

Abstract
Chromatin structure, dynamics, and function are being intensely investigated by a variety of methods, including microscopy, X-ray diffraction, nuclear magnetic resonance, biochemical crosslinking, chromosome conformation capture, and computation. A range of experimental techniques combined with modeling is clearly valuable to help interpret experimental data and, importantly, generate configurations and mechanisms related to the 3D organization and function of the genome. Contact maps, in particular, as obtained by a variety of chromosome conformation capture methods, are of increasing interest due to their implications on genome structure and regulation on many levels. In this perspective, using seven examples from our group’s studies, we illustrate how molecular modeling can help interpret such experimental data. Specifically, we show how computed contact maps related to experimental systems can be used to explain structures of nucleosomes, chromatin higher-order folding, domain segregation mechanisms, gene organization, and the effect on chromatin structure of external and internal fiber parameters, such as nucleosome positioning, presence of nucleosome free regions, histone posttranslational modifications, and linker histone binding. We argue that such computations on multiple spatial and temporal scales will be increasingly important for the integration of genomic, epigenomic, and biophysical data on chromatin structure and related cellular processes.
Graphical Abstract
Introduction: technology innovations as triggers of field advances
“I will ask you to mark again that rather typical feature of the development of our subject; how so much progress depends on the interplay of techniques, discoveries, and new ideas, probably in that order of decreasing importance.”
—Sydney Brenner (1927–2019), 1980
Technology and discovery
The above quote by the late Sydney Brenner, one of the molecular biology pioneers and a 2002 Nobel laureate in physiology or medicine, is often abbreviated to argue for the crucial role of techniques in scientific research, rather than the interplay of methodologies and ideas. However, in our 21st century scientific studies, where genomics and computing are key players, technological advances often allow us to see or discover phenomena or aspects that were not possible to observe before.
In biosciences, one of these technological advances was the invention of compound microscopes, with early contributions in the 16th and 17th centuries from Dutch Zaccharias Janssen and his son Hans Janssen to Anthony van Leeuwenhoek (1). Afterward, the pioneering work of Englishman Robert Hooke to improve the compound microscope led to the discovery of the basic unit of life, the cell. Hooke’s microscopic observations in Micrographia (2) revealed the benefit of using specially engineered instruments for a deeper understanding of nature. The need for higher magnification and greater resolution to observe smaller objects led to the invention of the electron microscope, with the first contribution coming in 1931 from German physicists Max Knoll and Ernst Ruska (3). Since the initial developments of light and electron microscopy, continuous efforts to improve image resolution and contrast, sample preparation, fluorescence labeling, and digital imaging have led to today’s sophisticated imaging techniques, like super-resolution light microscopy (4), fiber-optic imaging (5), and cryo electron microscopy (Cryo-EM) (6).
The technological innovation of microscopic techniques was fundamental for the progress of many fields of sciences. For example, optical microscopy advanced cell biology, allowing the visualization of cells, subcellular organelles, and molecules (7). Electron microscopy was crucial in the progress of bacteriology (8). And scanning tunneling and atomic force microscopy propelled the fields of nanoscience (9). The capability of microscopy techniques to visualize samples at unprecedented resolution has had a strong impact on biology and has led to innovative discoveries that have advanced molecular and cell biology, medicine, and material science.
Equally astounding and impactful on the biological sciences is the rise and wide availability of advanced computer technology. Proteomics, genomics, cellomics, and many more active fields of science today are all driven and made possible by bioinformatics and computational technology. Indeed, the 2013 Nobel Prize in Chemistry to Martin Karplus, Michael Levitt, and Arieh Warshel “for the development of multiscale models for complex chemical systems” (10) represents a significant turning point for computations in biology, chemistry, and allied fields, as “the prize provided a welcome seal of approval to a field that historically struggled behind experiment” (11).
X-ray and NMR
In the chromatin field, the earliest technological contributions came from X-ray crystallography. The elucidation of the double helical structure of DNA using this technique (12–14) led research toward the visualization of chromatin. Studies from the 1950s revealed that the nucleoprotein complex, formed by association of DNA and histones had a series of low angle diffraction rings that suggested a supercoiled superhelix of 120 Å pitch and 100 Å diameter (15, 16). More than a decade later, new X-ray diffraction studies showed a chromatin structure consisting of double helical DNA associated with histone units via an irregularly supercoil with an average pitch of 45 Å (17, 18). In the 1970s, the discovery, chemical characterization, and electron-microscopic visualization of nucleosomes (19–21) offered images of “beads on a string”, demonstrating that the chromatin structure consisted of a repeating unit of adjacent particles. The idea of a chromatin substructure was further supported by associations between histone proteins. Namely, the characterization of the H2A-H2B dimer (22, 23) and the (H3–H4)2 tetramer (19, 24) led to the proposal of an octamer model for the chromatin repeating unit (25). Around that time, nuclear magnetic resonance (NMR) studies of DNA led to a proposal for its interaction with the auxiliary linker histone (LH) (26) and showed its importance on chromatin condensation (27). Subsequently, improvements in diffractometry led to the determination of the crystal structure of the nucleosome at low resolution (7 Å) (28), followed by higher resolution (2.8 Å) (29).
Further studies with NMR and X-ray diffraction have been crucial for the understanding of the nucleosome core particle and chromatosome structure. For example, studies have revealed the configuration of histone tails, importance of water molecules in the interaction between DNA and histones (30), and the existence of different binding modes for LHs (31, 32).
Light microscopy, electron microscopy, and FRET
Improvements in microscopy techniques have provided important insights into the chromatin organization in situ and in vivo (33). The use of Cryo-EM to study chromatin fibers revealed that nucleosome compaction can be achieved by the interdigitation of nucleosomes from adjacent helical gyres (34) and by a LH-dependent left-handed twist of repeating tetranucleosomal structural units (35). Light microscopy, associated with fluorescence in situ hybridization (FISH) allowed fundamental discoveries, such as the existence of chromosome territories (36) and the clustering along the nuclear periphery of transcriptionally inactive regions of chromatin (37).
The association of light microscopy with Föster resonance energy transfer (FRET) also helped to uncover the structural dynamics of nucleosomes at bulk or single-molecule levels. FRET experiments contributed to the understanding of nucleosome dynamics through the study of DNA mobility (38), DNA dissociation (39), interactions with transcriptional factors (40), and the effects of histone posttranslational modifications (41) and core histone variants (42). More recently, the development of super-resolution fluorescence microscopy techniques extended the resolution level of light microscopy (4). The application of state-of-the-art super-resolution nanoscopy to study chromatin revealed that nucleosomes are organized as heterogeneous clusters of different sizes, dependent on cell type fundamentally (43, 44) and that different epigenetic states conduct to distinct chromatin packing (45).
Chromosome Capture Technology
The early 21st century saw the crucial rise of higher resolution Chromosome Conformation Capture (3C) technologies (46). These high-throughput methods help analyze genome organization by quantifying interactions between genomic loci as averages over large cell populations. The results are presented as heat or contact maps of pairwise intra and interchromosomal contact frequencies. High-frequency contacts represent DNA regions in close spatial proximity and can be used to suggest the 3D organization of chromatin (47, 48) along with gene regulation mechanisms (49, 50). The original 3C method, used to study interactions between specific pairs of sequences, was rapidly followed by the development of higher-throughput 3C-based methods and evolved into a family of techniques that differ by resolution, sensitivity, and type of interactions detected (51, 52). These include, 4C (47), 5C (53), Hi-C (54) (and variants single-cell Hi-C (55), Micro-C (56), Micro-C XL (57), and DNase Hi-C (58)), Capture-C (59), NG Capture-C (60), and two methods that incorporate chromatin immunoprecipitation: ChIA-PET (61) and ChIP-loop (62).
Overall, 3C studies have shed new light into the 3D organization of chromosomes inside the nucleus. The early works revealed the folding properties of the yeast chromosome III (46) and long-range gene regulation mechanisms in the b-globin locus (63), while later investigations focused on the study of the 3D architecture of chromosomes and whole genomes. Works have described the association between genome global organization and transcriptional regulation (64), existence of chromosome territories (54), and presence of chromosomal compartments formed by topologically associating domains (TADs) (65, 66). These TADs are considered the basic units of chromosome folding (65, 67) and are conserved structures across evolution and among species and cell types. They range in size from kilobases to millions of bases and are formed by smaller “sub-TADs” and, at a more local level, “loop regions”, which vary among cell types and appear to be related to regulatory events.
Though powerful, 3C techniques consider all existing associations, rather than discriminate between functional and nonfunctional associations, and usually reflect the interactions averaged over cell populations (68). The data are represented in a two-dimensional way, but for an accurate interpretation in its three-dimensional context in the nucleus, interpretation by computational models is crucial. Moreover, contact maps are not restricted to 3C techniques; they can be obtained with chromatin crosslinking methods, such as EMANIC, an electron microscopy-assisted nucleosome interaction capture technique (69) that produces nucleosome resolution contact maps. Together with mesoscale modeling, EMANIC has been used to show the coexistence of the two-start zig-zag and one-start solenoid conformations in heteromorphic chromatin fibers (69) and to describe the internal organization of condensed chromatin fibers (70). Experimental evidence of DNA DNA contacts has also been obtained with the recently developed technique ionizing radiation-induced spatially correlated cleavage of DNA with sequencing (RICC-seq) (71), which has a nucleosome resolution level.
Computational methods
The development of computational models for chromatin started in the 1970s, with contributions such as by Fuller and Benham on an elastic-rod and purely mechanical model for the analytical description of duplex DNA supercoiling (72, 73). It was soon realized that a numerical approach which considers thermal fluctuations was necessary for realistic description. The first computer simulation of DNA supercoiling by Monte Carlo (MC) methods was reported by Frank-Kamenetskii and coworkers in 1979 (74), followed by theoretical works by Le Bret (75) and Chen (76). In these studies, the DNA was described with the simple free-joint chain model, but subsequent MC simulations employed the worm-like chain model with excluded volume (77–80). In the 1980s, seminal contributions came from dynamics simulations of DNA. Allison and coworkers employed Brownian dynamics (BD) to simulate fluorescence anisotropy of linear DNA modeled as a worm-like chain polymer (81, 82), and Levitt presented the first all-atom molecular dynamics simulation of DNA (83). In the next decade, further insights came from dynamics simulations. Langevin dynamics applications by Schlick and Olson to DNA supercoiling and trefoil knotting revealed the large structural repertoire of DNA supercoils and knots (84, 85), and BD by Chirico and Langowski examined the twisting dynamics of supercoiled DNA molecules (86).
For simulating chromatin fibers, further coarse-grained models became necessary. Chromatin was simulated with BD by Langowski and coworkers in 1997 (87), where linker DNA was treated as a flexible polymer with stiff cylindrical segments and Debye-Hückel electrostatics, and nucleosomes were solid beads with excluded-volume interactions. Olson and coworkers used MC of a similar chromatin model that also included a term for nucleosome attraction (88). Later improvements by Wedemann and Langowski used oblate ellipsoids for nucleosomes and Gay-Berne potential for nucleosome nucleosome interactions (89). Our group developed a mesoscale chromatin model where initially nucleosomes were flat cylinders with core electrostatics described by a set of Debye-Hückel charges that reproduce the electric field around the atomistic nucleosome using the Poisson-Boltzmann potential; linker DNA was treated using Stigter’s worm-like chain model (90). Further improvements in our chromatin model included the explicit description of histone tails as coarse-grained beads (91), various LHs (92–94), epigenetic marks (95–97), implicit account of magnesium ions (98), and variable nucleosome positions (99–101). Validation against experimental data has shown that mesoscale models capture the essential physical properties of chromatin fibers (102), and thus contribute to our understanding of the different levels of DNA organization inside the cell and their relevance to regulation of gene expression (103). Many other groups have contributed to chromatin modeling, such as Papoian (104, 105), Nordenskiöld (98), and Ohyama (106), with all-atom and/or coarse-grained models of nucleosomes and/or chromatin fibers.
For chromosome investigations, polymer models became popular, thereby introducing an additional level of coarse graining. Contributions to this field have come from, for example, Nicodemi (107), Dekker (54), and Heermann (108). In these models, fibers are represented by a polymer made of flexibly connected monomer units with simple attractive and repulsive interactions, usually in the form of Lennard-Jones potentials (109). Polymer models of chromatin have helped in the interpretation of 3C derived data by quickly generating three-dimensional models of chromatin, chromosomes, or whole genomes. They have been used to study, for example, the internal organization of condensed chromatin (70), the structure of chromosome territories (108, 110, 111) and TADs (112), the coupling between chromatin folding and the epigenome (113), the internal organization of human chromosomes across the cell cycle (114), and the role of chromatin architecture in limb morphogenesis (115). Such models, while reaching large size scales, cannot treat all physical parameters like variable nucleosome positions, local binding of proteins such as LHs, and other factors shown to affect chromatin structure profoundly.
