

Abstract

The study of low-abundance proteins is a challenge to discovery-based proteomics. Mass spectrometry (MS) applications, such as thermal proteome profiling (TPP), face specific challenges in the detection of the whole proteome as a consequence of the use of nondenaturing extraction buffers. TPP is a powerful method for the study of protein thermal stability, but quantitative accuracy is highly dependent on consistent detection. Therefore, TPP can be limited in its amenability to study low-abundance proteins that tend to have stochastic or poor detection by MS. To address this challenge, we incorporated an affinity-purified protein complex sample at submolar concentrations as an isobaric trigger channel into a mutant TPP (mTPP) workflow to provide reproducible detection and quantitation of the low-abundance subunits of the cleavage and polyadenylation factor (CPF) complex. The inclusion of an isobaric protein complex trigger channel increased detection an average of 40× for previously detected subunits and facilitated detection of CPF subunits that were previously below the limit of detection. Importantly, these gains in CPF detection did not cause large changes in melt temperature (Tm) calculations for other unrelated proteins in the samples, with a high positive correlation between Tm estimates in samples with and without isobaric trigger channel addition. Overall, the incorporation of an affinity-purified protein complex as an isobaric trigger channel within a tandem mass tag (TMT) multiplex for mTPP experiments is an effective and reproducible way to gather thermal profiling data on proteins that are not readily detected using the original TPP or mTPP protocols.
Proteins are the functional units of a cell, carrying out and controlling processes at specific times and locations to maintain homeostasis and respond to external stimuli. As a consequence of functional changes, proteins can exist in a variety of biophysical states within cells as a consequence of variants in their primary sequence, post-translational modification (PTM) state, and/or subcellular localization. In many cases, a protein’s biophysical state is impacted by associations with other proteins, including both transient and stable protein–protein interactions. The characterization of protein–protein interactions (PPIs) is fundamental to gaining a full understanding of biological mechanisms. In fact, PPIs are so critical to proper protein function that gain or loss of interactions can lead to disease and/or cell death.1,2 Advances in mass spectrometry (MS)-based proteomics workflows continue to increase our ability to study protein complex dynamics and PPIs.3−8 MS-based approaches for protein interaction analysis rely on discovery-based proteomics performed using data-dependent acquisition (DDA). Generally in DDA, peptides with the most intense ions from MS1 are selected for fragmentation and MS2 analysis.9 This approach maximizes signal-to-noise levels and thereby increases confidence in the selection and subsequent identification of the peptide ions.
Challenges with the use of DDA include the selection of peptide ions from protein(s) of interest that are present at low relative abundance levels or when peptides of interest (such as PTM containing peptides) are present at low relative levels to their unmodified counterparts. Low-abundance peptides may be present at insufficient MS1 signal intensity levels to trigger fragmentation and MS2 analysis based on instrument settings for MS2 analysis. While fractionation and an extended high-performance liquid chromatography (HPLC) gradient help to spread out the elution of peptides into the mass spectrometer, many peptides may still coelute such that highly abundant ion species will outcompete those that are less abundant.10 A number of strategies have recently emerged to improve MS detection of low-abundance proteins and post-translational modifications (PTMs) for a variety of applications including single-cell proteomics.11−17 Although we will not discuss all of the recently established strategies here, one such strategy, boosting to amplify the signal with isobaric labeling (BASIL), has similarities that have informed the current work. Specifically, BASIL has been shown to successfully increase detection of low-abundance phosphopeptides through the addition of a boosting sample to a tandem mass tag (TMT)-based multiplex.18 TMTPro labeling allows for the multiplexing and relative quantitation of up to 16 samples.19−21 As each TMT label is isobaric, labeled peptides from the multiplexed samples elute into the mass spectrometer together and are analyzed simultaneously as one ion peak during MS1 scans which are distinguished in fragment ion scans during MSn (typically MS2 or MS3) analysis. By incorporating a phospho-enriched sample into a single channel in the TMT multiplex, Yi et al. increased ion abundance of phosphopeptides in the MS1 scan to the extent that MS2 was triggered for phosphopeptides that were typically below the level of detection in standard DDA approaches.18 BASIL allowed for the identification and quantification of phosphopeptides in other TMT channels, where enrichment had not been performed.18 The BASIL method has since been optimized for detection of phosphopeptides in single cells22 and similar approaches have been applied to phosphotyrosine-containing peptides,23 stable isotope labeling using amino acids in cell culture (SILAC)-labeled peptides,24 and using synthetic peptides to particular peptides of interest.25 BASIL and other similar methods that take advantage of isobaric carrier channels could have numerous applications in DDA-based quantitative workflows.
The challenges to studying low-abundance proteins in DDA proteomic experiments extend in particular to the mass spectrometry-based thermal proteome profiling (TPP) methods and are the focus of this study. TPP analysis takes advantage of TMT labeling technology to produce protein melt curves that can then be compared across conditions to measure alterations in protein thermal stability.26,27 Although TPP was originally developed to study drug and ligand binding, it has also been shown to be a robust approach to probe PPIs in a number of different applications (recently reviewed by Mateus et al.28). We recently developed a new application of TPP referred to as mutant TPP (mTPP), which is used to study the effects of protein missense mutations on the proteome at large with the ability to focus on specific protein complexes and their PPIs.29 mTPP analysis is advantageous over other methods for the study of PPIs in that it does not require antibodies, reagents such as crosslinkers, or production of fusion proteins. Additionally, mTPP can be performed with significantly less starting material than traditional affinity purification or enrichment approaches, making it applicable to a wider variety of sample types. Despite these advantages, we have encountered challenges associated with quantitative analysis of specific target proteins and their interaction partners. Detection of low-abundance target proteins is inherent to many DDA-based proteomic studies because of the large dynamic range of eukaryotic proteomes such that analysis of a global proteome results in excellent quantitation of high-abundance proteins, while the majority of the proteome is surveyed in a more stochastic manner at both the protein and peptide levels.30 One advantage of TMT- and isobaric tag for relative and absolute quantitation (iTRAQ)-based multiplexed workflows for global proteomics studies is that the pooling of multiple samples generates increased protein starting material that can then be subjected to extensive biochemical fractionation to facilitate deep proteome coverage.31−35 This advantage can be coupled with protein extraction methods using denaturants such as urea or sodium dodecyl sulfate (SDS) to isolate the full proteome of many cells and tissues.36 The workflow for TPP cannot exploit these advantages since (1) temperature treatment of lysates for TPP results in unequal levels of protein mixture across the multiplex that, in our hands, vary on average at least 10-fold from the lowest to the highest temperature treatment29 and (2) nondenaturing protein extraction buffers must be used to maintain protein structure, PPIs, and protein interactions with other molecules (including but not limited to lipids, metabolites, small molecules, and drugs).26−28 As a consequence, TPP workflows typically result in decreased proteome coverage relative to denaturant extracted proteomes even when equivalent amounts of starting material are used.29
We have developed a BASIL-like approach that expands proteome coverage for our mTPP workflow and increases the signal of low-abundance protein complexes and their representative peptides. Our mTPP experiment approach uses a protein complex affinity purification trigger channel in place of the phosphopeptide isobaric boosting channel used in BASIL.18 As a proof-of-concept, we investigated the ability of this approach to enhance detection of the relatively low-abundance protein complex cleavage and polyadenylation factor (CPF) complex in a mTPP workflow. Affinity-purified CPF that we previously characterized37−41 was incorporated as an isobaric trigger channel in our mTPP workflow at a ratio to the lowest heat-treated mTPP sample of ∼1:8 and ∼1:50. Using this approach, we observed a significant increase in the abundance of CPF complex members, including those that were not readily identified without the isobaric trigger channel. Importantly, the addition of an isobaric trigger channel into our mTPP workflow did not appear to have a significant impact on the calculated melt temperature (Tm) of proteins detected. Overall, the use of an isobaric trigger channel is a robust approach for prioritizing DDA selection of low-abundance proteins or peptides of interest such as missense mutant-containing proteins and their interaction partners, which are of particular focus within mTPP experiments.
