

Abstract
As a representative two-dimensional (2D) nanomaterial, graphene oxide (GO) has shown high potential in many applications due to its large surface area, high flexibility, and excellent dispersibility in aqueous solutions. These properties make GO an ideal candidate for bio-imaging, drug delivery, and cancer therapy. When delivered to the body, GO has been shown to accumulate in the liver, the primary accumulation site of systemic delivery or secondary spread from other uptake sites, and induce liver toxicity. However, the contribution of the GO physicochemical properties and individual liver cell types to this toxicity is unclear due to property variations and diverse cell types in the liver. Herein, we compare the effects of GOs with small (GO-S) and large (GO-L) lateral sizes in three major cell types in liver, Kupffer cells (KCs), liver sinusoidal endothelial cells (LSECs), and hepatocytes. While GOs induced cytotoxicity in KCs, they induced significantly less toxicity in LSECs and hepatocytes. For KCs, we found that GOs were phagocytosed that triggered NADPH oxidase mediated plasma membrane lipid peroxidation, which leads to PLC activation, calcium flux, mitochondrial ROS generation, and NLRP3 inflammasome activation. The subsequent caspase-1 activation induced IL-1β production and GSDMD-mediated pyroptosis. These effects were lateral size-dependent with GO-L showing stronger effects than GO-S. Amongst the liver cell types, decreased cell association and the absence of lipid peroxidation resulted in low cytotoxicity in LSECs and hepatocytes. Using additional GO samples with different lateral sizes, surface functionalities, or thickness, we further confirmed the differential cytotoxic effects in liver cells and the major role of GO lateral size in KUP5 pyroptosis by correlation studies. These findings delineated the GO effects on cellular uptake and cell death pathways in liver cells, and provide valuable information to further evaluate GO effects on the liver for biomedical applications.
Keywords: graphene oxide, phagocytosis, lipid peroxidation, NADPH oxidase, Phospholipase C (PLC), GSDMD-dependent pyroptosis
Graphical Abstract
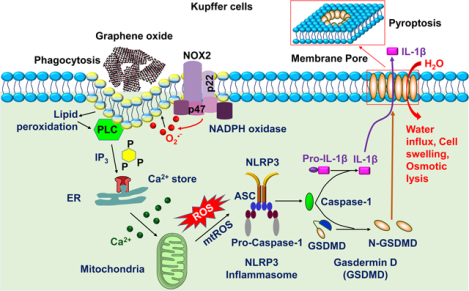
GOs induce lateral size-dependent toxicity to Kupffer cells but significantly less toxicity to LSECs and hepatocytes. GOs were taken up into Kupffer cells through phagocytosis, which triggered NADPH oxidase mediated plasma membrane lipid peroxidation, leading to PLC activation, calcium flux, mtROS generation, and NLRP3 inflammasome activation. Subsequent caspase-1 activation induced IL-1β production and GSDMD-mediated pyroptosis.
Introduction
Two-dimensional (2D) nanomaterials have revealed promising applications in energy, sensors, catalysis, biomedicine, and electronics [1–4]. Graphene oxide (GO, an oxidized graphene derivative) is a 2D material consisting of a single layer of carbon atoms arranged primarily in a regular hexagonal pattern and decorated with oxygen-containing functional groups [5,6]. As a representative 2D nanomaterial, GO exhibits large surface area, high flexibility [7], and excellent dispersibility in various solutions to render the material attractive for use in biomedicine [8], including tissue engineering [9,10], antimicrobial agents [11], bio-imaging [12], possible diagnosis and treatment to COVID-19 [13], drug delivery [14], and cancer therapy, particularly serving as a nanocarrier [15]. The liver is the primary target for nanocarriers after intravenous injection, acting as a biological filtration system that sequesters 30–99% of administered nanoparticles from the bloodstream [16]. The accumulation of GO in the liver has been shown to induce liver toxicity or profound changes at the transcriptional and epigenetic levels [17,18]. However, the effects of GO to the liver are under-researched due to the large physicochemical property variations across sample preparation and processing and the diverse cell types in the liver. Although there have been attempts to link physicochemical properties such as lateral size to the toxicity of GO [19,20], there are few studies on the mechanism of toxicity of GOs for liver cells.
