

Key Points
Low-cost transcriptional diagnostics can accurately classify lymphomas in LMICs.
Machine learning algorithms to classify gene expression could transform the diagnosis of lymphomas in LMICs.
Abstract
Inadequate diagnostics compromise cancer care across lower- and middle-income countries (LMICs). We hypothesized that an inexpensive gene expression assay using paraffin-embedded biopsy specimens from LMICs could distinguish lymphoma subtypes without pathologist input. We reviewed all biopsy specimens obtained at the Instituto de Cancerología y Hospital Dr. Bernardo Del Valle in Guatemala City between 2006 and 2018 for suspicion of lymphoma. Diagnoses were established based on the World Health Organization classification and then binned into 9 categories: nonmalignant, aggressive B-cell, diffuse large B-cell, follicular, Hodgkin, mantle cell, marginal zone, natural killer/T-cell, or mature T-cell lymphoma. We established a chemical ligation probe-based assay (CLPA) that quantifies expression of 37 genes by capillary electrophoresis with reagent/consumable cost of approximately $10/sample. To assign bins based on gene expression, 13 models were evaluated as candidate base learners, and class probabilities from each model were then used as predictors in an extreme gradient boosting super learner. Cases with call probabilities < 60% were classified as indeterminate. Four (2%) of 194 biopsy specimens in storage <3 years experienced assay failure. Diagnostic samples were divided into 70% (n = 397) training and 30% (n = 163) validation cohorts. Overall accuracy for the validation cohort was 86% (95% confidence interval [CI]: 80%-91%). After excluding 28 (17%) indeterminate calls, accuracy increased to 94% (95% CI: 89%-97%). Concordance was 97% for a set of high-probability calls (n = 37) assayed by CLPA in both the United States and Guatemala. Accuracy for a cohort of relapsed/refractory biopsy specimens (n = 39) was 79% and 88%, respectively, after excluding indeterminate cases. Machine-learning analysis of gene expression accurately classifies paraffin-embedded lymphoma biopsy specimens and could transform diagnosis in LMICs.
Visual Abstract

Introduction
Accurate diagnosis is an essential component of optimal cancer care.1 In high-income countries (HICs), this typically requires a pathologist to perform and review histology and immunohistochemistry (IHC). The World Health Organization (WHO) includes histopathology with IHC among the essential in vitro diagnostics for health care facilities with clinical laboratories.2 Yet, most patients in low- and middle-income countries (LMICs) do not have access because of both high cost and a dearth of pathologists.3,4
Many subtypes of lymphoma can be effectively treated with available therapies,5 including chemotherapies, monoclonal antibodies, or small molecule–targeted agents. As a result, there is a pressing need for inexpensive, accurate, and operator-independent diagnostics to guide therapeutic selection for patients with lymphoma. In many ways, this mimics the situation within LMICs in the late 1990s on the discovery of highly active antiretroviral therapy for HIV. Deployment of highly active antiretroviral therapy in LMICs absolutely required the validation and deployment of point-of-care diagnostics that were inexpensive and operator independent.6
Advancements in transcriptional profiling now allow for rapid assessment of the expression of multiple genes using formalin-fixed, paraffin-embedded (FFPE) samples.7,8 Several reports have identified gene signatures that can facilitate binary distinctions between subtypes of lymphoma (eg, Burkitt lymphoma vs diffuse large B-cell lymphoma [DLBCL]).9-12 These distinctions still require prior knowledge based on standard pathology to narrow between preselected comparators. We hypothesized that a single transcriptional assay could bin biopsy specimens obtained for suspicion of lymphoma into treatment-driven groups without prior knowledge and thereby decrease the need for pathologist review.
The Instituto de Cancerología y Hospital Dr. Bernardo Del Valle (INCAN) in Guatemala is the country’s only public cancer hospital. It serves a large urban population and rural indigenous Mayan communities. Approximately 100 patients present to INCAN with findings suspicious for lymphoma annually. Limited IHC is available for an out-of-pocket cost of approximately $450, which is beyond the means of most patients. As a result, biopsy specimens are commonly assessed solely by hematoxylin and eosin (H&E) staining, resulting in ambiguous diagnoses (eg, suspect large cell lymphoma or lymphoma-not otherwise specified [NOS]).
To address the feasibility of a transcriptional diagnostic for lymphoma, we collected FFPE biopsy specimens obtained at INCAN that were performed because of clinical suspicion of lymphoma over a 13-year period. We established diagnoses according to the WHO classification using standard-of-care pathology assessment.5 We then designed an assay with reagent and consumable costs of approximately $10 per sample to assess the expression of 37 genes and applied a machine learning–based platform to bin diagnoses. We show high accuracy for this approach across diagnostic subsets in validation cohorts.
Methods
Case selection
This study was approved by the institutional review boards of the Dana-Farber Cancer Institute and Stanford University and the ethics committee of La Liga Nacional Contra el Cáncer. Research was conducted in accordance with the Declaration of Helsinki. We reviewed medical records to identify all biopsy specimens collected at INCAN between 2006 and 2018 that were performed because of clinical suspicion for lymphoma (supplemental Table 1). This included 3015 tissue blocks from 1836 individual patients. To preserve tissue for future clinical needs, core biopsy specimens and cases with blocks <1 cm3 were excluded. Clinical data were collected by manual review of paper charts. Most biopsy specimens were from lymph nodes or secondary lymphoid tissue, but additional extranodal sites (eg, palate, testicle, eyelid, femur, thyroid, skin, mesentery, tongue, breast, lung) were included.