Experimental Section
Yeast Strains and Growth
All experiments were performed in Saccharomyces cerevisiae. The parental strain SMY73242 was obtained from the Mirkin Lab and used in the trigger experiments comparing technical replicates. For the biological replicate experiments, the wild-type strain used was BY4741 (Open Biosystems). The ssu72-2 temperature-sensitive mutant43 was from Euroscarf. The Pta1-FLAG strain was made via homologous recombination. The 3xFLAG tag DNA sequence was amplified from plasmids obtained from Funakoshi and Hochstrasser44 to insert the FLAG epitope tag into the genome at the 3′-end of the PTA1 gene in wild type (WT) (BY4741). Successful incorporation of the FLAG tag was confirmed via Western blot. For mTPP experiments, cells were grown as previously described.29
Sample Preparation
BY4741 and ssu72-2 samples for mTPP were prepared as described in Peck Justice et al.29 with an extended temperature range for the heat treatment. The lysate was treated at the following 10 temperatures: untreated, 25, 35, 46.2, 48.8, 51.2, 53.2, 55.2, 56.5, and 74.9 °C. A TMT 10plex kit (Thermo Scientific, Waltham, MA) was used to label each sample as shown in Figure 1. SMY732 lysate was treated at the following eight temperatures: 25, 35, 48.8, 51.2, 53.2, 55.2, 56.5, and 74.9 °C. A TMT 16plex kit (Thermo Scientific, Waltham, MA) was used to label peptide solutions derived from each temperature treatment, as shown in Figure S1. Note that some channels in the 16plex were used for other samples not described in this report. TMT labeling steps were performed according to manufacturer-provided instructions.
Figure 1.

Workflow overview for mTPP with isobaric trigger channel addition. (A) Equal amounts of protein from each lysate for every biological replicate sample were subjected to different temperature treatments to induce protein denaturation. The soluble fractions from each treatment as well as a Pta1-FLAG affinity purification sample were digested in solution with trypsin/Lys-C. The resulting peptides were labeled with isobaric mass tags (TMT 10plex) as shown and mixed prior to mass spectrometry (MS) analysis. Resulting tandem mass spectrometry (MS/MS) data were analyzed using Proteome Discoverer 2.4 to identify and quantify abundance levels of peptides for each temperature treatment and each biological replicate across genotypes. The dot plots of protein-abundance values for each protein detected in WT cells in technical replicates without (B) and with (C) the isobaric trigger channel (trigger) addition.
Affinity purification of native CPF via Pta1-FLAG was performed as described previously for Ssu72-FLAG purifications.37 The Pta1-FLAG affinity-purified sample was added at a ratio of 6.25 μg trigger to 50 μg of the lowest heat-treated sample (1:8 ratio) for the biological replicates. The untreated samples were removed from the multiplex from no trigger samples to accommodate for the isobaric trigger channel to be labeled with TMT126. Technical replicate samples were divided and multiplexed into two separate mixes. In one experiment, the set of combined labeled samples was analyzed with a ninth trigger channel (TMT126) at a ratio of 1 μg total isobaric trigger channel protein to 50 μg of the lowest heat-treated sample (1:50 ratio), which included the Pta1-FLAG affinity-purified material, while in the second experiment, the trigger was not added. Subsequent sample preparation steps were performed as described.28 The ratio of trigger channel protein to experimental samples (based on the lowest temperature sample concentration) was well below the 1:0.02–1:0.05 ratios reported to be successful for accurate quantitation for low cell/single-cell studies.45,46
LC-MS/MS Analysis
Following multiplex preparation, samples were subjected to high-pH reversed-phase fractionation.29 NanoLC-MS/MS analyses were performed on an Orbitrap Fusion Lumos mass spectrometer coupled to an EASY-nLC HPLC (Thermo Scientific, Waltham, MA). One-third of the fractions were loaded onto an Easy-Nano 25 cm column with 2 μm reversed-phase resin. The peptides were eluted using a 180 min gradient increasing from 95% buffer A (0.1% formic acid in water) and 5% buffer B (0.1% formic acid in acetonitrile) to 25% buffer B at a flow rate of 400 nL/min. The peptides were eluted using a 180 min gradient increasing from 95% buffer A (0.1% formic acid in water) and 5% buffer B (0.1% formic acid in acetonitrile) to 25% buffer B at a flow rate of 400 nL/min. MS data was acquired using data-dependent acquisition (DDA) using a top speed method following the first survey MS scan. During MS1, using a wide quadrupole isolation, survey scans were obtained with an Orbitrap resolution of 120k with vendor-defined parameters—m/z scan range, 375–1500; maximum injection time, 50; automatic gain control (AGC) target, 4 × 105; micro scans, 1; and RF lens (%), 30. During MS2, the following parameters were assigned to isolate and fragment the selected precursor ions: isolation window = 0.7; FirstMass = 120; activation type = higher-energy collisional dissociation (HCD); and collision energy (%) = 38; the data were recorded using Thermo Scientific Xcalibur (4.1.31.9) software.
Protein Identification and Quantification
The resulting RAW files were analyzed using Proteome Discoverer 2.4 (Thermo Scientific, Waltham, MA). The SEQUEST HT search engine was used to search against a yeast protein database from the UniProt sequence database containing 6279 yeast protein and common contaminant sequences (FASTA file used available on ProteomeXchange under accession PXD020689). Specific search parameters used were trypsin as the proteolytic enzyme, peptides with a max of two missed cleavages, precursor mass tolerance of 10 ppm, and a fragment mass tolerance of 0.02 Da. Static modifications were (1) carbamidomethylation on cysteine; (2) TMTsixplex label on lysine (K) and the N-termini of peptides. Dynamic modifications were the oxidation of methionine and acetylation of N-termini. Percolator false discovery rate (FDR) was set to a strict setting of 0.01. Values from both unique and razor peptides were used for quantification. The mass spectrometry proteomic data have been deposited to the ProteomeXchange Consortium via the PRIDE47 partner repository with the data set identifier PXD020689 and doi: 10.6019/PXD020689. The impurity adjustments supplied within the TMT kit from Thermo Scientific were also accounted for in the analysis to limit the impact of TMT channel crosstalk.
Data Analysis
Venn diagrams were created using Venny 2.1.48 Dot plots, scatter plots, and waterfall plots were created using ggplot249 in R Studio (R Studio for Mac, version 1.2.5001). Bar graphs were created in Excel (version 16.38). The TPP package (v3.12.0)50 in R Studio was used to generate normalized melt curves and to determine protein melt temperatures as described previously.27 Resulting data processing and analysis also occurred in R Studio. Changes in melt temperature (Tm), ΔTm values, were calculated by taking WT Tm -ssu72-2Tm, thereby limiting calculations to proteins detected in both WT and mutant. Further parsing was accomplished by limiting our data to melt curves with r2 values >0.9 and then by proteins that were detected in at least two of the three replicates. Changes in Tm that were outside of ±2σ (σ being the standard deviation) were considered statistically significant and identified as proteins destabilized or stabilized due to the mutations in SSU72. Gene ontology (GO) enrichment analysis was performed using the publicly available Gene Ontology Resource.51,52
Results and Discussion
Addition of an Affinity-Purified Isobaric Trigger Channel to mTPP Multiplexes Does Not Cause Large Changes in Peptide Coverage or Quantitation
We hypothesized that incorporation of a well-characterized affinity-purified sample isolated from our system of interest as an isobaric trigger channel would increase MS1 ion intensity of peptides of interest within the TMT multiplex. As a consequence, the identification of peptides from the affinity-purified native protein complex would boost the identification in the remaining experimental mTPP channels used for melt curve production and subsequent Tm calculation when comparing different experimental samples. The incorporation of an affinity-purified CPF complex purified from our system of interest has numerous potential advantages, similar to the approach used in BASIL,18 including native levels of CPF processing events, post-translational modifications, and protein interaction partners. Affinity purifications for the CPF complex were performed in a manner similar to mTPP using nondenaturing buffers to preserve PPIs. Qualitatively, the MS/MS fragment data for CPF complexes would also be improved from the inclusion of the isobaric trigger channel increasing the ion abundance of the fragments and therefore the probability of CPF identification at the peptide spectrum match (PSM) level. From a quantitative perspective, TMT126 information is obtained during data processing but is excluded for interpretation of the mTPP melt curves for each protein.
