

Abstract
The molecular mechanisms tuning cholinergic interneuron (CIN) activity, although crucial for striatal function and behavior, remain largely unexplored. Previous studies report that the Etv1/Er81 transcription factor is vital for regulating neuronal maturation and activity. While Er81 is known to be expressed in the striatum during development, its specific role in defining CIN properties and the resulting consequences on striatal function is unknown. We report here that Er81 is expressed in CINs and its specific ablation leads to prominent changes in their molecular, morphologic, and electrophysiological features. In particular, the lack of Er81 amplifies intrinsic delayed-rectifier and hyperpolarization-activated currents, which subsequently alters the tonic and phasic activity of CINs. We further reveal that Er81 expression is required for normal CIN pause and time-locked responses to sensorimotor inputs in awake mice. Overall, this study uncovers a new cell type-specific control of CIN function in the striatum which drives habit formation in adult male mice.
SIGNIFICANCE STATEMENT Although previous studies have shown that cholinergic interneurons drive striatal activity and habit formation, the underlying molecular mechanisms controlling their function are unknown. Here we reveal that key cholinergic interneuron physiological properties are controlled by Er81, a transcription factor regulating neuronal activity and development in a cell-specific manner. Moreover, our findings uncover a link between the Er81-dependent molecular control of cholinergic interneuron function and habit formation in mice. These insights will contribute to the future enhancement of our understanding of disorders that involve behavioral inflexibility, such as autism and addiction.
Keywords: interneuron, tuning, striatum, activity, habit
Introduction
Cholinergic interneurons (CINs) constitute only 1%-2% of all striatal neurons but are the main source of acetylcholine in the striatum and play a crucial role in regulating habitual behavior (Aoki et al., 2018). As the specific timing of CIN firing could be essential for habit regulation, it is crucial to understand how fine-tuned CIN activity contributes to striatal function. Unique morphological and electrophysiological features (Lim et al., 2014) underpin CIN function in controlling the striatal output neurons (Mamaligas and Ford, 2016; Gritton et al., 2019). CINs fire tonically and display phasic responses to stimuli, which consist of pauses preceded by a transient rise and followed by a “rebound” of CIN activity (Apicella, 2017). The pause in CIN firing is fundamental for striatal processing of information and behavior (Zucca et al., 2018). The tonic and phasic activity of CINs is governed by both synaptic inputs and the intrinsic inward (Z. Zhao et al., 2016) and delayed rectifier currents (Wilson and Goldberg, 2006), referred to as Ih and IsAHP, respectively. In particular, pause expression is mediated by the slow Kv7-dependent potassium current IKr in response to excitatory inputs and is regulated by dopamine (Straub et al., 2014; Zhang et al., 2018) and GABA (Lozovaya et al., 2018). The role of molecular factors in regulating the pause of CINs and in controlling the acquisition of these properties during maturation remains unexplored.
Recent findings suggest that developmental differentiation factors induce some degree of functional diversity among CINs, thus enabling them to acquire unique properties (Ahmed et al., 2019). Notably, the LIM homeodomain transcription factor Lhx6, which is required for GABAergic interneuron specification, segregates a subtype of GABAergic CIN with distinct functional properties (Lozovaya et al., 2018). The ETS transcription factor Etv1/Er81 plays specific roles in maturation (Abe et al., 2011; Ding et al., 2016), identity (Cave et al., 2010), and the establishment of synaptic connections (Arber et al., 2000; Hippenmeyer et al., 2005). Er81 also regulates cell excitability by controlling the expression of a potassium channel subunit in adult cortical interneurons (Dehorter et al., 2015). In the striatum, it is expressed in subclasses of interneurons (Mi et al., 2018; Nóbrega-Pereira et al., 2008), but its specific cellular distribution and function in CINs are unknown. We hypothesized that Er81 plays a fundamental role in determining key features of maturing striatal CINs and in controlling their function. Using molecular, electrophysiological, and behavioral approaches, we reveal that the Er81 transcription factor is necessary to set major functional properties of CINs. We unravel its role as a key contributing factor for pause expression, regulation of striatal activity, and habitual behavior.
Materials and Methods
Mice
We generated Er81+/+; ChAT-Cre (control; ChAT: choline acetyltransferase) and Er81flox/flox; ChAT-Cre (Er81 conditional KO [cKO]) mice by crossing Er81flox/flox mice (generous gift by Prof Marin at the MRC, London) with ChAT-Cre mice (#006410 ChAT-IRES-Cre from The Jackson Laboratory). We also generated Er81+/+; ChAT-Cre; RCE-GFP (control) and Er81flox/flox; ChAT-Cre; RCE-GFP (Er81 cKO) mice by crossing Er81+/+; ChAT-Cre or Er81flox/flox; ChAT-Cre with Er81+/+; RCE-GFP or Er81flox/flox; RCE-GFP (kindly supplied by Prof Marin at the MRC, London). We used the Er81+/+; Nkx2.1-CreER; RCE-GFP (control) and Er81flox/flox; Nkx2.1-Cre; RCE-GFP (Er81 cKO) mice for the analysis of the CIN morphology. Lhx6-Cre; RCE-GFP mice were used to analyze Er81 expression regarding the Lhx6 phenotype of CINs. All experiments were conducted with approval from the Australian National University Animal Experimentation Ethics Committee (protocol numbers A 2016/14, A2018/43, and A2018/66). All efforts were made to minimize suffering and reduce the number of animals. Only male mice were used in this study.
In vitro electrophysiological recordings
We used Er81+/+; ChAT-Cre; RCE-GFP (control) and Er81flox/flox; ChAT-Cre; RCE-GFP (Er81 mutant) mice at P6-7 or P30-P60 for slice electrophysiology experiments. Mice were deeply anesthetized with isoflurane and perfused with ice-cold oxygenated, ACSF containing the following (in mm): 248 sucrose, 3 KCl, 0.5 CaCl2, 4 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 1 glucose, saturated with 95% O2 and 5% CO2. The animals were then decapitated, and brain was removed and placed in ice cold oxygenated sucrose-based cutting solution. Sagittal slices of 400 μm were cut using a VT1200S vibratome (Leica Microsystems). Slices were then maintained at room temperature in ACSF containing the following (in mm): 124 NaCl, 3 KCl, 2 CaCl2, 1 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10 glucose saturated with 95% O2 and 5% CO2. For patch-clamp recordings in whole-cell configuration, slices were transferred to a chamber and continuously superfused with ACSF at 34°C. GFP-expressing cholinergic cells located in the dorsal striatum were visualized by infrared-differential interference optics with a 40× water-immersion objective. For targeting GFP expressing neurons, slices were illuminated by blue light through the objective. Microelectrodes (4-6 mΩ) were pulled from borosilicate glass (1.5 mm outer diameter × 0.86 inner diameter) using a vertical P10 puller (Narishige).
For voltage-clamp recordings, a cesium gluconate-based intracellular solution was used containing the following (in mm): 120 Cs-gluconate, 13 CsCl, 1 CaCl2, 10 HEPES, and 10 EGTA (pH 7.2-7.4, 275-285 mOsm). We used the cesium-gluconate solution to measure spontaneous and miniature GABAA currents at the reversal potential for glutamatergic (10 mV) events and glutamatergic currents at −60 mV. For current-clamp recordings, we used a potassium-gluconate-based intracellular solution containing the following (in mm): 140 K-gluconate, 10 HEPES, 2 NaCl, 4 KCl, 4 ATP, and 0.4 GTP. Neurobiotin (2-5 mg/ml) was added for postrecording immunocytochemistry. Electrophysiological signals were low-pass filtered online at 10 kHz with a Multiclamp 700B (Molecular Devices) amplifier and acquired at a 20 kHz sampling rate with a LIH 8 + 8 (HEKA) data acquisition board and WinWCP software (created by John Dempster, University of Strathclyde). Circuit capacitance was corrected after gigaseal formation. Series resistance and liquid junction potential were not corrected. Cell-attached spikes were recorded for at least 60 s, after gigaseal formation in voltage-clamp mode. Following break-in, the test pulse was monitored for a few seconds to ensure a stable, low access resistance (Ra < 20 mΩ). In whole-cell configuration, spontaneous firing of the CINs was recorded for 60 s and then a current pulse of 200 pA was delivered for 3 s to obtain slow afterhyperpolarization (sAHP). An additional 60 s of spontaneous activity was recorded following the pulse. Then, a current-steps protocol (−200 pA to 225 pA, 25 pA steps, 3 s duration and 7 s intervals) was applied to obtain membrane properties and excitability of the cells. In voltage-clamp mode, membrane potential was held at −60 mV. Hyperpolarizing voltage pulses (−120 mV to −70 mV, 10 mV steps, 3 s duration and 7 s intervals) were applied to measure Ih. To measure delayed rectifier or IsAHP currents, depolarizing voltage pulses were applied (−50 mV to −20 mV, 10 mV steps, 3 s duration and 7 s intervals). To measure postsynaptic currents (PSCs), the voltage was held at −60 mV for EPSCs or 10 mV for IPSCs. PSC rise and decay times were calculated as the intervals between 20% and 100% of the PSC peak before and after the peak, respectively.
Electrophysiological data were analyzed in MATLAB using custom-written codes. Spontaneous action potential (AP) before the first current pulse was taken to examine tonic activity of CINs. Threshold was detected based on the positive peaks occurring >10 mV/ms in the first derivative of the membrane potential trace. The AP width was defined as the interval between the first and second threshold crossings. The AHP amplitude and its peak delay were obtained at the minimum potential relative to the AP threshold within a 200 ms interval after the second threshold crossing of the AP. To plot the grand average APs, the traces were aligned to the AP threshold. A biexponential function was fit to the AHP to calculate rise and decay time constants. To characterize membrane potential dynamics following depolarization, APs were removed by linearly interpolating from 30 ms before the AP threshold to 100 ms after second threshold crossing point of the AP. The early component of sAHP was calculated by averaging 1 s of the Vm following the offset of the current pulse, and the late component was averaged from 1.5 to 4.5 s after the pulse offset, relative to the resting membrane potential. To plot the grand average trace of membrane potentials, the traces of the cells were aligned to the resting membrane potential. Input resistance (Rin) was calculated at the minimum of the voltage response (Vmin to −200 pA pulse) before the sag in Vm appeared. Sag ratio was calculated based on the following formula: Sag ratio = (Vmin – Vsteady)/(Vmin – Vrest), where Vmin was the minimum of the voltage during the pulse, Vsteady was the average of membrane potential at 2.5-3 s after the onset of the current pulse, and Vrest was the medium of the membrane potential within 500 ms preceding the onset of the current pulse. APs were converted to 0 and 1 arrays based on their peak time and then averaged across neurons in 40 µs bins and smoothed with a 40 ms window to obtain instantaneous AP rates (see Figs. 2B, 3C). AP rate was calculated as the average over the pulse (see Fig. 2C). Statistical tests on the instantaneous firing rates were applied on intervals where the effect was consistently maintained. Adaptation index was calculated as the range of the interspike intervals divided by their minimum.
Figure 2.

Alterations of the CIN properties in the absence of Er81. A, ChAT+ interneurons in P30 striatum of a control (ChAT-Cre; Er81+/+, left) and an Er81 cKO (ChAT-Cre; Er81f/f, right) mouse. Scale bar, 25 µm. B, CIN density in the dorsal striatum at P2 (top) and P30 (bottom) in the Er81 cKO compared with the control condition. C, Expression of key molecular markers of cholinergic cells in control and Er81 cKO conditions. Lhx6, LIM homeobox 6; Isl1, Islet1; slc5a7, choline transporter; ChAT, choline acetyltransferase. D, Morphologic reconstruction of soma and dendrites of control (black) and Er81 cKO (red) CINs at P30. E, Volume of dendritic spread in the control and the Er81 cKO CINs. F, Sholl analysis representing the complexity of the dendritic field in the control and the Er81 cKO. G, Total dendritic volume in the control and the Er81 cKO CINs. H, Number of dendritic branch points in the control and the Er81 cKO CINs. I, Left, ChAT+ neuropil. Right, Quantification of the area occupied by ChAT+ neuropil in the striatum. J, Grand average AHP waveforms (±SEM) in the control (black) and the Er81 cKO (red). Inset, The AP. Dashed line indicates the AP threshold. AHP delay and amplitude were determined from individual APs. K, AHP amplitude (left) and AHP peak delay (right) in the control and the Er81 cKO mice. Each dot represents the average AHP amplitude per cell. L, Average membrane potential (±SEM) during and after a 200 pA current injection. Individual traces were aligned to the resting membrane potential (dashed line). Gray shadings represent early and late phases of the slow AHP. M, Average of the early (left) and the late phase of the slow AHP (right) in the two conditions. *p < 0.05; **p < 0.01; ***p < 0.001.
Figure 3.

Reduced CIN excitability and enhanced delayed rectifier current IsAHP following Er81 deletion. A, Representative responses of P30 CINs to a 150 pA positive current pulse in the control and the Er81 cKO conditions. B, Average AP rate across all positive current pulses. Inset, Time-expanded view of the early response. C, Average AP rate during positive current steps. Stars represent genotype effect. D, Adaptation index as a function of the current step amplitude. Stars represent genotype effect. E, Example trace of the putative IsAHP following a depolarizing voltage pulse. F, Mean traces of IsAHP (±SEM) as a function of time at different holding potentials (−50 to −20 mV, 10 mV steps). G, Average IsAHP as a function of the holding potential. Stars represent genotype effect. H, Kcnq2 mRNA expression levels in the striatum of control and Er81 cKO mice. I, Representative image of a ChAT-GFP cell (green) stained for KCNQ2 protein (red) in control and Er81 cKO. Scale bar, 10 µm. J, Average KCNQ2 protein expression levels in control and Er81 cKO CINs. K, Grm5 mRNA expression levels in the striatum of the control and the Er81 cKO mice. L, Representative image of a ChAT-GFP cell (green) stained for mGluR5 protein (red) in control and Er81 cKO. Scale bar, 10 µm. M, Average mGluR5 protein expression levels in control and Er81 cKO CINs. *p < 0.05. **p < 0.01. ***p < 0.001.
In voltage-clamp mode, hyperpolarizing and depolarizing voltage pulses were delivered to characterize inward rectifier (Ih) and delayed rectifier (IsAHP) currents, respectively. To calculate IsAHP (tail current), the current at 4-5 s following the offset of the voltage step was subtracted and a 4 s window following the offset was averaged across time. Ih was quantified by subtracting the positive peak of the current and then averaging from this peak to up to 2.9 s. Postsynaptic currents were isolated using a manual threshold and applying principal component analysis in MATLAB.
In vivo recordings
Adult Er81f/f; ChAT-Cre and Er81+/+; ChAT-Cre mice (P60-P80) were used for in vivo extracellular array recordings. Animals were kept in individual cages and provided with ad libitum food and water. Before the surgical operation, anesthesia was induced by 3% isoflurane and the head was mounted on a stereotaxic device (Stoelting). Local anesthetic was applied to the scalp (Lignocaine, Lmx4) and eye gel (Viscotears from Novartis) to both eyes. The skull was exposed and cleared from fascia. A thin layer of tissue adhesive (Vetbond; 3M) was applied on the skull. A custom-made head bar was then glued to the skull and secured by dental cement. The cement was applied all over the skull, except the area of intended craniotomy, which was filled with silicon sealant (Kwik-Kast, WPI) at the end of the surgery. Animals were injected with 5 mg/kg of carprofen and 0.86 ml/kg of penicillin (intraperitoneal injections) and placed on a warm heating pad to recover from anesthesia. The animals were returned to their home cage and allowed 6 d of recovery from surgery. Then the mice were head-fixed for three sessions of 15-90 min over 3 d to habituate. On the following day, the mice were anesthetized with 3% isoflurane and two small craniotomies (<1 mm in diameter) were drilled (0.0-0.4 mm from bregma, 2.5 mm lateral) on both sides; and the dura was left intact, and the craniotomy was filled with silicon sealant. The first recording session started at least 3 h after the recovery from anesthesia. The recording probe was moved down to a depth of ∼1.2 mm (distance of the tip from the dura) followed by ∼0.1-0.2 mm advancement after each recording, with an active search for tonically active neurons when advancing the electrodes. This was repeated up to a depth of ∼4 mm. In the second recording session (24 h later), the opposite craniotomy was used for recording.
