

Abstract
Resiquimod is an immunopotent toll-like receptor 7/8 agonist with anti-tumor activity. Despite being potent against skin cancers, it’s poorly tolerated systemically due to toxicity. Integrating resiquimod into nanoparticles presents an avenue to circumvent the toxicity problem. Herein, the preparation of degradable nanoparticles with covalently bound resiquimod and their systemic application in cancer immunotherapy is reported. Dispersion in water of amphiphilic constructs integrating resiquimod covalently bound via degradable amide or ester linkages yields immune activating nanoparticles. The degradable agonist-nanoparticle bonds allow the release of resiquimod from the carrier nanoparticles. In vitro assays with antigen presenting cells demonstrate the nanoparticles retain the immunostimulatory activity of resiquimod. Systemic administration of the nanoparticles and checkpoint blockade (aPD-1) to a breast cancer mouse model with multiple established tumors triggers anti-tumor activity evidenced by suppressed tumor growth and enhanced CD8+ T-cell infiltration. Nanoparticles with ester links, which hydrolyze more readily, yield a stronger immune response with 75% of tumors eliminated when combined with aPD-1. The reduced tumor growth and presence of activated CD8+ T-cells across multiple tumors suggests the potential for treating metastatic cancer.
Keywords: Immunotherapy, metastatic breast cancer, nanotechnology, resiquimod, toll-like receptor agonist, vaccines
Graphical Abstract
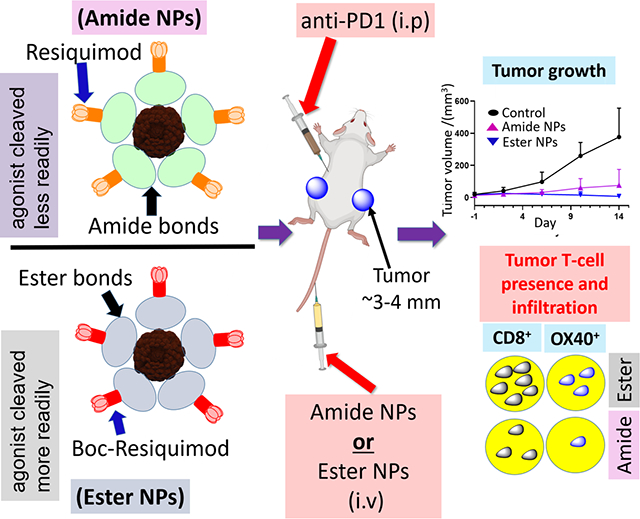
Nanoparticles exhibiting resiquimod’s toll-like receptor 7/8 anti-tumor and immunostimulating activity are reported. Polymeric conjugates with resiquimod linked to the polymer via hydrolysable bonds are translated into nanoparticles facilitating systemic administration of resiquimod for cancer immunotherapy. Intravenous administration of the nanoparticles to mice with pre-established breast cancer tumors triggers a strong immune response resulting in suppressed tumor growth and T-cell infiltration.
1. Introduction
Despite the development of numerous therapies for the treatment of metastatic breast cancer (MBC), the 5-year survival rates remain low necessitating the need for developing effective therapies.[1, 2] Major strategies for treating MBC until recently include radiation therapy, chemotherapy, endocrine therapy and hormone receptor (ERBB2+) targeted therapy in combination with chemotherapy or endocrine therapy.[3–5] Challenges associated with these approaches include the development of resistance, the possibility of developing infertility and toxicity-related side effects.[5]
Exploration of immunotherapy as an alternative standard modality of cancer treatment (including MBC) has gathered momentum in recent years. [6, 7] For instance, a phase 3 clinical trial of nab-paclitaxel in combination with the programmed death ligand 1 (PD-L1) inhibitor atezolizumab was observed to improve progression-free survival of MBC patients.[8, 9] Immunotherapy presents a powerful approach for treating metastatic cancers since it leverages the body’s immune system to generate a global antitumor response.[10, 11] Success of this strategy hinges on the generation of a fast, specific, and durable immune response typically through the use of antigens (peptide epitopes or whole protein) and/or immune potentiators such as toll-like receptor (TLR) agonists (e.g. CpG oligodeoxynucleotide, imidazoquinolines and poly (I:C)), sometimes with checkpoint blockade.[12–18] Thus, the efficient delivery of the immunomodulators to antigen presenting cells (APCs) in vivo is key to the realization of a systemic antitumor effect.[19] Parenteral administration of the immunomodulating agents in their native form is typically negatively impacted by issues such as poor solubility, systemic cytotoxicity, poor circulation and limited bioavailability.[20] As such, there is currently significant interest in the design of carrier vehicles that negate the aforementioned limiting factors and thus ensure the optimal in vivo delivery of immunomodulators to immune cells.
Among the approaches explored, the incorporation of immunomodulating agents into nanoparticles (NPs) is attracting significant attention.[19, 21, 22] To this end, NP carriers based on lipids, polymers and inorganic NPs are being investigated to overcome the issues associated with delivery of immunomodulating agents.[23–30] Advances in NP fabrication methods allow for the preparation of NPs of specific sizes and shapes which can therefore be tuned towards a desired application.[31–33] In the case of immunotherapy, fabrication of small NP sizes (<100 nm) is favored since they are known to effectively drain to the lymph nodes where most APCs reside and lead to effective delivery.[34] The formulation of immunomodulating nanocarriers can be via physical encapsulation of the immunomodulator(s) into NPs, through covalent attachment onto preformed NPs, or covalent attachment to surfactant-like molecules/polymers which are then transformed into NPs.[14, 21, 35–39]
Taking into account the need to retain the immunomodulating activity of the cargo, the encapsulation approach is advantageous in that there is no structural alteration; thus activity is typically not impacted. However, the encapsulation of some immunomodulating molecules (e.g. water soluble imidazoquinolines) can be quite challenging, leading to poor loading efficiencies, and moreover, the possibility of cargo leakage from the carrier. On the other hand, covalent attachment mitigates cargo leakage and also offers the possibility of tuning the amount of cargo loading and its rate of release.[40] The shortcomings of covalent attachment are the possible loss in activity of the immunomodulating agent due to alteration upon attachment, the limitation on reactions that can be imposed on the chemical functionalities involved and the conceivable need for intricate and laborious reactions/purifications.[30] Nevertheless, covalent attachment coupled with size control and NP stabilization (via crosslinking) is best suited for intravenous administration as it enables good cargo retention, circulation and delivery of the immunomodulator to APCs due to efficient draining to lymph nodes.
Herein, we report the preparation of a NP system in which resiquimod (Scheme 1, (1)), an immunomodulatory, small molecule, TLR7/8 agonist, is covalently attached to a biodegradable polymeric nanoplatform for the potential treatment of MBC in combination with PD-1 checkpoint blockade immunotherapy. Resiquimod is an immunopotent imidazoquinoline that has been typically used for topical treatment of skin lesions and skin cancers and its exploitation in cancer immunotherapy through parenteral administration routes has been attracting attention in recent years.[41, 42] Parenteral administration of resiquimod in humans is characterized by poor tolerability, poor circulation, reaction on injection site and flu-like symptoms which are indicative of large quantities of cytokines in the blood.[43–46] The incorporation of resiquimod (or other imidazoquinoline TLR agonists) into a NP system is anticipated to mitigate the aforementioned challenges thus improving its delivery through parenteral administration routes.[47, 48] Moreover, NPs improve uptake by APCs thus improving the potential of realizing a robust antitumor immune response.[49] Thus, several reports have demonstrated the integration of resiquimod into NPs and their efficacy as anti-tumor immunomodulators.[50] Most reports in literature however focus on the encapsulation of resiquimod rather than covalent conjugation primarily due to the concern of potential loss of activity due to chemical modification, as well as the limited reactivity of the hydroxyl and amine groups of the TLR agonist.[50–52]
Scheme 1.

Schematic illustration of the materials and strategy employed for the design of the resiquimod prodrug NPs and their application in treatment of mice with established bilateral MBC tumors. Conversion of resiquimod (1) to Boc-protected resiquimod (2) (A).[55] Coupling of (1) to azido-PEG12-activated ester to afford azido-PEG12-resiquimod (3) with resiquimod linked to the polymer via an amide link (B). Synthesis of azido-PEG12-resiquimod with PEG linked to resiquimod via an ester link (6) (C). Preparation of amphiphilic hyperbranched polymer-resiquimod conjugates via strain promoted azide-alkyne reactions of an alkyne functionalized hyperbranched degradable polymer with either (4), (5) or (6) to obtain amphiphiles (7), carboxylic acid decorated amphiphile, and (8), [56–58] polymer-resiquimod amphiphile with resiquimod linked to the polymer via amide links, and formation of amphiphilic polymer-resiquimod conjugate with resiquimod linked to the polymer via ester links (9), (D). Formation of micellar NPs from the hyperbranched polymer-resiquimod conjugates and their application in treatment of mice with bilateral breast cancer tumors (E). NPs from (8) are referred to as amide NPs and those from (9) are termed ester NPs.
There are limited examples of nanoplatforms wherein resiquimod is covalently incorporated. For example, Weissleder and co-workers prepared adamantane derivatives of resiquimod which were incorporated into NPs through host-guest interactions with cyclodextrin and administered intravenously in a murine model of colon adenocarcinoma tumors.[53] They observed a reduction in tumor growth and reduced systemic toxicity evidenced by an absence of rapid weight loss which is characteristic of mice injected with free resiquimod. Recently, Forrest et al. prepared a resiquimod-tocopherol conjugate which was formulated into a nanosuspension by incorporating it with hyaluronic acid modified with tocopherol.[45] The nanosuspensions were active against tumors in a murine model for head and neck cancers as well as against mast cell tumors in canines. In another study, resiquimod was covalently conjugated to a lipid which was integrated into cationic lipid NPs that were subsequently complexed with a tuberculosis antigen and administered to mice via intra muscular injection.[54] Although the liposomes migrated to lymph nodes (APC hubs) and did not dissipate quickly (as with free resiquimod), there was no significant difference in antibody response between the two treatments. The authors attributed this observation to reduced activity of resiquimod due to lipidation. However, another potential contributing factor, the impact of size of the liposomes (400–600 nm), was not discussed. Smaller NPs migrate to the lymph nodes more effectively while larger NPs are taken up by APCs away from lymph nodes which then migrate to the lymph node. Additionally, it is not apparent whether the liposomes were able to release the conjugated resiquimod to the cellular environment for recognition. Ilyinskii et al. fabricated PLGA NPs incorporating resiquimod attached by covalent conjugation which led to strong humoral and cellular immune responses with minimal systemic cytokine production in vivo.[49]
The present study demonstrates a NP system aimed at treating MBC via immunotherapy. Resiquimod was anchored onto a Boltorn H40 biodegradable hyperbranched polymer through hydrolytically degradable agonist-NP linkages (ester/amide) and formulated into NPs with sizes below 20 nm. The conjugation allows for control over the amount of resiquimod that can be loaded, while the ester bonds ensure it is released in its native form. It was envisioned these NPs could be administered through parenteral routes and interact with immune cells to elicit strong and robust immune responses while overcoming the aforementioned challenges associated with the administration of resiquimod.