To understand GO-induced liver toxicity, it is necessary to study how GO interacts with the major cell types in the liver. However, comparatively little information is available for the impact of GO on Kupffer cells (KCs), liver sinusoidal endothelial cells (LSECs), and hepatocytes. Kupffer cell, which constitutes 15% of liver cells or 80–90% of all the tissue macrophages in the body, is a major component of the mononuclear phagocyte system (MPS, a.k.a. the reticuloendothelial system or RES) [16,21,22]. KCs are responsible for phagocytosis of nanocarriers, endotoxin removal, and modulation of innate immune responses and also serves as the first line of defense for nanomaterials by phagocytic removal in the liver, which has a profound impact on the liver toxicity [16,21,23,24]. Although it has been shown that GOs could induce lipid peroxidation [25], oxidative stress, secretion of proinflammatory cytokines IL-1β and TNF-α [26], and cell death in macrophages [27], few studies have been performed on KCs. We have used an immortalized Kupffer cell line, KUP5, for studies on nanomaterial toxicity. Using a series of metal oxide nanoparticles (MOx) and rare earth oxides (REO), we found MOx induced differential cell death mechanisms, including apoptosis (ZnO, CuO, etc.) and a form of programmed necrosis, pyroptosis induced by REOs (Gd2O3, Eu2O3, etc.) [28,29]. The results were replicated in primary human Kupffer cells, suggesting KUP5 is a valid cell line for nanotoxicity studies. LSECs constitute approximately 3% of the total liver cell volume and are also an important part of the RES. The LSECs have high capacity clathrin-mediated endocytic activity and they play a central role in the clearance of blood-borne waste and innate immunity [30–33]. Although GOs have been shown to induce apoptosis in human umbilical vein endothelial cells (HUVECs) previously [34], no toxicity studies have been carried out on LSECs. Using an immortalized mouse hepatic sinusoidal endothelial cells-SV40 (LSECs) that are fully functional in response to an antigen-carrying PLGA nanoparticle in terms of antigen presentation and cytokine production, we can mimic the functions of LSECs in vivo [35]. Hepatocytes, which constitute as high as 60–80% of liver cells, perform important roles in metabolic, endocrine, and secretory functions [16,32]. Although studies on GO toxicity to hepatocytes are presented, the results are often conflicting. For example, GOs have been shown to induce cytotoxicity in human HepG2 hepatocytes and mouse Hepa 1–6 cells hepatocytes [19,26,36]. However, Qu et al. did not observe any impairment to cell growth and survival of HepG2 and Hepa 1–6 after GO exposure [27]. These seemingly conflicting results are likely due to the physicochemical property variations among different GO samples.
Herein, we study the effects of GOs on three major liver cell types, the KC (KUP5), LSEC, and hepatocyte (Hepa 1–6) cell. GOs were provided by the Nanomaterial Health Implications Research (NHIR) Consortium of the National Institute of Environmental Health Sciences (NIEHS), which were composed of two lateral sizes, GO-S (small) and GO-L (large). These GO samples had similar surface functional groups and compositions, which formed the basis for comparisons on the effect of lateral size. We determined the effects of GOs on cytotoxicity and found that GOs induced differential toxicity outcomes in the three liver cell types. We then explored the mechanisms that were responsible for the differential cytotoxic responses including cellular uptake, lipid peroxidation, NLRP3 inflammasome activation, and cell death. In addition, using three additional GO samples that differed from GO-S and GO-L in terms of lateral sizes, surface functionalities, or thickness, we showed that GO-induced effects were a universal feature to the liver cells tested in this study. We furtherly confirmed that lateral size played a key role in the GO-induced pyroptosis in KUP5 cells by evaluating the correlations between GO physicochemical properties and cellular response.