WHO guidelines–based pathology diagnosis
One-half of each FFPE block was shipped to Stanford University where H&E slides were generated from whole sections and reviewed by 2 expert hematopathologists (O.S. and Y.N.). Representative areas were selected, and 2 cores from each sample were included for tissue microarray (TMA) construction, as previously described.13 TMAs were sectioned at 4-μm thickness and subjected to IHC per routine protocol on automated immunostainers (Leica BOND-III, Leica Biosystems, Buffalo Grove, IL or BenchMark ULTRA, Roche/Ventana Medical Systems, Tucson, AZ). Epstein-Barr virus (EBV) was assessed by IHC for EBV-LMP1 and in situ hybridization for EBV-associated small RNAs by routine methods using the Ventana autostainer. Fluorescence in situ hybridization assays were performed on TMA sections to detect breakpoints in the MYC, BCL2, and BCL6 loci using 5′/3′ break-apart probes (ZytoVision, Bremerhaven, Germany), as previously described.14 All biopsy specimens were classified according to the 2016 WHO classification5 and then categorized into diagnostic bins (supplemental Table 2).
To ensure that the TMAs would accurately represent large tissue biopsy specimens, H&Es of each case were screened before the incorporation into the TMA to ensure that representative tumor areas were incorporated into the TMA. As a second validation, 80 cases (supplemental Table 3) were randomly selected using a uniform distribution [0,1] and stratified sampling to proportionally represent the 9 diagnostic bins and nondiagnostic categories. O.S. and Y.S. were blinded to the original diagnosis. H&Es from these cases were rereviewed after at least a 1-year washout period from prior review. A differential diagnosis was generated, and selected IHC stains were performed on whole sections.
Targeted expression profiling
Expression was quantified as previously described, with the addition of unique buffers for extraction from FFPE tissue.15 Briefly, 6-μm (multisite comparison) or 10-μm (training and validation sets) scrolls from paraffin-embedded tissue were cut from the entire tissue blocks without selecting for a minimum tumor percentage. Scrolls were added to individual wells of a 96-well polymerase chain reaction (PCR) reaction plate with 15 μL of 1× TE (10 mM Tris, 0.1 mM EDTA; pH 8.0), 50 μL of DxBuffer1 reaction buffer, 15 μL of Lymphoma RUO Mix A, 15 μL of Lymphoma RUO Mix B, and 5 μL of DirectMix C, followed by ligation for 5 minutes at 55°C, 10 minutes at 80°C, and 165 minutes at 55°C. Following ligation, 5 μL of Directbeads was added, followed by 15-minute incubation at 55°C.
Samples were placed on a magnetic plate and washed 3 times with DirectWash buffer. All liquid was removed, and 5 μL of DirectTaq and 5 μL of DxPrime were added. Samples were transferred to a PCR thermal cycler for hot start at 2 minutes at 95°C, followed by 30 cycles of denaturation for 10 seconds at 95°C, annealing for 20 seconds at 61°C, and extension for 20 seconds at 72°C, followed by 4°C hold. GeneScan 600LIZ Size Standard (0.5 μL) and 17.5 μL of formamide solution (Thermo Fisher Scientific) were combined with 2 μL of PCR reaction to a capillary electrophoresis plate. Capillary electrophoresis was run on an Applied Biosystems 3500 or SeqStudio Genetic Analyzer (Thermo Fisher Scientific). Control genes LIGC (ligation control) and PCRC (PCR control) were included in the assay. Two ubiquitously expressed normalizer genes (ISY1 and WDR55) were added to each reaction in each fluorescent channel. Probe sequences are available from the authors on request.
Model generation and validation
RStudio version 1.1.463 with R version 3.6.1 was used for analysis. Models were tuned and trained on a high-performance cluster. A model stacking approach, in which a “super learner” is trained on predicted class probabilities from several base learners, was used. The Classification and Regression Training package was used to select model training and validation sets. Normalized expression value of each gene was calculated by subtracting the mean of the log2 normalizer signals from the log2 signal of the response gene. Normalizer values are calculated independently for each fluorescent channel, and a floor of −5 was applied to all fragments. Diagnostic samples were split into training (70%) and validation (30%) sets, and their gene expression values were centered and scaled in the training set using the preProcess option in the train function. The createDataPartition function was used to maintain the ratio of diagnoses between the training and validation sets. The multiClassSummary and classProb options were selected in the trainControl function. Parameters for 14 candidate models were tuned on the training set using five repeats of 10-fold cross-validation and a logarithmic loss performance metric (supplemental Table 9). A grid search was used to tune models with 1 parameter. For models with 2 or more tuning parameters, an initial random search of at least 150 parameter settings was followed by a focused search over a smaller grid.
Base learners were selected by considering accuracy, sensitivity, specificity, and negative/positive predictive values for each diagnostic class in the validation set. Probabilities from these models were used as predictors for the extreme gradient booster super learner model, which determined the final class label for the first-stage model. Additional cohorts of relapsed samples and excluded samples were used as test sets.
Multisite comparison
Fifty-nine cases were randomly selected from the validation cohort in proportion to the incidence of the lymphoma subtype in the original cohort. Consecutive 6-μm sections were obtained from each block of tissue and either stayed at INCAN for testing or were shipped to DxTerity for testing. Technicians employed at each site ran the assay blinded. If a sample failed quality control on the first run, a second cut was used.
Results
WHO-based tumor classification
We assayed 670 banked FFPE samples from INCAN (Figure 1), including 650 that were collected because of clinical suspicion of lymphoma between 2006 and 2018 and 20 of normal tonsillar tissue that served as additional benign controls. Summarized clinical data for the 643 individual patients with suspected lymphoma is included in supplemental Table 4. Individual patient characteristics, diagnoses, and frontline treatments for the validation cohort are shown in supplemental Table 5. Biopsies were assayed by H&E on whole sections and then IHC (>30 000 individual stain assessments) on TMAs (supplemental Figure 1A) to establish a final diagnosis according to the 2016 WHO classification.5
Figure 1.
