

Abstract
In the last decade, nanoparticles (NPs) have become a key tool in medicine and biotechnology as drug delivery systems, biosensors and diagnostic devices. The composition and surface chemistry of NPs vary based on the materials used: typically organic polymers, inorganic materials, or lipids. Nanoparticle classes can be further divided into sub-categories depending on the surface modification and functionalization. These surface properties matter when NPs are introduced into a physiological environment, as they will influence how nucleic acids, lipids, and proteins will interact with the NP surface. While small-molecule interactions are easily probed using NMR spectroscopy, studying protein-NP interactions using NMR introduces several challenges. For example, globular proteins may have a perturbed conformation when attached to a foreign surface, and the size of NP-protein conjugates can lead to excessive line broadening. Many of these challenges have been addressed, and NMR spectroscopy is becoming a mature technique for in situ analysis of NP binding behavior. It is therefore not surprising that NMR has been applied to NP systems and has been used to study biomolecules on NP surfaces. Important considerations include corona composition, protein behavior, and ligand architecture. These features are difficult to resolve using classical surface and material characterization strategies, and NMR provides a complementary avenue of characterization. In this review, we examine how solution NMR can be combined with other analytical techniques to investigate protein behavior on NP surfaces.
Keywords: Protein surface interactions; NMR relaxation; surface structure, biomolecular corona
1. Introduction
Nanoparticles (NP) have been used to advance medicine and biotechnology, with applications ranging from clinical diagnosis, drug delivery, and biosensing.[1] Nanoparticle composition can vary significantly from metals to inorganic semiconductors.[2] In addition, there are a variety of surface modifications available depending on the desired physicochemical and biological activities. Identifying nanoparticle interfaces that facilitate both specific and non-specific interactions in biological media is a daunting task.[3–4] Nano-bio assemblies require the use of various analytical approaches to gain fundamental insight on these novel materials. Furthermore, when NPs are introduced into biological media composed of nucleic acids, lipids, and proteins will spontaneously interact forming a biomolecular corona.[5] Due to these phenomena, and given the broad medical applications of NPs, understanding NP-protein interactions is of paramount importance. It is hypothesized that transfer of a protein from aqueous medium to the NP corona can affect protein’s conformation and motions, which in turn can interfere with the NPs surface properties. Nanoparticle curvature is prone to induce conformational changes in both the secondary and tertiary structures of many proteins.[6–8] For example, Satzer et al. used circular dichroism (CD) spectroscopy to determine that α-helical content for both myoglobin and bovine serum albumin (BSA) decreased significantly in the presence of 150 nm gold nanoparticles (AuNPs).[9]
Different analytical tools have been employed to characterize biocorona formation on NP surfaces. These techniques include surface plasmon resonance (SPR),[10–13] transmission electron microscopy (TEM),[5,14–17] mass spectrometry (MS) based proteomics,[18–20] chromatography,[21] fluorescence spectroscopy,[22–26] CD spectroscopy,[6,26–28] electrophoresis (gel, capillary, and 2D electrophoresis),[13,29–30] dynamic light scattering (DLS),[16,23,31–32] isothermal titration calorimetry (ITC),[33–36] infrared spectroscopy (FTIR),[37–38] and nuclear magnetic resonance spectroscopy (NMR).[39–43] These techniques have been used to identify the structural and conformational changes of various proteins onto the NP surface.
Solution NMR spectroscopy has proven to be an outstanding tool to understand conformation, orientation, and dynamics of the molecules involved in biocorona formation on various NP surfaces, including AuNPs and silica nanoparticles (SiNPs). Also, solid-state NMR has been used for structural determination of peptides on hydroxyapatite surfaces.[44] However, solid-state NMR studies of the NP biocorona are less frequently found in the literature.
Solution NMR spectroscopy is unique among analytical techniques, because of its ability to probe local motions in the time frame of picoseconds to hours.[45] Effects of intermolecular interactions can be measured with high precision and mapped onto protein structures to understand the interaction sites of the protein.[46] Additional solution NMR experiments can be used to extract information about the protein, including steady-state kinetics, structural thermodynamics, and diffusion constants. Solution NMR techniques are generally limited by the sample’s molecular size (<35 kDa), though relaxation optimized NMR methods have opened the door to complexes reaching 1 MDa.[47] Nevertheless, acquiring information on NP-protein interactions using NMR is extremely challenging. In the past few years, NMR studies have contributed to new insights in the structure and function of biomolecule-NP conjugates. This review aims to summarize recent work on NP bioconjugates and explain how NMR can be used to understand the fundamentals of NP-protein interactions.
2. Non-NMR Approaches for Nanoparticle Studies
Under physiological conditions, biological molecules spontaneously adsorb onto NP surfaces.[48] This in turn changes the surface properties of the NP by the formation of a biocorona.[49] Understanding the biophysical characteristics of the NP biocorona can lead to new applications in the biological and medical fields. The biocorona consists of proteins, carbohydrates, lipids, and nucleic acids that either loosely (soft corona) or tightly (hard corona) associate with the NP surface. Measurement of binding affinities, binding capacities, association and dissociation rates, and stoichiometries of these biomolecules are needed for a complete understanding of the corona’s properties. Different analytical techniques have been employed to study NP-protein interactions, specifically UV-Vis, MS, DLS, ITC, and CD. Each technique has unique advantages and disadvantages when investigating NP-protein interactions.
2.1. Spectroscopic Approaches for Characterizing Binding (UV-Vis, Fluorescence, Raman, and CD)
In UV-Visible (UV-Vis) spectroscopy, the localized surface plasmon resonance (LSPR) phenomenon is used to characterize the metallic NPs, such as AuNP and silver nanoparticles (AgNPs), and its conjugates.[26,42,50–54] LSPR is only present in plasmonic NPs and cannot be used in non-metallic systems, such as liposomes and SiNPs. The LSPR peak shape and size is caused by a collective oscillation of free electrons of the metallic particle. A shift and broadening of the absorption spectra for the NP-protein complex will depend on the bioconjugate size, aggregation state, and the local dielectric environment.[52,55] Due to this phenomenon, UV-Vis is widely used to quantify metallic NPs and qualitatively measure conjugate binding. In a previous study, α-synuclein was titrated with various concentrations of AuNPs, and the plasmon peak shift (Δλ) versus α-synuclein concentration was graphed.[56] A Langmuir isotherm model was fit to the data to extract the equilibrium association constant (Ka=7.9±1.1 × 106). UV-Vis is a noninvasive method, where the integrity of the sample is not compromised, and is an inexpensive technique that requires little sample preparation. The major disadvantage of UV-Vis is the adsorption spectrum is highly influenced by solvent, pH, temperature, and high electrolyte concentration. Moreover, very little can be learned about biomolecular structure using UV-Vis spectroscopy alone.
Fluorescence, Raman, and CD spectroscopy can be effectively used to detect NP-protein binding.[26,55,57–64] These techniques’ advantage is that they do not require plasmonic NP systems. However, several factors can complicate each of these methods. For example, fluorescence spectroscopy typically requires natural fluorophores, such as tyrosine or tryptophan in proteins. [24,26,55,57–59,62–69] If these are not present, cysteine or amines can be labeled with fluorescent probes, such as fluorescein, to study their structural and dynamic properties.[70] Fluorescence correlation spectroscopy (FCS), a technique that measures the fluorescence bursts emitted by particles diffusing through small volumes, has been used to quantitatively study human serum albumin (HSA) adsorbed to polymer coated FePt and CdSe/ZnS nanoparticles. Autocorrelation analysis of fluorescence emission yields a characteristic diffusion time scale (τD) corresponding to the hydrodynamic radius (RH) of the NP bioconjugates.[23] Even though fluorescence based approaches are highly sensitive, the main disadvantage is the addition of fluorescent probes that can alter the NP-protein interaction. Also, the inner filter effect (IFE) and light scattering from the proteins or NPs may complicate the interpretation of fluorescence experiments.[71–74]
Some of the drawbacks of fluorescence spectroscopy are avoided by Raman spectroscopy, a non-destructive, label-free, highly specific technique that has a distinct fingerprint for solid and liquid solutions.[75–76] Raman spectroscopy measures the NP bioconjugate in aqueous solutions with great spectral resolution. Surface enhanced Raman spectroscopy (SERS) has improved the measurements of Raman spectroscopy with higher sensitivity and higher selectivity of chemical groups. SERS has been used to determine the structural and conformational changes of protein on metal NP surfaces.[76–80] For example, Szekeres et al. used SERS to identify structural changes in BSA molecules when attached to AuNPs, finding that BSA undergo structural changes which depends on the BSA concentration.[78] Apart from protein conformation data, the morphological changes in AuNP when interacting with protein are also detected through SERS spectra. One drawback is the intense laser heating the NP-protein conjugate, which can alter the structure and conformation, giving rise to misleading results.
Though neither fluorescence nor Raman spectroscopy can detect secondary or tertiary structure changes in proteins, CD is used extensively to determine the secondary structure of proteins and how these structures change upon binding to NP surfaces.[28,81] To exhibit a CD signal, a molecule must be chiral; however, NP surfaces are not typically chiral and will not generally interfere with signal and interpretation of data. In a previous study done by Deng et al., poly (acrylic acid)-coated gold nanoparticles (PAA–AuNP) were titrated with fibrinogen, a large protein that consists of both α-helices and β-sheets, and far-ultraviolet circular dichroism (UV-CD) was used to monitor protein structural changes upon NP interaction.[28] Indeed, fibrinogen secondary structure was lost with the addition of PAA-AuNP indicated by the progressive increase in ellipticity. They concluded that PAA-AuNP induced structural changes and exposed its C-terminus.[28] As a drawback, UV-CD provides only a rough estimation of conformational changes, since the unbound (native) protein is typically left in the cuvette when the NP-bound protein is measured, and the unbound protein often dominates the observed signal. Separating NP-bound protein by centrifugation, or performing a difference measurement are viable alternatives, but the signal originating solely from NP-bound proteins is often very weak.[37,54,64,82]
2.2. Non-Spectroscopic Approaches for Characterizing Binding (Light Scattering, Chromatography, Mass Spectrometry, and Calorimetry)
Other, non-spectroscopic approaches are also useful, and include dynamic light scattering, chromatography, mass spectrometry, and isothermal titration calorimetry. Dynamic light scattering (DLS) is a widely used technique to determine the hydrodynamic size of NPs in suspension.[16,83–84] DLS measures the scattering intensity fluctuations caused by the Brownian motion of NPs in solution and uses the Stokes-Einstein equation to relate the diffusion coefficient to the NP size. The measured hydrodynamic diameter reflects the dimensions of the NP as well as the corona layer bound to the NP surface in solution.[16] The hydrodynamic radius measured by DLS can also be used to determine the binding ratio of protein to NP. Woods et al. studied the monolayer formation of BCA and GB3 on AuNP using DLS.[85] The size increment when both proteins adsorbed to AuNP was similar to the values observed by TEM and the values predicted by the native protein structure.[85] DLS is a nonperturbative, fast, and accurate method, but the disadvantage is the need of a dust free and dilute sample that has a monodisperse population. Also, this method suffers from low sensitivity toward small particles and possible interference from light-absorbing species.
Light scattering can be extended in several ways, and two approaches in particular have been applied to nanoparticle-biomolecular interactions. Many DLS systems are capable of measuring electrophoretic light scattering, from which zeta potential can be determined.[80,86–87] Nanoparticles in solution have an electrostatic charge on their surface, and the zeta potential measures this potential of the electric double layer relative to the bulk solution. It is therefore related to surface charge, and particles with larger zeta potentials tend to form more stable suspensions.[88] Biomolecules in solution can adsorb onto the NP surface, changing the surface potential and nanoparticle stability. Dobrovolskaia et al. examined how NP size affected longevity in the bloodstream upon binding to plasma.[16] AuNPs of size 30 and 50 nm were incubated with plasma, which increased the zeta potential from −38.2 mV to −16.4 mV. A similar change in zeta potential was observed for 50 nm AuNP, and both changes corresponded to an increase in hydrodynamic radii as blood proteins were adsorbed. An alternative application to light scattering involves the coupling of static, multiangle light scattering (MALS) with chromatography. MALS differs from DLS in that scattering intensity is measured at several discrete angles. In ideal cases, both Mr (the relative molar mass) and the radius of gyration of nanoparticle can be determined,[89] and changes can be monitored in the presence and absence of biomolecules.[90] Overall, applications of light scattering have led to powerful tools for characterizing NPs, although relating MALS, DLS, and zeta potential protein tertiary structure on a NP surface can be very challenging.
Chromatographic methods, such as size exclusion chromatography (SEC) or gel filtration chromatography, are frequently used to separate complex mixtures of biological compounds based on their size. Gel filtration chromatography has been used to detect protein bound to NPs and determine the exchange rate by comparing the bound versus free protein elution profiles.[33,91–92] Cedervall et al. introduced a gel filtration-based method to both identify proteins attached to NPs and the exchange rate of plasma proteins.[93] Separation is based on the fact that NPs are too big to enter the pores of gel filtration media and will appear in the void volume. However, the free proteins are small enough to enter the pores and separate according to their molecular weight. One disadvantage is that, if the NP-protein interactions are weak, the large dilutions can disturb the NP-protein equilibrium. There are also inherent drawbacks due to the sensitivity of the SEC, including lower precision, accuracy, and longer experiment times. Another potential pitfall is the interaction of NPs with the stationary phase of the column.[94] In this case, an alternative approach, asymmetrical flow field-flow fractionation (AF4), can be employed. AF4 has no stationary phase, and a semipermeable membrane is used instead. During fractionation, a cross-flow is applied through the membrane, creating a concentration gradient that separates molecules based on their molecular size. Adsorption to the membrane can be problematic, but AF4 is thought to be more gentle than SEC,[95] and several excellent reviews discuss the strengths and weaknesses of AF4 applied to NPs.[95–97] Like SEC, AF4 can be used to separate complex mixtures of proteins and NPs before downstream analysis. When coupled with DLS or MALS, SEC and AF4 can be an extremely powerful tool for characterizing dispersions of polydisperse NP solutions.
