

Summary
Background
Old age and FMS-like tyrosine kinase 3 internal tandem duplication (FLIT3-ITD) mutations in patients with acute myeloid leukaemia are associated with early relapse and poor survival. Quizartinib is an oral, highly potent, and selective next-generation FLT3 inhibitor with clinical antileukaemic activity in relapsed or refractory acute myeloid leukaemia. We aimed to assess the efficacy and safety of single-agent quizartinib in patients with relapsed or refractory acute myeloid leukaemia.
Methods
We did an open-label, multicentre, single-arm, phase 2 trial at 76 hospitals and cancer centres in the USA, Europe, and Canada. We enrolled patients with morphologically documented primary acute myeloid leukaemia or acute myeloid leukaemia secondary to myelodysplastic syndromes and an Eastern Cooperative Oncology Group (ECOG) performance status of 0–2 into two predefined, independent cohorts: patients who were aged 60 years or older with relapsed or refractory acute myeloid leukaemia within 1 year after first-line therapy (cohort 1), and those who were 18 years or older with relapsed or refractory disease following salvage chemotherapy or haemopoietic stem cell transplantation (cohort 2). Patients with an FLT3-ITD allelic frequency of more than 10% were considered as FLT3-ITD positive, whereas all other patients were considered as FLT3-ITD negative. Patients received quizartinib once daily as an oral solution; the initial 17 patients received 200 mg per day but the QTcF interval was prolonged for more than 60 ms above baseline in some of these patients. Subsequently, doses were amended for all patients to 135 mg per day for men and 90 mg per day for women. The co-primary endpoints were the proportion of patients who achieved a composite complete remission (defined as complete remission + complete remission with incomplete platelet recovery + complete remission with incomplete haematological recovery) and the proportion of patients who achieved a complete remission. Efficacy and safety analyses included all patients who received at least one dose of quizartinib (ie, the intention-to-treat population). Patients with a locally assessed post-treatment bone marrow aspirate or biopsy were included in efficacy analyses by response; all other patients were considered to have an unknown response. This study is registered with ClinicalTrials.gov, number NCT00989261, and with the European Clinical Trials Database, EudraCT 2009-013093-41, and is completed.
Findings
Between Nov 19, 2009, and Oct 31, 2011, a total of 333 patients were enrolled (157 in cohort 1 and 176 in cohort 2). In cohort 1, 63 (56%) of 112 FLT3-ITD-positive patients and 16 (36%) of 44 FLT3-ITD-negative patients achieved composite complete remission, with three (3%) FLT3-ITD-positive patients and two (5%) FLT3-ITD-negative patients achieving complete remission. In cohort 2, 62 (46%) of 136 FLT3-ITD-positive patients achieved composite complete remission with five (4%) achieving complete remission, whereas 12 (30%) of 40 FLT3-ITD-negative patients achieved composite complete remission with one (3%) achieving complete remission. Across both cohorts (ie, the intention-to-treat population of 333 patients), grade 3 or worse treatment-related treatment-emergent adverse events in 5% or more of patients were febrile neutropenia (76 [23%] of 333), anaemia (75 [23%]), thrombocytopenia (39 [12%]), QT interval corrected using Fridericia’s formula (QTcF) prolongation (33 [10%]), neutropenia (31 [9%]), leucopenia (22 [7%]), decreased platelet count (20 [6%]), and pneumonia (17 [5%]). Serious adverse events occurring in 5% or more of patients were febrile neutropenia (126 [38%] of 333; 76 treatment related), acute myeloid leukaemia progression (73 [22%]), pneumonia (40 [12%]; 14 treatment related), QTcF prolongation (33 [10%]; 32 treatment related), sepsis (25 [8%]; eight treatment related), and pyrexia (18 [5%]; nine treatment related). Notable serious adverse events occurring in less than 5% of patients were torsades de pointes (one [<1%]) and hepatic failure (two [1%]). In total, 125 (38%) of 333 patients died within the study treatment period, including the 30-day follow-up. 18 (5%) patients died because of an adverse event considered by the investigator to be treatment related (ten [6%] of 157 patients in cohort 1 and eight [5%] of 176 in cohort 2.
Interpretation
Single-agent quizartinib was shown to be highly active and generally well tolerated in patients with relapsed or refractory acute myeloid leukaemia, particularly those with FLT3-ITD mutations. These findings confirm that targeting the FLT3-ITD driver mutation with a highly potent and selective FLT3 inhibitor is a promising clinical strategy to help improve clinical outcomes in patients with very few options. Phase 3 studies (NCT02039726; NCT02668653) will examine quizartinib at lower starting doses.
Funding
Ambit Biosciences/Daiichi Sankyo.
Introduction
Activating mutations of the FMS-like tyrosine kinase 3 (FLT3) gene are common in acute myeloid leukaemia.1 The most frequent mutation is an internal tandem duplication (ITD), occurring in approximately 25% of patients with acute myeloid leukaemia2 and characterised by high white blood cell and bone marrow blast counts as well as a high risk of relapse after standard chemotherapy.3-6 Patients with FLT3-ITD mutations (ie, FLT3-ITD-positive patients) are also less likely to respond to salvage chemotherapy and have shorter survival following relapse than FLT3-ITD-negative patients.3,7 Additionally, older patients similarly have poor responses, high risk of relapse, and shorter overall survival than younger patients, warranting the need for improved treatment options, especially for FLT3-ITD-positive patients or those unable to tolerate standard chemotherapy (eg, the elderly).8-10
Quizartinib is an oral, highly potent, and selective next-generation FLT3 inhibitor being investigated for the treatment of FLT3-ITD-positive acute myeloid leukaemia.11 In a phase 1 trial,12 quizartinib showed complete target inhibition in biomarker assays and clinical activity in FLT3-ITD-positive and FLT3-ITD-negative patients with relapsed or refractory acute myeloid leukaemia. Quizartinib was generally well tolerated with a manageable safety profile. The maximum tolerated dose was 200 mg per day, and the dose-limiting toxicity was grade 3 QT interval corrected using Fridericia’s formula (QTcF) prolongation.12 Therefore in this phase 2 study, we aimed to assess the efficacy and safety of quizartinib monotherapy in FLT3-ITD-positive and FLT3-ITD-negative patients with refractory or relapsed acute myeloid leukaemia.
Methods
Study design and participants
We did this open-label, multicentre, single-arm, phase 2 trial at 76 hospitals and cancer centres in the USA, Europe, and Canada (appendix pp 3, 4). We enrolled patients with morphologically documented primary acute myeloid leukaemia or acute myeloid leukaemia secondary to myelodysplastic syndromes, as defined by WHO classifications, and an Eastern Cooperative Oncology Group (ECOG) performance status of 0–2 into two predefined, independent cohorts. In cohort 1, we included patients 60 years or older who had refractory or relapsed acute myeloid leukaemia less than 1 year after any first-line therapy (previous transplant excluded). In cohort 2, we included patients 18 years or older (but excluded those aged >85 years) who relapsed or had refractory acute myeloid leukaemia after one salvage chemotherapy or after undergoing haemopoietic stem cell transplantation (although patients in cohort 2 were not allowed to have undergone haemopoietic stem cell transplantation within the previous 100 days). Patients with or without FLT3-ITD mutations were permitted in this study, and patients were allowed to have received previous treatment with other FLT3 inhibitors, excluding quizartinib. The use of chemotherapy or antileukaemic agents other than hydroxyurea was not permitted during this study; there was a washout period of 2 weeks or more for cytotoxic agents or five half-lives or more for non-cytotoxic agents including immunosuppressive therapy after haemopoietic stem cell transplantation, except for those with rapidly progressing disease. All eligible patients had to have adequate organ function (serum creatinine of ≤1·5 × the upper limit of normal [ULN]; glomerular filtration rate of >30 mL/min [calculated by the Cockcroft-Gault formula]; serum potassium, magnesium, and calcium concentrations at least within institutional normal limits; total serum bilirubin of ≤1·5 × ULN; and serum aspartate aminotransferase [AST] or alanine aminotransferase [ALT] ≤2·5 × ULN).
Exclusion criteria included age older than 85 years (except at the discretion of the investigator and with agreement of the sponsor), acute promyelocytic leukaemia, chronic myeloid leukaemia in blast phase, acute myeloid leukaemia that relapsed or was refractory after at least two (cohort 1) or three (cohort 2) previous lines of therapy, treatment-related myeloid neoplasms, and clinically active central nervous system (CNS) leukaemia (patients with controlled CNS leukaemia receiving intrathecal therapy could be enrolled at the investigator’s discretion). Radiotherapy or major surgery within 4 weeks of study entry and radiotherapy during the study period were also exclusion criteria. Patients with clinically significant cardiovascular disease were also excluded, defined as myocardial infarction within 12 months; uncontrolled angina within 6 months; current or past New York Heart Association class 3 or 4 congestive heart failure (unless a screening echocardiogram or multigated acquisition scan revealed a left ventricular ejection fraction of ≥45%); history of clinically significant ventricular arrhythmias; QTcF interval of 450 ms or more at screening; history of second-degree or third-degree heart block; uncontrolled hypertension; complete left bundle branch block; or atrial fibrillation. Furthermore, patients with uncontrolled infections (eg, HIV infection, or hepatitis B or C infections) were excluded, as were those with medical conditions, serious intercurrent illnesses, or other extenuating circumstances that could jeopardise patient safety or interfere with the study’s objectives.
The study protocol was approved by the respective institutional review boards or ethics committees at all participating centres, and all patients provided written informed consent in accordance with the Declaration of Helsinki and the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use of Good Clinical Practice guidelines.
Procedures
Patients received quizartinib once daily as an oral solution. Treatment was given in 28-day treatment cycles without any rest periods and continued until relapse, intolerance, or haemopoietic stem cell transplantation. 17 patients initially received 200 mg per day of quizartinib (ie, the maximum tolerated dose in the phase 1 study),12 but the QTcF interval was prolonged for more than 60 ms above baseline in some of these patients. Therefore, the protocol was subsequently amended on April 20, 2010, to starting doses of 135 mg per day for men and 90 mg per day for women in all other patients. These changes were made because of observations from the protocol-defined dose reductions in this initial group of patients, reportedly greater susceptibility to QTcF prolongation in women compared with men,12-14 observations from the phase 1 trial,12 and expert recommendations from the Data Monitoring Committee. Dose reductions were permitted in a stepwise fashion from 200 mg per day to 135 mg per day to 90 mg per day to 60 mg per day for one cycle or more at each step, and dose interruptions were permitted in patients who had grade 3 or 4 non-haematological treatment-related treatment-emergent adverse events that persisted for longer than 48 h without improvement to grade 2 or less. After sufficient resolution of these treatment-emergent adverse events, quizartinib could be restarted at the next lower dose. The appendix (p 1) describes additional details about dose reductions and interruptions due to QTcF prolongation. Concomitant use of strong CYP3A4 inhibitors, which increase quizartinib exposure, and medications known to cause QTcF prolongation were prohibited, but allowed with caution, if essential (eg, antibiotics, antifungals, and other antimicrobials to prevent or treat infections if absolutely essential for the care of the patient).