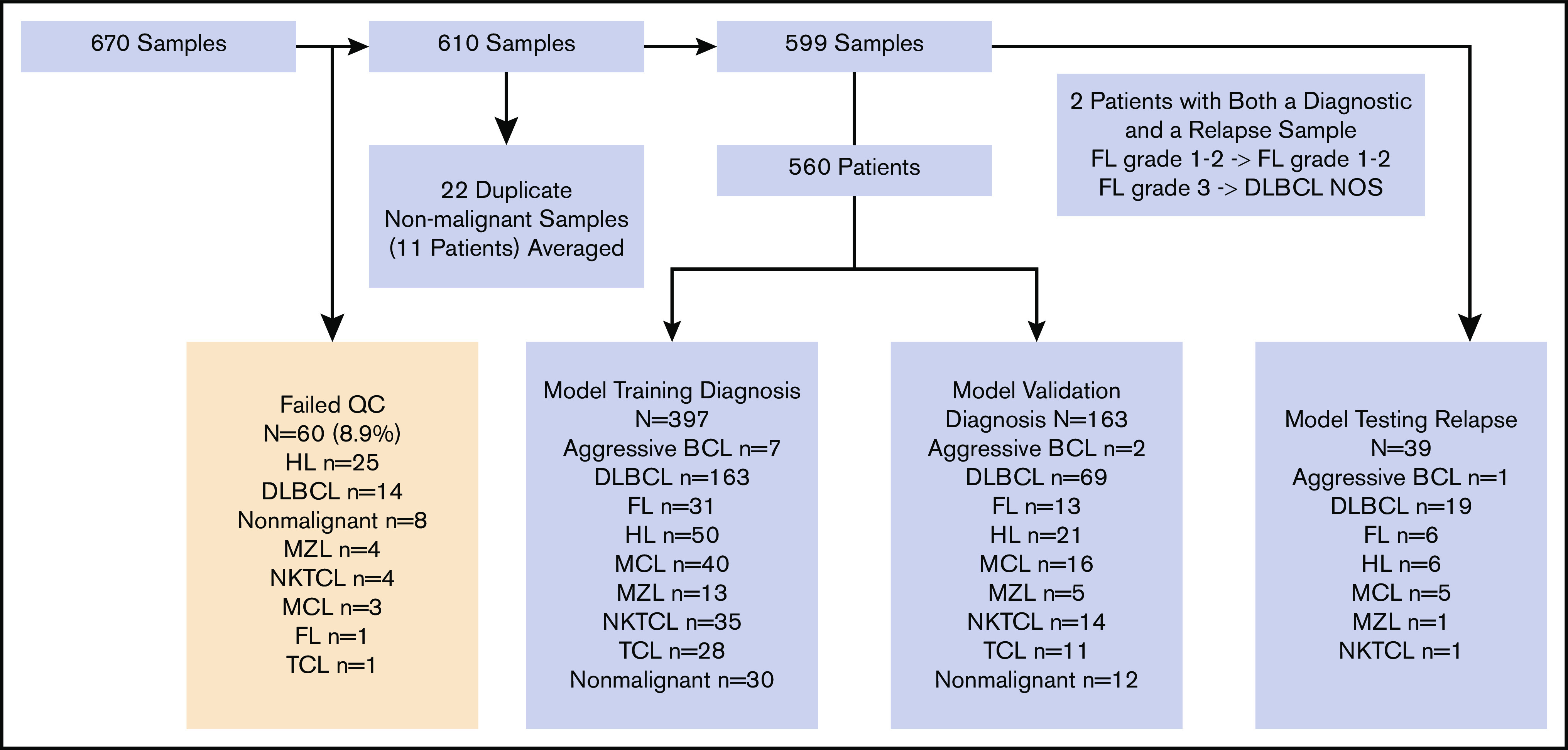
Schema of samples from INCAN. Of 670 FFPE biopsy specimens, 60 failed quality control based on expression of housekeeping genes. Among the remaining 610, 11 patients had 2 samples each, and the results from each pair were averaged. The remaining 599 samples (597 patients) consisted of 560 obtained before therapy and 39 after relapse. Two patients had biopsy specimens both before therapy and after relapse; 1 had FL grade 1 to 2 at both time points, and 1 had FL grade 3A before treatment and DLBCL-not otherwise specified (NOS) at relapse. Untreated biopsy specimens were divided into training (n = 397) and validation/testing (n = 163) cohorts.
To ensure that diagnosis using TMA of selected regions was adequate, we randomly sampled 80 cases proportional to the frequency of lymphoma subtypes in our cohort. Additional whole sections were cut from each case and subjected to repeat evaluation by IHC in a blinded manner. Two cases were excluded because of insufficient remaining tissue for IHC of whole sections. Diagnoses from the TMA and whole sections were concordant in 77 of 78 cases (98.7%) (supplemental Table 3), with a single diagnosis changed from peripheral T-cell lymphoma-NOS (PTCL-NOS) on the TMA to nodular lymphocyte predominant Hodgkin lymphoma (HL) after rare large CD20+ cells were noted on IHC of the whole section.
We then binned diagnoses into 9 therapeutically driven categories (Figure 1; supplemental Table 2A): (1) aggressive B-cell lymphoma including Burkitt lymphoma and B-lymphoblastic lymphoma; (2) DLBCL including high-grade B-cell lymphomas; (3) HL including nodular lymphocyte predominant HL; (4) marginal zone lymphoma (MZL); (5) mantle cell lymphoma (MCL); (6) follicular lymphoma (FL); (7) natural killer/T-cell lymphoma (NKTCL); (8) T-cell lymphoma (TCL) including cutaneous and peripheral TCL subtypes; and (9) nonmalignant including cases that lacked evidence of lymphoma or other malignancy.
