

Abstract
The development of a general method utilizing a hydroxymethyl directing group for asymmetric hydrogenation of 1,3-disubstituted isoquinolines to provide chiral 1,2,3,4-tetrahydroisoquinolines is reported. The reaction, which utilizes [Ir(cod)Cl]2 and a commercially available chiral xyliphos ligand, proceeds in good yield with high levels of enantioselectivity and diastereo-selectivity (up to 95% ee and >20:1 dr) on a range of differentially substituted isoquinolines. Directing group studies demonstrate that the hydroxymethyl functional group at the C1-position is more efficient at enabling hydrogenation than other substituents, although high levels of enantioselectivity were conserved across a variety of polar and non-polar functional groups. By utilizing the generated chiral β-amino alcohol as a functional handle, the synthetic utility is further highlighted via the synthesis of 1,2-fused oxazolidine, oxazolidinone, and morpholinone tetrahydroisoquinolines in one step. Additionally, a non-natural analog of the tetrahydroprotoberberine alkaloids was successfully synthesized.
Keywords: hydrogenation, asymmetric catalysis, heterocycles, isoquinolines, tetrahydroisoquinolines
Graphical Abstract

INTRODUCTION
The stereocontrolled synthesis of nitrogen-containing heterocycles remains a challenge of great importance, as it provides direct access to chiral compounds that are prevalent structural motifs in many biologically active molecules.1 As a result, the asymmetric hydrogenation of various heteroaromatic compounds has been extensively explored as a direct, efficient synthesis of enantiopure cyclic amines.2 Despite recent progress made toward the asymmetric hydrogenation of N-heterocycles such as quinolines, quinoxalines, and pyridines, the synthesis of 1,2,3,4-tetrahydroisoquinolines (THIQs) from isoquinolines remains significantly underdeveloped (Figure 1A).2 This is due in part to the stronger basicity and coordinating ability of the THIQ products compared to those of other heterocycles (e.g., quinolines), leading to catalyst deactivation, as well as the overall lower reactivity of isoquinoline substrates.3 Although a few effective strategies toward the asymmetric hydrogenation of substituted isoquinolines have been reported, these typically require preparation of the isoquinolinium salt, substrate activation with halogenides, and harsher hydrogenation reaction conditions (Figure 1B).4 Furthermore, previous to our research, there were only 2 catalytic systems describing efficient methods to access chiral 1,3-disubstituted tetrahydroisoquinolines,4c,e,g a more complex and sterically challenging system that generates two stereogenic centers. In addition, the limited substrate scope from these reports demonstrates the low tolerance of additional Lewis basic functionalities, such as alcohols or heteroaryl-substituted isoquinolines, which limit the applicability of these methodologies in synthesis. Since 1,3-disubstituted tetrahydroisoquinolines with Lewis basic moieties are ubiquitous motifs present in a wide range of natural products, such as the saframycin, naphthyridinomycin, and quinocarcin families,5 a general method for highly enantioselective and diastereoselective hydrogenation of neutral disubstituted isoquinolines under mild reaction conditions would be a significant advancement toward the preparation of chiral amine-containing cyclic molecules.
Figure 1.
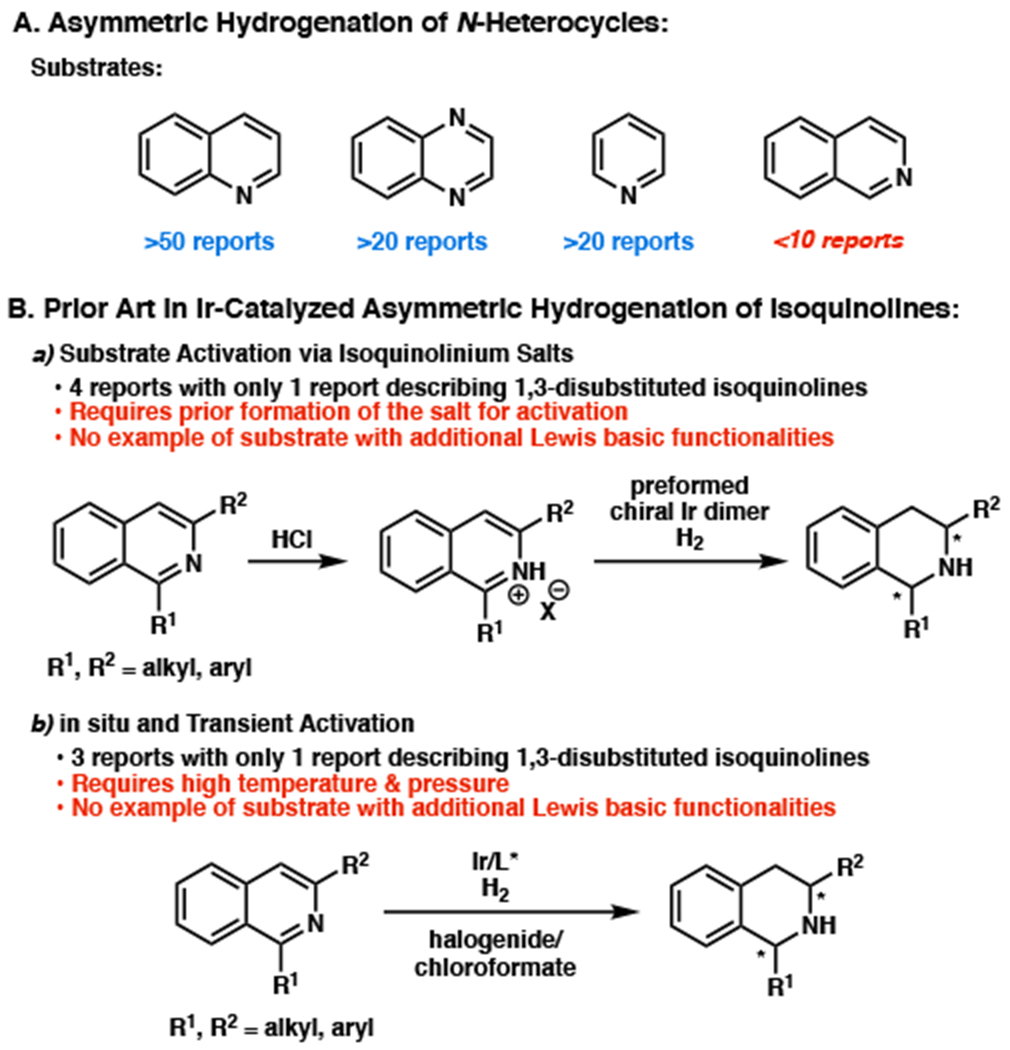
(a) Limitations in enantioselective hydrogenation of N-heterocycles. (b) Previous examples of iridium-catalyzed enantioselective and diastereoselective hydrogenation of mono-, and di-substituted isoquinolines.
