of transmission electron microscopy showing the cross-section arrangement of cilia from a healthy control (middle) and an image (right) of the main structures of the 9 + 2 motile axoneme, including the ODA and IDA. However, cross-sections from the respiratory cilia from III-2 showed a normal 9 + 2 architecture with absence of outer and inner dynein arms (red arrowheads). Scale bars = 100 nm. (e) Sperm of healthy men and III-2 were labeled with anti-PIH1D3 antibodies (red). The proteins are localized in the tails of sperm of controls. In contrast, PIH1D3 expression was absent or severely reduced in sperm from III-2. Scale bars = 10 μm. PIH1D3: PIH1 domain containing 3; PCD: primary ciliary dyskinesia; ODA: outer dynein arms; IDA: inner dynein arms.
X-linked PIH1D3 is a protein-coding gene. It is a highly conserved protein that is present only in species with motile cilia/flagella containing dynein arms and those that require intraflagellar transport (IFT) for ciliary assembly.6 In the Human Protein Atlas, human PIH1D3 is reported to be expressed in ciliated tissues, and its expression is the highest in the lung, fallopian tube, and testis.7 PIH1D3 is a cytoplasmic protein that is involved in early axonemal dynein arm assembly.8 Thus, PID1D3 plays an important role in IFT-related dynein arm assembly. Mutations in PIH1D3 lead to PCD, and two previous studies have identified 11 different mutations in PIH1D3, including 3 genomic deletion mutations (1.93-Mb, 3.27-Mb, and 3.73-Mb deletions in DCP603/DCP1747, DCP1337 and DCP855, respectively).6,8
Here, we reported (GenBank: NM_001169154.1, 185-kb, 106295582–106481871del) the deletion of the PIH1D3 gene in two cousins with PCD (Figure 1c). Analysis of the sperm sample from the proband (III-2) revealed the presence of largely immotile sperm, but >4% of sperm exhibited normal morphology. Next, we analyzed the transmission electron microscopy cross-sections from respiratory cilia from the proband (III-2) and found a normal 9 + 2 architecture without outer and inner dynein arms (Figure 1d), indicating that PIH1D3 is vital for sperm motility. Bioinformatics prediction and amino acid conservation analysis suggested that the deletion mutation (185-kb) is a pathogenic mutation. This mutation was not found in the East Asian population of the Genome AD exome, 1000 Genome, or exome aggregation consortium (ExAC) databases, and this mutation was not found in 100 healthy Chinese men with normal fertility, confirming that the mutation is a novel deletion mutation. Immunofluorescence analysis of sperm obtained from III-2 and healthy men revealed that the PIH1D3 gene was absent in or severely downregulated in the III-2 sperm sample (Figure 1e).
This mutation was also present in his family. We investigated the pedigree to explore the possibility of an X-linked recessive inheritance mode in this patient's family (Figure 1a). The proband (III-2) had a cousin (III-3) and an uncle (II-3) with similar respiratory symptoms and a history of subfertility or infertility. Copy number variation (CNV) analysis confirmed the mutation in III-2, III-3, and II-3 and in one of the alleles of the mother of individuals III-2, III-3, and II-3. The proband had a son and daughter by assisted reproductive technology. CNV analysis also confirmed the presence of this mutation in one of the alleles of his daughter (IV-2), confirming an X-linked recessive inheritance mode (Figure 1a and Supplementary Table 1). We informed the proband for the necessity of genetic counseling for his daughter prior to conception.
Supplementary Table 1.