Original diagnoses from INCAN were also binned into these categories. For 244 (38%; 95% confidence interval [CI]; 34%-42%) of 643 unique patients with biopsy specimens evaluated at both centers, the diagnoses at INCAN and Stanford resulted in similar classification into 1 of the 9 groups (supplemental Table 6). In the remaining cases, the diagnoses either resulted in separate grouping or diagnosis at INCAN was incomplete (eg, lymphoma NOS).
Lymphoma classification based on gene expression
We established a chemical ligation probe-based assay (CLPA) that quantifies the expression of 37 genes plus 2 controls from FFPE biopsy specimens using standard capillary electrophoresis equipment (supplemental Figure 1B). The genes were selected from previous publications based on (1) lineage- or subtype-specific expression, (2) prognostic value, or (3) therapeutic relevance (supplemental Table 7). We performed the CLPA on scrolls cut from each biopsy specimen. Of the 670, 60 (8.9%) failed quality control metrics based on expression of control genes (Figure 1). Older age of tissue biopsy specimen was associated with significantly higher failure rate, with only 4 (2%) of 194 biopsy specimens from 2015 or later failing quality control (supplemental Table 8). After averaging 11 duplicate runs of nonmalignant samples, we established a cohort of 599 samples from 597 patients (Figure 1). The 560 samples obtained before treatment were divided 70%:30% into training and validation cohorts (Figure 1).
Distributions of gene expression for each gene and correlations between genes are included in supplemental Figure 2. Unsupervised clustering of 560 cases based on expression of the 37 genes demonstrated notable clustering (Figure 2), in some cases because of the expression of very few genes. For example, expression of CCND1 alone clustered nearly all cases of MCL, whereas expression of EBER1, NCAM1, CD244, and TBX21 clustered cases of NKTCL (Figure 2).
Figure 2.

Unsupervised hierarchical clustering using Spearman correlation with complete linkage of the 560 biopsy specimens obtained before therapy based on normalized gene expression across the 37 genes in the CLPA. Diagnosis is according to IHC-based classification. Agg BCL, aggressive B-cell lymphoma; Nonmal, nonmalignant.
Thirteen models were evaluated as candidate base learners, and the class probabilities from each model were then used as predictors in an extreme gradient boosting super learner to assign class labels for each sample (Figure 3; supplemental Table 9). After training on 397 cases, the validation cohort of 163 cases was assessed and compared with the IHC-based diagnosis. Overall accuracy for the assay was 86% (140 of 163), with ≥90% accuracy for DLBCL, HL, MCL, and NKTCL (Figure 4A; supplemental Table 10). There were no significant differences in classification accuracy (excluding samples that failed quality control) based on biopsy specimen age (supplemental Table 11A) or between nodal/secondary lymphoid tissue and extranodal biopsy specimens (supplemental Table 11B).
Figure 3.

Schematic outlining the 13 base learner models used by the XGB Super Learner to determine classification. This involves categorization into 1 of 9 diagnostic bins. GBM, stochastic gradient boosting; HDDA, high dimensional discriminant analysis; KNN, k-nearest neighbors; MDA, mixture discriminant analysis; MULTINOM, penalized multinomial regression; NB, naïve Bayes; NN, neural network; PAM, nearest shrunken centroids; PDA, penalized discriminant analysis; PLS, partial least squares; RF, random forest; SVMPOLY, support vector machine with polynomial kernel; SVMRAD, support vector machine with radial kernel with radial kernel; XGB, eXtreme gradient boosting.
Figure 4.
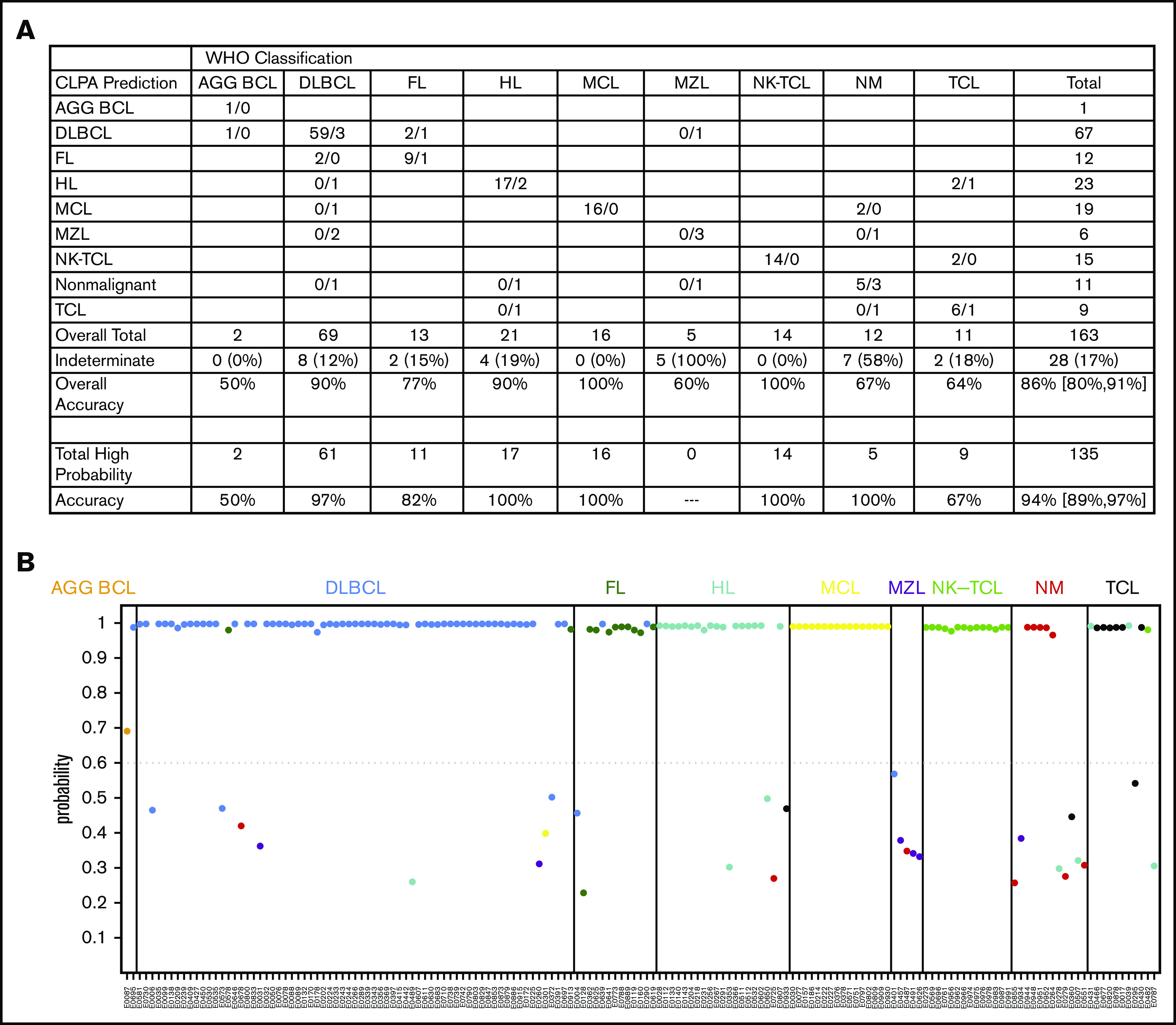
CLPA accuracy. (A) Predicted calls among each diagnostic bin. Cases predicted within diagnostic bins are separated by “/” with the first number representing calls that met the ≥60% probability threshold and the number following the “/” representing cases that did not meet the ≥60% probability threshold. Overall Accuracy includes all 163 samples. Total High Probability indicates the number of cases that were classified with >60% probability, with accuracy among these cases indicated. (B) Probabilities for each call among the 163 biopsy specimens in the validation cohort. Bins based on standard pathology are listed along the top. Dots are colored based on the CLPA call. Additional metrics are in supplemental Table 9.