At the end of second recording session, the mice were perfused transcardially and the brain was removed for histologic verification.
To obtain sensory driven responses in striatum, a bilateral train of air puffs (10 pulses of 200 ms/cycle) was applied to the whisker arrays during each recording using a Pico spritzer (Parker Instruments) device. The pressure was adjusted to generate visible movements of the whiskers allowing the puffers to be placed in front of the animal, out of the reach of the whiskers. The puff train was repeated every 10 s for 20 trials per recording. Electrophysiological data were acquired using 32 channel NeuroNexus double linear arrays (A1x32-Poly2-10 mm-50s-177, NeuroNexus) coupled with CerePlex Direct data acquisition system (BlackRock Microsystems). A high-speed imaging system (Mikrotron) was used to film the top view of the head at 250 frame/s during electrophysiological acquisition.
The depth of recording at each electrode site was calculated using the probe guidelines (A1x32-Poly2-10mm-50s-177). The whole span of recording depth for individual channels across animals was 0.37-4.20 mm. Based on histology data, we estimated the boundary between cortex and striatum to be at ∼2 mm. We limited our analysis of striatal cells to 2.0-3.0 mm of depth. Cells were sorted and analyzed using custom written codes in MATLAB as follows. For every channel, 1.5 ms event waveforms were detected at 1 ms spaced peaks which exceeded 5 times root-mean-square of the high-pass filtered signal (at 300 Hz). Mean spike rates across trials were smoothed by a 16.7 ms averaging window. The width of the average spike waveforms of each cell was calculated at half-amplitude to exclude 10% widest sorted waveforms from control and Er81 cKO conditions. For classification of different cell types, putative FS cells defined by a narrow spike waveform (maximum width = 0.475 ms) (Dorst et al., 2020) were discarded alongside artifacts by excluding the narrowest 20%. Spiny projection neurons (SPNs) and CIN were both defined by wider waveforms (Dorst et al., 2020). CINs were separated from SPNs by their average spontaneous firing rate in quiet state (with a threshold at 1 Hz, to distinguish low and high spontaneous activity) (English et al., 2011), their evoked response (i.e., the direction of the change in firing rate during 2 s of air puff stimulation compared with the spontaneous activity) and the proportion of long ISIs (i.e.,. ISI > 2 s, PropISI), as described by Benhamou et al. (2014). CINs typically respond to sensorimotor inputs (Benhamou et al., 2014; Gritton et al., 2019) through suppression or pause of their ongoing tonic activity with cortical and thalamic inputs (Ding et al., 2010; Doig et al., 2014) or with salient sensory stimuli (Aosaki et al., 1994; Apicella et al., 1997, 2009; Ravel et al., 2003; Morris et al., 2004; Thorn and Graybiel, 2014). Moreover, CIN responses to sensorimotor inputs can also be heterogeneous (Benhamou et al., 2014; Gritton et al., 2019). These criteria divided the units into four groups: (1) excited SPNs with spontaneous firing rate < 1 Hz, (2) inhibited SPNs with spontaneous firing rate < 1 Hz, (3) inhibited CINs with spontaneous firing rate ≥ 1 Hz and small propISIs (<30%), and (4) excited CINs/other interneurons (Beatty et al., 2012) with spontaneous firing rate ≥ 1 Hz and small propISIs (<30%). All ISIs were computed based on the spontaneous activity (i.e., activity during quiet states). We sorted the data in each group based on the extent of change in firing rate (δ firing rate) during stimulus presentation (see Fig. 6D,E). We then segregated the sorted responses into 10 equal-sized fractions based on the total cell number in each group, to ensure that similarly responsive neurons are considered when comparing the changes in firing rate between the control and Er81 cKO conditions. In order to include the head and whisker motions in the analysis, MATLAB's foreground detection algorithm was used to obtain a motion index (average of the image in binary foreground images). Spike triggered averaging (STA) of the motion index was then performed for each cell and the resulting STA was normalized within the interval (−3 to 3 s). Correlations between firing rate and the air puff and motion were calculated using cross-correlation analysis in MATLAB. For a smooth estimation of the firing rate, spike times were convolved by a leaky integrator function (). Onsets of the air puffs were also convolved by the same function. The peaks of cross-correlations were chosen within a ±2 s lag. Since there was no genotype effect on the lag of the peak, we used the peak cross-correlation to analyze the strength of correlations between the variables.
Figure 6.
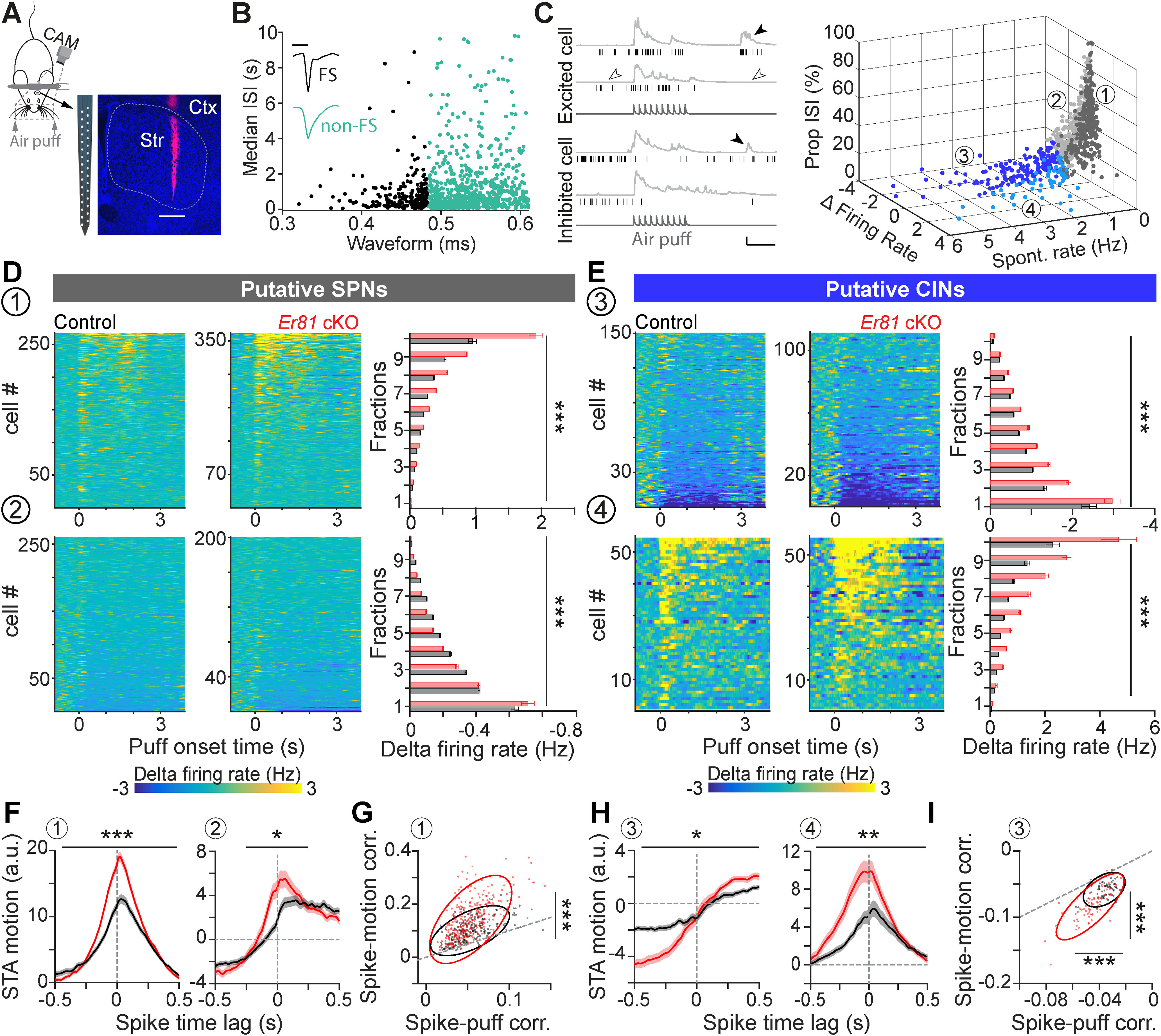
Enhanced inhibitory response of striatal CINs in the absence of Er81. A, Head-fixed awake recording by multielectrode arrays. Track of the recording array marked by DiI (magenta). Str, Striatum; Ctx, cortex. Scale bar, 0.5 mm. B, Units classified into FS (black) and non-FS cells (teal) based on their spike waveform and median ISIs. Inset, Representative traces (scale bars, 0.5 ms; normalized amplitudes). C, Left, Example trials showing spike raster (black) and the combined movements of the whiskers and nose quantified as motion index (light gray) for one excited cell and one inhibited cell. Dark gray traces at the bottom represent air puff trains. Filled arrowheads indicate the bouts of voluntary movement. Empty arrowheads indicate quiet states (scale bars, 1 s, 10 a.u.). Right, 3D scatter plot separating striatal neuron subtypes into four groups (1-4), based on spontaneous firing rate (spontaneous rate, quantified in quiet states), evoked firing rate (firing rate during air puff presentation, normalized to the activity before the stimulus) and proportion of ISIs >2 s (propISIs). Threshold for δ firing rate: 0; threshold for spontaneous rate: 1 Hz. D, Top, Normalized spiking activity in Group 1 (putative SPNs; left, control; middle, Er81 cKO; right). Bar plots represent average (±SEM) of the evoked activity within fractions of the sorted data for each genotype (gray, control; red, Er81 cKO). Bottom, Normalized spiking activity in Group 2 (SPNs and other cell types, bar plots as in top). Stars represent genotype effect. E, Top, Normalized spiking activity of the putative CINs (Group 3, bar plots as in D). Bottom, Normalized spiking activity of Group 4 (putative CINs and other interneurons, bar plots as in D). Stars represent genotype effect. F, STA (average STA across cells ± SEM, normalized within 3 s from the spike time) of the motion index corresponding to the putative SPNs (left, Group 1) and Group 2 (right). G, Strength of correlations between the spiking activity of the putative SPNs and the air puff (x axis) or motion (y axis). Dots represent individual cells with positive correlation regarding both variables. The ellipses are the first contours of the fitted Gaussian mixture models. Dashed line indicates the line of equality. H, Average STA across the putative CINs (left, Group 2) and Group 4 (right). Vertical dashed lines indicate the spike time. Horizontal dashed lines extend zeros. I, Strength of correlations between the spiking activity of putative CINs and the air puff (x axis) or motion (y axis). Dots represent individual cells with a negative correlation regarding both variables. Ellipses represent the first contours of the fitted Gaussian mixture models. Dashed line indicates the line of equality. *p < 0.05. **p < 0.01. ***p < 0.001.
Immunohistochemistry
Animals were perfused transcardially with 0.01 m PBS to eliminate blood and extraneous material followed by 4% PFA under isoflurane anesthesia. Brains were left incubated for 2-5 h in PFA. The fixative was then removed from tissues by 3 washes in PBS. Tissues were sectioned at 60 µm using a Leica 1000S vibratome and kept in a cryoprotective ethylene glycol solution at −20˚C until processed for immunofluorescence.
Sections were first washed and permeabilized; then nonspecific binding sites were blocked by immersing the tissue in 10% normal donkey serum, 2% BSA in PBS-T for 2 h. Tissues were then stained using the following primary antibodies overnight: mouse anti-β galactosidase (1:1000; Promega), rabbit anti-Er81 (1:5000; generous gift from Prof Silvia Arber; KO-validated) (Dehorter et al., 2015), chicken anti-GFP (1:3000; Aves Lab), mouse anti-parvalbumin (PV, 1:3000; Sigma Millipore), goat anti-ChAT (1:200; Merck), guinea pig anti-vGluT1 and -vGluT2 (1:2000; Chemicon) and anti-vGluT3 (1:2000; Merck), mouse anti-Lhx6 (1:X; Santa Cruz Biotechnology), rabbit anti-mgluR5 (1:50; Alomone Labs), sheep anti-Neuropeptide Y (1:1000; Merck), rabbit anti-KCNQ2 and anti-HCN2 (1:200; Thermo Fisher Scientific), mouse anti-c-Fos (1:500; Santa Cruz Biotechnology) or rabbit anti-p-Ser240-244-S6rp (1:500; Cell Signaling Technology). After 3 washes with 15 min intervals, we added anti-rabbit, anti-chicken, anti-mouse, anti-goat Alexa-488, -555, or -647 (1:200; Invitrogen) secondary antibodies for 2-3 h or anti-guinea pig biotinylated (1:200; Jackson ImmunoResearch Laboratories), anti-sheep biotinylated (1:200; Invitrogen) followed by streptavidin 555 or 647 for 1. After 3 washes with 15 min intervals, slices were stain for 10 min with DAPI (5 μm; Sigma Millipore), mounted on Livingstone slides, and then covered with Mowiol (Sigma Millipore) and coverslip (Thermo Fisher Scientific).
After patch-clamp recordings, slices were immediately fixed in 4% PFA for 2-5 h, rinsed in PBS (3 times, 30 min intervals), and kept overnight at 4°C. The same procedure as described above was performed for immunostaining. Images were acquired by a Nikon A1 confocal laser scanning microscope. Stained sections of control and mutant mice were imaged during the same imaging session; and laser power, photomultiplier gain, pinhole, and detection filter settings were kept constant. Immunofluorescence signals were quantified using ImageJ or MATLAB using routine particle analysis procedures to obtain soma or nuclear masks. The signal within masks was normalized to the background. The background masks were obtained by segmentation of thick fibers passing the striatum. To quantify the percent of βGal-positive cells, the Cartesian distances between the weighted centroids of the ChAT+ nuclei masks and βGal masks (threshold at mean + 0.4 SD) were calculated. If the nucleus mask was within 5 µm of a βGal mask, the cell was considered as ChAT+ βGal+.
Western blot
Brains were dissected in ACSF, and the striatum and cortex were extracted. Tissues were placed in lysis buffer with 1% β-mercaptoethanol (Sigma Millipore) before storage at −80°C. Total protein was extracted using the AllPrep DNA/RNA/Protein Mini Kit (QIAGEN, #80004) according to the manufacturer's instructions, including DNase treatment. The pellets were suspended in ALO buffer and then stored at −20°C. We extracted the striatum and cortex and homogenized by handheld homogenizer, at 1:5 g/ml (50 mm Tris-HCl, pH 7.4, 0.32 m sucrose, 5 mm EDTA, 1% Triton X-100, 1% protease inhibitor cocktail) and then stored at −80°C; 20 μl of each sample was run on 8%-16% Mini-PROTEAN TGX Stain-Free Gels (200V, 30 min), transferred to a PVDF membrane (340 mA, 70 min). The membrane was then blocked (1H, 3% BSA in TBS-T), incubated overnight with primary antibody overnight with the guinea-pig anti-vGluT3 (Merck; 1:2000), goat anti-ChAT (Merck; 1:500), mouse anti-Lhx6 (Santa Cruz Biotechnology; 1:500), then washed (3 washes with 5 min intervals, TBS-T) before incubation with HRP-conjugated secondary antibody for 2 h (Bio-Rad, 1:1000). After 3 washes with 5 min intervals with TBS-T, we developed with Clarity Western ECL Blotting Substrates (Bio-Rad). Membranes were imaged by ChemiDoc MP System (Bio-Rad). For reprobing, blots were stripped with harsh stripping buffer for 30 min, then reblocked and probed β-tubulin for 1 h with the mouse anti-β-tubulin (1:1000; Sigma Millipore). Bands were analyzed by area under curve relative to tubulin loading controls (Image Lab software).