Chromatin discovery by technology
To appreciate the impact of various methods for the study of chromatin, we have conducted a literature search over a scientific database to measure the volume and impact of frequently used techniques to study chromatin structure and function over the past five decades. Specifically, we use Scopus, Elsevier’s abstract and citation database of peer-reviewed literature (scientific journals, books, and conference proceedings), to search for original articles and reviews on the subject of chromatin studies using different techniques. The techniques were divided into eight categories: (a) NMR; (b) X-ray crystallography; (c) medium resolution methods, such as Cryo-EM, transmission electron microscopy, atomic force microscopy, small angle X-ray scattering (SAXS) and electron spectroscopic imaging; (d) light microscopy techniques, such as fluorescence recovery after photobleaching, fluorescence anisotropy, continuous photobleaching, single particle tracking, fluorescence correlation spectroscopy, FRET, fluorescence photoactivation, FISH, fluorescence cross correlation spectroscopy, and fluorescence-lifetime imaging; (e) super-resolution microscopy techniques, such as structured illumination microscopy, single-molecule localization microscopy, reversible structural optical fluorescence transition, stochastic optical reconstruction microscopy photoactivation localization, and stimulated emission depletion microscopy; (f) computational techniques, such as molecular dynamics, Monte Carlo, and other modeling/simulations; (g) crosslinking techniques, such as EMANIC; and (h) Chromosome Conformation Capture techniques.
Specifically, we searched for a combination of method and topic in the search command. Thus, for example, the keywords for the techniques were used in combination with the subject “chromatin, nucleosome, and nucleosomes”; specific details are presented in Table S1 in Supplemental Information.
We plot the data in two ways: number of articles by year (Figure 1A), and a weighted number of articles (Figure 1B), where the total impact of each year’s article is plotted and divided over the years since published (to provide a “yearly impact” figure). Overall, our analysis shows that computational methods (red curve) are both high impact and high volume, although the number is declining at present (but this could be explained, in part, by a data lag of getting computational articles into the Scopus database). Other impactful and voluminous techniques are: light microscopy (blue curve), medium resolution methods (violet curve), crosslinking (turquoise curve), and 3C technologies (black curve).
Figure 1.
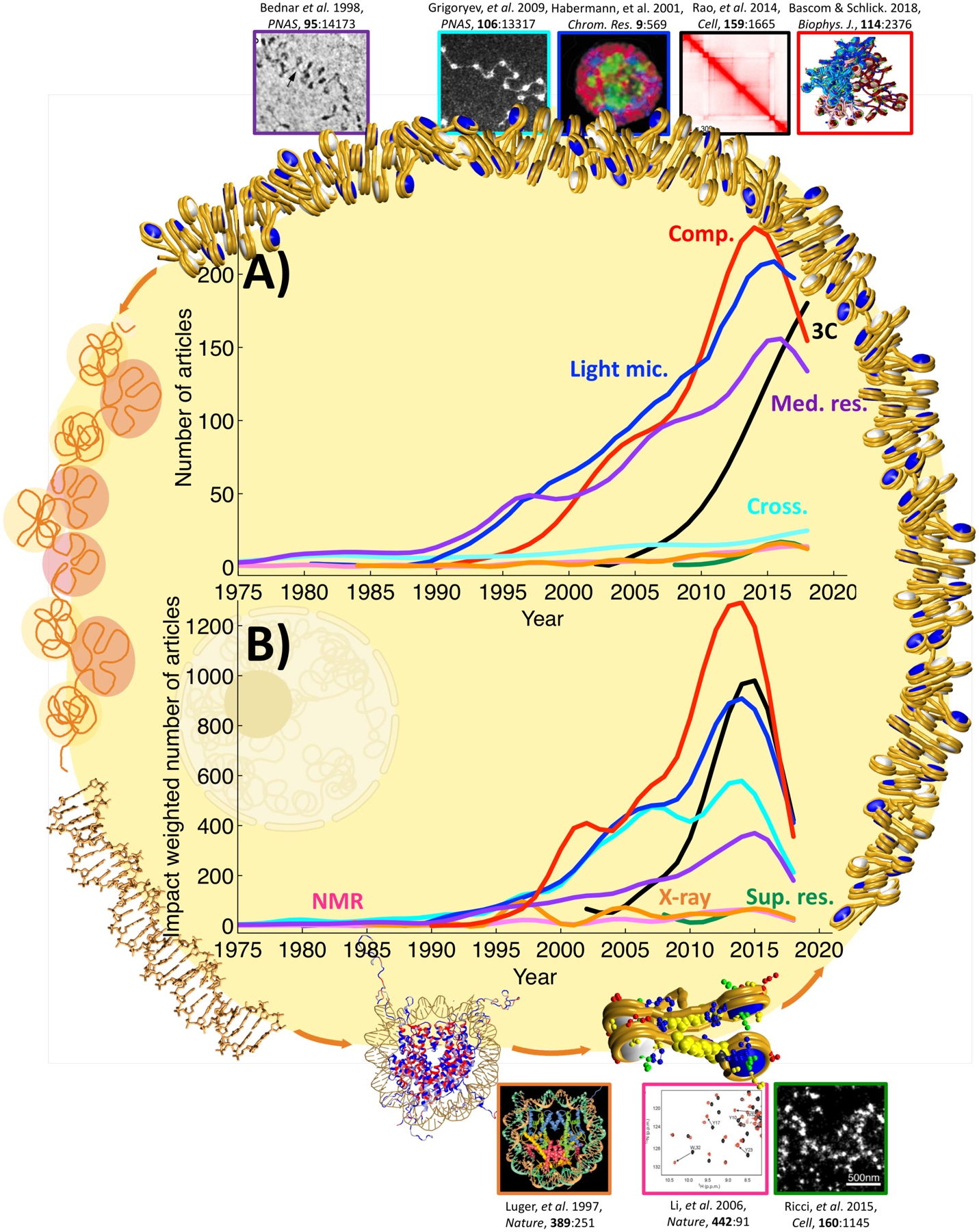
The impact of various techniques on the study of chromatin structure and function obtained with the Scopus database and a specific combination of keywords for technique and topic, as detailed in Table S1. A) Number of articles per year. B) Impact weighted number of articles; for each article: the total number of citations to date, normalized by number of years since publication. “Comp.” (red curve): computational methods, “Light mic.” (blue): light microscopy techniques, “3C” (black): chromosome conformation capture techniques, “Med. res.” (violet): medium resolution methods, “Cross.” (turquoise): crosslinking techniques, “NMR” (pink): nuclear magnetic resonance techniques, “X-ray” (orange): x-ray crystallography techniques, and “Sup. res.” (green): super-resolution microscopy techniques. Boundary images illustrate chromatin organization levels, from DNA to the nucleosome, nucleosome chains, fibers, and chromosomes. Top and bottom images mark key discoveries obtained by techniques in each category, where the border colors correspond to the technique. Top images, from left to right: Cryo-EM image showing chromatin repeating subunit, adapted with permission from (199); transmission EM image of in situ cross-linked nucleosome chains from metaphase chromatin (70); FISH image showing distribution of chromosome territories in a neuron nucleus, adapted with permission from (200); contact map of 1 Mb segment of human Chr14 at 25 kb resolution, adapted with permission from (191); chromatin fiber of 100 nucleosomes constructed with alternating 25 nucleosomes with acetylated tails and 25 nucleosomes with wild type tails repeated twice, adapted with permission form (97). Bottom images, from left to right: X-ray crystal structure of the nucleosome core particle at 2.8 Å resolution, adapted with permission form (29); 1H,15N NMR spectra showing amide resonances of a transcription factor (BPTF PHD) in the absence (black) and presence (red) of a methylated H3 peptide, adapted with permission form (201); and super-resolution microscopy image of H2B in human fibroblast nucleus showing localization of nucleosomes, adapted with permission from (43).
In more detail, Figure 1A shows that computational methods present the highest volume, followed very closely by light microscopy, medium resolution methods, and 3C techniques. Next come crosslinking, X-ray (orange curve), NMR (pink curve), and super-resolution microscopy (green curve). In the 1990s, the number of papers resulting from medium resolution techniques, computational methods, and light microscopy grew exponentially. Computational methods show a marked increase after 2000, which might be a consequence of the increased usage of Graphics Processing Units (GPUs) for scientific computations. Medium resolution methods show a less pronounced exponential growth and two peaks, in 1996 and 2006, which are result of a high number of studies using transmission electron microscopy in those years. In 2002, 3C techniques were developed and immediately started to experience a marked exponential growth, which persists today. After only 15 years, 3C techniques have reached almost the same volume as computational methods, medium resolution methods, and light microscopy, which have been used to study chromatin for two to four decades. The remaining techniques, namely super-resolution microscopy, crosslinking, X-ray crystallography, and NMR have maintained mostly constant the number of published articles per year since their development.
Figure 1B shows that computational methods (red curve), light microscopy (blue curve), and 3C techniques (black curve) appear to have the largest impact, followed by crosslinking techniques and (turquoise curve) medium resolution methods (violet curve). The large volume that medium resolution techniques showed in Figure 1A is not reflected in their impact. In contrast, crosslinking techniques appear to have a large impact, although their volume is much smaller. The curve for the X-ray technique (orange) shows two peaks, in 1997 and 2004, corresponding to seminal papers on the nucleosome core particle (29) and the 53BP1 checkpoint protein implicated in sensing DNA double-strand breaks through his binding to methylated histone H3 (116). The crosslinking technique curve shows peaks in 2008 and 2012 associated with reviews and protocols published in high impact journals. For 3C techniques, we note a marked exponential growth around 2005. Computational methods show a marked increase between 2010 and 2015, likely reflecting a growing acceptance by the scientific community at large, as discussed in the introduction and in this article (11).
Overview: molecular modeling to interpret genome contact and related experimental data
In this perspective, we illustrate the importance of using molecular modeling to better interpret 3C derived and other experimental data to help relate frequency patterns among DNA regions to the 3D organization of chromatin. Molecular modeling offers an important bridge between structure and function (117). Specifically, we illustrate results from our group’s studies that help add key insights into the organization of the chromatin structure. We discuss how studies shed light into mechanisms of higher-order fiber and chromosome folding, the effect of LH subtype and binding mode on chromatin condensation, the regulation of chromatin structure by DNA linker length and nucleosome free regions, effects of epigenetic marks (histone acetylation) on chromatin structure, implications of higher-order folding in gene silencing, mechanisms of chromatin compartmentalization, and the effect of epigenetic factors (histone acetylation and LH binding) in controlling chromatin architecture.
As our focus is on coarse-grained models of chromatin, we refer readers to reviews of recent advances in multiscale modeling of DNA and chromatin by Dans et al. (118) and by Ozer and Schlick (103). For specific descriptions of mesoscale models, see Langowski and Heermann (119), a recent chapter book focused on the development and applications of our mesoscale model (120), and a review about the status of coarse-grained computer simulations of chromatin (121). For a broad perspective of the progress made in nuclear organization and function by polymer models of DNA and chromatin, see Amitai et al. (122). For a complete treatise of physical modeling approaches to study genome folding, we refer readers to a recent book edited by Tiana and Giorgetti (123). Finally, for a historical perspective on multiscale coarse-grained modeling of chromatin, see Korolev, Nordenskiöld, and Lyubartsev (124).
Regarding experimental methods, for a comparison of 3C technologies, see Denker and de Laat (52), and for an overview on advanced microscopy methods to visualize and analyze chromatin fiber organization, see Ricci et al. (125).
For a perspective on recent advances on higher-order structures of chromatin and their implications on the regulation of gene expression, see Bascom and Schlick (100), Grigoryev (126, 127), and Rowley and Corces (3C technologies) (128). Noteworthy biological perspectives on histone post translational modifications are by Andrews et al. (129) and Azad et al. (130) and on LH binding modes and their roles in the regulation of chromatin structure and function by Öztürk et al. (31) and Fyodorov et al. (131).
In the next section, we focus on studies from our group; for other excellent examples illustrating productive interplay between modeling and experiment, we refer readers to the following perspective articles or reviews: Haddad, Jost, and Vaillant about the usage of polymer models to study nuclear compartments and their intrinsic connection to 3C derived data (132); Nicodemi and coworkers illustrating how polymer models help connect chromatin folding with Hi-C experiments (133); Stasiak and coworkers on numerical simulations of chromatin supercoiling (134); and Olson and coworkers on studies of chromatin/protein interactions and chromatin architecture (135). Undoubtedly, these reviews are not exhaustive given the great amount of excellent work in this exciting field. We apologize for all unintentional omissions.
Illustrative Studies: mesoscale modeling to help interpret experimental data and genome folding
1. Hierarchical looping as a chromatin higher-order folding mechanism
A challenge in the chromatin field has been the elucidation of chromatin secondary and tertiary organization, namely, whether the secondary structure involves a solenoid or zigzag topologies, or others (136), and how higher-order chromatin fibers are further organized in 3D. Now it is generally accepted that chromatin fibers are heteromorphic and fluid, with presence of both zigzag and solenoid secondary topologies (69, 137–139), though the zigzag is persistent (69, 70, 140, 141). The focus has now shifted to understanding chromatin’s higher-order folding and implications in the regulation of gene expression (126).