Pta1-3xFLAG affinity purifications were digested with Lys-C/trypsin and labeled with TMT126 for inclusion within the mTPP multiplex. mTPP quantitative analysis and curve generation was performed using the remaining channels as described in the methods (Figure 1A). The mTPP samples were subjected to eight or nine different temperatures and then centrifuged to separate soluble and insoluble material as previously described.29 For samples with eight temperature points no 46.2° treatment sample was included. Samples were then processed and subjected to LC-MS/MS analysis using an MS2-based fragmentation and TMT quantitation workflow (Figure 1). Between 1750 and 3150, proteins were detected and quantified depending on the replicate (Table S1) when using SEQUEST HT and Proteome Discoverer 2.4 for qualitative and quantitative analysis. Replicates are designated as preparation 1, 2, 3 (hence p1, p2, p3). The p1 replicate had fewer IDs overall but p2 and p3 had very similar peptide detection levels (Table S1). Dot plots were generated to show the abundance value for each quantified protein and to gain insights into general trends with the quantitative data (Figures 1B,C and S2). Consistent with previous mTPP experiments,29 there was an overall decrease in protein abundance as the temperature at which the sample was treated increased. Importantly, incorporation of a protein complex isobaric trigger channel into the multiplex did not alter the overall trend of decreasing protein abundance with increased temperature or have a significant effect on the number of proteins detected. The average ion abundance at each temperature treatment also remained consistent between samples with and without the isobaric trigger channel suggesting that there were no significant levels of TMT channel crosstalk from the inclusion of the CPF trigger (compare abundance distributions between Figure 1B,C). The average quantitative ratio of the isobaric trigger channel to the mTPP experimental sample processed at 25 °C remains consistent at a 1:50 (Figure 1C) or 1:8 (Figure S2), reflecting the ratios used for mixing of the multiplex.
The impact of the trigger on mTPP analysis was investigated using both technical replicates and biological replicates so that we could evaluate differences in our workflow and their impact on qualitative and quantitative parameters. Technical replicate analyses showed very similar numbers of detected PSMs, peptides, and proteins suggesting that the addition of the trigger channel at a ratio of 1:50 has little impact on overall LC-MS/MS detection (Figure 2A, yellow). While there was not an obvious effect on the overall abundance of proteins in the samples, it is possible that the trigger could affect the detection and identification of proteins by biasing the mass spectrometer toward proteins present in the affinity purification. Comparisons of MS-based measurements across the technical replicates showed that the trigger channel incorporation did not have a significant impact on protein identification and quantification (Figure 2A). The biological replicates showed more variation across samples which is attributed to their separate processing for TPP in addition to variation that could occur from trypsin digestion and other processing steps.53,54 Trigger p1 in the biological replicate study did have overall lower levels of proteins detected, but this was not likely a consequence of trigger channel addition considering that trigger p2 and trigger p3 samples had similar detection levels to the no trigger sample (Figure 2A, green). Direct comparison of proteins quantified in the no trigger vs trigger samples showed an 80% overlap in quantified proteins with unique proteins present in all individual data sets (Figure 2B,C). Overall, these data suggest that the addition of an isobaric trigger channel has little to no impact on overall proteome detection outside of the inherent variability seen in independent sample processing (for the biological replicates) and LC-MS/MS runs.
Figure 2.
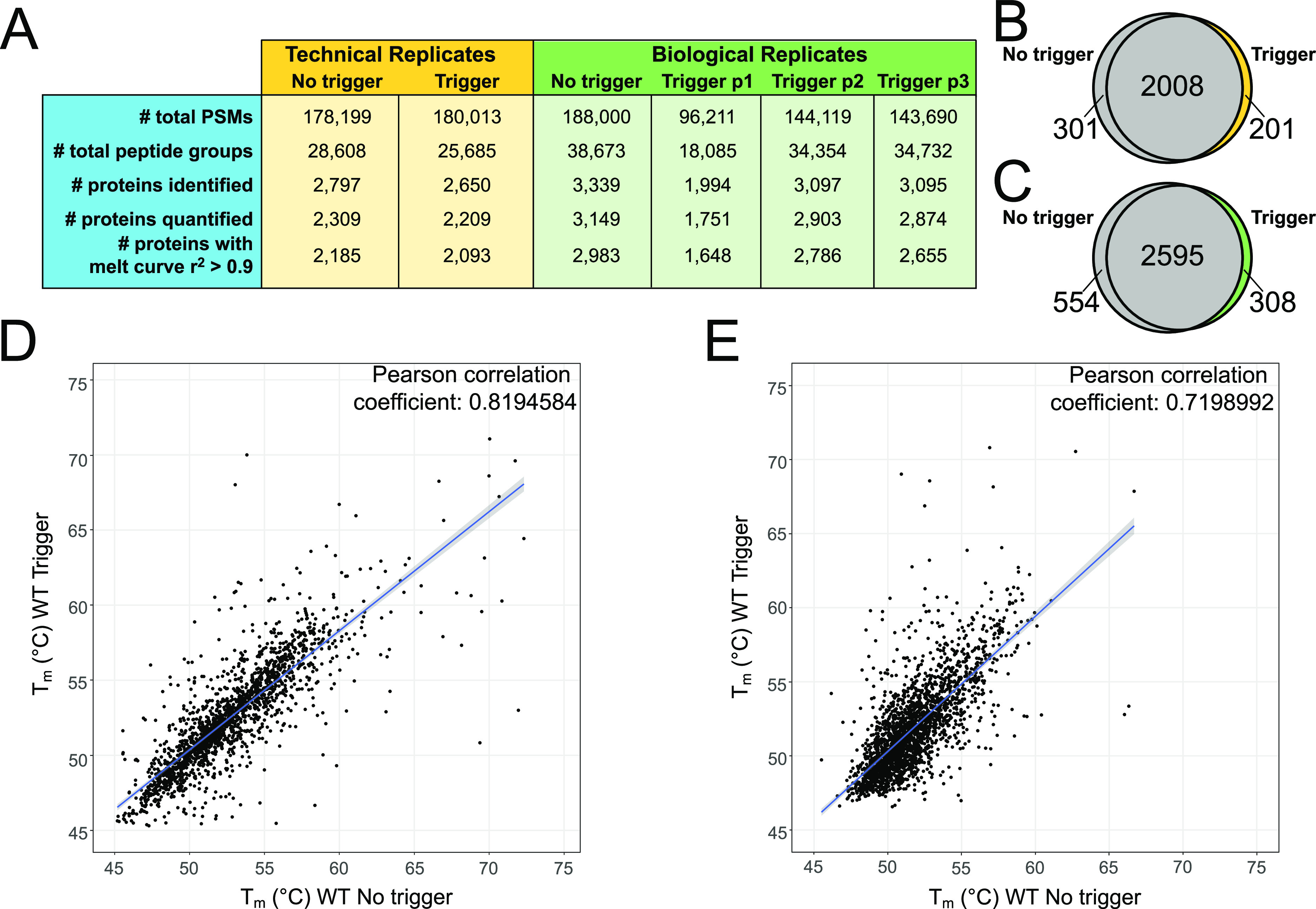
Data set comparisons from isobaric trigger channel addition. (A) Summary of LC-MS/MS data in technical and biological replicates with and without isobaric trigger channel addition. Venn diagrams comparing quantified proteins in no trigger (gray) vs trigger (yellow/green) in (B) technical replicates and (C) biological replicate using trigger p2. Correlation plot of the calculated Tms in no trigger vs trigger in (D) technical replicates and (E) biological replicates. The blue line represents the linear fit of the data.
A critical feature of mTPP analysis is the ability to accurately calculate melt temperature (Tm) from the resulting melt curves. To ensure that incorporation of the trigger did not have major impacts on Tm calculation of proteins outside of the CPF complex, we performed Pearson correlation analysis of the protein melt temperatures detected in both the no trigger and trigger samples (Figure 2D, Tm data from the TPP package in Table S2). From these, we can see a high degree of correlation of 0.82 between the no trigger and trigger samples for proteins, which met the criteria for quantitation in our mTPP data analysis workflow (including the number of proteins with melt curves having an r2 greater than or equal to 0.9). Additionally, even across biological replicates, there is a strong positive correlation of 0.72 between Tm calculations in the no trigger vs trigger samples (Figure 2E, Tm data from the TPP package in Table S2). The ability to make comparisons using biological replicate data would be beneficial in scenarios with limiting samples where technical replicates may not be feasible. Biological replicates are also important for rigorous statistical analysis.
Isobaric Trigger Channel Facilitates mTPP Analysis of the Cleavage and Polyadenylation Factor Complex
CPF and its accessory factors cleavage factor IA and IB play major roles in RNA processing. CPF is responsible for efficient and specific cleavage and polyadenylation of messenger RNAs55,56 and has been shown to have important roles in termination of RNA Polymerase II transcription.57,58 The CPF complex is currently described as having 14 subunits (Figure 3A) which provide the complex with numerous activities including endonuclease, polyadenylation, and phosphatase functions.59 Ssu72, which is mutated in the ssu72-2 yeast strain, is an integral subunit of CPF (Figure 3A, indicated with a star). Performing mTPP according to the established protocol29 resulted in limited detection of the CPF (Figure 3C–F). One notable exception to the low detection of CPF when no trigger was used was the subunit Glc7. Along with its presence in CPF, Glc7 is also the catalytic subunit of PP160 and thereby functions in many other protein complexes in eukaryotic cells (reviewed in refs (61) and (62)), where it plays roles in cell cycle regulation and nutrient regulation.60,63 Glc7 has a higher global abundance than other CPF subunits and is thereby more readily detected.