Recently, our group has successfully completed the total synthesis of jorumycin and jorunnamycin A, two bistetrahydroisoquinoline natural products that exhibit potent antiproliferative activity, as well as strong Gram-positive and Gram-negative antibiotic character.6 Through an unprecedented, nonbiomimetic synthetic route, we were successful in harnessing catalysis to allow expedient access to these natural products, as well as a diverse range of non-natural analogs that are otherwise inaccessible using prior biomimetic synthetic approaches. One of the key steps of this synthesis involves a catalytic enantioselective hydrogenation of bis-isoquinoline 1 to afford the THIQ motif, a crucial intermediate that forms the pentacyclic carbon skeleton 2 in one step by further hydrogenation of the second isoquinoline and eventual amide ring closure (Figure 2). Considering the ubiquity of the hydroxymethyl group at the C1 position, and the amino alcohol functionality in a large number of natural products, we envision that this synthetic method could access a wide variety of THIQs as well as chiral amino alcohols, both highly valuable pharmacophores. Following the successful development of our asymmetric hydrogenation technology for bis-isoquinolines, herein we disclose a mild, general method for the enantioselective and diastereoselective hydrogenation of 1,3-disubstituted isoquinolines.
Figure 2.
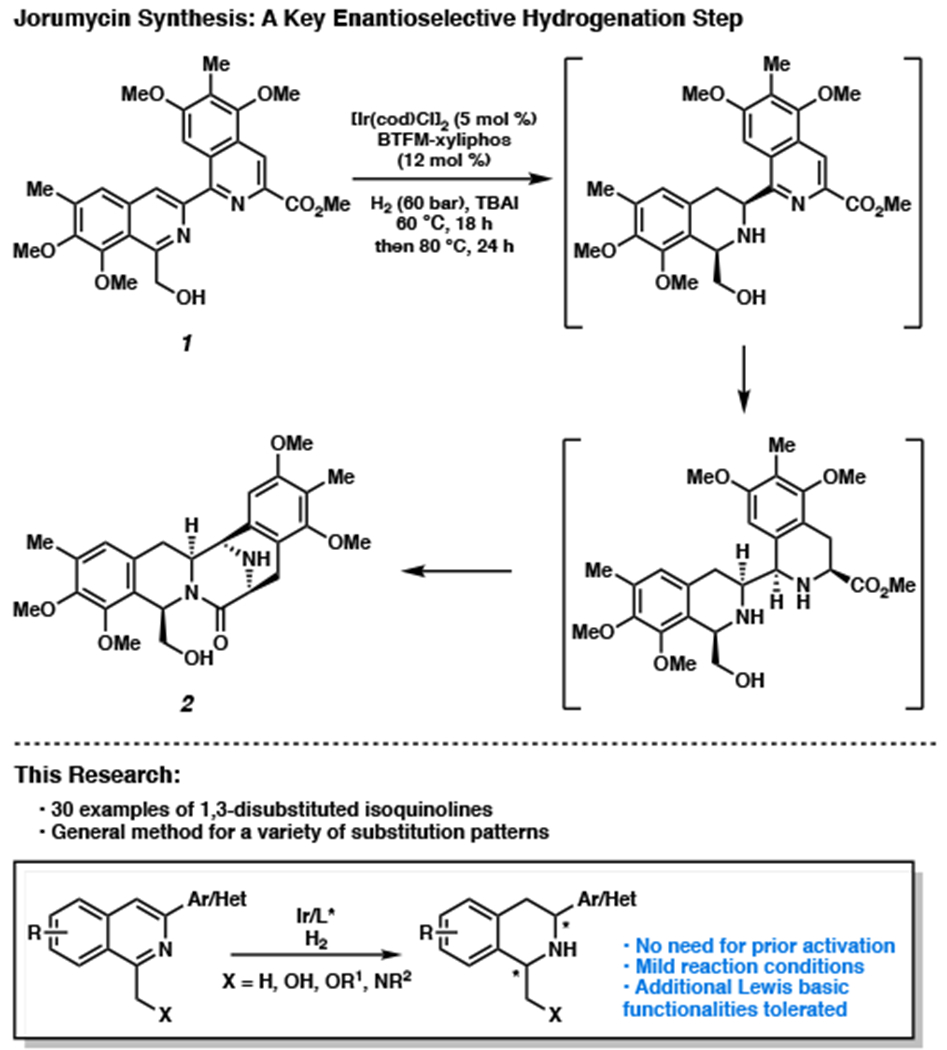
Our research on iridium-catalyzed enantioselective and diastereoselective hydrogenation of 1,3-disubstituted isoquinolines.
RESULTS AND DISCUSSION
Substrate Syntheses.
Due to a limited number of methods for the syntheses of 1,3-disubstituted isoquinolines,7 we first established a simple and divergent sequence to access a wide variety of 1-(hydroxymethyl)-3-arylisoquinoline substrates (i.e., 7, Scheme 1). Utilizing Pd-catalyzed arylation of ester enolates reported by Donohoe and coworkers,8 mono-arylated tert-butyl acetate 4 was isolated in an excellent 92% yield. Cyclization to isoquinoline triflate 5 was then achieved via hydrolysis of the ketal, followed by isoquinoline annulation with aqueous ammonium hydroxide, and alcohol triflation.9 At this stage, different aryl or heteroaryl groups could be coupled with intermediate 5 using Suzuki coupling conditions to deliver a wide range of 1,3-disubstituted isoquinolines (i.e., 6), highlighting the divergent synthesis of our synthetic route. Finally, SeO2 oxidation to the aldehyde and subsequent NaBH4 reduction provided our desired isoquinoline starting materials 7a–r. It is worth noting that this sequence allows for an introduction of various aryl and heteroaryl groups at the C3-position of isoquinolines, as well as different substituents with varied electronics on the isoquinoline carbocycle (e.g., 9a–f, vide infra, Scheme 4). Currently in the literature, the 1,3-disubstituted isoquinoline motif is typically accessed via transition-metal-catalyzed tandem C–H activation/annulation of arenes with alkynes.10 These methods have shown limited success in producing C3-heteroaryl isoquinolines.11
Scheme 1. Syntheses of 1-(hydroxymethyl)-3-aryl Isoquinoline Substratesa.
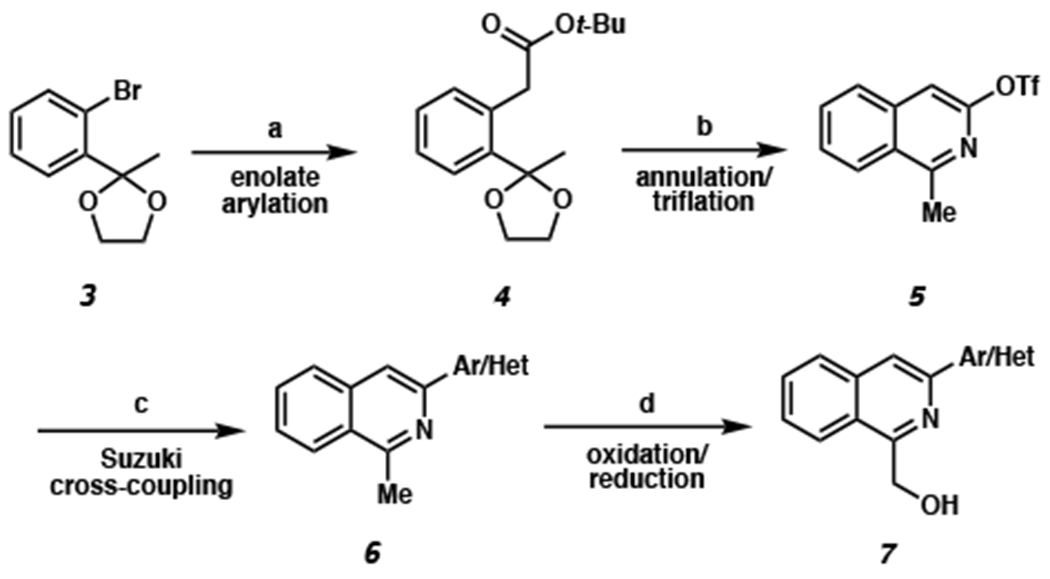
aConditions: (a) t-butyl acetate (2 equiv), LiHMDS (2.5 equiv), Pd2(dba)3 (5 mol %), P(t-Bu3)•HBF4 (10 mol %), toluene, 23 °C, 18 h, 92% yield. (b) 33% TFA in CH2Cl2, 23 °C, 2 h; then aq. NH4OH, MeCN, 70 °C, 12 h; then Tf2O (2 equiv), pyridine, CH2Cl2, 0 °C, 1 h, 38% yield over 3 steps. (c) aryl/heteroaryl boronic acid (1.5 equiv), XPhos Pd G3 (2 mol %), 2:1 K3PO4:THF, 40 °C, 2 h, 77–95% yield. (d) SeO2 (2 equiv), 1,4-dioxane, 110 °C, 2 h; then NaBH4 (1 equiv), 4:1 CH2Cl2:MeOH, 10 min, 25–97% yield.