Material and methods
Materials
GO-S, GO-L, and GO-1 were provided by Engineered Nanomaterials Resource and Coordination Core, part of NIEHS Nanomaterials Health Implications Research (NHIR) Consortium. GO-2 and GO-3 synthesized from graphite (Asbury, 3061 Grade) using a modified Hummers’ method following the literature precedent [37]. The mouse Kupffer cell line, KUP5, was purchased from RIKEN Cell Bank (Japan). The immortalized mouse liver sinusoidal endothelial cells-SV40 (LSECs), Prigrow medium, and flasks for growing LSECs were purchased from Applied Biological Materials (Vancouver, BC, Canada). The mouse hepatocyte cell line, Hepa 1–6, was purchased from ATCC. Low-endotoxin fetal bovine serum (FBS) was purchased from Gemini Bio-Products (West Sacramento, CA, USA). The CellTiter 96 aqueous one solution cell proliferation assay (MTS) was purchased from Promega (Madison, WI, USA). Hoechst 33342, Fluorescein Isothiocyanate-labeled bovine serum albumin (FITC-BSA), Alexa Fluor 594-conjugated wheat germ agglutinin (WGA), and Image-iT lipid peroxidation kit were purchased from Life Technologies (Grand Island, NY, USA). The FAM-FLICA Caspase-1 and Caspase 3/7 Assay Kits were purchased from ImmunoChemistry Technologies, LLC (Bloomington, MN). The lipopolysaccharide (LPS), 1-Methyl-3-(3-(dimethylamino)propyl)carbodiimide (EDC), N-hydroxysuccinimide (NHS), wortmannin (WM), cytochalasin D (Cyto D), Pitstop 2, dibenziodolium chloride (DPI), U-73122, BAPTA-AM, CA-074-Me, necrostatin-1 (NEC-1), and p22phox, p47phox, and gasdermin D (GSDMD) siRNAs were purchased from Sigma (St. Louis, MO). EnzChek Direct Phospholipase C Assay Kit, MitoSOX indicator, and Fluo-4, AM were purchased from Invitrogen (Carlsbad, CA). CLI-095 was purchased from InvivoGen (San Diego, CA). The ELISA kits for mouse IL-1β and TNF-α were purchased from R&D Systems (Minneapolis, MN).
Physicochemical characterizations of GO samples
The primary sizes and morphologies of GO-S and GO-L were characterized by transmission electron microscopy (TEM, JEOL 1200 EX transmission electron microscope) without further purification or modification. The height and size distribution of GOs were assessed by atomic force microscopy (AFM). Briefly, The Si wafers functionalized with a monolayer of 2.5 mM (3-aminopropyl)-triethoxysilane (APTES) on the surface were rinsed with filtered DI water and dried under N2. The GOs were placed on the wafer and underwent a heat treatment at 250 °C for 0.5 h. AFM images were obtained with the same tip and scanning conditions by an Asylum Research Cypher ES AFM. All images were taken at random locations on the sample and showed little variation.
The stock solutions at a concentration of approximately 500 μg/mL in deionized water (DI water) were prepared and sonicated in a Branson 2510 Ultrasonic Cleaner sonicator (Branson Ultrasonics, Danbury, CT) for 15 min to characterize the GO in suspension. Then, these suspensions were diluted to a final concentration of 50 μg/mL in DI water or cell culture media, followed by further sonication for 15 min. The hydrodynamic diameters, polydispersity index (PDI), and zeta potential of GO suspensions were determined using a ZetaPALS instrument (Brookhaven Instrument, Holtsville, NY).
To investigate the chemical state and calculate the atomic concentration of functional groups on the GO surface, suspensions of GO were vacuum filtered onto alumina membranes with a pore size of 0.1 μm and dried at room temperature. X-ray photoelectron spectroscopy (XPS) analysis was performed using a Thermo Scientific ESCALAB 250Xi with a monochromatic Al Kα X-ray source at Northwestern University. The collected spectra were analyzed using a Smart background correction and peak fitted using XPSPEAK41 software.
Cell culture
KUP5 cells were cultured in high-glucose Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% FBS, 250 μM 1-thioglycerol, 10 μg/mL bovine insulin, and 100 U/mL/100 μg/mL of penicillin-streptomycin (Gibco, Waltham, MA). LSECs were cultured in the Prigrow medium supplemented with 5% FBS and 100 U/mL/100 μg/mL of penicillin-streptomycin. Hepa 1–6 cells were cultured in high-glucose DMEM medium supplemented with 10% FBS and 100 U/mL/100 μg/mL penicillin-streptomycin. All cells were cultured under a humidified environment of 95% O2, 5% CO2, and 37 °C.