Figure 3.
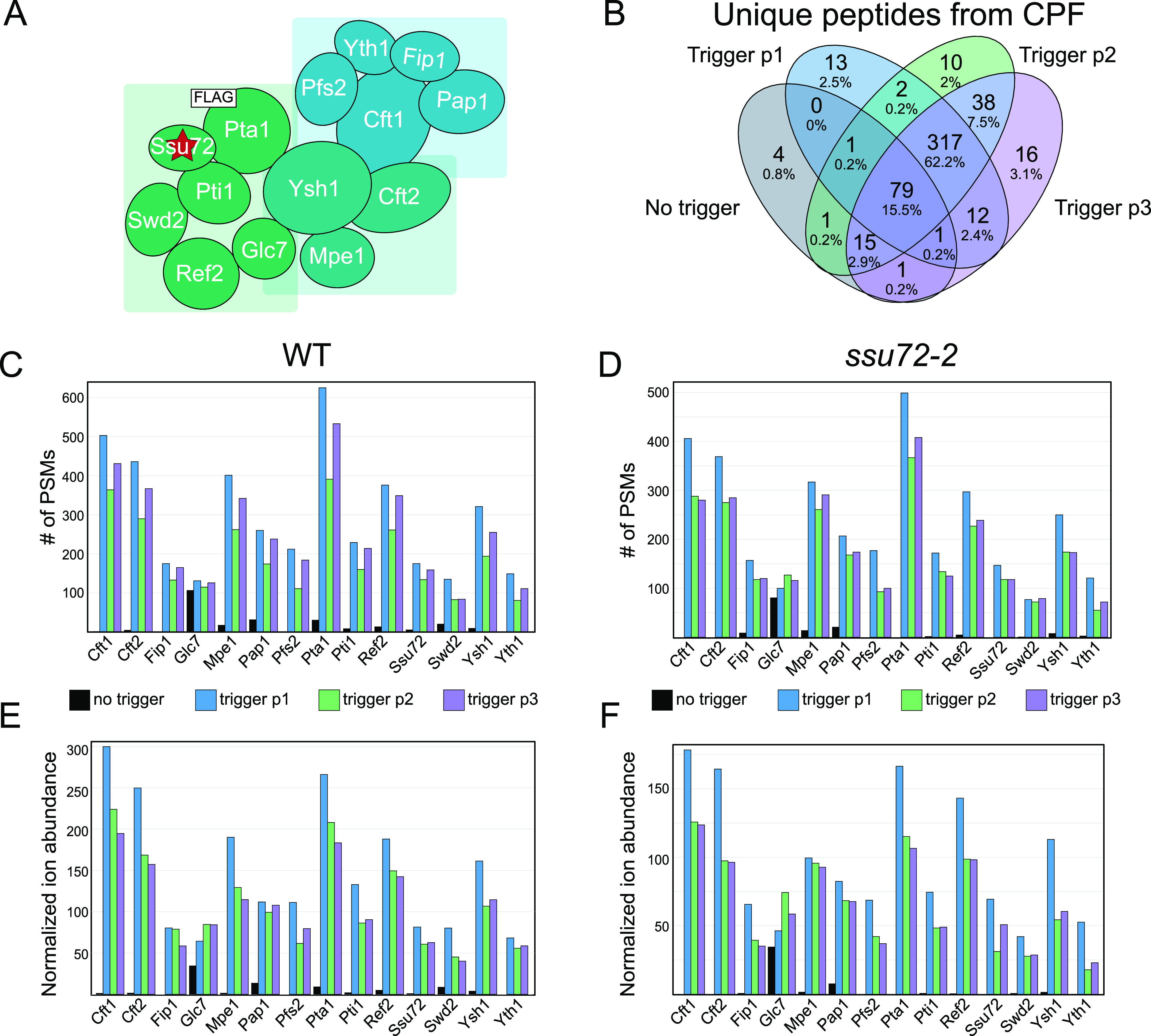
Peptide detection and quantitation for subunits of the cleavage and polyadenylation factor complex present in the Pta1-FLAG isobaric trigger channel. (A) Model of CPF adapted from Casañal et al.59 The red star denotes the mutant protein used in these studies, ssu72-2; the white square denotes the FLAG-tagged subunit used for the trigger channel affinity purification, Pta1. (B) Venn diagram showing the unique peptides detected for CPF subunits across each WT biological replicate. Number of PSMs for CPF subunits in each (C) WT and (D) ssu72-2 replicate experiment. Ion abundance for CPF subunits normalized to the abundance of Pgk1 (1000×) in each (E) WT and (F) ssu72-2 replicate experiment.
We have previously shown that PSM level detection of affinity-purified protein complexes results in highly reproducible quantitation of protein complexes in label-free quantitation workflows.40,41 This prior work found that RNA polymerase II complex digestions result in the generation of a number of highly detectable peptides and it is likely that this would also be the case for CPF affinity purifications.41 If these findings hold true, there should be a significant overlap in unique peptide identifications across the independent LC-MS/MS runs for biological replicates. As shown in Figure 3B, a significant overlap of unique peptides from CPF subunits was identified across the three biological replicates containing the isobaric CPF trigger (peptide data provided in Table S4). These findings clearly show that the use of a high-purity affinity purification as an isobaric trigger channel can facilitate reproducible quantitation of over 300 unique peptides. Considering that DDA of eukaryotic proteomes can often lead to stochastic peptide selection from low-abundance proteins, these data show that the CPF trigger channel can increase analytical measurement precision through reproducible peptide selection for MS2. From an individual subunit perspective, incorporation of the isobaric Pta1-FLAG trigger channel significantly increased the identification of most CPF subunits substantially (Figure 3C–F). Some CPF subunits that were previously not detected in no trigger samples (such as Cft1, Cft2, and Pfs2) were represented by hundreds of PSMs by utilizing the isobaric CPF trigger channel (Figure 3C,D). The increased level of PSM detection was accompanied by increased normalized ion abundance (Figure 3E,F).
Of note, Pta1-FLAG purifications are isolated from yeast that have been engineered to express Pta1-FLAG at native levels resulting in the purification of CPF complexes which have biologically relevant stoichiometry, protein processing, and protein post-translational modification(s). The CPF trigger channel also facilitated mTPP analysis of CPF subunit cotranslational modifications such as N-terminal methionine cleavage and acetylation of the new N-terminus (serine 2 in the protein database, Figure 4). Post-translational modification events such as phosphorylation of Pti1 at serine 272 were also reproducibly measured in CPF trigger mTPP experiments (Figure S5). These data show that the use of native protein complex purifications as an isobaric trigger channel has the unique advantage of triggering tandem MS analysis of peptides representing various biologically relevant proteoforms. Additionally, manual inspection of the ratio of the trigger channel abundance (126) to the lowest temperature abundance within the mTPP channels revealed that the amount of CPF peptides needed for boosting to allow for reproducible quantitation ranged between <2 and 5-fold (Figures 4 and S5).
Figure 4.

CPF trigger channel allows for reproducible detection of protein processing and amino acid modifications. MS2 fragment ion spectrum for Pta1 N-terminus. TMT reporter ions are indicated with a star (left) with a close-up view of the CPF trigger channel signal relative to the mTPP experimental data shown to the right.
Mutations in ssu72-2 Do Not Impact the Thermal Stability of the CPF Protein Complex
The CPF complex contains two protein phosphatases, Glc7 and Ssu72. Ssu72 is an integral component of CPF and its function is required for proper termination and 3′-end processing of RNAs.64−67 Additionally, its interactions with TFIIB have shown to be critical for the formation of gene loops, which regulate gene expression by linking transcription termination and initiation factors.68−71 Much of the characterization of Ssu72 has been accomplished through studies using the ssu72-2 mutant yeast strain.43,64 The ssu72-2 TS mutant contains a single mutation, R129A, that confers temperature sensitivity at 37 °C. This mutation impairs the catalytic activity of Ssu72, leading to a decrease in transcription elongation efficiency and defects in gene looping.69,71 Whether or not the missense mutation in the ssu72-2 cells affects the thermal stability of the Ssu72/CPF complex has not been previously examined and is required to make a conclusion about if the ssu72-2 causes a protein-specific change in activity that causes the phenotype or if it causes a protein complex-specific change (such as instability or poor assembly) that could alter the activity or recruitment of other CPF subunits.