Discontinuation criteria were withdrawal of consent, non-compliance, use of prohibited concomitant medications without approval by the investigator, erroneous inclusion into the study, use of antileukaemic therapy (other than hydroxyurea) or intrathecal therapy for CNS leukaemia in remission, elective haemopoietic stem cell transplantation, development of a concurrent disease that results in the investigator determining that continued quizartinib treatment and study participation were not in the best interest of the patient, intolerable treatment-related adverse events or serious adverse events, progressive disease or relapse without clinical benefit, and pregnancy. Patients who were bridged to haemopoietic stem cell transplantation were not permitted to restart quizartinib following transplantation.
We did bone marrow assessments based on local morphology at screening (within 14 days of quizartinib treatment), on day 15 of the first treatment cycle, and on day 1 of the second, third, and fourth cycles, unless the patient achieved a composite complete remission. If a patient achieved a composite complete remission, these assessments were then done every three cycles and at the end of treatment. Best responses throughout the course of treatment were classified according to the local derivation.
We also did physical examinations, laboratory analyses (including serum chemistry, urinalysis, and haematology assessments), and assessments of vital signs and adverse events throughout the study. Laboratory results and assessments of adverse events or serious adverse events were evaluated at screening (laboratory analyses only); during cycle 1 on day 1, day 2 (assessments of adverse events or serious adverse events only), day 8 (range 7–9), and day 15 (range 14–16); during cycle 2 on day 1 (range −2 to 4) and day 15 (range 14–16); during subsequent cycles on day 1 (range −2 to 4) and every 14 days (plus or minus 1 day) thereafter; and at the end-of-treatment visit (within 7 days [plus or minus 1 day] of the final study drug dose). Treatment-emergent adverse events and serious adverse events were also assessed at the 30-day follow-up visit. We graded adverse events with the National Cancer Institute’s Common Terminology Criteria for Adverse Events (version 4.0) and coded them using the Medical Dictionary for Regulatory Activities terminology. The appendix (p 1) provides details about monitoring and management of QTcF prolongation.
FLT3-ITD genotyping was assessed in peripheral blood or bone marrow, or both, by PCR at central laboratories (Genoptix Medical Laboratory [now Navigate BioPharma Services], Carlsbad, CA, USA; and University of Dresden, Dresden, Germany).15 Genoptix Medical Laboratory used an assay sensitivity of 1% and the University of Dresden’s laboratory used an assay sensitivity of 0·5%. FLT3-ITD allelic frequency was calculated as the percentage of FLT3-ITD to total FLT3 (ie, FLT3-ITD and wild-type FLT3 without correction for blast percentage. Patients with an allelic frequency higher than 10% were considered FLT3-ITD positive whereas all other patients were considered FLT3-ITD negative. The threshold for FLT3-ITD-positive mutational status was selected to allow reliably quantifiable determination of mutational status with the methods used at the time the study was designed.
Outcomes
The co-primary endpoints were the proportion of patients who achieved a composite complete remission (defined as complete remission + complete remission with incomplete platelet recovery + complete remission with incomplete haematological recovery) and the proportion of patients who achieved a complete remission. Responses to quizartinib were based on the Cheson criteria,16 but were modified for complete remission with incomplete haematological recovery and partial remission. The modified criteria for complete remission with incomplete haematological recovery were as follows: less than 5% bone marrow blasts, 1% or less of peripheral blood blasts if available, no Auer rods, no evidence of extramedullary disease, with residual neutropenia (≤1 × 109/L) with or without platelet recovery, and no requirement for transfusion independence. Patients satisfying the criteria for complete remission or complete remission with incomplete platelet recovery, but who were not transfusion independent, were also classified as having complete remission with incomplete haematological recovery. The modified criteria for partial remission were defined as a decrease in bone marrow blasts of 50% or more from baseline to total bone marrow blasts of 5–25%, no evidence of extramedullary disease, and no requirement for transfusion independence. Patients were classified according to their best response, which was defined as the best measured response status by disease assessment (ie, in descending order of best response: complete remission, complete remission with incomplete platelet recovery, complete remission with incomplete haematological recovery, and partial remission). The appendix (p 1) shows the additional details about classifications of response to quizartinib treatment and definitions for relapsed disease following quizartinib treatment.
Secondary endpoints reported here were duration of composite complete remission, the proportion of patients who achieved an overall response, leukaemia-free survival, overall survival, effect of quizartinib treatment on bridging to haemopoietic stem cell transplantation (ie, the proportion of patients bridged to transplantation), and safety and tolerability. All other secondary endpoints will be reported separately and were as follows: duration of remission, treatment induction and post-induction treatment-related mortality, time to treatment response, pharmacokinetics and pharmacodynamics of quizartinib, and effect of quizartinib on haematological improvement, duration of disease control, blood and platelet transfusions, infections, number of hospitalisation days, and ECOG performance status.
Statistical analysis
We originally determined the sample sizes of 90 patients for an FLT3-ITD mutational positive subset per cohort and 60 for an FLT3-ITD mutational negative subset to yield approximately 89% statistical power at a one-sided α of 0·025. Per recommendations from the Data Monitoring Committee, the planned sample size was increased to approximately 300 patients in total. Efficacy analyses included all enrolled patients who received at least one dose of quizartinib (ie, the intention-to-treat population) and were analysed by cohort to accurately capture the nuances of each unique, clinically relevant, and predefined cohort. Patients with a locally assessed post-treatment bone marrow aspirate or biopsy were included in efficacy analyses by response; all other patients were considered to have an unknown response. We estimated the proportion of patients with composite complete remission and complete remission with two-sided 95% CIs, and we summarised the primary and secondary efficacy endpoints using descriptive statistics. We used Kaplan-Meier methods to summarise time-to-event data. In patients achieving a composite complete remission who then relapsed, the duration of composite complete remission was measured from the start of the first observed response to the date of documented relapse; in those patients not relapsing, the duration of composite complete remission was censored at the last assessment visit at which the patient was known to be relapse-free or at the time of bridging to haemopoietic stem cell transplantation.
Overall survival was measured in the intention-to-treat population from the first study dose until the date of death. Patients without a mortality date in the death record were censored at the latest of the following: treatment discontinuation date, last dosing administration date, last disease assessment date, or the last follow-up visit in which the patient was known to be alive. We did a prespecified sensitivity analysis of overall survival censoring patients who discontinued treatment to receive a haemopoietic stem cell transplantation using the date of treatment discontinuation as the censoring time. If the time of death occurred after treatment discontinuation, it was not included in the sensitivity analysis. Additionally, we did 28-day landmark analyses for overall survival stratified by achieved response, including only patients alive at the end of the first cycle of treatment. Similarly, we determined the leukaemia-free survival and duration of composite complete remission for only those who achieved a composite complete remission.
For the safety analyses, we included all enrolled patients who received at least one dose of quizartinib (intention-to-treat population), and we summarised adverse events using descriptive statistics.
We did all analyses using SAS (version 9.1 or later). This trial is registered with ClinicalTrials.gov, number NCT00989261, and with the European Clinical Trials Database, EudraCT 2009-013093-41.
Role of the funding source
The sponsor and investigators designed the study. The investigators collected the data and the sponsor’s biostatisticians, in consultation with the investigators, analysed the data. The authors had a role in data interpretation and writing and reviewing of the report. Editorial services were provided by medical writers contracted by the funder. GG, DT, and DL had full access to all the raw data. The corresponding author had full access to all the data and had final responsibility to submit for publication.
Results
Between Nov 19, 2009, and Oct 31, 2011, we enrolled a total of 333 patients: 157 in cohort 1 and 176 in cohort 2 (figure 1). Data cutoff was Sept 28, 2012, when all but four (1%) of 333 patients (two [1%] of 157 in cohort 1 and two [1%] of 176 in cohort 2) had discontinued study treatment. The median follow-up for all patients in the intention-to-treat population was 24·6 weeks (IQR 14·0–41·4). Median ages were 69 years (IQR 66–73; 155 [99%] of 157 patients aged ≥60 years) in cohort 1 and 51 years (40–60; 44 [25%] of 176 aged ≥60 years) in cohort 2. There were more patients with acute myeloid leukaemia secondary to myelodysplastic syndrome in cohort 1 than in cohort 2 (32 [20%] of 157 vs 11 [6%] of 176). The majority of patients in both cohorts had intermediate cytogenetic risk at baseline (70 [80%] of 88 in cohort 1 and 69 [78%] of 89 in cohort 2; table 1).
Figure 1: Trial profile.
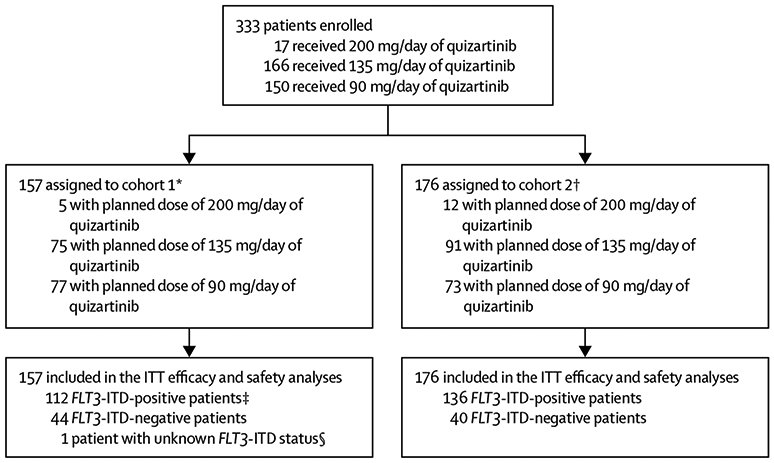
FLT3-ITD=FMS-like tyrosine kinase 3 internal tandem duplication. ITT=intention to treat. *Patients in cohort 1 were aged 60 years or older who relapsed after less than 1 year of, or were refractory to, first-line chemotherapy. †Patients in cohort 2 were aged 18 years or older with relapsed or refractory acute myeloid leukaemia following one salvage chemotherapy or haemopoietic stem cell transplantation. ‡Two younger FLT3-ITD-positive patients were enrolled to cohort 1 and are considered as protocol deviations. They were only excluded from any age-specific analyses. §This patient was not included in the mutation level-specific analyses.