Electrophoresis is another useful technique to separate complex NP-protein mixtures that provides qualitative and quantitative analysis. Polyacrylamide gel electrophoresis (PAGE) is one of the most widely used methods to separate NP-protein complexes.[3,58,93,98–100] Macromolecules are differentiated according to their electrophoretic mobility, which is a function of the molecule’s length, conformation and charge. For proteins, sodium dodecyl sulfate (SDS) is used to denature proteins and give them a uniform charge/size ratio. García et al. used SDS-PAGE gel electrophoresis to identify the adsorbed proteins onto several different classes of AuNPs after incubation with fetal bovine serum.[101] The data suggested that cationic and anionic AuNPs adsorbed a larger amount of proteins compared to AuNPs coated with polyethylene glycol (PEG) and glycans. Even though SDS-PAGE is an effective tool to identify the composition of the protein corona, it suffers from poor protein separation if the protein mixture is too complex resulting in comigration of several proteins with similar size.
Mass spectrometry (MS) is a high throughput, sensitive analytical technique used to monitor larger proteins (up to ~100 kDa) interacting with NPs. The two main ionization methods used to investigate biomolecules are matrix-assisted laser desorption ionization (MALDI) and electrospray ionization (ESI). Protein samples are often digested using proteolytic enzymes into smaller fragments that are more suitable for the mass range of instruments. MS can provide both qualitative and quantitative information regarding the protein mixtures present on NP surfaces, and MS can be used in parallel with chromatographic and gel-based methods to identify the composition of the protein corona.[3,65,93,100,102–104] Importantly, the use of protein fragments enables one to investigate structural questions on the NP surface. For example, Shrivastava et al. identified that cytochrome c had a specific orientation on SiNPs using post translational modification and MS. Cytochrome c lysine residues were acetylated in solution after complete protein saturation of the SiNP surface. Then, a pepsin digest was used to identify and compare the intensity ratios from MALDI-TOF (time of flight) for acetylated cytochrome c free and bound states in solution. Results showed that the lysine residues bound to SiNP surface had a lower intensity, resulting from the steric occlusion of lysine residues during acetylation when bound to SiNPs. Even though MS is a destructive method, it provides qualitative and quantitative values that reflect the protein abundance in the protein corona, and it often requires comparatively much smaller amounts of sample.
Isothermal titration calorimetry (ITC) can be used to directly measure the enthalpy change when proteins interact with NPs.[33,35–36,64] In general, to measure the enthalpy change, the protein of interest is gradually added to a solution containing NPs and the evolved heat of binding is measured. These heats are calculated using the power required to maintain isothermal conditions. Fitting a thermodynamic model to the heats produces parameters like stoichiometry and the enthalpy of binding. For example, Cedervall et al. added HSA into copolymer nanoparticles to measure the stoichiometry, affinity, and enthalpy of the NP-protein interaction.[93] The equilibrium association constant was calculated to be 2×106 for the interaction between HSA and the NP surface.[33] They effectively used ITC to demonstrate the thermodynamic nature between NP-protein complex formation and protein conformational changes upon binding. Several drawbacks exist when using ITC to study NP-protein binding. For one, the method requires high sample concentrations (0.1–1 mM) and volumes (~1 mL). An additional challenge is that adsorption may not produce a measurable heat, even when NPs are quite concentrated. Finally, if multiple steps occur, e.g. binding and unfolding, ITC thermograms may be challenging to interpret. Despite these challenges, ITC remains a useful tool for understanding the strength of biomolecule-NP interactions.
In summary, each of the analytical methods mentioned above has their own advantages and disadvantages that coincides with the physical properties being measured. In the next section, we discuss how NMR spectroscopy can complement the techniques described above. NMR has many unique advantages, as it is non-destructive and uses relevant aqueous solvents for biological samples. NMR techniques can be used to monitor fine structural changes, owing to its ability to probe individual residues. This information is particularly useful when a molecule is interacting with a ligand or NP. Finally, NMR can be applied to a wide variety of samples in the solution and the solid-state, providing both direct and indirect measurements of molecular dynamics.
3. Solid-State NMR Approaches for Nanoparticle Studies
While the focus of the current review is solution NMR, solid-state NMR is an emerging analytical tool for understanding protein behavior on NP surfaces. Despite this, it has been infrequently used thus far to study NP-protein interactions.[41] High-resolution solid-state NMR techniques, both newly developed and traditional, can provide structural and motional details at the atomic and molecular level. The interfacial interactions revealed by NMR are straightforward, and interpretations are separated into chemical and physical principles that underlie the binding of the molecules to the surface. Solid-state NMR can be used to extract data from biomolecules directly interacting with mesogenic and amorphous solids, where conventional methods, like X-ray crystallography and solution NMR, fail. One drawback using solid-state NMR is its inherently low sensitivity (though dynamic nuclear polarization methods to boost signal are rapidly advancing),[105] and its requirement that surface adsorption should be relatively long-lived. Long et al. studied the structural and dynamic characterization of salivary statherin adsorbed to hydroxyapatite surface using solid-state NMR. This data suggested the interaction of statherin was mediated by the N-terminus, where two negatively charged phosphoserines and three carboxylate-containing side chains were located.[44] In a similar study, ultrafast magic-angle-spinning (MAS) 1H NMR and multinuclear and multidimensional (13C and 15N) NMR were used to determine the structure of the large protein assembly conjugated to PEGylated AuNP.[106] They focused on the E. coli asparaginase II (ANSII) protein, which was covalently tethered to AuNPs. Two-dimensional 1H-15N CP-HSQC (cross-polarization heteronuclear single-quantum coherence) solid state NMR spectra were collected for both free ANSII and bound to PEGylated AuNPs. The spectra were superimposable indicating the retention of native structure on the AuNP surface. Chemical shift perturbation analysis of the 2D 1H-15N CP-HSQC spectrum of ANSIIAuNPs revealed that the largest variations involved the residues located on the protein surface or on the loops. Even though the covalent tether was in a sense artificial, these data suggest heteronuclear solid-state NMR spectra can be effectively used to understand the structural and dynamics of biomolecules on NPs.
In another study, Bower et al. employed solid state NMR to obtain the structure of surface-immobilized peptide (LKα14) onto AuNPs.[107] These peptides covalently bind to alkanethiolates and self-assemble as monolayers on the colloidal AuNPs. Ramachandran angles (ϕ and ψ) for the immobilized peptides were determined by measuring the distances between backbone carbonyl 13C spins. A double-quantum filtered dipolar coupling with a windowless sequence (DQDRAWS) experiment was used, and additional information was obtained by determining the relative orientation of chemical shift anisotropy tensors of 13C carbonyl spins on adjacent peptide planes. Solid state NMR structural measurements indicated a slight conformational change in the backbone torsion angles when the peptide adsorbed onto the NP surface. This change might be a consequence of the peptides adsorbing on an immobile, two-dimensional surface rather than interacting with other flexible peptides to reach an energy minimum. They concluded that the LKα14 peptide adopts helical structure on the NP, because this conformation exposes the maximum number of lysine residues for attachment. While not widely used in NP-protein interaction studies, solid state NMR has great potential to reveal the structural details of surface adsorbed proteins, and advances like dynamic nuclear polarization[108] may accelerate the development of solid state NMR in this area.
4. Solution NMR Approaches for Nanoparticle Studies
Solution NMR spectroscopy exploits the non-zero spin properties of nuclei, mainly 1H, 13C, and 15N, to determine the chemical shifts, relative intensities, and linewidths for NMR-visible spin systems. These parameters depend on the dynamics and conformation of the free and bound macromolecule (often proteins). The local chemical environment for each nucleus results in a unique chemical shift, whereas the signal intensity depends on the number of nuclei resonating at that chemical shift. Finally, linewidth is related to how quickly the nuclei relax (R2, s−1). R2 is influenced by many factors, including protein rotational correlation time (τc), local motion, and chemical (e.g. conformational, binding) exchange. When nanoparticles are added to a solution containing protein, the protein signals are expected to be perturbed depending on the nature of the NP-protein interaction. The most significant effect happens when rotational diffusion is slowed. This occurs when large NP-protein assemblies are formed, resulting in decreased signal intensity and significant line broadening (Figure 1).
Figure 1.
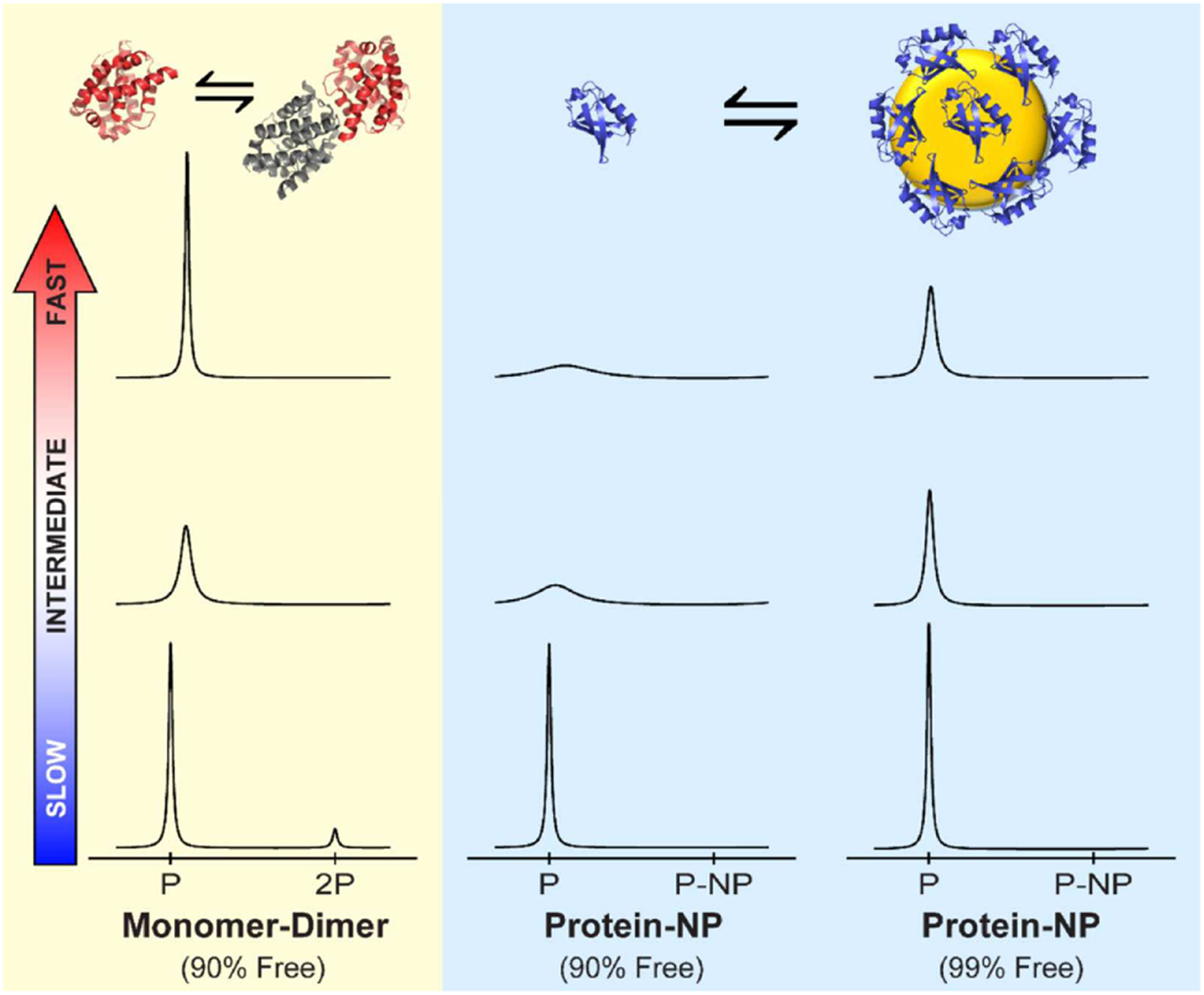
Comparison of exchange between a protein binding to a similarly-sized molecular target traditional protein-protein binding and nanoparticle-protein binding. For a monomer (P) to dimer (2P) exchange (yellow background, left panels), individual NMR signals are observed for both species in the slow exchange regime (bottom), and a weighted average signal is observed for fast-exchanging species (top). Intermediate exchange broadens the signal. Nanoparticle binding differs because of extremely slow rotational correlation time (τc) in the bound state (blue background, center and right panels). For example, protein τc values typically range from 2–20 ns, whereas an 80 nm (diameter) NP has a τc of approximately 105 ns, and τc for a 10 nm NP is 100 ns. In the slow exchange limit, binding to a NP decreases the signal intensity as before, but no signals are observed from the nanoparticle-bound species because of its slow rotational correlation time (bottom middle and bottom left panels). As the conformational exchange rate increases, the nanoparticle-bound state continues to broaden the free protein signals, complicating detection. With 10% of protein bound, the signal becomes undetectably broad in the fast exchange limit (top middle). Even with only 1% of bound protein, the effect on the free protein signal is substantial (top right). Spectra are simulated for 15N resonances at 800 MHz using 20 nm NPs in water for kex = 2.5 s−1 (slow), kex = 2.5×103 s−1 (intermediate) and kex = 2.5×106 s−1 (fast).