Scheme 4. Substrate Scope of IQ Backbone Substituentsa.
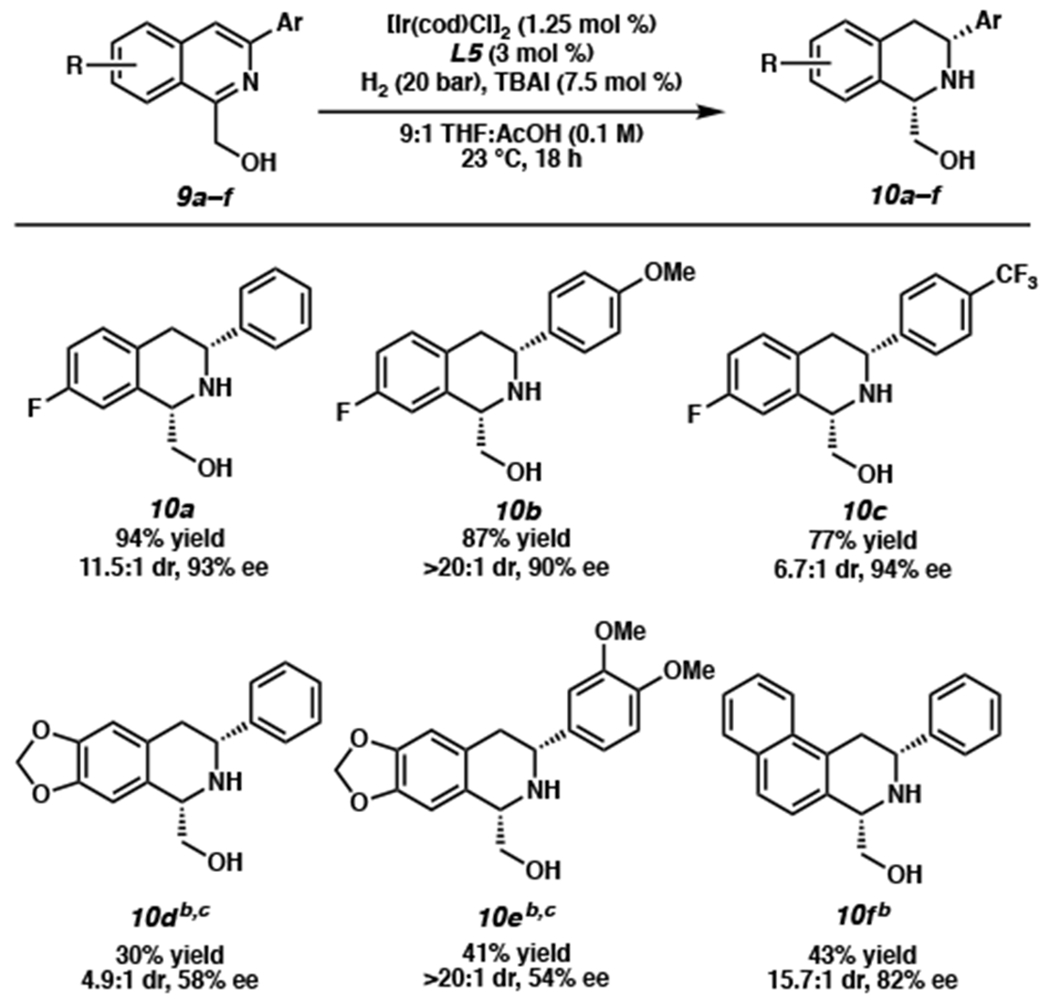
aReactions performed on a 0.2 mmol scale. bReaction performed at 60 °C and 60 bar H2. cCH2Cl2 cosolvent used to improve substrate solubility.
Reaction Optimization.
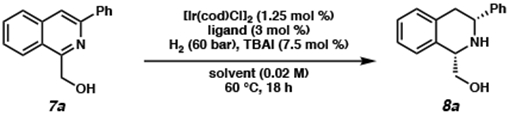
With a divergent sequence to access 1,3-disubstituted isoquinolines, we began our hydrogenation studies with 1-(hydroxymethyl)-3-phenylisoquinoline (7a) as our model substrate, using slightly modified conditions from our previously reported hydrogenation on the bis-isoquinoline system (Table 1). An initial experiment, employing 1.25 mol % [Ir(cod)Cl]2 and 3 mol % of the BTFM-xyliphos ligand (L1), gave high conversion of the substrate but surprisingly modest enantioselectivity (49% ee, entry 1). Seeking to improve the ee, we surveyed a wide variety of chiral ligand scaffolds (see Supporting Information) and found the xyliphos ligand framework to be optimal. By exploring different electronics of this ligand scaffold, we observed that replacing the 3,5-bistrifluoromethylphenyl (BTFM) with more electron-rich aryl groups provided the product with both excellent conversion and higher enantioselectivity (entries 1–5). Ligand L5, which features 4-methoxy-3,5-dimethylphenyl (DMM) substituted phosphine delivers the product with the highest ee of 89%, albeit with a low 2:1 diastereoselectivity (entry 5). Interestingly, a background reaction was observed in the absence of the chiral ligand, providing the product in excellent conversion and diastereoselectivity (entry 6). From this finding, we obtained all racemic hydrogenated products by simply performing the hydrogenation in the absence of ligand, affording the cis-product as a single diastereomer.
Table 1.