Detection of CPF with and without the trigger channel resulted in similar numbers of CPF subunit PSMs in ssu72-2 as in WT, which facilitates mTPP analysis of CPF complex thermal stability from a quantitative perspective (Figure 4C,D). Protein melt curve analysis using the TPP R package (Figure 5A, mTPP result data in Table S3) showed no obvious changes in any of the 14 CPF subunits in ssu72-2 relative to WT. We have defined statistically significant changes in protein thermal stability as any ΔTm, which fall at least two standard deviations above or below the average ΔTm across the three ssu72-2 replicates relative to WT. Whole proteome analysis of ΔTm using mTPP found statistically significant decreases in the thermal stability of 59 proteins and increases in the thermal stability of 69 proteins in ssu72-2 cells (Figure 5B and Table S5). GO term analysis72 of proteins that had a significant change in thermal stability in ssu72-2 showed a 2.40-fold enrichment in proteins involved in the nucleobase-containing compound biosynthetic process with a p-value of 4.14 × 10–5. These results suggest that the defects in transcription caused by the disrupted catalytic activity of Ssu72 in this mutant strain are not due to impacts on the stability of Ssu72 or stability or the assembly of the CPF complex. Secondary effects of ssu72-2 have been associated with changes in the Nrd1–Nab3–Sen1 complex activity that impacts a variety of processes including GTP production.67,73,74 The temperature sensitivity of this strain is more likely to be a result of a need for efficient transcription at higher temperatures to respond to heat stress.75,76 A deeper investigation into the proteins with changes in thermal stability will help to further elucidate the impacts of this catalytic mutant on gene expression.
Figure 5.

Effects of ssu72-2 on CPF complex stability and the global proteome. (A) mTPP is normalized CPF subunit melt curves. Plots for each of the CPF subunits are normalized by the TPP package for a representative replicate, trigger p2. The curves shown in gray are WT and turquoise are ssu72-2. Each line represents one of the 14 CPF subunits. Replicates for (A) are provided in Figure S5. (B) Waterfall plots visualizing whole proteome changes in melt temperature (Tm), WT- ssu72-2. Median values are shown for proteins that are quantified in at least two replicates. The dotted lines signify a confidence interval of 95%. There are significant decreases in the thermal stability of 59 proteins and significant increases in 69 proteins. Change in Tm and median values are provided in Table S5.
Conclusions
The integration of an isobaric affinity-purified protein complex trigger channel increased our ability to analyze the thermal stability of the low-abundance protein complex CPF via mTPP. Our analysis did not observe major changes on the Tm estimates of unrelated proteins, suggesting that an affinity-purified isobaric trigger channel is a robust analytical approach for measurement of low-abundance protein mTPP analysis. CPF protein complex digestion results in the detection of a highly reproducible peptide population which will support precise measurement of low-abundance protein melt curves for proteins of interest while still obtaining survey information about the proteome at large. The use of natively expressed purifications from the system of interest, however, has distinct advantages including native protein processing, post-translational modifications, and protein interaction partners.
The use of isobaric purified protein complex trigger channels in TPP studies, and potentially other global proteomics applications, will improve the ability to perform proteomic analysis of low-abundance protein complexes with analytical reproducibility and precision to measure systems-level perturbations due to genetic variation(s). The potential for this method to be used across different organisms, even those that are difficult to get large amounts of protein from, is further supported by the adaptation of BASIL for single-cell phosphoproteomics.22 As many biologically relevant, as well as disease relevant, protein complexes are of relatively low abundance in the cell,77 improvements in the reproducible detection of such proteins in proteomics experiments would be beneficial to increasing our understanding of the critical cellular mechanisms in normal and disease states (Supporting Information).
Acknowledgments
We would like to thank all of the members of the Mosley Lab and IUSM Proteomics Core for their helpful discussions and support for this project. Funding was provided by NIH R01 GM099714 (A.L.M.), NIH T32 HL007910 (S.A.P.J.), and the Showalter Research Trust (A.L.M.). N.A.M. was supported in part by the IU Diabetes and Obesity Research Training Program, DeVault Fellowship. This project was also supported, in part, with support from the Indiana Clinical and Translational Sciences Institute, which is funded by Award number UL1TR002529 from the NIH, NCATS Award. Acquisition of the IUSM Proteomic core instrumentation used for this project was provided by the Indiana University Precision Health Initiative. Some of the TMT reagents were graciously provided via the Thermo Scientific TMT Research Award (S.A.P.J.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.1c00012.
TMT channels used for each sample (Figure S1); WT protein-abundance dot plots (Figure S2); ssu72-2 protein-abundance dot plots (Figure S3); unique CPF peptide detection (Figure S4); Pti1 phosphorylation detection with CPF trigger channel (Figure S5); PSMs and ion-abundance measurements for CPF subunits in WT technical replicates (Figure S6); and Replicate CPF melt curves (Figure S7) (PDF)
mTPP data (Table S1) (XLSX)
TPP package results, WT no trigger vs WT trigger (Table S2) (XLSX)
TPP package results, WT vs ssu72-2 (Table S3) (XLSX)
Peptide groups for CPF subunits in WT and ssu72-2 (Table S4) (XLSX)
Changes in Tm and median changes (Table S5) (XLSX)
Author Present Address
† Department of Biology, Taylor University, Upland, Indiana 46989, United States.
Author Present Address
‡ Translational Genomics Research Institute, Phoenix, Arizona 85005, United States.
Author Contributions
S.A.P.J. designed and performed mTPP experiments, analyzed data, prepared the figures, and wrote the manuscript. N.A.M. performed mTPP experiments and contributed to the manuscript. J.F.V. incorporated affinity-purified CPF and confirmed purification via AP-MS (data shown elsewhere). A.B.W. contributed to the design of experiments. A.L.M. oversaw various aspects of the project, designed mTPP experiments, provided funding, provided direction on data analysis, and wrote the manuscript. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Sahni N.; Yi S.; Taipale M.; Fuxman Bass J. I.; Coulombe-Huntington J.; Yang F.; Peng J.; Weile J.; Karras G. I.; Wang Y.; Kovacs I. A.; Kamburov A.; Krykbaeva I.; Lam M. H.; Tucker G.; Khurana V.; Sharma A.; Liu Y. Y.; Yachie N.; Zhong Q.; Shen Y.; Palagi A.; San-Miguel A.; Fan C.; Balcha D.; Dricot A.; Jordan D. M.; Walsh J. M.; Shah A. A.; Yang X.; Stoyanova A. K.; Leighton A.; Calderwood M. A.; Jacob Y.; Cusick M. E.; Salehi-Ashtiani K.; Whitesell L. J.; Sunyaev S.; Berger B.; Barabasi A. L.; Charloteaux B.; Hill D. E.; Hao T.; Roth F. P.; Xia Y.; Walhout A. J. M.; Lindquist S.; Vidal M. Widespread macromolecular interaction perturbations in human genetic disorders. Cell 2015, 161, 647–660. 10.1016/j.cell.2015.