In the context of nanoparticle binding, chemical exchange occurs when association with the nanoparticle causes a modulation in a nuclei’s chemical environment. The change in environment will generally induce both a change in chemical shift (|Δν|) and a change in relaxation, because proteins may alter their conformation and because nanoparticle τc values are significantly greater than those for soluble proteins. A two-state model is often used to describe binding, and for fixed NP and protein concentrations, the on and off rates are each associated with a rate constant, and koff. Because the concentration of NP binding sites is often ambiguous, the exchange rate kex is defined as , where is an apparent first order rate constant. Traditionally, chemical exchange is classified into three categories: slow (kex ≪ |Δν|), intermediate kex ≈ |Δν|), and fast (kex ≫|Δν|). In NP systems, and additional contribution – lifetime line broadening – is also present, distinct from the line broadening caused by chemical exchange on the intermediate chemical shift time scale.[109] This lifetime line broadening occurs because the NP relaxation rate is extremely fast, owing to the size of the NP and its slower rotational correlation time (the τc of a NP can be hundreds of ns or more, whereas protein τc values typically range from 2–20 ns). Lifetime line broadening is different from intermediate chemical shift timescale exchange broadening because it can occur even when there is no chemical shift change (Δv = 0). Under these conditions, the exchange regime is determined by the difference in relaxation rates, where and are the R2 rates of the NP-protein complex and the free protein, respectively. Then, slow exchange is intermediate exchange is , and fast exchange is .109 Under slow exchange (typically s–ms), separate signals are observed for the two states, but the apparent R2 of the small, easily visible state will be the sum of intrinsic R2 of that state plus As the rate increases, the bound and free signals gradually broaden and coalesce under intermediate exchange. Eventually, the average signal sharpens as exchange becomes fast. The exchange contribution – either from chemical exchange or lifetime line broadening – can be quantified in part by measuring the difference in relaxation rate in the presence and absence of nanoparticles, where the observed relaxation rate with NPs present is and is defined as . The observed difference in relaxation rates () will have different functional forms depending on whether exchange is fast, intermediate, or slow. This behavior has been discussed extensively for binding of two protein-sized macromolecules.[109–110]
Figure 1 (left column) demonstrates exchange graphically for the dimerization of two similarly-sized protein species. Under slow exchange, when the dimeric form is populated at 10% total protein, a small but visible signal is observed corresponding to the dimer (bottom panel). This species will have a somewhat broader linewidth, resulting from the increased size of the dimeric species. The peak corresponding to the monomer will be quantitatively reduced in proportion to the amount of dimeric species. Moreover, the integral of each peak will be strictly proportional to the number of molecules (nuclear spins) in the monomeric and dimeric states. Intermediate and fast exchange (upper two panels) have similar features for a 10% dimer population: only a single peak is observed. However, in intermediate exchange (middle left panel), the monomeric peak broadens significantly because of the dimer-monomer exchange, even though the dimer represents a small fraction of the ensemble. In fast exchange, the average peak sharpens and has shifted according to the population weighted chemical shift (top left panel).
The picture described above changes significantly when one of the binding partners is a nanoparticle. The central column in Figure 1 demonstrates the behavior when a protein binds to a 20 nm nanoparticle (10% protein bound). Under slow exchange, the bound species peak is not observed at all due to the extremely slow rotational diffusion of the nanoparticle. Using a non-interacting reference, one can still quantify the amount of bound protein by measuring the diminished peak intensity (bottom center panel).[111] However, direct observation of the nanoparticle-associated species is impossible because of the nanoparticle’s large size. As the exchange rate increases from slow to fast, the free protein peak becomes significantly broader, eventually becoming impossible to observe (top center panel). Because of the large difference in R2 values between the bound and free states, very little nanoparticle binding is needed in the fast exchange regime to elicit a significant effect. The rightmost column in Figure 1 shows the effect when only 1% of protein is in the nanoparticle-associated state. While the change is imperceptible under slow exchange, the large nanoparticle R2 causes significant broadening under fast exchange, far more than would be expected for a protein binding to a similarly-sized molecular target.
Different nanoparticle surfaces exhibit different exchange behaviors. In our experience, citrate-coated spherical AuNPs always exhibit extremely slow exchange, with no line broadening observed.[112] On the other hand, other nanoparticle chemistries, like SiNP exhibit faster exchange, leading to a marked increase in the observed protein R2 values.[113–115] Either way, the surface-associated protein is not directly detectable, because it experiences roughly the same τc (i.e. extremely slowly tumbling) as the bare nanoparticle. In the remainder of this review, we will discuss how solution NMR spectroscopy can be used to probe the nanoparticle-associated state indirectly. In general, one of two strategies are used to accomplish this. For slowly exchanging nanoparticle surfaces, protein concentrations are chosen so that a significant fraction of the total protein concentration is associated with the nanoparticle (Figure 1, bottom center panel). Provided that , bound protein can be quantified using the reduction in the non-associated signal, and the protein can be displaced using small molecules (e.g. mercaptobenzimidizole for AuNPs).[111] For rapidly exchanging nanoparticle surfaces, conditions are chosen so that only a small fraction of the total protein concentration interacts with the nanoparticle. Then, relaxation behavior and other properties are used to study the bound state,[43,116–119] offering the possibility of performing numerous experiments that can address a variety of features in studying NP-protein interactions.
As an example, we have measured 15N R2values for the small GB3 protein using 15 nm, citrate-coated AuNPs and 30 nm spherical SiNPs (Figure 2). For this small, globular protein, all residues exhibit similar behavior. 15N CPMG experiments for residue 24 reveal a nearly flat response with no apparent relaxation dispersion for either AuNPs or SiNPs. For GB3, which is in slow exchange with the AuNP-bound state, no dispersion is observed () because only the free state is visible, and there is effectively no exchange on the timescale of the NMR experiment.[112] This is confirmed by the behavior of GB3 in the absence and presence of AuNPs: the slow exchange behavior results in nearly identical R2 values in the presence and absence of AuNPs (Figure 2A). For SiNPs, different behavior is observed (Figure 2B). While the relaxation dispersion profile is still flat, the difference in R2 values with and without NPs is significant (), even when the nanoparticle concentration is extremely small relative to the protein concentration (500 μM GB3 and 0.25 μM SiNPs). The is large because the protein experiences exchange, and therefore the observed value is influenced by lifetime line broadening (compare to Figure 1, top right panel).[110] For GB3 on SiNPs, the relaxation dispersion profile is flat as the large nanoparticle R2 precludes refocusing at any of the applied CPMG frequencies. This is in stark contrast to GB3 on AuNPs, where the profile is flat because exchange is very slow.
Figure 2.

The CPMG and DEST profiles for 15N GB3, residue E24. GB3 CPMG profiles binding with (A) AuNP surface and (B) SiNP surface. No relaxation dispersion is observed for GB3 on either AuNPs or SiNPs, as evidenced by the flat response of R2 versus CPMG frequency. (C) 500 μM GB3 DEST profile binding with 0.25 μM SiNPs. Two saturation fields are used, 500 Hz (red points) and 350 Hz (blue points). These saturation fields selectively saturate the resonances of the NP-bound protein, resulting in a decrease in the intensity of observable resonances of the protein free in solution. This decrease occurs because the nanoparticle-bound spins are in chemical exchange with the observable spins in solution. The saturation behavior can be fit (red and blue lines) to a continuous function to determine the kinetic rate constants for association and dissociation. In this case for protein association, is 4 s−1 and koff is 19.8 s−1.
4.1. 1D Proton NMR, Quantitative NMR, and Half-Filter Experiments
In 1D proton (1H) NMR, the protein amide and aliphatic regions are clearly distinguishable. This signal can be used to quantify the bound protein concentration to NP when the interaction is in slow regime, where line broadening is not observed in the presence of NPs. Protein (typically at concentrations of 20–50 μM) is titrated with various concentrations of NPs to identify how the protein interacts with NP using 1D NMR. The 1D 1H signal intensity decreases as the NP concentration increases. The signal loss can be directly related to the bound protein concentration,[111] which increases as the NP concentration increases. When the bound protein concentration is plotted against the total NP concentration, one can estimate the binding capacity of the NP, provided that binding is sufficiently tight (Kd <1 μM).[111,120] Binding capacity reflects the total number of proteins bound to the nanoparticle, regardless of whether the proteins form a single layer or multiple layers on the NP surface, and regardless of whether the protein unfolds on the surface. This is true because, in the slow exchange regime (Figure 1, bottom panels), the free peak simply represents the protein fraction not associated with the NP. Normally, nothing can be understood about directly the surface-bound protein; however, using this novel, yet simple method, we found a good agreement between the binding capacity measured by NMR and the binding capacity predicted assuming a folded monolayer for six different proteins on a 15 nm AuNP.[111]
1D half-filter experiment[121] (Figure 3A) can be used to extend 1D 1H quantitation to monitor the competitive binding of multiple proteins to a NP surface. NMR spectra are obtained, and the half-filter NMR experiment is used for signal filtering of 15N and 13C attached protons. This experiment requires that proteins be isotopically enriched with either 13C or 15N, but not both. For each data point, three spectra are recorded: one with no filter (used for comparison with a reference standard, either external or internal), one with a 13C filter (used to select 13C-attached protons), and one with a 15N filter (used to select 15N-attached protons). Relative concentrations are calculated with respect to the standard, which can be compared to a sample with no NPs in order to obtain absolute concentrations. This method was used to measure the competition between the wild-type (WT) GB3 protein and all of its lysine to alanine variants, allowing an investigation of which lysine residues matter most in NP binding.[39] The advantage of the 1D half-filter experiments is its ability to detect two protein-NP mixtures in situ in the same sample. However, its limitation is that it requires distinct isotopic labeling with good peak dispersion, making it challenging to investigate complex mixtures of three or more proteins.
Figure 3.

(A) The use of 1D half filter experiment to quantify binding of two proteins in same solution with AuNPs. GB3 (13C labeled) and ubiquitin (Ubq, 15N labeled) are in the same solution, and 80 nM AuNPs are added. The 15N and 13C filter can be applied to differentiate proton signals originating from each protein independently, and quantitative NMR approaches can be used to measure binding. The residual water signal has been removed for clarity. (B) Backbone amide 1H-15N heteronuclear single-quantum coherence (HSQC) experiment to quantify two proteins isotopically labeled the same in the nanoparticle solution. 15N labeled GB3 and 15N labeled Ubq are mixed in the same solution with 80 nM AuNP. The peak intensity perturbation measured for 25 μM GB3 and 25 μM Ubq in the presence of AuNP (I, red spectra) compared to the absence of AuNP (Io, blue spectra). (C) The 1H-15N HSQC experiment can be used to determine the binding capacity for each protein by plotting the bound concentration versus various AuNP concentrations. The HSQC spectral intensities are used to determine the bound concentration relative to an external standard. Here, 20 μM GB3 is mixed with 0, 20, 40, 60, and 80 nM AuNP to identify how much is bound to the NP surface. Observed concentrations for GB3 (blue circles) are plotted against the expected values for a folded monolayer of protein on the NP surface (black circles and solid line).
4.2. Two-Dimensional NMR Methods
The heteronuclear single-quantum coherence spectrum (1H-15N HSQC) of a protein can be regarded as the fingerprint for the protein backbone.[122] By monitoring 2D NMR spectra, one can identify perturbations in backbone amide nitrogen and proton chemical shifts, linewidths, or intensities upon binding to a NP. These perturbations are subject to the effects shown in Figure 1, and a range of behaviors can be observed depending on the timescale of exchange and the size of the NPs. To date, 2D NMR has been used to identify and map residues involved in binding to NP, as well as identify the orientation and conformational changes of proteins upon binding (vide infra). HSQC is widely used to study proteins up to 35 kDa,[123] and transverse relaxation optimized spectroscopy (TROSY)[124] can be used to extend this size limitation up to 70–80 kDa or even larger.[125] However, even with protein perdeuteration methods,[126] backbone and methyl TROSY methods are not currently able to overcome the relaxation challenges encountered by proteins on NP surfaces. In addition, care must be taken to ensure that any chemical shift perturbations observed are actually caused by NP adsorption and not buffer mismatch between the protein and NP solutions. This is particularly important when using commercially prepared NPs, where the NP storage buffer might not be precisely known.
As with 1D experiments, quantitative HSQC spectra can be used to measure the binding capacity of NPs provided the system is in slow exchange and the line shape is unaffected. The relative signal intensity will decrease upon addition of NPs, corresponding to protein binding to the NP surface. As before, the binding capacity can be determined by identifying the bound concentration when protein interacts with NPs in solution (Figure 3C). Using two dimensional methods also has the advantage of simplifying competition experiments, provided that resonances in the two-dimensional plane do not overlap. For example, Figure 3B shows GB3 and Ubq 15N HSQC resonances when mixed with AuNPs, an analogous situation to what is shown as a one-dimensional spectrum in Figure 3A. When exchange is fast enough so that line broadening does not dominate, it is possible that traditional chemical shift perturbation experiments can be observed.[127] The requirement of fast exchange limits this application of HSQC spectra to the soft corona, or to NPs where the surface chemistry prevents long-lived biomolecular binding. This is because proteins and nucleic acids must be weakly associated and in rapid exchange for chemical shift perturbations to be observed as they would in a traditional protein-protein interaction study.