Optimization of the Enantioselective Hydrogenation of Isoquinolines to afford THIQsa
 | |||||
|---|---|---|---|---|---|
| entry | ligand | Solvent | % conversionb | cis:transb | % ee of cisc |
| 1 | L1 | PhMe:AcOH | >95 | >20:1 | −49d |
| 2 | L2 | PhMe:AcOH | >95 | 3.2:1 | 84 |
| 3 | L3 | PhMe:AcOH | 66 | 4.5:1 | 63 |
| 4 | L4 | PhMe:AcOH | 92 | 4.9:1 | 3 |
| 5 | L5 | PhMe:AcOH | >95 | 2.0:1 | 89 |
| 6 | – | PhMe:AcOH | >95 | >20:1 | 0 |
| 7 | L5 | CH2Cl2:AcOH | >95 | 1.5:1 | 84 |
| 8 | L5 | dioxane:AcOH | 66 | 5.5:1 | 87 |
| 9 | L5 | THF:AcOH | >95 | 9.7:1 | 90 |
| 10 | L5 | CPME:AcOH | 38 | >20:1 | 88 |
| 11 | L5 | 2-MeTHF:AcOH | >95 | 9.5:1 | 90 |
| 12e | L5 | THF:AcOH | >95 | 15.7:1 | 92 |
 | |||||
Conditions: 0.04 mmol 7a, 1.25 mol % [Ir(cod)Cl]2, 3 mol % ligand, 7.5 mol % TBAI, 60 bar H2 in 2.0 mL 9:1 solvent:AcOH.
Determined from crude 1H NMR using 1,3,5-trimethoxybenzene as standard.
Determined by chiral SFC analysis of Cbz-protected product.
Opposite enantiomer of ligand used.
Reaction performed on a 0.2 mmol scale at 23 °C, 20 bar H2, and 0.1 M concentration of 7a.
Investigation of different solvents with L5 as the optimal ligand reveals that while the use of CH2Cl2 provided similar results to toluene (entry 7), the diastereoselectivity could be improved with the use of ethereal solvents (entries 8–11). Although bulkier ethereal solvents proved to worsen conversion (entries 8 and 10), we were delighted to find that at THF and the more sustainable solvent 2-MeTHF delivered the product in excellent conversion with high levels of diastereoselectivity and enantioselectivity (entries 9 and 11). The absence of AcOH resulted in low conversion,12 while further exploration of different additives (e.g., LiI, NaI, KI, etc.) demonstrated that TBAI is the optimal additive (see Supporting Information). Finally, we were excited to observe that lowering the temperature to 23 °C and H2 pressure to 20 bar maintained excellent levels of conversion, diastereoselectivity, and enantioselectivity (entry 12). To the best of our knowledge, these are the mildest reaction conditions reported for isoquinoline hydrogenation to afford chiral THIQs to date.4
Substrate Scope.
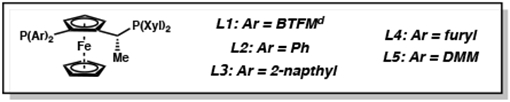
With optimized reaction conditions identified, we explored the general substrate scope for this transformation (Scheme 2). Gratifyingly, a wide variety of aryl substituents at the 3-position of the isoquinoline are well tolerated under the mild reaction conditions of 20 bar H2 at ambient temperature. Substitution at the para-position of the 3-aryl ring delivered the hydrogenated products 8a–g in consistently high yields and enantioselectivity. Electron-rich substrates such as the 3-(p-tert-butylphenyl)isoquinoline (7b) and the p-methoxyphenyl (7d) afforded chiral THIQs with excellent yields, diastereoselectivity, and enantioselectivity, similar to 7a. Interestingly, however, a general trend of lower diastereoselectivity was observed with electron-withdrawing substituents both at the para- and meta- positions (8f–i). We envision that the observed lower diastereoselectivity arises from the weaker coordinating ability of the nitrogen to the iridium catalyst in electron-poor substrates, discouraging coordination to the catalyst in a bidentate fashion,13 and thus resulting in poorer facial selectivity in the second hydride addition step. However, at this time, we still cannot rule out the possibility that epimerization in situ also influences the trend seen in diastereoselectivity.14 Investigation of steric effects revealed that more sterically encumbered isoquinolines such as the 3-napthyl and 3-xylyl substrates furnished the products (8j–k) in excellent isolated yields with similarly high enantioselectivity (95% and 92% ee, respectively) as a single diastereomer. The most sterically demanding substrate 7m, bearing an ortho-tolyl substituent, provided product 8m in a modest 64% yield with lower enantioselectivity (49% ee), albeit still with a high 10.1:1 diastereoselectivity. Additionally, we were pleased to find that the nitrile and nitro functional groups as well as the napthyl substituent were not reduced (8g, 8h, and 8j), highlighting the chemoselectivity of this catalytic process.
Scheme 2. Substrate Scope of Different Aryl Substituentsa.
aReactions performed on a 0.2 mmol scale. bReaction performed at 60 °C and 60 bar H2. cCH2Cl2 cosolvent used to improve substrate solubility.
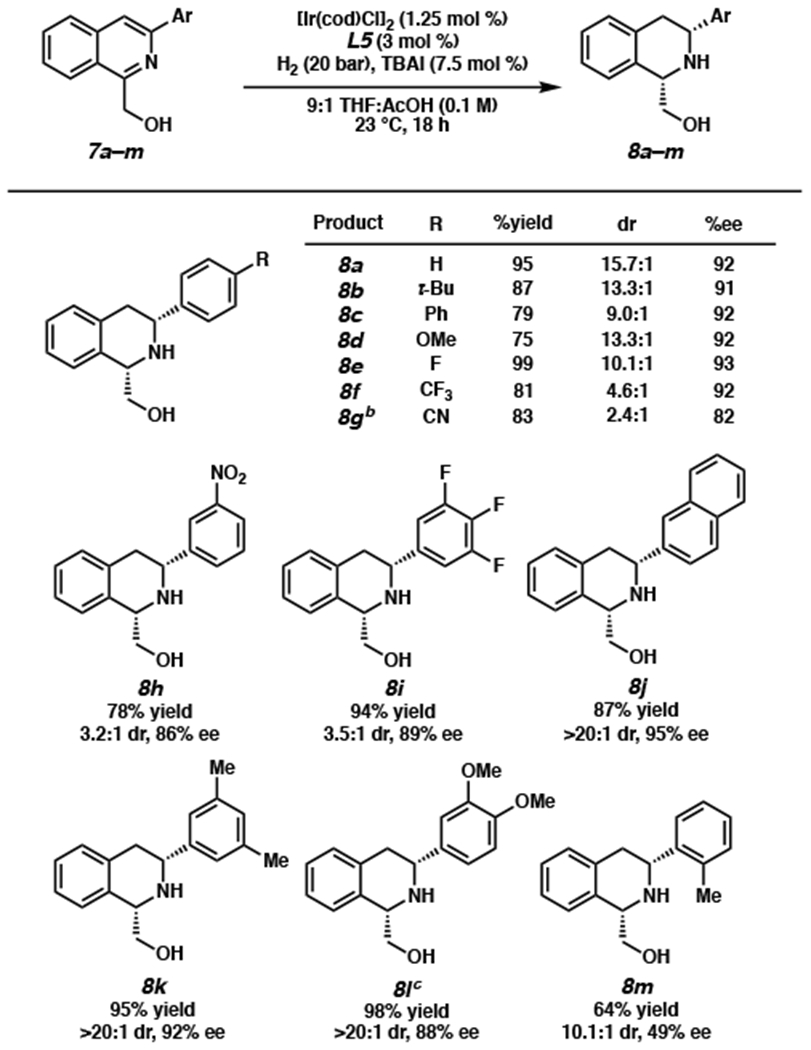
Pleased to find the reaction tolerable to a range of 3-aryl substituted isoquinolines, we sought to further extend the scope of the transformation by exploring heteroaryl-substituted isoquinolines (Scheme 3). Although performing the hydrogenation at 23 °C and 20 bar H2 resulted in lower conversion, partially due to solubility issues, we found that heterocyclic substituents including furan, thiophene, pyrazole, and pyridine were well tolerated at 60 °C and under higher pressure of 60 bar H2, producing THIQs 8n–r. We also observed that the substitution pattern on the heteroaryl groups strongly affects the reaction conversion. For instance, an isoquinoline with a 3-substituted thiophene proceeded with a significantly lower conversion than the 2-thiophene substrate (8o and 8p). Similarly, no conversion was observed with 3- and 4-pyridyl substrates, whereas 2-pyridyl THIQ 8r was isolated in 48% yield under the same reaction conditions. We speculate that this may be due to the competitive binding of the catalyst by the more distal heteroatom of 3- and 4-substituted heterocycles that inhibits directed hydrogenation of the isoquinoline ring. From product 8p we were successful in obtaining an X-ray crystal structure to confirm the relative and absolute stereochemistry of our hydrogenation product.