2. Results and Discussion
2.1. Synthesis of amphiphilic prodrug conjugates
Our approach to the design of the polymer-resiquimod conjugates and NPs is illustrated in Scheme 1. The strategy is inspired by our previous works employing highly efficient, metal free, ‘click’ chemistry reactions to generate hybrid nanoconstructs from Boltorn H40 unimicelles with sizes in the range of 10–30 nm.[58, 59] First, we prepared the azide-PEG resiquimod conjugates in which the imidazoquinoline was conjugated via the available reactive functional groups (amine and hydroxyl) affording amide (-CONH-agonist link, blue colored link in (8), Scheme 1D) and ester (-COO-agonist link, purple colored link in (9), Scheme 1D) linkages, respectively (Scheme 1). Both linkages have the potential to undergo hydrolytic degradation and thus should release resiquimod (via degradation of the amide, blue (8) or ester, purple links (9) Scheme 1D, E) when taken up by APCs leading to immune system activation through the TLR 7/8 pathway. [60] In addition to being hydrolytically labile, the linkages are stable enough in circulation to ensure negligible cleavage and agonist release which would lead to toxicity. The amide-linked azido PEG resiquimod conjugate (4) was a product of coupling the activated ester (3) with resiquimod (1) via the amine group of the agonist under basic conditions. Formation of the amide-PEG agonist product was confirmed by mass spectrometry (m/z 940 Da) following isolation by HPLC (Figure S1, Supporting information (SI)). For the ester-PEG agonist conjugate, the amine group of the agonist was first Boc protected to afford (2) prior to conjugation with azido-PEG12 acid in order to prevent the amine from reacting. [55] Characterization of (2) was achieved via 1H NMR and mass spectrometry (Figure S2, SI). The carboxylic acid of azido-PEG12 acid (5) was linked to (2) through the tertiary hydroxyl group in the presence of Boc anhydride (Boc2O) and dimethylaminopyridine (DMAP), yielding the ester link (6) similar to the esterification reaction in Scheme 1D. [56] Product formation was also confirmed via 1H NMR and mass spectrometry (m/z 1040 Da) (Figure S3, SI).
Next, the amphiphilic polymer-resiquimod conjugates were prepared via strain promoted azide-alkyne click chemistry (SPAAC) by conjugation of (4) to obtain (8), or (6) to obtain (9) (direct route) (scheme 1D). The formation of (9) could also be accomplished indirectly by functionalization of (4) with carboxylic acid moieties using (5) to yield (7) which can undergo esterification with (2) to afford (9) (scheme 1D). Preparation and characterization of the hyperbranched alkyne functionalized polymer was previously reported by us. [58] SPAAC is a powerful tool for preparing conjugates due to its versatility in terms of functionalities that can be accommodated and high reaction yields under mild reaction conditions. Conjugation of (4) to the hyperbranched alkyne polymer afforded the amphiphilic agonist conjugate (8) which was characterized by size exclusion chromatography (SEC) and 1H NMR. By SEC, a shift in peak elution time to shorter elution time was observed after conjugation of (4) which was indicative of an increase in molar mass of the starting alkyne functionalized polymer. Prior to analysis of the purified product by 1H NMR, removal of the free azido-PEG agonist conjugate was confirmed by FTIR in which the peak of the azide (2109 cm−1) was absent.[58] Subsequent analysis by 1H NMR showed peaks due to the presence of -CH2- of PEG at 3.7 ppm and those of the aryl protons of resiquimod (7.7–8.5 ppm) confirming the presence of the azido PEG agonist in the amphiphile (Figure S4, SI). Following the same amphiphile synthesis approach, (9) was obtained by coupling the alkyne functionalized hyperbranched polymer with (6). In the alternative approach to obtain (9), (5) was conjugated to the alkyne functionalized hyperbranched polymer followed by conjugation of (2). Both approaches provided the desired product as ascertained by SEC and 1H NMR (1H NMR is shown Figure S4, SI).
2.2. Hydrolytic degradation of the agonist-NP links and NP formation
Upon uptake by cells, the amide/ester links between the polymer and the agonist are expected to cleave releasing resiquimod which is the immunoactive component. Indeed, when the azido-PEG-agonist conjugates were incubated in aqueous buffer pH 7.4 (phosphate buffered saline, PBS), free resiquimod was observed for both conjugates (amide/ester) (Figure 1A–B). Degradation was faster for the ester conjugate compared to the amide conjugate, indicating the ester link was more labile. [60] The degradation of both conjugates may however be faster in cells due to enzymatic activity and low pH of endosomal compartments. Nonetheless, the results show that the agonist-polymer links bear hydrolytic degradability, and resiquimod can be freed from the NPs to activate the immune system. Aqueous solubilization of the amphiphilic polymer-resiquimod conjugates in water gave core-shell like nanostructures with the agonist on the surface. This observation is in line with our previous reports on similar amphiphilic materials. [58, 59] Herein, for brevity, NPs from (8) are referred to as amide NPs and those from (9) are termed ester NPs. The NP diameter was 10 ± 0.3 nm (amide NPs) and 15 ± 0.9 nm (ester NPs) by dynamic light scattering (DLS) (Figure 1C). The observed sizes allow for improved circulation of the agonist and efficient permeation and accumulation into lymph nodes where APCs are in abundance. Moreover, metastasis typically happens via the lymphatic system and thus the resiquimod is able to reach the migrating/migrated tumors thereby effecting therapy. The zeta potentials of the NP solutions were −54 ± 4 mV (amide NPs) and −9 ± 1 mV (ester NPs) and there were no signs of precipitation upon preparation and isolation implying good stability. The NPs had an irregular spherical-ellipsoid morphology as ascertained by transmission electron microscopy (TEM) 7 ± 3 nm (amide NPs) and 14 ± 5 nm (ester NPs) (Figure 1C and Figure S5).
Figure 1.

HPLC chromatograms showing hydrolysis of (6) in PBS at 37 °C leads to ‘release’ of resiquimod (A). Calculated amount of resiquimod released via hydrolysis of (6) and (4) in PBS at 37 °C over time (B). Dynamic light scattering (DLS) measured sizes of amide NPs (10 ± 0.3 nm) and ester NPs (15 ± 0.9 nm) formulated from (8) and (9), respectively. For DLS, N=5 measurements and for TEM, the average diameter and standard deviation were calculated based on at least 50 NPs from different parts of the grid. TEM images showing the morphology of the NPs are inserted (C) (larger TEM images are available in supporting information, Figure S5). NP immune activity in vitro was determined (i) using RAW Blue cells by looking at the NF-KB activity measured via secreted embryonic alkaline phosphatase (SEAP) assay (N=3) (D), and (ii) by determining the ability of the NPs to cause maturation of BMDCs (N=3) (E). In (D), n.s. = not significant and * represents p < 0.05 compared to the polymer NPs at a specific concentration, as analyzed using ANOVA with a Tukey post hoc test. The EC50 values in (D) were estimated using the dose response (stimulation) fitting in GraphPad. The composite maturation index (CMI) reported in (E) represents an unweighted average of the expression of CD80, CD86, and MHCII normalized to the iDC (untreated) population. iDC: immature DCs; DMSO aq: dimethylsulfoxide in PBS buffer (the resiquimod was dissolved in a small amount of DMSO (<10% v/w) prior to dilution with PBS thus DMSO aq. was a control); LPS: lipopolysaccharide (* represents p < 0.05 compared to the iDC population, as analyzed using ANOVA with a Tukey post hoc test).
Since hydrolytic degradation of the agonist-NP amide and ester links is expected over time, studying the stability over time does not give true reflection of the NP stability. Removal of the resiquimod moieties over time results in PEG-carboxylic acid decorated NPs remaining (NPs of (7)), which are stable in aqueous solution. Thus, formulation of the NPs is recommended to be conducted just prior to application. It is noteworthy that an attempt to physically encapsulate the resiquimod into the hyperbranched acid decorated polymer gave very poor loading, (<0.1 % w/w). Similarly, attempts to load resiquimod into PEG-cholesterol amphiphilic conjugates also resulted in very poor loading, (<0.5% w/w), indicating the difficulty associated with encapsulation of resiquimod into micelles. Our observation is congruent to that made by Duong and co-workers. [51] We suspect resiquimod bears an amphipathic like behavior partitioning itself between the aqueous phase and hydrophobic cores resulting in losses in steps of preparation (e.g. dialysis) leading to poor loading efficiency. Conjugation mitigates such losses and leads to a higher loading, which is estimated as 15 % w/w in this work.
2.3. NP immunoactivity in vitro
Subsequently, we investigated the ability of the resiquimod PEG conjugates and NPs to activate the immune system via TLR 7/8 signaling in vitro. First, we carried out an NF-κB reporter assay using RAW-blue macrophages expressing the NF-κB/AP-1-inducible secreted embryonic alkaline phosphatase (SEAP) reporter gene. We compared NF-κB activation in untreated cells with various treatments in a dose dependent manner to determine the fold increase in NF-κB activation at each concentration.