In collaboration with Grigoryev and his EMANIC cross-linking/EM approach, we focused on describing the nature of condensed interphase and metaphase chromatin. By constructing different models that mimic chromatin fibers studied experimentally, we helped interpret in situ EMANIC results that revealed the presence of persistent zigzag motifs in both interphase and metaphase chromatin as well as an increase of nucleosome long-range interactions only in metaphase chromatin (70). Specifically, by simulating with our mesoscale model chromatin fibers of 96 nucleosomes with different LH densities (0, 0.5, and 1 LH per nucleosome) and relevant nucleosome repeat lengths (NRLs), we suggested a mechanism that reconciles the increased long-range nucleosome contacts without the loss of the zigzag motifs in metaphase chromatin. Our mesoscale model, presented in Figure 2, is described in detail recently by Bascom and Schlick (120). Arya and Schlick (92), Luque et al. (94), and Collepardo-Guevara and Schlick (142) further summarize validations performed against experimental data. In summary, our model coarse grains different chromatin constituents as follows: nucleosomes are treated as rigid bodies by Debye-Hückel pseudo-charges (Figure 2A) (90); histone tails (Figure 2A) and folded tails carrying epigenetic marks (Figure 2B) are parametrized with the Levitt-Warshel united-atom bead model (95, 143); linker DNA is modeled as an elastic worm-like chain with charges derived by the Stigter’s procedure for elastic rods (144) (Figure 2C); and two LHs models are available (H1E and H1C), with coarse-grained beads for the globular head (GH) and the C-terminal domain (CTD) (Figure 2D). For LH binding, both on and off-dyad binding are handled (Figure 2E), as well as variable LH densities, from 0 to 1.6, by describing some chromatosomes with 2 LHs (93) (Figure 2F). The potential energy function of the mesoscale chromatin model includes: bending, stretching, and torsional terms for linker DNA beads; bending and stretching terms for histone tails and linker histone beads; and electrostatic and excluded volume interaction terms for all beads and points charges, modeled by Debye-Hückel and Lennard-Jones potentials, respectively (92). The equilibrium thermal ensemble of chromatin fiber configurations is sampled with the classical Metropolis-Monte Carlo algorithm (145), employing efficient and tailored local and global moves. In the study of interphase and metaphase chromatin, we reproduced EMANIC nucleosome nucleosome contacts for interphase and metaphase systems (see bar graph in Figure 3, blue slice) by associating fiber with different LH densities (see fiber structures in Figure 3, blue slice). The folded fibers without LH revealed the presence of zigzag motifs plus stacked, hierarchical loops that enhance long-range interactions, in agreement with the EMANIC contact probability profiles of metaphase chromatin. On the other hand, contact maps of fibers with sub-stoichiometric levels of LH (e.g., 1 LH per 2 nucleosomes) show presence of medium-range contacts but absence of long-range interactions, resembling the experimentally-measured contact probability profiles for interphase chromatin. Our proposed hierarchical looping folding mechanism refers to loops of loops, formed by folding and stacking different sized loops in three-dimensional space, similar to “rope flaking” used in mountaineering. Hierarchical loops emerge clearly in the contact maps, indicated by the presence of straight regions parallel to the main diagonal, marking interactions between nucleosomes belonging to different loops (see contacts colored in blue in the contact map in Figure 3, blue slice). Similarly, the presence of hairpin regions is depicted in the contact maps by straight regions perpendicular to the main diagonal (see contacts colored in green in the contact map in Figure 3, blue slice).
Figure 2.
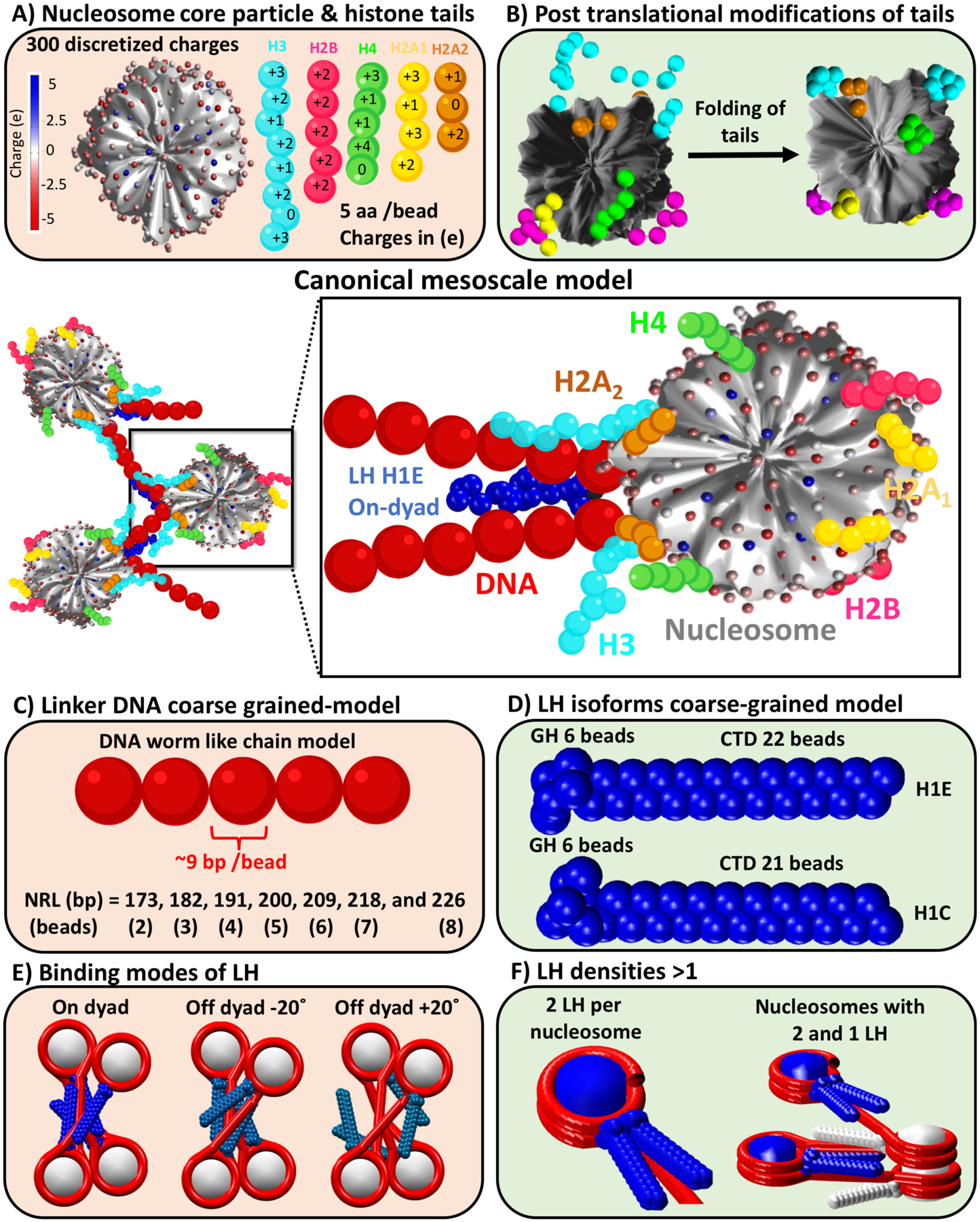
Our canonical chromatin mesoscale model at center with all its components detailed as follows: A) Rigid nucleosome core particle (NCP) modeled by 300 discrete charges determined with the DiSCO algorithm (90), along with flexible histone tails (H3 N-tail in cyan, H2B N-tail in magenta, H4 N-tail in green, H2A N-tail in yellow, and H2A C-tail in orange), coarse-grained as 5 amino acids per bead with charges also determined by DiSCO (90, 173); B) Nucleosome with wild type (left) and with folded histone tails (right) containing lysine acetylation modeled by increased stretching, bending, and torsional intertail-bead force constants by a factor of 100 (95); C) Linker DNA modeled as a worm-like chain polymer, coarse-grained as ~9 bp per bead. The nucleosome repeat lengths (NRLs) of 147 bp plus linker DNA length in bp are modeled by 2, 3, 4, 5, 6, 7, and 8 beads to mimic NRLs = 173, 182, 191, 200, 209, 218, and 226 bp or DNA linker lengths = 26, 35, 44, 53, 62, 71, and 79 bp, respectively (202); D) Linker histone (LH) isoforms H1E and H1C, modeled with 22 and 21 beads, respectively (5 amino acids per bead) for the CTDs and 6 beads for GHs with their charges determined by DiSCO (90, 93, 94); E) On and off-dyad (+20° and −20°) binding modes for the LHs (93); and F) Chromatosome with 2 LH bound (left) and tetranucleosome fiber with a density of 1.5 LH per nucleosome (right), where nucleosomes with 2 LH are colored blue and with 1 LH are colored white.
Figure 3.
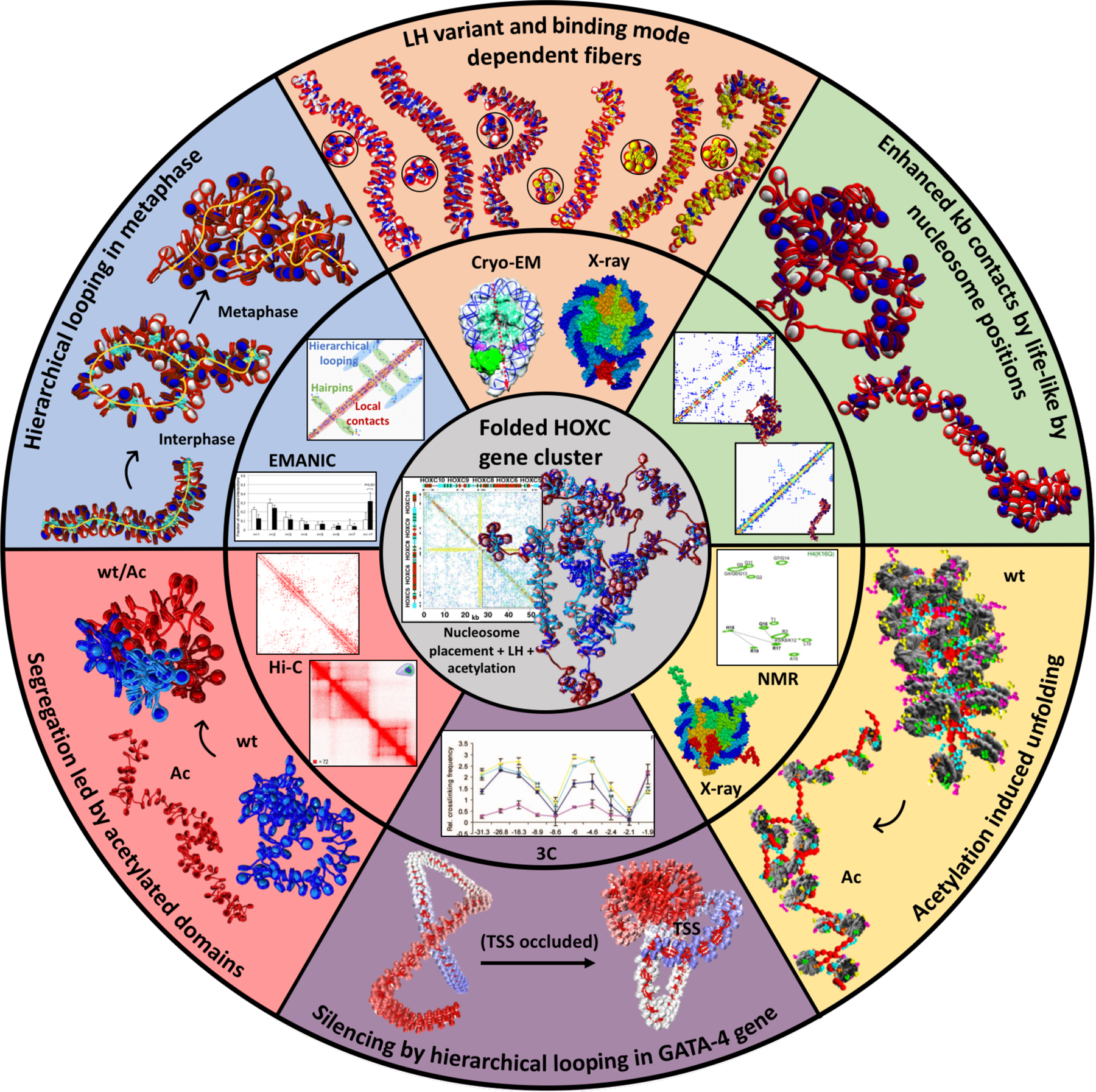
Mesoscale modeling studies emphasizing the interplay between experiment (middle ring) and modeling (outer ring), along with crucial internal/external fiber parameters that direct gene folding, as demonstrated for the HOXC gene cluster (center). Clockwise from blue slice: Hierarchical looping in metaphase chromosomes. Computed fiber structures for terminally differentiated cells (1 LH per nucleosome), interphase chromatin (0.5 LH per nucleosome), and metaphase chromatin (no LH), with LHs in turquoise explain nucleosome contacts determined by the EM-assisted nucleosome interaction capture (EMANIC) technique for metaphase (black bars) and interphase (white bars) chromatin in situ, by the hierarchical looping folding motif, also evident in the accompanying computed contact matrix (70). Peach slice: LH variant and binding mode dependent fibers. The chromatin fiber topologies (top and side views) are sensitive to different combinations of LH variant, binding mode, and density. The six fibers, from left to right, correspond to: ρ = 1, 100 H1E on-dyad; ρ = 1, 100 H1E −20°; ρ = 1, 100 H1E +20°; ρ = 1.3, 100 H1E +20° and 30 H1C on-dyad; ρ = 1.6, 100 H1E +20° and 60 H1C on-dyad; and ρ = 1.6, 40 H1E +20°, 60 H1E +20°, and 60 H1C −20°, where nucleosomes containing two LH bound are colored in yellow (93). The crystal structures of a chromatosome with LH (in red) bound on-dyad (PDBID: 4QLC (203)) and Cryo-EM chromatosome with LH bound off-dyad (in green) were used to generate our two binding modes. The cryo-EM image was adapted with permission from (35). Green slice: Enhanced kb contacts by life-like nucleosome positions. Fibers with life-like linker lengths and nucleosomes free regions (NFRs) generate many more long-range kb contacts compared to uniform linker-length fibers. This is evident by the folded fibers and the associated contact maps, which indicate hierarchical looping in life-like fibers compared to mostly short-range interactions in uniform fibers (99). Yellow slice: Acetylation induced unfolding. Compared to a wild type chromatin (wt), the fiber with acetylated histone tails (Ac) drives global unfolding due to lack of stabilizing internucleosome interactions (95). The experimental NMR spectra of the core histone H4 K16Q were used to validate the acetylated tail structures obtained by all-atom molecular dynamics simulations. Adapted with permission from (204). The crystal structure of the nucleosome (PDB: 1KX5 (30)) was used to construct dinucleosomes in our multiscale modeling (95). Violet slice: Gene silencing by hierarchical looping in the GATA-4 gene. The experimental 3C contacts in 4 different cells (blue, UT cells; violet, DT cells; yellow, HCT116 cells; and light blue, DKO cells) were used as constraints in our GATA-4 gene model, with image adapted from (181). The representative unfolded (left) and folded (right) GATA-4 gene structures suggest how folding by hierarchical looping would silence the transcription start site (TSS) of the gene (178). Pink slice: Segregation induced by acetylated domains. Intrinsic compartments of acetylated (Ac)/wild type (wt) segments form at the kb level for a mixed fiber construct (50% wt and 50% Ac) compared to fibers with 100% wt (blue) and 100% Ac (red). Both the Hi-C contact map of a segment of human Chr3 and our computed contact map of the alternating fiber construct show segregation patterns. The Hi-C contact map image was adapted with permission from (96). Gray inner portion: Folded HOXC gene cluster. All previous internal/external fiber parameters, including nucleosome positions, LH binding positions, and acetylation islands are combined to fold in silico the HOXC gene cluster and reveal a contact hub (101); see also commentary on our work in (198). The contact map of the folded HOXC gene cluster unravels a central interaction between two domains: LH-rich (top left of contact map) and acetylation-rich regions (bottom right).