FACS
Mice were rapidly decapitated, and brain slices were prepared as for electrophysiological recordings. Dorsal striatum was dissected and placed in ACSF, in a 15 ml tube with 10 ml digestion buffer (0.5 × g Trehalose and 10 mg Pronase, 1 mg DNase I, 50 μl MgCl2 1 m in oxygenated ACSF; Sigma Millipore) at 37°C for 30 min with inversion every 10 min. Tissues were allowed to settle on ice for 1 min, digestion buffer was removed, and the pellet was washed with 2 ml washing buffer (10 ml ACSF, 0.5 × g Trehalose, 1 mg DNase I, 50 μl MgCl2 1 m in oxygenated ACSF) at 4°C. To dissociate the cells, we resuspended the tissue in 1 ml wash buffer and triturated 12-15 times with 1 ml pipette. Tubes were placed on ice, large tissue pieces were allowed to settle, and ∼500 μl of cloudy suspension containing dissociated cells was transferred to a 1.5 ml tube on ice. This process was repeated for the remaining 500 μl suspension with 200 μl pipette 12-15 times and pooled into the 1.5 ml tubes on ice. The last cells were removed by adding 200 μl to the original tube. The cell suspension was filtered through a 70-μm-tick mesh filter into a snap cap tube and 200 μl DAPI 5 μm was added to exclude dead cells. FACS Melody software was used for cell sorting. Cells were collected in 350 μl of lysis buffer with 1% β-mercaptoethanol before storage at −80°C for RNA extraction.
qRT-PCR
Total RNA was extracted using the RNeasy Micro kit (QIAGEN) according to the manufacturer's instructions, including DNase treatment. RNA concentration and purity were determined using the Nanodrop spectrophotometer (Thermo Fisher Scientific). RNA was reverse-transcribed into cDNA using SuperScriptIII First Strand Synthesis System (Invitrogen) with random hexanucleotides according to the manufacturer's instructions. cDNA was analyzed using real time qPCR, in technical triplicates using TaqManGene Expression Assay. Reactions were performed on a MicroAmp Optical 384-Well Reaction Plate with Bar code (Applied Biosystems). Each 10 μl reaction included 5 μl of TaqMan 2× Universal PCR Master Mix (Applied Biosystems), 0.5 μl of TaqMan probes gene expression assay, and 4.5 μl of cDNA (5 ng total). PCRs were monitored using the 7900HT real time PCR system (Applied Biosystems). Amplification conditions were activation of AmpErase UNG for 2 min at 50°C, activation of AmpliTaq Gold Enzyme for 10 min at 95°C, followed by 40 cycles of denaturation at 95°C for 15 s, annealing at 60°C for 1 min, and extension at 60°C for 1 min. Relative quantification of mRNA level was performed using the comparative cycle threshold (CT) following the manufacturer's instructions (ABI). Results were normalized relative to the level of GAPDH (Mm99999915_g1 from Thermo Fisher Scientific) using 2–ΔΔCT method. Primers used are as follows: Gapdh (Mm99999915_g1); Er81/Etv1 (Mm00514804_m1); Lhx6 (Mm01333348_m1); Isl1 (Mm00517585_m1); ChAT (Mm01221880_m1); Slc5a7 (Mm00452075_m1); Grm5 (Mm00690332_m1); Kcnq2 (Mm00440080_m1); Hcn2 (Mm00468538_m1); Gad2 (Mm00484623_m1); Zic4 (Mm00657066_m1); Ache (Mm00477274_g1); and Lhx7 (Mm00802919_m1). The changes in the level of gene expression by age were quantified as relative mRNA expression (ΔCT) normalized to P30. The fold expression value of the genes of Er81 cKO is obtained by deducting the Er81 cKO value with the average of the control value of the same age. Two-sample t test was used for statistical analysis.
Morphology and synaptic bouton analysis
Er81+/+; Nkx2.1-CreER; RCE-GFP and Er81F/F; Nkx2.1-CreER; RCE-GFP P0 mouse pups were injected with low-titer tamoxifen (Sigma Millipore, 25 µl of 10 mg/ml solution in corn oil, i.p.) to induce GFP expression and Er81 cKO in MGE-derived interneurons Nkx2.1. Brains were removed after deep anesthesia under 3%-4% isoflurane, and PBS followed by 4% PFA was transcardially perfused. We then proceeded to the immunohistochemistry to select Nkx2.1-GFP/ChAT+ interneurons. Images were acquired using a Nikon A1 Confocal microscope and the Nikon Instruments Elements software. Image stacks were acquired for morphologic reconstruction with 20× objective. Images of neurons were acquired using with 60× objective and 2× scanning zoom for bouton analysis (stacks of 20-25 µm with 1 µm resolution) on the soma, proximal dendrites (<50 μm from the soma), and distal dendrites (>90 µm from the soma) (Hjorth et al., 2020). Parameters were kept consistent across WT and cKO slices. Morphologic quantification was done using the Surface and Filament tools in the IMARIS software. The automatic component of the filament tool reconstructed the dendritic field of the neuron with the dendrite beginning point set to 15 µm and the end point set to 1 µm. The volume of dendritic spread was found by using the Convex Hull Xtension of IMARIS. Bouton analysis was done using the Surface and Spots tools in IMARIS. The surface of the neuron soma or dendrite was reconstructed. A colocalization channel was created to identify individual boutons. This channel was then passed by a threshold to filter weak colocalizations. The brightest 30% of colocalized particles were chosen as boutons. Spots outside 1.5 µm of the neuron surface were also excluded. Bouton density was calculated as the number of boutons divided by the surface area of the soma.
Behavioral task
Six-week-old ChAT-Cre; Er81+/+ (control) and ChAT-Cre; Er81flox/flox (Er81 cKO) mice were caged in standardized room conditions under a 12:12 light: dark cycle with ad libitum food and water. Starting 5 d before the behavioral task and throughout the training, mice were maintained under a mild water restriction (2 h ON/22 h OFF) with ad libitum food access. Mice were then trained in an arena (40 cm length × 27 cm width × 25 cm height) made of transparent Plexiglas, divided into three 9 cm corridors, and placed on top of a DigiGait apparatus (Mouse Specifics) where a camera recorded the ventral view of the animal in the middle corridor. The outer corridors were equipped with dispensers to deliver a drop of 10% sucrose in correct trials. We used custom-written MATLAB programs to automatically deliver rewards on lick detection through capacitance sensors, to monitor mouse behavior and to extract the data.
At the beginning of a session, mice were placed at the starting point of the middle corridor and had to go through one of the one-way gates to choose between the two outer corridors. If the choice was correct, the mouse could collect the reward automatically delivered by the dispenser. The mouse could then go back to the start area through a second one-way gate to start the next trial. Before acquisition, mice were habituated to the apparatus over a period of 6 d during which they were allowed to move freely in the maze, with both dispensers delivering the reward. To complete the habituation session, mice had to complete 10 trials, one trial being defined as running from the start site to one of the outer corridors, then back to the start site. If 10 trials were not completed after 40 min, the session was terminated.
During the acquisition phase, each mouse was required to complete 20 trials per session and one session a day for 3 d. Only one side contained the reward. The rewarding side (left or right) was homogenized within an experimental group and balanced between groups. On day 4, corresponding to reversal 1, the side containing the reward was switched for each mouse. Mice were then exposed to the new side for 10 consecutive sessions (day 4 to day 13). From day 14, corresponding to reversal 2, the reward sides were switched again. Mice were tested for 3 d. After the training in day 14, some of the mice were removed from training for immunohistochemistry analyses. These mice displayed similar performance to those that fully completed the task (day 14: n = 5 control and 4 Er81 cKO mice; behavior vs cFos groups: F(3,21) = 1.5 p = 0.244; one-way ANOVA; data not shown). Performance was defined as the percentage choice of the reward side, regardless of reward collection by the animal.
Statistical analysis
All statistical tests were conducted in MATLAB (The MathWorks), GraphPad Prism 8, and R, except for behavior (see below). When comparing two groups, data were passed to Shapiro–Wilk normality test to determine suitability for a parametric test. If the data passed this test, an independent two-tailed t test was applied; otherwise, a Wilcoxon rank sum test was performed. Pearson's correlation strength (ρ) and p values were computed using MATLAB's corr function with its inbuilt t test. p values < 0.05 were considered statistically significant. We used one-way ANOVAs to analyze three or more groups of data. Two-way ANOVAs were used to analyze the interactions between two independent variables. All these tests were followed by Bonferroni's multiple comparisons test for pairwise comparisons when dealing with three or more groups. A random permutation test was performed by shuffling the STAs of the cells in the control and Er81 cKO groups to compute 95% CI and determine the statistical significance of the differences between STAs in the control and the Er81 cKO condition. For behavior, statistical analyses were performed with the StatView software, using two-way ANOVA to investigate statistical significance of interaction between repeated measures of daily performance and genotype, followed by Bonferroni's multiple comparisons test when appropriate. Statistical significance of performance against chance level (50%) was analyzed using two-tailed one-sample analyses.
Data are presented as mean ± SEM. Exact p values are reported in the text. Individual points in each graph represent the samples used for the statistical analysis.
Results
Er81 is expressed in striatal cholinergic interneurons
We first analyzed the expression of the Er81 transcription factor from birth to adulthood. We found that both mRNA (n = 8 mice per group, F(5) = 31.39, p = 7.38 × 10−11, one-way ANOVA; P6 vs P30: p = 4.54 × 10−9, Bonferroni's multiple comparisons test; Fig. 1A) and protein levels (P6; n = 4 mice vs P30; n = 6 mice, t(8) = 5.10, p = 9.28 × 10−4, two-sample t test; Fig. 1B) significantly drop from postnatal day 6 (P6) to P30 in the total striatum. In CINs specifically, we observed a similar decrease in Er81 protein levels from P6 to P30 (P6; n = 12 mice vs P30; n = 10 mice, t(20) = 4.49, p = 2.23 × 10−4, two-sample t test; Fig. 1C). To further determine the proportion of CINs expressing Er81 at any stage during development, we utilized β-galactosidase staining, which perdures long after being synthesized (Dehorter et al., 2015). We found that most CINs (62 ± 7%, n = 106 cells) in the Er81nlsLacZ/+ mice express β-galactosidase (and therefore Er81) in the striatum, unlike cholinergic cells in the basal forebrain (6 ± 2%, n = 89, z = 6.79 p = 1.48 × 10−11, Wilcoxon rank sum test; Fig. 1D,E). As it has been shown that the LIM homeodomain transcription factor Lhx6 segregates GABAergic CINs (Lozovaya et al., 2018), we next examined whether Er81 expression was based on Lhx6 segregation. We found that Er81 is expressed in both Lhx6-positive and Lhx6-negative CINs (Fig. 1F,G), and was expressed at similar levels (Lhx6+; n = 154 cells, Lhx6–; n = 110 cells, 5 mice, z = 0.31, p = 0.755, Wilcoxon rank sum test; data not shown). Thus, Er81 and Lhx6 expressions segregate different subgroups of CINs. To reveal a potential role of Er81 in CIN maturation and/or function, we specifically removed Er81 using cKO (ChAT-Cre; Er81f/f and control mice: ChAT-Cre; Er81+/+; Fig. 2A). We did not observe any change in cholinergic cell density within the striatum at P2 (while cells are migrating) and P30 (when cells are established in the structure) between control and Er81 cKO conditions, suggesting that the specific deletion of Er81 does not affect CIN neurogenesis and migration (P2 control; n = 4011 cells, 6 mice, P2 Er81 cKO; n = 1050 cells, 6 mice, t(10) = 0.17, p = 0.871, P30 control; n = 1084 cells, 6 mice, P30 Er81 cKO; n = 1050 cells, 6 mice, t(10) = 0.28, p = 0.784, two-sample t tests; Fig. 2B).
Figure 1.

Expression of Er81 in cholinergic interneurons. A, Postnatal expression of Er81 mRNA from P0 to P120 in the dorsal striatum. Data normalized to P30. Stars represent Bonferroni's test result. B, Er81 protein levels in P6 and P30 in the dorsal striatum quantified by Western blot. C, Er81 protein levels in CINs of the striatum at P6 and P30. Er81 immunofluorescence (IF) is normalized to the background of the images. D, Er81 expression in ChAT+ neurons in the striatum marked with βGal at P30 (top row). Lack of Er81 expression in ChAT+ neurons in basal forebrain (bottom row). Scale bars: 50 µm; Inset, 25 µm. E, Cumulative distribution of the βGal-expressing ChAT+ cells in the striatum and basal forebrain. F, Immunostaining for ChAT (blue), Lhx6 (GFP, green), and Er81 (red). Scale bar, 30 µm. G, Classification of CINs into four groups based on Er81 and Lhx6 expressions. ***p < 0.001.
Cholinergic interneuron molecular and morphological properties change in the absence of Er81
We assessed the molecular properties of the CINs related to the presence of Er81 in the striatum. First, we quantified the percentage of CINs expressing Lhx6 protein (control; 49 ± 4%, n = 213 cells, 5 mice, Er81 cKO; 41 ± 7%, n = 196 cells, 5 mice, z = 0.42, p = 0.676, Wilcoxon rank sum test; data not shown) as well as the Lhx6 protein levels (control; 0.48 ± 0.049 a.u., n = 213 cells, 5 mice; Er81 cKO; 0.50 ± 0.078 a.u., n = 196 cells, 5 mice, t(8) = 0.18, p = 0.858, two-sample t test; data not shown) and mRNA levels (control; n = 5 mice, Er81 cKO; n = 6 mice, t(9) = 0.37, p = 0.716, two-sample t test; Fig. 2C) and found that they were not different between the control and the Er81 cKO CINs at P30. This result reinforces the idea that striatal cholinergic population is molecularly diverse, and that Er81 and Lhx6 expression marks different CIN subgroups. We then analyzed other key markers to determine the effect Er81 may have, as a transcriptional regulator, on CIN GABAergic identity throughout development. We found no change in GAD2 (glutamic acid decarboxylase 2) and Lhx6 mRNA at P6 (Lhx6, control: n = 8 mice, Er81 cKO: n = 6; z = 1.87, p = 0.061, Wilcoxon rank sum test; GAD2, control: n = 6 mice, Er81 cKO; n = 5 mice, t(9) = 0.43, p = 0.674, two-sample t test; data not shown). These results suggest that Er81 deletion does not affect the GABAergic cell identity of the CINs. We then quantified the expression of genes associated with striatal cholinergic cell identity, using FACS to select GFP-expressing CINs, followed by qPCR. We found only a select number of genes with altered expression following Er81 ablation. At developmental stages, only the LIM homeodomain transcription factor Islet1 (Isl1), which is necessary for CIN identity (Allaway and Machold, 2017), displayed an increase in mRNA expression (P6; 5 mice per group, t(8) = 2.44, p = 0.040; Fig. 2C; P30; 5 mice per group, t(8) = 0.12, p = 0.906, two-sample t tests; data not shown). Other factors, such as the LIM homeodomain transcription factor Lhx7 (Lopes et al., 2012) (P6; control; n = 9 mice, Er81 cKO; n = 4 mice, t(11) = 0.29, p = 0.780, two-sample t test; data not shown), Zic4 (Magno et al., 2017) (P6; n = 12 mice per group, t(22) = 0.04, p = 0.966, two-sample t test; data not shown), acetylcholine esterase Ache (P6; control; n = 11 mice; Er81 cKO; n = 9 mice, t(18) = 0.190, p = 0.851, two-sample t test; data not shown), and choline transporter Slc5a7 (P6; control; n = 6 mice; Er81 cKO; n = 5 mice, t(9) = 0.16, p = 0.878, two-sample t test; data not shown) mRNA levels showed no developmental changes following the KO. However, in adult stages, only Slc5a7 expression was upregulated in the cKO condition (P30; control; n = 5 mice, Er81 cKO; n = 6 mice, t(9) = 2.40, p = 0.040, two-sample t test; Fig. 2C). The main marker of cholinergic identity, choline acetyltransferase (ChAT), displayed marked changes in expression during maturation. ChAT mRNA and protein expressions significantly increased in the Er81 cKO at P6 (mRNA; control; n = 10 mice, Er81 cKO; n = 9 mice; 2.03 ± 0.19, t(17) = 4.84, p = 1.52 × 10−4, protein; control; n = 4 mice, Er81 cKO; n = 5 mice, t(7) = 4.0, p = 0.005, two-sample t test; data not shown). On the other hand, ChAT mRNA level was lower (control; n = 3 mice, Er81 cKO; n = 6 mice, t(7) = 4.3, p = 0.004, two-sample t test; Fig. 2C) and ChAT protein levels were unchanged at P30 in the Er81 cKO conditions (control; n = 4 mice; Er81 cKO; n = 4 mice, z = 0.14, p = 0.886, Wilcoxon rank sum test; data not shown), indicative of early maturation of the CINs. As Er81 incites a number of molecular changes during developmental stages and in particular in dendritogenesis (Abe et al., 2011; Willardsen et al., 2014; Ding et al., 2016), we analyzed CIN dendritic properties and ChAT+ neuropil area. While the overall spread and the number of dendritic intersections measured by Sholl analysis were unchanged (control; n = 8 cells, 3 mice, Er81 cKO; n = 7 cells, 3 mice, spread volume: t(13) = 0.55, p = 0.591, two-sample t test; Sholl: two-way ANOVA, F(1,13, genotype) = 2.68, p = 0.126, F(150,1950, interaction) = 0.61, p = 0.999; Fig. 2D–F), the dendritic volume and number of branch points showed a significant reduction in the absence of Er81 (dendrite volume; t(13) = 3.18, p = 0.007, number of branch points; t(13) = 2.37, p = 0.034; Fig. 2G and H). In addition, CIN axonal projections were significantly decreased in the Er81 cKO condition (ChAT neuropil area; control; n = 4 mice, Er81 cKO; n = 3 mice, t(5) = 7.06, p = 8.8 × 10−4; Fig. 2I). Together, these results show that Er81 regulates key developmental genes and the morphologic complexity of the striatal CINs.