Determination of GO effects on cell viability
Cytotoxicity assays were performed in KUP5, LSEC, and Hepa 1–6 cells using the MTS assay. Before exposure to GO samples, cells were seeded at a density of 3 × 104/well in 96-well plates (Corning, NY, USA). GO suspensions were freshly prepared in complete cell culture media with different concentrations of 0–100 μg/mL. Following the exposure of the cells to GO suspensions for 24 h, the cell culture media was removed, followed by the addition of one hundred microliters of culture media containing 16.7% MTS stock solution for 0.5 h in a humidified 5% CO2 incubator. The cell culture media were centrifuged at 2000 rpm for 10 min in an NI Eppendorf 5430 microcentrifuge to spin down the cell debris and GO. 80 μL of the supernatant was collected from each well and transferred into a new 96-well plate. The absorbance of formed formazan was read at 490 nm on a SpectraMax M5e microplate reader (Molecular Devices, Sunnyvale, CA). Non-treated control cells are considered to be 100% in cell viability and percent viability of treated cells is calculated against control cells.
Labeling of GO by FITC-BSA
FITC-BSA-labeled GO samples were prepared by a diimide-activated amidation reaction as described before [25]. Briefly, 5 mg EDC and 10 mg NHS were dissolved in 2 mL GO suspensions (100 μg/mL) in DI water, followed by stir for 2 h at room temperature. The GO pellets were collected by centrifugation at 20000 rpm for 0.5 h, followed by reaction with 1 mL of 0.1 mg/mL of the FITC-BSA solution while being stirred for 2 h. The FITC-labeled samples were centrifuged at 20000 rpm for 0.5 h, washed three times to remove free FITC-BSA, suspended in 400 μL DI water, and stored at 4 °C in a refrigerator for further use.
GO cellular uptake assessment through confocal microscopy, flow cytometry, and TEM
To assess GO cellular localization by confocal imaging, KUP5, LSEC, and Hepa 1–6 cell suspensions at densities of 1 × 105 /well were seeded into 8-well chambers (Nunc Lab-Tek II chambered coverglass, Sigma-Aldrich), respectively. The cells were exposed to 12.5 μg/mL FITC-BSA-GO suspensions for 16 h, followed by three washes in PBS. Cell membranes and nuclei were stained with 5 μg/mL WGA and 8 μM Hoechst 33342 for 15 min, respectively. The cells were visualized under a Leica Confocal SP8-SMD confocal microscope (Leica, Germany). High-magnification images were obtained under the 63× objective. The cell association of GO with KUP5, LSEC, and Hepa 1–6 cells were quantified by flow cytometry (FACS) analysis on Side Scattering (SSC) values of cells treated with non-FITC-labeled GO based on the established method [25].
TEM was used to evaluate the cellular uptake of GOs. After exposure to 50 μg/mL GO for 16 h, the cells were washed and fixed 2.5% glutaraldehyde and 3% paraformaldehyde in PBS buffer for TEM analysis. Following staining in 1% osmium tetroxide (OsO4) in PBS for 1 h at room temperature, the cells were dehydrated in a graded series of ethanol and then treated with propylene oxide before being embedded the pellets in fresh Epon resin in the mold. Approximately 50–70 nm thick sections were cut on a Reichert-Jung Ultracut E ultramicrotome and picked up on Formvar-coated copper grids. The sections were stained with uranyl acetate and Reynolds lead citrate and examined on a JEOL transmission electron microscope at 80 kV [25].
To determine the cellular uptake mechanism of GO, before GO exposure, the KUP5, LSEC, and Hepa 1–6 cells were treated with inhibitors including 1 μM wortmannin for 0.5 h, 5 μg/mL cytochalasin D for 1 h, and 20 μM Pitstop 2 for 0.5 h, respectively. The dose of each inhibitor was chosen based on the previous studies to make sure non-toxicity to the aforementioned cells [20,38,39]. FITC-GOs at 12.5 μg/mL dose were used to treat the cells for 16 hours and the localization of GO in cells was visualized by a confocal microscope. To quantify the FITC-GO association with cells in the presence of inhibitors [38], KUP5, LSECs, and Hepa 1–6 cells seeded at a density of 3 × 104/well in 96-well black plates were incubated with various inhibitors and FITC-GO, respectively. Then, the culture media were removed, followed by three washes in PBS. The fluorescence intensity of FITC was excited at 494 nm and measured with a 520 nm by SpectraMax M5e microplate reader. The control cells were treated with FITC-GOs without inhibitors.
siRNA knockdown in KUP5 cells
The knockdown of NADPH oxidase in KUP5 cells was performed as described before [40]. We focused on NOX2, which is the primary source of ROS generation in macrophages. We used siRNA to knockdown NADPH oxidase p22phox and p47phox subunits as well as GSDMD that plays a major role in pyroptosis in KUP5 cells using electroporation method at the Integrated Molecular Technologies Core Facility (University of California, Los Angeles) [29,40]. Briefly, 10 μL of 200 ng/mL of each of the siRNA in buffer was electroporated into 1 × 106 KUP5 cells. After electroporation, cells were maintained in complete media for another 48 h before further use.