4.3. Hydrogen-Deuterium Exchange (HDX)
NMR-based HDX experiments monitor the amide protons as they exchange with deuterated solvent. This exchange is extremely sensitive to structural changes and other environmental factors, such as pH, temperature, and primary sequence.[128–129] By monitoring deuteration state, HDX has the potential to alleviate the relaxation challenges depicted in Figure 1. This is because, while transverse nuclear magnetization relaxes rapidly on the surface of a NP, chemical substitution of H to D is a structural probe that will persist throughout adsorption and desorption from the surface. HDX measurements can be measured in several ways in the context of biomolecule-NP interactions. For example, real-time NMR HDX experiments monitor the exchange of H to D directly after the buffer solution is rapidly exchanged from H2O to D2O.[129] Two-dimensional NMR spectra are recorded continuously, and the spectral signal gradually decays as protons are exchanged for deuterons, which are not observed in a 1H-15N HSQC experiment. Acquisition of real-time HDX has been facilitated by rapid-pulse methodologies like SOFAST[130] and BEST,[131] which can produce two dimensional spectra in as little as a few minutes. However, the low sample concentrations used in NP binding experiments[112] can affect sensitivity, making fast acquisition challenging. In systems where binding to the surface is dynamic (seconds or faster, i.e. the soft corona), perturbations in the HDX rate are expected if the chemical environment changes significantly during attachment to the NP surface. Slower timescale binding, such as that observed for the hard corona, may be more difficult to observe using HDX. This is because there may not be an appreciable exchange between the bound and unbound states. Real-time HDX measurements are limited to amide protons exchanging on the timescale of minutes to hours, making this approach practical only for compact globular proteins, where many protons are buried and protected from exchange by secondary structures.
An alternative experiment, NH/D SOLEXSY (Solvent Exchange Spectroscopy) can be used to detect HDX exchange rates occurring at faster time scales (0.2–2 s−1) allowing the investigation of HDX in the protein loop regions.[132] This experiment is performed in a mixture of 50% H2O-D2O, and HDX occurs continuously throughout the experiment. The peak intensities acquired through SOLEXSY give rise to a decay curve that can be fit to extract the exchange rate.[132] The measurement of protein HDX using SOLEXSY in the presence of NPs can provide information on HDX in fast-exchanging loops or exposed amide protons, provided the nanoparticle interaction is sufficiently fast so that HDX can be observed (i.e., fast-exchanging soft corona proteins). Prior experiments on AuNPs found that HDX-rates determined using SOLEXSY differed little between proteins in the presence and absence of NPs.[112] However, this is likely because AuNP binding forms a hard corona, with little evidence for soft corona exchange. Consequently, if proteins do not desorb during the SOLEXSY mixing time, no difference in the HDX rates will be observed even if significant structural change is present while the protein is on the NP surface.
Faster measurements of HDX using NMR are possible, in particular the WEX-II,[133] the CLEANEX-PM[134–135] and the WEX-III TROSY[136] experiments. However, as these approaches measure exchange between the bulk water and the amide backbone, they are strictly speaking hydrogen exchange experiments, not hydrogen-deuterium exchange experiments. In NP binding, this is important because each of these approaches uses saturation of water to monitor transfer of magnetization from the protein to the bulk solvent or vice-versa. Because this saturation rapidly dephases when the protein is on the NP surface, these experiments are not generally suitable for studying protein binding to NPs.
HDX rates in the presence of NPs, relative to rates without NPs, can be used to quantify structural changes that occur on the NP surface. Protection factors are presented as the log value of ratio between the exchange rate of protein in solution and protein bound to a NP. It is known that individual protection factors are related to local protein stability.[137–138] HDX studies by Engel et al. have shown that a beta-lactoglobulin (BLA) protein folding intermediate is oriented on polystyrene NP in a site-specific manner.[139] These researchers determined that unfolding is initiated by local interactions of protein with polystyrene NPs. The adsorption-initiated partial unfolding of BLA is faster on NPs (74 s−1) than global protein unfolding. HDX has also been used to monitor conformational changes of GB3 and Ubq in the presence of 15 nm AuNPs.[112] Using real-time HDX, it was found that the presence of AuNPs does not significantly alter exchange rates for either GB3 or Ubq. This finding strongly suggests that the hard protein corona on AuNPs does not experience significant desorption, even after hours have passed. In addition, the fast exchanging soft-corona (if present), does not appear to perturb HDX rates for these two globular proteins.[112] Thus, provided accurate HDX rates can be measured, HDX can be an extremely useful tool when studying biomolecule-NP binding, both when adsorption is fast as well as when it is slow.
4.4. Saturation Transfer and Relaxation Methods
Larger NP-protein systems tend to have slow rotational diffusion, enhancing the fast transverse spin relaxation (R2) leading to broadened or even undetectable signals (Figure 1). Reversible binding equilibria can be exploited to study adsorption, allowing the measurement of kinetic rate constants and other relevant parameters. There are two primary ways for interpreting relaxation in the context of nanoparticle binding: saturation transfer and quantitative analysis of the R2 rates. In the dark-state exchange saturation transfer (DEST) experiment,[140–142] longitudinal 15N magnetization (which decays slowly in slow moving species like NP-bound proteins) of the observable species is transferred by chemical exchange from the corresponding invisible state back to the visible species. This approach was originally applied to characterize oligomers in amyloid β peptides, and it is generally applicable any time a low molecular weight species is in exchange with a much larger one.[140] The large transverse relaxation rates () of the supramolecular entity (i.e. the NP-bound state) preclude direct observation but allow for efficient partial saturation of the longitudinal magnetization by a weak radio frequency field, even at large offsets where the signals of the visible species are unaffected. Partial saturation is recorded as an attenuation of the signals of the visible species. The combination of DEST and two-dimensional 1H 15N HSQC experiments allows for single-residue resolution of dynamic information on the NP-bound protein (the NMR-invisible state) to be obtained.[141] This information, combined with measurements of the relaxation rate in the presence and absence of NPs (), can be used to determine the kinetic rate of exchange (koff and ; Figure 2C).
Exchange processes accessible by DEST generally occur on timescales ranging from ~1 ms to ~1 s, but the specifics will depend on the size of the nanoparticle and the rate of exchange, as these two parameters directly influence the linewidth in the nanoparticle-associated state. In addition, under certain circumstances (i.e. when of the NP-bound state is smaller than the saturation field used), additional relaxation data are needed to resolve the ambiguity in the population of bound protein (pB) and the absolute rate.[119] These additional relaxation measurements ()[143] can be used to determine pB, making it straightforward to estimate . Stated differently, for NPs smaller than 30 nm, DEST and can only determine the product of without the collection of . Thus, in practice, DEST has been applied to liposomal NPs and ceria NPs,[43,144–145] but it cannot be used for the proteins tested with AuNPs thus far, as the exchange rate is too slow.[112]
Measuring relaxation under multiple conditions can also be a useful tool to identify biomolecular properties on nanoparticle surfaces. This approach takes advantage of the fact that relaxation is related to rotational diffusion through the spectral density function. The key parameter in this experiment is , the difference between the relaxation observed in the presence and absence of NPs, typically when only a low fraction of protein is associated with the NP (). As a reminder, note that is not the of the NP-associated state, which is typically large and precludes direct observation; instead, it is the apparent relaxation rate in the presence of exchange between the free state and a small population of the NP-associated state. An extended Lipari-Szabo approach,[146] can be used to separate the spectral density function into contributions from nanoparticle rotational diffusion and biomolecular rotational diffusion on the NP surface. Assuming that the timescale of NP binding is separable from the timescale of motion on the NP surface, a model can be fit to the observed ΔR2 rates in the presence of NPs. This model includes the kinetic on and off rates, an order parameter for binding, and protein rotation axis angles relative to the NP surface. Ceccon et al. developed and applied this approach to Ubq on liposomal NPs, where exchange is fast.[43] Importantly, two sizes of NPs were needed (103 nm and 27 nm) to unambigiously determine the model parameters, and it was assumed that Ubq interaction parameters were independent of nanoparticle size.
An alternative approach for disordered proteins can be employed as well. However, for disordered proteins, globally defined axes of rotational diffusion are not well defined. Because of this, alternative models must be used to interpret the ΔR2 values. More direct relaxation approaches are also possible. For example, Xie et al. used an interaction model built up from individual amino acids to describe the interaction of the transactivation domain of p53 with SiNPs.[113] This model, termed the free residue interaction model (FRIM), can be used to determine which regions in a disordered protein are most favored to interact with a NP surface. Because ΔR2 values must be observable, this approach is again only suitable for monitoring proteins interacting with NPs in fast exchange and is therefore limited to soft corona interactions. Nevertheless, intrinsically disordered proteins (IDPs) and IDP regions make up a significant fraction of the proteome,[147–152] and the FRIM approach makes an important contribution in understanding how these proteins interact with surfaces.
4.5. Paramagnetic Relaxation Enhancement (PRE)
Paramagnetic Relaxation Enhancement (PRE) has emerged as an alternative, powerful tool to investigate various dynamic processes involving macromolecules. The PRE-arises from a dipolar coupling between nuclear spins and unpaired electrons, and it increases nuclear R2 relaxation with an r−6 distance dependence between the paramagnetic center and the nucleus of interest.[153] Typically, the nuclear spin of interest is a proton, as 1H nuclei are the most sensitive to the relaxation enhancement. Quantitatively, the observed R2 relaxation (R2,obs) can be interpreted as , where R2 is the relaxation rate in the absence of PRE and is the enhancement term. In the case of biomolecule-NP association, the observed R2 value is ensemble averaged, complicating a strict interpretation of the r−6 distance dependence.[153–154] For a static interaction, the PRE interaction range is limited to 25 Å, but this value will be influenced by the population of NP-associated vs. unassociated proteins, as well as the density of unpaired electrons on the NP surface. In practice, an intensity ratio is measured, comparing the intensity of protein peaks with and without paramagnetically-doped NPs (Ipara/Idia, Figure 4); in this case, Ipara/Idia, plotted against residue number, is interpreted semi-quantitatively, and it is assumed that lower intensity ratios have a strong interaction with the NP surface. The PRE can only be detected when the exchange is in the fast regime due to the intrinsic line broadening that occurs upon long-lived adsorption to the NP.
Figure 4.
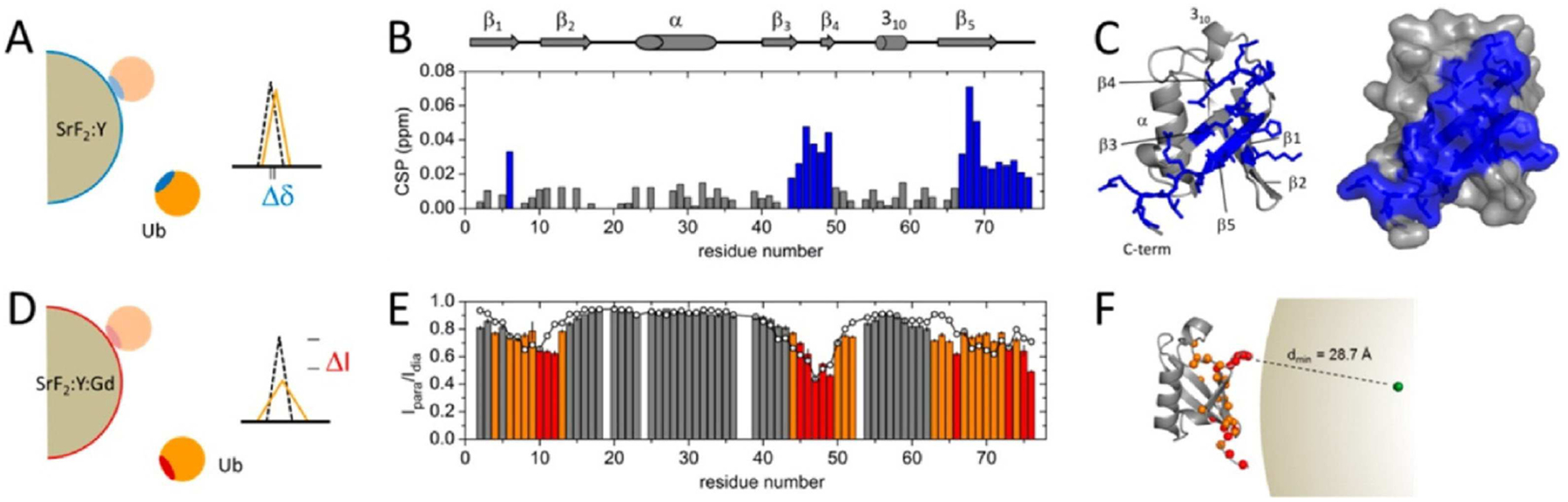
Use of PRE and chemical shift perturbation to investigate the orientation and binding site of Ubq on paramagnetically doped fluoride-based SrF2 NPs. (A) The site-specific chemical shift perturbation (CSP) observed for Ubq upon transient adsorption to diamagnetic NPs. The diagram indicates that, for weakly associating proteins in the soft corona, only residues near the interaction site are perturbed. (B) Residue-specific CSPs observed for Ubq upon interaction with NPs. (CSP>0.02 ppm are highlighted in blue). (C) Large CSPs mapped on to the native structure of Ubq, revealing the interaction surface in blue. (D) In the presence of a paramagnetically doped NP, proximity to the stable radical electron induces relaxation in 1H spins. The diagram indicates that, in addition to experiencing CSP, such spins experience additional line broadening. (E) Intensity ratio (Ipara/Idia) of peaks in the presence and absence of a paramagnetic spin label. Lower intensities occur because of proximity to the NP surface, corresponding to the interaction site identified by PRE. (F) Graphical representation of Ubq Binding to SrF2 NPs. Reprinted with permission from Zanzoni et al.,[127] copyright 2016 American Chemical Society.