Er81 deletion in cholinergic interneurons induces an increase in Ih and IsAHP currents
To determine how the Er81 transcription factor regulates CIN functional properties, we performed in vitro whole-cell patch-clamp recordings from GFP-labeled ChAT-positive (ChAT+) neurons in control (ChAT-cre; RCE-GFP; Er81+/+) and Er81 cKO mice (ChAT-Cre; RCE-GFP; Er81f/f mice). While we could not find any correlation between the expression of Er81 and most of the CIN basic membrane properties at P6 and P30 in control conditions (Table 1), we did observe changes after Er81 deletion from CINs at P6 (Table 2) and P30 (Table 3). In particular, the AHP phase of the APs was altered in the Er81 cKO condition at P30, with significantly slower AHP kinetics (Table 3) compared with the control (P30; control; n = 13 cells, 4 mice, Er81 cKO; n = 31 cells, 4 mice, AHP amplitude; z = 1.4, p = 0.150, Wilcoxon rank sum test, AHP peak delay; z = 2.28, p = 0.023; Fig. 2J,K) but not at P6 (AHP amplitude; control; n = 13 cells, 4 mice, Er81 cKO; n = 10 cells, 3 mice, z = 0.28, p = 0.780, AHP peak delay; z = 0.71, p = 0.476, Wilcoxon rank sum tests; data not shown). The absence of Er81 also led to a significant amplification of the early component of the induced sAHP, whereas the late component amplitude was not affected by Er81 deletion (P30; control; n = 14 cells, 4 mice, Er81 cKO; n = 33 cells, 4 mice; two-way ANOVA, F(1,45, genotype) = 0.56, p = 0.459, F(1,45, interaction) = 40.2, p = 9.7 × 10−8, followed by Bonferroni's multiple comparisons test; early sAHP; p = 0.003, late sAHP; p = 0.126; Fig. 2L,M). Analysis of sAHP at P6 stage did not reveal any difference between the control and the Er81 cKO conditions (control; n = 13 cells, 4 mice, Er81 cKO; n = 9 cells, 3 mice, two-way ANOVA, F(1,20, genotype) = 0.005, p = 0.944, F(1,20, interaction) = 0.011, p = 0.918, followed by Bonferroni's multiple comparisons test; early sAHP; p > 0.999, late sAHP; p > 0.999; data not shown). The preceding depolarization was not significantly different between the two groups at either stage (Tables 2, 3). To further analyze CIN physiological properties, we recorded the evoked firing responses and the tail currents responsible for sAHP (Wilson and Goldberg, 2006). CINs exhibited less persistent evoked firing in the Er81 cKO mice (Fig. 3A). While the control CINs maintained a high rate of activity during stimulation, the instantaneous firing rate of the P30 Er81 cKO cells quickly dropped (control; n = 16 cells, 5 mice, Er81 cKO; n = 31 cells, 4 mice, two-way ANOVA; F(1,45, genotype) = 5.7, p = 0.021, F(1,45, interaction) = 3.1, p = 0.084, followed by Bonferroni's multiple comparison test; 0-0.1 s; p = 0.470, 0.1-3 s; p = 0.008; Fig. 3B). We did not observe such a difference at P6 (control; n = 16 cells, 5 mice, Er81 cKO; n = 17 cells, 4 mice, two-way ANOVA; F(1,31, genotype) = 0.87, p = 0.358, F(1,31, interaction) = 0.12, p = 0.728, followed by Bonferroni's multiple comparison test; 0-0.1 s; p = 0.696, 0.1-3 s; p > 0.999; data not shown). Furthermore, the average firing rate of the CINs remained unchanged at P6 (control; n = 16 cells, 5 mice, Er81 cKO; n = 17 cells, 4 mice, two-way ANOVA; F(1,31, genotype) = 0.52, p = 0.477, F(9,279, interaction) = 0.81, p = 0.611; data not shown) but was consistently lower in the Er81 cKO conditions for all current intensities at P30 (control; n = 16 cells, 5 mice, Er81 cKO; n = 31 cells, 4 mice, two-way ANOVA; F(1,45, genotype) = 13.7, p = 5.7 × 10−4, F(9,405, interaction) = 8.69, p = 5.7 × 10−12; Fig. 3C). Together, our results indicate that the initial responsiveness of the CINs is not altered, but their overall excitability is reduced because of stronger firing rate adaptations in the absence of Er81 at P30 (control; n = 16 cells, 5 mice, Er81 cKO; n = 31 cells, 4 mice, two-way ANOVA; F(1,45, genotype) = 8.6, p = 0.005, F(9,405, interaction) = 6.7, p = 5.9 × 10−9; Fig. 3D) but not at P6 (control; n = 16 cells, 5 mice, Er81 cKO n = 17 cells, 4 mice, two-way ANOVA; F(1,31, genotype) = 0.43, p = 0.515, F(9,279, interaction) = 0.48, p = 0.884; data not shown). It has been shown that CIN firing rate is controlled by IsAHP, which is composed of a large calcium-dependent delayed rectifier current (Wilson and Goldberg, 2006; Zhang et al., 2018) and the cadmium-insensitive, voltage-dependent IKr (Zhang et al., 2018). Therefore, the observed differences (Fig. 3B) might be because of an increase in IsAHP in the Er81 cKO conditions. To test this, we applied a depolarizing pulse in voltage-clamp mode to activate IsAHP with both of its components (Wilson and Goldberg, 2006), which was maintained for a few seconds after the voltage pulse (Fig. 3E). We found that the amplitude of IsAHP remains unchanged at P6 (control; n = 9 cells, 5 mice, Er81 cKO; n = 10 cells, 3 mice, two-way ANOVA; F(1,17, genotype) = 0.16, p = 0.693, F(3,51, interaction) = 0.45, p = 0.721; data not shown), but is significantly larger in the Er81 cKO compared with the control CINs at P30 (control; n = 14 cells, 3 mice, Er81 cKO; n = 22 cells, 4 mice, two-way ANOVA; F(1,34, genotype) = 6.0, p = 0.019, F(9,405, interaction) = 6.7, p = 5.9 × 10−9; Fig. 3F,G). In order to decipher the molecular mechanisms behind this regulation, we first analyzed the expression of previously identified IKr mediators in CINs, such as the KCNQ2/3 hetero-tetramer channels (Zhang et al., 2018) and the metabotropic glutamate receptor 5 (mGluR5), which can amplify IsAHP by raising intracellular calcium concentration (Reiner and Levitz, 2018). We found a significant decrease in Kcnq2 mRNA but not the channel protein levels (mRNA; control; n = 6 mice, Er81 cKO; n = 5 mice, t(9) = 4.8, p = 9.6 × 10−4, two-sample t test, Fig. 3H; protein; control; n = 5 mice, 260 CINs, Er81 cKO; n = 4 mice, CINs, t(7) = 1.1, p = 0.302, two-sample t test, Fig. 3I,J). Interestingly, Grm5 mRNA levels were also decreased, whereas the mGluR5 protein level was increased in the Er81 cKO (mRNA; control; n = 7 mice, Er81 cKO; n = 6 mice, t(11) = 4.14, p = 0.002, two-sample t test, Fig. 3K; protein; control; n = 5 mice, 264 CINs, Er81 cKO; n = 4 mice, 211 CINs, t(7) = 6.2, p = 4.3 × 10−4, two-sample t test, Fig. 3L,M). These results demonstrate that Er81 deletion could alter IsAHP current via elevated mGluR5 at P30. Er81 deletion did not cause any significant change in AHPs, IsAHP, or their associated properties at P6. We conclude that the ablation of Er81 expression has an impact on cell maturation, which leads to permanent changes in cell properties in mature state. This is different from direct transcriptional control observed in PV interneurons (Dehorter et al., 2015).
Table 1.
Correlation between CIN physiological parameters and the Er81 expression levels at P6 and P30a
| rho | P6 |
P30 |
||||
|---|---|---|---|---|---|---|
| p | n | rho | p | n | ||
| Resting membrane potential (mV) | −0.533 | 0.113 | 10 | 0.280 | 0.405 | 11 |
| Membrane capacitance (pF) | −0.206 | 0.500 | 13 | 0.286 | 0.302 | 15 |
| Membrane resistance (mΩ) | 0.174 | 0.570 | 13 | −0.327 | 0.234 | 15 |
| AP threshold (mV) | −0.712 | 0.021* | 10 | 0.337 | 0.310 | 11 |
| AP amplitude (mV) | 0.303 | 0.395 | 10 | 0.008 | 0.982 | 11 |
| AP rise to peak (ms) | −0.377 | 0.283 | 10 | −0.217 | 0.522 | 11 |
| AP fall to threshold | 0.195 | 0.589 | 10 | 0.102 | 0.766 | 11 |
| AHP amplitude (mV) | 0.162 | 0.654 | 10 | 0.137 | 0.688 | 11 |
| AHP peak delay (ms) | 0.279 | 0.435 | 10 | 0.280 | 0.405 | 11 |
| AHP rise time constant (ms) | 0.634 | 0.049* | 10 | −0.187 | 0.582 | 11 |
| AHP decay time constant (ms) | −0.158 | 0.662 | 10 | 0.029 | 0.932 | 11 |
| Spontaneous AP rate (whole-cell) | 0.683 | 0.029* | 10 | 0.353 | 0.287 | 11 |
| Spontaneous AP rate (cell-attached) | 0.345 | 0.569 | 5 | 0.442 | 0.321 | 7 |
| Evoked AP rate (Hz) | 0.109 | 0.765 | 10 | 0.469 | 0.146 | 11 |
| First AP latency (ms) | 0.269 | 0.452 | 10 | −0.162 | 0.634 | 11 |
| Early sAHP amplitude (mV) | −0.174 | 0.631 | 10 | 0.406 | 0.215 | 11 |
| Late sAHP amplitude (mV) | −0.202 | 0.575 | 10 | 0.437 | 0.179 | 11 |
| AP recovery time (s) | −0.332 | 0.382 | 10 | −0.531 | 0.141 | 11 |
| sEPSC frequency (Hz) | −0.254 | 0.426 | 12 | −0.138 | 0.542 | 22 |
| sEPSC amplitude (pA) | −0.357 | 0.254 | 12 | 0.067 | 0.767 | 22 |
| sIPSC frequency (Hz) | −0.257 | 0.445 | 11 | −0.250 | 0.486 | 10 |
| sIPSC amplitude (pA) | −0.234 | 0.489 | 11 | 0.474 | 0.166 | 10 |
| Ih (pA) | 0.375 | 0.256 | 11 | −0.172 | 0.574 | 13 |
| IsAHP (pA) | −0.324 | 0.434 | 8 | 0.633 | 0.251 | 5 |
aIh, Hyperpolarization-activated current; IsAHP, delayed rectifier current.
*p < 0.05.
Table 2.
Intrinsic properties of striatal CINs in the Er81 cKO and the control mice at P6
| P6 | Ctl | n | Er81 cKO | nrepresent Wilcoxon rank sum test, and | Statistica | p |
|---|---|---|---|---|---|---|
| Resting membrane potential (mV) | −49.18 ± 1.03 | 13 | −47.10 ± 1.49 | 9 | t(20) = 1.19 | 0.248 |
| Membrane capacitance (pF) | 63.06 ± 3.76 | 16 | 63.22 ± 4.41 | 17 | t(31) = 0.03 | 0.979 |
| Membrane resistance (mΩ) | 209.72 ± 13.37 | 16 | 217.19 ± 15.24 | 17 | t(31) = 0.37 | 0.716 |
| AP threshold (mV) | −37.95 ± 1.03 | 13 | −34.82 ± 1.36 | 9 | z = 1.90 | 0.062 |
| AP amplitude (mV) | 60.24 ± 2.53 | 13 | 62.94 ± 3.15 | 9 | t(20) = 0.67 | 0.508 |
| AP rise to peak (ms) | 1.15 ± 0.07 | 13 | 1.14 ± 0.11 | 9 | t(20) = 0.09 | 0.929 |
| AP fall to threshold (ms) | 3.07 ± 0.13 | 13 | 3.14 ± 0.24 | 9 | z = 0.20 | 0.841 |
| AHP rise time constant (ms) | 3.79 ± 0.45 | 13 | 3.28 ± 0.44 | 9 | t(20) = 0.77 | 0.449 |
| AHP decay time constant (ms) | 737.2 ± 184.6 | 13 | 381.0 ± 75.1 | 9 | z = 0.67 | 0.504 |
| Spontaneous AP rate (Hz, whole-cell) | 1.29 ± 0.19 | 13 | 3.34 ± 0.50 | 9 | t(20) = 4.32 | 3.3 × 10−4 |
| Spontaneous AP rate (Hz, cell-attached) | 1.04 ± 0.28 | 8 | 1.07 ± 0.23 | 16 | z = 0.09 | 0.927 |
| First AP latency (ms) | 3.79 ± 0.21 | 13 | 3.93 ± 0.49 | 9 | z = 0.67 | 0.503 |
| AP recovery time (s) | 7.91 ± 2.77 | 12 | 4.01 ± 1.03 | 9 | z = 1.31 | 0.189 |
| Preceding depolarization (mV) | 29.27 ± 2.63 | 13 | 25.92 ± 3.18 | 9 | t(20) = 0.81 | 0.425 |
az statistics represent Wilcoxon rank sum test, and t statistics represent two-sample t test.
Table 3.