Overall, the combined experimental and computational results showed that LH density can regulate the transition between interphase and metaphase chromatin by controlling higher-order chromatin structure through a hierarchical looping mechanism. The observation that intrinsic hierarchical looping appears to be restricted by the binding of LH is consistent with the recognized facts that: (1) LHs are not necessary to condense mitotic chromatin (146), (2) LH binding/unbinding dynamics peaks at the metaphase cell stage (147), (3) proliferating cells usually contain sub-stoichiometric levels of LH (148), (4) LHs have lower affinity during mitosis due to phosphorylation (149), and (5) Cryo-EM structures of 30 nm fibers are generally obtained for saturated levels of LH (34, 35). The rapid confirmation of our conclusions regarding zigzag nucleosome interactions were also cited in high-profile genome-wide studies (56, 71).
Importantly, the results provide an explanation for apparently conflicting experimental observations. While in situ Cryo-EM (150, 151) and SAXS (152) experiments have suggested that chromatin fibers are disordered and flexible, in vivo and in situ studies obtained with super-resolution microscopy and FISH techniques indicate the existence of compact domains formed by the clustering of nucleosomes (43, 44) and the presence of sharp and well defined chromosomal boundaries separating translocated chromosomal regions (153). By highlighting the hierarchical looping mechanism, canonical zigzag and higher-order structures that differ from the 30 nm fibers emerge and their shapes can easily be controlled by LH levels and other factors. Thus, self-associating zigzag loops contain both regular secondary features and more random tertiary elements. This mechanism is also consistent with Micro-C results that have shown a hierarchical “beads on a string” architecture in the yeast genome, with zigzag geometry but absence of any regular secondary structure (56). Furthermore, Cryo-EM tomography experiments have revealed that chromatin chains of proliferating cells are open and disordered with 5–24 nm diameter (151, 154), consistent with this description.
2. The crucial role of linker histone binding mode and density on chromatin fiber structure
The auxiliary LH protein, disordered in isolation, is known to fold in part when binding to nucleosomes (155, 156). Its role on the structure and function of chromatin has gained attention recently due to evidence showing the importance of LH in chromatin organization and several biological functions, such as regulation of genome stability, DNA replication, and DNA repair (131). The existence of an ensemble of chromatosomes due to the presence of different binding modes of LH has also brought for the interesting structural features (31). The density (ρ) of LH in the chromatin fiber can vary significantly, from 0 to 1.6 LH per nucleosome (157), and ρ > 1 are known to be implicated in chromatin structure and vision disorders (158).
Our group has modeled LH early on by using a three beads coarse-grained model of the rat H1e bound on the dyad, where 1 bead represented the GH and the 2 other beads the CTD (92). Later, to describe the dynamics and flexibility of the CTD and the non-uniform charge distribution of the GH, our LH model was refined to 22 beads for the CTD and 6 beads for the GH (94). Our results revealed that LH binding is dynamic, that dynamic binding reduces fiber stiffness (159), and that the CTD spontaneously folds upon binding to the nucleosome via a dynamic charge neutralization mechanism (94).
To further understand the role of different binding modes, densities, and isoforms of LHs, we have recently incorporated the H1c variant in addition to H1e, as well as two off-dyad binding modes (+20° and −20°) constructed based on Cryo-EM structures (35) and other molecular modeling studies (32, 160) and the option to model chromatosomes with 2 bound LHs to incorporate LH densities greater than unity (Figure 2D, 2E, and 2F) (93). By constructing systems with different combination of these parameters (e.g., LH variant, density, and off vs. on-dyad binding) (see fiber structures in Figure 3, peach slice), we determined their effect on the fiber architecture. Overall, our results revealed a heterogeneous ensemble of chromatin fibers that depend on LH variant, binding mode or modes, and density. Specifically, we found that in fibers with LH density ρ = 1, the off-dyad −20° binding mode is better chromatin condenser (highest packing ratios) than others binding modes, such as on-dyad and off-dyad +20°, which produce less compact but more bendable fibers (longer persistence lengths). Interestingly, in fibers with ρ > 1, we found that chromatosomes best accommodate 2 LH by combining off and on-dyad binding modes rather than two off-dyad LHs, as might be expected. Furthermore, densities ρ > 1 do not generally lead to greater overall condensation than ρ = 1; by encouraging bending, self-associating fiber interactions are increased instead.
The role of LH in tuning the chromatin fiber architecture, balancing compaction and bendability, underscores how this auxiliary protein offers an additional epigenetic regulator for gene expression. By combining different LH variants, binding modes, and densities, compact fibers necessary for repression or bendable fibers necessary for looping and folding can be produced.
Our results help support experimental evidence from Cryo-EM and X-ray structures suggesting that the off-dyad binding mode is a better condenser (35, 161, 162). Importantly, we propose for the first time the structure of chromatosomes containing 2 LH bound and the best combination of binding modes to achieve highest compaction. Further studies that incorporate the dynamic binding/unbinding of LH and the possibility of interchange among different binding modes will be key to further understand LH function.
3. The increase of long-range chromatin fiber self-contacts by life-like nucleosome positions
Nucleosome placement along the fiber determines the length of DNA between adjacent nucleosomes, usually measured as the NRL, and the presence of nucleosome free regions (NFR). Both these variables have central roles in gene expression by affecting chromatin structure (163). Chemical mapping of nucleosome positioning at base-pair resolution for two yeast species (164, 165) and mouse embryonic stem cells (166) has shown that linker length distributions follow a specific pattern, but their effect over the chromatin fiber structure and the regulation of gene expression is not clearly known. It is believed that NRLs and NFRs are closely related to the contact domains and loops on the fiber and chromosomal levels. Thus, these factors produce different interactions among distant DNA elements (long-range or kilobase range contacts) through the formation of chromatin loops (167), thereby regulating gene expression through different levels of structural groups (168–170).
While heterogeneous distributions of nucleosome positions have generally been used in coarse-grained models, we began investigating the role of nucleosome positioning on chromatin fiber architecture by varying linker lengths and including NFRs (99). For comparison, we analyzed folding patterns in 100-nucleosome fibers with uniform linker lengths, combinations of two linker lengths positioned in alternation or randomly, “life-like” fibers with a realistic distribution of linker lengths (165) with and without the presence of short and long NFRs, and “gene encoding-like” fibers that mimic a 2 kb segment in yeast’s Chromosome 9 (56).
The resulting contact maps and other analyzed geometric features clarified how important these nonuniform nucleosome distributions are: kb-range interacting domains emerge naturally in fibers with “life-like” nucleosome positions with NFRs, contributing to chromatin spatial compartmentalization. In contrast, uniform linker lengths showed little long-range interactions (see fiber structures in Figure 3, green slice). The computed contact maps of these fibers (Figure 3, green slice) further indicate the presence of hierarchical looping at the kb-range, triggered by an increase in fiber flexibility and self-association.
Overall, this study underscored the role of nucleosome positioning in the chromatin fiber architecture and gene regulation. Computed contact maps at nucleosome resolution by mesoscale modeling provide complementary information to the experimentally derived data, such as contact maps obtained by Micro-C (56), nucleosome interactions determined by EMANIC (69), and nucleosome distributions determined by super-resolution nanoscopy (43). Most 3C techniques offer the opportunity to study chromatin interactions at the kb-resolution level; however, data interpretation and the identification of interactions are far from straightforward. Usually, the identified interactions must be verified by other independent methods, such as high-resolution FISH studies or further modeling.
4. The mechanisms underlying fiber unfolding by tail acetylation
Histone tails are essential chromatin components that contribute to its compaction/unfolding through mediation of internucleosome interactions. Numerous post-translational modifications (PTMs) apply to the histone tails, altering histone DNA interactions and nucleosome dynamics (171). Thus, PTMs have a profound impact on gene regulation, contributing to the precise repression or activation of specific regions of the genome (172).
Our group has been studying the role of histone tails, initially using rigid tails models (91) and later incorporating flexibility (173). Specifically, we have determined that H4 tails are the most important in mediating internucleosome interactions, while H3 tails crucially screen the repulsion between DNA linkers, stabilizing the chromatin fiber. On the other hand, H2A and H2B tails facilitate fiber/fiber cross interactions, and are thus expected to be important in self associating segments (92, 174). These interaction patterns between histone tails and chromatin components are sensitive to NRLs and LH density (175), suggesting that these combined parameters are tightly regulated in living systems as needed to control transcription (148).
In collaboration with Orozco’s group, we extended our study of histone tails by including the effect of PTMs, specifically lysine acetylation, in a multiscale approach that combined all-atom molecular dynamics (MD) simulations with mesoscale modeling (95). On the first level, we simulated atomistic isolated tails in their native state with and without PTMs; on a second level, the roles of histone tails in internucleosome interactions were studied by atomistic MD simulations of dinucleosomes; the third level incorporated the modified tails into our mesoscale model by altered rigidities to study their influence on chromatin fiber architecture (Figure 2B).
The MD simulations of isolated tails revealed that epigenetic modifications affect tail flexibility. Namely, there is an increase in secondary structure content and persistence length of the histone proteins, which produces a less flexible tail overall. The study of dinucleosomes showed how this decrease in tail flexibility and disorder impairs internucleosome interactions, as H4 tails cannot interact with the acidic patch of the neighboring nucleosome. Furthermore, when the folded tails were incorporated into our mesoscale model, chromatin unfolding followed due to the loss of key stabilizing internucleosome interactions (see fiber structures in Figure 3, yellow slice).
Our results were further supported by a recent metadynamics study that shows how H4 Lys16 acetylation impairs internucleosome interactions due to an altered conformation and a weaker binding to the acidic patch (176). Along the same lines, recent Cryo-EM studies have shown that nucleosome stacking and chromatin compaction can be regulated by the interaction between H4 tail and H2A acidic patch (177).
Overall, this study not only revealed the mechanism by which epigenetic modifications of histone tails can regulate gene expression through chromatin fiber structure; it opened the way to multiscale modeling of chromatin by bridging local to global effects on several levels of chromatin.