PRE has been used on numerous occasions to identify protein orientation on NP surfaces. Zanzoni et al. determined the Ubq interaction interface on paramagnetically doped fluoride-based (SrF2:Y3+,Gd3+) NPs using PRE. The residue intensities perturbed in the presence paramagnetically labeled NPs were predominantly located in the stretches of residues 4–13 (β1 and β2), 44–55 (β3 and β4), and 63–76 (β5) (Figure 4E).[127] In a similar study, Ceccon et al. used PRE to investigate Ubq on lipid-based nanoparticles (POPG-LUV), and they were able map the binding site of Ubq on these Gd3+-paramagnetically tagged lipid-based NPs.[43] They concluded that the Ubq central hydrophobic region mainly interacts with the negatively charged surface of POPG-LUV. Both groups identified similar binding sites Ubq on fairly different NPs, suggesting that Ubq may have a common mode for binding to surfaces.
5. Results from Different Nanoparticle Systems
5.1. Noble Metal Nanoparticles (Gold/Silver)
Gold nanoparticles (AuNPs) are frequently used in NP-protein studies due to their low toxicity and unique optical, chemical, and physical properties.[155] Functionalization of AuNPs is straightforward, leading to a broad diversity of surface behaviors and properties. Because of this, it is challenging to generalize the behavior of biomolecule-AuNP association, and for any NP system, subtle changes to surface chemistry can lead to large changes in binding behavior, altering both the soft and hard corona composition and dynamics. Most proteins bind to AuNPs spontaneously without causing any aggregation,[48] and when aggregation does occur, hydrogels can be used to maintain NPs in suspension.[145] Thus, AuNPs provide a good platform to study NP-protein interactions to gain insight on orientation, structure, and function of the protein.[40,111] In this and the remaining sections of this review, we will briefly summarize several key applications where NMR has been leveraged to understand how and why proteins interact with NPs, starting with AuNPs.
AuNPs have displayed a variety of behaviors depending on surface functionalization, but generally globular proteins interact with AuNPs in slow exchange with minimal structural perturbation as measured by NMR. In the presence of 1.5 nm AuNPs, fibroblast growth factor (FGF1) exhibited a minimal perturbation of its secondary structure[156] as monitored by NMR; however, Ubq had a few residues in the N-terminus that exhibited chemical shift perturbation upon binding, suggesting that, while both proteins interact with small nanospheres, Ubq exhibits a site-specific preference in doing so.[157] For larger AuNPs, early studies on Ubq suggested that chemical shift perturbations were also present in the presence of 12 nm particles.[40] However, chemical shift perturbations should be associated with an increase in linewidth (Figure 1), and this was not observed. It was later proposed that an additional protein contamination or buffer mismatch influenced the Ubq NMR spectra.[111] Because AuNPs interact with DNA as well as many proteins, molecules besides the target protein can influence NMR measurements of NP binding. For studies of specific protein interactions, it is therefore important that the proteins used are the highest purity possible and that the buffer conditions are well controlled.
Characterization by both 1D and 2D NMR spectroscopy suggests that Ubq and GB3 have a site-specific orientation on 15 nm citrate AuNPs.[39,111] In both of these cases, binding is in very slow exchange, allowing the quantification of adsorbed protein.[111] It would appear that, for many stable globular proteins interacting with medium size AuNPs (10–80 nm),[85] monolayer binding on the AuNP surface gives rise to a stable hard corona with little evidence for a dynamic, soft corona. Unstable, non-globular proteins[85] and intrinsically disordered proteins (IDPs)[56,85] behave differently, suggesting that these proteins have altered conformations on the AuNP surface. In particular, Lin et al. used HSQC spectra to determine that the orientation of α-synuclein, an IDP, can be flipped depending on the surface charge of the AuNP.[56] Meng et al. investigated the potential difference between MTAB ligands at the ends and the sides of gold nanorods (AuNRs) using chemical shift perturbation and T2 relaxation.[158] As compared to the spherical AuNPs, MTAB-AuNRs have two distinctly different chemical environments (elongated sides and spherical ends) due to the rod-like nature of the nanoparticle. Both T2 and relaxation times were compared for different aspect ratios of MTAB-AuNRs.[158] When the aspect ratio increased, the difference between T2 and also increased, and from this data, the authors concluded that the MTAB headgroup mobility depends on the nanorod aspect ratio.
Understanding protein conformational change on AuNP surfaces is a major issue yet to be resolved. NMR, when combined with molecular simulations, is an extremely powerful tool and has produced promising results. For example, a combined molecular-dynamics NMR approach was used to characterize the interaction between the fibrillogenic protein β2-microglobulin (B2m) and 5 nm citrate-capped AuNPs. Peak intensity analysis of HSQC data revealed the presence of CSPs for specific amides, especially those close to the N-terminus, when attaching to the AuNP.[159] The small size of the AuNPs in this study allowed a direct comparison between simulations and NMR CSPs, even though the binding was found to be in slow exchange. Ultimately, the B2m protein attachment to AuNP did not disrupt the secondary structure nor lead to amyloid aggregation,[159] although simulations did identify that the citrate ions can alter stability by changing the local pH.
Silver nanoparticles (AgNP) are another common noble nanoparticle with antibacterial and antifungal properties.[160–162] Protein interaction with AgNPs has been characterized through CSP, NMR titrations, and relaxation-based experiments.[163–164] AgNPs without any surface functionalization were used in Ubq to observe several CSP changes in the amino acids of the β-sheet region, suggesting this may be its interaction site.[164] Brahmkhatri et al. demonstrated that a conjugate of AgNP with Ubq is highly stable over a wide range of pH values compared to the bare AgNP.[165] Chemical shift perturbation was used to identify the Ubq residues responsible for binding with AgNP and the NMR titrations were carried out to measure the dissociation constant (KD).[165] Pal et al. carried out a similar study to understand the structural and dynamic changes of antimicrobial peptide Odorranain-A-OA1 (OA1) upon binding to AgNP.[163] For this peptide, the 1D 1H spectrum indicated chemical shift perturbations upon binding indicating the peptide interaction with AgNP. The specific residues interacting with AgNP were identified by acquiring 2D 13C-1H HSQC spectra.[163] Most notably, the Cys 15 residue was perturbed due to the direct interaction with AgNP. Generally, silver nanoparticles appear to have weaker protein binding compared to AuNPs, and AgNPs can gradually undergo oxidative dissolution in biological fluids, eventually dissolving while releasing soluble Ag+ ions.[166–167] This reduced stability appears to influence protein binding, leading to a more dynamic corona when observed by NMR.[165]
5.2. Silica Nanoparticles
Silica nanoparticles (SiNPs) are inorganic nanoparticles with many applications ranging from biosensors (optical properties) to drug delivery vectors (encapsulation capacity).[168–172] The characteristic features of SiNPs are their low cost, low toxicity, and biocompatibility.[173] Several groups have used NMR to investigate SiNP-protein interactions using a variety of proteins, including lysozyme,[174] carbonic anhydrase,[42,175–176] fibrinogen,[177] BSA,[178] and cytochrome c.[179] While a hard corona may form on SiNPs,[180] NMR studies generally find that proteins also exhibit fast exchange behavior in the presence of SiNPs. Because of this fast exchange behavior, solution NMR techniques have been used to monitor the CSPs in the presence of low concentrations of SiNPs.[176] This has enabled the identification of residues interacting with SiNP surfaces. Silica surfaces are generally negative,[181–182] as terminal silanol groups dissociate to produce a negative charge. SiNPs have a negative zeta potential,[183] and it is not surprising therefore that positively charged residues interact with the negatively charged SiNP surface. This has been confirmed using the NMR-derived FRIM model, discussed above.[113]
While proteins may retain some native structure on SiNPs, NMR studies have suggested that proteins tend to deform on these surfaces. For example, acylphosphatase (AcP) was examined in the presence of 4 nm and 15 nm SiNPs, and HSQC spectra were used to examine structural changes.[184] It was found that, for both sizes, interaction occurred preferentially at the α-helices in AcP. HSQC spectra indicated that only 15% of the peaks are affected by SiNPs, and these residues were common to both 4 and 15 nm SiNPs. At the same time, enzymatic activity experiments revealed that the protein lost activity on SiNP surfaces, and more activity was lost on the larger 15 nm SiNPs. This suggests that a specific interaction is present, but that the protein deforms once bound. A 2004 study focusing on carbonic anhydrase observed significant broadening of the protein NMR signals in the presence of 6, 9, and 15 nm SiNPs. This broadening increased over time and could be fit with a biexponential time dependence. The time constants of the fast and slow phases were 5 h and 45 h, respectively, suggesting that, while the protein interacts with the SiNP surface in fast NMR exchange, the nature of that interaction may change over time.[42] It was suggested that this slow change occurs during the formation of the SiNP hard corona. Using supporting spectroscopic techniques, this study also found a trend where increasing SiNP size resulted in an increasing perturbation to the protein secondary structure.
5.3. Carbon Based Nanoparticles
There are several different carbon-based nanoparticles, carbon quantum dots, carbon nanotubes, and fullerenes.[185–187] Under standing how biomolecules interact with carbon nanostructures is important for a number of applications, including biosensing, and NMR has seen some use in probing protein-surface interactions in this area. The principal challenge in using NMR to study protein interactions with carbon-based NPs is the solubility in aqueous media. One study was able to overcome this challenge by studying the small fullerene C60 (1 nm) as it interacts with lysozyme.[188] Lysozyme solubilizes C60, forming a stable 1:1 complex in aqueous solution. Because C60 is comparable in size to a small peptide, NMR spectra can be acquired directly with minimal line broadening, and CSPs can be measured in the complex. Amino acid residues experience the largest perturbations close to the catalytic site of lysozyme, suggesting that the protein interacts with C60 at that location. Another study overcame the solubility issue by using polyhydroxylation of fullerenes. Zanzoni et al. used NMR to observe the binding of Ubq to hydroxylated fullerenes.[189] Based on CSP data and the intensity change upon binding, the binding site of Ubq was identified a hydrophobic patch centered around Leu8, Ile44, and Val70. 15N-R2 spin relaxation rate changes and DEST were used to characterize the reversible formation of soluble aggregates upon protein binding to fullerene surfaces. No other studies to our knowledge have explored biomolecular interactions with carbon nanomaterials using NMR, but it is likely that these examples cover two extremes: On the one hand, bare carbon nanomaterials are hydrophobic and will likely exhibit slow exchange behavior, as is seen with lysozyme. On the other, adding solubilizing groups will make the surface more hydrophilic, promoting faster exchange at the surface. It remains to be seen whether these observations from NMR spectroscopy can be generalized to other structures, such as carbon nanotubes, and this remains an area of active investigation.[190]
5.4. Polystyrene and Polymeric Nanoparticles
Polystyrene and other polymeric nanoparticles represent an extremely broad class of NPs, and the variety of materials and surface functionalizations used make generalizations challenging. Here, we focus on polystyrene latex spheres, which are typically functionalized with either amidine (basic) or carboxyl (acidic) groups to prevent aggregation in aqueous media. The first notable example applying NMR to polystyrene NPs was in 2004 by Engel, et al.[139] These investigators examined the behavior of bovine alpha-lactalbumin (BLA) using HDX and found that the protein adopts a molten-globular like structure on the surface of polystyrene NPs. They found that the hydrophobic NP surface produced a stable hard corona, allowing HDX measurements to occur on the NP surface itself. The observed NP-bound HDX rates were compared to the molten globule rates for BLA, and the rates were observed to correlate. While this would suggest a hard corona and slow exchange behavior, a recent study by Yang et al. used saturation-transfer difference (STD) NMR to investigate the association between amino acids and the polystyrene surface.[191] STD NMR[192–194] is similar to DEST in that off-resonance saturation is used to monitor the strength of binding in an invisible, NP-bound state. Aromatic amino acids exhibited the strongest STD effect, suggesting that significant π-π interactions can occur between amino acid side chains and the NP surface. This hypothesis was confirmed using sum frequency generation spectroscopy, a nonlinear optical method used to probe surfaces. Together, these results suggest that polystyrene NPs strongly interact with proteins, causing their deformation upon adsorption. Nevertheless, adsorption is likely characterized in its early stages by faster interactions as individual residues rapidly sample the hydrophobic surface. Thus, NMR studies suggest that biomolecules on polystyrene NPs initially form a dynamic soft corona that eventually hardens in to a hard corona under (very) slow exchange – an observation that agrees with prior work on polystyrene and polymeric nanospheres.[3]
5.5. Liposomal Nanoparticles
Protein interactions with liposomes, lipid vesicles, and micelles have been extensively studied using NMR spectroscopy. Liposomal NPs are relevant because of their similarity to the plasma membrane and their applications in drug delivery.[195–196] Protein-liposome interactions are generally fast and dynamic on the NMR timescale and give rise to a measurable ΔR2, enabling the application of DEST and other relaxation based methods.[119,144] As described above, Ceccon et al. studied the interaction of Ubq with negatively charged and zwitterionic liposomes, and relaxation fitting was used to identify the orientation axis on the NP surface.[43] This work was later extended to multidomain proteins, focusing on a covalent Ubq dimer. Additional theoretical considerations are needed to handle binding in this case, and it was found that the linker connecting Ubq molecules could influence binding for the distal Ubq domain. DEST studies of protein-liposome interactions have also been informative. Investigations of fatty acid binding protein have also been used to identify the interaction site with 90 nm liposomes consisting of a 1:1 ratio of palmitoyl-oleoyl-phosphatidylglycerol (POPG) and palmitoyl-oleoyl-phosphatidylcholine (POPC).[43]
IDPs or partially disordered proteins are also prone to lipid binding. The α-synuclein protein (αS) is a water soluble, natively unfolded protein involved in Parkinson’s Disease, and αS is in equilibrium between water soluble and membrane-associated states.[197–199] Similarly, huntingtin is a protein involved in Huntington’s Disease,[200] and the poly-Q tails of this protein are implicated in membrane association.[201] NMR experiments have been used to study the αS bound to small unilamellar vesicles (SUVs).[198] Bodner et al. observed a decrease in peak intensities, revealing that the N-terminal end of αS preferentially associates with the SUV.[198] A related study examined the NMR signals originating from 31P nuclei in the vesicle and identified a binding model with different distinct bound species of αS.[202] More recently, investigations on huntingtin-derived peptides have used chemical exchange saturation transfer (CEST) methods to identify chemical shift changes in an otherwise invisible lipid micelle-bound state.[116] These data have been combined with experiments using pulsed EPR to study dimerization on the NP surface, and kinetic rates for dimerization have been extracted for different lengths of poly-Q tails. Thus, while introduction of protein disorder adds complexity to NP association, under appropriate conditions NMR can be used to monitor surface folding and molecular interactions.