Table 1:
Baseline patient characteristics
| Cohort 1 |
Cohort 2 |
|||
|---|---|---|---|---|
|
FLT3-ITD positive (n=112) |
FLT3-ITD negative (n=44) |
FLT3-ITD positive (n=136) |
FLT3-ITD negative (n=40) |
|
| Sex | ||||
| Women | 56 (50%) | 23 (52%) | 67 (49%) | 16 (40%) |
| Men | 56 (50%) | 21 (48%) | 69 (51%) | 24 (60%) |
| Median age, years | 69 (66–73) | 69 (66–72) | 50 (39–59) | 54 (44–61) |
| Number of patients aged 60 years or older* | 110 (98%) | 44 (100%) | 32 (24%) | 12 (30%) |
| Relapsed after previous treatment†‡ | 68 (61%) | 23 (52%) | 49 (36%) | 10 (25%) |
| Refractory to previous treatment†‡ | 42 (38%) | 21 (48%) | 87 (64%) | 30 (75%) |
| Previous high-intensity therapy§ | 93 (83%) | 35 (80%) | 132 (97%) | 38 (95%) |
| Previous allogeneic HSCT¶ | 0 | 0 | 40 (29%) | 11 (28%) |
| ECOG performance score∥ | ||||
| 0–1 | 94 (84%) | 41 (93%) | 111 (82%) | 38 (95%) |
| 2 | 15 (13%) | 3 (7%) | 24 (18%) | 2 (5%) |
| Secondary acute myeloid leukaemia | 17 (15%) | 15 (34%) | 10 (7%) | 1 (3%) |
| Cytogenetic risk** | ||||
| Favourable | 1 (2%) | 0 | 0 | 1 (5%) |
| Intermediate | 54 (84%) | 15 (65%) | 54 (79%) | 15 (71%) |
| Poor | 9 (14%) | 8 (35%) | 14 (21%) | 5 (24%) |
| Median baseline bone marrow blast count, %†† | 78 (51–90) | 35 (18–80) | 81 (56–90) | 48 (21–70) |
| Median white blood cell counts at baseline, × 109/L‡‡ | 15·1 (5·0–37·0) | 3·4 (1·5–10·0) | 10·9 (3·9–31·1) | 2·2 (0·8–5·9) |
| FLT3-ITD allelic frequency | ||||
| ≤10% | 0 | 44 (100%) | 0 | 40 (100%) |
| Detectable | ·· | 12 (27%) | ·· | 14 (35%) |
| Undetectable | ·· | 32 (73%) | ·· | 26 (65%) |
| >10% to <25% | 26 (23%) | 0 | 28 (21%) | 0 |
| ≥25% to ≤50% | 54 (48%) | 0 | 62 (46%) | 0 |
| >50% | 32 (29%) | 0 | 46 (34%) | 0 |
Data are n (%) or median (IQR). One patient with unknown FLT3-ITD status was enrolled in cohort 1, but was not included in the mutation level-specific analyses and therefore is not included in this table. Two younger patients who had FLT3-ITD-positive status were enrolled in cohort 1 and are considered protocol deviations. FLT3-ITD=FMS-like tyrosine kinase 3 internal tandem duplication. HSCT=haemopoietic stem cell transplantation. ECOG=Eastern Cooperative Oncology Group.
In cohort 1, 54 (48%) of 112 patients who were FLT3-ITD positive and 21 (48%) of 44 who were FLT3-ITD negative were aged 70 years or older.
Data for response to previous treatment were not available for two FLT3-ITD-postive patients in cohort 1.
In cohort 1, relapsed or refractory refers to response to first-line therapy. In cohort 2, relapsed or refractory refers to response to last line of therapy, or the latter of first-line or second-line therapy, including salvage chemotherapy or HSCT; in patients who received other lines of therapy subsequent to second-line therapy, relapsed or refractory refers to response to second-line therapy.
Previous high-intensity therapies include anthracyclines and related agents (eg, daunorubicin, idarubicin, or mitoxantrone).
In cohort 2, most (35 [69%] of 51) previous allogeneic HSCTs were done as first-line therapy (n=28 were FLT3-ITD positive, and n=7 were FLT3-ITD negative).
ECOG performance score was missing for one FLT3-ITD-positive patient in each cohort.
Cytogenetic risk was determined for 64 FLT3-ITD-positive patients, 23 FLT3-ITD-negative patients, and one patient with unknown FLT3-ITD mutational status in cohort 1; and 68 FLT3-ITD-positive and 21 FLT3-ITD-negative patients in cohort 2. The patient with unknown cytogenetic risk in cohort 1 had intermediate cytogenetic risk at baseline.
Median baseline bone marrow blast counts were determined for 110 FLT3-ITD-positive and 43 FLT3-ITD-negative patients in cohort 1, and 131 FLT3-ITD-positive and 39 FLT3-ITD-negative patients in cohort 2.
White blood cell counts at baseline were missing for one patient in the FLT3-ITD-positive group in cohort 1.
In cohort 1, 112 (71%) of 157 patients were FLT3-ITD positive, 44 (28%) were FLT3-ITD negative, and one patient (<1%) had unknown FLT3-ITD mutation status and was not included in mutation level-specific analyses; in cohort 2, 136 (77%) of 176 were FLT3-ITD positive and 40 (23%) were FLT3-ITD negative. Of those who were FLT3-ITD negative, 32 (73%) of 44 in cohort 1 and 26 (65%) of 40 in cohort 2 had a true undetectable mutation. Within each cohort, most baseline characteristics were generally similar between FLT3-ITD-positive and FLT3-ITD-negative patients, with some exceptions (table 1).
In cohort 1, 63 (40%) of 157 patients were refractory to first-line treatment, and 92 (59%) had relapsed after first-line treatment. In cohort 2, 117 (66%) of 176 were refractory to their last line of therapy, and 59 (34%) had relapsed after their last line of therapy. In cohort 2, 104 (77%) of 136 FLT3-ITD-positive patients and 31 (78%) of 40 FLT3-ITD-negative patients had achieved composite complete remission during any previous therapy. 51 (29%) of 176 patients (40 [78%] FLT3-ITD-positive patients and 11 [22%] FLT3-ITD-negative patients) in cohort 2 had relapsed after any previous allogeneic haemopoietic stem cell transplantation, 69% (35 of 51 patients) of which occurred as part of their first-line therapy (28 [70%] of 40 FLT3-ITD-positive and seven [64%] of 11 FLT3-ITD-negative patients; table 1).
In cohort 1, the median duration of treatment was 14·2 weeks (IQR 7·5–23·1) in FLT3-ITD-positive patients and 9·6 weeks (4·2–25·9) in FLT3-ITD-negative patients. The most common reasons for treatment discontinuation were relapsed disease, lack of response or disease progression, and adverse events (table 2). In the 32 patients who discontinued quizartinib because of adverse events, the most common reasons were infection (12 [38%]), haemorrhage (four [13%]), and QTcF prolongation (three [9%]). 13 (8%) of 157 patients (11 [7%] FLT3-ITD positive, one [1%] FLT3-ITD negative, and one [1%] unknown FLT3-ITD mutational status) discontinued for haemopoietic stem cell transplantation: eight (62%) of 13 while in composite complete remission and two (15%) while in partial remission.
Table 2:
Patient disposition
| Cohort 1 |
Cohort 2 |
|||
|---|---|---|---|---|
|
FLT3-ITD positive (n=112) |
FLT3-ITD negative (n=44) |
FLT3-ITD positive (n=136) |
FLT3-ITD negative (n=40) |
|
| Total number of patients who discontinued study treatment | 110 (98%)* | 44 (100%) | 134 (99%)* | 40 (100%) |
| Disease relapse† | 44 (39%) | 11 (25%) | 29 (21%) | 2 (5%) |
| Lack of response or disease progression† | 21 (19%) | 19 (43%) | 28 (21%) | 15 (38%) |
| Adverse events | 25 (22%) | 7 (16%) | 20 (15%) | 3 (8%) |
| QTcF prolongation | 3 (3%) | 0 | 2 (1%) | 1 (3%) |
| Elective haemopoietic stem cell transplantation | 11 (10%) | 1 (2%) | 47 (35%) | 14 (35%) |
| Deaths‡ | 6 (5%) | 4 (9%) | 5 (4%) | 3 (8%) |
| Patient withdrew consent | 2 (2%) | 0 | 0 | 0 |
| Patient non-compliant with dosing schedule | 1 (1%) | 0 | 2 (1%) | 0 |
| Other reasons | 0 | 2 (5%) | 3 (2%) | 3 (8%) |
Data are n (%). One patient with unknown FLT3-ITD status was enrolled in cohort 1, but was not included in the mutation level-specific analyses and therefore is not included in this table. Patients with more than one reason for treatment discontinuation were only counted once and included in the most relevant category. FLT3-ITD=FMS-like tyrosine kinase 3 internal tandem duplication. QTcF=QT interval corrected using Fridericia′s correction formula.
At the time of data cutoff, four patients were alive and receiving quizartinib treatment (two in cohort 1 and two in cohort 2).
These numbers were based on investigator assessment and do not reflect the definition of relapse from quizartinib as defined in the appendix (p 1). Disease relapse was a predefined category with no further text specifications. Conversely, patients included in the no response or disease progression category were based on specific text descriptions provided by the investigators if the adverse event or other category was selected at the time of study treatment discontinuation.
Cause of death in cohort 1: pneumonia (n=2), bacterial sepsis (n=1), sepsis (n=1), subdural haematoma (n=1), systemic inflammatory response syndrome (n=1), cerebral haemorrhage (n=1), myocardial infarction (n=1), haemorrhagic stroke (n=1), and acute hepatic failure (n=1). Cause of death in cohort 2: acute myeloid leukaemia progression (n=3), bacterial sepsis (n=1), haemorrhage (n=1), cardiac arrest (n=1), lung infection (n=1), and sepsis (n=1).