Indeed, in a follow-up work, when we compared acetylation of different tails individually, we found that whereas acetylation of H3, H2A, or H2B tails alone does not affect fiber folding significantly, H4 tail acetylation decreases local nucleosome contacts markedly, producing an opening of the fiber. Moreover, when all tails are acetylated, the effect is stronger and long-range interactions are also impaired, though LH binding in non-acetylated regions can counteract this effect (97).
5. Hierarchical looping as a gene silencing mechanism, exemplified by the GATA-4 gene locus
Chromatin looping is a mechanism that brings into close physical proximity pairs of genomic sites that are located far away from each other along the linear genome sequence. This mechanism mediates the higher-order architecture of chromatin and is implicated in the regulation of gene expression, site-specific recombination, and DNA replication (71).
Our study of the GATA-4 gene locus incorporating long-range 3C derived data showed the role of such folding in gene silencing (178). GATA-4 gene is involved in the differentiation of embryonic stem cells (179), as well as development and differentiation of endoderm-derive organs (180). The experimental study determined five loops for this gene (181) (see 3C data in Figure 3, violet slice). The transcription start site (TSS), located between loops 3 and 4, is thought to be enclosed by the formation of a multi-loop complex, down-regulating gene expression.
Our constructed mesoscale model for this system with the five loops determined experimentally modeled as harmonic restraints suggested that hierarchical looping provides a mechanism to enclose the TSS and inhibit GATA-4 transcription (see fiber structures in Figure 3, violet slice). By incorporating linker histones and divalent ions, we showed how the presence of LHs and Mg2+ and the loop size can regulate long-range interactions and fiber stiffness. We found that the smaller the loop length, the more flexible the fiber is. LHs and Mg2+ ions act to increase the fiber packing ratio and stabilize the fiber without affecting its flexibility. The higher-order structure of the GATA-4 gene obtained by our MC sampling of the mesoscale model (see fiber structure in Figure 3, violet slice) suggests that the silencing mechanism implicated is the formation of hierarchical loops that occlude the TSS, inhibiting transcription.
Thus, our incorporation of experimentally derived data help interpret gene’s folding mechanism. In this synergistic way, experiments and modeling collaborate to obtain a more complete picture of the relationship between chromatin structure and function.
Chromatin looping can be studied with biochemical methods (182–184), direct visualization with EM (185), FISH techniques (186, 187), and 3C technologies (46, 188–192). However, such methods can potentially disrupt the native higher-order structure of chromatin. In this sense, mesoscale models provide complementary structural information while incorporating detailed parameter variations of internal fiber features from first principles.
6. The role of epigenetic marking on chromatin domain definitions
Loop domains and compartments in chromatin establish functional domains and are therefore important to model (46, 54). In collaboration with the Liberman Aiden group, we explored loop domains and chromatin compartmentalization patterns in fibers marked by acetylation. The experimental in situ Hi-C contact maps obtained for all pair of loci across the human genome showed cohesin-independent compartmentalization of chromatin intervals containing similar patterns of histone marks (96) (see contact map in Figure 3, pink slice).
To investigate whether chromatin compartmentalization could be directed by interactions between tails with similar patterns of histone marks on the kb level, we compared behavior for three chromatin fiber systems of 100 nucleosomes: 100% native tails, 100% acetylated tails, and constructs with alternating 25 native tails and 25 acetylated tails repeated twice. The latter were modeled by more rigid tails, following our all-atom and multiscale study of histone acetylation (95), as shown in Figure 2B. The computed contact maps and folded fibers (Figure 3, pink slice) highlighted the role of histone tails in directing chromatin folding at the nucleosome level. While fibers with native tails led to compact structures with hierarchical loops and fibers with acetylated tails unfolded due to the absence of tail-directed stabilizing internucleosome interactions, the alternating fiber constructs showed segregated interactions of the two nucleosome types (see fiber structures in Figure 3, pink slice). Namely, nucleosomes with native histone tails clustered together and nucleosomes with acetylated tails clustered separately. The densest regions in the contact map account for native/native local and nonlocal interactions, while less dense regions come from local acetylated/acetylated and native/acetylated interactions (see contact map in Figure 3, pink slice).
These results demonstrate the intrinsic segregation propensity led by histone tail types in chromatin structure on the kb level and how acetylation mediates local and global condensation. This mechanism has been further supported by our investigations of different types of epigenetically marked histone tails (97) and by MC simulations of chromatin fibers containing regions with high level of histone methylation, showing phase segregation of dense heterochromatin and less dense euchromatin (193).
This folding study driven by epigenetic marks represents another example where computed contact maps obtained by mesoscale modeling help interpret experimentally derived data and to develop new mechanisms. Thus, segregation and kb-range interactions can be driven by nonuniform nucleosome positions, NFRs, and epigenetic modulation of histone tails, all important internal fiber parameters that regulate internucleosome interactions and hence our genome’s three-dimensional architecture.
7. Directed folding of the HOXC gene cluster by epigenetic factors and other basic nucleosome variables
All these features of directed folding were combined into our recent folding of the HOXC gene cluster to reveal a functionally important contact hub (101).
Namely, we combined specific position of nucleosomes (166, 194), LH binding (195), and acetylations (196) to “fold” in silico the 55-kb HOXC gene cluster from internal physical parameters (101). Specifically, we incorporated into our model epigenetic features as a function of genome positions based on Chip-seq data (196), nucleosome free regions (NFRs) based on MNase-seq data (194), linker lengths based on chemical mapping data (166), and specific LHs positions based on high resolution mapping data (195). The combined model includes a total of 284 nucleosomes, 55 kb of DNA, 5 genes containing 11 exons, and 5 acetylation islands.
When all these factors are combined, the HOXC gene spontaneously forms a dynamic connection hub, establishing a series of compact hierarchical loops that bring together promoters and exon/intron junctions (see folded HOXC gene structure in Figure 3, gray inner portion). Significantly, the computed contact map (Figure 3, gray inner portion) shows an increase in contact density and the presence of two contact domains corresponding to LH rich and acetylation rich regions that are brought together.
These results are consistent with a segregation mechanism in which same-type nucleosomes are clustered together, as was observed before with the alternating fiber constructs of wild type/acetylated tails (96). Indeed, the first Hi-C study in human cells showed that the genome is partitioned in domains that segregate into nuclear subcompartments (A and B) associated with different patterns of histone modifications (54). And such separation of chromatin into structural types has also been supported by polymer models that showed TADs of the same epigenomic state interacting dynamically with each other (113, 197).
Even though the role of epigenetic marks on chromatin structure can be studied using a large spectrum of techniques, mesoscale modeling of chromatin fiber, such as demonstrated for the HOXC gene cluster, can provide information at the nucleosome resolution level from “first principles”. As emphasized in the accompany commentary (198), this work opens the way to modeling chromosomal regions at the nucleosome resolution based on the constituent information: nucleosome positions, NFRs, linker histone binding, and epigenetic marks. More work is needed to develop appropriate models for other types of epigenetic marks, include the effect of remodeling proteins, and scale up the models to larger units.
Conclusion
Chromatin structure is intrinsically related to its function. A battery of excellent techniques is now available to study fibers, gene elements, and chromosomes at different levels of resolution, providing a variety of complementary data. The Sydney Brenner quote at the beginning of this article underscores the importance of technological innovation in the advancement of science. Our statistical analysis of publications on chromatin suggests that both computational methods and chromosome conformation capture techniques have exhibited the largest exponential growth in recent years. In this perspective, we illustrated their interplay as a way to explore chromatin architecture and interpret how chromatin higher-order structures regulates gene expression by fundamental mechanisms. The seven examples we described emphasize how configurations of fibers and associated computed measurements including nucleosome interaction maps obtained from chromatin fibers ranging from 100-nucleosome systems to genes of order of 55 kb help interpret 3C derived data and build realistic folded models of gene loci and clusters to propose/test/predict mechanisms.
Numerous further challenges lie ahead for both modeling and experimentally of chromatin and many new technologies will undoubtedly be available for genome structure interrogation. But one need not be overly optimistic to expect “folded” models of entire genomes in the not-too-distant future.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health, National Institute of General Medical Sciences awards R01-GM055264 and R35-GM122562, and in part by Phillip-Morris USA Inc. and Phillip-Morris International, to T.S.
References
- 1.Masters BR (2008) History of the Optical Microscope in Cell Biology and Medicine. eLS, doi: 10.1002/9780470015902.a0003082. [DOI] [Google Scholar]
- 2.Hooke R (1665) Micrographia, or, Some physiological descriptions of minute bodies made by magnifying glasses : with observations and inquiries thereupon London: Printed by Martyn J and Allestry J. [Google Scholar]
- 3.Ruska E (1987) The Development of the Electron Microscope and of Electron Microscopy (Nobel Lecture). Angew. Chemie Int. Ed. English, 26, 595–605. [Google Scholar]
- 4.Huang B, Bates M and Zhuang X (2009) Super resolution fluorescence microscopy. Annu. Rev. Biochem, 78, 993–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hell SW (2007) Far-field optical nanoscopy. Science, 316, 1153–1158. [DOI] [PubMed] [Google Scholar]
- 6.Schröder RR (2015) Advances in electron microscopy: A qualitative view of instrumentation development for macromolecular imaging and tomography. Arch. Biochem. Biophys, 581, 25–38. [DOI] [PubMed] [Google Scholar]
- 7.Bradbury S (2018) Landmarks in biological light microscopy. J. Microsc, 155, 281–305. [Google Scholar]
- 8.Marton L (1941) The Electron Microscope: A New Tool for Bacteriological Research. J. Bacteriol, 41, 397–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schaming D and Remita H (2015) Nanotechnology: from the ancient time to nowadays. Found. Chem, 17, 187–205. [Google Scholar]
- 10.The Royal Swedish Academy of Sciences (2013. October 9) The Nobel Prize in Chemistry 2013 [press release]. (2013). [Google Scholar]
- 11.Schlick T (2013) The 2013 Nobel Prize in Chemistry Celebrates Computations in Chemistry and Biology. SIAM News, 46, 1–4. [Google Scholar]
- 12.Franklin RE and Gosling RG (1953) Molecular Configuration in Sodium Thymonucleate. Nature, 171, 740–741. [DOI] [PubMed] [Google Scholar]
- 13.Watson JD and Crick FH (1953) Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature, 171, 737–738. [DOI] [PubMed] [Google Scholar]
- 14.Wilkins MHF, Stokes AR and Wilson HR (1953) Molecular structure of deoxypentose nucleic acids. Nature, 171, 738–740. [DOI] [PubMed] [Google Scholar]
- 15.Luzzati V and Nicolaieff A (1959) Etude par diffusion des rayons X aux petits angles des gels d’acide désoxyribonucléique et de nucléoprotéines : (note préliminaire). J. Mol. Biol, 1, 127–133. [Google Scholar]
- 16.Wilkins MHF, Zubay G and Wilson HR (1959) X-ray diffraction studies of the molecular structure of nucleohistone and chromosomes. J. Mol. Biol, 1, 179–185. [Google Scholar]
- 17.Bram S and Ris H (1971) On the structure of nucleohistone. J. Mol. Biol, 55, 325–336. [DOI] [PubMed] [Google Scholar]