Activation was observed from ~ 20 nm - 5000 nm for free resiquimod and the ester NPs whilst, for the amide NPs, activation was only observed from ~1000 nm - 5000 nm (Figure 1D). The fold change in NF-κB activation from 20 nm is much higher for the ester NPs than that of the amide NPs suggesting the ester NPs are more potent. The fold change for the amide NPs is only significantly different from that of the polymer NPs and dextrose controls at the highest concentration tested (5000 nM) (p<0.05, ANOVA) whereas, for the ester NPs, the difference is significant starting from 62 nM. Indeed, an estimation of the EC50 revealed the EC50 of the amide NPs (7118 nM) was significantly greater than that of the ester NPs (more than 20 times that of the ester NPs). This observation is linked to how labile each of the polymer-agonist bonds are (ester versus amide links), and congruent with the agonist-NP hydrolytic degradation study which showed the NPs with the ester links degraded and released resiquimod faster than the NPs with the amide links (Figure 1B). Another possibility could be an effect of linker length as reported by Westcott et. al. wherein activity of amide-linked resiquimod conjugates was observed to be affected by the choice of linker and length. [61] Interestingly, (2) was observed to cause immune system activation implying the protecting group does not render the resiquimod ineffective, possibly because the Boc group comes off in endosomal compartments due to low pH and enzymatic activity (esterases) thus “releases” resiquimod. As expected, free resiquimod (1) showed the highest fold increase in NF-κB activation at all concentrations above 10 nm. NPs fabricated from (7) did not show any activation nor did dextrose (5% w/v) and PBS buffer pH 7.4 which are typically employed for dispersing samples prior to in vivo injections.
We also investigated the activation of bone marrow-derived dendritic cells (BMDCs) by the NPs. DCs are key APCs for triggering the adaptive immune system, thus evaluating their activation by the designed NPs is critical. We co-cultured different concentrations of the NPs with BMDCs and then assessed the expression of the co-stimulatory molecules (CD80 and CD86) and MHCII which are determinative of the extent of maturation. This was reported as a simplified metric - the composite maturation index (CMI). [62] The CMI represents an unweighted average of the expression of CD80, CD86, and MHCII normalized to the immature DCs iDC (untreated) population. Thus, the CMI provides an indication of the immunogenicity of a molecule/NP system. Cells treated with PBS buffer pH 7.4, aqueous dextrose (5% w/v) and NPs fabricated from the PEG-carboxylic acid amphiphile did not increase maturation in comparison to the iDC negative control, indicating they do not activate BMDCs (Figure 1E). The positive control treatment, lipopolysaccharide (LPS, TLR4 agonist), showed a high CMI, as did the resiquimod-treated cells, which is expected as resiquimod is a strong immunopotentiator. The DC activation was dose dependent with CMI values decreasing with a decrease in concentration. Ester NPs also activated BMDCs in a dose dependent manner with significantly greater CMIs (p<0.05) compared to iDCs observed above the lowest concentration. On the other hand, amide NPs also showed dose dependent BMDC activation but the CMIs were not significantly greater than the iDCs CMI. These results further confirm that the ester NPs are a more effective immunopotentiator compared to the amide NPs due to the former bearing more labile polymer-agonist links than the latter. It is noteworthy that enzymatic activity of proteases (amide NPs) and esterases (ester NPs) encountered in biological milieu in vivo can accelerate hydrolytic degradation of the agonist-NP links, improving release of resiquimod for both NPs. [63, 64] The flow cytometry gating strategy for the maturation experiment is shown in Figure S6 (with examples showing iDCs (A) and DCs incubated with resiquimod (B)).
2.4. Cellular uptake
The TLR 7/8 receptor is resident within the endosomal compartment and thus the NPs need to be internalized by APCs to impart immune system activation. [65] We therefore studied the internalization of the NPs by RAW Blue macrophages and murine breast cancer cells (NDLs) using NPs labelled with fluorescent pHrodo-red dye obtained using a previously described approach. [58, 59] The pHrodo-red dye is an intracellular pH indicator showing strong fluorescence at low pH (lysosomes) and poor fluorescence at neutral-basic pH. NPs derived from these fluorescent conjugates were incubated with either RAW Blue macrophages or NDL mammary carcinoma cells at 4 °C and at 37 °C and uptake was determined after 4 hr by flow cytometry and fluorescence microscopy. Endocytosis is expected to occur at 37 °C while it is constrained at 4 °C. Albeit uptake is restricted at 4 °C, NPs adhere to the surface of the cells and thus signal due to non-endocytosed NPs should be observed. [58] In Figure 2A, uptake was clearly observed by fluorescence microscopy for the NPs with the ester-agonist link in both cell lines at 37 °C while no signal was observed for the samples incubated at 4 °C and the control samples (PBS pH 7.4). The merged images of the fluorescent NPs for both cell lines at 37 °C show colocalization of the fluorescence signal (endosomal compartments) with that of the cells as observed in the bright field images. The results indicate the NPs were trafficked into endosomal compartments. Similar results were also observed for the fluorescently-labeled NPs with the amide links (Figure S7). Flow cytometry also confirmed the signal of the fluorescently-labeled NPs (ester and amide) was significantly greater than that of the control for both cell lines affirming the cells internalized the NPs (Figure 2B, C). The absence of signal, both via microscopy and flow cytometry, for the samples incubated at 4 °C indicates there is no contribution to the observed fluorescence signals due to particles adhered to the cell surfaces. For both NPs at 37 °C, cell uptake was observed to be greater for the RAW Blue cell line compared to the NDL cell line based on fluorescence signal intensity observed in both microscopy and flow cytometry results. Additionally, the uptake of ester NPs was greater than that of amide NPs by both cell lines at 37 °C. The superior uptake of ester NPs can be attributed to the differences in surface chemistry of the two NPs. The ester NPs have a more apolar surface than amide NPs due to the presence of the Boc groups on the surface. Apolar moieties tend to facilitate better cell membrane adhesion and destabilization and thus likely lead to the observed superior uptake. [58]
Figure 2.

Uptake of pHrodo-red labeled NPs by NDL cells (N=3) and RAW Blue macrophages (N=3) obtained at 4 hr (37 °C and 4 °C) via microscopy with NPs visible (red signal) in the fluorescence channel and merged channel. The pHrodo-red fluoresces in lysosomal compartments. Image scale bars are 50 μm (A). Flow cytometry results illustrating the uptake of the pHrodo-red labelled NPs in NDL cancer cells (B) and RAW Blue macrophages (C). * represents p < 0.05 as analyzed using ANOVA with a Tukey post hoc test. Cell viability of RAW Blue macrophages, BMDCs (N=3) and NDL (N=3) cell lines obtained at 24 hr after incubation with different concentrations of ester NPs (D). Microscopy results for uptake of pHrodo red labeled amide NPs are shown in Figure S7.
2.5. Cytotoxicity
Prior to preclinical studies, we evaluated the toxicity of the agonist-bearing NPs in vitro. In drug delivery, it is desirable for the carrier material to have minimal or no toxicity. The carrier material in this case, the PEG-acid functionalized branched polymer (which does not have the agonist), was observed in a previous study to have no noticeable toxicity both in vitro and in vivo. [58, 59] We proceeded to study the cytotoxicity of the drug-bearing NPs in order to determine if the presence of the agonist resulted in substantial toxicity which would negatively impact in vivo administration. In addition, we also observed the direct antitumor effect of the agonist on cancer cells. Cytotoxicity was determined via the MTT cell viability assay wherein the NPs were assessed for toxicity against BMDCs, RAW Blue macrophages and the NDL cell line. The ester NPs were employed for this study since these are more hydrolytically labile, leading to release of larger amounts of resiquimod. The concentration range was chosen based on the amount of resiquimod intended for in vivo administration (4 μg resiquimod per gram of mouse), and thus a maximum of 1 mg mL−1 of NPs corresponding to ~150 μg mL−1 of resiquimod was selected. Cell viability generally decreased with increasing concentration of material for all three cell lines with viabilities dropping to nearly 50% for BMDCs and macrophages suggesting high amounts of resiquimod lead to toxicity (Figure 2D). The viability of the NDL cells decreased with increasing concentration of NPs (hence resiquimod) down to ca. 90% at the highest concentration suggesting accumulation of the NPs in tumors would have some antitumor effect. From our previous studies, we have observed that BMDCs and RAW macrophages are more sensitive to increasing amounts of polymer NP material than cancer cells. [59] This observation can be attributed to high level of material uptake by BMDCs and macrophages compared to cancer cells (Figure 2).
2.6. Biodistribution of the agonist nanoparticles in mice with breast cancer tumors
Biodistribution of the NPs was evaluated in vivo using mice with bilateral implanted orthotopic breast tumors (NDL cells, ~4 mm). Similar to the NP uptake study, the NPs employed were fluorescently labelled with the Cy7 dye to allow in vivo fluorescence imaging by SPECTRAL LAGO X. NPs were administered via the tail vein and circulation in vivo was confirmed via fluorescence imaging wherein strong Cy7 signal was observed throughout the body shortly after injection (Figure 3A). For comparison, a group of control mice was injected with saline and fluorescence was not detected when imaged on SPECTRAL LAGO X confirming that the signal in the non-control group came from Cy7. Similar NPs without the Cy7 had the same fluorescence profile as saline showing no signal by fluorescence spectroscopy when excited at λ 750 nm hence saline was employed as a control. The fluorescence signal diminished after 4 hr with the remaining signal observed near clearance organs (liver, spleen, kidney and intestine) (Figure 3A and Figure S8, SI). Taking into account that some hydrolytic degradation of the agonist-NP links of the NPs was observed at pH 7.4, the reduction in circulation at 4 hr reduces the potential for sustained systemic toxicity. Indeed, the tumors, liver, spleen, kidneys, heart, skin and hindlimb muscle were collected for ex vivo imaging and, in all cases, the fluorescence intensity in organs from mice injected with fluorescent NPs was greater than that of organs from mice injected with dye-free NPs (Figure 3B–C). It is noteworthy that the results in (C) are not normalized for organ weights and are therefore semi-quantitative comparisons of the controls versus the mice injected with fluorescent NPs.
Figure 3.