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragoza R.; Das J.; Wierbowski S. D.; Liang J.; Tran T. N.; Liang S.; Beltran J. F.; Rivera-Erick C. A.; Ye K.; Wang T. Y.; Yao L.; Mort M.; Stenson P. D.; Cooper D. N.; Wei X.; Keinan A.; Schimenti J. C.; Clark A. G.; Yu H. Extensive disruption of protein interactions by genetic variants across the allele frequency spectrum in human populations. Nat. Commun. 2019, 10, 4141 10.1038/s41467-019-11959-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin E. L.; Bruckner R. J.; Paulo J. A.; Cannon J. R.; Ting L.; Baltier K.; Colby G.; Gebreab F.; Gygi M. P.; Parzen H.; Szpyt J.; Tam S.; Zarraga G.; Pontano-Vaites L.; Swarup S.; White A. E.; Schweppe D. K.; Rad R.; Erickson B. K.; Obar R. A.; Guruharsha K. G.; Li K.; Artavanis-Tsakonas S.; Gygi S. P.; Harper J. W. Architecture of the human interactome defines protein communities and disease networks. Nature 2017, 545, 505–509. 10.1038/nature22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chick J. M.; Munger S. C.; Simecek P.; Huttlin E. L.; Choi K.; Gatti D. M.; Raghupathy N.; Svenson K. L.; Churchill G. A.; Gygi S. P. Defining the consequences of genetic variation on a proteome-wide scale. Nature 2016, 534, 500–505. 10.1038/nature18270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin A. C.; Bosche M.; Krause R.; Grandi P.; Marzioch M.; Bauer A.; Schultz J.; Rick J. M.; Michon A. M.; Cruciat C. M.; Remor M.; Hofert C.; Schelder M.; Brajenovic M.; Ruffner H.; Merino A.; Klein K.; Hudak M.; Dickson D.; Rudi T.; Gnau V.; Bauch A.; Bastuck S.; Huhse B.; Leutwein C.; Heurtier M. A.; Copley R. R.; Edelmann A.; Querfurth E.; Rybin V.; Drewes G.; Raida M.; Bouwmeester T.; Bork P.; Seraphin B.; Kuster B.; Neubauer G.; Superti-Furga G. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 2002, 415, 141–147. 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- Lambert J. P.; Ivosev G.; Couzens A. L.; Larsen B.; Taipale M.; Lin Z. Y.; Zhong Q.; Lindquist S.; Vidal M.; Aebersold R.; Pawson T.; Bonner R.; Tate S.; Gingras A. C. Mapping differential interactomes by affinity purification coupled with data-independent mass spectrometry acquisition. Nat. Methods 2013, 10, 1239–1245. 10.1038/nmeth.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go C. D.; Knight J. D. R.; Rajasekharan A.; Rathod B.; Hesketh G. G.; Abe K. T.; Youn J.-Y.; Samavarchi-Tehrani P.; Zhang H.; Zhu L. Y.; Popiel E.; Lambert J.-P.; Coyaud É.; Cheung S. W. T.; Rajendran D.; Wong C. J.; Antonicka H.; Pelletier L.; Raught B.; Palazzo A. F.; Shoubridge E. A.; Gingras A.-C. A proximity biotinylation map of a human cell. bioRxiv 2019, 1–31. 10.1101/796391. [DOI] [Google Scholar]
- Rolland T.; Tasan M.; Charloteaux B.; Pevzner S. J.; Zhong Q.; Sahni N.; Yi S.; Lemmens I.; Fontanillo C.; Mosca R.; Kamburov A.; Ghiassian S. D.; Yang X.; Ghamsari L.; Balcha D.; Begg B. E.; Braun P.; Brehme M.; Broly M. P.; Carvunis A. R.; Convery-Zupan D.; Corominas R.; Coulombe-Huntington J.; Dann E.; Dreze M.; Dricot A.; Fan C.; Franzosa E.; Gebreab F.; Gutierrez B. J.; Hardy M. F.; Jin M.; Kang S.; Kiros R.; Lin G. N.; Luck K.; MacWilliams A.; Menche J.; Murray R. R.; Palagi A.; Poulin M. M.; Rambout X.; Rasla J.; Reichert P.; Romero V.; Ruyssinck E.; Sahalie J. M.; Scholz A.; Shah A. A.; Sharma A.; Shen Y.; Spirohn K.; Tam S.; Tejeda A. O.; Trigg S. A.; Twizere J. C.; Vega K.; Walsh J.; Cusick M. E.; Xia Y.; Barabasi A. L.; Iakoucheva L. M.; Aloy P.; De Las Rivas J.; Tavernier J.; Calderwood M. A.; Hill D. E.; Hao T.; Roth F. P.; Vidal M. A proteome-scale map of the human interactome network. Cell 2014, 159, 1212–1226. 10.1016/j.cell.2014.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aebersold R.; Mann M. Mass-spectrometric exploration of proteome structure and function. Nature 2016, 537, 347–355. 10.1038/nature19949. [DOI] [PubMed] [Google Scholar]
- Altelaar A. F.; Munoz J.; Heck A. J. Next-generation proteomics: towards an integrative view of proteome dynamics. Nat. Rev. Genet. 2013, 14, 35–48. 10.1038/nrg3356. [DOI] [PubMed] [Google Scholar]
- Meier F.; Geyer P. E.; Virreira Winter S.; Cox J.; Mann M. BoxCar acquisition method enables single-shot proteomics at a depth of 10,000 proteins in 100 minutes. Nat. Methods 2018, 15, 440–448. 10.1038/s41592-018-0003-5. [DOI] [PubMed] [Google Scholar]
- Potel C. M.; Lin M.-H.; Heck A. J. R.; Lemeer S. Defeating Major Contaminants in Fe3+- Immobilized Metal Ion Affinity Chromatography (IMAC) Phosphopeptide Enrichment. Mol. Cell. Proteomics 2018, 17, 1028–1034. 10.1074/mcp.TIR117.000518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey S. J.; Azimifar S. B.; Mann M. High-throughput phosphoproteomics reveals in vivo insulin signaling dynamics. Nat. Biotechnol. 2015, 33, 990–995. 10.1038/nbt.3327. [DOI] [PubMed] [Google Scholar]
- Specht H.; Slavov N. Optimizing Accuracy and Depth of Protein Quantification in Experiments Using Isobaric Carriers. J. Proteome Res. 2020, 880–887. 10.1021/acs.jproteome.0c00675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavov N. Single-cell protein analysis by mass spectrometry. Curr. Opin. Chem. Biol. 2021, 60, 1–9. 10.1016/j.cbpa.2020.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y.; Scheibinger M.; Ellwanger D. C.; Krey J. F.; Choi D.; Kelly R. T.; Heller S.; Barr-Gillespie P. G. Single-cell proteomics reveals changes in expression during hair-cell development. Elife 2019, e50777 10.7554/eLife.50777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budnik B.; Levy E.; Harmange G.; Slavov N. SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol. 2018, 19, 161 10.1186/s13059-018-1547-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi L.; Tsai C. F.; Dirice E.; Swensen A. C.; Chen J.; Shi T.; Gritsenko M. A.; Chu R. K.; Piehowski P. D.; Smith R. D.; Rodland K. D.; Atkinson M. A.; Mathews C. E.; Kulkarni R. N.; Liu T.; Qian W. J. Boosting to Amplify Signal with Isobaric Labeling (BASIL) Strategy for Comprehensive Quantitative Phosphoproteomic Characterization of Small Populations of Cells. Anal. Chem. 2019, 91, 5794–5801. 10.1021/acs.analchem.9b00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister G. C.; Huttlin E. L.; Haas W.; Ting L.; Jedrychowski M. P.; Rogers J. C.; Kuhn K.; Pike I.; Grothe R. A.; Blethrow J. D.; Gygi S. P. Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal. Chem. 2012, 84, 7469–7478. 10.1021/ac301572t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A.; Schafer J.; Kuhn K.; Kienle S.; Schwarz J.; Schmidt G.; Neumann T.; Hamon C. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- Thompson A.; Wolmer N.; Koncarevic S.; Selzer S.; Bohm G.; Legner H.; Schmid P.; Kienle S.; Penning P.; Hohle C.; Berfelde A.; Martinez-Pinna R.; Farztdinov V.; Jung S.; Kuhn K.; Pike I. TMTpro: Design, Synthesis, and Initial Evaluation of a Proline-Based Isobaric 16-Plex Tandem Mass Tag Reagent Set. Anal. Chem. 2019, 91, 15941–15950. 10.1021/acs.analchem.9b04474. [DOI] [PubMed] [Google Scholar]