- 18.Pardon JF, Richards BM and Cotter RI (1974) X-ray diffraction studies on oriented nucleohistone gels. Cold Spring Harb. Symp. Quant. Biol, 38, 75–81. [DOI] [PubMed] [Google Scholar]
- 19.Kornberg RD and Thomas JO (1974) Chromatin structure; oligomers of the histones. Science, 184, 865–868. [DOI] [PubMed] [Google Scholar]
- 20.Olins AL and Olins DE (1974) Spheroid chromatin units (v bodies). Science (80-.)., 183, 330–332. [DOI] [PubMed] [Google Scholar]
- 21.Woodcock CL (1973) Ultrastructure of inactive chromatin. J. Cell Biol, 59, 43–55. [Google Scholar]
- 22.Kelley RI (1973) Isolation of a histone IIb1–IIb2 complex. Biochem. Biophys. Res. Commun, 54, 1588–1594. [DOI] [PubMed] [Google Scholar]
- 23.D’Anna JA and Isenberg I (1974) Interactions of histone LAK (f2a2) with histones KAS (f2b) and GRK (f2a1). Biochemistry, 13, 2098–2104. [DOI] [PubMed] [Google Scholar]
- 24.Roark DE, Geoghegan TE and Keller GH (1974) A two-subunit histone complex from calf thymus. Biochem. Biophys. Res. Commun, 59, 542–547. [DOI] [PubMed] [Google Scholar]
- 25.Kornberg RD (1974) Chromatin Structure: A Repeating Unit of Histones and DNA. Science (80-.)., 184, 868–871. [DOI] [PubMed] [Google Scholar]
- 26.Gourevitch M, Puigdomenech P, Cave A, Etienne G, Mery J and Parello J (1974) Model studies in relation to the molecular structure of chromatin. Biochimie, 56, 967–985. [DOI] [PubMed] [Google Scholar]
- 27.Puigdomenechenech P, Martinez P, Cabare O, Palau J, Bradbury EM and Crane-Robinson C (1976) Studies on the Role and Mode of Operation of the Very-Lysine-Rich Histones in Eukaryote Chromatin: Nuclear-Magnetic-Resonance Studies on Nucleoprotein and Histone Ψ1-DNA Complexes from Marine Invertebrate Sperm. Eur. J. Biochem, 65, 357–363. [DOI] [PubMed] [Google Scholar]
- 28.Richmond TJ, Finch JT, Rushton B, Rhodes D and Klug A (1984) Structure of the nucleosome core particle at 7 Å resolution. Nature, 311, 532. [DOI] [PubMed] [Google Scholar]
- 29.Luger K, Mader AW, Richmond RK, Sargent DF and Richmond TJ (1997) Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature, 389, 251–260. [DOI] [PubMed] [Google Scholar]
- 30.Davey CA, Sargent DF, Luger K, Maeder AW and Richmond TJ (2002) Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 Å resolution. J. Mol. Biol, 319, 1097–1113. [DOI] [PubMed] [Google Scholar]
- 31.Öztürk MA, Cojocaru V and Wade RC (2018) Toward an Ensemble View of Chromatosome Structure: A Paradigm Shift from One to Many. Structure, 26, 1050–1057. [DOI] [PubMed] [Google Scholar]
- 32.Öztürk MA, Cojocaru V and Wade RC (2018) Dependence of Chromatosome Structure on Linker Histone Sequence and Posttranslational Modification. Biophys J, 114, 2363–2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Woodcock CL and Ghosh RP (2010) Chromatin higher-order structure and dynamics. Cold Spring Harb. Perspect. Biol, 2, a000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robinson PJJ, Fairall L, Huynh VAT and Rhodes D (2006) EM measurements define the dimensions of the ‘30-nm’ chromatin fiber: Evidence for a compact, interdigitated structure. Proc. Natl. Acad. Sci, 103, 6506–6511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song F, Chen P, Sun D, Wang M, Dong L, Liang D, Xu R-M, Zhu P and Li G (2014) Cryo-EM study of the chromatin fiber reveals a double helix twisted by tetranucleosomal units. Science (80-.)., 344, 376–380. [DOI] [PubMed] [Google Scholar]
- 36.Cremer T and Cremer C (2001) Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat. Rev. Genet, 2, 292–301. [DOI] [PubMed] [Google Scholar]
- 37.Shopland LS, Lynch CR, Peterson KA, Thornton K, Kepper N, Hase J. von, Stein S, Vincent S, Molloy KR, Kreth G, et al. (2006) Folding and organization of a contiguous chromosome region according to the gene distribution pattern in primary genomic sequence. J. Cell Biol, 174, 27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li G and Widom J (2004) Nucleosomes facilitate their own invasion. Nat. Struct. Mol. Biol, 11, 763–769. [DOI] [PubMed] [Google Scholar]
- 39.Tomschik M, Zheng H, van Holde K, Zlatanova J and Leuba SH (2005) Fast, long-range, reversible conformational fluctuations in nucleosomes revealed by single-pair fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. U. S. A, 102, 3278–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.White CL and Luger K (2004) Defined Structural Changes Occur in a Nucleosome upon Amt1 Transcription Factor Binding. J. Mol. Biol, 342, 1391–1402. [DOI] [PubMed] [Google Scholar]
- 41.Simon M, North JA, Shimko JC, Forties RA, Ferdinand MB, Manohar M, Zhang M, Fishel R, Ottesen JJ and Poirier MG (2011) Histone fold modifications control nucleosome unwrapping and disassembly. Proc. Natl. Acad. Sci. U. S. A, 108, 12711–12716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoch DA, Stratton JJ and Gloss LM (2007) Protein-protein Förster resonance energy transfer analysis of nucleosome core particles containing H2A and H2A.Z. J. Mol. Biol, 371, 971–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ricci MA, Manzo C, García-Parajo MF, Lakadamyali M and Cosma MP (2015) Chromatin Fibers Are Formed by Heterogeneous Groups of Nucleosomes In Vivo. Cell, 160, 1145–1158. [DOI] [PubMed] [Google Scholar]
- 44.Nozaki T, Imai R, Tanbo M, Nagashima R, Tamura S, Tani T, Joti Y, Tomita M, Hibino K, Kanemaki MT, et al. (2017) Dynamic Organization of Chromatin Domains Revealed by Super-Resolution Live-Cell Imaging. Mol. Cell, 67, 282–293. [DOI] [PubMed] [Google Scholar]
- 45.Boettiger AN, Bintu B, Moffitt JR, Wang S, Beliveau BJ, Fudenberg G, Imakaev M, Mirny LA, Wu C and Zhuang X (2016) Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature, 529, 418–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dekker J, Rippe K, Dekker M and Kleckner N (2002) Capturing Chromosome Conformation. Science (80-.)., 295, 1306–1311. [DOI] [PubMed] [Google Scholar]
- 47.Simonis M, Klous P, Splinter E, Moshkin Y, Willemsen R, de Wit E, van Steensel B and de Laat W (2006) Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nat. Genet, 38, 1348–1354. [DOI] [PubMed] [Google Scholar]
- 48.Andrey G, Montavon T, Mascrez B, Gonzalez F, Noordermeer D, Leleu M, Trono D, Spitz F and Duboule D (2013) A switch between topological domains underlies HoxD genes collinearity in mouse limbs. Science, 340, 1234167. [DOI] [PubMed] [Google Scholar]
- 49.Duan Z, Andronescu M, Schutz K, McIlwain S, Kim YJ, Lee C, Shendure J, Fields S, Blau CA and Noble WS (2010) A three-dimensional model of the yeast genome. Nature, 465, 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Montavon T, Soshnikova N, Mascrez B, Joye E, Thevenet L, Splinter E, de Laat W, Spitz F and Duboule D (2011) A Regulatory Archipelago Controls Hox Genes Transcription in Digits. Cell, 147, 1132–1145. [DOI] [PubMed] [Google Scholar]
- 51.Davies JOJ, Oudelaar AM, Higgs DR and Hughes JR (2017) How best to identify chromosomal interactions: a comparison of approaches. Nat. Methods, 14, 125–135. [DOI] [PubMed] [Google Scholar]
- 52.Denker A and de Laat W (2016) The second decade of 3C technologies: detailed insights into nuclear organization. Genes Dev, 30, 1357–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dostie J, Richmond TA, Arnaout RA, Selzer RR, Lee WL, Honan TA, Rubio ED, Krumm A, Lamb J, Nusbaum C, et al. (2006) Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res, 16, 1299–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. (2009) Comprehensive mapping of long range interactions reveals folding principles of the human genome. Science, 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, Laue ED, Tanay A and Fraser P (2013) Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature, 502, 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hsieh T-HS, Weiner A, Lajoie B, Dekker J, Friedman N and Rando OJ (2015) Mapping nucleosome resolution chromosome folding in yeast by Micro-C. Cell, 162, 108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hsieh T-HS, Fudenberg G, Goloborodko A and Rando OJ (2016) Micro-C XL: assaying chromosome conformation from the nucleosome to the entire genome. Nat. Methods, 13, 1009–1011. [DOI] [PubMed] [Google Scholar]
- 58.Ma W, Ay F, Lee C, Gulsoy G, Deng X, Cook S, Hesson J, Cavanaugh C, Ware CB, Krumm A, et al. (2015) Fine-scale chromatin interaction maps reveal the cis-regulatory landscape of human lincRNA genes. Nat. Methods, 12, 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hughes JR, Roberts N, McGowan S, Hay D, Giannoulatou E, Lynch M, De Gobbi M, Taylor S, Gibbons R and Higgs DR (2014) Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nat. Genet, 46, 205–212. [DOI] [PubMed] [Google Scholar]
- 60.Davies JOJ, Telenius JM, McGowan SJ, Roberts NA, Taylor S, Higgs DR and Hughes JR (2016) Multiplexed analysis of chromosome conformation at vastly improved sensitivity. Nat. Methods, 13, 74–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed Y. Bin, Orlov YL, Velkov S, Ho A, Mei PH, et al. (2009) An oestrogen-receptor-alpha-bound human chromatin interactome. Nature, 462, 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fullwood MJ and Ruan Y (2009) ChIP-based methods for the identification of long-range chromatin interactions. J. Cell. Biochem, 107, 30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tolhuis B, Palstra RJ, Splinter E, Grosveld F and de Laat W (2002) Looping and interaction between hypersensitive sites in the active beta-globin locus. Mol. Cell, 10, 1453–1465. [DOI] [PubMed] [Google Scholar]
- 64.Tanaka A, Tanizawa H, Capizzi JR, Lee M, Iwasaki O, Wickramasinghe P, Fu Z and Noma K (2010) Mapping of long-range associations throughout the fission yeast genome reveals global genome organization linked to transcriptional regulation. Nucleic Acids Res, 38, 8164–8177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, et al. (2012) Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature, 485, 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Phillips-Cremins JE, Sauria MEG, Sanyal A, Gerasimova TI, Lajoie BR, Bell JSK, Ong C-T, Hookway TA, Guo C, Sun Y, et al. (2013) Architectural protein subclasses shape 3-D organization of genomes during lineage commitment. Cell, 153, 1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS and Ren B (2012) Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature, 485, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dekker J (2005) The three ‘C’ s of chromosome conformation capture: controls, controls, controls. Nat. Methods, 3, 17–21. [DOI] [PubMed] [Google Scholar]
- 69.Grigoryev SA, Arya G, Correll S, Woodcock CL and Schlick T (2009) Evidence for heteromorphic chromatin fibers from analysis of nucleosome interactions. Proc. Natl. Acad. Sci, 106, 13317–13322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Grigoryev SA, Bascom G, Buckwalter JM, Schubert MB, Woodcock CL and Schlick T (2016) Hierarchical Looping of Zigzag Nucleosome Chains in Metaphase Chromosomes. Proc. Natl. Acad. Sci. USA, 113, 1238–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Risca VI, Denny SK, Straight AF and Greenleaf WJ (2017) Variable chromatin structure revealed by in situ spatially correlated DNA cleavage mapping. Nature, 541, 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Benham CJ (1977) Elastic model of supercoiling. Proc. Natl. Acad. Sci. U. S. A, 74, 2397–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fuller FB (1971) The writhing number of a space curve. Proc. Natl. Acad. Sci. U. S. A, 68, 815–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vologodskii AV, Anshelevich VV, Lukashin AV and Frank-Kamenetskii MD (1979) Statistical mechanics of supercoils and the torsional stiffness of the DNA double helix. Nature, 280, 294–298. [DOI] [PubMed] [Google Scholar]
- 75.Le Bret M (1980) Monte carlo computation of the supercoiling energy, the sedimentation constant, and the radius of gyration of unknotted and knotted circular DNA. Biopolymers, 19, 619–637. [DOI] [PubMed] [Google Scholar]
- 76.Chen Y (1981) Monte Carlo study of freely jointed ring polymers. III. The generation of undistorted perfect ring polymers. J. Chem. Phys, 75, 5160–5163. [Google Scholar]
- 77.Frank-Kamenetskii MD, Lukashin AV, Anshelevich VV and Vologodskii AV (1985) Torsional and bending rigidity of the double helix from data on small DNA rings. J. Biomol. Struct. Dyn, 2, 1005–1012. [DOI] [PubMed] [Google Scholar]
- 78.Shimada J and Yamakawa H (1988) Moments for DNA topoisomers: The helical wormlike chain. Biopolymers, 27, 657–673. [DOI] [PubMed] [Google Scholar]
- 79.Klenin KV, Vologodskii AV, Anshelevich VV, Klishko VY, Dykhne AM and Frank-Kamenetskii MD (1989) Variance of Writhe for Wormlike DNA Rings with Excluded Volume. J. Biomol. Struct. Dyn, 6, 707–714. [DOI] [PubMed] [Google Scholar]
- 80.Hao MH and Olson WK (1989) The global equilibrium configurations of supercoiled DNA. Macromolecules, 22, 3292–3303. [Google Scholar]
- 81.Allison S, Austin R and Hogan M (1989) Bending and twisting dynamics of short linear DNAs. Analysis of the triplet anisotropy decay of a 209 base pair fragment by Brownian simulation. J. Chem. Phys, 90, 3843–3854. [Google Scholar]