6. Conclusions and Outlook
Solution NMR spectroscopy is a powerful tool for characterizing biomolecule-NP association, owing to its ability to monitor structure, thermodynamics, and dynamics, often simultaneously in the same experiment. When combined with other analytical and computational techniques, protein surface interactions can be studied in great detail. Even simple 1D 1H NMR spectra in the presence and absence of NPs provide information on protein interactions, and straightforward experiments involving quantitative NMR have been used effectively to characterize the behavior of proteins upon adsorption. More sophisticated experiments involving chemical exchange, HDX, and relaxation can complement this information, providing a near-complete picture of adsorbed protein behavior. At present, the main challenge is understanding how NP surface properties influence the type of behavior observed. For example, liposomal and silica NPs exhibit dynamic protein binding behavior, suggestive of a soft corona, whereas other NPs like citrate-capped gold and polystyrene NPs, appear to trap proteins on the surface for extended periods of time, a key property of the hard corona. Each of these behaviors (fast vs. slow vs. no exchange) will influence which set of experiments is most appropriate for studying the system of interest. Thus, while current NMR approaches cannot in general measure signals for surface-bound biomolecules, the development of indirect methods reviewed here has made the study of protein-NP interactions quite tractable. As nanoparticles continue to proliferate in biosensing and drug delivery applications, NMR will undoubtedly continue to be useful in understanding how NPs interact with the components of biological fluids.
Acknowledgements
We thank Nick Anthis, Nick Fawzi, and Vincenzo Venditti for helpful discussions on DEST and relaxation experiments. This work was supported in part by the National Science Foundation under grant 1818090. This work was also supported by in part the National Institutes of Allergies and Infectious Disease of the National Institutes of Health under award number R01AI139479. No nongovernmental sources were used to fund this work.
Biographies
Yasiru Randika Perera received his B.S. from the Institue of Chemistry, Sri Lanka in 2012. He is pursuing his Ph.D. in Chemistry at Mississippi State University under the mentorship of Prof. Nicholas Fitzkee. In 2018, he was awarded the most outstanding graduate researcher. His research interest includes studying the structure and dynamics of proteins on metal nanoparticles.

Rebecca A. Hill is currently pursuing her Ph.D. under the direction of Professor Nicholas Fitzkee at Mississippi State University. Since starting graduate school, she has been focusing on quantitatively investigating bacterial metabolites and protein interactions on various nanoparticle surfaces using NMR techniques. In 2019, she was awarded the most outstanding graduate researcher. Prior to starting her graduate work, she received her B.S. degree in chemistry from University of West Florida in 2015. After obtaining her Ph.D. in Chemistry, she plans on pursuing a career in industry.

Dr. Nicholas Fitzkee is an associate professor in the Department of Chemistry at Mississippi State University. He earned his Ph.D. in Biophysics from Johns Hopkins University, and he completed a postdoc at the National Institutes of Health. Dr. Fitzkee is broadly interested in using NMR spectroscopy to study the molecular basis of protein adsorption to surfaces. His group works to develop novel, NMR-based techniques for understanding protein-surface interactions. A major goal of his research is developing predictive physical models for understanding protein adsorption.

References
- [1].Lin W, Chem. Rev 2015, 115, 10407–10409. [DOI] [PubMed] [Google Scholar]
- [2].Pelaz B, Jaber S, de Aberasturi DJ, Wulf V, Aida T, de la Fuente JM, Feldmann J, Gaub HE, Josephson L, Kagan CR, Kotov NA, Liz-Marzán LM, Mattoussi H, Mulvaney P, Murray CB, Rogach AL, Weiss PS, Willner I, Parak WJ, ACS Nano. 2012, 6, 8468–8483. [DOI] [PubMed] [Google Scholar]
- [3].Lundqvist M, Stigler J, Elia G, Lynch I, Cedervall T, Dawson KA, Proc. Natl. Acad. Sci. USA 2008, 105, 14265–14270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Saptarshi SR, Duschl A, Lopata AL, J. Nanobiotechnol 2013, 11, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Nel AE, Mädler L, Velegol D, Xia T, Hoek EMV, Somasundaran P, Klaessig F, Castranova V, Thompson M, Nat. Mater 2009, 8, 543. [DOI] [PubMed] [Google Scholar]
- [6].Wangoo N, Suri CR, Shekhawat G, Appl. Phys. Lett 2008, 92, 133104. [Google Scholar]
- [7].Mukhopadhyay A, Basu S, Singha S, Patra HK, Research. 2018, 2018, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kurylowicz M, Paulin H, Mogyoros J, Giuliani M, Dutcher JR, J. R. Soc. Interface 2014, 11, 20130818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Satzer P, Svec F, Sekot G, Jungbauer A, Eng. Life Sci 2016, 16, 238–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Jensen TR, Malinsky MD, Haynes CL, Van Duyne RP, J. Phys. Chem. B 2000, 104, 10549–10556. [Google Scholar]
- [11].Endo T, Kerman K, Nagatani N, Hiepa HM, Kim D-K, Yonezawa Y, Nakano K, Tamiya E, Anal. Chem 2006, 78, 6465–6475. [DOI] [PubMed] [Google Scholar]
- [12].Nath N, Chilkoti A, Anal. Chem 2002, 74, 504–509. [DOI] [PubMed] [Google Scholar]
- [13].Aggarwal P, Hall JB, McLeland CB, Dobrovolskaia MA, McNeil SE, Adv. Drug Delivery Rev 2009, 61, 428–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Nehl CL, Liao H, Hafner JH, Nano Lett. 2006, 6, 683–688. [DOI] [PubMed] [Google Scholar]
- [15].Elechiguerra JL, Burt JL, Morones JR, Camacho-Bragado A, Gao X, Lara HH, Yacaman MJ, J. Nanobiotechnol 2005, 3, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Dobrovolskaia MA, Patri AK, Zheng J, Clogston JD, Ayub N, Aggarwal P, Neun BW, Hall JB, McNeil SE, Nanomed. Nanotechnol. Biol. Med 2009, 5, 106–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gupta AK, Curtis ASG, J. Mater. Sci. Mater. Med 2004, 15, 493–496. [DOI] [PubMed] [Google Scholar]
- [18].Lai ZW, Yan Y, Caruso F, Nice EC, ACS Nano. 2012, 6, 10438–10448. [DOI] [PubMed] [Google Scholar]
- [19].Patil S, Sandberg A, Heckert E, Self W, Seal S, Biomaterials. 2007, 28, 4600–4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lundqvist M, Stigler J, Cedervall T, Berggård T, Flanagan MB, Lynch I, Elia G, Dawson K, ACS Nano. 2011, 5, 7503–7509. [DOI] [PubMed] [Google Scholar]
- [21].Sun T, Qing G, Adv. Mater 2011, 23, H57–H77. [DOI] [PubMed] [Google Scholar]
- [22].Xie J, Zheng Y, Ying JY, J. Am. Chem. Soc 2009, 131, 888–889. [DOI] [PubMed] [Google Scholar]
- [23].Röcker C, Pötzl M, Zhang F, Parak WJ, Nienhaus GU, Nat. Nanotechnol 2009, 4, 577. [DOI] [PubMed] [Google Scholar]
- [24].You C-C, Miranda OR, Gider B, Ghosh PS, Kim I-B, Erdogan B, Krovi SA, Bunz UHF, Rotello VM, Nat. Nanotechnol 2007, 2, 318. [DOI] [PubMed] [Google Scholar]
- [25].Gogoi SK, Gopinath P, Paul A, Ramesh A, Ghosh SS, Chattopadhyay A, Langmuir. 2006, 22, 9322–9328. [DOI] [PubMed] [Google Scholar]
- [26].Shang L, Wang Y, Jiang J, Dong S, Langmuir. 2007, 23, 2714–2721. [DOI] [PubMed] [Google Scholar]
- [27].Laera S, Ceccone G, Rossi F, Gilliland D, Hussain R, Siligardi G, Calzolai L, Nano Lett. 2011, 11, 4480–4484. [DOI] [PubMed] [Google Scholar]
- [28].Deng ZJ, Liang M, Monteiro M, Toth I, Minchin RF, Nat. Nanotechnol 2010, 6, 39. [DOI] [PubMed] [Google Scholar]
- [29].Gessner A, Lieske A, Paulke BR, Müller RH, Eur. J. Pharm. Biopharm 2002, 54, 165–170. [DOI] [PubMed] [Google Scholar]
- [30].Deng ZJ, Mortimer G, Schiller T, Musumeci A, Martin D, Minchin RF, Nanotechnology. 2009, 20, 455101. [DOI] [PubMed] [Google Scholar]
- [31].Liu X, Dai Q, Austin L, Coutts J, Knowles G, Zou J, Chen H, Huo Q, J. Am. Chem. Soc 2008, 130, 2780–2782. [DOI] [PubMed] [Google Scholar]
- [32].Reinhard BM, Siu M, Agarwal H, Alivisatos AP, Liphardt J, Nano Lett. 2005, 5, 2246–2252. [DOI] [PubMed] [Google Scholar]
- [33].Cedervall T, Lynch I, Lindman S, Berggård T, Thulin E, Nilsson H, Dawson KA, Linse S, Proc. Natl. Acad. Sci. USA 2007, 104, 2050–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Baier G, Costa C, Zeller A, Baumann D, Sayer C, Araujo PHH, Mailänder V, Musyanovych A, Landfester K, Macro-mol. Biosci 2011, 11, 628–638. [DOI] [PubMed] [Google Scholar]
- [35].Joshi H, Shirude PS, Bansal V, Ganesh KN, Sastry M, J. Phys. Chem. B 2004, 108, 11535–11540. [Google Scholar]
- [36].De M, You C-C, Srivastava S, Rotello VM, J. Am. Chem. Soc 2007, 129, 10747–10753. [DOI] [PubMed] [Google Scholar]
- [37].Xu Y, Sherwood J, Qin Y, Crowley D, Bonizzoni M, Bao Y, Nanoscale. 2014, 6, 1515–1524. [DOI] [PubMed] [Google Scholar]
- [38].Wang M, Fu C, Liu X, Lin Z, Yang N, Yu S, Nanoscale. 2015, 7, 15191–15196. [DOI] [PubMed] [Google Scholar]
- [39].Wang A, Perera YR, Davidson MB, Fitzkee NC, J. Phys. Chem. C 2016, 120, 24231–24239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Calzolai L, Franchini F, Gilliland D, Rossi F, Nano Lett. 2010, 10, 3101–3105. [DOI] [PubMed] [Google Scholar]
- [41].Marbella LE, Millstone JE, Chem. Mater 2015, 27, 2721–2739. [Google Scholar]
- [42].Lundqvist M, Sethson I, Jonsson B-H, Langmuir. 2004, 20, 10639–10647. [DOI] [PubMed] [Google Scholar]
- [43].Ceccon A, Tugarinov V, Bax A, Clore GM, J. Am. Chem. Soc 2016, 138, 5789–5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Long JR, Shaw WJ, Stayton PS, Drobny GP, Biochemistry. 2001, 40, 15451–15455. [DOI] [PubMed] [Google Scholar]
- [45].Kleckner IR, Foster MP, Biochim. Biophys. Acta 2011, 1814, 942–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Cala O, Guillière F, Krimm I, Anal. Bioanal. Chem 2014, 406, 943–956. [DOI] [PubMed] [Google Scholar]
- [47].Ruschak AM, Kay LE, Proc. Natl. Acad. Sci. USA 2012, 109, 20184–20185. [Google Scholar]
- [48].De Roe C, Courtoy PJ, Baudhuin P, J. Histochem. Cytochem 1987, 35, 1191–1198. [DOI] [PubMed] [Google Scholar]
- [49].Monopoli MP, Pitek AS, Lynch I, Dawson KA, in Nanomaterial Interfaces in Biology: Methods and Protocols, (Eds.: Bergese P, Hamad-Schifferli K), Humana Press, Totowa, NJ, 2013, pp. 137–155. [Google Scholar]