Biodistribution studies of Cy7-labelled NPs following injection via the mouse tail vein (N=4 each, control (saline) or dye-labeled NPs). Fluorescence images acquired using the SPECTRAL LAGO X, 3 min post injection and 4 hr later (A). Ex-vivo fluorescence images of harvested organs as visualized on the SPECTRAL LAGO X 4 hr post injection (B). Assessment of accumulation of the NPs in different organs through quantification of the fluorescence signals (C). The results in (C) are not normalized for organ weights and are therefore semi-quantitative comparisons of the controls versus the mice injected with fluorescent NPs. Comparisons were performed via the Welch’s t-test (**p ≤0.01, ***p ≤ 0.001, and **** p ≤ 0.0001).
Accumulation in tumors was significant which is important for the antitumor activity of resiquimod.[14] Not surprisingly, the fluorescence signal was greatest in the kidneys, which act as a primary organ of clearance. Notably, the experiment showed the NPs had a longer circulation time compared to resiquimod, which we observed in a recent study to have a very short residence time of less than 20 min. [66]
2.7. Preclinical evaluation of the TLR 7/8 agonist nanoparticles in treatment of breast cancer
The efficacy of the NPs and free resiquimod with and without a checkpoint blockade antibody anti-PD-1 (aPD-1) was evaluated in mice that had pre-established breast cancer tumors. To mimic metastases, NDL breast cancer tumor chunks were transplanted bilaterally into the left and right flanks of mice and allowed to grow to ~3–4 mm before treatment began (Figure 4A). Mice were treated with either resiquimod (with/without aPD-1), the ester NPs (with/without aPD-1), or amide NPs (with/without aPD-1) or saline for the untreated mice (no treatment control, NTC). We have previously reported and also observed in various studies in our laboratory that the aPD-1 antibody alone generally has no significant effect on suppression of tumor growth for the NDL tumor model. [16] Results from a recent study (unpublished) in our lab that are congruent with our previous report are shown herein (Figure S9, SI). However, the presence of the aPD-1 can augment the efficacy of the NPs by curtailing immunosuppressive activity. [67–70] Albeit the amide NPs exhibited weak immunogenicity in vitro, these studies do not fully capture the complex nature of an in vivo system. [49] We reasoned that various enzymes potentially resident in immune cells or encountered in tumors and different organs of the body may accelerate degradation of the agonist-NP links resulting in improved efficacy. NPs were administered intravenously while the aPD-1 was given intraperitoneally as detailed in the treatment protocol in Figure 4A. We opted for the intraveneous route since not all tumors are easily accessible for intratumoral injection or removal by surgery especially when there is metastasis. The administration route employed also ensures that the resiquimod interacts directly with tumors (accumulates) imparting its anti-tumor effect resulting in improved efficacy. Thus, this treatment approach would afford a global anti-tumor response.
Figure 4.

In vivo efficacy of the amide NPs and ester NPs compared with resiquimod with/without checkpoint blockade (aPD-1). Treatment protocol for preclinical studies with mice bearing pre-established bilateral NDL breast cancer tumors (N=4 animals per group) (A). The treatment began when tumor sizes were ca. 4 mm in diameter. Average percentage changes in tumor volumes ± standard deviations (pooled, average of all tumors per group per time point) (B, C) and the corresponding weight change (pooled) (D, E). Figures (C) and (E) are extracted from the selected areas in (B) and (D) respectively. Statistical significance was determined via one-way ANOVA (per time point) with a Tukey post-test (n.s = not significant, *p ≤ 0.05). Plots of the change in tumor volumes of individual tumors and body weights are available in SI (Figure S10).
The NP effectiveness in the presence and absence of aPD-1 was first assessed. The change in average tumor volume for mice treated with either NPs or free resiquimod (with or without aPD-1) indicate agonist-containing treatments significantly suppressed tumor growth compared to the NTC cohort (Figure 4B, C). No differences in tumor sizes across all cohorts were observed at day 3 suggesting the treatments had not yet taken effect (Figure 4B, C). However, post day 3, differences in the treatments emerged with both NP-treated groups outperforming the NTC cohort in arresting tumor growth. Tumor regression was enhanced with the ester NPs + aPD-1 and complete tumor regression occurred in most (3 out of 4) of the mice treated, highlighting superiority over the amide NPs + aPD-1 (Figure S10, SI). The average increase in tumor volume for both NP-treated groups was less than for the NTC group from days 10 to 14, underscoring the efficacy of the TLR 7/8 NPs in treatment of breast cancer. A slight increase in tumor volume was observed for the mice treated with amide NPs + aPD-1 indicating that the ester NPs + aPD-1 treatment is more effective than the amide NPs + aPD-1, which can be related to the hydrolytic stability of the agonist-polymer links.[60] As previously mentioned, the ester NPs show more propensity to release resiquimod intracellularly than the amide NPs and thus make available more agonist upon cell uptake in different organs compared to the amide NPs. Notably, where tumor regression was observed, the tumor size reductions occurred for both the left and right flank tumors after systemic injection (for both NPs), suggesting the NPs + aPD-1 are potentially effective for MBC treatment. Though effective at slowing tumor growth, the NP treatments without aPD-1 did not eliminate tumors and therefore the results also highlight the importance of including aPD-1 in the treatments.
Treatment with free resiquimod+aPD-1 also reduced and eliminated 75% of the tumors by day 14 (Figure S10, SI). There was no significant difference in tumor volume reduction for the cohorts treated with free resiquimod (with or without aPD-1) as compared with the NPs. For the treatment with free resiquimod without aPD-1, although there was tumor shrinkage for some of the animals, tumor elimination was only observed in 30%. These results further suggest the presence of aPD-1 improves the efficacy of the treatment with resiquimod in this tumor model.
We also monitored mouse weight, since toxicity is of concern when administering resiquimod systemically. Weight loss of about 10% (average of all mice per cohort) was observed for the free resiquimod-treated cohorts (with and without aPD-1) whereas the NP treatments and the NTC cohorts did not show weight loss (Figure 4D, E and Figure S10B, SI).[53] This observation indicates that there is negligible hydrolytic degradation of the agonist-NP links in circulation. The weight loss exhibited by the resiquimod-treated mice was significant in comparison to the NTC cohort. Thus, despite being effective at eliminating tumors, the free resiquimod based treatments exhibit toxicity which is not observed for the NP based treatments.
2.8. Assessment of tumor T-cell infiltration via histological analysis
At the end of the preclinical efficacy study, the tumors were harvested and the presence of immune cells indicating effectiveness of the different treatments towards breast cancer was assessed via immunohistochemistry. The ultimate goal of the treatments is immune system activation and production of active cytotoxic T-cells (primarily CD8+ cells, OX40+).[68] As shown in Figure 5A, tumors from the NTC group were viable with a typical diameter of ~6–7 mm as evidenced from hematoxylin and eosin stained images (H&E). Very few CD8+ T-cells (brown punctate staining) were observed in the NTC tumors, with little to no infiltration by the cytotoxic T-cells due to lack of/poor immune system activation and hence the continued rapid growth of the tumors.
Figure 5.

Histology of hematoxylin and eosin (H&E)-stained tumor tissues (purple-pink), CD8+ T-cell stained tissues (brown spots) and OX40 ligand stained tissues (activated T-cells, brown spots) from mice in the no-treatment (NTC) cohort (A) and the NP-treated cohorts (B-D). Tumors from mice treated with amide NPs + aPD-1 are shown in (B) and those from mice treated with ester NPs + aPD-1 (two mice), are shown in (C-D). In B-D, the faint pink-purple color around the tumors in H&E is dead/dying tumor tissue which appears as faint gray in the CD8+ T-cell and OX40 images. The dotted outlines represent areas that are magnified in the subsequent series of figures illustrated by the roman numerals next to the black arrows. Black scale bars are 3 mm while red scale bars are 0.5 mm. Quantification of the number of immune cells in tumors is shown in (E). Statistical significance in (E) was determined via one-way ANOVA with a Tukey post-test (****p ≤ 0.0001).
Moreover, a comparison of images of tumors stained for CD8+ T-cells (e.g. Figure 5A, ii–iv) with those stained for the OX40 ligand (indicating activated CD8+ T-cells, Figure 5A, v–vii) shows that only a fraction of the CD8+ T-cells were activated. Tumors from mice treated with the amide NPs were smaller than those isolated from NTC mice and tumor cell death was observed from the H&E images.
For the agonist-containing treatments, the remaining viable tumors were observed to be surrounded by dead/dying tumor cells indicating the effectiveness of the treatments (Figure 5B–D and Figures S11–S14, SI). Infiltration of tumors by CD8+ T-cells was evident throughout the tumors in all cases, albeit the stain indicative of activated CD8+ cells (OX40) was slightly less than that observed for CD8+ T-cells. Since our in vivo results indicated that the most effective and non-toxic treatment was that of the combination of NPs with aPD-1, further discussion on immunohistochemistry is thus focused on the ester NPs + aPD-1 and amide NPs + aPD-1 cohorts.
For ester NPs + aPD-1, few viable tumor cells remained, as observed on the H&E images (Figure 5C–D and Figure S15, SI) which is in line with tumor growth measurements. The remaining areas of viable tumor were heavily infiltrated with CD8+ T-cells as shown in Figure 5C–D (ii–iii), and most importantly, a large proportion of the CD8+ T-cells were activated (Figure 5C–D, (iv–vi)). The density of CD8+ T-cells and activated CD8+ T-cells (OX40) based on the respective stains indicates the ester NPs generated more active CD8+ T-cells than the amide NPs (Figure S16) and hence is the more potent of the two (see quantification in Figure 5E). The CD8+ T-cell area coverage was on average ~8% and OX40+ coverage was on average ~4% for the ester NPs whilst the coverage for amide NPs and the saline control was below 2% for both CD8+ T-cells and OX40+ cells. The superior activity of the ester NPs over the amide NPs is linked to the lower hydrolytic stability of the ester links compared to the amide links. Since both NPs activate the immune system to generate CD8+ T-cells and diminish tumor growth, it is most likely that, with additional treatments or slightly higher dosages, results observed with the ester NPs can be achieved with the amide NPs.