- Tsai C. F.; Zhao R.; Williams S. M.; Moore R. J.; Schultz K.; Chrisler W. B.; Pasa-Tolic L.; Rodland K. D.; Smith R. D.; Shi T.; Zhu Y.; Liu T. An Improved Boosting to Amplify Signal with Isobaric Labeling (iBASIL) Strategy for Precise Quantitative Single-cell Proteomics. Mol. Cell. Proteomics 2020, 19, 828–838. 10.1074/mcp.RA119.001857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua X. Y.; Mensah T.; Aballo T. J.; Mackintosh S. G.; Edmondson R. D.; Salomon A. R. Tandem Mass Tag approach utilizing pervanadate BOOST channels delivers deeper quantitative characterization of the tyrosine phosphoproteome. Mol. Cell. Proteomics 2020, 730–743. 10.1074/mcp.TIR119.001865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klann K.; Tascher G.; Munch C. Functional Translatome Proteomics Reveal Converging and Dose-Dependent Regulation by mTORC1 and eIF2alpha. Mol. Cell 2020, 77, 913.e4–925.e4. 10.1016/j.molcel.2019.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto W. R.; Bone R. N.; Sohn P.; Syed F.; Reissaus C. A.; Mosley A. L.; Wijeratne A. B.; True J. D.; Tong X.; Kono T.; Evans-Molina C. Endoplasmic reticulum stress alters ryanodine receptor function in the murine pancreatic beta cell. J. Biol. Chem. 2019, 294, 168–181. 10.1074/jbc.RA118.005683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitski M. M.; Reinhard F. B.; Franken H.; Werner T.; Savitski M. F.; Eberhard D.; Martinez Molina D.; Jafari R.; Dovega R. B.; Klaeger S.; Kuster B.; Nordlund P.; Bantscheff M.; Drewes G. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 2014, 346, 1255784 10.1126/science.1255784. [DOI] [PubMed] [Google Scholar]
- Franken H.; Mathieson T.; Childs D.; Sweetman G. M.; Werner T.; Togel I.; Doce C.; Gade S.; Bantscheff M.; Drewes G.; Reinhard F. B.; Huber W.; Savitski M. M. Thermal proteome profiling for unbiased identification of direct and indirect drug targets using multiplexed quantitative mass spectrometry. Nat. Protoc. 2015, 10, 1567–1593. 10.1038/nprot.2015.101. [DOI] [PubMed] [Google Scholar]
- Mateus A.; Kurzawa N.; Becher I.; Sridharan S.; Helm D.; Stein F.; Typas A.; Savitski M. M. Thermal proteome profiling for interrogating protein interactions. Mol. Syst. Biol. 2020, 16, e9232 10.15252/msb.20199232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peck Justice S. A.; Barron M. P.; Qi G. D.; Wijeratne H. R. S.; Victorino J. F.; Simpson E. R.; Vilseck J. Z.; Wijeratne A. B.; Mosley A. L. Mutant thermal proteome profiling for characterization of missense protein variants and their associated phenotypes within the proteome. J. Biol. Chem. 2020, 16219–16238. 10.1074/jbc.RA120.014576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraj N.; Wisniewski J. R.; Geiger T.; Cox J.; Kircher M.; Kelso J.; Paabo S.; Mann M. Deep proteome and transcriptome mapping of a human cancer cell line. Mol. Syst. Biol. 2011, 7, 548. 10.1038/msb.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batth T. S.; Francavilla C.; Olsen J. V. Off-line high-pH reversed-phase fractionation for in-depth phosphoproteomics. J. Proteome Res. 2014, 13, 6176–6186. 10.1021/pr500893m. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Yang F.; Gritsenko M. A.; Wang Y.; Clauss T.; Liu T.; Shen Y.; Monroe M. E.; Lopez-Ferrer D.; Reno T.; Moore R. J.; Klemke R. L.; Camp D. G. 2nd; Smith R. D. Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics 2011, 11, 2019–2026. 10.1002/pmic.201000722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertins P.; Tang L. C.; Krug K.; Clark D. J.; Gritsenko M. A.; Chen L.; Clauser K. R.; Clauss T. R.; Shah P.; Gillette M. A.; Petyuk V. A.; Thomas S. N.; Mani D. R.; Mundt F.; Moore R. J.; Hu Y.; Zhao R.; Schnaubelt M.; Keshishian H.; Monroe M. E.; Zhang Z.; Udeshi N. D.; Mani D.; Davies S. R.; Townsend R. R.; Chan D. W.; Smith R. D.; Zhang H.; Liu T.; Carr S. A. Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography-mass spectrometry. Nat. Protoc. 2018, 13, 1632–1661. 10.1038/s41596-018-0006-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogrebe A.; von Stechow L.; Bekker-Jensen D. B.; Weinert B. T.; Kelstrup C. D.; Olsen J. V. Benchmarking common quantification strategies for large-scale phosphoproteomics. Nat. Commun. 2018, 9, 1045 10.1038/s41467-018-03309-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilar M.; Olivova P.; Daly A. E.; Gebler J. C. Orthogonality of separation in two-dimensional liquid chromatography. Anal. Chem. 2005, 77, 6426–6434. 10.1021/ac050923i. [DOI] [PubMed] [Google Scholar]
- Ludwig K. R.; Schroll M. M.; Hummon A. B. Comparison of In-Solution, FASP, and S-Trap Based Digestion Methods for Bottom-Up Proteomic Studies. J. Proteome Res. 2018, 17, 2480–2490. 10.1021/acs.jproteome.8b00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victorino J. F.; Fox M. J.; Smith-Kinnaman W. R.; Peck Justice S. A.; Burriss K. H.; Boyd A. K.; Zimmerly M. A.; Chan R. R.; Hunter G. O.; Liu Y.; Mosley A. L. RNA Polymerase II CTD phosphatase Rtr1 fine-tunes transcription termination. PLoS Genet. 2020, 16, e1008317 10.1371/journal.pgen.1008317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard L. G.; Dronamraju R.; Kerschner J. L.; Hunter G. O.; Axley E. D.; Boyd A. K.; Strahl B. D.; Mosley A. L. Quantitative Analysis of Dynamic Protein Interactions during Transcription Reveals a Role for Casein Kinase II in Polymerase-associated Factor (PAF) Complex Phosphorylation and Regulation of Histone H2B Monoubiquitylation. J. Biol. Chem. 2016, 291, 13410–13420. 10.1074/jbc.M116.727735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-Kinnaman W. R.; Berna M. J.; Hunter G. O.; True J. D.; Hsu P.; Cabello G. I.; Fox M. J.; Varani G.; Mosley A. L. The interactome of the atypical phosphatase Rtr1 in Saccharomyces cerevisiae. Mol. BioSyst. 2014, 10, 1730–1741. 10.1039/C4MB00109E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosley A. L.; Hunter G. O.; Sardiu M. E.; Smolle M.; Workman J. L.; Florens L.; Washburn M. P. Quantitative proteomics demonstrates that the RNA polymerase II subunits Rpb4 and Rpb7 dissociate during transcriptional elongation. Mol. Cell. Proteomics 2013, 12, 1530–1538. 10.1074/mcp.M112.024034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosley A. L.; Sardiu M. E.; Pattenden S. G.; Workman J. L.; Florens L.; Washburn M. P. Highly reproducible label free quantitative proteomic analysis of RNA polymerase complexes. Mol. Cell. Proteomics 2011, 10, M110 000687 10.1074/mcp.M110.000687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinty R. J.; Puleo F.; Aksenova A. Y.; Hisey J. A.; Shishkin A. A.; Pearson E. L.; Wang E. T.; Housman D. E.; Moore C.; Mirkin S. M. A Defective mRNA Cleavage and Polyadenylation Complex Facilitates Expansions of Transcribed (GAA)n Repeats Associated with Friedreich’s Ataxia. Cell Rep. 2017, 20, 2490–2500. 10.1016/j.celrep.2017.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappas D. L.; Hampsey M. Functional Interaction between Ssu72 and the Rpb2 Subunit of RNA Polymerase II in Saccharomyces cerevisiae. Mol. Cell. Biol. 2000, 20, 8343–8351. 10.1128/MCB.20.22.8343-8351.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funakoshi M.; Hochstrasser M. Small epitope-linker modules for PCR-based C-terminal tagging in Saccharomyces cerevisiae. Yeast 2009, 26, 185–192. 10.1002/yea.1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung T. K.; Lee C. Y.; Bayer F. P.; McCoy A.; Kuster B.; Rose C. M. Defining the carrier proteome limit for single-cell proteomics. Nat. Methods 2021, 18, 76–83. 10.1038/s41592-020-01002-5. [DOI] [PubMed] [Google Scholar]
- Dou M.; Clair G.; Tsai C. F.; Xu K.; Chrisler W. B.; Sontag R. L.; Zhao R.; Moore R. J.; Liu T.; Pasa-Tolic L.; Smith R. D.; Shi T.; Adkins J. N.; Qian W. J.; Kelly R. T.; Ansong C.; Zhu Y. High-Throughput Single Cell Proteomics Enabled by Multiplex Isobaric Labeling in a Nanodroplet Sample Preparation Platform. Anal. Chem. 2019, 91, 13119–13127. 10.1021/acs.analchem.9b03349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Riverol Y.; Csordas A.; Bai J.; Bernal-Llinares M.; Hewapathirana S.; Kundu D. J.; Inuganti A.; Griss J.; Mayer G.; Eisenacher M.; Pérez E.; Uszkoreit J.; Pfeuffer J.; Sachsenberg T.; Yılmaz Ş.; Tiwary S.; Cox J.; Audain E.; Walzer M.; Jarnuczak A. F.; Ternent T.; Brazma A.; Vizcaíno J. A. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveros J. C.Venny. An Interactive Tool for Comparing Lists with Venn’s Diagrams, 2007–2015.