Scheme 3. Substrate Scope of Heterocyclic Substituentsa.
aReactions performed on a 0.2 mmol scale
Furthermore, we were interested in exploring the substrate scope of isoquinolines with different electronics and substitution patterns on the isoquinoline carbocycle (Scheme 4). Electron-poor fluorinated isoquinolines with varied electronics at the 3-position provided THIQs in high yields (77–94%) with high diastereoselectivity and enantioselectivity under our standard conditions (10a–c). It should be noted that these electron-poor THIQs would be difficult to access utilizing electrophilic aromatic substitution strategies, a classical method for the syntheses of THIQs. On the other hand, the electron-rich dioxolane-appended isoquinolines (9d–e) afforded lowered conversion under the same reaction conditions, due in part to the poor solubility of the substrates in THF. Nevertheless, executing the reaction at 60 °C and 60 bar H2 with CH2Cl2 as cosolvent improved the solubility and conversion to yield products 10d–e with high diastereoselectivity.15 Interestingly, we observed significantly lower enantioselectivity for the dioxolane THIQs, which is observed in other reports as well.4a,g Finally, the napthyl-fused THIQ 10f was obtained with high diastereoselectivity and enantioselectivity, despite its extended aromatic system and larger steric hindrance. Although we observed modest conversion for these highly decorated isoquinolines, we were pleased to see that we were able to isolate unreacted starting material after column chromatography to obtain 80%, 99%, and 79% yield, respectively, of 10d–f based on recovered starting material. Consistent with our results for THIQs 8j and 8r, we observed that only the ring with the least degree of aromatic stabilization was reduced for 10f.
Directing Group Studies.
Having demonstrated that this transformation is general for a wide variety of 1,3-disubstituted isoquinolines, we then turned our attention to investigate the effects of different “directing” groups at the C1-position (Scheme 5). Isoquinolines bearing other polar groups such as an ester (12a), ethers (12b–c), and a Boc-protected amine (12d) delivered the products in lower yields than the hydroxy-directed substrate at 23 °C and 20 bar H2. However, to our delight, by increasing the temperature and H2 pressure, these yields could be improved with no erosion of enantioselectivity and diastereoselectivity.
Scheme 5. Substrate Scope of Different Directing Groupsa.
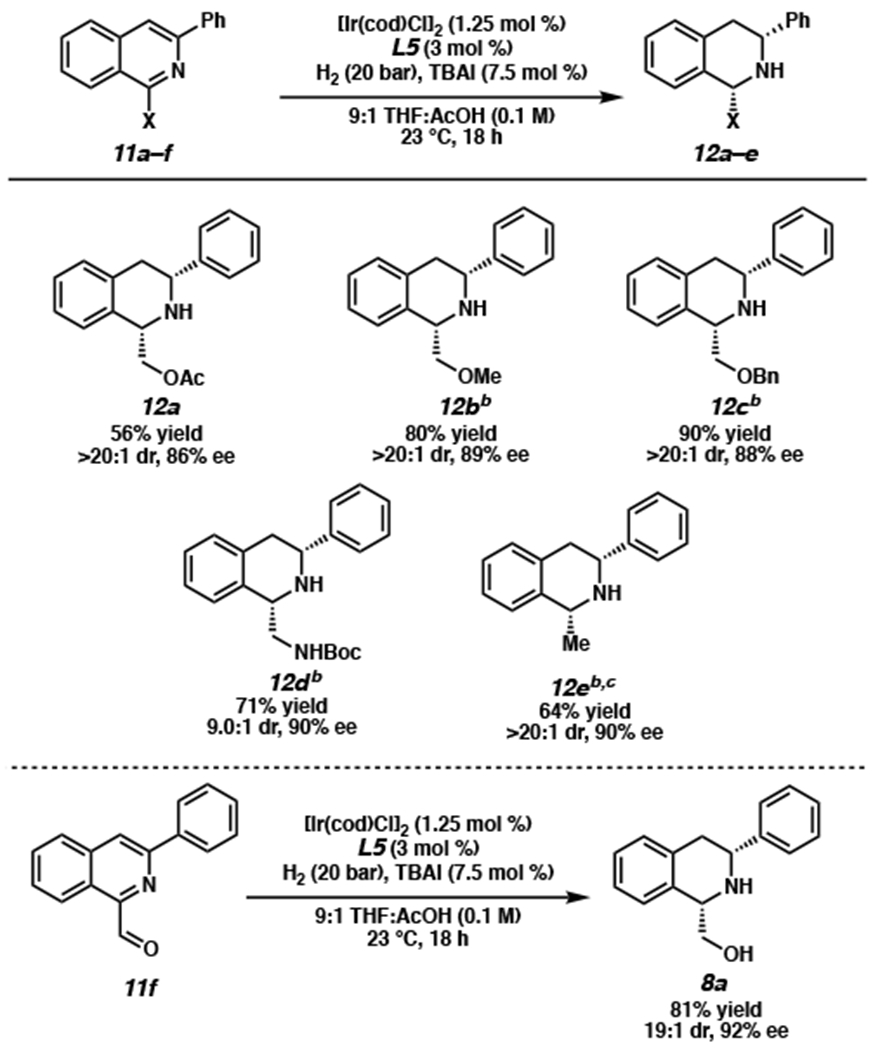
aReactions performed on a 0.2 mmol scale. bReaction performed at 60 °C and 60 bar H2. cRelative and absolute stereochemistry determined by experimental and computed VCD and optical rotation, see Supporting Information.16
Additionally, we found that aldehyde 11f was reduced to the alcohol in situ, affording the hydroxymethyl THIQ 8a in comparable yield, enantioselectivity, and diastereoselectivity to that of the hydroxy-directing substrate 7a (vide supra). Interestingly, an isoquinoline lacking a potential directing group (12e) also afforded chiral THIQs with no erosion of enantioselectivity (90% ee),16 although elevated temperature and pressure are needed to obtain a synthetically useful yield (64%). While the hydroxy-directing aspect is the enabling feature in the context of our total synthesis of jorumycin, we are pleased to find that we can obviate this requirement in our developed hydrogenation technology. Nevertheless, surveying a variety of different directing groups demonstrates the importance of a functional group for directed hydrogenation, with the hydroxy functionality acting as the best directing group for mild and efficient asymmetric hydrogenation. Notably, this is the first asymmetric hydrogenation method of isoquinolines in which additional Lewis basic functionalities are tolerated. It is also the first report investigating the effects of different directing groups in enantioselective hydrogenation of isoquinolines.