Intrinsic properties of striatal CINs in the Er81 cKO and the control mice at P30
| P30 | Control | n | Er81 cKO | n | Statistica | p |
|---|---|---|---|---|---|---|
| Resting membrane potential (mV) | −48.20 ± 1.32 | 14 | −48.90 ± 0.79 | 33 | t(45) = 0.52 | 0.608 |
| Membrane capacitance (pF) | 111.81 ± 11.85 | 16 | 121.03 ± 8.80 | 31 | z = 0.82 | 0.412 |
| Membrane resistance (mΩ) | 92.39 ± 6.66 | 16 | 86.57 ± 4.79 | 31 | z = 0.94 | 0.346 |
| AP threshold (mV) | −39.14 ± 0.81 | 13 | −38.90 ± 0.80 | 32 | z = 0.14 | 0.890 |
| AP amplitude (mV) | 64.18 ± 2.38 | 13 | 66.22 ± 1.47 | 32 | z = 1.01 | 0.310 |
| AP rise to peak (ms) | 0.70 ± 0.06 | 13 | 0.68 ± 0.03 | 32 | z = 0.30 | 0.764 |
| AP fall to threshold (ms) | 2.88 ± 0.26 | 13 | 2.71 ± 0.13 | 32 | t(43) = 0.66 | 0.512 |
| AHP rise time constant (ms) | 3.70 ± 0.26 | 13 | 5.52 ± 0.57 | 32 | z = 2.09 | 0.037* |
| AHP decay time constant (ms) | 257.90 ± 30.82 | 13 | 670.88 ± 127.88 | 32 | z = 1.46 | 0.143 |
| Spontaneous AP rate (Hz, whole-cell) | 3.69 ± 1.03 | 13 | 2.91 ± 0.44 | 32 | z = 0.23 | 0.822 |
| Spontaneous AP rate (Hz, cell-attached) | 7.09 ± 1.72 | 11 | 5.43 ± 0.80 | 16 | t(25) = 0.97 | 0.341 |
| First AP latency (ms) | 4.98 ± 0.90 | 14 | 4.34 ± 0.35 | 33 | z = 0.05 | 0.963 |
| AP recovery time (s) | 5.23 ± 1.35 | 11 | 9.41 ± 2.03 | 27 | z = 0.71 | 0.479 |
| Preceding depolarization (mV) | 9.88 ± 0.96 | 14 | 10.37 ± 0.60 | 33 | z = 0.00 | 1.000 |
az statistics represent Wilcoxon rank sum test, and t statistics represent two-sample t test.
*p < 0.05.
A large inward rectifier current (Ih) is associated with a longer pause in CIN firing and a reduction in spontaneous activity (Wilson and Goldberg, 2006). However, despite a larger IsAHP current in Er81 cKO condition (Fig. 2F), we saw no difference in the AP recovery time and baseline firing rates (Table 3), suggesting that the overall duration of the pause in CIN firing is unlikely to be affected by Er81 deletion. Consistently, we also observed that a counter current is engaged to balance CIN activity in the absence of Er81, as the late sAHP showed no difference between the two groups (Fig. 2L). We examined the prominent inward rectifying Ih current that contributes to rebound activity following hyperpolarization and the tonic firing of the CINs (Bennett et al., 2000). While we could not see any difference between control and Er81 cKO conditions in CINs at P6 (sag ratio: control; n = 16 cells, 5 mice; Er81 cKO; n = 17 cells, 4 mice; two-way ANOVA; F(1,31, genotype) = 0.002, p = 0.960, F(7,217, interaction) = 0.35, p = 0.927; rebound firing: two-way ANOVA, F(1,31,genotype) = 0.037, p = 0.849, F(1,31, interaction) = 0.52, p = 0.475, followed by Bonferroni's multiple comparisons test; 3-3.23 s; p > 0.999, 3.23-5 s; p > 0.999; data not shown), we found that both sag ratio and rebound firing were enhanced at P30 in Er81 cKO CINs (control; n = 16 cells, 5 mice, Er81 cKO; n = 31 cells, 4 mice, sag ratio: F(1,45, genotype) = 7.1, p = 0.010, F(7,315, current) = 9.5, p = 2.2 × 10−4, F(7,315, interaction) = 5.1, p = 1.7 × 10−5, two-way ANOVA; rebound firing: F(1,45, genotype) = 2.4, p = 0.125, F(1,45, interaction) = 6.0, p = 0.0178, two-way ANOVA, followed by Bonferroni's multiple comparisons test: 3-3.23 s; p = 1.000, 3.23-5 s; p = 0.029; Fig. 4A–C). Consistently, we found the Ih current amplitude unchanged in the Er81 cKO compared with the control CINs at P6 (control; n = 13 cells, 5 mice, Er81 cKO; n = 11 cells, 4 mice, two-way ANOVA; F(1,22, genotype) = 0.17, p = 0.678, F(5,110, interaction) = 0.08, p = 0.996, followed by Bonferroni's multiple comparisons test; p > 0.999 at all holding potentials; data not shown) and increased at P30 (control; n = 18 cells, 4 mice, Er81 cKO; n = 22 cells, 4 mice, two-way ANOVA; F(1,38, genotype) = 1.6, p = 0.215, F(7,315, interaction) = 5.1, p = 1.7 × 10−5, followed by Bonferroni's multiple comparisons test; V hold = −80 mV; p = 0.012; V hold = −70 mV; p = 0.003; Fig. 4D–F). We next analyzed the expression of the main channel subunits underlying the enhanced Ih current, that is, the hyperpolarization activated cyclic nucleotide-gated potassium and sodium channel 1 and 2 (HCN1 and HCN2) (Z. Zhao et al., 2016). However, we did not find any difference in the expression of either HCN2 or HCN1 between the Er81 cKO and the control CINs (Hcn2 mRNA; control; n = 5 mice, Er81 cKO; n = 4 mice, t(7) = 0.9, p = 0.391; HCN2 protein; control; n = 5 mice, 350 cells, Er81 cKO; n = 4 mice, 270 cells, t(7) = 0.9, p = 0.391, two-sample t test; HCN1 protein; control; n = 5 mice, 60 cells, Er81 cKO; n = 4 mice, 55 cells, z = 0.9, p = 0.391, Wilcoxon rank sum test; Fig. 4G–J). These results show that, rather than a quantitative change in the channel subunits underlying Ih, their function is likely enhanced in the absence of Er81. Together, we demonstrate that the absence of Er81 affects the two major intrinsic currents IsAHP and Ih in the striatal CINs, through mGluR5 upregulation.
Figure 4.

Increase in the Ih in CINs following Er81 deletion. A, Representative responses of P30 CINs to a negative current pulse in the control and the Er81 cKO conditions. Arrowheads indicate the voltage sag. B, Average sag ratio at different current steps. Stars represent genotype effect. C, Rebound firing at the end of negative current steps. D, Example voltage-clamp trace showing the slowly activated Ih (arrow) during a hyperpolarizing voltage pulse. E, Traces of Ih averaged across CINs at different holding potentials (−70 to −110 mV, 10 mV steps). F, Time-averaged Ih across CINs at different holding potentials. Stars represent Bonferroni's test result. G. Hcn2 mRNA expression levels in the striatum of control and Er81 cKO mice. H, Representative image of a ChAT-GFP cell (green) expressing HCN2 protein in control and Er81 cKO. Scale bar, 10 µm. I, Expression of HCN2 protein in the CINs of control and the Er81 cKO group. J, Expression of HCN1 protein in the CINs of control and the Er81 cKO group. a.u.: arbitrary units. *p < 0.05. **p < 0.01.
Rewiring of cholinergic interneurons in the absence of Er81
Changes in the functional properties of CINs are likely to affect their synaptic integration within the striatal microcircuit. To test whether the deletion of Er81 alters CIN synaptic profile, we performed in vitro patch-clamp recordings of the sEPSCs and sIPSCs onto CINs at P6 and P30. We did not observe any significant changes in the frequency and amplitude of sEPSCs in the absence of Er81 at either postnatal stage (P6, control; n = 12 cells, 6 mice, Er81 cKO; n = 14, 3 mice, rate; z = 0.9, p = 0.368, Wilcoxon rank sum test, amplitude; t(24) = 0.5, p = 0.624, two-sample t test, rise time; z = 0.3, p = 0.777, decay time; z = 0.7, p = 0.456, Wilcoxon rank sum tests; data not shown; P30, control; n = 22 cells, 9 mice, Er81 cKO; n = 20 cells, 4 mice, rate; z = 1.8, p = 0.076, Wilcoxon rank sum test, amplitude; t(40) = 0.8, p = 0.451, two-sample t test, rise time; z = 1.2, p = 0.222, decay time; z = 1.1, p = 0.284, Wilcoxon rank sum tests; Fig. 5A–D). As CINs receive excitatory inputs from both the cortex and thalamus (Ding et al., 2010), we examined whether pathway-specific inputs were modified in the absence of Er81. Bouton analyses revealed no significant changes in the density of the cortical vesicular glutamate transporter type 1 (vGluT1) and thalamic vesicular glutamate transporter type 2 (vGluT2) positive boutons (Doig et al., 2014) on the somas of CINs in the absence of Er81 at either postnatal stages (P30 somas; vGluT1; control; n = 15 cells, 3 mice, Er81 cKO; n = 15 cells, 3 mice, t(28) = 1.98, p = 0.058, vGluT2; control; n = 10 cells, 3 mice, Er81 cKO; n = 10 cells, 3 mice, t(18) = 0.10, p = 0.924, Fig. 5E–H, P6 somas; vGluT1; control; n = 12 cells, 3 mice, Er81 cKO,; t(22) = 1.09, p = 0.288 n = 12 cells, 3 mice in each condition, vGluT2; control; n = 14 cells, 3 mice, Er81 cKO = 10 cells, 3 mice, t(22) = 1.05, p = 0.306, two-sample t tests; data not shown). In addition, we found a homogeneous distribution of both transporters across proximal and distal segments of CIN dendrites in P30 WT conditions (vGluT1: n = 16 proximal, 9 distal dendrites, vGluT2: n = 13 proximal, 10 distal dendrites, 6 mice; two-way ANOVA: F(1,20, Transporter) = 0.90, p = 0.355, F(1,20, Dendrite segment) = 0.01, p = 0.913, F(1,20, Interaction) = 1.36, p = 0.258; data not shown), and this was unchanged in the KO (WT vs cKO, n = 4 mice; vGluT1: n = 17 proximal dendrites, t(8) = 0.87, p = 0.406; 8 distal dendrites, t(8) = 0.18, p = 0.860; vGluT2: n = 14 proximal dendrites, t(8) = 0.97, p = 0.362; 6 distal dendrites, t(8) = 0.73, p = 0.487; two-sample t tests; data not shown). Recordings of sIPSCs showed similar frequency but significantly larger amplitude and decay time in the Er81 cKO compared with the control cells at P30 (control; n = 16 cells, 4 mice, Er81 cKO; n = 24 cells, 5 mice, rate; z = 0.37, p = 0.709, amplitude; z = 3.55, p = 3.9 × 10−4, rise time; z = 0.94, p = 0.348, Wilcoxon rank sum test, decay time; t(38) = 3.70, p = 6.8 × 10−4, two-sample t test; Fig. 5I–L), with no changes at P6 (control; n = 12 cells, 3 mice, Er81 cKO; n = 12 cells, 3 mice, rate; z = 0.20, p = 0.840, amplitude; z = 0.38, p = 0.707, rise time; t(22) = 1.05, p = 305, decay time; z = 0.26, p = 0.795, Wilcoxon rank sum tests; data not shown). Since interneurons expressing the neuropeptide Y (NPY+) are the principal inhibitory sources to CINs (English et al., 2011; Szydlowski et al., 2013; Straub et al., 2016) and PV neurons are the most common type of inhibitory interneuron in the striatum, we determined whether NPY+ and PV+ bouton densities onto CINs were altered in the absence of Er81. We observed a higher density of NPY+ boutons in the Er81 cKO compared with control mice at P6 (control; n = 14 cells, 3 mice, Er81 cKO; n = 11 cells, 3 mice, t(23) = 2.43, p = 0.023, two-sample t test; data not shown) with no change in NPY+ boutons on the soma or dendrites at P30 (soma, control; n = 15 cells, 4 mice; Er81 cKO; n = 14 cells, 3 mice, t(27) = 0.58, p = 0.567, two-sample t tests; Fig. 5M,N; dendrites, control; n = 9 cells, 3 mice, Er81 cKO; n = 9 cells, 3 mice, t(16) = 0.46, p = 0.650, two-sample t tests; data not shown). We also found denser PV+ boutons on the Er81 cKO CIN somas compared with controls at P30 (control; n = 12 cells, 3 mice, Er81 cKO; n = 12 cells, 3 mice, t(22) = 2.18, p = 0.040; Fig. 5O,P), but not in dendrites (control; n = 12 cells, 3 mice; Er81 cKO; n = 12 cells, 3 mice, t(22) = 0.34, p = 0.738; data not shown). As Er81 deletion affects cholinergic neuropil (Fig. 2F), we expected that CIN output to other striatal cell types would also be altered. We found that the density of boutons expressing the specific CIN synaptic terminal marker vGluT3 (Gras et al., 2008; Higley et al., 2011; Nelson et al., 2014) was increased at P30 (soma, control; n = 14 cells, 3 mice, Er81 cKO; n = 15 cells, 3 mice, t(27) = 2.24, p = 0.033; Fig. 5Q,R; dendrites, control; n = 12 cells, 3 mice, Er81 cKO; n = 12 cells, 3 mice, t(22) = 1.53, p = 0.141, two-sample t tests; data not shown) but not at P6 (control; n = 16 cells, 3 mice, Er81 cKO; n = 7 cells, 3 mice, t(21) = 0.13, p = 0.899, two-sample t tests; data not shown) on neighbor CINs in the absence of Er81. On the contrary, vGluT3 bouton density onto NPY+ interneurons was reduced at P6 (control; n = 17 cells, 3 mice, Er81 cKO; n = 12 cells, 3 mice, t(27) = 2.47, p = 0.020, two-sample t tests; data not shown) but not at P30 (control; n = 21 cells, 4 mice, Er81 cKO; n = 16 cells, 3 mice, z = 0.11, p = 0.915; Fig. 5S) and was significantly reduced onto P30 PV-expressing interneurons in the Er81 cKO condition (control; n = 12 cells, 3 mice, Er81 cKO; n = 12 cells, 3 mice, t(22) = 2.29, p = 0.032, two-sample t test; Fig. 5T). Overall, these results suggest that the increase in IPSC amplitude of mature CINs lacking Er81 is because of a stronger connection with PV and/or CIN interneurons. Such changes in the circuitry are likely to influence the in vivo activity of these neurons and the striatal network.
Figure 5.

Rewiring of the CINs in the absence of Er81. A, Left, Excitatory inputs to CINs from cortex (ctx, dark blue) and thalamus (thal, light blue). Right, Average sEPSC traces of a control and Er81 cKO CIN. B-D. Rate (B), amplitude (C), rise time (D, left), and decay time (D, right) of sEPSCs in control and Er81 cKO CINs. E, Top, Control and Er81 cKO CINs (ChAT, gray) with vesicular glutamate transporter 1 (vGluT1, dark blue, cortical boutons). Bottom, 3D reconstruction of control and Er81 cKO CINs soma (gray) and vGluT1 boutons (spots). F, vGluT1 bouton density on control and Er81 cKO CINs. G, Control and Er81 cKO CINs (ChAT, gray) with vGluT2 boutons (light blue, thalamic boutons). Details as in E. H, vGluT2 bouton density on control and Er81 cKO CINs. I, Left, Schematic of local inhibitory inputs to CINs from NPY+ (yellow) and PV+ (magenta) interneurons. Right, Average spontaneous sIPSC traces of a control and Er81 cKO CIN. J-L. Rate (J), amplitude (K), rise time (L, left), and decay time (L, right) of sIPSCs in control and Er81 cKO CINs. M, Control and Er81 cKO CINs with NPY+ boutons (yellow). Details as in E. N, NPY+ bouton density on control and Er81 cKO CINs. O, Control and Er81 cKO CINs with PV+ boutons (magenta). Details as in E. P, PV+ bouton density on control and Er81 cKO CINs. Q, Control and Er81 cKO CINs with vGluT3+ boutons (green, cholinergic boutons). Details as in E. R, vGluT3+ bouton density on control and Er81 cKO CINs. S, ChAT+ bouton density on NPY+ interneurons in control and Er81 cKO conditions. T, ChAT bouton density on PV+ interneurons in control and Er81 cKO conditions. Diamonds represent averages. Circles represent individual neurons. Scale bars, 5 µm. *p < 0.05. **p < 0.01. ***p < 0.001.