In cohort 2, the median duration of treatment was 9·2 weeks (IQR 6·1–14·3) in FLT3-ITD-positive patients and 8·1 weeks (5·7–11·6) in FLT3-ITD-negative patients. The most common reasons for treatment discontinuation in this cohort were elective haemopoietic stem cell transplantation, lack of response or disease progression, relapsed disease, and adverse events (table 2). In the 25 patients who discontinued quizartinib because of adverse events, the most common reasons were infection (11 [44%]), QTcF prolongation (three [12%]), and myelo-suppression (two [8%]). Of the 61 patients (47 [77%] FLT3-ITD-positive patients and 14 [23%] FLT3-ITD-negative patients) who discontinued treatment for haemopoietic stem cell transplantation, 29 (48%) were in composite complete remission and 22 (36%) were in partial remission. Among those bridging to haemopoietic stem cell transplantation, quizartinib was discontinued at a median of 8 days (IQR 4–14) before the conditioning regimen. The median time from the start of quizartinib to discontinuation for haemopoietic stem cell transplantation was 65 days (IQR 51–83).
In cohort 1, the proportion of FLT3-ITD-positive patients who achieved composite complete remission was 63 (56%) of 112 (three [3%] had complete remission, four [4%] had complete remission with incomplete platelet recovery, and 56 [50%] had complete remission with incomplete haematological recovery) and 23 (21%) had partial remission, resulting in an overall response in 86 (77%) patients (table 3). Of the 42 FLT3-ITD-positive patients in this cohort who were refractory to first-line therapy, 20 (48%) achieved composite complete remission and 11 (26%) achieved partial remission with quizartinib. In the 68 FLT3-ITD-positive patients in this cohort who relapsed after achieving composite complete remission following first-line therapy, 41 (60%) achieved composite complete remission and 12 (18%) achieved partial remission with quizartinib (appendix p 5). A prespecified exploratory subgroup analysis of responses by FLT3-ITD allelic frequency in cohort 1 is in the appendix (p 7). The proportion of FLT3-ITD-negative patients in cohort 1 who achieved composite complete remission was 16 (36%) of 44 (two [5%] had complete remission, one [2%] had complete remission with incomplete platelet recovery, and 13 [30%] had complete remission with incomplete haematological recovery) and four (9%) had partial remission, resulting in an overall response in 20 (45%) patients. In FLT3-ITD-negative patients with low but detectable FLT3-ITD allelic frequency (≤10%), a composite complete remission was reported in seven (58%) of 12 patients compared with nine (28%) of 32 in those with undetectable FLT3-ITD mutational status. 34 (54%) of 63 FLT3-ITD-positive patients and six (38%) of 16 FLT3-ITD-negative patients who achieved composite complete remission did so after one cycle of quizartinib (table 3). Median duration of composite complete remission was 12·1 weeks (95% CI 6·3–15·7) in FLT3-ITD-positive patients and 16·4 weeks (8·1–30·4) in FLT3-ITD-negative patients (figure 2A). Similarly, leukaemia-free survival was 12·1 weeks (95% CI 6·1–14·3) in FLT3-ITD-positive patients and 16·4 weeks (8·1–26·1) in FLT3-ITD-negative patients. None of the five patients who achieved complete remission in this cohort were bridged to haemopoietic stem cell transplantation.
Table 3:
Cumulative response assessment*
| Cohort 1 |
Cohort 2 |
|||
|---|---|---|---|---|
|
FLT3-ITD positive (n=112) |
FLT3-ITD negative (n=44) |
FLT3-ITD positive (n=136) |
FLT3-ITD negative (n=40) |
|
| Best response | ||||
| Composite complete remission | 63 (56%) | 16 (36%) | 62 (46%) | 12 (30%) |
| Complete remission | 3 (3%) | 2 (5%) | 5 (4%) | 1 (3%) |
| Complete remission with incomplete platelet recovery | 4 (4%) | 1 (2%) | 2 (1%) | 1 (3%) |
| Complete remission with incomplete haematological recovery | 56 (50%) | 13 (30%) | 55 (40%) | 10 (25%) |
| Partial remission | 23 (21%) | 4 (9%) | 39 (29%) | 6 (15%) |
| No response | 20 (18%) | 17 (39%) | 24 (18%) | 16 (40%) |
| Unknown | 6 (5%) | 7 (16%) | 11 (8%) | 6 (15%) |
| Overall response† | 86 (77%) | 20 (45%) | 101 (74%) | 18 (45%) |
| Composite complete remission after one cycle | 34/63 (54%) | 6/16 (38%) | 39/62 (63%) | 8/12 (67%) |
Data are n (%) or n/N (%). One patient with unknown FLT3-ITD status was enrolled in cohort 1, but was not included in the mutation level-specific analyses; this patient achieved a composite complete remission. FLT3-ITD=FMS-like tyrosine kinase 3 internal tandem duplication.
Best response was determined using response data from all quizartinib cycles. Of the 44 FLT3-ITD-negative patients in cohort 1, 32 had no detectable mutation (nine [28%] of whom achieved a composite complete remission and three [9%] of whom achieved a partial remission) and 12 had a detectable but low FLT3-ITD mutation (seven [58%] of whom achieved a composite complete remission and one [8%] of whom achieved a partial remission). Of the 40 FLT3-ITD-negative patients in cohort 2, 26 had an undetectable FLT3-ITD allelic frequency (seven [27%] of whom achieved a composite complete remission and three [12%] achieved a partial remission) and 14 had a low but detectable expression of the FLT3-ITD mutation (five [36%] of whom achieved a composite complete remission and three [21%] achieved a partial remission).
Defined as composite complete remission plus partial remission.
Figure 2: Kaplan-Meier plots of duration of composite complete remission by FLT3-ITD mutational status in patients from cohort 1 (A) and cohort 2 (B).

FLT3-ITD=FMS-like tyrosine kinase 3 internal tandem duplication. *One patient in cohort 1 with an unknown FLT3-ITD mutation status is not included in this analysis; however, that patient had a duration of composite complete remission equal to 2·9 weeks.
In all patients in cohort 1, regardless of FLT3-ITD status, the proportions of patients who achieved composite complete remission were similar in patients aged 60–69 years (41 [51%] of 80) and those aged 70 years or older (39 [52%] of 75; appendix p 9). Median durations of composite complete remission were 8·1 weeks (95% CI 6·1–17·4) in those aged 60–69 years and 13·9 weeks (8·1–18·4) in those aged 70 years or older.
In cohort 2, the proportion of FLT3-ITD-positive patients who achieved composite complete remission was 62 (46%) of 136 (five [4%] had complete remission, two [2%] had complete remission with incomplete platelet recovery, and 55 [40%] had complete remission with incomplete haematological recovery) and 39 (29%) had partial remission, resulting in an overall response in 101 (74%) patients (table 3). Of the 87 FLT3-ITD-positive patients in this cohort who were refractory to their last line of therapy, 41 (47%) achieved composite complete remission and 24 (28%) achieved partial remission with quizartinib (appendix p 6). In the 49 FLT3-ITD-positive patients in this cohort who relapsed after achieving composite complete remission following their last line of therapy, 21 (43%) achieved composite complete remission and 15 (31%) achieved partial remission with quizartinib (appendix p 6). A prespecified exploratory subgroup analysis of responses by FLT3-ITD allelic frequency in cohort 2 is in the appendix (p 8). The proportion of FLT3-ITD-negative patients who achieved composite complete remission was 12 (30%) of 40 (one [3%] had complete remission, one [3%] had complete remission with incomplete platelet recovery, and ten [25%] had complete remission with incomplete haematological recovery) and six (15%) had partial remission, resulting in an overall response in 18 (45%) patients. 47 (64%) of 74 patients who achieved a composite complete remission in the cohort (39 [63%] of 62 FLT3-ITD-positive patients and eight [67%] of 12 FLT3-ITD-negative patients) did so at the end of one cycle of quizartinib. Five (36%) of 14 FLT3-ITD-negative patients with low but detectable FLT3-ITD allelic frequency (ie, ≤10%) achieved a composite complete remission and three (21%) achieved partial remission. By contrast, seven (27%) of 26 patients with undetectable FLT3-ITD achieved a composite complete remission and three (12%) achieved a partial remission (table 3). The median durations of composite complete remission were 10·6 weeks (95% CI 8·1–16·1) in all FLT3-ITD-positive patients and 7·0 weeks (4·1–8·1) in all FLT3-ITD-negative patients (figure 2B). Similar findings were recorded for leukaemia-free survival, which was 12·1 weeks (95% CI 8·3–19·1) in FLT3-ITD-positive patients and 6·6 weeks (5·0–8·1) in FLT3-ITD-negative patients.
In cohort 2, quizartinib enabled 61 (35%) of 176 patients to bridge to haemopoietic stem cell transplantation (47 [35%] of 136 FLT3-ITD-positive patients and 14 [35%] of 40 TLT3-ITD-negative patients; table 2), of which 45 (96%) of 47 FLT3-ITD-positive patients (appendix p 10) and 13 (93%) of 14 FLT3-ITD-negative patients achieved a composite complete remission or partial remission on quizartinib. One (17%) of six patients who achieved a complete remission in this cohort was bridged to haemopoietic stem cell transplantation.
Overall survival results in cohort 1 are shown in figure 3A. Results from a sensitivity analysis of overall survival censoring patients who discontinued treatment for haemopoietic stem cell transplantation were similar (data not shown) to the primary analysis and confirm the robustness of these data. FLT3-ITD-negative patients with low but detectable allelic frequency had a median overall survival of 21·4 weeks (95% CI 19·0–43·4), and those with an undetectable allelic frequency had a median overall survival of 18·2 weeks (7·6–29·4). A landmark analysis of overall survival in the 106 FLT3-ITD-positive patients alive at day 28 is in the appendix (p 23) and a subgroup analysis of overall survival by age group in FLT3-ITD-positive patients in this cohort is in the appendix (p 25). Overall, in this cohort, 16 (14%) of 112 FLT3-ITD-positive patients who responded to quizartinib survived for 1 year or longer (table 4).
Figure 3: Kaplan-Meier plots of overall survival by FLT3-ITD mutational status in all patients in cohort 1 (A) and cohort 2 (B).

FLT3-ITD=FMS-like tyrosine kinase 3 internal tandem duplication. *One patient in cohort 1 with an unknown FLT3-ITD mutation status is not included in this analysis; however, that patient had an overall survival equal to 24·0 weeks.