- Wickham H.ggplot2: Elegant Graphics for Data Analysis; Springer-Verlag: New York, 2016. [Google Scholar]
- Childs D.; Kurzawa N.; Franken H.; Doce C.; Savitski M.; Huber W.. TPP: Analyze Thermal Proteome Profiling (TPP) Experiments, R package version 3.10.0, 2018.
- The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. 10.1093/nar/gkaa1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M.; Ball C. A.; Blake J. A.; Botstein D.; Butler H.; Cherry J. M.; Davis A. P.; Dolinski K.; Dwight S. S.; Eppig J. T.; Harris M. A.; Hill D. P.; Issel-Tarver L.; Kasarskis A.; Lewis S.; Matese J. C.; Richardson J. E.; Ringwald M.; Rubin G. M.; Sherlock G. Gene ontology: tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walmsley S. J.; Rudnick P. A.; Liang Y.; Dong Q.; Stein S. E.; Nesvizhskii A. I. Comprehensive analysis of protein digestion using six trypsins reveals the origin of trypsin as a significant source of variability in proteomics. J. Proteome Res. 2013, 12, 5666–5680. 10.1021/pr400611h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhart J. M.; Schumbrutzki C.; Wortelkamp S.; Sickmann A.; Zahedi R. P. Systematic and quantitative comparison of digest efficiency and specificity reveals the impact of trypsin quality on MS-based proteomics. J. Proteomics 2012, 75, 1454–1462. 10.1016/j.jprot.2011.11.016. [DOI] [PubMed] [Google Scholar]
- Chen J.; Moore C. Separation of factors required for cleavage and polyadenylation of yeast pre-mRNA. Mol. Cell. Biol. 1992, 12, 3470–3481. 10.1128/MCB.12.8.3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler M. M.; Zhao J.; Moore C. L. Purification of the Saccharomyces cerevisiae cleavage/polyadenylation factor I. Separation into two components that are required for both cleavage and polyadenylation of mRNA 3′ ends. J. Biol. Chem. 1996, 271, 27167–27175. 10.1074/jbc.271.43.27167. [DOI] [PubMed] [Google Scholar]
- Proudfoot N. J. Transcriptional termination in mammals: Stopping the RNA polymerase II juggernaut. Science 2016, 352, aad9926 10.1126/science.aad9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton J. D.; Davidson L.; Bauer D. L. V.; Natsume T.; Kanemaki M. T.; West S. Xrn2 accelerates termination by RNA polymerase II, which is underpinned by CPSF73 activity. Genes Dev. 2018, 32, 127–139. 10.1101/gad.308528.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casañal A.; Kumar A.; Hill C. H.; Easter A. D.; Emsley P.; Degliesposti G.; Gordiyenko Y.; Santhanam B.; Wolf J.; Wiederhold K.; Dornan G. L.; Skehel M.; Robinson C. V.; Passmore L. A. Architecture of eukaryotic mRNA 3′-end processing machinery. Science 2017, 358, 1056–1059. 10.1126/science.aao6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z. H.; Wilson S. E.; Peng Z. Y.; Schlender K. K.; Reimann E. M.; Trumbly R. J. The Yeast Glc7-Gene Required for Glycogen Accumulation Encodes a Type-1 Protein Phosphatase. J. Biol. Chem. 1991, 266, 23796–23801. 10.1016/S0021-9258(18)54353-2. [DOI] [PubMed] [Google Scholar]
- Martín R.; Stonyte V.; Lopez-Aviles S. Protein Phosphatases in G1 Regulation. Int. J. Mol. Sci. 2020, 21, 395 10.3390/ijms21020395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moura M.; Conde C. Phosphatases in Mitosis: Roles and Regulation. Biomolecules 2019, 9, 55 10.3390/biom9020055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy N. T.; Li L.; Khalil M.; Cannon J. F. Regulation of yeast glycogen metabolism and sporulation by Glc7p protein phosphatase. Genetics 1998, 149, 57–72. 10.1093/genetics/149.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dichtl B.; Blank D.; Ohnacker M.; Friedlein A.; Roeder D.; Langen H.; Keller W. A Role for SSU72 in Balancing RNA Polymerase II Transcription Elongation and Termination. Mol. Cell 2002, 10, 1139–1150. 10.1016/S1097-2765(02)00707-4. [DOI] [PubMed] [Google Scholar]
- Nedea E.; He X.; Kim M.; Pootoolal J.; Zhong G.; Canadien V.; Hughes T.; Buratowski S.; Moore C. L.; Greenblatt J. Organization and Function of APT, a Subcomplex of the Yeast Cleavage and Polyadenylation Factor Involved in the Formation of mRNA and Small Nucleolar RNA 3′-Ends. J. Biol. Chem. 2003, 278, 33000–33010. 10.1074/jbc.M304454200. [DOI] [PubMed] [Google Scholar]
- Steinmetz E. J.; Brow D. A. Ssu72 Protein Mediates Both Poly(A)-Coupled and Poly(A)-Independent Termination of RNA Polymerase II Transcription. Mol. Cell. Biol. 2003, 23, 6339–6349. 10.1128/MCB.23.18.6339-6349.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D. W.; Mosley A. L.; Ramisetty S. R.; Rodriguez-Molina J. B.; Washburn M. P.; Ansari A. Z. Ssu72 phosphatase-dependent erasure of phospho-Ser7 marks on the RNA polymerase II C-terminal domain is essential for viability and transcription termination. J. Biol. Chem. 2012, 287, 8541–8551. 10.1074/jbc.M111.335687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari A.; Hampsey M. A role for the CPF 3′-end processing machinery in RNAP II-dependent gene looping. Genes Dev. 2005, 19, 2969–2978. 10.1101/gad.1362305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allepuz-Fuster P.; O’Brien M. J.; Gonzalez-Polo N.; Pereira B.; Dhoondia Z.; Ansari A.; Calvo O. RNA polymerase II plays an active role in the formation of gene loops through the Rpb4 subunit. Nucleic Acids Res. 2019, 47, 8975–8987. 10.1093/nar/gkz597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh B. N.; Hampsey M. A transcription-independent role for TFIIB in gene looping. Mol. Cell 2007, 27, 806–816. 10.1016/j.molcel.2007.07.013. [DOI] [PubMed] [Google Scholar]
- Tan-Wong S. M.; Zaugg J. B.; Camblong J.; Xu Z.; Zhang D. W.; Mischo H. E.; Ansari A. Z.; Luscombe N. M.; Steinmetz L. M.; Proudfoot N. J. Gene loops enhance transcriptional directionality. Science 2012, 338, 671–675. 10.1126/science.1224350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H.; Huang X.; Muruganujan A.; Tang H.; Mills C.; Kang D.; Thomas P. D. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017, 45, D183–D189. 10.1093/nar/gkw1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem C.; Devaux F.; Torchet C.; Jacq C.; Quevillon-Cheruel S.; Labesse G.; Facca C.; Faye G. Ssu72 is a phosphatase essential for transcription termination of snoRNAs and specific mRNAs in yeast. EMBO J. 2003, 22, 1588–1598. 10.1093/emboj/cdg141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loya T. J.; O’Rourke T. W.; Reines D. A genetic screen for terminator function in yeast identifies a role for a new functional domain in termination factor Nab3. Nucleic Acids Res. 2012, 40, 7476–7491. 10.1093/nar/gks377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahat D. B.; Salamanca H. H.; Duarte F. M.; Danko C. G.; Lis J. T. Mammalian Heat Shock Response and Mechanisms Underlying Its Genome-wide Transcriptional Regulation. Mol. Cell 2016, 62, 63–78. 10.1016/j.molcel.2016.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte F. M.; Fuda N. J.; Mahat D. B.; Core L. J.; Guertin M. J.; Lis J. T. Transcription factors GAF and HSF act at distinct regulatory steps to modulate stress-induced gene activation. Genes Dev. 2016, 30, 1731–1746. 10.1101/gad.284430.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho B.; Baryshnikova A.; Brown G. W. Unification of Protein Abundance Datasets Yields a Quantitative Saccharomyces cerevisiae Proteome. Cell Syst. 2018, 6, 192.e3–205.e3. 10.1016/j.cels.2017.12.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.