Changes in spike timing of striatal cholinergic interneurons alter striatal sensorimotor processing
CINs have a key role in striatal function by controlling the output neurons (Mamaligas and Ford, 2016) and modulating their GABAergic (English et al., 2011), glutamatergic (Mamaligas et al., 2019), and dopaminergic inputs (Threlfell et al., 2012; Kosillo et al., 2016; Brimblecombe et al., 2018). It is unknown how fine-tuning of the CIN firing impacts sensorimotor processing. We anticipated that alterations in CIN properties will have a crucial impact on striatal function. To address that, we performed in vivo multielectrode array recordings from the dorsal striatum, which receives somatotopically organized sensory and motor inputs from the cortex (Robbe, 2018). We coupled our recordings to whisker stimulation in awake adult mice (Fig. 6A), taking advantage of the whiskers as mobile sensors to study movement-related neuronal activities in a head-fixed configuration. In the first step, we excluded from analysis the putative fast spiking cells characterized by narrow spikes (Mallet et al., 2005; Lee et al., 2017; Dorst et al., 2020) and potential artifacts (see Materials and Methods; Fig. 6B, black). This exclusion based on waveform resulted in 731 cells from 3 control and 732 cells from 4 Er81 cKO mice (Fig. 6B, teal). We then identified putative SPNs and CINs by the following three criteria: spontaneous activity (<1 Hz or >1 Hz, respectively) (English et al., 2011; Sharott et al., 2012), phasic responses to sensorimotor stimuli, as captured by δ firing rate (Inokawa et al., 2010), and the proportion of ISIs > 2 s (PropISI >30% or < 30%, respectively; Fig. 6C, right) (Benhamou et al., 2014). Figure 6D, E illustrates the response profile of all recorded cells based on these segregations. Group 1 (n = 267 cells in control and 366 cells in Er81 cKO mice) represents putative SPNs (pSPNs, Fig. 6D) and Group 3 (n = 150 cells in control and 111 cells in Er81 cKO mice) represents putative CINs (pCINs; Fig. 6E). Group 2 (n = 260 cells in control and 200 cells in Er81 cKO mice) and Group 4 (n = 54 cells in control and 55 cells in Er81 cKO mice) represent more heterogeneous cell populations of pSPNs and pCINs, respectively, with other putative interneurons, such as low-threshold spiking interneurons (Sharott et al., 2009; Beatty et al., 2012).
In the Er81 cKO mice, we found significantly larger responses to the stimulus in Group 1 compared with the control (pSPNs; n = 267 control cells; n = 366, Er81 cKO cells; two-way ANOVA, F(1,623, genotype) = 138.1, p = 6.11 × 10−29, F(9,623, interaction) = 35.9, p = 3.57 × 10−51; Fig. 6D, top) and a weaker response in Group 2 (pSPNs and other interneurons; n = 260 control cells, Er81 cKO; n = 200 Er81 cKO cells, two-way ANOVA, F(1,458, genotype) = 14.6, p = 1.49 × 10−4, F(9,458, interaction) = 6.058, p = 4.72 × 10−8; Fig. 6D, bottom). In contrast, cells in Group 3 (pCINs) exhibited a stronger inhibition/pause responses in the Er81 cKO condition, compared with the control (n = 150 control cells; n = 111 Er81 cKO cells; two-way ANOVA, F(1,255, genotype) = 70.5, p = 3.26 × 10−15, F(9,255, interaction) = 5.3, p = 1.42 × 10−6; Fig. 6E, top). Cells in group 4 (pCINs and other putative interneurons) displayed a stronger response to stimulus in the Er81 cKO compared with the control condition (n = 54 control cells; n = 55 Er81 cKO cells; two-way ANOVA, F(1,107, genotype) = 108.4, p = 5.95 × 10−18, F(9,107, interaction) = 11.0, p = 5.18 × 10−12; Fig. 6E, bottom).
Interestingly, the quiet state firing rate was significantly reduced in Groups 1 and 2 (pSPNs; Group 1: control; 0.27 ± 0.018 Hz, Er81 cKO; 0.20 ± 0.013 Hz, two-way ANOVA; F(1,623 genotype) = 10.4, p = 0.001, F(9,623, interaction) = 0.9, p = 0.494; Group 2: control; 0.45 ± 0.050, Er81 cKO; 0.34 ± 0.020; two-way ANOVA; F(1,458, genotype) = 22.8, p = 2.44 × 10−6, F(9,458, interaction) = 0.7, p = 0.729; data not shown), while the quiet state firing rate of the putative CINs was higher in the absence of Er81 (Group 3: control; 2.19 ± 0.261 Hz, Er81 cKO; 2.90 ± 0.180 Hz, two-way ANOVA; F(1,255, genotype) = 16.4, p = 6.88 × 10−5, F(9,255, interaction) = 1.1, p = 0.352; Group 4: control; 1.86 ± 0.122, Er81 cKO; 2.45 ± 0.217, two-way ANOVA; F(1,107, genotype) = 5.5, p = 0.021, F(9,107, interaction) = 0.6, p = 0.799; data not shown).
These results reveal that an increase in pCIN inhibition (Group 3) in the absence of Er81 leads to enhanced excitation of the pSPNs (Group 1) during stimulation, and an elevated firing rate of pCINs during quiet states (Group 3) leads to a reduced firing rate of pSPNs (Group 1). This is consistent with the fact that CINs are known to suppress SPN activity (English et al., 2011; Zucca et al., 2018).
To further investigate the activity profile of these four groups, we analyzed neuronal activity relative to the movement. As head-fixed awake mice voluntarily move (Ranjbar-Slamloo and Arabzadeh, 2019), striatal cell firing could be affected by movement as well as the sensory stimulus. We thus analyzed how the snout and whiskers behaved during the course of stimulation. We found that the overall motion index was different, revealing a higher motor activity of the Er81 cKO mice compared with control (n = 21 recordings from 3 control mice; 1.76 ± 5 × 10−4 a.u.; n = 23 recordings from 4 Er81 cKO mice; 2.16 ± 7 × 10−4 a.u., t(42) = 4.5, p = 0.003, two-sample t test; data not shown). We then performed a reverse correlation analysis to examine the coupling between spikes and movements. The STA motions showed a larger peak at spike time in the putative SPN groups of the Er81 cKO mice compared with control (Group 1, n = 267 control cells; n = 366 Er81 cKO cells, F(1,631, genotype) = 13.3, p = 2.86 × 10−4; Group 2, n = 260 control cells; n = 200 Er81 cKO cells; F(1,458, genotype) = 6.3, p = 0.012, two-way ANOVA; Fig. 6F). This indicates enhanced temporal precision of the putative output neuron activity encoding motion. Magnitude of the STA near spike time was also larger in the putative CINs, indicating a better temporal alignment of the spikes to the onset of the movement in the Er81 cKO mice (Group 3; n = 150 control cells; n = 111 Er81 cKO cells; F(1,259, genotype) = 4.0, p = 0.045, Fig. 6H, left; Group 4; n = 54 control cells; n = 55 Er81 cKO cells, F(1,107, genotype) = 10.0, p = 2.0 × 10−3, two-way ANOVA, Fig. 6H, right). Together with the enhanced suppression (i.e., more reliable pause) during stimulus, these results show that striatal neurons are more effectively recruited by the sensorimotor inputs in the absence of Er81.
To further examine the relative contributions of the stimulus and the motion in generating spikes in the absence of Er81, we performed cross-correlation analysis between the firing rate, motion, and puff stimulation. In putative SPNs (Group 1), positive spike-motion correlations were on average stronger in the absence of Er81, whereas the corresponding spike-puff correlations were not affected (control; n = 233 cells, Er81 cKO; n = 317 cells, spike-puff correlations; z = 0.37, p = 0.714, spike-motion correlations; z = 6.6, p = 4.9 × 10−11, Wilcoxon rank sum tests; Fig. 6G). On the other hand, negative correlations in the putative CINs (Group 3) were significantly larger in the Er81 cKO compared with the control mice (control; n = 74 cells, Er81 cKO; n = 69 cells, spike-puff correlations; z = 5.5, p = 3.0 × 10−8, Wilcoxon rank sum test, spike-motion correlations; t(141) = 8.0, p = 5.1 × 10−13, two-sample t test; Fig. 6I). Interestingly, we also found that spike-motion correlations were consistently higher than spike-puff correlations (Fig. 6G,I). Together, our results suggest that neurons were mostly tuned to the voluntary movements, rather than the stimulus. They indicate that physiological changes in the Er81 cKO mice lead to higher motor activity and a more effective modulation of cell activity in the striatum. These data imply that the putative CIN population producing phasic responses during movement are more responsive to sensorimotor inputs in the absence of Er81, and cause a higher excitation of the putative output neurons.
Er81 is a molecular regulator of habit formation
Striatal CINs are crucial for cognitive flexibility (Okada et al., 2014, 2018; Aoki et al., 2018). To assess the impact of Er81 on striatal-dependent behaviors, we designed a reversal learning task, allowing the concomitant assessment of cognitive flexibility and habit formation in the Er81 cKO mice. In the first part of the training, both control and Er81 cKO mice were able to discriminate the rewarded corridor of the maze (day 3: control; 66.4% of correct answers, n = 17 mice, Er81 cKO; 68.9% of correct answers, n = 17 mice; two-way ANOVA, F(2,64), days 1-3 × genotype = 0.567; p = 0.570; Fig. 7A), and switch to the opposite side after a first reversal on day 4 (day 4: control; 42.8%, Er81 cKO: 43.5%; n = 17; two-way ANOVA, F(1,32), days 3-4 × genotype = 0.762; p = 0.9509; Fig. 7A). Er81 cKO mice hence displayed normal learning capacities (days 1-3) as well as intact cognitive flexibility (days 4-13; two-way ANOVA, F(9,207), days 4-13 × genotype = 1.559; p = 0.1294). After they reached their performance plateau, mice were then overtrained for 5 d, to induce habit formation (Balleine et al., 2009; Lingawi and Balleine, 2012) (day 13: control; 85.6%, n = 12 mice, Er81 cKO; 83.6%, n = 13 mice; one-way ANOVA, F(1,23, genotype) = 0.19, p = 0.669; Fig. 7A). As the strategy used (goal-directed or habitual) cannot be revealed during training (Balleine and Ostlund, 2007), we assessed whether mice were engaged in goal-directed or habitual behavior by performing a second reversal (day 14). This step aims to change the action-outcome contingency to probe behavior: an animal persisting on choosing the nonrewarding side would reveal its previous engagement in an automatic, habitual behavior. During this session, control mice displayed performance significantly below chance level (controls: 25.4%; controls vs 50%: t(11) = 6.6, p = 2.1 × 10−5; one-sample analysis; n = 12 mice; Fig. 7A, right) and significantly lower compared with the first reversal (day 4; one-way ANOVA, F(1,11), day 4 vs day 14 in controls = 10.1, p = 0.009). Together, these results confirm that control mice were successfully engaged in habitual behavior, through their inability to flexibly shift to the new rewarded side (Dickinson et al., 1995; Yin et al., 2004). Interestingly, Er81 cKO mice performed significantly higher compared with controls during the second reversal (day 14; one-way ANOVA, F(1,23, genotype) = 4.3, p = 0.045, controls: 25.4%; n = 12 and Er81 cKO; 34.7%; n = 13; Fig. 7A, right). To further analyze this session, we compared the first five trials to the last five trials of this session. While both groups performed similarly at the beginning of the session (controls: 13.3% and Er81 cKO: 20.0%; one-way ANOVA, F(1,23, genotype) = 1.5, p = 0.234; Fig. 7B, Trials 1-5), only control mice remained significantly below chance level at the end of the session (controls: 31.7%; controls vs 50%, one-sample analysis: t(11) = −2.6, p = 0.027; Fig. 7B, Trials 16-20). In contrast, Er81 cKO mice displayed rapid improvement of their performance, reaching chance level by the end of the session (Er81 cKO: 49.2%; n = 13). This result was further confirmed by the following session (day 15) during which Er81 cKO mice performed significantly above chance level, unlike controls (Er81 cKO vs 50%; n = 9, one-sample analysis; t(8) = 3.6, p = 0.005; Fig. 7A, right). Moreover, analysis of the performance across the second reversal (days 13-16) demonstrates a significant difference between the control and Er81 cKO mice (two-way ANOVA, F(3,39), days 13-16 × genotype) = 3.0; p = 0.040), unlike the first reversal (two-way ANOVA, F(3,69), days 3-6 × genotype = 0.4; p = 0.727). Together, these results demonstrate that mice lacking Er81 display intact goal-directed actions and an impairment of habit formation. These mice also showed general hyperactivity as they move faster in the middle corridor (n = 13 control mice; n = 12 Er81 cKO mice; z = 4.8, p = 1.05 × 10−6, Wilcoxon rank sum test; Fig. 7C), which is consistent with the higher motion index of the snout and whiskers in the head-fixed experiments. Considering the differential role of the dorsomedial and dorsolateral striatum in cognitive flexibility and habit formation, respectively (Packard and Knowlton, 2002), we then analyzed the changes in striatal activity associated with the paradigm. Expression of the cellular activity marker cFos in the dorsal striatum revealed an alteration of the task-induced activity of the output neurons in the Er81 cKO mice (Fig. 7D). While training had no effect on cFos expression in controls, the Er81 deletion induced a drastic hypoactivation of the striatum in trained mice, compared with nontrained mice (two-way ANOVA, F(1,13, interaction) = 6.8; p = 0.022; day 14: F(1,7, genotype) = 7.5; p = 0.029). This result suggests that the striatum in Er81 cKO does not adapt properly to the behavioral demand, which could underlie the mice inability to form habits. This is further supported by the analysis of the cFos expression in the dorsomedial striatum (DMS) versus dorsolateral striatum (DLS). We found a significant decrease of cFos expression in the DLS of Er81 cKO mice compared with controls (day 14; one-way ANOVA, F(1,6,genotype) = 7.3; p = 0.036) and compared with day 4 (day 4 vs day 14 in Er81 cKO: one-way ANOVA, F(1,8) = 8.5; p = 0.019; data not shown). This specific DLS hypoactivation could therefore underpin the observed lack of habit formation; conversely, the intact DMS activity could explain the use of cognitive flexibility in Er81 cKO. Overall, our results indicate that the fine-tuned activity of CINs by Er81 is crucial for striatal function and habit formation.
Figure 7.

Er81 cKO mice exhibit disrupted habitual behavior. A, Reversal learning task in a 3 corridors arena (top). Dashed curve indicates one-way gate. Blue droplet represents sucrose reward. Performance of the mice in each session (bottom; control in black vs Er81 cKO in red). Arrows indicate the days of reversal. ##p < 0.01, statistical comparison with the chance level (performance at 50%, dashed line). Horizontal line indicates interaction between performance and genotype. B, Average performance across the first 5 trials and last 5 trials of day 14. #p < 0.05, statistical comparison with the chance level (performance at 50%, dashed line). C, Average locomotion speed of control and Er81 cKO mice in the middle corridor. D, Fraction of the cells expressing cFos in nontrained (NT) and day 14 of training (D14) in the control and the Er81 cKO. *p < 0.05. ***p < 0.001.
Discussion
Here, by using in vitro and in vivo assessments, we find that the Er81 transcription factor has an age-dependent effect on some CIN physiological properties, thus demonstrating that Er81 contributes to CIN microcircuit formation and fine-tuned activity. We also show that Er81 controls in vivo striatal neuron responses and their correlation to sensorimotor inputs. Finally, we reveal that proper CIN maturation through Er81 function is required for normal habit acquisition in mice.
Control of the cholinergic interneuron identity
The functional implications of molecular heterogeneity in striatal CINs are at present not well understood (Magno et al., 2017; Lozovaya et al., 2018; Munoz-Manchado et al., 2018). CINs originate from distinct areas of the subpallium which give rise to heterogeneous populations of cholinergic cells in the forebrain (Ahmed et al., 2019). These areas are further divided into subdomains, based on the combinatorial expression of multiple transcription factors, such as Lhx6, Lhx7, Isl1, and Er81 (Flames et al., 2007; He et al., 2016). The temporal order of expression of these transcription factors and their interactions is crucial for normal development of cholinergic neurons (Y. Zhao et al., 2003; Allaway and Machold, 2017; Ahmed et al., 2019). In this study, we identified Er81 as a molecular controller of key CIN properties and described how its expression affects other transcription factors. Future fate-mapping investigations will determine more specifically the proportion of Er81-expressing striatal CINs that may originate from different sources (i.e., MGE, POa, and septum).