- 82.Allison SA (1986) Brownian dynamics simulation of wormlike chains. Fluorescence depolarization and depolarized light scattering. Macromolecules, 19, 118–124. [Google Scholar]
- 83.Levitt M (1983) Computer simulation of DNA double-helix dynamics. Cold Spring Harb. Symp. Quant. Biol, 47, 251–262. [DOI] [PubMed] [Google Scholar]
- 84.Schlick T and Olson WK (1992) Supercoiled DNA energetics and dynamics by computer simulation. J. Mol. Biol, 223, 1089–1119. [DOI] [PubMed] [Google Scholar]
- 85.Schlick T and Olson WK (1992) Trefoil Knotting Revealed by Molecular Dynamics Simulations of Supercoiled DNA. Science (80-.)., 257, 1110–1115. [DOI] [PubMed] [Google Scholar]
- 86.Chirico G and Langowski J (1994) Kinetics of DNA supercoiling studied by Brownian dynamics simulation. Biopolymers, 34, 415–433. [Google Scholar]
- 87.Ehrlich L, Munkel C, Chirico G and Langowski J (1997) A Brownian dynamics model for the chromatin fiber. Comput. Appl. Biosci, 13, 271–279. [DOI] [PubMed] [Google Scholar]
- 88.Katritch V, Bustamante C and Olson WK (2000) Pulling chromatin fibers: computer simulations of direct physical micromanipulations. J. Mol. Biol, 295, 29–40. [DOI] [PubMed] [Google Scholar]
- 89.Wedemann G and Langowski J (2002) Computer simulation of the 30-nanometer chromatin fiber. Biophys. J, 82, 2847–2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Beard DA and Schlick T (2001) Modeling salt-mediated electrostatics of macromolecules: the discrete surface charge optimization algorithm and its application to the nucleosome. Biopolymers, 58, 106–115. [DOI] [PubMed] [Google Scholar]
- 91.Sun J, Zhang Q and Schlick T (2005) Electrostatic mechanism of nucleosomal array folding revealed by computer simulation. Proc. Natl. Acad. Sci. U. S. A, 102, 8180–8185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Arya G and Schlick T (2009) A Tale of Tails: How Histone Tails Mediate Chromatin Compaction in Different Salt and Linker Histone Environments. J. Phys. Chem. A, 113, 4045–4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Perišić O, Portillo-Ledesma S and Schlick T (2019) Sensitive effect of linker histone binding mode and subtype on chromatin condensation. 10.1093/nar/gkz234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Luque A, Collepardo-Guevara R, Grigoryev S and Schlick T (2014) Dynamic Condensation of Linker Histone C-terminal Domain Regulates Chromatin Structure. Nucleic Acids Res, 42, 7553–7560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Collepardo-Guevara R, Portella G, Vendruscolo M, Frenkel D, Schlick T and Orozco M (2015) Chromatin Unfolding by Epigenetic Modifications Explained by Dramatic Impairment of Internucleosome Interactions: A Multiscale Computational Study. J. Am. Chem. Soc, 137, 10205–10215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rao SSP, Huang S-C, Glenn St Hilaire B, Engreitz JM, Perez EM, Kieffer-Kwon K-R, Sanborn AL, Johnstone SE, Bascom GD, Bochkov ID, et al. (2017) Cohesin Loss Eliminates All Loop Domains. Cell, 171, 305–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bascom GD and Schlick T (2018) Chromatin Fiber Folding Directed by Cooperative Histone Tail Acetylation and Linker Histone Binding. Biophys. J, 114, 2376–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fan Y, Korolev N, Lyubartsev AP and Nordenskiöld L (2013) An Advanced Coarse-Grained Nucleosome Core Particle Model for Computer Simulations of Nucleosome-Nucleosome Interactions under Varying Ionic Conditions. PLoS One, 8, e54228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bascom G, Kim T and Schlick T (2017) Kilobase Pair Chromatin Fiber Contacts Promoted by Living-System-Like DNA Linker Length Distributions and Nucleosome Depletion. J. Phys. Chem. B, 121, 3882–3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bascom G and Schlick T (2017) Linking Chromatin Fibers to Gene Folding by Hierarchical Looping. Biophys. J, 112, 434–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bascom G, Myers C and Schlick T (2018) Mesoscale modeling reveals formation of an epigenetically driven hoxc gene hubs. Proc. Natl. Acad. Sci. USA, 116, 4955–4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schlick T, Hayes J and Grigoryev S (2012) Toward convergence of experimental studies and theoretical modeling of the chromatin fiber. J. Biol. Chem, 287, 5183–5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ozer G, Luque A and Schlick T (2015) The Chromatin Fiber:Multiscale Problems and Approaches. Curr. Opin. Struc. Biol, 31, 124–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Potoyan DA and Papoian GA (2012) Regulation of the H4 tail binding and folding landscapes via Lys-16 acetylation. Proc. Natl. Acad. Sci. U. S. A, 109, 17857–17862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Potoyan DA and Papoian GA (2011) Energy Landscape Analyses of Disordered Histone Tails Reveal Special Organization of Their Conformational Dynamics. J. Am. Chem. Soc, 133, 7405–7415. [DOI] [PubMed] [Google Scholar]
- 106.Kimura H, Shimooka Y, Nishikawa J, Miura O, Sugiyama S, Yamada S and Ohyama T (2013) The genome folding mechanism in yeast. J. Biochem, 154, 137–147. [DOI] [PubMed] [Google Scholar]
- 107.Barbieri M, Chotalia M, Fraser J, Lavitas L-M, Dostie J, Pombo A and Nicodemi M (2012) Complexity of chromatin folding is captured by the strings and binders switch model. Proc. Natl. Acad. Sci. U. S. A, 109, 16173–16178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bohn M and Heermann DW (2010) Diffusion-Driven Looping Provides a Consistent Framework for Chromatin Organization. PLoS One, 5, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tark-Dame M, van Driel R and Heermann DW (2011) Chromatin folding - from biology to polymer models and back. J. Cell Sci, 124, 839–845. [DOI] [PubMed] [Google Scholar]
- 110.Cook PR and Marenduzzo D (2009) Entropic organization of interphase chromosomes. J. Cell Biol, 186, 825–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mateos-Langerak J, Bohn M, de Leeuw W, Giromus O, Manders EMM, Verschure PJ, Indemans MHG, Gierman HJ, Heermann DW, van Driel R, et al. (2009) Spatially confined folding of chromatin in the interphase nucleus. Proc. Natl. Acad. Sci. U. S. A, 106, 3812–3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhan Y, Giorgetti L and Tiana G (2017) Modelling genome-wide topological associating domains in mouse embryonic stem cells. Chromosom. Res, 25, 5–14. [DOI] [PubMed] [Google Scholar]
- 113.Jost D, Carrivain P, Cavalli G and Vaillant C (2014) Modeling epigenome folding: formation and dynamics of topologically associated chromatin domains. Nucleic Acids Res, 42, 9553–9561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Naumova N, Imakaev M, Fudenberg G, Zhan Y, Lajoie BR, Mirny LA and Dekker J (2013) Organization of the mitotic chromosome. Science, 342, 948–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kragesteen BK, Spielmann M, Paliou C, Heinrich V, Schöpflin R, Esposito A, Annunziatella C, Bianco S, Chiariello AM, Jerković I, et al. (2018) Dynamic 3D chromatin architecture contributes to enhancer specificity and limb morphogenesis. Nat. Genet, 50, 1463–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Huyen Y, Zgheib O, DiTullio RA Jr, Gorgoulis VG, Zacharatos P, Petty TJ, Sheston EA, Mellert HS, Stavridi ES, Halazonetis TD, et al. (2004) Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature, 432, 406–411. [DOI] [PubMed] [Google Scholar]
- 117.Schlick T (2010) Molecular Modeling and Simulation: An Interdisciplinary Guide 2nd ed. Springer-Verlag, New York, NY. [Google Scholar]
- 118.Dans PD, Walther J, Gómez H and Orozco M (2016) Multiscale simulation of DNA. Curr. Opin. Struct. Biol, 37, 29–45. [DOI] [PubMed] [Google Scholar]
- 119.Langowski J and Heermann DW (2007) Computational modeling of the chromatin fiber. Semin. Cell Dev. Biol, 18, 659–667. [DOI] [PubMed] [Google Scholar]
- 120.Bascom G and Schlick T (2018) 5 - Mesoscale Modeling of Chromatin Fibers. In Lavelle C, Victor J-M. (eds), Translational Epigenetics. Academic Press, Boston, Vol. 2, pp. 123–147. [Google Scholar]
- 121.Korolev N, Fan Y, Lyubartsev AP and Nordenskiöld L (2012) Modelling chromatin structure and dynamics: status and prospects. Curr. Opin. Struct. Biol, 22, 151–159. [DOI] [PubMed] [Google Scholar]
- 122.Amitai A and Holcman D (2017) Polymer physics of nuclear organization and function. Phys. Rep, 678, 1–83. [Google Scholar]
- 123.Tiana G. and Giorgetti L. eds. (2019) Modeling the 3D Conformation of Genomes 1st ed. Boca Raton: CRC Press, 10.1201/9781315144009. [DOI] [Google Scholar]
- 124.Korolev N, Nordenskiöld L and Lyubartsev AP (2016) Multiscale coarse-grained modelling of chromatin components: DNA and the nucleosome. Adv. Colloid Interface Sci, 232, 36–48. [DOI] [PubMed] [Google Scholar]
- 125.Ricci MA, Cosma MP and Lakadamyali M (2017) Super resolution imaging of chromatin in pluripotency, differentiation, and reprogramming. Curr. Opin. Genet. Dev, 46, 186–193. [DOI] [PubMed] [Google Scholar]
- 126.Grigoryev SA (2018) Chromatin Higher-Order Folding: A Perspective with Linker DNA Angles. Biophys. J, 114, 2290–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Grigoryev SA and Schubert M (2019) Unraveling the multiplex folding of nucleosome chains in higher order chromatin. Essays Biochem, 63, 109–121. [DOI] [PubMed] [Google Scholar]
- 128.Rowley MJ and rdan and Corces VG (2018) Organizational principles of 3D genome architecture. Nat. Rev. Genet, 19, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Andrews FH, Strahl BD and Kutateladze TG (2016) Insights into newly discovered marks and readers of epigenetic information. Nat. Chem. Biol, 12, 662–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Azad GK, Swagatika S, Kumawat M, Kumawat R and Tomar RS (2018) Modifying Chromatin by Histone Tail Clipping. J. Mol. Biol, 430, 3051–3067. [DOI] [PubMed] [Google Scholar]
- 131.Fyodorov DV, Zhou B-R, Skoultchi AI and Bai Y (2018) Emerging roles of linker histones in regulating chromatin structure and function. Nat. Rev. Mol. Cell. Biol, 19, 192–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Haddad N, Jost D and Vaillant C (2017) Perspectives: using polymer modeling to understand the formation and function of nuclear compartments. Chromosom. Res, 25, 35–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Annunziatella C, Chiariello AM, Esposito A, Bianco S, Fiorillo L and Nicodemi M (2018) Molecular Dynamics simulations of the Strings and Binders Switch model of chromatin. Methods, 142, 81–88. [DOI] [PubMed] [Google Scholar]
- 134.Racko D, Benedetti F, Dorier J and Stasiak A (2018) Are TADs supercoiled? Nucleic Acids Res, 47, 521–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Olson WK, Clauvelin N, Colasanti AV, Singh G and Zheng G (2012) Insights into Gene Expression and Packaging from Computer Simulations. Biophys. Rev, 4, 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tremethick DJ (2007) Higher-order structures of chromatin: the elusive 30 nm fiber. Cell, 128, 651–654. [DOI] [PubMed] [Google Scholar]
- 137.Collepardo-Guevara R and Schlick T (2014) Chromatin fiber polymorphism triggered by variations of DNA linker lengths. Proc. Natl. Acad. Sci, 111, 8061–8066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Maeshima K, Imai R, Tamura S and Nozaki T (2014) Chromatin as dynamic 10-nm fibers. Chromosoma, 123, 225–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Fussner E, Ching RW and Bazett-Jones DP (2011) Living without 30nm chromatin fibers. Trends Biochem. Sci, 36, 1–6. [DOI] [PubMed] [Google Scholar]
- 140.Horowitz RA, Agard DA, Sedat JW and Woodcock CL (1994) The three-dimensional architecture of chromatin in situ: electron tomography reveals fibers composed of a continuously variable zig-zag nucleosomal ribbon. J. Cell Biol, 125, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Scheffer MP, Eltsov M and Frangakis AS (2011) Evidence for short-range helical order in the 30-nm chromatin fibers of erythrocyte nuclei. Proc. Natl. Acad. Sci. USA, 108, 16992–16997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Collepardo-Guevara R and Schlick T (2011) The effect of linker histone’s nucleosome binding affinity on chromatin unfolding mechanisms. Biophys. J, 101, 1670–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Levitt M and Warshel A (1975) Computer simulation of protein folding. Nature, 253, 694–698. [DOI] [PubMed] [Google Scholar]
- 144.Stigter D (1977) Interactions of highly charged colloidal cylinders with applications to double-stranded. Biopolymers, 16, 1435–1448. [DOI] [PubMed] [Google Scholar]
- 145.Metropolis N and Ulam S (1949) The Monte Carlo Method. J. Am. Stat. Assoc, 44, 335–341. [DOI] [PubMed] [Google Scholar]