3. Conclusions
In summary, polymeric NPs incorporating resiquimod covalently bound via degradable/hydrolytically labile bonds (amide/ester) were prepared and demonstrated to have antitumor activity when administered systemically along with aPD-1 in mice with established breast tumors. In the absence of aPD-1, the NPs slowed tumor growth but did not eliminate tumors. Combined with aPD-1, NPs with ester-resiquimod linkages were more effective at slowing tumor growth and tumor elimination compared to NPs with amide-resiquimod linkages due to the ester links being more hydrolytically labile/easily degradable (most likely accelerated by activity of esterases in cells) thus releasing the agonist more readily. Both NPs combined with aPD-1 initiated a strong immune response with CD8+ T-cells and OX40 ligand associated T-cells (activated T-cells) which was observed via immunohistochemistry. CD8+ T-cell concentration was enhanced in tumors from the NP-treated cohorts suggesting the antitumor immunoactivity was systemic and thus the NPs are potentially applicable for the treatment of MBC along with checkpoint blockade. When the NP treatments were administered, the toxicity-related effect (weight loss) observed upon systemic application of free resiquimod was not detected, implying the integration of resiquimod into NPs helped mitigate the toxicity issue and thus the NPs can potentially be translated to the clinic.
4. Experimental section
Preparation of Boc-protected resiquimod (2):
We adapted the approach reported by Vilaivan which gave high yields.[55] Resiquimod (10 mg, 32 mmol) and Boc anhydride (17 mg, 78 mmol) were weighed into a glass vial that was sealed with a rubber septum and vented with a needle. To the vial was added tert-butanol (2 mL) and the reaction mixture was placed on a heating block maintained at 37 °C for 2 days with agitation to ensure complete dissolution of the contents. At the end of the reaction, the product was purified via HPLC (acetonitrile/water, 10% to 90% acetonitrile over 60 min, λ 254 nm and 318 nm) and the collected fractions were concentrated via lyophilization to afford a white solid product (11 mg, 83%). ESI ([M+H]+ = m/z 415 Da).
Synthesis of azido PEG resiquimod amide (4):
Azido PEG12 succinimidyl ester (3) (10 mg, 1 eq.) was dissolved in dry DMF (500 μL) followed by the addition of (2) (1 eq) dissolved in dry DMF (500 μL) then DIPEA (25 μL). The resulting solution was left on a shaker at room temperature for 24 hr and an aliquot (50 μL). The reaction was stopped and the product was purified via semi-preparative HPLC (acetonitrile/water, 10% to 90% acetonitrile over 60 min, λ 254 nm and 318 nm) to afford the product as a colorless gel (azide peak observed at 2100 cm−1). The compound mass was confirmed by mass spectrometry (m/z=940.19 Da [M+H]+). Yield =3.2 mg, 25%. Higher yields (ca. 60%) were achieved using 10 eq of (3) and keeping all other reactants/solvents constant.
Synthesis of azido PEG resiquimod ester (6):
We adapted the synthetic approach reported by Zipse and co-workers. [56] In a glovebag under nitrogen, (2) (1 eq.) was weighed into a dry glass vial (flame dried) and dissolved in anhydrous tetrahydrofuran (100 μL). Stock solutions of azido-PEG12-acid (5) (1 eq.), DMAP (0.1 eq) and distilled triethylamine in dry tetrahydrofuran were prepared in separate dry vials and the desired amounts of each reagent were aliquoted into the glass vial containing (2) (azido-PEG12-acid (5) (1 eq.), DMAP (0.1 eq) and distilled triethylamine (3 eq.)). The vial was sealed and purged with a slow flow of nitrogen to lower the volume of the contents to ca. 100 μL followed by cooling down to −20 °C over a period of 10 minutes. To the resulting solution was added molten Boc2O (5 eq.) and the reaction solution was left to stir at room temperature in the dark for 48 hr. [56] The solvent was removed using a stream of nitrogen gas and the residue was purified by HPLC (C18 column, acetonitrile/water (with 0.1% NH4OH), 10% to 90% acetonitrile over 60 min, λ 254 nm and 318 nm). ESI ([M+H]+) = m/z 1040 Da, [M-N2+H]+ = m/z 1012 Da, [M-Boc+H]+ = m/z 940 Da.
Preparation of amphiphilic hyperbranched polymer decorated with carboxylic acid moeities (7):
To the alkyne functionalized hyperbranched polymer (1 eq of alkyne units) in DMF was added the azido-PEG12-acid polymer (5) (3 eq) dissolved in DMF. The resulting solution was left on a shaker at room temperature for 3 days and the reaction was assumed to have reached the maximum conversion. The crude product was purified by dialysis against water for 3 days (MWCO 3.5 kDa) and lyophilized to obtain a pale yellow-brown solid which was analyzed by FTIR and 1H NMR.
Synthesis of amphiphilic hyperbranched polymer-resiquimod conjugate with amide polymer-agonist links (8):
To the alkyne functionalized Boltorn H40 hyperbranched polymer (4 mg, 4 eq of alkyne units) in DMF was added the (4) (5 eq) dissolved in DMF. The resulting solution was left on a shaker at room temperature for 3 days and the reaction was assumed to have reached the maximum conversion. An aliquot (50 μL) was withdrawn and analyzed by HPLC to determine the extent of the reaction. The crude product was purified via dialysis (SpectraPor, MWCO 3.5 kDa) and lyophilized to obtain a pale-brown gel which was analyzed by FTIR and 1H NMR. Yield=7 mg. The amount of (4) remaining in solution as quantified by HPLC was used to estimate the amount of (4) attached to the hyperbranched alkyne polymer by difference. It was assumed that the mass of the hyperbranched core did not change since it was far greater than the membrane MWCO. The extent of resiquimod loading was thus estimated by calculating the amount of resiquimod based on moles of (4) attached which was divided by the total mass of recovered material and expressed as a percentage. Estimated agonist loading, 15% w/w.
Synthesis of amphiphilic hyperbranched polymer-resiquimod conjugate with ester polymer-agonist links (9) (direct method, similar to synthesis of (8)):
The same method detailed above for the synthesis of (8) was followed with (6) being used instead of (4). The crude product was purified via dialysis for 6 hours (SnakeSkin, MWCO 10 kDa) and lyophilized to obtain an off white gel-solid which was analyzed by FTIR and 1H NMR. A known amount of the product was dissolved in aqueous TFA (2 mL, 1% w/v) and placed on a shaker at 37 °C and left for 48 hr to effect hydrolysis and the amount of resiquimod released (hence loading) was estimated by HPLC.
Synthesis of amphiphilic hyperbranched polymer-resiquimod conjugate with ester polymer-agonist links (9) (alternate method, Scheme 1D)
(i). Using the DMAP/Boc2O/TEA system:
The procedure used for the synthesis of (6) is followed substituting (5) with (7). [56] At the end of the reaction, the solvent was removed using a stream of nitrogen gas and the residue was dissolved in dry dichloromethane followed by precipitation into hexane/ether (1:1) twice. Product recovery was achieved via centrifugation at 4000 rpm for 10 min and decanting the solvent. The product was dissolved in a small amount of DMSO and diluted with water followed by purification purified via dialysis against deionized water for 6 hours under rapid stirring (SnakeSkin, MWCO 10 kDa) with the water being changed every two hours. Lyophilization afforded the product as an off white gel-solid which was analyzed by FTIR and 1H NMR.
(ii). Using the COMU/MTBD system:
In a glovebag under nitrogen, (2) (5 mg, 12 eq.) was weighed into a dry glass vial and dissolved in anhydrous DMF (500 μL). (7) (10 eq.), COMU (10 eq.) and MTBD (20 eq) in separate vials were dissolved in dry DMF (500 μL each) and added to the glass vial with the solution of (2).[57] The vial was sealed and left to stir at room temperature in the dark for 48 hr. The solvent removed using a stream of nitrogen gas and the residue was dissolved in dry dichloromethane followed by precipitation into hexane/ether (1:1). Product recovery was achieved via centrifugation at 4000 rpm for 10 min and decanting the solvent. The product was dissolved in a small amount of DMSO and diluted with water followed by purification purified via dialysis against deionized water for 6 hours under rapid stirring (SnakeSkin, MWCO 10 kDa) with the water being changed every two hours. Lyophilization afforded the product as an off white gel-solid which was analyzed by FTIR and 1H NMR.
NB:
Purification could also be achieved by passing the aqueous solution through a size exclusion column (Sephadex G75) and eluting the product with water followed by lyophilization of the collected fractions. A known amount of the product was dissolved in aqueous TFA (2 mL, 1% w/v) and placed on a shaker at 37 °C and left for 48 hr to effect hydrolysis and the amount of resiquimod released (hence loading) was estimated by HPLC. The loading was estimated based on the mass of resiquimod released and the mass of the starting product. Agonist loading, ca.13 % w/w.
Aqueous solubilization of (8)/(9) to obtain nanoparticles:
The polymer-agonist amphiphile (4 mg) was dissolved in degassed DMSO (50 μL) then previously degassed acetone (1 mL) and left to stir for 15 min. Filtered (0.2 μm sterile filter), deoxygenated water (2 mL) was then added to the solution using a syringe pump (0.1 mL min−1) while stirring at ca. 500 rpm. The acetone was removed by bubbling nitrogen gas through the solution while it was continuously stirred. Bubbling of the gas was continued till the volume was reduced to ~0.8 mL and the concentration of the NPs was then adjusted accordingly using filtered water. An aliquot of the solution was obtained for analysis, diluted to ca. (0.5 mg mL−1) and mixed with (25 μL) of PBS buffer (pH 7.4, 25 mmol NaCl) then analyzed via DLS (size).
Hydrolysis of (4)/(6) (resiquimod “release”):
A known amount of the conjugate (3 mg) was weighed into a glass vial and dissolved in (3 mL) of PBS buffer (pH 7.4, 25 mmol NaCl). The vial was placed on a heating block at 37 °C that was shaking at 500 rpm and aliquots (150 mL) were withdrawn at predetermined time intervals and analysed by HPLC (C12 column, acetonitrile/water (0.05 % TFA)), 10% to 90% acetonitrile over 60 min, λ 254 nm and 318 nm). The amount of resiquimod in solution was determined by HPLC based on a standard curve generated using pure resiquimod. The percentage of resiquimod “released” via the hydrolytic degradation process was then determined based on the mass of released resiquimod in solution and the calculated mass of resiquimod in the starting conjugate. Since the hydrolysis of (4) was very slow, we attempted to replace PBS with acetate buffer (pH 5, 0.1 m), to see if its hydrolysis is accelerated in lysosomal condition but the difference in hydrolysis was only marginally faster.