We noted that 135 of 163 (83%) calls from the validation set had >90% confidence probability (Figure 4B). Thus, we chose a conservative cutoff probability value of ≥60% and reclassified all cases with <60% probability value as indeterminate. Of the remaining 136, 128 (94%) were classified correctly (Figure 4A; supplemental Table 10). Among the 8 mischaracterized cases with probability ≥60% were as follows: n = 1 called DLBCL by CLPA but B-lymphoblastic lymphoma by IHC, n = 2 called FL by CLPA but DLBCL by IHC, n = 2 called DLBCL by CLPA but FL by IHC, n = 2 called HL by CLPA but anaplastic large cell lymphoma or PTCL-NOS by IHC, and n = 1 called NKTCL by CLPA but PTCL-NOS by IHC (supplemental Table 12). Rereview of 1 case called PTCL-NOS by IHC but HL by CLPA showed rare CD30+, CD15+ (subset), PAX5+ (variable), MUM1+, EBV-negative cells consistent with classical HL, indicating that the CLPA made the accurate call. There were no significant changes on rereview of the remaining cases.
Approximately 7.5% of the biopsies performed for suspicion of lymphoma at INCAN were categorized as nonmalignant by IHC (Figure 1). Many of these patients were incorrectly diagnosed with lymphoma at INCAN and treated with cytotoxic agents. In contrast, the CLPA called all nonmalignant cases either nonmalignant or indeterminate (Figure 4A). At the same time, only 1 of 234 samples classified as malignant lymphoma by standard pathology in the 3 validation cohorts was classified with high confidence as nonmalignant by CLPA.
Analyses to validate assay performance
To test the reliability of the assay across independent laboratories, we selected 59 cases from the validation cohort, including cases with <60% or ≥60% probability calls. Consecutive sections were used for CLPA testing at DxTerity in the United States and by INCAN laboratory staff in Guatemala City. Of the 59, 58 cases passed quality control at both sites, and the remaining case failed at both sites. Thirty-seven of 58 cases reached 60% diagnostic probability at both sites, and the diagnosis at both sites was concordant in 36 of 37 cases (97%). The single discordant call was called FL at INCAN and DLBCL at DxTerity. In 9 of 58 cases, only 1 site reached the 60% probability threshold (5 at INCAN and 4 at DxTerity); 8 of the 9 were concordant with IHC-based diagnosis. The remaining 12 cases were indeterminate at both sites.
As an additional test cohort, we assayed the 39 cases from patients with relapsed disease. Two of the biopsy specimens were from patients included in the initial 560 cases (both in the training cohort): 1 with the same diagnosis and 1 who relapsed with DLCBL after presenting with FL (Figure 1). Overall accuracy of CLPA-based classification compared with IHC was 79% (95% CI: 64%-91%). After excluding the 5 cases with probability values <0.6, overall accuracy increased to 88% (95% CI: 73%-97%; supplemental Table 13).
Our initial review of cases suspected to be lymphoma included 32 cases with diagnoses that were not included within the 9 bins (supplemental Table 2B). These included chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL; n = 14), plasma cell neoplasms (n = 7), T-lymphoblastic lymphoma (n = 4), blastic plasmacytoid dendritic cell neoplasm (n = 1), carcinoma (n = 3), plasmablastic lymphoma (n = 1), and neuroectodermal tumor (n = 1). We hypothesized that these biopsy specimens would be classified as indeterminate by the CLPA. Indeed 25 of 32 (78%) cases had probability <0.6 and were correctly classified as indeterminate. Of the 7 cases with probability >0.6, n = 2 were called DLBCL by CLPA but CLL/SLL by IHC, n = 1 was called HL by CLPA and plasmablastic lymphoma by IHC, n = 1 was called non-malignant but CLL/SLL by IHC, and n = 1 was called TCL by CLPA but T-lymphoblastic lymphoma by IHC.