Table 4:
Relation between best response to quizartinib and overall survival in all FLT3-ITD-positive patients in cohort 1 and between response to quizartinib, HSCT, and overall survival in all FLT3-ITD-positive patients in cohort 2
| Cohort 1 |
Cohort 2 |
||||||
|---|---|---|---|---|---|---|---|
| Number of FLT3-ITD-positive patients (n=112) |
Median overall survival, weeks (95% CI) |
Overall survival of 1 year or more, n/N (%) |
Number of FLT3-ITD-positive patients (n=136) |
Median overall survival, weeks (95% CI) |
Bridged to HSCT and overall survival of 1 year or more, n/N (%) |
Not bridged to HSCT and overall survival of 1 year or more, n/N (%) |
|
| Composite complete remission | 63 (56%) | 29·7 (24·9–34·7) | 12/63 (19%) | 62 (46%) | 31·1 (24·4–41·7) | 10/62 (16%) | 8/62 (13%) |
| Complete remission | 3 (3%) | 68·9 (30·6–74·3) | 2/3 (67%) | 5 (4%) | 77·4 (43·0–NR) | 0 | 4/5 (80%) |
| Complete remission with incomplete platelet recovery | 4 (4%) | 54·4 (32·9–NR) | 2/4 (50%) | 2 (1%) | NA* | 2/2 (100%) | 0 |
| Complete remission with incomplete haematological recovery | 56 (50%) | 25·9 (23·7–33·9) | 8/56 (14%) | 55 (40%) | 27·1 (23·3–34·4) | 8/55 (15%) | 4/55 (7%) |
| Partial remission | 23 (21%) | 36·4 (25·–39·9) | 4/23 (17%) | 39 (29%) | 25·9 (19·3–33·0) | 7/39 (18%) | 1/39 (3%) |
| No response or unknown response | 26 (23%) | 10·7 (7·1–15·9) | 0 | 35 (26%) | 9·0 (6·1–17·9) | 0 | 1/35 (3%) |
Data are n (%), unless otherwise stated. FLT3-ITD=FMS-like tyrosine kinase 3 internal tandem duplication. HSCT=haemopoietic stem cell transplantation. NR=not reached. NA=not applicable.
Only two FLT3-ITD-positive patients in cohort 2 had a best response of complete remission with incomplete platelet recovery: one patient died at 83 weeks and the other patient was censored at 105 weeks.
Overall survival results in cohort 2 are shown in figure 3B. Results from a sensitivity analysis of overall survival censoring patients who discontinued treatment for haemopoietic stem cell transplantation were similar (data not shown) and confirm the robustness of these data. A landmark analysis of overall survival in the 129 FLT3-ITD-positive patients alive at 28 days is in the appendix (p 24); of these patients, 45 (35%) bridged to haemopoietic stem cell transplantation after achieving composite complete remission or partial remission with quizartinib. In this cohort, 27 (20%) of 136 FLT3-ITD-positive patients survived for 1 year or longer; of these patients, 17 (63%) were bridged to haemopoietic stem cell transplantation following treatment with quizartinib (table 4).
Quizartinib was generally well tolerated with a manageable safety profile; 30-day all-cause mortality was 6% (nine of 157) in cohort 1 and 5% (eight of 176) in cohort 2. Table 5 summarises the treatment-emergent adverse events that occurred in this study. In all 333 enrolled patients (ie, the intention-to-treat population), grade 3 or worse treatment-related treatment-emergent adverse events that were reported in 5% or more patients consisted of febrile neutropenia (76 [23%] of 333), anaemia (75 [23%]), thrombocytopenia (39 [12%]), QTcF prolongation (33 [10%]), neutropenia (31 [9%]), leucopenia (22 [7%]), decreased platelet count (20 [6%]), and pneumonia (17 [5%]). All patients in this study had at least one adverse event, and safety data per cohort are shown in the appendix (pp 11, 12). Dose reductions occurred in 107 (32%) of 333 patients in the intention-to-treat population and were evenly distributed across cohorts (52 [33%] of 157 patients in cohort 1 and 55 [31%] of 176 in cohort 2). Treatment-related treatment-emergent adverse events of any grade reported in 20% or more of patients across both cohorts were nausea (130 [39%] of 333), QTcF prolongation (96 [29%]), fatigue (88 [26%]), vomiting (87 [26%]), anaemia (84 [25%]), febrile neutropenia (78 [23%]), diarrhoea (76 [23%]), and dysgeusia (69 [21%]; appendix p 18). Across both cohorts (ie, the intention-to-treat population), treatment-emergent adverse events leading to discontinuations occurred in 105 (32%) of 333 patients; treatment-related treatment-emergent adverse events leading to discontinuations occurred in 43 (13%) patients. In cohort 1, treatment-related treatment-emergent adverse events were similar in patients aged 70 years or older and those aged 60–69 years; also, those leading to discontinuations were infrequent and similar between age groups. In cohort 1, 30-day all-cause mortality was higher in patients aged 70 years or older compared with those aged 60–69 years (seven [9%] of 75 vs two [3%] of 80; appendix p 11).
Table 5:
Treatment-emergent adverse events in all patients* that occurred in 10% or more of patients (grade 1–2) or 2% or more of patients (grade ≥3)
| Grade 1–2 | Grade 3 | Grade 4 | Grade 5 | |
|---|---|---|---|---|
| Febrile neutropenia | ·· | 124 (37%) | 13 (4%) | 0 |
| Anaemia | ·· | 76 (23%) | 11 (3%) | 0 |
| Acute myeloid leukaemia | ·· | 4 (1%) | 5 (2%) | 67 (20%) |
| Thrombocytopenia | ·· | 10 (3%) | 39 (12%) | 0 |
| Pneumonia | ·· | 31 (9%) | 6 (2%) | 7 (2%) |
| Neutropenia | ·· | 9 (3%) | 25 (8%) | 1 (<1%) |
| Electrocardiogram QT prolonged | 63 (19%) | 34 (10%) | 1 (<1%) | 0 |
| Platelet count decreased | ·· | 3 (1%) | 24 (7%) | 0 |
| Leucopenia | ·· | 7 (2%) | 18 (5%) | 0 |
| Sepsis | ·· | 6 (2%) | 10 (3%) | 9 (3%) |
| Asthenia | 42 (13%) | 24 (7%) | 0 | 0 |
| Fatigue | 95 (29%) | 17 (5%) | 1 (<1%) | 0 |
| Hypokalaemia | 45 (14%) | 13 (4%) | 3 (1%) | 0 |
| Diarrhoea | 122 (37%) | 14 (4%) | 0 | 0 |
| Neutrophil count decreased | ·· | 2 (1%) | 12 (4%) | 0 |
| Pyrexia | 89 (27%) | 12 (4%) | 0 | 1 (<1%) |
| Device-related infection | ·· | 12 (4%) | 0 | 0 |
| Alanine aminotransferase increased | ·· | 11 (3%) | 1 (<1%) | 0 |
| Pancytopenia | ·· | 6 (2%) | 4 (1%) | 1 (<1%) |
| Vomiting | 120 (36%) | 10 (3%) | 1 (<1%) | 0 |
| Lung infection | ·· | 9 (3%) | 1 (<1%) | 1 (<1%) |
| White blood cell count decreased | ·· | 4 (1%) | 7 (2%) | 0 |
| Gastrointestinal haemorrhage | ·· | 7 (2%) | 3 (1%) | 0 |
| General physical health deterioration | ·· | 7 (2%) | 1 (<1%) | 2 (1%) |
| Bacteraemia | ·· | 8 (2%) | 1 (<1%) | 1 (<1%) |
| Dyspnoea | 44 (13%) | 7 (2%) | 3 (1%) | 0 |
| Hypotension | ·· | 10 (3%) | 0 | 0 |
| Abdominal pain | 36 (11%) | 9 (3%) | 0 | 0 |
| Nausea | 169 (51%) | 9 (3%) | 0 | 0 |
| Cellulitis | ·· | 8 (2%) | 0 | 1 (<1%) |
| Fungal pneumonia | ·· | 5 (2%) | 1 (<1%) | 3 (1%) |
| Decreased appetite | 81 (24%) | 9 (3%) | 0 | 0 |
| Hyponatraemia | ·· | 9 (3%) | 0 | 0 |
| Atrial fibrillation | ·· | 8 (2%) | 0 | 0 |
| Urinary tract infection | ·· | 8 (2%) | 0 | 0 |
| Hyperglycaemia | ·· | 8 (2%) | 0 | 0 |
| Back pain | ·· | 8 (2%) | 0 | 0 |
| Pain | ·· | 7 (2%) | 0 | 0 |
| Clostridium difficile colitis | ·· | 6 (2%) | 1 (<1%) | 0 |
| Septic shock | ·· | 2 (1%) | 2 (1%) | 3 (1%) |
| Blood bilirubin increased | ·· | 6 (2%) | 1 (<1%) | 0 |
| Dehydration | ·· | 6 (2%) | 1 (<1%) | 0 |
| Epistaxis | 51 (15%) | 7 (2%) | 0 | 0 |
| Headache | 41 (12%) | 5 (2%) | 0 | 0 |
| Petechiae | 57 (17%) | 4 (1%) | 0 | 0 |
| Peripheral oedema | 88 (26%) | 3 (1%) | 0 | 0 |
| Constipation | 68 (20%) | 2 (1%) | 0 | 0 |
| Dyspepsia | 53 (16%) | 2 (1%) | 0 | 0 |
| Pain in extremity | 38 (11%) | 2 (1%) | 0 | 0 |
| Hypomagnesaemia | 35 (11%) | 0 | 1 (<1%) | 0 |
| Rash | 47 (14%) | 1 (<1%) | 0 | 0 |
| Dizziness | 45 (14%) | 0 | 0 | 0 |
| Dysgeusia | 78 (23%) | 0 | 0 | 0 |
| Cough | 63 (19%) | 0 | 0 | 0 |
For patients with multiple events of the same type, only the maximum grade is reported. A list of grade 1–2 treatment-emergent adverse events occurring in 10% or more of patients and all grade 3–5 treatment-emergent adverse events, regardless of relation to treatment, is provided in the appendix (pp 13-17).
Treatment-emergent adverse events across both cohorts (n=333), regardless of relation to treatment.
Serious adverse events occurring in 5% or more of the intention-to-treat population were febrile neutropenia (126 [38%] of 333), acute myeloid leukaemia progression (73 [22%]), pneumonia (40 [12%]), QTcF prolongation (33 [10%]), sepsis (25 [8%]), and pyrexia (18 [5%]). Of these serious adverse events, febrile neutropenia (76 [23%] of 333), QTcF prolongation (32 [10%]), pneumonia (14 [4%]), pyrexia (nine [3%]), and sepsis (eight [2%]) were considered treatment related. Notable serious adverse events occurring in less than 5% of patients were torsades de pointes (one [<1%]) and hepatic failure (two [1%]).