Modified QUANTI-Blue assay with RAW Blue Cells:
The in vitro immunogenicity of the NPs was assessed via a modified QUANTI-Blue Assay. RAW-Blue macrophages in growth media comprising of DMEM (Gibco) supplemented with 10% heat-inactivated fetal bovine serum, 1% Penicillin/Streptomycin and (200 μg mL−1) Zeocin were maintained in a humidified CO2 (5%) incubator at 37 °C. Prior to plating in a 96 well plate for the analysis, the cells were suspended in the media following mechanical detachment. The cell suspension was centrifuged followed by replacement of the supernatant with fresh media (37 °C) and resuspension of the cells. The cells were counted and plated on a 96 well tissue culture plate (1 × 105) cells per well in 100 μL of media to which the desired treatments were then added in triplicate (total volume 200 μL/well). Following a 24 hr incubation period, 50 μL from each well was aliquoted and transferred to a new 96 well tissue culture plate containing 150 μL QUANTI-Blue solution in each well. After 3 hr of incubation of the new plate, the absorbance was read with a TECAN microplate reader (λ=625 nm) to determine the NF-kB induced secreted embryonic alkaline phosphatase (SEAP).
Dendritic Cell Phenotype and Maturation:
NPs were added to mouse BMDC cultures at 37 °C for a period of 24 hr prior to analysis. NPs were then cultured at the desired concentrations. Dendritic cell immunophenotype was quantified by measuring cell-surface marker expression by flow cytometry. Following NP incubation, DCs were lifted by incubating with a 5 mM Na2EDTA (Fisher Scientific) in PBS solution at 37 °C for 10 min. DCs were then washed with 1% fetal bovine serum in PBS and incubated with antibodies against CD16/CD32 (Fcγ III/II Receptor) (clone 2.4G2, IgG2b, k); (BD Pharmingen, CA) for 15 min at 4 °C to block Fcγ receptors on DCs. Cells were washed and then stained with antibodies against CD80 (clone 16–10A1, IgG2, k), CD86 (clone GL1, IgG2a, k), MHCII (I-A/I-E, clone M5/114.15.2, IgG2b, k), and CD11c (clone HL3, IgG1, l2) for 30 min at 4 °C. Data acquisition was performed using flow cytometry (Attune NxT). Flow cytometry gating strategy for the dendritic cell (DC) maturation experiment is shown in the SI (examples shown in Figure S6, immature DCs (A) and DCs incubated with resiquimod (B)). The cell populations were gated based on singlets then only live cells were selected using UV Zombie dye staining. Cells were further chosen according to the cell markers CD11c for DCs. DC populations were then analyzed for other surface expressions. All of the positive staining gates were set according to Fluorescence Minus One (FMO) controls. Statistical analyses were performed using general linear model ANOVA followed by post-hoc pair-wise comparisons using the Tukey test. Differences were considered significant when p ≤ 0.05 using Prism (Version 7, GraphPad, La Jolla, CA)
Fluorescently labelled analogs of (8):
(i). pHrodo red labelled analog for uptake:
To the alkyne functionalized hyperbranched polymer (1 eq of alkyne units) in DMF was added the azido-PEG3-pHrodo red dye (0.15 eq) dissolved in DMF. The resulting solution was left on a shaker for 24 hr at room temperature in the dark. An aliquot (50 μL) was withdrawn and analyzed by HPLC to determine the extent of reaction. Maximum conversion was achieved based on the absence of the peak due to the pure dye in the HPLC chromatogram. To the reaction solution was added (4) (2 eq based on starting alkyne units (excess)) dissolved in DMF. The resulting solution was left on a shaker for 24 hr at room temperature in the dark. The reaction was deemed to have reached the maximum conversion and the red coloured crude product was purified with a centrifugal filter (Millipore, MWCO 3 kDa) and lyophilized to obtain a dark green gel.
(ii). Cy7 labelled analog for biodistribution:
To the alkyne functionalized hyperbranched polymer (1 eq of alkyne units) in DMF was added the sulfo azido Cy7 dye (0.15 eq) dissolved in DMF. The resulting solution was left on a shaker for 24 hr at room temperature in the dark. An aliquot (50 μL) was withdrawn and analyzed by HPLC to determine the extent of reaction. Maximum conversion was achieved based on the absence of the peak due to the pure dye in the HPLC chromatogram. To the reaction solution was added (4) (2 eq based on starting alkyne units (excess)) dissolved in DMF. The resulting solution was left on a shaker for 24 hr at room temperature in the dark. The reaction was deemed to have reached the maximum conversion and the green coloured crude product was purified with a centrifugal filter (Millipore, MWCO 3 kDa) and lyophilized to obtain a dark green gel.
Fluorescently labelled analogs of (9):
The procedures detailed above for the synthesis of the fluorescently labelled analogs of (8) was followed as above substituting (4) with (6) to obtain the fluorescently labelled analogs of (9). To check for the presence of the agonist in the product, a small the product was subjected to hydrolysis in TFA (2 mL, 1% w/v) as described above.
Nanoparticle formation:
For in vitro experiments, pHrodo red NPs/Cy7-labelled NPs were prepared following the aqueous solubilization procedure described above. To obtain the nanoparticle solutions in dextrose (5% w/v) at the desired concentrations, nanoparticle solutions were concentrated to reduce the volume below the intended final volume. A pre-weighed amount of dextrose was added followed by dilution with filtered water (0.2 μm) to the desired volume which gave the desired concentration of NPs and dextrose.
Cellular Internalization Studies:
(a). Flow cytometry:
Cells (BMDCs or RAW Blue macrophages) were plated at 2 × 105 cells/well in 24-well tissue culture plates for flow cytometry or at 4 × 105 cells/dish in 35-mm dishes for microscopy, 24 hr prior to experiments. Particles were incubated with cells continuously at 37 °C in complete media (100 μg mL−1 in 1 mL for imaging dishes and (300 μL) for 24-well plates) for up to 24 hr (triplicate). For flow cytometry, cells were rinsed once with PBS and collected at 0, 4 and 24 hr in (170 μL) TrypLE Express dissociation buffer (ThermoFisher Scientific, Waltham MA). Samples were then analyzed on a BD FACScan flow cytometer.
(b). Fluorescence Microscopy:
Internalization of NPs was confirmed via fluorescent microscopy using a custom upright fluorescence microscope (Mikron, San Marcos, CA) with a digital Cascade 512b camera (Photometrics, Tucson, AZ) and a 63 × water-immersion objective (Achroplan, Zeiss, NY) driven with SimplePCI 6 software. At day 6 of culture, cells were plated on a 35 mm tissue-culture dish. 24 hr later, media was removed and cells were incubated with media containing NPs at (100 μg mL−1) for 4 hr. After 4 hr, cells were subsequently washed twice with PBS and imaged.
Cell Viability of NDL Cancer cells:
Cells were plated at 2000 cells in (100 μL) media per well in 96-well tissue culture plates 24 hr prior to peptide/particle addition. Nanoparticles were added to each well in (100 μL) complete media (triplicate). Cells were incubated continuously with NPs (1, 0.5, 0.25, and 0.1 mg mL−1) for 24 hr at 37 °C in a 5% CO2 incubator. MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) reagent (Invitrogen, Carlsbad, CA) was added to media at a concentration of (0.5 mg mL−1), and cells were incubated 2 hr at 37 °C in a 5% CO2 incubator. Media was removed, and formazan crystals dissolved in DMSO (100 μL/well, Sigma Aldrich, St. Louis, MO). Absorbance was measured using a TECAN (San Jose, CA) Infinite® M1000 microplate reader.
Cell Viability of antigen presenting cells:
Cells (BMDCs or RAW Blue macrophages) were plated at 2500 cells per well in 96-well tissue culture plates 24 hr prior to particle addition. Nanoparticles were added to each well in 100 μL complete media (triplicate). Cells were incubated continuously with NPs (1, 0.5, 0.25, and 0.1 mg mL−1) for 24 hr at 37 °C in a 5% CO2 incubator. MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) reagent (Invitrogen, Carlsbad, CA) was added to media at a concentration of (0.5 mg mL−1), and cells were incubated 2 hr at 37 °C in a 5% CO2 incubator. Media was removed, and formazan crystals dissolved in DMSO (100 μL/well, Sigma Aldrich, St. Louis, MO). Absorbance was measured using a Tecan (San Jose, CA) Infinite® M1000 microplate reader. The viability was determined based on equation 1 below.
| (1) |
Animal experiments:
All animal experiments were conducted under a protocol approved by Stanford University or the University of California, Davis, Institutional Animal Care and Use Committee (IACUC). To obtain the subcutaneous NDL metastatic tumor model of breast cancer, 6–9 weeks old female FVB mice were used (Charles River, Willmington, MA). NDL tumor chunks obtained from a donor mouse that was sacrificed were surgically implanted bilaterally into the left and right flank fat pad of each mouse. At the time of surgery, animals were maintained anesthetized with 3% isoflurane (in oxygen, flow rate: 2 L min−1). Tumors were allowed to grow to ~3–4 mm before the therapeutic treatment protocol below was started.
Biodistribution:
Nanoparticles for the biodistribution study were prepared following the self-assembly procedure described for the preparation of Cy7 labelled NPs used for the cell uptake studies. The volume was adjusted to achieve the desired concentration using a stream of nitrogen gas above the solution to induce evaporation. Dextrose was then added to obtain a suspension of the NPs in a 5% (w/v) solution of dextrose. For administration and imaging, mice were shaved to remove fur prior to commencing the experiment. Fluorescent NPs were intravenously injected via the tail vein. Following the injection of NPs, whole body fluorescence images of the mice were taken under 3% isoflurane (in oxygen, flow rate: 2 L min−1) anesthesia at different time points using a SPECTRAL LAGO X Imaging System (Spectral Instruments Imaging; Tucson, AZ, USA) with an excitation and emission wavelength of 745 and 790 nm, respectively. The exposure time was 1 sec and the field of view (FOV) was 25 × 25 cm. Aura Imaging Software was used for data analysis. The whole-body images were scanned at 5 min and 4 hr post I.V injection. At 4 hr post injection, mice were euthanized and various organs/tissues (tumors, heart, kidneys, spleen, skin and liver) were dissected and imaged ex vivo simultaneously. Another group of mice that were intravenously injected with saline as negative control were included and imaged using the same protocol.