Finally, we performed a cost analysis based on our experience purchasing supplies in Guatemala. The cost of manufacturing assay-specific reagents is approximately $5 per sample. Running 95 samples per week with appropriate controls resulted in a cost of $6.76 per sample for reagents plus consumables (supplemental Table 14). Cost increases with decreasing test volume but does not exceed $15 per sample for 16 tests per week.
Discussion
The lack of access to adequate pathology services is a critical roadblock that limits improvements in health care across LMICs.16 Several groups from the United States and Europe have established partnerships with medical centers in LMICs; these typically use telemedicine, build critical infrastructure, and enhance the training of local clinicians. Although such efforts can markedly increase capacity at partner sites, they fail to improve outcomes for most patients across the world.17
We were inspired by the remarkable success in treatment of chronic myelogenous leukemia orchestrated by the Max Foundation (https://www.themaxfoundation.org/). In collaboration with pharmaceutical companies, the Max Foundation has extended approximately 10 million doses of imatinib and other high-cost drugs to patients who have a confirmed BCR-ABL rearrangement across > 70 countries. Those diagnoses were enabled by the development of a low-cost BCR-ABL assay on the GeneXpert platform,18 which is widely available across LMICs. We similarly designed our assay to use equipment that is available in many LMICs; based on an informal survey of potential partners in Latin America, most large centers have access to a PCR machine and capillary electrophoresis instruments, either within their hospital/university or by collaborating with private laboratories.
We hypothesized that a more extensive assay that quantifies expression across a larger gene set could classify lymphomas into treatment-directed bins. Our findings indicate that, even in complex diagnostic settings like lymphoma, gene expression–based testing can be both more accurate and less expensive than currently available strategies in LMICs. The CLPA accurately classified biopsy specimens, including FFPE blocks that were stored at room temperature in a tropical climate for more than 10 years into 9 bins. Assay failure among biopsy specimens from 2015 or later (ie, <3 years before CLPA testing) was only 2%.
The CLPA assay was performed directly from FFPE sections and did not require prereview to ensure high tumor cell content. In addition to binning lymphomas, the CLPA quantified the expression of specific transcripts that could guide targeted therapies, such as MS4A1/CD20 and MME/CD30 for monoclonal antibodies or ALK for anaplastic lymphoma kinase (ALK) inhibitor therapy. It is worth noting that many of the genes included in the panel have also been associated with specific diagnoses and could be used to further subcategorize within bins (eg, SOX8, MME, BCL6, TBX21, ICOS, and GATA3 for TCLs19) or are associated with outcomes (eg, MKI6720).
The overall concordance between the CLPA and IHC-based diagnosis ranged between 79% and 94%, depending on the cohort and stringency of calling. However, the true accuracy of the assay is somewhat unclear for 2 reasons. First, clinical pathology testing is a highly problematic standard for benchmarking accuracy. Discordance rates between expert hematopathologists in the diagnosis of lymphoma are typically 10% in high-income countries17,21 and exceed 30% in LMICs.22 Many academic centers in high-income countries require that lymphoma pathology be rereviewed at the time of second opinion because of the high rates of misdiagnosis. We addressed this by using 2 independent hematopathologists to confirm all diagnoses.
The second reason that clinical pathology testing is a problematic standard relates to interpatient heterogeneity in lymphoma biology. Gene expression and sequencing studies have consistently reported that a fraction of lymphomas diagnosed as 1 entity based on IHC cluster more closely in unsupervised analyses with a separate entity (eg, cases of DLBCL that are Burkitt-like12). Considering these factors, prospective treatment studies using CLPA for diagnosis will be required to clarify whether subsets of patients are truly misclassified in ways that compromise outcome. Adding clinical characteristics (eg, site of presentation, duration of symptoms), patient demographics (age, sex, HIV status), and other available information (eg, local epidemiology) to the machine learning model for CLPA-based binning could further improve the accuracy of the assay.
Many types of lymphoma are either curable or highly responsive to therapeutic regimens that are accessible within LMICs, including CVP (cyclophosphamide, vincristine, and prednisone) or CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) chemotherapy for follicular, marginal zone, mantle cell, peripheral T-cell, and DLBCL, CODOX-m (cyclophosphamide, vincristine, doxorubicin, and methotrexate) or hyper-CVAD (hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone) chemotherapy for Burkitt or B-lymphoblastic lymphoma, ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine) chemotherapy for HL, and SMILE (dexamethasone, methotrexate, ifosfamide, L-asparaginase, and etoposide) chemotherapy for extranodal NKTCL.23,24 Generic versions of some targeted agents and biosimilars for rituximab are also available, whereas others (eg, BTK inhibitors, venetoclax, phosphatidylinositol 3-kinase inhibitors, brentuximab) could be made available, as ABL inhibitors have been for chronic myelogenous leukemia and gastrointestinal stromal tumors. It will simply require forward-thinking pharmaceutical companies and appropriate advocacy.