Synthetic Utility of the Hydrogenated Products.
Overall, the broad application of our hydrogenation technology to afford chiral THIQs provides access to a range of decorated analogs that are difficult to synthesize via biomimetic approaches (e.g., Pictet-Spengler, Bischler-Napieralski). These often require electron-rich substrates to undergo cyclization (eq 1).17
 |
(1) |
With the scope of the transformation established, we sought to demonstrate the synthetic utility of the produced chiral THIQs toward more complex scaffolds that could be applicable to natural products. Additionally, we envisioned taking advantage of the chiral β-amino alcohol that is generated in our product as a building block to forge more complex enantioenriched heterocyclic scaffolds (Scheme 6).18
Scheme 6. Derivatization of a Hydrogenated Producta.
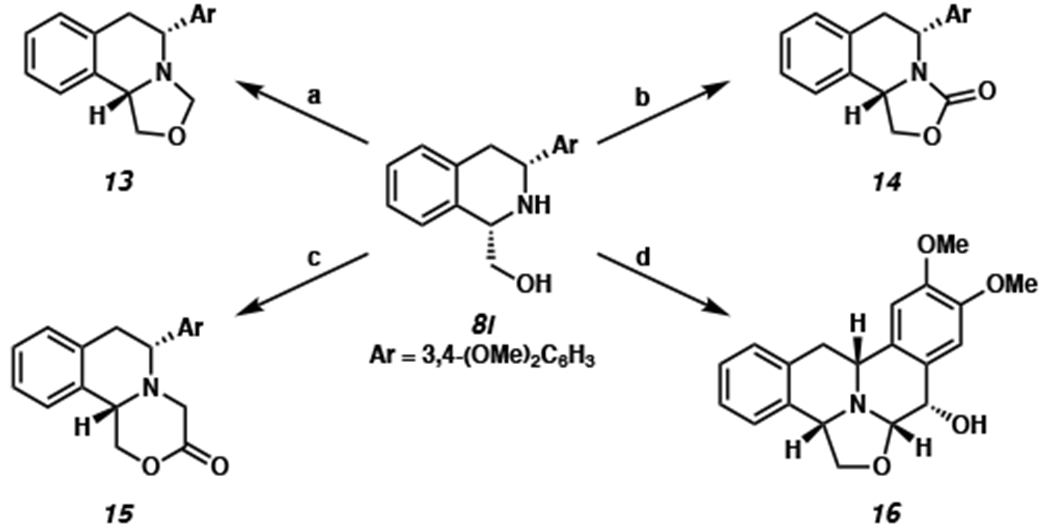
aConditions: (a) 37% aq. CH2O (1.85 equiv), DCE, 23 °C, 15 min, 93% yield. (b) CDI (4 equiv), THF, 50 °C, 15 h, 84% yield. (c) 40% aq. glyoxal (20 equiv), DCE, 23 °C, 18 h, 41% yield. (d) 60% aq. 2,2-dimethoxyacetaldehyde (1.85 equiv), DCE, 23 °C, 18 h; then Eaton’s Reagent (4 equiv), CH2Cl2, 23 °C, 3 h, 38% yield.
Prior to our investigation into the synthetic utilitiy of THIQ products, we performed the hydrogenation of isoquinoline 7l on a larger scale (1 mmol). We were pleased to find that the hydrogenated product 8l was still obtained in good yield (91% isolated yield) with excellent selectivity (>20:1 dr, 88% ee). With an ample amount of 8l in hand, we subjected THIQ 8l to aqueous fomaldehyde solution and found that the tricyclic 1,2-fused oxazolidine THIQ 13 was formed rapidly via the cyclization of the alcohol onto the iminium generated in situ. Furthermore, the reaction of amino alcohol 8l with carbonyldiimidazole (CDI) afforded oxazolidinone-fused THIQ 14 in 84% yield.19 To our delight, we found that these 6,6,5-tricyclic systems are conserved structural motifs in a number of natural products such as quinocarcin, tetrazomine, and bioxalomycin.5 Lastly, a different tricyclic scaffold containing fused morpholinone (15) can be isolated in 41% yield by the addition of excess glyoxal to 8l at room temperature.20 Being able to access a variety of complex heterocyclic scaffolds in one step from our hydrogenated THIQs further highlights the advantages of the hydroxymethyl functionality at the C1 position, beyond directing hydrogenation.
In addition to the tricyclic scaffold, we were pleased to find that an analog of tetrahydroprotoberberine alkaloids, a family of natural products with a tetracyclic bis-THIQ core,21 can be synthesized via a 2-step sequence. First, reaction of 8l with glyoxal dimethyl acetal delivered the oxazolidine-fused intermediate with a dimethoxy acetal substituent at the carbinol-amine carbon. Subsequently, exploration of both Bronsted and Lewis acid-mediated Pomeranz-Fritsch reaction revealed that the use of Eaton’s reagent22 delivered pentacyclic THIQ 16 in 38% yield as a single diastereomer. This complex scaffold could be of medicinal interest, as previous studies have shown that tetrahydroprotobeberine derivatives possess a wide array of interesting biological activities.17,23
CONCLUSIONS
We have developed a general, efficient enantioselective hydrogenation reaction of 1,3-disubstituted isoquinolines toward the syntheses of chiral THIQs. Key to the success of this reaction is the installation of a directing group at the C1-position that facilitates hydrogenation to reduce a variety of isoquinolines under mild reaction conditions. The developed method affords chiral THIQs in good yields, with high levels of diastereoselectivity and enantioselectivity. The reaction conditions tolerate a wide range of substitution on the 1-, 3-, 6-, 7-, and 8-position of the isoquinoline core. To date, this report represents the broadest scope and highest tolerance of Lewis-basic functionality of any asymmetric isoquinoline reduction technology currently known. Furthermore, this method is amenable to the production of electron-deficient THIQs that are difficult to obtain through the classical Pictet-Spengler approach. In order to demonstrate the synthetic utility of the hydrogenated products, we utilize the hydroxyl directing group as a functional handle for further synthetic manipulations. As a result, we have completed the syntheses of various tricyclic and pentacyclic skeletons that are of potential medicinal interest. Further exploration of the mechanism, and other applications of this technology are currently underway.
Supplementary Material
ACKNOWLEDGMENT
We thank NIH-NIGMS (R01GM127972A) and Caltech for the support of our research program. A. N. thanks the Royal Thai Government Scholarship Program. E. R. W. was supported by a Postdoctoral Fellowship (PF-16-011-01-CDD) from the American Cancer Society. C.U.G. was supported by a Feodor Lynen Research Fellowship from the Alexander von Humboldt Foundation. We thank Dr. David VanderVelde (Caltech) and Maximilian B. Kaiser for NMR expertise, and Dr. Michael Takase (Caltech) for assistance with X-ray analysis.