In the mature cortex, Er81 directly regulates the functional diversity of cortical PV-expressing interneurons (Dehorter et al., 2015). While Er81 expression is strong and mostly nuclear in the cortical PV interneurons, it remains weak and cytoplasmic in the CINs of the striatum. This suggests that Er81 works through different mechanisms in the CINs than in the PV interneurons (Dehorter et al., 2015), likely through protein-protein interactions or regulation of translational processes. However, while the molecular targets may be different, Er81 seems to have a common role in neuronal function across different cell types, regulating excitability and spike timing (i.e., the phasic activity of the striatal CINs and the firing latency in the cortical [Dehorter et al., 2015] and striatal PV interneurons [unpublished data]). Moreover, the role of Er81 in the dopaminergic (Flames and Hobert, 2009; Cave et al., 2010) and serotoninergic cell fates (Lloret-Fernández et al., 2018) and here, in cholinergic cell identity, suggests a general regulatory mechanism of Er81 on the emergence of functional neuromodulatory systems in the developing brain. CINs are neurochemically complex as they corelease GABA (Saunders et al., 2015; Granger et al., 2016) and glutamate (Higley et al., 2011) alongside acetylcholine, to provide both potent inhibition (Zucca et al., 2018) or excitation (Koos and Tepper, 2002; Tepper et al., 2018) within the striatum. As we found changes in the molecular properties of the CINs (acetylcholine transporter, Isl1, and ChAT), corelease with other neurotransmitters might be also affected by Er81 deletion and ultimately alter the neurochemical function of the CINs. Our study therefore emphasizes the need to further investigate the molecular factors determining the identity and the functional diversity of striatal CINs.
Mechanisms regulating tonic and phasic activity of the striatal cholinergic interneurons
Intrinsic electrophysiological properties are responsible for maintaining tonic activity of striatal CINs (Bennett et al., 2000). Together with synaptic inputs (Franklin and Frank, 2015; Klug et al., 2018), they also contribute to the phasic response to stimuli (Zhang et al., 2018). The role of molecular factors in the maturation of these properties was unknown. For the first time, our findings link the Er81 transcription factor with intrinsic currents and plasticity-related receptors, such as mGluR5, and provide a basis to determine which molecular factors contribute to the emergence of the unique properties of striatal CINs. Elevated IsAHP and Ih currents, together with larger inhibitory inputs on CINs, explain the sharper firing dynamics and higher correlations of the putative CINs activity with sensorimotor inputs in vivo in the absence of Er81 (Bennett et al., 2000; Wilson and Goldberg, 2006; Zhang et al., 2018). KCNQ2/3 and HCN2 channels in CINs underlie the IsAHP and the Ih, respectively. While our results show no difference in their expression in the absence of Er81, these voltage-sensitive channels could be modulated by second messengers, such as phosphatidylinositol-4,5-bisphosphate and calcium for KCNQ2/3 (Wilson and Goldberg, 2006; Falkenburger et al., 2010; Kim et al., 2016), and cAMP for HCN2 (Z. Zhao et al., 2016; Alvarez-Baron et al., 2018). We propose that the enhanced IsAHP in the Er81 cKO cells is because of a higher intracellular calcium concentration mediated through mGluR5 (Niswender and Conn, 2010). It is also likely that different potassium channel subtypes underlie IsAHP as the current can be divided into two calcium-sensitive components (Wilson and Goldberg, 2006) and a calcium-insensitive component (Zhang et al., 2018). We also suggest that enhanced cAMP signaling leads to the increase in the Ih current. Lower CIN excitability in the absence of Er81 can indeed result in a decreased acetylcholine recruitment at the CIN membrane, reduced M2 muscarinic receptor activity and consequently, to higher levels of intracellular cAMP (Z. Zhao et al., 2016; Alvarez-Baron et al., 2018). We propose that the changes in CIN activity in the absence of Er81 modify their connectivity and morphology via homeostatic compensations during maturation.
Cholinergic interneuron activity modulates striatal sensorimotor plasticity and learning
To probe striatal deficits in Er81 cKO mice, we designed a mixed, striatum-dependent behavioral paradigm, consisting of a within-subject reversal learning task. We first assessed cognitive flexibility (day 4) and then overtrained mice in the second stage (day 9 to day 13) to force habit formation (Lingawi and Balleine, 2012). This was followed by a second reversal (day 14), allowing the assessment of the strategy used by the mice during the second stage. Overall, extensive training leads to automatic choice of the designated reward side, and the reversal of the rewarded side induces a change in action-outcome contingency. Insensitivity to such a change reveals habit behavior (Balleine et al., 2009; Bergstrom et al., 2018). Here, control mice displayed an inability to shift to the new rewarded side after the second reversal, demonstrating that they were engaged in habitual behavior. In contrast, Er81 cKO mice displayed rapid improvement of their performance after the second reversal, demonstrating that they displayed intact cognitive flexibility and were not engaged in habit formation during the second stage. While our results support that Er81 is required for habitual behavior, differences in reversal between control and Er81 cKO mice may not exclusively originate from an inability to form habits and may also be explained by differences in reward sensitivity, persistence, or general cognitive flexibility. Future studies should specifically target habit formation or cognitive flexibility to further understand the impact of Er81.
Our study also reveals that developmental alterations of striatal CINs strongly impact the process of integration of sensorimotor information, which is critical for the acquisition and the update of adaptive actions (Markowitz et al., 2018; Robbe, 2018). It also highlights the significance of cholinergic signaling for movement control and learning. Putative striatal CINs better encode motion and display enhanced phasic responses in the Er81 cKO mice. In particular, at the onset of movement, spikes are more time-locked to the events and less jittered. This also enhanced the timing of putative output neurons which directly encode CIN firing (Mamaligas and Ford, 2016). Enhanced timing of the striatal cells indicates that spike timing-dependent plasticity could be affected by the Er81 deletion (Cui et al., 2018), via a metabotropic glutamate receptor-induced LTD or a NMDA receptor-induced LTP (Fino et al., 2008). However, the mechanisms of spike timing-dependent plasticity have been poorly investigated in the CINs as of yet (Perrin and Venance, 2019). More investigation is therefore required to better understand how CIN alterations impact intrinsic, synaptic, and structural plasticity and overall, the striatal function. CINs control habit formation via the synchronization and strengthening of the activity of the striatal output neurons (O'Hare et al., 2016; Gritton et al., 2019). Strong pauses have been shown to emerge during learning in response to reward-associated stimuli but not to neutral stimuli (Morris et al., 2004). Adaptable CIN responses to sensorimotor inputs (Aosaki et al., 1994) are required for suppression of competing actions and the expression of habitual behavior. We reveal a notable increase in the recruitment of CINs following the ablation of the Er81 transcription factor. Previous studies showed that manipulation of CINs in both the DMS and DLS modulates cognitive flexibility after several days of training (Bradfield et al., 2013; Okada et al., 2014; Aoki et al., 2015). In particular, the activity of the CINs in the DLS is necessary for habit substitution (Aoki et al., 2018). Consistently, we have found that elevated CIN function in the DLS (increased quiet state firing and increased inhibition in response to sensorimotor inputs in the cKO) is correlated with decreased habit formation. We uncover a crucial molecular mechanism underlying this relationship and demonstrate, for the first time, that the Er81 transcription factor underlies the functional tuning of CIN activity and habitual behavior. However, the involvement of the DMS in these conditions is yet to be elucidated. Moreover, it would be of major interest to study the Er81 transcription factor as a potential target to manipulate set-shifting capacities, known to contribute to the pathophysiology and cognitive defects commonly observed in disorders where repetitive behaviors are debilitating (Lewis and Kim, 2009), such as autism (Karvat and Kimchi, 2014), obsessive-compulsive disorder (Xu et al., 2015; Martos et al., 2017), and addiction (Graybiel and Grafton, 2015).
Footnotes
The authors declare no competing financial interests.
This work was supported by National Health and Medical Research Council Grant APP1144145 and the Australian National University. We thank Prof. Silvia Arber and Prof. Oscar Marin for the Er81 antibody and Er81 conditional mutant mice; and Prof. John Bekkers and Prof. Bernard Balleine for critical comments on the manuscript.
References
- Abe H, Okazawa M, Nakanishi S (2011) The Etv1/Er81 transcription factor orchestrates activity-dependent gene regulation in the terminal maturation program of cerebellar granule cells. Proc Natl Acad Sci USA 108:12497–12502. 10.1073/pnas.1109940108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed NY, Knowles R, Dehorter N (2019) New insights into cholinergic neuron diversity. Front Mol Neurosci 12:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allaway KC, Machold R (2017) Developmental specification of forebrain cholinergic neurons. Dev Biol 421:1–7. 10.1016/j.ydbio.2016.11.007 [DOI] [PubMed] [Google Scholar]
- Alvarez-Baron CP, Klenchin VA, Chanda B (2018) Minimal molecular determinants of isoform-specific differences in efficacy in the HCN channel family. J Gen Physiol 150:1203–1213. 10.1085/jgp.201812031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki S, Liu AW, Zucca A, Zucca S, Wickens JR (2015) Role of striatal cholinergic interneurons in set-shifting in the rat. J Neurosci 35:9424–9431. 10.1523/JNEUROSCI.0490-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki S, Liu AW, Akamine Y, Zucca A, Zucca S, Wickens JR (2018) Cholinergic interneurons in the rat striatum modulate substitution of habits. Eur J Neurosci 47:1194–1205. 10.1111/ejn.13820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aosaki T, Tsubokawa H, Ishida A, Watanabe K, Graybiel AM, Kimura M (1994) Responses of tonically active neurons in the primate's striatum undergo systematic changes during behavioral sensorimotor conditioning. J Neurosci 14:3969–3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apicella P (2017) The role of the intrinsic cholinergic system of the striatum: what have we learned from TAN recordings in behaving animals? Neuroscience 360:81–94. 10.1016/j.neuroscience.2017.07.060 [DOI] [PubMed] [Google Scholar]
- Apicella P, Legallet E, Trouche E (1997) Responses of tonically discharging neurons in the monkey striatum to primary rewards delivered during different behavioral states. Exp Brain Res 116:456–466. 10.1007/pl00005773 [DOI] [PubMed] [Google Scholar]
- Apicella P, Deffains M, Ravel S, Legallet E (2009) Tonically active neurons in the striatum differentiate between delivery and omission of expected reward in a probabilistic task context. Eur J Neurosci 30:515–526. 10.1111/j.1460-9568.2009.06872.x [DOI] [PubMed] [Google Scholar]
- Arber S, Ladle DR, Lin JH, Frank E, Jessell TM (2000) ETS gene Er81 controls the formation of functional connections between group Ia sensory afferents and motor neurons. Cell 101:485–498. 10.1016/s0092-8674(00)80859-4 [DOI] [PubMed] [Google Scholar]
- Balleine BW, Ostlund SB (2007) Still at the choice-point: action selection and initiation in instrumental conditioning. Ann NY Acad Sci 1104:147–171. 10.1196/annals.1390.006 [DOI] [PubMed] [Google Scholar]
- Balleine BW, Liljeholm M, Ostlund SB (2009) The integrative function of the basal ganglia in instrumental conditioning. Behav Brain Res 199:43–52. [DOI] [PubMed] [Google Scholar]
- Beatty JA, Sullivan MA, Morikawa H, Wilson CJ (2012) Complex autonomous firing patterns of striatal low-threshold spike interneurons. J Neurophysiol 108:771–781. 10.1152/jn.00283.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benhamou L, Kehat O, Cohen D (2014) Firing pattern characteristics of tonically active neurons in rat striatum: context dependent or species divergent? J Neurosci 34:2299–2304. 10.1523/JNEUROSCI.1798-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BD, Callaway JC, Wilson CJ (2000) Intrinsic membrane properties underlying spontaneous tonic firing in neostriatal cholinergic interneurons. J Neurosci 20:8493–8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergstrom HC, Lipkin AM, Lieberman AG, Pinard CR, Gunduz-Cinar O, Brockway ET, Taylor WW, Nonaka M, Bukalo O, Wills TA, Rubio FJ, Li X, Pickens CL, Winder DG, Holmes A (2018) Dorsolateral striatum engagement interferes with early discrimination learning. Cell Rep 23:2264–2272. 10.1016/j.celrep.2018.04.081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradfield LA, Bertran-Gonzalez J, Chieng B, Balleine BW (2013) The thalamostriatal pathway and cholinergic control of goal-directed action: interlacing new with existing learning in the striatum. Neuron 79:153–166. 10.1016/j.neuron.2013.04.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brimblecombe KR, Threlfell S, Dautan D, Kosillo P, Mena-Segovia J, Cragg SJ (2018) Targeted activation of cholinergic interneurons accounts for the modulation of dopamine by striatal nicotinic receptors. eNeuro 5:ENEURO.0397-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cave JW, Akiba Y, Banerjee K, Bhosle S, Berlin R, Baker H (2010) Differential regulation of dopaminergic gene expression by Er81. J Neurosci 30:4717–4724. 10.1523/JNEUROSCI.0419-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Prokin I, Mendes A, Berry H, Venance L (2018) Robustness of STDP to spike timing jitter. Sci Rep 8:8139. 10.1038/s41598-018-26436-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehorter N, Ciceri G, Bartolini G, Lim L, del Pino I, Marín O (2015) Tuning of fast-spiking interneuron properties by an activity-dependent transcriptional switch. Science 349:1216–1220. 10.1126/science.aab3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson A, Balleine B, Watt A, Gonzalez F, Boakes RA (1995) Motivational control after extended instrumental training. Anim Learn Behav 23:197–206. 10.3758/BF03199935 [DOI] [Google Scholar]