Across both cohorts (ie, the intention-to-treat population), a total of 125 (38%) of 333 patients died within the study treatment period, including the 30-day follow-up. 71 (21%) patients died from disease progression of acute myeloid leukaemia and 54 (16%) died from other reported causes, which were consistent with the population being studied who had late-stage, pretreated acute myeloid leukaemia and age-associated comorbidities. All causes of death are listed in the appendix (p 19). A total of 18 (5%) patients died because of an adverse event considered by the investigator to be treatment related: ten (6%) of 157 patients in cohort 1 and eight (5%) of 176 in cohort 2. Of these deaths, only pneumonia (two [1%] of 333) and sepsis (two [1%]) occurred in two or more patients.
Five (2%) of 331 patients with post-baseline measurements (one [1%] of 156 in cohort 1 and four [2%] of 175 in cohort 2) had liver chemistry abnormalities within Hy’s range (ALT or AST >3 × ULN and total bilirubin >2 × ULN; appendix p 20). Two additional patients in cohort 2 who had liver chemistry assessments within Hy’s range were reported directly to the sponsor and, thus, were not included in the investigator-assessed safety population. All patients had alternative confounding factors; therefore, no abnormalities were considered quizartinib related. Additional details regarding these liver chemistry laboratory values are included in the appendix (pp 1, 20). There were two (1%) of 333 patients in the intention-to-treat population who had a serious adverse event of hepatic failure. However, neither of these patients had liver chemistry abnormalities within Hy’s range (elevated bilirubin only but ALT and AST were within normal limits). Moreover, each of these cases occurred in the context of multiple organ failure or sepsis and disease progression of acute myeloid leukaemia.
The 17 initial patients who enrolled into this study received 200 mg quizartinib daily; however, 12 (71%) of these patients had a QTcF interval of more than 480 ms and 14 (82%) had an increased QTcF interval of 60 ms above baseline. In response to the QTcF prolongation recorded in these patients, doses were subsequently reduced for all patients (135 mg per day in men and 90 mg per day in women). Across both cohorts, the maximum QTcF change from baseline was more than 60 ms in 14 (82%) of the 17 patients with a planned dose of 200 mg per day of quizartinib compared with 61 (41%) of 150 women with a planned dose of 90 mg per day and 62 (37%) of 166 men with a planned dose of 135 mg per day. A QTcF interval of more than 500 ms (averaged from triplicate electrocardiograms [ECGs]) was recorded in six (35%) of 17 patients with starting doses of 200 mg per day, 25 (15%) of 166 men with starting doses of 135 mg per day, and 26 (17%) of 150 women with starting doses of 90 mg per day. ECG data for QTcF prolongation by starting dose are included in the appendix for each cohort (pp 21, 22). QTcF prolongation was reversible and successfully managed by treatment interruptions or dose reductions, or both; only six (2%) of 333 patients (three in each cohort) discontinued quizartinib because of QTcF prolongation. However, grade 3 or worse treatment-related treatment-emergent adverse events of QTcF prolongation were reported more frequently in the older age group of cohort 1 (aged ≥70 years) than in the younger age group (aged 60–69 years; 13 [17%] of 75 vs four [5%] of 80). Nevertheless, in the intention-to-treat population, only one (<1%) case of grade 4 QTcF prolongation (ie, torsades de pointes) was reported in a woman aged 63 years assigned to cohort 1 and who was receiving 90 mg per day of quizartinib. However, it resolved after treatment discontinuation. Importantly, this patient was acutely sick from Klebsiella bacteraemia or sepsis with episodes of respiratory arrest before torsades de pointes, and had hypocalcaemia immediately before torsades de pointes. Additionally, she had a history of ongoing atrial fibrillation. No other arrhythmias associated with QTcF prolongation were observed in this patient.
Discussion
In this phase 2 trial of single-agent quizartinib, we showed that this oral, highly potent, and selective next-generation FLT3 inhibitor for the treatment of FLT3-ITD-positive acute myeloid leukaemia is highly active, resulting in a high proportion of responders across many patient types with relapsed or refractory acute myeloid leukaemia. Quizartinib also enabled approximately a third of patients in cohort 2 to bridge to haemopoietic stem cell transplantation and is generally well tolerated, with a manageable safety profile. The findings of this study are presented in two predefined, independent cohorts. All efficacy data are reported by cohort to accurately capture the nuances of each unique, clinically relevant, and predefined cohort.
The observed number of FLT3-ITD-positive patients achieving composite complete remission (63 [56%] of 112 in cohort 1 and 62 [46%] of 136 in cohort 2) and overall response (86 [77%] in cohort 1 and 101 [74%] in cohort 2) seem to be substantially greater than the single-agent responses previously reported with other FLT3 inhibitors, including midostaurin,17 lestaurtinib,18 sorafenib,19 and crenolanib, although such cross-trial comparisons must be made with caution because of population differences.20,21 These tyrosine-kinase inhibitors of varying selectivity offer their own merits and appear to be active in patients with FLT3-mutated acute myeloid leukaemia, although their effect is stronger in combination with chemotherapy. Notably, midostaurin in combination with chemotherapy was recently approved by the US Food and Drug Administration and European Commission for patients with newly diagnosed FLT3-mutated acute myeloid leukaemia.22,23 However, no therapies specifically targeting FLT3-ITD mutations are currently approved for patients with relapsed or refractory disease—a patient population with a particularly poor prognosis.7 All four of these agents, along with the newer FLT3 and AXL inhibitor gilteritinib, were recently reviewed elsewhere.24 The responses observed in our study also compare favourably to those reported in the latest gilteritinib study, which used similar response criteria.25 Importantly, the standard Cheson criteria (developed to characterise responses to chemotherapy) were modified in our study to more appropriately classify responses to quizartinib, which is a pharmacologically different compound compared with standard chemotherapeutic agents. In particular, quizartinib is administered daily in an outpatient setting and has a long half-life of about 3 days26 that results in long-term, continuous activity.12 These fundamental differences could contribute to delayed neutrophil and platelet recovery, and account for the modification to the response criteria. Nevertheless, the numbers of responses recorded in our study represent a true clinical benefit to patients treated with quizartinib, as shown by the improved median overall survival of responders compared with non-responders and, in cohort 2, the ability of many patients to bridge to haemopoietic stem cell transplantation. Additionally, by comparison with standard chemotherapy, the responses to quizartinib were much greater than the approximately 25% composite complete remissions reported with salvage chemotherapy in similar patient populations,7,27 and the 13% reported for unselected second-salvage patients.28 The large proportion of pretreated FLT3-ITD-positive patients in both cohorts who achieved a response to quizartinib is consistent with the observation that leukaemia cells at relapse are highly dependent on FLT3-ITD signalling.29 Furthermore, drug-resistant mutations in FLT3-ITD have been observed in FLT3-ITD-positive patients who stop responding to quizartinib,30 which also suggests dependence of leukaemia cells on FLT3-ITD signalling. The response to quizartinib occurred rapidly in FLT3-ITD-positive patients, with the majority of responses achieved within one cycle (median time to composite complete remission was approximately 4 weeks in each cohort). Among FLT3-ITD-negative patients with low but detectable FLT3-ITD allelic frequency (≤10%) in this study, composite complete remission was achieved in seven (58%) of 12 in cohort 1 and five (36%) of 14 in cohort 2; and in those with undetectable FLT3-ITD mutations, composite complete remission was achieved in nine (28%) of 32 in cohort 1 and seven (27%) of 26 in cohort 2. Together, these findings corroborate previous reports from the phase 1 quizartinib trial12 and indicate that patients with low allelic frequency might benefit from quizartinib similarly to those with more than 10% allelic frequency. The mechanism by which quizartinib induced remission in a considerable proportion of patients with undetectable FLT3-ITD mutations is unknown and requires further investigation; however, the observed activity might be due to partial inhibition of c-Kit by quizartinib at the doses used in this study.11,31 Additionally, inhibition of wild-type FLT3, which is frequently overexpressed in acute myeloid leukaemia32 (although overexpression was not assessed in this study), might also contribute to the observed activity.
Median overall survival of FLT3-ITD-positive patients was consistent across both cohorts (25·4 weeks in cohort 1 vs 24·0 weeks in cohort 2). These survival durations were longer than previously reported with standard therapy in FLT3-ITD-positive patients in first relapse (13 weeks)7 and in second salvage (1·5 months).28 Median overall survival in quizartinib-treated FLT3-ITD-positive patients was also similar to or greater than that in FLT3-ITD-negative patients across cohorts (19·1 weeks in cohort 1 and 25·1 weeks in cohort 2), indicating that quizartinib might partially overcome the negative effect that the FLT3-ITD mutation has on survival.
Because patients with FLT3-ITD-positive, relapsed or refractory disease have a very poor prognosis, allogeneic haemopoietic stem cell transplantation is typically the best option; however, some level of response to therapy is generally required to allow a reasonable probability of a favourable outcome after transplantation.33 Achievement of remission and haemopoietic stem cell transplantation are the two most important factors influencing long-term survival after relapse.34 Multiple studies have shown that patients with circulating peripheral blood blasts or higher bone marrow blasts before haemopoietic stem cell transplantation have worse outcomes than those with lower counts.34-36 Therefore, FLT3-ITD-positive patients are typically ineligible for haemopoietic stem cell transplantation at the time of diagnosis or relapse, and require treatment to achieve at least a haematological remission and ideally deeper remissions (eg, without measurable residual disease).33 Our study of cohort 2 showed the ability of quizartinib to reduce bone marrow blasts to less than 5% (ie, composite complete remission) in 46% of FLT3-ITD-positive patients and to less than 25% (ie, composite complete remission plus partial remission) in 74% of FLT3-ITD-positive patients, and to enable 35% of those patients to bridge to haemopoietic stem cell transplantation—an important factor in improving overall survival. Indeed, most of the FLT3-ITD-positive patients in cohort 2 who survived for 1 year or more were those who bridged to transplant after responding to quizartinib. These findings contrast with the low remission in first or second salvage, thus enabling the possibility of transplant for patients who received quizartinib.33
Old age is also a poor prognostic factor for response and overall survival in acute myeloid leukaemia.9 Despite this poor prognosis, patients in cohort 1 who were 70 years or older responded similarly well to quizartinib as those aged 60–69 years (composite complete remission of 52% vs 51%, respectively), albeit with an expected lower median overall survival as shown in FLT3-ITD-positive patients (22·7 vs 28·7 weeks, respectively). Nevertheless, median overall survival in these patients exceeded published data for much younger FLT3-ITD-positive patients in first relapse,27,37 suggesting that quizartinib could offer clinical benefit to older patients with acute myeloid leukaemia as well.