Assessment of plasma membrane lipid peroxidation
KUP5, LSEC, and Hepa 1–6 cells at a density of 2 × 105/well in 8-well Lab-Tek chamber slide were incubated with 12.5 μg/mL GO for 16 h or 10 μM cumene hydroperoxide (CH, positive control) for 1 h. After being washed three times in PBS, the cells were incubated with 10 μM Image-iT lipid peroxidation sensor for 0.5 h and Hoechst 33342 for 15 min, respectively. The stained cells were washed and used for confocal microscopy to visualize the reduced and oxidized fluorescent dye at excitation/emission wavelengths of 581/591 nm (Texas Red filter set) and 488/510 nm (traditional FITC filter), respectively. The flow cytometry analysis on a BD LSRFortessa X-20 flow cytometer (Franklin Lakes, NJ) was performed to quantify for the percentage of cells with lipid peroxidation.
We determined if the plasma membrane lipid peroxidation in KUP5 cells was mediated by NADPH oxidases. Before GO exposure, the KUP5 cells were treated with NADPH oxidase inhibitors, 10 μM diphenylene iodonium (DPI) for 0.5 h [41], or 100 μM apocynin for 12 h. The dose of the inhibitors was chosen to make sure its non-toxicity to cells. In addition, we used siRNA to knockdown the p22phox and p47phox subunits of NOX2 in KUP5 cells. The membrane lipid peroxidation was assessed using confocal microscopy and flow cytometry.
Assessment of PLC activity in KUP5
To assess phospholipase C (PLC) activity in KUP5 cells, the cells were seeded at a density of 3 × 104/well in 96-well black plates and treated with 12.5 μg/mL GO for 16 h. PLC activity was evaluated by using the commercial EnzChek Direct Phospholipase C assay kit, according to the kit’s protocols provided and with the aid of a microplate reader [42,43]. To confirm the ability of GO to activate PLC, KUP5 cells were treated with 10 μM U-73122, a pharmacological PLC inhibitor, for 4 h. In addition, other inhibitors including an NADPH oxidase inhibitor DPI and phagocytosis inhibitor WM were used to evaluate their effects on GO-induced PLC activation.
Measurement of the cytoplasmic calcium flux
KUP5 cells were cultured in 8-well Lab-Tek chamber slide and loaded with the plasma membrane permeable calcium indicator Fluo-4 AM ester (5 μM) in a Pluronic F-127-buffered DMSO solution for 1 h at 37 °C. Then, cells were washed in an indicator-free medium to remove any dye that is nonspecifically associated with the cell surface, followed by the incubation for a further 0.5 h to allow complete de-esterification of intracellular AM esters. Finally, the cells were fixed with 4 % paraformaldehyde in PBS, stained with Hoechst 33342, and imaged using a Leica Confocal SP8-SMD microscope. The fluorescence intensity was monitored at excitation/emission wavelengths of 494/516 nm. To confirm the role of calcium flux, KUP5 treated with a calcium chelator BAPTA-AM (10 μM) or other inhibitors, including a PLC inhibitor U-73122, an NADPH oxidase inhibitor DPI, and a phagocytosis inhibitor WM, for comparison purposes.
Determination of mitochondrial ROS generation
KUP5 cells were treated with GOs for 16 h, then the cells were washed three times with PBS and treated with 5 μM MitoSOX in HBSS at 37 °C for 10 min. The cells were fixed with 4 % paraformaldehyde in PBS, stained with Hoechst 33342, and imaged using a Leica Confocal SP8-SMD microscope. The fluorescence intensity was monitored as the rate of oxidation of the dye in the cells at excitation/emission wavelengths of 510/580 nm. Before GO exposure, KUP5 cells were treated with various inhibitors, including a calcium chelator BAPTA-AM, a PLC inhibitor U-73122, an NADPH oxidase inhibitor DPI, and a phagocytosis inhibitor WM, for comparison purposes.