We used available treatment guidance and the epidemiology of lymphoma subtypes to design our diagnostic bins. As treatment options evolve, refinements to the binning of cases and selection of genes will undoubtedly be required to maximize clinical utility. For example, we did not use fluorescence in situ hybridization results to distinguish DLBCL from high grade B-cell lymphoma with MYC, BCL2, and/or BCL6 rearrangements (double/triple hit lymphoma). Although more aggressive regimens may improve outcomes for patients with MYC rearrangement,25,26 recent classifications suggest that some non–MYC-rearranged DLBCLs have outcomes and biology that highly overlap with MYC-rearranged cases.27 Thus, it remains appropriate (and standard of care in many LMICs) to treat these cases with CHOP-based therapy.23
Our assay has multiple limitations that must be carefully considered. First is the inclusion of B-cell lymphoblastic lymphoma and Burkitt lymphoma in the same diagnostic bin. Together, these accounted for only 10 cases diagnosed by lymph node biopsy at INCAN over the previous 12 years. On the rare occasion that this call is made, H&E and IHC staining for terminal deoxynucleotide transferase could guide the final diagnosis. The 15% to 20% of all cases, including biopsy specimens from diseases not binned using our current assay, that have a probability score < 0.6 would also require additional assessments, including IHC. Among the latter, CLL/SLL and plasma cell neoplasms are more frequently diagnosed from blood or bone marrow at INCAN than by lymph node biopsy, but this may vary across institutions. Of note, the number of genes using standard capillary electrophoresis equipment and our current chemistry can be easily expanded to 55. Thus, we are redesigning the assay to distinguish Burkitt, B-cell lymphoblastic lymphoma, and plasma cell neoplasms. We are also testing additional cohorts in other LMICs to improve the diagnostic accuracy of lymphoma subtypes that were infrequent at INCAN (eg, marginal zone lymphoma, TCL), to ensure assay performance across populations with different genetic backgrounds and coexisting pathology (eg, tuberculosis, HIV) and to quantify assay performance on needle biopsies. For the latter, it is worth noting that the CLPA training and validation testing described above was performed on a single 10-μm section from excisional biopsy specimens that ranged in size from 5 × 10 mm2 to 20 × 20 mm2. This results in a tissue volume of 0.5 to 4.0 mm3, which is comparable to one-half or less of a needle biopsy. Core biopsies are frequently used for clinical and research sequencing, consistent with the conclusion that they contain an adequate quantity of genetic material for diagnosis.
Based on the assay performance at INCAN, approximately 75% of patient biopsy specimens would be accurately binned, approximately 17% would be called indeterminate, 6% would be incorrectly binned, and 2% would fail the assay. As a proof-of-principle, this performance markedly exceeds the status quo in many LMIC settings and thus could improve diagnosis as a primary test for lymphoma. Limited resources could then be allocated to patients with indeterminate calls. The medicolegal considerations for an assay that incorrectly bins 6% of patients will vary greatly by country, but as for all laboratory tests, the results should always be considered within the context of an individual patient’s presentation and the ordering physician’s clinical judgement.
We initiated a prospective study of CLPA-based diagnosis that extends across centers in Guatemala, El Salvador, and Belize. The assay is performed entirely at INCAN. Turnaround from biopsy to reporting can be performed at INCAN in <24 hours when urgent results are needed, although this does increase cost. Finally, we are establishing an open-access R Shiny App that allows anyone on Earth to input CLPA data and receive a diagnosis and probability score from the machine-learning model. Similar gene sets and calling algorithms can easily be established for other cancer types and thereby guide optimal therapeutic selection for patients in desperate need.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Ani A. Avanian for assistance with the visual abstract.
These studies were supported by an American Society of Hematology Global Research Award (F.V.), a Fulbright Scholar Award (E.L.B.), an American Society of Clinical Oncology Conquer Cancer Foundation Young Investigator Award (E.L.B.), a Leukemia and Lymphoma Society New Idea Award (D.M.W.), the Celgene Cancer Care Links Program (D.M.W.), and National Institutes of Health, National Cancer Institute grant R35CA231958 (D.M.W.).
Footnotes
Probe sequences are available from the corresponding author (davidm_weinstock@dfci.harvard.edu) upon request.
Authorship
Contribution: E.S.-O., M.M.S.T., T.G., N.L., F.L., C.C.C.A., and S.L.D. designed and performed experiments; M.P. and K.E.S. analyzed data and drafted the manuscript; F.V., O.S., Y.N., and E.L.B. designed and performed experiments, analyzed data, and drafted the manuscript; and R.T. and D.M.W. designed experiments and drafted the manuscript.
Conflict-of-interest disclosure: T.G., S.L.D., and R.T. are employees of DxTerity Diagnostics. D.M.W. is a cofounder of Travera, Ajax, and Root Diagnostics; receives consulting or advisory board fees from Magnetar, Bantam, ASELL, Daiichi Sankyo, Secura Bio, and AstraZeneca; and receives research funding from Daiichi Sankyo, Abcuro, and Verastem. The remaining authors declare no competing financial interests.
Correspondence: David Weinstock, Dana-Farber Cancer Institute, 450 Brookline Ave, Dana 510B, Boston, MA 02215; e-mail: davidm_weinstock@dfci.harvard.edu.
References
- 1.Cazap E, Magrath I, Kingham TP, Elzawawy A. structural barriers to diagnosis and treatment of cancer in low- and middle-income countries: the urgent need for scaling up. J Clin Oncol. 2016;34(1):14-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization . Second WHO model list of essential in vitro diagnostics. https://www.who.int/docs/default-source/nutritionlibrary/complementary-feeding/second-who-model-list-v8-2019.pdf?sfvrsn=6fe86adf_1. Accessed 1 June 2020.