Toxicity was consistent with the phase 1 data12 and was generally well managed by dose interruptions or reductions, or both. Grade 3 or worse treatment-related treatment-emergent adverse events were most commonly associated with myelosuppression and QTcF prolongation. Of note, in most instances, myelosuppression was possibly due to acute myeloid leukaemia itself. However, treatment with 200 mg per day of quizartinib at the start of this trial (phase 1 maximum tolerated dose12) yielded a higher number of patients with QTcF prolongation than expected. Lower doses were associated with substantially lower numbers of patients with QTcF prolongation, while maintaining high levels of efficacy. Notably, only one case of grade 4 QTcF prolongation occurred in a female patient from cohort 1 with multiple confounding factors, including electrolyte abnormalities, sepsis with episodes of respiratory arrest, and atrial fibrillation. Thus, attributing causality was hindered because of these substantial confounding factors. Additionally, across phase 1 and 2 quizartinib studies in patients with acute myeloid leukaemia, only the previously mentioned case of grade 4 QTcF prolongation was reported; no grade 5 QTcF prolongation has been recorded (data on file). Moreover, QTcF prolongation was reversible and successfully managed with protocol-specified ECG monitoring, maintenance of normal serum electrolytes, avoidance (when feasible) of concomitant medications that might alter the QTcF interval, and dose reductions or interruptions or both. A follow-on phase 2b study with lower doses of quizartinib (30 mg per day or 60 mg per day) showed similar responses with reduced numbers of reported QTcF prolongation.38,39 Furthermore, in this phase 2b study, QTcF prolongation was determined to be concentration dependent with sex not being a significant covariate.39 Additional analyses are planned for ongoing phase 3 trials to further characterise the effect that sex, bodyweight, and concomitant medications have on drug exposure and QTcF prolongation.
The main limitation of this phase 2 trial was the non-randomised, single-arm, open-label study design, which might limit the generalisability of these findings. Furthermore, because the 90 mg dose was only administered to women, interpretation of the sex or dose effect was confounded. Nevertheless, the large number of responders and improved median overall survival in these patients compared with non-responders, as shown by the landmark analyses (appendix pp 23, 24), suggest that quizartinib has great therapeutic potential and served as the impetus for the large, phase 3, randomised controlled trial of quizartinib versus salvage chemotherapy (QuANTUM-R; NCT02039726 or EudraCT 2013-004890-28). Additionally, in this phase 3 trial, both men and women will be administered a 60 mg dose of quizartinib, with a 30 mg lead-in; therefore, the sex and dose effect will not be confounded and will allow for robust subgroup analyses. Lastly, although common for all potent FLT3 inhibitors, the majority of the responses observed with quizartinib were complete remission with incomplete haematological recovery. The clinical benefit of this response is controversial; however, although not powered to assess it formally, our data showed improved median overall survival from treatment with quizartinib, suggesting that the large proportion of patients with a response indeed translated to clinical benefit. Further examination into the level and significance of measurable residual disease following treatment with FLT3 inhibitors, including quizartinib, might help elucidate these observations.
In conclusion, these results suggest that quizartinib might be a valuable treatment option for patients with FLT3-ITD-positive relapsed or refractory acute myeloid leukaemia and warrant further investigation in phase 3 clinical trials. To that end, the QuANTUM-R phase 3, randomised controlled trial designed to evaluate the benefit of a lower dose of quizartinib versus salvage chemotherapy in patients with FLT3-ITD-positive relapsed or refractory acute myeloid leukaemia is underway. The QuANTUM-R study also allows for the reinitiation of quizartinib therapy after haemopoietic stem cell transplantation. Additional ongoing phase 3 studies are investigating quizartinib combined with standard induction chemotherapy and consolidation, followed by single-agent quizartinib, in patients aged 18–75 years with newly diagnosed acute myeloid leukaemia (QuANTUM-First; NCT02668653 or EudraCT 2015-004856-24) and combined with low-dose cytarabine in elderly patients (NCRI-LI1 trial; EudraCT 2011-000749-19).
Supplementary Material
Research in context.
Evidence before this study
We searched PubMed for published studies of FMS-like tyrosinekinase 3 (FLT3) inhibitors for the treatment of relapsed or refractory acute myeloid leukaemia. We used the search terms ″FLT3 inhibitor″ OR ″FLT3-ITD inhibitor″ AND ″relapsed or refractory AML″ AND ″clinical trial″. We did not restrict the searches by date or language. The current evidence shows that patients with relapsed or refractory acute myeloid leukaemia have a poor prognosis marked by limited response to standard salvage chemotherapy and short survival following relapse, especially in those with FLT3 internal tandem duplication (ITD)-positive disease or individuals of old age, or both. Treatment options for patients with acute myeloid leukaemia are limited to standard chemotherapy, which is largely ineffective. At the time of this submission, no FLT3 inhibitors were approved for the treatment of relapsed or refractory, FLT3-mutated acute myeloid leukaemia. However, the recent US Food and Drug Administration and European Commission approval of midostaurin, a multikinase inhibitor, in combination with standard chemotherapy provides a new treatment option for patients with newly diagnosed FLT3-mutated acute myeloid leukaemia, yet does not fulfil the unmet need in relapsed or refractory, FLT3-mutated acute myeloid leukaemia. Furthermore, elderly patients with this disease, particularly those who are unable to tolerate chemotherapy, still need more effective and tolerable therapeutic options.
Added value of this study
Quizartinib is an oral, highly potent, and selective next-generation FLT3 inhibitor. This phase 2 trial included elderly patients who relapsed within 1 year after first-line chemotherapy or were primary refractory to first-line treatment (ie, cohort 1) and those 18 years or older who were refractory to or relapsed after salvage chemotherapy or after haemopoietic stem cell transplantation (ie, cohort 2). This study showed that quizartinib is highly active and well tolerated as a single agent in patients with FLT3-ITD-positive, relapsed or refractory acute myeloid leukaemia and is active in some patients with FLT3-wild type acute myeloid leukaemia.
Implications of all the available evidence
FLT3-ITD is a driver mutation that presents with high leukaemic burden and is associated with poor prognosis. In these patient populations, treatment with quizartinib resulted in a high proportion of responders, including in elderly patients (cohort 1), and allowed approximately a third of patients to bridge to haemopoietic stem cell transplantation (cohort 2). These findings warrant further evaluation of quizartinib as an oral, highly potent, and selective next-generation FLT3 inhibitor, including in this setting and in combination with induction chemotherapy.
Acknowledgments
This study was sponsored, funded, and supported by Ambit Biosciences, which was acquired by Daiichi Sankyo in November, 2014. Research funding was provided by Daiichi Sankyo. This work was supported by the National Cancer Institute Leukaemia SPORE grant P50 CA100632 (to ML, HK, and JC) and Core Grant P30 CA016672 (to JC). Editorial assistance was provided by Mark Rocco of SciMentum and was funded by Daiichi Sankyo. The results of this study were previously presented at the American Society of Clinical Oncology Annual Meeting in 2012 and 2013, the American Society for Hematology Annual Meeting in 2011 and 2012, and the European Hematology Association congresses in 2011–13.
Footnotes
Declaration of interests
JC has received grants and personal fees from Astellas/Daiichi Sankyo during the conduct of this study; grants and personal fees from Astellas, Ariad, and Novartis; and grants from Arog Pharmaceuticals and Forma Therapeutics outside the submitted work. AEP has received personal fees and non-financial support from Ambit Biosciences/Daiichi Sankyo, Astellas US Pharma, Arog Pharmaceuticals, and Novartis outside the submitted work. HDoh has received grants and personal fees from Novartis and grants from Arog Pharmaceuticals outside the submitted work. BS has received other fees from Ambit Biosciences/Daiichi Sankyo during the conduct of this study and non-financial support from Novartis Pharma AG, Amgen GmbH, GlaxoSmithKline GmbH, PharmaMar, and Astellas Pharma GmbH outside the submitted work. HDom has received personal fees from Ambit Biosciences/Daiichi Sankyo during the conduct of this study; grants and personal fees from Roche/Genentech, Amgen, Ariad, Jazz Pharma, and Kite Pharma; and personal fees from Pfizer, Novartis, Celgene, Agios, Sunesis, Karyopharm, Menarini, Astellas, Janssen, Servier, and Seattle Genetics outside the submitted work. SS has received personal fees from Astellas, Daiichi Sankyo, Novartis, and Sunesis; and other fees from Alexion, Boehringer Ingelheim, CTI Biopharma, Baxalta, Sunesis, and Tolero outside the submitted work. JKA has received non-financial support from Northwestern University during the conduct of this study; personal fees from Immune Pharmaceuticals and Celgene; personal and other fees from Syros, Janssen Pharmaceuticals, NCCN, Novartis, Seattle Genetics, Spectrum, BMS, and Ariad; and other fees from MethylGene, Boehringer Ingelheim, Astellas, Agios Pharmaceuticals, CSL, Cyclacel, Epizyme, Genentech, Pfizer, BioLineRX, and Talon Therapeutics outside the submitted work. NR has received non-financial support from Merck Sharp & Dohme and personal fees from Jazz Pharmaceuticals, Sunesis, Novartis, and Pfizer outside the submitted work. NPS has received grants from Ambit Biosciences/Daiichi Sankyo during the conduct of this study. CCS has received grants from Astellas during the conduct of this study. GG and DT are former employees of Ambit Biosciences/ Daiichi Sankyo. DL has received personal fees from Daiichi Sankyo during the conduct of this study, and personal fees and other fees from Daiichi Sankyo outside the submitted work. ML has received grants from the National Cancer Institute, USA, during the conduct of this study and served as a consultant for Astellas, Daiichi Sankyo, Novartis, and Arog Pharmaceuticals outside the submitted work. HK, GM, TK, PR, EE, CDB, AB, AK, ESW, and NI declare no competing interests.
Contributor Information
Jorge Cortes, Department of Leukemia, The University of Texas MD Anderson Cancer Center, Houston, TX, USA.
Alexander E Perl, Division of Hematology and Oncology, Abramson Cancer Center of the University of Pennsylvania, Philadelphia, PA, USA.
Hartmut Döhner, Department of Internal Medicine III, Ulm University Hospital, Ulm, Germany.
Hagop Kantarjian, Department of Leukemia, The University of Texas MD Anderson Cancer Center, Houston, TX, USA.
Giovanni Martinelli, Istituto Scientifico Romagnolo per lo Studio e la Cura dei Tumori (IRST) Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS), Meldola, Italy.
Tibor Kovacsovics, Center for Hematologic Malignancies, Oregon Health & Science University, Portland, OR, USA.