In vivo preclinical evaluation of the TLR 7/8 agonist nanoparticles in treatment of breast cancer:
Nanoparticles for in vivo preclinical evaluation of the TLR 7/8 agonist nanoparticles in treatment of breast cancer experiments were prepared following the aqueous solubilization procedure described above (for (8)/(9)). The volume was adjusted to achieve the desired concentration using a stream of nitrogen gas above the solution to induce evaporation. Dextrose was then added to obtain a suspension of the NPs in a 5% (w/v) solution of dextrose. Therapeutic treatment with NPs (equivalent to ca.80–100 μg resiquimod) was intraveneously injected in (100–180 μL) of solution to achieve a dose of (4 μg g−1) of mouse. Each mouse was also administered with aPD-1 (200 μg) intraperitoneal (i.p.) injection. At the time of injection, animals were maintained anesthetized with 3% isoflurane (in oxygen, flow rate: 2 L min−1). Treatments were repeated two more times on days 4 and 7. Ultrasound imaging was used to obtain the volume size of tumors twice per week using an Acuson Sequoia® 512 system (Siemens Medical Solution USA, Inc., Issaquah, WA). Mice were euthanized 7 days after the last treatment and tumors harvested for histology. For mice that were treated with resiquimod (with or without aPD-1), a solution of the agonist was prepared by dissolving the agonist in DMSO (10 mg/mL) which was diluted to (0.6 mg/mL) using PBS pH 7.4 (-Ca, -Mg). In the NTC cohort, animals were injected intravenously with saline only (100 μL). For the study shown in SI (Figure S8), the treatment protocol was similar to that described herein and animals were injected with aPD-1 (aPD-1 cohort, N=4, i.p. injection, 200 mg) or not subjected to any treatment (no treatment cohort, N=4). Tumors sizes were measured via ultrasound and the tumor size (mm3) was calculated based on equation 2 below.
| (2) |
In equation 2, l, w, and h are the lengths of the major, minor, or vertical axis, respectively. The percentage change in tumor volume was calculated using equation 3.
| (3) |
In equation 3, Vt is the tumor volume recorded at time (t) and V0 is the initial volume of the tumor when treatments commenced.
The change in mouse mass was calculated using equation 4 below wherein, mt is the average mouse mass of a cohort recorded at time (t) and m0 is the average mouse mass of a cohort recorded when treatments commenced.
| (4) |
Immunohistochemistry:
Tumor samples were fixed in neutral buffered formalin for 24 hr or longer followed with being transferred into ethanol (70 % v/v) and kept in ethanol overnight before further processing. A Tissue-Tek VIP autoprocessor (Sakura, Torrance, CA) was used to process tumors which were then embedded in Paraplast paraffin (melting temperature 56–60 °C), sectioned to 4 μm and mounted on glass slides. Tumor sections were then stained using Mayer’s hematoxylin and eosin (H&E) to facilitate histology and morphology evaluation. To observe the presence of CD8+ T-cells, staining was performed with a rat anti-mouse CD8a primary antibody (1:500;14–0808 e Biosciences). For immune cells quantification, the histology images were analyzed with “Analyze Particles” function in ImageJ, and presented as the area percentages covered by either CD8+ or Ox40+ cells.
Statistical Analysis
Statistical analyses were performed using Prism (Version 7, GraphPad, La Jolla, CA) with p≤0.05 being considered as statistically significant. All data were represented as mean ± standard deviation (minimum N=3). Sample sizes/replicates are indicated for the experiments are in figure legends. Comparisons of two or more groups were performed via the Welch’s t-test or one-way analysis of variance (ANOVA) with a Tukey post-test. No pre-processing of data was performed prior to statistical analysis.
Supplementary Material
Acknowledgements
We gratefully acknowledge the support of the National Institutes of Health (NIHR01CA112356, NIHR01CA210553, R01CA253316, and R01CA250557). The authors thank Dr. Qian (Jane) Chen with the UC Davis Pathology Department for assistance with tumor transplant surgeries and acquisition of immunohistochemistry images and Caitlyn Miller for assistance with RAW Blue cell lines.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Supporting Information (SI)
Supporting Information is available from the Wiley online library or from the author.
Contributor Information
Hamilton Kakwere, Molecular Imaging Program at Stanford (MIPS), Department of Radiology, Stanford University, Palo Alto, CA 94305, USA.
Hua Zhang, Molecular Imaging Program at Stanford (MIPS), Department of Radiology, Stanford University, Palo Alto, CA 94305, USA.
Elizabeth S. Ingham, Department of Biomedical Engineering, University of California (Davis), Davis, CA 95616, USA
Marina Nura-Raie, Molecular Imaging Program at Stanford (MIPS), Department of Radiology, Stanford University, Palo Alto, CA 94305, USA.
Spencer K. Tumbale, Molecular Imaging Program at Stanford (MIPS), Department of Radiology, Stanford University, Palo Alto, CA 94305, USA
Riley Allen, Department of Biomedical Engineering, University of California (Davis), Davis, CA 95616, USA.
Sarah M. Tam, Department of Biomedical Engineering, University of California (Davis), Davis, CA 95616, USA
Bo Wu, Molecular Imaging Program at Stanford (MIPS), Department of Radiology, Stanford University, Palo Alto, CA 94305, USA.
Cheng Liu, Molecular Imaging Program at Stanford (MIPS), Department of Radiology, Stanford University, Palo Alto, CA 94305, USA.
Azadeh Kheirolomoom, Molecular Imaging Program at Stanford (MIPS), Department of Radiology, Stanford University, Palo Alto, CA 94305, USA.
Brett Z. Fite, Molecular Imaging Program at Stanford (MIPS), Department of Radiology, Stanford University, Palo Alto, CA 94305, USA
Asaf Ilovitsh, Molecular Imaging Program at Stanford (MIPS), Department of Radiology, Stanford University, Palo Alto, CA 94305, USA.
Jamal S. Lewis, Department of Biomedical Engineering, University of California (Davis), Davis, CA 95616, USA
Katherine W. Ferrara, Molecular Imaging Program at Stanford (MIPS), Department of Radiology, Stanford University, Palo Alto, CA 94305, USA
References
- [1].Mariotto AB, Etzioni R, Hurlbert M, Penberthy L and Mayer M, Cancer Epidemiol. Biomark. Prev, 2017, 26, 809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lobbezoo DJ, van Kampen RJ, Voogd AC, Dercksen MW, van den Berkmortel F, Smilde TJ, van de Wouw AJ, Peters FP, van Riel JM, Peters NA, de Boer M, Peer PG and Tjan-Heijnen VC, Br. J. Cancer, 2015, 112, 1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Waks AG and Winer EP, JAMA, 2019, 321, 288. [DOI] [PubMed] [Google Scholar]
- [4].Patel TA and Perez EA, Metastatic Breast Cancer. In Management of Breast Diseases, Jatoi I; Kaufmann M, Eds. Springer Berlin Heidelberg: Berlin, Heidelberg, 2010; pp 425. [Google Scholar]
- [5].Harbeck N, Penault-Llorca F, Cortes J, Gnant M, Houssami N, Poortmans P, Ruddy K, Tsang J and Cardoso F, Nat. Rev. Dis. Primers, 2019, 5, 66. [DOI] [PubMed] [Google Scholar]
- [6].Yang W, Zhou Z, Lau J, Hu S and Chen X, Nano Today, 2019, 27, 28. [Google Scholar]
- [7].Borst J, Ahrends T, Bąbała N, Melief CJM and Kastenmüller W, Nat. Rev. Immunol, 2018, 18, 635. [DOI] [PubMed] [Google Scholar]
- [8].Page DB, Bear H, Prabhakaran S, Gatti-Mays ME, Thomas A, Cobain E, McArthur H, Balko JM, Gameiro SR, Nanda R, Gulley JL, Kalinsky K, White J, Litton J, Chmura SJ, Polley M-Y, Vincent B, Cescon DW, Disis ML, Sparano JA, Mittendorf EA and Adams S, npj Breast Cancer, 2019, 5, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Diéras V, Hegg R, Im S-A, Shaw Wright G, Henschel V, Molinero L, Chui SY, Funke R, Husain A, Winer EP, Loi S and Emens LA, N. Engl. J. Med, 2018, 379, 2108. [DOI] [PubMed] [Google Scholar]
- [10].Farkona S, Diamandis EP and Blasutig IM, BMC Med, 2016, 14, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Min Y, Roche KC, Tian S, Eblan MJ, McKinnon KP, Caster JM, Chai S and Herring LE, Nature Nanotechnology, 2017, 12, 877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gutjahr A, Papagno L, Nicoli F, Lamoureux A, Vernejoul F, Lioux T, Gostick E, Price DA, Tiraby G, Perouzel E, Appay V, Verrier B and Paul S, J Immunol, 2017, 198, 4205. [DOI] [PubMed] [Google Scholar]
- [13].Tran TH, Tran TTP, Truong DH, Nguyen HT, Pham TT, Yong CS and Kim JO, Acta Biomater, 2019, 94, 82. [DOI] [PubMed] [Google Scholar]