ABBREVIATIONS
- ee
enantiomeric excess
- dr
diastereomeric ratio
- THIQ
1,2,3,4-tetrahydroisoquinoline
- BTFM
3,5-bistrifluoromethylphenyl
- DMM
4-methoxy-3,5-dimethylphenyl
- TBAI
tetrabutylammonium iodide
- CPME
cyclopentyl methyl ether
- Boc
tert-butoxycarbonyl
- DCE
1,2-dichloroethane
- CDI
carbonyldiimidazole
- VCD
vibrational circular dichroism
Footnotes
Supporting Information Available. Detailed experimental procedures, compound characterization data, and computational analysis. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).(a) Tayor RD; MacCoss M; Lawson ADG Rings in Drugs. J. Med. Chem 2014, 57, 5845. [DOI] [PubMed] [Google Scholar]; (b) Vitaku E; Smith DT; Njardarson JT Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]; (c) Lovering F; Bikker J; Humblet C Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem 2009, 52, 6752–6756. [DOI] [PubMed] [Google Scholar]; (d) Lovering F Escape from Flatland 2: Complexity and Promiscuity. Med. Chem. Commun 2013, 4, 515–519. [Google Scholar]; (e) Roughley SD; Jordan AM The Medicinal Chemist’s Toolbox: An Analysis of Reactions Used in the Pursuit of Drug Candidates. J. Med. Chem 2011, 54, 3451–3479. [DOI] [PubMed] [Google Scholar]
- (2).(a) Zhou Y-G Asymmetric Hydrogenation of Heteroaromatic Compounds. Acc. Chem. Res 2007, 40, 1357–1366. [DOI] [PubMed] [Google Scholar]; (b) Wang D-S; Chen Q-A; Lu S-M; Zhou Y-G Asymmetric Hydrogenation of Heteroarenes and Arenes. Chem. Rev 2012, 112, 2557–2590. [DOI] [PubMed] [Google Scholar]
- (3).(a) Zhao D; Glorius F Enantioselective Hydrogenation of Isoquinolines. Angew. Chem. Int. Ed 2013, 52, 9616–9618. [DOI] [PubMed] [Google Scholar]; (b) Wiedner ES; Chambers MB; Pitman CL; Bullock RM; Miller AJM; Appel AM Thermodynamic Hydricity of Transition Metal Hydrides. Chem. Rev 2016, 116, 8655–8692. [DOI] [PubMed] [Google Scholar]
- (4).For Ir-catalyzed asymmetric hydrogenation of isoquinolines, see; (a) Lu SM; Wang YQ; Han XW; Zhou Y-G Asymmetric Hydrogenation of Quinolines and Isoquinolines Activated by Chloroformates. Angew. Chem. Int. Ed 2006, 45, 2260–2263. [DOI] [PubMed] [Google Scholar]; (b) Shi L, Ye Z-S; Cao LL; Guo RN; Hu Y; Zhou Y-G Enantio-selective Iridium-Catalyzed Hydrogenation of 3,4-Disubstituted Isoquinolines. Angew. Chem. Int. Ed 2012, 51, 8286–8289. [DOI] [PubMed] [Google Scholar]; (c) Iimuro A; Yamaji K; Kandula S; Nagano T; Kita Y; Mashima K Asymmetric Hydrogenation of Isoquinolinium Salts Catalyzed by Chiral Iridium Complexs: Direct Synthesis for Optically Active 1,2,3,4-Tetrahydroisoquinolines. Angew. Chem. Int. Ed 2013, 52, 2046–2050. [DOI] [PubMed] [Google Scholar]; (d) Ye Z-S; Guo R-N; Cai X-F; Chen M-W; Shi L; Zhou Y-G Enantioselective Iridium-Catalyzed Hydrogenation of 1- and 3-Substituted Isoquinolinium Salts. Angew. Chem. Int. Ed 2013, 52, 3685–3689. [DOI] [PubMed] [Google Scholar]; (e) Kita Y; Yamaji K; Higashida K; Sathaiah K; Iimuro A; Mashima K Enhancing Effects of Salt Formation on Catalytic Activity and Enantioselectivity for Asymmetric Hydrogenation of Isoquinolinium Salts by Dinuclear Halide-Bridged Iridium Complexes Bearing Chiral Diphosphine Ligands. Chem. Eur. J 2015, 21, 1915–1927. [DOI] [PubMed] [Google Scholar]; (f) Guo R-N; Cai X-F; Shi L; Ye Z-S; Chen M-W; Zhou Y-G An Efficient Route to Chiral N-Heterocycles Bearing a C-F Stereogenic Center via Asymmetric Hydrogenation of Fluorinated Isoquinolines. Chem. Commun 2013, 49, 8537–8539. [DOI] [PubMed] [Google Scholar]; (g) Chen M-W; Ji Y; Wang J; Chen Q-A; Shi L; Zhou Y-G Asymmetric Hydrogenation of Isoquinolines and Pyridines Using Hydrogen Halide Generated in Situ as Activator. Org. Lett 2017, 19, 4988–4991. For Ru-Catalyzed Enatioselective Hydrogenation of Isoquinolines, see [DOI] [PubMed] [Google Scholar]; (h) Wen J; Tan R; Liu S; Zhao Q; Zhang X Strong Bransted Acid Promoted Asymmetric Hydrogenation of Isoquinolines and Quinolines Catalyzed by a Rh-Thiourea Chiral Phosphine Complex via Anion Binding. Chem. Sci 2016, 7, 3047–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Scott JD; Williams RM Chemistry and Biology of Tetrahydroisoquinoline Antitumor Antibiotics. Chem. Rev 2002, 102, 1669–1730. [DOI] [PubMed] [Google Scholar]; (b) Siengalewicz P; Rinner U; Mulzer, Recent Progress in the Total Synthesis of Napthyridinomycin and Lemonomycin Tetrahydroisoquinoline Antitumor Antibiotics (TAAs). J. Chem. Soc. Rev 2008, 37, 2676–2690. [DOI] [PubMed] [Google Scholar]
- (6).Welin ER; Ngamnithiporn A; Klatte M; Lapointe G; Pototschnig GM; McDermott MSJ; Conklin D; Gilmore CD; Tadross PM; Haley CK; Negoro K; Glibstrup E; Grünanger CU; Allan KM; Virgil SC; Slamon DJ; Stoltz BM Concise Total Syntheses of (−)-Jorunnamycin A and (−)-Jorumycin Enabled by Asymmetric Catalysis. Science. 2019, 363, 270–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).For C–H activation/annulation strategy, see; (a) Zhang Z-W; Lin A; Yang J Methyl Ketone Oxime Esters as Nucleophilic Coupling Partners in Pd-Catalyzed C–H Alkylation and Application in the Synthesis of Isoquinolines. J. Org. Chem 2014, 79, 7041–7050. [DOI] [PubMed] [Google Scholar]; For ketone enolate-arylation/annulation, see; (b) Donohoe TJ; Pilgrim BS; Jones GR; Bassuto JA Synthesis of Substituted Isoquinolines Utilizaing Palladium-Catalyzed ✓-Arylation of Ketones. Proc. Natl. Acad Sci. U.S.A 2012, 109, 11605–11608. [DOI] [PMC free article] [PubMed] [Google Scholar]; For benz-annulation of isocoumarins, see; (c) Manivel P Probakaran K; Khan FN; Jin JS Facile Benzannulation of Isocoumarins in the Efficient Synthesis of Diversified 1,3-Disubstituted Isoquinolines. Res. Chem. Intermed 2012, 38, 347–357. [Google Scholar]