- [50].Aubin-Tam M-E, Hamad-Schifferli K, Langmuir. 2005, 21, 12080–12084. [DOI] [PubMed] [Google Scholar]
- [51].Chen B-X, Wilson SR, Das M, Coughlin DJ, Erlanger BF, Proc. Natl. Acad. Sci. USA 1998, 95, 10809–10813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Delfino I, Cannistraro S, Biophys. Chem 2009, 139, 1–7. [DOI] [PubMed] [Google Scholar]
- [53].Edri E, Regev O, Anal. Chem 2008, 80, 4049–4054. [DOI] [PubMed] [Google Scholar]
- [54].Jiang X, Jiang J, Jin Y, Wang E, Dong S, Biomacromolecules. 2005, 6, 46–53. [DOI] [PubMed] [Google Scholar]
- [55].Kathiravan A, Renganathan R, Anandan S, Polyhedron. 2009, 28, 157–161. [Google Scholar]
- [56].Lin W, Insley T, Tuttle MD, Zhu L, Berthold DA, Král P, Rienstra CM, Murphy CJ, J. Phys. Chem. C 2015, 119, 21035–21043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Yongli C, Xiufang Z, Yandao G, Nanming Z, Tingying Z, Xinqi S, J. Colloid Interface Sci 1999, 214, 38–45. [DOI] [PubMed] [Google Scholar]
- [58].Lee IS, Lee N, Park J, Kim BH, Yi Y-W, Kim T, Kim TK, Lee IH, Paik SR, Hyeon T, J. Am. Chem. Soc 2006, 128, 10658–10659. [DOI] [PubMed] [Google Scholar]
- [59].Mu Q, Liu W, Xing Y, Zhou H, Li Z, Zhang Y, Ji L, Wang F, Si Z, Zhang B, Yan B, J. Phys. Chem. C 2008, 112, 3300–3307. [Google Scholar]
- [60].Zhou H, Mu Q, Gao N, Liu A, Xing Y, Gao S, Zhang Q, Qu G, Chen Y, Liu G, Zhang B, Yan B, Nano Lett. 2008, 8, 859–865. [DOI] [PubMed] [Google Scholar]
- [61].Kapur A, Aldeek F, Ji X, Safi M, Wang W, Del Cid A, Steinbock O, Mattoussi H, Bioconjugate Chem. 2017, 28, 678–687. [DOI] [PubMed] [Google Scholar]
- [62].Pramanik S, Banerjee P, Sarkar A, Bhattacharya SC, J. Lumin 2008, 128, 1969–1974. [Google Scholar]
- [63].Sahoo B, Goswami M, Nag S, Maiti S, Chem. Phys. Lett 2007, 445, 217–220. [Google Scholar]
- [64].Xiao Q, Huang S, Qi Z-D, Zhou B, He Z-K, Liu Y, Biochim. Biophys. Acta 2008, 1784, 1020–1027. [DOI] [PubMed] [Google Scholar]
- [65].Belgorodsky B, Fadeev L, Ittah V, Benyamini H, Zelner S, Huppert D, Kotlyar AB, Gozin M, Bioconjugate Chem. 2005, 16, 1058–1062. [DOI] [PubMed] [Google Scholar]
- [66].Fischer NO, Verma A, Goodman CM, Simard JM, Rotello VM, J. Am. Chem. Soc 2003, 125, 13387–13391. [DOI] [PubMed] [Google Scholar]
- [67].Karlsson M, Carlsson U, Biophys. J 2005, 88, 3536–3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Macaroff PP, Oliveira DM, Lacava ZGM, Azevedo RB,Lima ECD, Morais PC, Tedesco AC, IEEE Trans. Magn 2004, 40, 3027–3029. [Google Scholar]
- [69].De M, Rana S, Akpinar H, Miranda OR, Arvizo RR, Bunz UHF, Rotello VM, Nat. Chem 2009, 1, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Toseland CP, J. Chem. Biol 2013, 6, 85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Nettles CB, Zhou Y, Zou S, Zhang D, Anal. Chem 2016, 88, 2891–2898. [DOI] [PubMed] [Google Scholar]
- [72].Xu JX, Vithanage BCN, Athukorale SA, Zhang D, Analyst (Cambridge UK). 2018, 143, 3382–3389. [DOI] [PubMed] [Google Scholar]
- [73].Siriwardana K, Vithanage BCN, Zou S, Zhang D, Anal. Chem 2017, 89, 6686–6694. [DOI] [PubMed] [Google Scholar]
- [74].Xu JX, Siriwardana K, Zhou Y, Zou S, Zhang D, Anal. Chem 2018, 90, 785–793. [DOI] [PubMed] [Google Scholar]
- [75].Zhang D, Neumann O, Wang H, Yuwono VM, Barhoumi A, Perham M, Hartgerink JD, Wittung-Stafshede P, Halas NJ,Nano Lett. 2009, 9, 666–671. [DOI] [PubMed] [Google Scholar]
- [76].Cao YC, Jin R, Nam J-M, Thaxton CS, Mirkin CA, J. Am. Chem. Soc 2003, 125, 14676–14677. [DOI] [PubMed] [Google Scholar]
- [77].Reymond-Laruinaz S, Saviot L, Potin V, Mar-arco de Lucas M. d. C., Appl. Surf. Sci 2016, 389, 17–24. [Google Scholar]
- [78].Szekeres GP, Kneipp J, Front. Chem 2019, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Sathuluri RR, Yoshikawa H, Shimizu E, Saito M, Tamiya E, PLoS One 2011, 6, e22802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Athukorale S, Leng X, Xu JX, Perera YR, Fitzkee NC, Zhang D, Front. Chem 2019, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Johnson WC Jr, Proteins Struct. Funct. Bioinf 1990, 7, 205–214. [DOI] [PubMed] [Google Scholar]
- [82].Karajanagi SS, Vertegel AA, Kane RS, Dordick JS, Langmuir. 2004, 20, 11594–11599. [DOI] [PubMed] [Google Scholar]
- [83].Pecora R, J. Nanopart. Res 2000, 2, 123–131. [Google Scholar]
- [84].Martínez-Castañón GA, Niño-Martínez N, Martínez-Gutierrez F, Martínez-Mendoza JR, Ruiz F, J. Nanopart. Res 2008, 10, 1343–1348. [Google Scholar]
- [85].Woods KE, Perera YR, Davidson MB, Wilks CA, Yadav DK, Fitzkee NC, J. Phys. Chem. C 2016, 120, 27944–27953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Boulos SP, Davis TA, Yang JA, Lohse SE, Alkilany AM, Holland LA, Murphy CJ, Langmuir. 2013, 29, 14984–14996. [DOI] [PubMed] [Google Scholar]
- [87].Bhattacharjee S, J. Controlled Release 2016, 235, 337–351. [DOI] [PubMed] [Google Scholar]
- [88].Skoglund S, Hedberg J, Yunda E, Godymchuk A, Blomberg E, Odnevall Wallinder I, PLoS One 2017, 12, e0181735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Cho TJ, Hackley VA, Anal. Bioanal. Chem 2010, 398, 2003–2018. [DOI] [PubMed] [Google Scholar]
- [90].Tsai D-H, DelRio FW, Keene AM, Tyner KM, MacCuspie RI, Cho TJ, Zachariah MR, Hackley VA, Langmuir. 2011, 27, 2464–2477. [DOI] [PubMed] [Google Scholar]
- [91].Fan R, Chew SW, Cheong VV, Orner BP, Small. 2010, 6, 1483–1487. [DOI] [PubMed] [Google Scholar]
- [92].Bayburt TH, Grinkova YV, Sligar SG, Nano Lett. 2002, 2, 853–856. [Google Scholar]
- [93].Cedervall T, Lynch I, Foy M, Berggård T, Donnelly SC, Cagney G, Linse S, Dawson KA, Angew. Chem. Int. Ed 2007, 46, 5754–5756; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2007, 119, 5856–5858. [Google Scholar]
- [94].Pitkänen L, Striegel AM, Trends. Anal. Chem 2016, 80, 311–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Wagner M, Holzschuh S, Traeger A, Fahr A, Schubert US, Anal. Chem 2014, 86, 5201–5210. [DOI] [PubMed] [Google Scholar]
- [96].Fraunhofer W, Winter G, Coester C, Anal. Chem 2004, 76, 1909–1920. [DOI] [PubMed] [Google Scholar]
- [97].Zattoni A, Rambaldi DC, Reschiglian P, Melucci M, Krol S, Garcia AMC, Sanz-Medel A, Roessner D, Johann C, J. Chromatogr. A 2009, 1216, 9106–9112. [DOI] [PubMed] [Google Scholar]
- [98].Peng ZG, Hidajat K, Uddin MS, J. Colloid Interface Sci 2004, 271, 277–283. [DOI] [PubMed] [Google Scholar]
- [99].Dutta D, Sundaram SK, Teeguarden JG, Riley BJ, Fifield LS, Jacobs JM, Addleman SR, Kaysen GA, Moudgil BM, Weber TJ, Toxicol. Sci 2007, 100, 303–315. [DOI] [PubMed] [Google Scholar]
- [100].Nagayama S, Ogawara K.-i., Fukuoka Y, Higaki K, Kimura T, Int. J. Pharm 2007, 342, 215–221. [DOI] [PubMed] [Google Scholar]
- [101].García I, Sánchez-Iglesias A, Henriksen-Lacey M, Grzelczak M, Penadés S, Liz-Marzán LM, J. Am. Chem. Soc 2015, 137, 3686–3692. [DOI] [PubMed] [Google Scholar]
- [102].Kim HR, Andrieux K, Gil S, Taverna M, Chacun H, Desmaële D, Taran F, Georgin D, Couvreur P, Biomacromolecules. 2007, 8, 793–799. [DOI] [PubMed] [Google Scholar]
- [103].Mu Q, Li Z, Li X, Mishra SR, Zhang B, Si Z, Yang L, Jiang W, Yan B, J. Phys. Chem. C 2009, 113, 5390–5395. [Google Scholar]
- [104].Bayraktar H, You C-C, Rotello VM, Knapp MJ, J. Am. Chem. Soc 2007, 129, 2732–2733. [DOI] [PubMed] [Google Scholar]
- [105].Rankin AGM, Trébosc J, Pourpoint F, Amoureux J-P, Lafon O, Solid State Nucl. Magn. Reson 2019, 101, 116–143. [DOI] [PubMed] [Google Scholar]
- [106].Giuntini S, Cerofolini L, Ravera E, Fragai M, Luchinat C, Sci. Rep 2017, 7, 17934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Bower PV, Louie EA, Long JR, Stayton PS, Drobny GP, Langmuir. 2005, 21, 3002–3007. [DOI] [PubMed] [Google Scholar]
- [108].Lilly Thankamony AS, Wittmann JJ, Kaushik M, Corzilius B, Prog. Nucl. Magn. Reson. Spectrosc 2017, 102–103, 120–195. [DOI] [PubMed] [Google Scholar]
- [109].Anthis NJ, Clore GM, Q. Rev. Biophys 2015, 48, 35–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Cavanagh J, Fairbrother WJ, Palmer AG, Skelton NJ, Protein NMR Spectroscopy Principles and Practice, 2nd ed., Academic Press, Burlington, 2007, p. 885. [Google Scholar]
- [111].Wang A, Vangala K, Vo T, Zhang D, Fitzkee NC, J. Phys. Chem. C 2014, 118, 8134–8142. [Google Scholar]
- [112].Wang A, Vo T, Le V, Fitzkee NC, J. Phys. Chem. B 2014, 118, 14148–14156. [DOI] [PubMed] [Google Scholar]
- [113].Xie M, Hansen AL, Yuan J, Brüschweiler R, J. Phys. Chem. C 2016, 120, 24463–24468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Xie M, Li D-W, Yuan J, Hansen AL, Brüschweiler R, Chem. Eur. J 2018, 24, 16997–17001. [DOI] [PubMed] [Google Scholar]
- [115].Lesniak A, Fenaroli F, Monopoli MP, Åberg C, Dawson KA, Salvati A, ACS Nano. 2012, 6, 5845–5857. [DOI] [PubMed] [Google Scholar]
- [116].Ceccon A, Schmidt T, Tugarinov V, Kotler SA, Schwieters CD, Clore GM, J. Am. Chem. Soc 2018, 140, 6199–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Dal Cortivo G, Wagner GE, Cortelletti P, Padmanabha Das KM, Zangger K, Speghini A, Dell’Orco D, Meyer NH, Sci. Rep 2018, 8, 3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Ceccon A, Tugarinov V, Clore GM, J. Am. Chem. Soc 2019, 141, 94–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Ceccon A, Clore GM, Tugarinov V, J. Phys. Chem. B 2018, 122, 11271–11278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Hill RA, Hunt J, Sanders E, Tran M, Burk GA, Mlsna TE, Fitzkee NC, Environ. Sci. Technol 2019, 53, 2635–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Freeman R, Mareci TH, Morris GA, J. Magn. Reson 1981, 42, 341–345. [Google Scholar]
- [122].Bax A, Griffey RH, Hawkins BL, J. Magn. Reson 1983, 55, 301–315. [Google Scholar]
- [123].Wüthrich K, Nat. Struct. Biol 1998, 5, 492. [DOI] [PubMed] [Google Scholar]
- [124].Pervushin K, Riek R, Wider G, Wüthrich K, Proc. Natl. Acad. Sci. USA 1997, 94, 12366–12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Tugarinov V, Muhandiram R, Ayed A, Kay LE, J. Am. Chem. Soc 2002, 124, 10025–10035. [DOI] [PubMed] [Google Scholar]
- [126].Venters RA, Farmer Ii BT, Fierke CA, Spicer LD, J. Mol. Biol 1996, 264, 1101–1116. [DOI] [PubMed] [Google Scholar]
- [127].Zanzoni S, Pedroni M, D’Onofrio M, Speghini A, Assfalg M, J. Am. Chem. Soc 2016, 138, 72–75. [DOI] [PubMed] [Google Scholar]