- [14].Zhang L, Jing D, Wang L, Sun Y, Li JJ, Hill B, Yang F, Li Y and Lam KS, Nano Lett, 2018, 18, 7092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Nagato T, Lee Y-R, Harabuchi Y and Celis E, Clin. Cancer. Res, 2014, 20, 1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Silvestrini MT, Ingham ES, Mahakian LM, Kheirolomoom A, Liu Y, Fite BZ, Tam SM, Tucci ST, Watson KD, Wong AW, Monjazeb AM, Hubbard NE, Murphy WJ, Borowsky AD and Ferrara KW, JCI Insight, 2017, 2, e90521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Heo MB and Lim YT, Biomaterials, 2014, 35, 590. [DOI] [PubMed] [Google Scholar]
- [18].Conniot J, Scomparin A, Peres C, Yeini E, Pozzi S, Matos AI, Kleiner R, Moura LIF, Zupančič E, Viana AS, Doron H, Gois PMP, Erez N, Jung S, Satchi-Fainaro R and Florindo HF, Nat. Nanotech, 2019, 14, 891. [DOI] [PubMed] [Google Scholar]
- [19].Goldberg Michael S. Cell, 2015, 161, 201. [DOI] [PubMed] [Google Scholar]
- [20].Aichhorn S, Linhardt A, Halfmann A, Nadlinger M, Kirchberger S, Stadler M, Dillinger B, Distel M, Dohnal A, Teasdale I and Schöfberger W, Chem. Eur. J, 2017, 23, 17721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Moon JJ, Huang B and Irvine DJ, Adv. Mater, 2012, 24, 3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang C and Pu K, Chem. Soc. Rev, 2020, 49. [DOI] [PubMed] [Google Scholar]
- [23].Kapadia CH, Perry JL, Tian S, Luft JC and DeSimone JM, J. Control. Release, 2015, 219, 167. [DOI] [PubMed] [Google Scholar]
- [24].Hanson MC, Crespo MP, Abraham W, Moynihan KD, Szeto GL, Chen SH, Melo MB, Mueller S and Irvine DJ, J. Clin. Investig, 2015, 125, 2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liang C, Xu L, Song G and Liu Z, Chem. Soc. Rev, 2016, 45, 6250. [DOI] [PubMed] [Google Scholar]
- [26].Luo M, Samandi LZ, Wang Z, Chen ZJ and Gao J, J. Control. Release, 2017, 263, 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kwong B, Liu H and Irvine DJ, Biomaterials, 2011, 32, 5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Jia YP, Shi K, Yang F, Liao JF, Han RX, Yuan LP, Hao Y, Pan M, Xiao Y, Qian ZY and Wei XW, Adv. Funct. Mater, 2020, 30, 2001059. [Google Scholar]
- [29].Huang C-H, Mendez N, Echeagaray OH, Weeks J, Wang J, Vallez CN, Gude N, Trogler WC, Carson DA, Hayashi T and Kummel AC, ACS Appl. Mater. Interfaces, 2019, 11, 26637. [DOI] [PubMed] [Google Scholar]
- [30].Kim H, Niu L, Larson P, Kucaba TA, Murphy KA, James BR, Ferguson DM, Griffith TS and Panyam J, Biomaterials, 2018, 164, 38. [DOI] [PubMed] [Google Scholar]
- [31].Rao JP and Geckeler KE, Prog. Polym. Sci, 2011, 36, 887. [Google Scholar]
- [32].Giner-Casares JJ, Henriksen-Lacey M, Coronado-Puchau M and Liz-Marzán LM, Mater. Today, 2016, 19, 19. [Google Scholar]
- [33].Dave V, Tak K, Sohgaura A, Gupta A, Sadhu V and Reddy KR, J. Microbiol. Methods, 2019, 160, 130. [DOI] [PubMed] [Google Scholar]
- [34].Saeed M, Gao J, Shi Y, Lammers T and Yu H, Theranostics, 2019, 9, 7981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhang Y, Lin S, Wang X-Y and Zhu G, WIREs Nanomed. Nanobi,, 2019, 11, 1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].de Titta A, Ballester M, Julier Z, Nembrini C, Jeanbart L, van der Vlies AJ, Swartz MA and Hubbell JA, Proc. Natl. Acad. Sci. U. S. A, 2013, 110, 19902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Nuhn L, Vanparijs N, De Beuckelaer A, Lybaert L, Verstraete G, Deswarte K, Lienenklaus S, Shukla NM, Salyer AC, Lambrecht BN, Grooten J, David SA, De Koker S and De Geest BG, Proc. Natl. Acad. Sci. U. S. A, 2016, 113, 8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kockelmann J, Stickdorn J, Kasmi S, De Vrieze J, Pieszka M, Ng DYW, David SA, De Geest BG and Nuhn L, Biomacromolecules, 2020, 21, 2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kim WG, Choi B, Yang H-J, Han J-A, Jung H, Cho H, Kang S and Hong SY, Bioconjugate Chem, 2016, 27, 2007. [DOI] [PubMed] [Google Scholar]
- [40].Tom JK, Dotsey EY, Wong HY, Stutts L, Moore T, Davies DH, Felgner PL and Esser-Kahn AP, ACS Central Science, 2015, 1, 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Tomai MA and Vasilakos JP, Chapter 8 - Toll-Like Receptor 7 and 8 Agonists for Vaccine Adjuvant Use. In Immunopotentiators in Modern Vaccines (Second Edition), Schijns VEJC; O’Hagan DT, Eds. Academic Press: 2017; pp 149. [Google Scholar]
- [42].Chi H, Li C, Zhao FS, Zhang L, Ng TB, Jin G and Sha O, Front. Pharmacol, 2017, 8, 304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Rodell CB, Arlauckas SP, Cuccarese MF, Garris CS, Li R, Ahmed MS, Kohler RH, Pittet MJ and Weissleder R, Nat. Biomed. Eng, 2018, 2, 578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].De Vrieze J, Louage B, Deswarte K, Zhong Z, De Coen R, Van Herck S, Nuhn L, Kaas Frich C, Zelikin AN, Lienenklaus S, Sanders NN, Lambrecht BN, David SA and De Geest BG, Angew. Chem. Int. Ed, 2019, 58, 15390. [DOI] [PubMed] [Google Scholar]
- [45].Lu R, Groer C, Kleindl PA, Moulder KR, Huang A, Hunt JR, Cai S, Aires DJ, Berkland C and Forrest ML, J. Control. Release, 2019, 306, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Pockros PJ, Guyader D, Patton H, Tong MJ, Wright T, McHutchison JG and Meng TC, J. Hepatol, 2007, 47, 174. [DOI] [PubMed] [Google Scholar]
- [47].Widmer J, Thauvin C, Mottas I, Nguyen VN, Delie F, Allémann E and Bourquin C, Int. J. Pharm, 2018, 535, 444. [DOI] [PubMed] [Google Scholar]
- [48].Dowling DJ, ImmunoHorizons, 2018, 2, 185. [DOI] [PubMed] [Google Scholar]
- [49].Ilyinskii PO, Roy CJ, O’Neil CP, Browning EA, Pittet LA, Altreuter DH, Alexis F, Tonti E, Shi J, Basto PA, Iannacone M, Radovic-Moreno AF, Langer RS, Farokhzad OC, von Andrian UH, Johnston LPM and Kishimoto TK, Vaccine, 2014, 32, 2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Thauvin C, Widmer J, Mottas I, Hocevar S, Allémann E, Bourquin C and Delie F, Eur. J. Pharm. Biopharm, 2019, 139, 253. [DOI] [PubMed] [Google Scholar]
- [51].Duong AD, Sharma S, Peine KJ, Gupta G, Satoskar AR, Bachelder EM, Wyslouzil BE and Ainslie KM, Mol. Pharm, 2013, 10, 1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lu Q, Qi S, Li P, Yang L, Yang S, Wang Y, Cheng Y, Song Y, Wang S, Tan F and Li N, J. Mater. Chem. B, 2019, 7, 2499. [DOI] [PubMed] [Google Scholar]
- [53].Rodell CB, Ahmed MS, Garris CS, Pittet MJ and Weissleder R, Theranostics, 2019, 9, 8426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wilkinson A, Lattmann E, Roces CB, Pedersen GK, Christensen D and Perrie Y, J. Control. Release, 2018, 291, 1. [DOI] [PubMed] [Google Scholar]
- [55].Vilaivan T, Tetrahedron Lett, 2006, 47, 6739. [Google Scholar]
- [56].Held I, von den Hoff P, Stephenson DS and Zipse H, Adv. Synth. Catal, 2008, 350, 1891. [Google Scholar]
- [57].Twibanire J.-d. A. K. and Grindley TB, Org. Lett, 2011, 13, 2988. [DOI] [PubMed] [Google Scholar]
- [58].Kakwere H, Ingham ES, Allen R, Mahakian LM, Tam SM, Zhang H, Silvestrini MT, Lewis JS and Ferrara KW, Bioconjugate Chem, 2017, 28, 2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kakwere H, Ingham ES, Allen R, Mahakian LM, Tam SM, Zhang H, Silvestrini MT, Lewis JS and Ferrara KW, Biomater. Sci, 2018, 6, 2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Noverraz F, Montanari E, Pimenta J, Szabó L, Ortiz D, Gonelle-Gispert C, Bühler LH and Gerber-Lemaire S, Bioconjugate Chem, 2018, 29, 1932. [DOI] [PubMed] [Google Scholar]
- [61].Westcott MM, Clemens EA, Holbrook BC, King SB and Alexander-Miller MA, Vaccine, 2018, 36, 1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Lewis JS, Roche C, Zhang Y, Brusko TM, Wasserfall CH, Atkinson M, Clare-Salzler MJ and Keselowsky BG, J. Mater. Chem. B, 2014, 2, 2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Dong H, Pang L, Cong H, Shen Y and Yu B, Drug Deliv, 2019, 26, 416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Vandooren J, Opdenakker G, Loadman PM and Edwards DR, Adv Drug Deliv Rev, 2016, 97, 144. [DOI] [PubMed] [Google Scholar]
- [65].Takeda K and Akira S, Int. Immunol, 2005, 17, 1. [DOI] [PubMed] [Google Scholar]
- [66].Zhang H, Tang W-L, Kheirolomoom A, Fite BZ, Wu B, Lau K, Baikoghli M, Raie MN, Tumbale SK, Foiret J, Ingham ES, Mahakian LM, Tam SM, Cheng RH, Borowsky AD and Ferrara KW, J. Control. Release, 2020. doi 10.1016/j.jconrel.2020.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Chowdhury PS, Chamoto K and Honjo T, J. Intern. Med, 2018, 283, 110. [DOI] [PubMed] [Google Scholar]
- [68].Zhang R, Zhu Z, Lv H, Li F, Sun S, Li J and Lee C-S, Small, 2019, 15, 1903881. [DOI] [PubMed] [Google Scholar]
- [69].Hayakawa T, Yaguchi T and Kawakami Y, Cancer Sci, 2020, 111, 4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Leclerc M, Voilin E, Gros G and Corgnac S, Front. Immunol, 2019, 10, 3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.