- 3.Eniu AE, Martei YM, Trimble EL, Shulman LN. Cancer care and control as a human right: recognizing global oncology as an academic field. Am Soc Clin Oncol Educ Book. 2017;37:409-415. [DOI] [PubMed] [Google Scholar]
- 4.Farmer P, Frenk J, Knaul FM, et al. Expansion of cancer care and control in countries of low and middle income: a call to action. Lancet. 2010;376(9747):1186-1193. [DOI] [PubMed] [Google Scholar]
- 5.Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375-2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maheu-Giroux M, Marsh K, Doyle CM, et al. National HIV testing and diagnosis coverage in sub-Saharan Africa: a new modeling tool for estimating the “first 90” from program and survey data. AIDS. 2019;33(suppl 3):S255-S269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carey CD, Gusenleitner D, Chapuy B, et al. Molecular classification of MYC-driven B-cell lymphomas by targeted gene expression profiling of fixed biopsy specimens. J Mol Diagn. 2015;17(1):19-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Geiss GK, Bumgarner RE, Birditt B, et al. Direct multiplexed measurement of gene expression with color-coded probe pairs [published correction appears in Nat Biotechnol. 2008;26:709]. Nat Biotechnol. 2008;26(3):317-325. [DOI] [PubMed] [Google Scholar]
- 9.Bobée V, Ruminy P, Marchand V, et al. Determination of molecular subtypes of diffuse large B-cell lymphoma using a reverse transcriptase multiplex ligation-dependent probe amplification classifier: a CALYM study. J Mol Diagn. 2017;19(6):892-904. [DOI] [PubMed] [Google Scholar]
- 10.Mareschal S, Ruminy P, Bagacean C, et al. Accurate classification of germinal center B-cell-like/activated B-cell-like diffuse large B-cell lymphoma using a simple and rapid reverse transcriptase-multiplex ligation-dependent probe amplification assay: a CALYM study. J Mol Diagn. 2015;17(3):S1525-1578(15)00046-X. [DOI] [PubMed] [Google Scholar]
- 11.Mottok A, Wright G, Rosenwald A, et al. Molecular classification of primary mediastinal large B-cell lymphoma using routinely available tissue specimens. Blood. 2018;132(22):2401-2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dave SS, Fu K, Wright GW, et al. ; Lymphoma/Leukemia Molecular Profiling Project . Molecular diagnosis of Burkitt’s lymphoma. N Engl J Med. 2006;354(23):2431-2442. [DOI] [PubMed] [Google Scholar]
- 13.Marinelli RJ, Montgomery K, Liu CL, et al. The Stanford Tissue Microarray Database. Nucleic Acids Res. 2008;36(database issue):D871-D877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wright G, Tan B, Rosenwald A, Hurt EH, Wiestner A, Staudt LM. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc Natl Acad Sci USA. 2003;100(17):9991-9996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim CH, Abedi M, Liu Y, et al. A novel technology for multiplex gene expression analysis directly from whole blood samples stabilized at ambient temperature using an RNA-stabilizing buffer. J Mol Diagn. 2015;17(2):118-127. [DOI] [PubMed] [Google Scholar]
- 16.Wilson ML, Fleming KA, Kuti MA, Looi LM, Lago N, Ru K. Access to pathology and laboratory medicine services: a crucial gap. Lancet. 2018;391(10133):1927-1938. [DOI] [PubMed] [Google Scholar]
- 17.Laurent C, Baron M, Amara N, et al. Impact of expert pathologic review of lymphoma diagnosis: study of patients from the French Lymphopath Network. J Clin Oncol. 2017;35(18):2008-2017. [DOI] [PubMed] [Google Scholar]
- 18.Dufresne SD, Belloni DR, Levy NB, Tsongalis GJ. Quantitative assessment of the BCR-ABL transcript using the Cepheid Xpert BCR-ABL Monitor assay. Arch Pathol Lab Med. 2007;131(6):947-950. [DOI] [PubMed] [Google Scholar]
- 19.Iqbal J, Wright G, Wang C, et al. ; Lymphoma Leukemia Molecular Profiling Project and the International Peripheral T-cell Lymphoma Project . Gene expression signatures delineate biological and prognostic subgroups in peripheral T-cell lymphoma. Blood. 2014;123(19):2915-2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He X, Chen Z, Fu T, et al. Ki-67 is a valuable prognostic predictor of lymphoma but its utility varies in lymphoma subtypes: evidence from a systematic meta-analysis. BMC Cancer. 2014;14(1):153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strobbe L, van der Schans SA, Heijker S, et al. Evaluation of a panel of expert pathologists: review of the diagnosis and histological classification of Hodgkin and non-Hodgkin lymphomas in a population-based cancer registry. Leuk Lymphoma. 2014;55(5):1018-1022. [DOI] [PubMed] [Google Scholar]
- 22.Özkaya N, Başsüllü N, Demiröz AS, Tüzüner N. Discrepancies in lymphoma diagnosis over the years: a 13-year experience in a tertiary center. Turk J Haematol. 2017;34(1):81-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zelenetz AD, Gordon LI, Abramson JS, et al. NCCN guidelines insights: B-cell lymphomas, version 3.2019. J Natl Compr Canc Netw. 2019;17(6):650-661. [DOI] [PubMed] [Google Scholar]
- 24.Horwitz SM, Ansell SM, Ai WZ, et al. NCCN guidelines insights: T-cell lymphomas, version 2.2018. J Natl Compr Canc Netw. 2018;16(2):123-135. [DOI] [PubMed] [Google Scholar]
- 25.Dunleavy K, Fanale MA, Abramson JS, et al. Dose-adjusted EPOCH-R (etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, and rituximab) in untreated aggressive diffuse large B-cell lymphoma with MYC rearrangement: a prospective, multicentre, single-arm phase 2 study. Lancet Haematol. 2018;5(12):e609-e617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bartlett NL, Wilson WH, Jung SH, et al. Dose-adjusted EPOCH-R compared with R-CHOP as frontline therapy for diffuse large B-cell lymphoma: clinical outcomes of the phase III intergroup trial alliance/CALGB 50303. J Clin Oncol. 2019;37(21):1790-1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chapuy B, Stewart C, Dunford AJ, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes [published corrections appear in Nat Med. 2018;24:1292 and Nat Med. 2018;24:1290-1291]. Nat Med. 2018;24(5):679-690. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.