Determination of caspases-1 and 3/7 activation
KUP5 cells cultured in an 8-well Lab-Tek chamber slide at 2 × 105 cells/well were primed with LPS (1 μg/mL) for 4 h and incubated with GO for 16 h. Cells were washed with PBS and stained with FAM-FLICA Caspase-1 or Caspase 3/7 substrates for 1 h at 37 °C based on the manufacturer’s procedure. Then, the fixed cells with 4% paraformaldehyde in PBS were stained with Hoechst 33342 and imaged using a Leica Confocal SP8-SMD microscope. The fluorescence intensity in the cells was monitored at excitation/emission wavelengths of 492/520 nm. Various inhibitors were also used to pretreat KUP5 cells for comparison purposes.
Determination of IL-1β and TNF-α production
KUP5 cells seeded at a density of 3 × 104 cells per well in a 96-well plate were primed by replacing the tissue culture medium with fresh medium containing 1 μg/mL LPS for 4 h. The primed cells were exposed to 12.5 μg/mL GO suspension containing 0.1 μg/mL LPS for 24 h. The cellular supernatants were collected for IL-1β or TNF-α quantification by enzyme-linked immunosorbent assay (ELISA) according to the manufacturer’s instructions. Various inhibitors were also used to pretreat KUP5 cells for comparison purposes.
Assessment of morphological changes under optical microscopy
KUP5, LSEC, and Hepa 1–6 cells were exposed to 12.5 μg/mL GO for 16 h in a 12-well plate, respectively. The morphology of the cell was monitored using a Zeiss Optical Microscope (Carl Zeiss, Inc. Peabody, MA, USA). For the KUP5 cells, the cells were pretreated with 1 μg/mL LPS for 4 h before GO treatment, the morphology of wild-type cells treated in the presence or absence of inhibitors as well as the cells with GSDMD knockdown was examined by optical microscopy. The percentage of swollen cells was calculated based on the cells displaying at least two giant blebs [29].
Statistical analysis
All the values are the mean and standard deviation of three independent experiments. Statistical significance was determined by two-tailed Student’s t-test for two-group analysis or one-way ANOVA for multiple group comparisons. A P-value of less than 0.05 was considered statistically significant. A two-tailed Pearson correlation coefficient was performed to determine the correlations between GO physicochemical properties and the cellular response in KUP5 cells.
Results
Physicochemical characterization of GO-S and GO-L