- [128].Zahn R, Liu A, Lührs T, Riek R, von Schroetter C, López García F, Billeter M, Calzolai L, Wider G, Wüthrich K, Proc. Natl. Acad. Sci. USA 2000, 97, 145–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Krishna MMG, Hoang L, Lin Y, Englander SW, Methods. 2004, 34, 51–64. [DOI] [PubMed] [Google Scholar]
- [130].Schanda P, Brutscher B, J. Am. Chem. Soc 2005, 127, 8014–8015. [DOI] [PubMed] [Google Scholar]
- [131].Schanda P, Van Melckebeke H, Brutscher B, J. Am. Chem. Soc 2006, 128, 9042–9043. [DOI] [PubMed] [Google Scholar]
- [132].Chevelkov V, Xue Y, Krishna Rao D, Forman-Kay JD, Skrynnikov NR, J. Biomol. NMR 2010, 46, 227–244. [DOI] [PubMed] [Google Scholar]
- [133].Mori S, Abeygunawardana C, van Zijl PCM, Berg JM, J. Magn. Reson. Ser. B 1996, 110, 96–101. [DOI] [PubMed] [Google Scholar]
- [134].Hwang T-L, Mori S, Shaka AJ, van Zijl PCM, J. Am. Chem. Soc 1997, 119, 6203–6204. [Google Scholar]
- [135].Hwang T-L, van Zijl PCM, Mori S, J. Biomol. NMR 1998, 11, 221–226. [DOI] [PubMed] [Google Scholar]
- [136].Fitzkee NC, Torchia DA, Bax A, Protein Sci. 2011, 20, 500–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Bai Y, Englander SW, Proteins Struct. Funct. Bioinf 1996, 24, 145–151. [DOI] [PubMed] [Google Scholar]
- [138].Ragona L, Fogolari F, Romagnoli S, Zetta L, Maubois JL, Molinari H, J. Mol. Biol 1999, 293, 953–969. [DOI] [PubMed] [Google Scholar]
- [139].Engel MFM, Visser AJWG, van Mierlo CPM, Proc. Natl. Acad. Sci. USA 2004, 101, 11316–11321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Fawzi NL, Ying J, Ghirlando R, Torchia DA, Clore GM, Nature. 2011, 480, 268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Fawzi NL, Ying J, Torchia DA, Clore GM, J. Am. Chem. Soc 2010, 132, 9948–9951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Libich DS, Fawzi NL, Ying J, Clore GM, Proc. Natl. Acad. Sci. USA 2013, 110, 11361–11366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Yuwen T, Brady JP, Kay LE, J. Am. Chem. Soc 2018, 140, 2115–2126. [DOI] [PubMed] [Google Scholar]
- [144].Ceccon A, Lelli M, D’Onofrio M, Molinari H, Assfalg M, J. Am. Chem. Soc 2014, 136, 13158–13161. [DOI] [PubMed] [Google Scholar]
- [145].Egner TK, Naik P, Nelson NC, Slowing II, Venditti V, Angew. Chem. Int. Ed 2017, 56, 9802–9806; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2017, 129, 9934–9938. [Google Scholar]
- [146].Clore GM, Szabo A, Bax A, Kay LE, Driscoll PC, Gronenborn AM, J. Am. Chem. Soc 1990, 112, 4989–4991. [Google Scholar]
- [147].Oldfield CJ, Dunker AK, Annu. Rev. Biochem 2014, 83, 553–584. [DOI] [PubMed] [Google Scholar]
- [148].Theillet F-X, Kalmar L, Tompa P, Han K-H, Selenko P, Dunker AK, Daughdrill GW, Uversky VN, Intrinsically Disord. Prot 2013, 1, e24360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Dunker AK, Babu MM, Barbar E, Blackledge M, Bondos SE, Dosztányi Z, Dyson HJ, Forman-Kay J, Fuxreiter M, Gsponer J, Han K-H, Jones DT, Longhi S, Metallo SJ, Nishikawa K, Nussinov R, Obradovic Z, Pappu RV, Rost B, Selenko P, Subramaniam V, Sussman JL, Tompa P, Uversky VN, Intrinsically Disord. Prot 2013, 1, e24157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Uversky VN, Biochim. Biophys. Acta 2013, 1834, 932–951. [DOI] [PubMed] [Google Scholar]
- [151].Uversky VN, Front. Phys 2019, 7. [Google Scholar]
- [152].Habchi J, Tompa P, Longhi S, Uversky VN, Chem. Rev 2014, 114, 6561–6588. [DOI] [PubMed] [Google Scholar]
- [153].Iwahara J, Tang C, Marius Clore G, J. Magn. Reson 2007, 184, 185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Tang C, Louis JM, Aniana A, Suh J-Y, Clore GM, Nature. 2008, 455, 693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Giljohann DA, Seferos DS, Daniel WL, Massich MD, Patel PC, Mirkin CA, Angew. Chem. Int. Ed 2010, 49, 3280–3294; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2010, 122, 3352–3366. [Google Scholar]
- [156].Kogot JM, Parker AM, Lee J, Blaber M, Strouse GF, Logan TM, Bioconjugate Chem. 2009, 20, 2106–2113. [DOI] [PubMed] [Google Scholar]
- [157].Brancolini G, Kokh DB, Calzolai L, Wade RC, Corni S, ACS Nano. 2012, 6, 9863–9878. [DOI] [PubMed] [Google Scholar]
- [158].Wu M, Vartanian AM, Chong G, Pandiakumar AK, Hamers RJ, Hernandez R, Murphy CJ, J. Am. Chem. Soc 2019, 141, 4316–4327. [DOI] [PubMed] [Google Scholar]
- [159].Brancolini G, Corazza A, Vuano M, Fogolari F, Mimmi MC, Bellotti V, Stoppini M, Corni S, Esposito G, ACS Nano. 2015, 9, 2600–2613. [DOI] [PubMed] [Google Scholar]
- [160].Rai M, Yadav A, Gade A, Biotechnol. Adv 2009, 27, 76–83. [DOI] [PubMed] [Google Scholar]
- [161].Panáček A, Kolář M, Večeřová R, Prucek R, Soukupová J, Kryštof V, Hamal P, Zbořil R, Kvítek L, Biomaterials. 2009, 30, 6333–6340. [DOI] [PubMed] [Google Scholar]
- [162].Prucek R, Tuček J, Kilianová M, Panáček A, Kvítek L, Filip J, Kolář M, Tománková K, Zbořil R, Biomaterials. 2011, 32, 4704–4713. [DOI] [PubMed] [Google Scholar]
- [163].Pal I, Brahmkhatri VP, Bera S, Bhattacharyya D, Quirishi Y, Bhunia A, Atreya HS, J. Colloid Interface Sci 2016, 483, 385–393. [DOI] [PubMed] [Google Scholar]
- [164].Mangini V, Dell’Aglio M, Stradis AD, Giacomo AD, Pascale OD, Natile G, Arnesano F, Chem. Eur. J 2014, 20, 10745–10751. [DOI] [PubMed] [Google Scholar]
- [165].Brahmkhatri VP, Chandra K, Dubey A, Atreya HS, Nanoscale. 2015, 7, 12921–12931. [DOI] [PubMed] [Google Scholar]
- [166].Liu J, Hurt RH, Environ. Sci. Technol 2010, 44, 2169–2175. [DOI] [PubMed] [Google Scholar]
- [167].Molleman B, Hiemstra T, Langmuir. 2015, 31, 13361–13372. [DOI] [PubMed] [Google Scholar]
- [168].Bharti C, Nagaich U, Pal AK, Gulati N, Int. J. Pharm. Invest 2015, 5, 124–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Vallet-Regí M, Colilla M, Izquierdo-Barba I, Manzano M, Molecules. 2018, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Mamaeva V, Rosenholm JM, Bate-Eya LT, Bergman L, Peuhu E, Duchanoy A, Fortelius LE, Landor S, Toivola DM, Lindén M, Sahlgren C, Mol. Ther 2011, 19, 1538–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Zhou Y, Quan G, Wu Q, Zhang X, Niu B, Wu B, Huang Y, Pan X, Wu C, Acta Pharmaceutica Sinica B. 2018, 8, 165–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Mathelié-Guinlet M, Gammoudi I, Beven L, Moroté F, Delville M-H, Grauby-Heywang C, Cohen-Bouhacina T, Procedia Eng. 2016, 168, 1048–1051. [Google Scholar]
- [173].Tang L, Cheng J, Nano Today. 2013, 8, 290–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Vertegel AA, Siegel RW, Dordick JS, Langmuir. 2004, 20, 6800–6807. [DOI] [PubMed] [Google Scholar]
- [175].Lundqvist M, Sethson I, Jonsson B-H, Biochemistry. 2005, 44, 10093–10099. [DOI] [PubMed] [Google Scholar]
- [176].Lundqvist M, Sethson I, Jonsson B-H, Langmuir. 2005, 21, 5974–5979. [DOI] [PubMed] [Google Scholar]
- [177].Deng ZJ, Liang M, Toth I, Monteiro MJ, Minchin RF, ACS Nano. 2012, 6, 8962–8969. [DOI] [PubMed] [Google Scholar]
- [178].Roach P, Farrar D, Perry CC, J. Am. Chem. Soc 2006, 128, 3939–3945. [DOI] [PubMed] [Google Scholar]
- [179].Shang W, Nuffer JH, Muñiz-Papandrea VA, Colón W, Siegel RW, Dordick JS, Small. 2009, 5, 470–476. [DOI] [PubMed] [Google Scholar]
- [180].Vidaurre-Agut C, Rivero-Buceta E, Romaní-Cubells E, Clemments AM, Vera-Donoso CD, Landry CC, Botella P, ACS Omega. 2019, 4, 8852–8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Lowe BM, Skylaris C-K, Green NG, J. Colloid Interface Sci 2015, 451, 231–244. [DOI] [PubMed] [Google Scholar]
- [182].Boehm H-P, Angew. Chem 1980, 92, 328–328; [Google Scholar]; Angew. Chem. Int. Ed 1980, 19, 230–230. [Google Scholar]
- [183].Sikora A, Shard AG, Minelli C, Langmuir. 2016, 32, 2216–2224. [DOI] [PubMed] [Google Scholar]
- [184].Shrivastava S, McCallum SA, Nuffer JH, Qian X, Siegel RW, Dordick JS, Langmuir. 2013, 29, 10841–10849. [DOI] [PubMed] [Google Scholar]
- [185].Al-Jumaili A, Alancherry S, Bazaka K, Jacob MV, Materials. 2017, 10, 1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Hu Y, Shenderova OA, Hu Z, Padgett CW, Brenner DW, Rep. Prog. Phys 2006, 69, 1847–1895. [Google Scholar]
- [187].Liu H, Zhang L, Yan M, Yu J, J. Mater. Chem. B 2017, 5, 6437–6450. [DOI] [PubMed] [Google Scholar]
- [188].Calvaresi M, Arnesano F, Bonacchi S, Bottoni A, Calò V, Conte S, Falini G, Fermani S, Losacco M, Montalti M, Natile G, Prodi L, Sparla F, Zerbetto F, ACS Nano. 2014, 8, 1871–1877. [DOI] [PubMed] [Google Scholar]
- [189].Zanzoni S, Ceccon A, Assfalg M, Singh RK, Fushman D, D’Onofrio M, Nanoscale. 2015, 7, 7197–7205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Marchesan S, Prato M, Chem. Commun 2015, 51, 4347–4359. [DOI] [PubMed] [Google Scholar]
- [191].Zhang Y, Casabianca LB, J. Phys. Chem. Lett 2018, 9, 6921–6925. [DOI] [PubMed] [Google Scholar]
- [192].Viegas A, Manso J, Nobrega FL, Cabrita EJ, J. Chem. Educ 2011, 88, 990–994. [Google Scholar]
- [193].Szczygiel A, Timmermans L, Fritzinger B, Martins JC, J. Am. Chem. Soc 2009, 131, 17756–17758. [DOI] [PubMed] [Google Scholar]
- [194].Mayer M, Meyer B, Angew. Chem. Int. Ed 1999, 38, 1784–1788; [DOI] [PubMed] [Google Scholar]; Angew. Chem 1999, 111, 1902–1906. [Google Scholar]
- [195].Malam Y, Loizidou M, Seifalian AM, Trends Pharmacol. Sci 2009, 30, 592–599. [DOI] [PubMed] [Google Scholar]
- [196].Allen TM, Cullis PR, Adv. Drug Delivery Rev 2013, 65, 36–48. [DOI] [PubMed] [Google Scholar]
- [197].Spillantini MG, Schmidt ML, Lee VMY, Trojanowski JQ, Jakes R, Goedert M, Nature. 1997, 388, 839–840. [DOI] [PubMed] [Google Scholar]
- [198].Bodner CR, Dobson CM, Bax A, J. Mol. Biol 2009, 390,775–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Bodner CR, Maltsev AS, Dobson CM, Bax A, Biochemistry. 2010, 49, 862–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Andresen JM, Gayán J, Djoussé L, Roberts S, Brocklebank D, Cherny SS, The USVCRG, The HDMCRG, Cardon LR, Gusella JF, MacDonald ME, Myers RH, Housman DE, Wexler NS, Ann. Hum. Genet 2007, 71, 295–301. [DOI] [PubMed] [Google Scholar]
- [201].Jayaraman M, Kodali R, Sahoo B, Thakur AK, Mayasundari A, Mishra R, Peterson CB, Wetzel R, J. Mol. Biol 2012, 415, 881–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Madine J, Doig AJ, Middleton DA, Biochemistry. 2006, 45, 5783–5792. [DOI] [PubMed] [Google Scholar]