- 146.Hashimoto H, Takami Y, Sonoda E, Iwasaki T, Iwano H, Tachibana M, Takeda S, Nakayama T, Kimura H and Shinkai Y (2010) Histone H1 null vertebrate cells exhibit altered nucleosome architecture. Nucleic Acids Res, 38, 3533–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Chen D, Dundr M, Wang C, Leung A, Lamond A, Misteli T and Huang S (2005) Condensed mitotic chromatin is accessible to transcription factors and chromatin structural proteins. J. Cell Biol, 168, 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Woodcock CL, Skoultchi AI and Fan Y (2006) Role of linker histone in chromatin structure and function: H1 stoichiometry and nucleosome repeat length. Chromosom. Res, 14, 17–25. [DOI] [PubMed] [Google Scholar]
- 149.Bleher R and Martin R (1999) Nucleo-cytoplasmic translocation of histone H1 during the HeLa cell cycle. Chromosoma, 108, 308–316. [DOI] [PubMed] [Google Scholar]
- 150.Eltsov M, Maclellan KM, Maeshima K, Frangakis AS and Dubochet J (2008) Analysis of cryo-electron microscopy images does not support the existence of 30-nm chromatin fibers in mitotic chromosomes in situ. Proc. Natl. Acad. Sci. U. S. A, 105, 19732–19737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ou HD, Phan S, Deerinck TJ, Thor A, Ellisman MH and O’Shea CC (2017) ChromEMT: Visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science, 357, eaag0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Nishino Y, Eltsov M, Joti Y, Ito K, Takata H, Takahashi Y, Hihara S, Frangakis AS, Imamoto N, Ishikawa T, et al. (2012) Human mitotic chromosomes consist predominantly of irregularly folded nucleosome fibres without a 30-nm chromatin structure. EMBO J, 31, 1644–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Daban J-R (2015) Stacked thin layers of metaphase chromatin explain the geometry of chromosome rearrangements and banding. Sci. Rep, 5, 14891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Mahamid J, Pfeffer S, Schaffer M, Villa E, Danev R, Kuhn Cuellar L, Förster F, Hyman AA, Plitzko JM and Baumeister W (2016) Visualizing the molecular sociology at the HeLa cell nuclear periphery. Science (80-.)., 351, 969–972. [DOI] [PubMed] [Google Scholar]
- 155.Fang H, Clark DJ and Hayes JJ (2012) DNA and nucleosomes direct distinct folding of a linker histone H1 C-terminal domain. Nucleic Acid. Res, 40, 1475–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Roque A, Iloro I, Ponte I, Arrondo JLR and Suau P (2005) DNA-induced secondary structure of the carboxyl-terminal domain of histone H1. J. Biol. Chem, 280, 32141–32147. [DOI] [PubMed] [Google Scholar]
- 157.Woodcock CL, Skoultchi AI and Fan Y (2006) Role of linker histone in chromatin structure and function: H1 stoichiometry and nucleosome repeat length. Chromosom. Res., 14, 17–25. [DOI] [PubMed] [Google Scholar]
- 158.Popova EY, Grigoryev SA, Fan Y, Skoultchi AI, Zhang SS and Barnstable CJ (2013) Developmentally Regulated Linker Histone H1c Promotes Heterochromatin Condensation and Mediates Structural Integrity of Rod Photoreceptors in Mouse Retina. J. Biol. Chem, 288, 17895–17907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Collepardo-Guevara R and Schlick T (2012) Crucial Role of Dynamic Linker Histone Binding and Divalent Ions for DNA Accessibility and Gene Regulation Revealed by Mesoscale Modeling of Oligonucleosomes. Nucleic Acids Res, 40, 8803–8817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Öztürk MA, Pachov GV, Wade RC and Cojocaru V (2016) Conformational selection and dynamic adaptation upon linker histone binding to the nucleosome. Nucleic Acids Res, 44, 6599–6613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Bednar J, Garcia-Saez I, Boopathi R, Cutter AR, Papai G, Reymer A, Syed SH, Lone IN, Tonchev O, Crucifix C, et al. (2017) Structure and Dynamics of a 197 bp Nucleosome in Complex with Linker Histone H1. Mol. Cell, 66, 384–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Garcia-Saez I, Menoni H, Boopathi R, Shukla MS, Soueidan L, Noirclerc-Savoye M, Roy A. Le, Skoufias DA, Bednar J, Hamiche A, et al. (2018) Structure of an H1-Bound 6-Nucleosome Array Reveals an Untwisted Two-Start Chromatin Fiber Conformation. Mol. Cell, 72, 902–915.e7. [DOI] [PubMed] [Google Scholar]
- 163.Bai L and Morozov AV (2010) Gene regulation by nucleosome positioning. Trends Genet, 26, 476–483. [DOI] [PubMed] [Google Scholar]
- 164.Moyle-Heyrman G, Zaichuk T, Xi L, Zhang Q, Uhlenbeck OC, Holmgren R, Widom J and Wang J-P (2013) Chemical map of Schizosaccharomyces pombe reveals species-specific features in nucleosome positioning. Proc. Natl. Acad. Sci, 110, 20158–20163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Brogaard K, Xi L, Wang J-PP and Widom J (2012) A map of nucleosome positions in yeast at base-pair resolution. Nature, 486, 496–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Voong LN, Xi L, Sebeson AC, Xiong B, Wang J-P and Wang X (2016) Insights into Nucleosome Organization in Mouse Embryonic Stem Cells through Chemical Mapping. Cell, 167, 1555–1570.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kadauke S and Blobel GA (2009) Chromatin loops in gene regulation. Biochim. Biophys. Acta- Gene Regul. Mech, 1789, 17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Dekker J and Misteli T (2015) Long-range chromatin interactions. Cold Spring Harb. Perspect. Biol, 7, a019356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Mifsud B, Tavares-Cadete F, Young AN, Sugar R, Schoenfelder S, Ferreira L, Wingett SW, Andrews S, Grey W, Ewels PA, et al. (2015) Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat. Genet, 47, 598–606. [DOI] [PubMed] [Google Scholar]
- 170.Sanyal A, Lajoie BR, Jain G and Dekker J (2012) The long-range interaction landscape of gene promoters. Nature, 489, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Bowman GD and Poirier MG (2015) Post-translational modifications of histones that influence nucleosome dynamics. Chem. Rev, 115, 2274–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Cruickshank MN, Besant P and Ulgiati D (2010) The impact of histone post-translational modifications on developmental gene regulation. Amino Acids, 39, 1087–1105. [DOI] [PubMed] [Google Scholar]
- 173.Arya G, Zhang Q and Schlick T (2006) Flexible histone tails in a new mesoscopic oligonucleosome model. Biophys. J, 91, 133–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Arya G and Schlick T (2006) Role of histone tails in chromatin folding revealed by a mesoscopic oligonucleosome model. Proc. Natl. Acad. Sci, 103, 16236–16241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Luque A, Ozer G and Schlick T (2016) Correlation among DNA Linker Length, Linker Histone Concentration, and Histone Tails in Chromatin. Biophys. J, 110, 2309–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Zhang R, Erler J and Langowski J (2017) Histone Acetylation Regulates Chromatin Accessibility: Role of H4K16 in Inter-nucleosome Interaction. Biophys. J, 112, 450–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Chen Q, Yang R, Korolev N, Liu CF and Nordenskiöld L (2017) Regulation of Nucleosome Stacking and Chromatin Compaction by the Histone H4 N-Terminal Tail–H2A Acidic Patch Interaction. J. Mol. Biol, 429, 2075–2092. [DOI] [PubMed] [Google Scholar]
- 178.Bascom GD, Sanbonmatsu KY and Schlick T (2016) Mesoscale Modeling Reveals Hierarchical Looping of Chromatin Fibers near Gene Regulatory Elements. J. Phys. Chem. B, 120, 8642–8653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Fujikura J, Yamato E, Yonemura S, Hosoda K, Masui S, Nakao K, Miyazaki Ji J and Niwa H (2002) Differentiation of embryonic stem cells is induced by GATA factors. Genes Dev, 16, 784–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Laverriere AC, MacNeill C, Mueller C, Poelmann RE, Burch JB and Evans T (1994) GATA-4/5/6, a subfamily of three transcription factors transcribed in developing heart and gut. J. Biol. Chem, 269, 23177–23184. [PubMed] [Google Scholar]
- 181.Tiwari VK, McGarvey KM, Licchesi JDF, Ohm JE, Herman JG, Schübeler D and Baylin SB (2008) PcG Proteins, DNA Methylation, and Gene Repression by Chromatin Looping. PLOS Biol, 6, e306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Dunn TM, Hahn S, Ogden S and Schleif RF (1984) An operator at −280 base pairs that is required for repression of araBAD operon promoter: addition of DNA helical turns between the operator and promoter cyclically hinders repression. Proc. Natl. Acad. Sci. U. S. A, 81, 5017–5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kramer H, Niemoller M, Amouyal M, Revet B, von Wilcken-Bergmann B and Muller-Hill B (1987) lac repressor forms loops with linear DNA carrying two suitably spaced lac operators. EMBO J, 6, 1481–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Mukherjee S, Erickson H and Bastia D (1988) Enhancer-origin interaction in plasmid R6K involves a DNA loop mediated by initiator protein. Cell, 52, 375–383. [DOI] [PubMed] [Google Scholar]
- 185.Griffith J, Hochschild A and Ptashne M (1986) DNA loops induced by cooperative binding of lambda repressor. Nature, 322, 750–752. [DOI] [PubMed] [Google Scholar]
- 186.Fraser J, Williamson I, Bickmore WA and Dostie J (2015) An Overview of Genome Organization and How We Got There: from FISH to Hi-C. Microbiol. Mol. Biol. Rev, 79, 347–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Gerasimova TI, Byrd K and Corces VG (2000) A chromatin insulator determines the nuclear localization of DNA. Mol. Cell, 6, 1025–1035. [DOI] [PubMed] [Google Scholar]
- 188.Souaid C, Bloyer S and Noordermeer D (2018) 19 - Promoter–Enhancer Looping and Regulatory Neighborhoods: Gene Regulation in the Framework of Topologically Associating Domains. In Lavelle C, Victor J-M. (eds), Translational Epigenetics. Academic Press, Boston, Vol. 2, pp. 435–456. [Google Scholar]
- 189.Hou C, Zhao H, Tanimoto K and Dean A (2008) CTCF-dependent enhancer-blocking by alternative chromatin loop formation. Proc. Natl. Acad. Sci. U. S. A, 105, 20398–20403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Phillips-Cremins JE and Corces VG (2013) Chromatin insulators: linking genome organization to cellular function. Mol. Cell, 50, 461–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. (2014) A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell, 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Rowley MJ and Corces VG (2016) Capturing native interactions: intrinsic methods to study chromatin conformation. Mol. Syst. Biol, 12, 897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.MacPherson Q, Beltran B and Spakowitz AJ (2018) Bottom-up modeling of chromatin segregation due to epigenetic modifications. Proc. Natl. Acad. Sci. U. S. A, 115, 12739–12744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Yazdi PG, Pedersen BA, Taylor JF, Khattab OS, Chen YY-H, Chen YY-H, Jacobsen SE and Wang PH (2015) Increasing Nucleosome Occupancy Is Correlated with an Increasing Mutation Rate so Long as DNA Repair Machinery Is Intact. PLoS One, 10, e0136574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Cao K, Lailler N, Zhang Y, Kumar A, Uppal K, Liu Z, Lee EK, Wu H, Medrzycki M, Pan C, et al. (2013) High-Resolution Mapping of H1 Linker Histone Variants in Embryonic Stem Cells. PLOS Genet., 9, e1003417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Karolchik D, Baertsch R, Diekhans M, Furey TS, Hinrichs A, Lu YT, Roskin KM, Schwartz M, Sugnet CW, Thomas DJ, et al. (2003) The UCSC Genome Browser Database. Nucleic Acids Res, 31, 51–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Di Pierro M, Cheng RR, Lieberman Aiden E, Wolynes PG and Onuchic JN (2017) De novo prediction of human chromosome structures: Epigenetic marking patterns encode genome architecture. Proc. Natl. Acad. Sci. U. S. A, 114, 12126–12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Di Pierro M (2019) Inner workings of gene folding. Proc. Natl. Acad. Sci. U. S. A, 116, 4774–4775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Bednar J, Horowitz RA, Grigoryev SA, Carruthers LM, Hansen JC, Koster AJ and Woodcock CL (1998) Nucleosomes, Linker DNA, and Linker Histone form a Unique Structural Motif that Directs the Higher-Order Folding and Compaction of Chromatin. Proc. Natl. Acad. Sci. U. S. A, 95, 14173–14178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Habermann FA, Cremer M, Walter J, Kreth G, von Hase J, Bauer K, Wienberg J, Cremer C, Cremer T and Solovei I (2001) Arrangements of macro- and microchromosomes in chicken cells. Chromosom. Res, 9, 569–584. [DOI] [PubMed] [Google Scholar]
- 201.Li H, Ilin S, Wang W, Duncan EM, Wysocka J, Allis CD and Patel DJ (2006) Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature, 442, 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Perišić O, Collepardo-Guevara R and Schlick T (2010) Modeling Studies of Chromatin Fiber Structure as a Function of DNA Linker Length. J. Mol. Biol, 403, 777–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Zhou B-R, Jiang J, Feng H, Ghirlando R, Xiao TS and Bai Y (2015) Structural Mechanisms of Nucleosome Recognition by Linker Histones. Mol. Cell, 59, 628–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Zhou B-R, Feng H, Ghirlando R, Kato H, Gruschus J and Bai Y (2012) Histone H4 K16Q mutation, an acetylation mimic, causes structural disorder of its N-terminal basic patch in the nucleosome. J. Mol. Biol, 421, 30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.