Philippe Rousselot, Service d′Hématologie et Oncologie, Hôpital de Versailles, Université Versailles Saint-Quentin-en-Yvelines Paris-Saclay U1173, Le Chesnay, France.
Björn Steffen, Department of Medicine II, Hematology/Oncology, Goethe University, Frankfurt, Germany.
Hervé Dombret, University Hospital Saint-Louis, Assistance Publique-Hôpitaux de Paris (AP-HP), University Paris Diderot, Paris, France.
Elihu Estey, Seattle Cancer Care Alliance, University of Washington School of Medicine, Seattle, WA, USA.
Stephen Strickland, Vanderbilt-Ingram Cancer Center, Nashville, TN, USA.
Jessica K Altman, Department of Medicine, Division of Hematology and Oncology, Robert H Lurie Comprehensive Cancer Center of Northwestern University, Chicago, IL, USA.
Claudia D Baldus, Charité-Universitätsmedizin Berlin, Campus Benjamin Franklin, Hematology and Oncology, Berlin, Germany.
Alan Burnett, Department of Haematology, Cardiff University, Cardiff, Wales, UK.
Alwin Krämer, Klinische Kooperationseinheit Molekulare Hämatologie/ Onkologie, Medizinische Klinik V, Universität Heidelberg and German Cancer Research Center (DKFZ), Heidelberg, Germany.
Nigel Russell, Department of Haematology, Nottingham University Hospital, Nottingham, UK.
Neil P Shah, Department of Medicine, Division of Hematology and Oncology, University of California at San Francisco, San Francisco, CA, USA.
Catherine C Smith, Department of Medicine, Division of Hematology and Oncology, University of California at San Francisco, San Francisco, CA, USA.
Eunice S Wang, Department of Immunology, Roswell Park Comprehensive Cancer Center, Buffalo, NY, USA.
Norbert Ifrah, Service des Maladies du Sang, Centre Hospitalier Universitaire d′Angers, Angers, France.
Guy Gammon, Daiichi Sankyo, San Diego, CA, USA.
Denise Trone, Daiichi Sankyo, San Diego, CA, USA.
Deborah Lazzaretto, Daiichi Sankyo, San Diego, CA, USA.
Mark Levis, The Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University, Baltimore, MD, USA.
References
- 1.Patel JP, Gonen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 2012; 366: 1079–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levis M, Small D. FLT3: ITDoes matter in leukemia. Leukemia 2003; 17: 1738–52. [DOI] [PubMed] [Google Scholar]
- 3.Boissel N, Cayuela JM, Preudhomme C, et al. Prognostic significance of FLT3 internal tandem repeat in patients with de novo acute myeloid leukemia treated with reinforced courses of chemotherapy. Leukemia 2002; 16: 1699–704. [DOI] [PubMed] [Google Scholar]
- 4.Whitman SP, Maharry K, Radmacher MD, et al. FLT3 internal tandem duplication associates with adverse outcome and gene-and microRNA-expression signatures in patients 60 years of age or older with primary cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. Blood 2010; 116: 3622–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mrozek K, Marcucci G, Paschka P, Whitman SP, Bloomfield CD. Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukemia with normal cytogenetics: are we ready for a prognostically prioritized molecular classification? Blood 2007; 109: 431–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gale RE, Green C, Allen C, et al. The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood 2008; 111: 2776–84. [DOI] [PubMed] [Google Scholar]
- 7.Ravandi F, Kantarjian H, Faderl S, et al. Outcome of patients with FLT3-mutated acute myeloid leukemia in first relapse. Leuk Res 2010; 34: 752–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lazenby M, Gilkes AF, Marrin C, Evans A, Hills RK, Burnett AK. The prognostic relevance of flt3 and npm1 mutations on older patients treated intensively or non-intensively: a study of 1312 patients in the UK NCRI AML16 trial. Leukemia 2014; 28: 1953–59. [DOI] [PubMed] [Google Scholar]
- 9.Cancer and Leukemia Group B 8461, Farag SS, Archer KJ, et al. Pretreatment cytogenetics add to other prognostic factors predicting complete remission and long-term outcome in patients 60 years of age or older with acute myeloid leukemia: results from Cancer and Leukemia Group B 8461. Blood 2006; 108: 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klepin HD, Rao AV, Pardee TS. Acute myeloid leukemia and myelodysplastic syndromes in older adults. J Clin Oncol 2014; 32: 2541–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zarrinkar PP, Gunawardane RN, Cramer MD, et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood 2009; 114: 2984–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cortes JE, Kantarjian H, Foran JM, et al. Phase I study of quizartinib administered daily to patients with relapsed or refractory acute myeloid leukemia irrespective of FMS-like tyrosine kinase 3-internal tandem duplication status. J Clin Oncol 2013; 31: 3681–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burke JH, Ehlert FA, Kruse JT, Parker MA, Goldberger JJ, Kadish AH. Gender-specific differences in the QT interval and the effect of autonomic tone and menstrual cycle in healthy adults. Am J Cardiol 1997; 79: 178–81. [DOI] [PubMed] [Google Scholar]
- 14.Benton RE, Sale M, Flockhart DA, Woosley RL. Greater quinidine-induced QTc interval prolongation in women. Clin Pharmacol Ther 2000; 67: 413–18. [DOI] [PubMed] [Google Scholar]
- 15.Murphy KM, Levis M, Hafez MJ, et al. Detection of FLT3 internal tandem duplication and D835 mutations by a multiplex polymerase chain reaction and capillary electrophoresis assay. J Mol Diagn 2003; 5: 96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheson BD, Bennett JM, Kopecky KJ, et al. Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J Clin Oncol 2003; 21: 4642–49. [DOI] [PubMed] [Google Scholar]
- 17.Fischer T, Stone RM, Deangelo DJ, et al. Phase IIB trial of oral midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J Clin Oncol 2010; 28: 4339–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith BD, Levis M, Beran M, et al. Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood 2004; 103: 3669–76. [DOI] [PubMed] [Google Scholar]
- 19.Metzelder SK, Schroeder T, Finck A, et al. High activity of sorafenib in FLT3-ITD-positive acute myeloid leukemia synergizes with allo-immune effects to induce sustained responses. Leukemia 2012; 26: 2353–59. [DOI] [PubMed] [Google Scholar]
- 20.Randhawa JK, Kantarjian HM, Borthakur G, et al. Results of a phase II study of crenolanib in relapsed/refractory acute myeloid leukemia patients (Pts) with activating FLT3 mutations. Blood 2014; 124: 389 (abstr). [Google Scholar]
- 21.Cortes JE, Kantarjian HM, Kadia TM, et al. Crenolanib besylate, a type I pan-FLT3 inhibitor, to demonstrate clinical activity in multiply relapsed FLT3-ITD and D835 AML. Proc Am Soc Clin Oncol 2016; 34: 7008 (abstr). [Google Scholar]
- 22.Rydapt. Midostaurin: prescribing information. East Hanover, NJ: Novartis Pharmaceuticals Corporation, 2017 [Google Scholar]
- 23.Rydapt. Midostaurin: summary of product characteristics. Camberley, UK: Novartis Europharm, 2017. [Google Scholar]
- 24.Fathi AT, Chen YB. The role of FLT3 inhibitors in the treatment of FLT3-mutated acute myeloid leukemia. Eur J Haematol 2017; 98: 330–36. [DOI] [PubMed] [Google Scholar]
- 25.Perl AE, Altman JK, Cortes J, et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first-in-human, open-label, phase 1–2 study. Lancet Oncol 2017; 18: 1061–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanga M, James J, Marini J, Gammon G, Hale C, Li J. An open-label, single-dose, phase 1 study of the absorption, metabolism and excretion of quizartinib, a highly selective and potent FLT3 tyrosine kinase inhibitor, in healthy male subjects, for the treatment of acute myeloid leukemia. Xenobiotica 2017; 47: 856–69. [DOI] [PubMed] [Google Scholar]
- 27.Levis M, Ravandi F, Wang ES, et al. Results from a randomized trial of salvage chemotherapy followed by lestaurtinib for patients with FLT3 mutant AML in first relapse. Blood 2011; 117: 3294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giles F, O’Brien S, Cortes J, et al. Outcome of patients with acute myelogenous leukemia after second salvage therapy. Cancer 2005; 104: 547–54. [DOI] [PubMed] [Google Scholar]
- 29.Pratz KW, Sato T, Murphy KM, Stine A, Rajkhowa T, Levis M. FLT3-mutant allelic burden and clinical status are predictive of response to FLT3 inhibitors in AML. Blood 2010; 115: 1425–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith CC, Wang Q, Chin CS, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature 2012; 15: 260–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Galanis A, Levis M. Inhibition of c-Kit by tyrosine kinase inhibitors. Haematologica 2015; 100: e77–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carow CE, Levenstein M, Kaufmann SH, et al. Expression of the hematopoietic growth factor receptor FLT3 (STK-1/Flk2) in human leukemias. Blood 1996; 87: 1089–96. [PubMed] [Google Scholar]
- 33.Forman SJ, Rowe JM. The myth of the second remission of acute leukemia in the adult. Blood 2013; 121: 1077–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kurosawa S, Yamaguchi T, Miyawaki S, et al. Prognostic factors and outcomes of adult patients with acute myeloid leukemia after first relapse. Haematologica 2010; 95: 1857–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Armistead PM, de Lima M, Pierce S, et al. Quantifying the survival benefit for allogeneic hematopoietic stem cell transplantation in relapsed acute myelogenous leukemia. Biol Blood Marrow Transplant 2009; 15: 1431–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duval M, Klein JP, He W, et al. Hematopoietic stem-cell transplantation for acute leukemia in relapse or primary induction failure. J Clin Oncol 2010; 28: 3730–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sudhindra A, Smith CC. FLT3 inhibitors in AML: are we there yet? Curr Hematol Malig Rep 2014; 9: 174–85. [DOI] [PubMed] [Google Scholar]
- 38.Cortes JE, Tallman MS, Schiller G, et al. Results of a phase 2 randomized, open-label, study of lower doses of quizartinib (AC220; ASP2689) in subjects with FLT3-ITD positive relapsed or refractory acute myeloid leukemia (AML). Blood 2013; 122: 494 (abstr). [Google Scholar]
- 39.Levis MJ, Cortes JE, Gammon GM, Trone D, Kang D, Li J. Laboratory and clinical investigations to identify the optimal dosing strategy for quizartinib (AC220) monotherapy in FLT3-ITD-positive (+) relapsed/refractory acute myeloid leukemia (AML). Blood 2016; 128: 4042 (abstr). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.