- Ding B, Cave JW, Dobner PR, Mullikin-Kilpatrick D, Bartzokis M, Zhu H, Chow CW, Gronostajski RM, Kilpatrick DL (2016) Reciprocal autoregulation by NFI occupancy and ETV1 promotes the developmental expression of dendrite-synapse genes in cerebellar granule neurons. Mol Biol Cell 27:1488–1499. 10.1091/mbc.E15-07-0476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding JB, Guzman JN, Peterson JD, Goldberg JA, Surmeier DJ (2010) Thalamic gating of corticostriatal signaling by cholinergic interneurons. Neuron 67:294–307. 10.1016/j.neuron.2010.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doig NM, Magill PJ, Apicella P, Bolam JP, Sharott A (2014) Cortical and thalamic excitation mediate the multiphasic responses of striatal cholinergic interneurons to motivationally salient stimuli. J Neurosci 34:3101–3117. 10.1523/JNEUROSCI.4627-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorst MC, Tokarska A, Zhou M, Lee K, Stagkourakis S, Broberger C, Masmanidis S, Silberberg G (2020) Polysynaptic inhibition between striatal cholinergic interneurons shapes their network activity patterns in a dopamine-dependent manner. Nat Commun 11:5113. 10.1038/s41467-020-18882-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- English DF, Ibanez-Sandoval O, Stark E, Tecuapetla F, Buzsaki G, Deisseroth K, Tepper JM, Koos T (2011) GABAergic circuits mediate the reinforcement-related signals of striatal cholinergic interneurons. Nat Neurosci 15:123–130. 10.1038/nn.2984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkenburger BH, Jensen JB, Hille B (2010) Kinetics of PIP2 metabolism and KCNQ2/3 channel regulation studied with a voltage-sensitive phosphatase in living cells. J Gen Physiol 135:99–114. 10.1085/jgp.200910345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fino E, Deniau JM, Venance L (2008) Cell-specific spike-timing-dependent plasticity in GABAergic and cholinergic interneurons in corticostriatal rat brain slices. J Physiol 586:265–282. 10.1113/jphysiol.2007.144501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flames N, Hobert O (2009) Gene regulatory logic of dopamine neuron differentiation. Nature 458:885–889. 10.1038/nature07929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flames N, Pla R, Gelman DM, Rubenstein JL, Puelles L, Marin O (2007) Delineation of multiple subpallial progenitor domains by the combinatorial expression of transcriptional codes. J Neurosci 27:9682–9695. 10.1523/JNEUROSCI.2750-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin NT, Frank MJ (2015) A cholinergic feedback circuit to regulate striatal population uncertainty and optimize reinforcement learning. Elife 4:e12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granger AJ, Mulder N, Saunders A, Sabatini BL (2016) Cotransmission of acetylcholine and GABA. Neuropharmacology 100:40–46. 10.1016/j.neuropharm.2015.07.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gras C, Amilhon B, Lepicard EM, Poirel O, Vinatier J, Herbin M, Dumas S, Tzavara ET, Wade MR, Nomikos GG, Hanoun N, Saurini F, Kemel ML, Gasnier B, Giros B, El Mestikawy S (2008) The vesicular glutamate transporter VGLUT3 synergizes striatal acetylcholine tone. Nat Neurosci 11:292–300. 10.1038/nn2052 [DOI] [PubMed] [Google Scholar]
- Graybiel AM, Grafton ST (2015) The striatum: where skills and habits meet. Cold Spring Harb Perspect Biol 7:a021691. 10.1101/cshperspect.a021691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritton HJ, Howe WM, Romano MF, DiFeliceantonio AG, Kramer MA, Saligrama V, Bucklin ME, Zemel D, Han X (2019) Unique contributions of parvalbumin and cholinergic interneurons in organizing striatal networks during movement. Nat Neurosci 22:586–597. 10.1038/s41593-019-0341-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M, Tucciarone J, Lee S, Nigro MJ, Kim Y, Levine JM, Kelly SM, Krugikov I, Wu P, Chen Y, Gong L, Hou Y, Osten P, Rudy B, Huang ZJ (2016) Strategies and tools for combinatorial targeting of GABAergic neurons in mouse cerebral cortex. Neuron 92:555. 10.1016/j.neuron.2016.10.009 [DOI] [PubMed] [Google Scholar]
- Higley MJ, Gittis AH, Oldenburg IA, Balthasar N, Seal RP, Edwards RH, Lowell BB, Kreitzer AC, Sabatini BL (2011) Cholinergic interneurons mediate fast VGluT3-dependent glutamatergic transmission in the striatum. PLoS One 6:e19155. 10.1371/journal.pone.0019155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hippenmeyer S, Vrieseling E, Sigrist M, Portmann T, Laengle C, Ladle DR, Arber S (2005) A developmental switch in the response of DRG neurons to ETS transcription factor signaling. PLoS Biol 3:e159. 10.1371/journal.pbio.0030159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjorth JJJ, Kozlov A, Carannante I, Frost Nylen J, Lindroos R, Johansson Y, Tokarska A, Dorst MC, Suryanarayana SM, Silberberg G, Hellgren Kotaleski J, Grillner S (2020) The microcircuits of striatum in silico. Proc Natl Acad Sci USA 117:9554–9565. 10.1073/pnas.2000671117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inokawa H, Yamada H, Matsumoto N, Muranishi M, Kimura M (2010) Juxtacellular labeling of tonically active neurons and phasically active neurons in the rat striatum. Neuroscience 168:395–404. 10.1016/j.neuroscience.2010.03.062 [DOI] [PubMed] [Google Scholar]
- Karvat G, Kimchi T (2014) Acetylcholine elevation relieves cognitive rigidity and social deficiency in a mouse model of autism. Neuropsychopharmacology 39:831–840. 10.1038/npp.2013.274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KS, Duignan KM, Hawryluk JM, Soh H, Tzingounis AV (2016) The voltage activation of cortical KCNQ channels depends on global PIP2 levels. Biophys J 110:1089–1098. 10.1016/j.bpj.2016.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klug JR, Engelhardt MD, Cadman CN, Li H, Smith JB, Ayala S, Williams EW, Hoffman H, Jin X (2018) Differential inputs to striatal cholinergic and parvalbumin interneurons imply functional distinctions. Elife 7:e35657. 10.7554/eLife.35657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koos T, Tepper JM (2002) Dual cholinergic control of fast-spiking interneurons in the neostriatum. J Neurosci 22:529–535. 10.1523/JNEUROSCI.22-02-00529.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosillo P, Zhang YF, Threlfell S, Cragg SJ (2016) Cortical control of striatal dopamine transmission via striatal cholinergic interneurons. Cereb Cortex 26:4160–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Holley SM, Shobe JL, Chong NC, Cepeda C, Levine MS, Masmanidis SC (2017) Parvalbumin interneurons modulate striatal output and enhance performance during associative learning. Neuron 93:1451–1463. 10.1016/j.neuron.2017.02.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis M, Kim SJ (2009) The pathophysiology of restricted repetitive behavior. J Neurodev Disord 1:114–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SA, Kang UJ, McGehee DS (2014) Striatal cholinergic interneuron regulation and circuit effects. Front Synaptic Neurosci 6:22. 10.3389/fnsyn.2014.00022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingawi NW, Balleine BW (2012) Amygdala central nucleus interacts with dorsolateral striatum to regulate the acquisition of habits. J Neurosci 32:1073–1081. 10.1523/JNEUROSCI.4806-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloret-Fernández C, Maicas M, Mora-Martínez C, Artacho A, Jimeno-Martín Á, Chirivella L, Weinberg P, Flames N (2018) A transcription factor collective defines the HSN serotonergic neuron regulatory landscape. Elife 7:e32785. 10.7554/eLife.32785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes R, Verhey van Wijk N, Neves G, Pachnis V (2012) Transcription factor LIM homeobox 7 (Lhx7) maintains subtype identity of cholinergic interneurons in the mammalian striatum. Proc Natl Acad Sci USA 109:3119–3124. 10.1073/pnas.1109251109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozovaya N, Eftekhari S, Cloarec R, Gouty-Colomer LA, Dufour A, Riffault B, Billon-Grand M, Pons-Bennaceur A, Oumar N, Burnashev N, Ben-Ari Y, Hammond C (2018) GABAergic inhibition in dual-transmission cholinergic and GABAergic striatal interneurons is abolished in Parkinson disease. Nat Commun 9:1422. 10.1038/s41467-018-03802-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magno L, Barry C, Schmidt-Hieber C, Theodotou P, Häusser M, Kessaris N (2017) NKX2-1 is required in the embryonic septum for cholinergic system development, learning, and memory. Cell Rep 20:1572–1584. 10.1016/j.celrep.2017.07.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet N, Le Moine C, Charpier S, Gonon F (2005) Feedforward inhibition of projection neurons by fast-spiking GABA interneurons in the rat striatum in vivo. J Neurosci 25:3857–3869. 10.1523/JNEUROSCI.5027-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamaligas AA, Ford CP (2016) Spontaneous synaptic activation of muscarinic receptors by striatal cholinergic neuron firing. Neuron 91:574–586. 10.1016/j.neuron.2016.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamaligas AA, Barcomb K, Ford CP (2019) Cholinergic transmission at muscarinic synapses in the striatum is driven equally by cortical and thalamic inputs. Cell Rep 28:1003–1014. 10.1016/j.celrep.2019.06.077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz JE, Gillis WF, Beron CC, Neufeld SQ, Robertson K, Bhagat ND, Peterson RE, Peterson E, Hyun M, Linderman SW, Sabatini BL, Datta SR (2018) The striatum organizes 3D behavior via moment-to-moment action selection. Cell 174:44–58.e17. 10.1016/j.cell.2018.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martos YV, Braz BY, Beccaria JP, Murer MG, Belforte JE (2017) Compulsive social behavior emerges after selective ablation of striatal cholinergic interneurons. J Neurosci 37:2849–2858. 10.1523/JNEUROSCI.3460-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi D, Li Z, Lim L, Li M, Moissidis M, Yang Y, Gao T, Hu TX, Pratt T, Price DJ, Sestan N, Marin O (2018) Early emergence of cortical interneuron diversity in the mouse embryo. Science 360:81–85. 10.1126/science.aar6821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G, Arkadir D, Nevet A, Vaadia E, Bergman H (2004) Coincident but distinct messages of midbrain dopamine and striatal tonically active neurons. Neuron 43:133–143. 10.1016/j.neuron.2004.06.012 [DOI] [PubMed] [Google Scholar]
- Munoz-Manchado AB, Bengtsson Gonzales C, Zeisel A, Munguba H, Bekkouche B, Skene NG, Lonnerberg P, Ryge J, Harris KD, Linnarsson S, Hjerling-Leffler J (2018) Diversity of interneurons in the dorsal striatum revealed by single-cell RNA sequencing and PatchSeq. Cell Rep 24:2179–2190.e2177. 10.1016/j.celrep.2018.07.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson AB, Bussert TG, Kreitzer AC, Seal RP (2014) Striatal cholinergic neurotransmission requires VGLUT3. J Neurosci 34:8772–8777. 10.1523/JNEUROSCI.0901-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender CM, Conn PJ (2010) Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol 50:295–322. 10.1146/annurev.pharmtox.011008.145533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nóbrega-Pereira S, Kessaris N, Du T, Kimura S, Anderson SA, Marín O (2008) Postmitotic Nkx2-1 controls the migration of telencephalic interneurons by direct repression of guidance receptors. Neuron 59:733–745. 10.1016/j.neuron.2008.07.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hare JK, Ade KK, Sukharnikova T, Van Hooser SD, Palmeri ML, Yin HH, Calakos N (2016) Pathway-specific striatal substrates for habitual behavior. Neuron 89:472–479. 10.1016/j.neuron.2015.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada K, Nishizawa K, Fukabori R, Kai N, Shiota A, Ueda M, Tsutsui Y, Sakata S, Matsushita N, Kobayashi K (2014) Enhanced flexibility of place discrimination learning by targeting striatal cholinergic interneurons. Nat Commun 5:3778. 10.1038/ncomms4778 [DOI] [PubMed] [Google Scholar]
- Okada K, Nishizawa K, Setogawa S, Hashimoto K, Kobayashi K (2018) Task-dependent function of striatal cholinergic interneurons in behavioural flexibility. Eur J Neurosci 47:1174–1183. 10.1111/ejn.13768 [DOI] [PubMed] [Google Scholar]
- Packard MG, Knowlton BJ (2002) Learning and memory functions of the basal ganglia. Annu Rev Neurosci 25:563–593. 10.1146/annurev.neuro.25.112701.142937 [DOI] [PubMed] [Google Scholar]
- Perrin E, Venance L (2019) Bridging the gap between striatal plasticity and learning. Curr Opin Neurobiol 54:104–112. 10.1016/j.conb.2018.09.007 [DOI] [PubMed] [Google Scholar]
- Ranjbar-Slamloo Y, Arabzadeh E (2019) Diverse tuning underlies sparse activity in layer 2/3 vibrissal cortex of awake mice. J Physiol 597:2803–2817. 10.1113/JP277506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravel S, Legallet E, Apicella P (2003) Responses of tonically active neurons in the monkey striatum discriminate between motivationally opposing stimuli. J Neurosci 23:8489–8497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A, Levitz J (2018) Glutamatergic signaling in the central nervous system: ionotropic and metabotropic receptors in concert. Neuron 98:1080–1098. 10.1016/j.neuron.2018.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbe D (2018) To move or to sense? Incorporating somatosensory representation into striatal functions. Curr Opin Neurobiol 52:123–130. 10.1016/j.conb.2018.04.009 [DOI] [PubMed] [Google Scholar]
- Saunders A, Granger AJ, Sabatini BL (2015) Corelease of acetylcholine and GABA from cholinergic forebrain neurons. Elife 52:e06412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharott A, Moll CK, Engler G, Denker M, Grun S, Engel AK (2009) Different subtypes of striatal neurons are selectively modulated by cortical oscillations. J Neurosci 29:4571–4585. 10.1523/JNEUROSCI.5097-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharott A, Doig NM, Mallet N, Magill PJ (2012) Relationships between the firing of identified striatal interneurons and spontaneous and driven cortical activities in vivo. J Neurosci 32:13221–13236. 10.1523/JNEUROSCI.2440-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub C, Tritsch NX, Hagan NA, Gu C, Sabatini BL (2014) Multiphasic modulation of cholinergic interneurons by nigrostriatal afferents. J Neurosci 34:8557–8569. 10.1523/JNEUROSCI.0589-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub C, Saulnier JL, Begue A, Feng DD, Huang KW, Sabatini BL (2016) Principles of synaptic organization of GABAergic interneurons in the striatum. Neuron 92:84–92. 10.1016/j.neuron.2016.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szydlowski SN, Pollak Dorocic I, Planert H, Carlen M, Meletis K, Silberberg G (2013) Target selectivity of feedforward inhibition by striatal fast-spiking interneurons. J Neurosci 33:1678–1683. 10.1523/JNEUROSCI.3572-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepper JM, Koos T, Ibanez-Sandoval O, Tecuapetla F, Faust TW, Assous M (2018) Heterogeneity and diversity of striatal GABAergic interneurons: update 2018. Front Neuroanat 12:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorn CA, Graybiel AM (2014) Differential entrainment and learning-related dynamics of spike and local field potential activity in the sensorimotor and associative striatum. J Neurosci 34:2845–2859. 10.1523/JNEUROSCI.1782-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threlfell S, Lalic T, Platt NJ, Jennings KA, Deisseroth K, Cragg SJ (2012) Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron 75:58–64. 10.1016/j.neuron.2012.04.038 [DOI] [PubMed] [Google Scholar]
- Willardsen M, Hutcheson DA, Moore KB, Vetter ML (2014) The ETS transcription factor Etv1 mediates FGF signaling to initiate proneural gene expression during Xenopus laevis retinal development. Mech Dev 131:57–67. 10.1016/j.mod.2013.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CJ, Goldberg JA (2006) Origin of the slow afterhyperpolarization and slow rhythmic bursting in striatal cholinergic interneurons. J Neurophysiol 95:196–204. 10.1152/jn.00630.2005 [DOI] [PubMed] [Google Scholar]
- Xu M, Kobets A, Du JC, Lennington J, Li L, Banasr M, Duman RS, Vaccarino FM, DiLeone RJ, Pittenger C (2015) Targeted ablation of cholinergic interneurons in the dorsolateral striatum produces behavioral manifestations of Tourette syndrome. Proc Natl Acad Sci USA 112:893–898. 10.1073/pnas.1419533112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HH, Knowlton BJ, Balleine BW (2004) Lesions of dorsolateral striatum preserve outcome expectancy but disrupt habit formation in instrumental learning. Eur J Neurosci 19:181–189. 10.1111/j.1460-9568.2004.03095.x [DOI] [PubMed] [Google Scholar]
- Zhang YF, Reynolds JN, Cragg SJ (2018) Pauses in cholinergic interneuron activity are driven by excitatory input and delayed rectification, with dopamine modulation. Neuron 98:918–925.e913. 10.1016/j.neuron.2018.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Marin O, Hermesz E, Powell A, Flames N, Palkovits M, Rubenstein JL, Westphal H (2003) The LIM-homeobox gene Lhx8 is required for the development of many cholinergic neurons in the mouse forebrain. Proc Natl Acad Sci USA 100:9005–9010. 10.1073/pnas.1537759100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Zhang K, Liu X, Yan H, Ma X, Zhang S, Zheng J, Wang L, Wei X (2016) Involvement of HCN channel in muscarinic inhibitory action on tonic firing of dorsolateral striatal cholinergic interneurons. Front Cell Neurosci 10:71. 10.3389/fncel.2016.00071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucca S, Zucca A, Nakano T, Aoki S, Wickens J (2018) Pauses in cholinergic interneuron firing exert an inhibitory control on striatal output in vivo. Elife 7:e32510. 10.7554/eLife.32510 [DOI] [PMC free article] [PubMed] [Google Scholar]