The interstitial deletions of the X chromosome carried by affected individuals III2, III3, II3, II5, II7, I2 and IV2
| ID | chrom | loc.start | loc.end | r.len | cytoband.region | cnv.info |
|---|---|---|---|---|---|---|
| III-2 | chrX | 106220001 | 106500000 | 280000 | q22.3 | del(X)(q22.3).seq[GRCh37/hg19]X1 |
| III-3 | chrX | 106220001 | 106500000 | 280000 | q22.3 | del(X)(q22.3).seq[GRCh37/hg19]X1 |
| II-3 | chrX | 106180001 | 106500000 | 320000 | q22.3 | del(X)(q22.3).seq[GRCh37/hg19]X1 |
| II-5 | chrX | 106220001 | 106500000 | 280000 | q22.3 | del(X)(q22.3).seq[GRCh37/hg19]X1 |
| II-7 | chrX | 106220001 | 106500000 | 280000 | q22.3 | del(X)(q22.3).seq[GRCh37/hg19]X1 |
| I-2 | chrX | 106220001 | 106500000 | 280000 | q22.3 | del(X)(q22.3).seq[GRCh37/hg19]X1 |
| IV-2 | chrX | 106220001 | 106460000 | 240000 | q22.3 | del(X)(q22.3).seq[GRCh37/hg19]X1 |
In conclusion, this is the first report of two cousins with PCD conceived by X-linked deletion of the PIH1D3 gene. Our study demonstrated that a novel deletion mutation (GenBank: NM_001169154.1, 185-kb, 106295582–106481871del) of the PIH1D3 gene may cause severe PCD, leading to infertility, possibly by affecting early axonemal dynein arm assembly. This finding will aid researchers and clinicians in the development of approaches for patient counseling and management, especially in cases of nonmotile sperm syndrome.
AUTHOR CONTRIBUTIONS
CH and NCL identified the case, conducted the genetic studies, and drafted the manuscript. XBW carried out the laparoscopy. BHG, JXZ and LZ participated in the genetic analysis and coordinated to draft the manuscript. ZL conceived of the study and reviewed and edited the manuscript. All authors read and approved the final manuscript.
COMPETING INTERESTS
All authors declared no competing interests.
ACKNOWLEDGMENTS
This work was supported by the National Key R&D Program of China (No. 2017YFC1002003) and the China Postdoctoral Science Foundation (No. 2019M661521).
Supplementary Information is linked to the online version of the paper on the Asian Journal of Andrology website.
REFERENCES
- 1.Carlsen E, Giwercman A, Keiding N, Skakkebaek NE. Evidence for decreasing quality of semen during past 50 years. BMJ. 1992;305:609–13. doi: 10.1136/bmj.305.6854.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Noone PG, Leigh MW, Sannuti A, Minnix SL, Carson JL, et al. Primary ciliary dyskinesia: diagnostic and phenotypic features. Am J Respir Crit Care Med. 2004;169:459–67. doi: 10.1164/rccm.200303-365OC. [DOI] [PubMed] [Google Scholar]
- 3.Bush A, Cole P, Hariri M, Mackay I, Phillips G, O'callaghan C, et al. Primary ciliary dyskinesia: diagnosis and standards of care. Eur Respir J. 1998;12:982–8. doi: 10.1183/09031936.98.12040982. [DOI] [PubMed] [Google Scholar]
- 4.Leigh MW, Pittman JE, Carson JL, Ferkol TW, Dell SD, et al. Clinical and genetic aspects of primary ciliary dyskinesia/Kartagener syndrome. Genet Med. 2009;11:473–87. doi: 10.1097/GIM.0b013e3181a53562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shapiro AJ, Davis SD, Ferkol T, Dell SD, Rosenfeld M, et al. Laterality defects other than situs inversus totalis in primary ciliary dyskinesia: insights into situs ambiguus and heterotaxy. Chest. 2014;146:1176–86. doi: 10.1378/chest.13-1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olcese C, Patel MP, Shoemark A, Kiviluoto S, Legendre M, et al. X-linked primary ciliary dyskinesia due to mutations in the cytoplasmic axonemal dynein assembly factor PIH1D3. Nat Commun. 2017;8:1–15. doi: 10.1038/ncomms14279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Uhlén M, Björling E, Agaton C, Szigyarto CA, Amini B, et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol Cell Proteomics. 2005;4:1920–32. doi: 10.1074/mcp.M500279-MCP200. [DOI] [PubMed] [Google Scholar]
- 8.Paff T, Loges NT, Aprea I, Wu K, Bakey Z, et al. Mutations in PIH1D3 cause X-linked primary ciliary dyskinesia with outer and inner dynein arm defects. Am J Hum Genet. 2017;100:160–8. doi: 10.1016/j.ajhg.2016.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]