GOs with two lateral sizes, small (GO-S) and large (GO-L), were prepared. Their physicochemical characterization is detailed in Fig. 1. The difference in lateral size was determined by AFM analysis in Fig. 1a. GO samples contain sheets with an average height of approximately 1 nm, indicating that the majority of GO sheets are monolayers. The average lateral size calculated as square root area of the GO nanosheets is 91 ± 79 and 583 ± 343 nm for GO-S and GO-L (Table 1), respectively. Representative TEM images in Fig. 1b confirmed that both GO samples showed a typical sheet-like structure.
Fig. 1.
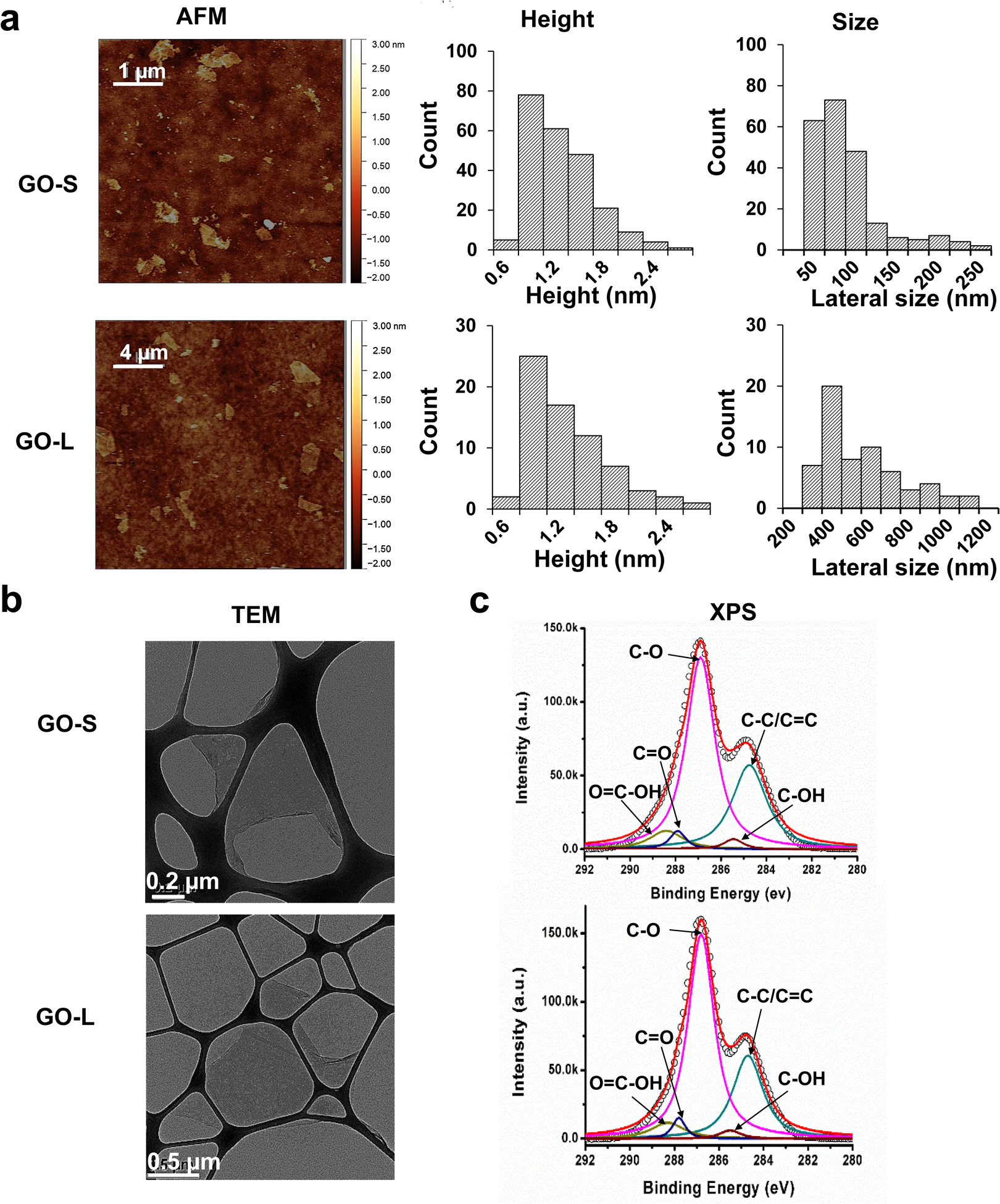
Physiochemical characterization of GOs. (a) Characterization of GO-S and GO-L by AFM. The left row shows the AFM topography images of GOs. Scale bar for GO-S is 2 μm; Scale bar for GO-L is 4 μm. The middle row shows the histogram of GO height distribution. The average height of GO-S is 1.3 ± 0.9 nm and the average height of GO-L is 1.2 ± 0.5 nm. The right row shows the histogram of GO size distribution. The average lateral size of GO-S is 91 ± 79 nm and the average lateral size of GO-L is 583 ± 343 nm. (b) TEM images of GO-S and GO-L. The images were captured through a JEOL 1200-EX TEM with an accelerating voltage of 80K eV. Scale bar in the upper panel is 0.2 μm; Scale bar in the lower panel is 0.5 μm. (c) C 1s XPS spectra of GO-S (upper panel) and GO-L (lower panel) surface composition.
Tab. 1.
Quantification of lateral size, thickness (height), and surface functional groups for GO-S and GO-S
| GO samples | GO-S | GO-L | |
|---|---|---|---|
| AFM | Lateral size (nm) | 97 ± 79 | 583 ± 343 |
| Height (nm) | 1.3 ± 0.9 | 1.2 ± 0.5 | |
| At.% from XPS | C-C/C=C | 30.8 ± 0.1 | 29.2 ± 0.1 |
| C-OH | 2.0 ± 0.0 | 2.0 ± 0.0 | |
| C-O | 58.1 ± 0.2 | 59.9 ± 0.2 | |
| C=O | 2.9 ± 0.0 | 3.0 ± 0.1 | |
| O=C-OH | 6.2 ± 0.1 | 6.0 ± 0.1 |
Additionally, the surface chemical properties of GOs were examined by XPS to ensure that the GO samples had similar physicochemical properties, which is important to isolate the role of lateral sizes in their cellular responses. The XPS results displayed similar surface functional groups on both GO samples, with major characteristic peaks at 284.6, 285.5, 286.6, 287.8, and 288.6 eV, representing C-C/C=C, C-OH, C-O, C=O, and O=C-OH groups, respectively (Fig. 1c). The quantitative analysis of each group showed that GO-S and GO-L possessed similar atomic percentages of surface functional groups such as oxygen content (~59%), hydroxyl groups (~2%), and carboxyl groups (~6%) (Table 1). Taken together, these characterization data demonstrate that GO-S and GO-L samples showed comparable physicochemical properties other than lateral size differences, which allows the study to determine the role of lateral size in GO-induced cellular effects.