- (8).Pilgrim BS; Gatland AE; Esteves CHA; McTernan CT; Jones GR; Tatton MR Procopiou, P. A.; Donohoe, T. J. Palladium-Catalyzed Enolate Arylation as a Key C–C Bond-Forming Reaction for the Synthesis of Isoquinolines. Org. Biomol. Chem 2016, 14, 1065–1090. [DOI] [PubMed] [Google Scholar]
- (9).Allan KM; Hong BD; Stoltz BM; Expedient Synthesis of 3-Hydroxyisoquinolines and 2-Hydroxy-1,4-Napthoquinones via One-Pot Aryne Acyl-Alkylation/Condensation. Org. Biomol. Chem 2009, 7, 4960–4964. [DOI] [PubMed] [Google Scholar]
- (10).For example of transition-metal-catalyzed tandem C–H activation/annulation of arenes and alkynes, see; (a) Zhu Z; Tang X; Li X; Wu W; Deng G; Jiang H Palladium-Catalyzed C–H Functionalization of Aromatic Oximes: A Strategy for the Synthesis of Isoquinolines. J. Org. Chem 2016, 81, 1401–1409. [DOI] [PubMed] [Google Scholar]; (b) Zhou S; Wang M; Wang L; Chen K; Wang J; Song C; Zhu J Bidentate Directing-Enabled, Traceless Heterocycle Synthesis: Cobalt-Catalyzed Access to Isoquinolines. Org. Lett 2016, 18, 5632–5635. [DOI] [PubMed] [Google Scholar]; (c) Chinnagolla RK; Pimparkar S; Jeganmohan M Ruthenium-Catalyzed Highly Regioselective Cyclization of Ketoximes with Alkynes by C–H Bond Activation: A Practical Route to Synthesize Substituted Isoquinolines. Org. Lett 2012, 14, 3032–3035. [DOI] [PubMed] [Google Scholar]; (d) Zhao D; Lied F; Glorius F Rh(III)-Catalyzed C–H Functionalization/Aromatization Cascade with 1,3-Dienes: A Redox Neutral and Regioselective Access to Isoquinolines. Chem. Sci 2014, 5, 2869–2873. [Google Scholar]
- (11).(a) Chu H; Sun S; Yu J-T; Cheng J Rh-Catalyzed Sequential Oxidative C–H Activation/Annulation with Geminal-Substituted Vinyl Acetates to Access Isoquinolines. Chem. Commun 2015, 51, 13327. [DOI] [PubMed] [Google Scholar]; (b) Arambasic M; Hooper JF; Willis MC Activating Group Recycling in Action: A Rhodium-Catalyzed Carbothiolation Route to Substituted Isoquinolines. Org. Lett 2013, 15, 5162–5165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).See Supporting Information for additional results using other acids. Although further studies are required to fully understand the role of acid, we speculate that the acid helps promoting a) the tautomerization of enamine to imine prior to the second reduction and b) the dissociation of THIQ product from Ir-complex through protonation.
- (13).In a similar Ir-xyliphos system, it is reported that the alpha-alkoxy imine binds to the catalyst complex in a bidentate fashion, see; (a) Dorta R; Broggini D; Stoop R; Rüegger H Spindler F; Togni A Chiral Xyliphos Complexes for the Catalytic Imine Hydrogenation Leading to the Metolachlor Herbicide: Isolation of Catalyst-Substrate Adducts. Chem. Eur. J 2004, 10, 267–278. [DOI] [PubMed] [Google Scholar]; (b) Dorta R; Broggini D; Kissner R; Togni A Iridium-Imine and –Amine Complexes Relevant to the (S)-Metolachlor Process: Structures, Exchanges Kinetics, and C–H Activation by IrI Causing Racemization. Chem. Eur. J 2004, 10, 4546–4555. [DOI] [PubMed] [Google Scholar]
- (14).Hopmann KH; Bayer A On the Mechanism of Iridium-Catalyzed Asymmetric Hydrogenation of Imines and Alkenes: A Theoretical Study. Organometallics. 2011, 30, 2483–2497. [Google Scholar]
- (15).Increasing the catalyst loading to 2.5 mol % of [Ir(cod)Cl]2 and 6 mol % of L5 does not improve the conversion any further.
- (16).The absolute stereochemistry of product 12e was determined via the combination of measured and computed vibrational circular dichroism (VCD) spectra and optical rotations. The configuration of 12e (R, R) was found to be analogous to that determined for hydroxymethyl product 8a and also observed crystallographically for 8p. See Supporting Information.
- (17).(a) Chrzanowska M Grajewska A; Rozwadowska MD Asymmetric Synthesis of Isoquinoline Alkaloids: 2004–2015. Chem. Rev 2016, 116, 12369–12465. [DOI] [PubMed] [Google Scholar]; (b) Carrillo L; Badia D; Dominguez E; Anakabe E; Osante I; Tellitu I; Vicario JL Chiral Amino Alcohols As Intermediates in the Stereocontrolled Synthesis of 3-Disubstiuted Tetrahydroisoquinolines and Protoberbertines. J. Org. Chem 1999, 64, 115–1120. [Google Scholar]
- (18).Haftchenary S; Nelson SD; Furst L; Dandapani S; Ferrara SJ; Bošković ZV; Lazú SF; Guerrero AM; Serrano JC; Crews DK; Brackeen C; Mowat J; Brumby T; Bauser M; Schreiber SL; Phillips AJ Efficient Routes to a Diverse Array of Amino Alcohol-Derived Chiral Fragments. ACS. Comb. Sci 2016, 18, 569–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Alternatively, the oxazolidinone-fused product 7b could be synthesized utilizing a 2-step sequence. First is the Boc-protection of the amine. The Boc-protected product was subsequently cyclized to afford oxazolidinone-fused THIQ 7b under the Appel reaction conditions. See Supporting Information for experimental details.
- (20).Zhang G-L; Chen C; Xiong Y; Zhang L-H; Ye J; Ye X-S Synthesis of N-Substituted Iminosugar Derivatives and Their Immunosuppresive Activities. Carbohydrate Research. 2010, 345, 780–786. [DOI] [PubMed] [Google Scholar]
- (21).Gadhiya SV; Giri R; Cordone P; Karki A; Harding WW Diverse Approaches and Recent Advances in the Synthesis of Tetrahydroprotoberberines. Curr. Org. Chem 2018, 22, 1893–1905. [Google Scholar]
- (22).Thimmaiah S; Ningegowda M; Shivananju NS; Ningegowda R; Siddaraj R; Priya BS Eaton’s Reagent Catalysed Alacritous Synthesis of 3-Benzazepinones. Eur. J. Chem 2016, 7, 391–396. [Google Scholar]
- (23).Guo D; Li J; Lin H; Zhou Y; Chen Y; Sun H; Zhang D; Li H; Shoichet BK; Shan L; Xie X; Jiang H; Liu H Design, ynthesis, and Biological Evaluation of Novel Tetrahydroprotoberberine Derivatives (THPBs) as Selective ✓1A-Adrenoceptor Antagonists. J. Med. Chem 2016, 59, 9489–9502. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.