Because GOs were used for in vitro cellular studies, endotoxin levels were determined by the LAL assay to rule out bacterial contaminations. The results showed that the GOs had low levels of endotoxin levels at approximately 0.05 EU/ml, lower than FDA’s 0.5 EU/ml limit for medical devices (Fig. S1). Furthermore, the dispersibility of GO in aqueous solutions was assessed by hydrodynamic size, PDI, and zeta potential in DI water and cell culture media (Table 2). As expected, GO-L showed larger hydrodynamic sizes than GO-S across different media. The PDI of GOs was 0.2 in water. This value increased to 0.3–0.4 when GOs were suspended in cell culture media, suggesting decreased dispersity. The hydrodynamic sizes of GOs were generally smaller in water than in cell culture media, especially for GO-S. GO-S showed a hydrodynamic radius of 246 nm, while the hydrodynamic radii became larger in cell culture media with size a range of 336–459 nm. GO-L showed a hydrodynamic radius of 586 nm, while the hydrodynamic radii became larger in KUP5 cell culture media with a hydrodynamic radius of 687 nm. These hydrodynamic sizes can be explained by further agglomeration and adsorption of serum proteins or the formation of protein corona on GOs in cell culture media [44,45]. In addition, as expected, the GOs carry negative surface charges because of the presence of carboxyl and hydroxyl groups on the surface, with higher values of −40–43 mV in water than that in culture media −7–11 mV, which was due to high ionic strength and protein corona formation in cell culture media, respectively.
Tab. 2.
Hydrodynamic size, polydispersity index, and zeta potential of GOs
| Media | Sample | Hydrodynamic size (nm) | Polydispersity index (PDI) | Z-potential (mV) |
|---|---|---|---|---|
| DI H2O | GO-S | 246.3 ± 0.7 | 0.2 | −43.6 ± 2.0 |
| GO-L | 585.6 ± 20.9 | 0.2 | −40.6 ± 3.8 | |
| DMEM (KUP5) | GO-S | 336.7 ± 7.9 | 0.3 | −10.4 ± 1.0 |
| GO-L | 686.5 ± 11.3 | 0.3 | −9.9 ± 1.6 | |
| Prigrow medium (LSEC) | GO-S | 394.9 ± 5.4 | 0.3 | −11.4 ± 2.6 |
| GO-L | 591.7 ± 9.7 | 0.4 | −9.3 ± 1.7 | |
| DMEM (Hepa 1–6) | GO-S | 459.9 ± 6.3 | 0.3 | −9.6 ± 0.8 |
| GO-L | 553.1 ± 12.3 | 0.3 | −7.4 ± 1.0 |
GO induced differential cytotoxic responses in KUP5, LSEC, and Hepa 1–6 cells
Cell viability studies were undertaken by the MTS assay to obtain provisional toxicological profiling of the GOs in KUP5, LSEC, and Hepa 1–6 cell lines (Fig. 2), which represent three of the principal liver cell types impacted by nanomaterials. The MTS results demonstrated that there were differential response profiles as a reflection of GOs in all cell types over a dose range of 0–100 μg/mL (Fig. 2a). KUP5 cells were more sensitive to the cytotoxic effects of GOs, while LSECs and Hepa 1–6 cells showed no cytotoxicity, except for LSECs at the highest dose of 100 μg/mL. GO toxicity to KUP5 was dose-dependent after 24 h exposure and GO-L exhibited higher toxicity than GO-S and toxicity became significant at 25 μg/mL, while GO-S induced toxicity at 50 μg/mL. GO-L-induced toxicity was significantly higher than GO-S at 100 μg/mL. The heat map in Fig. 2b displays the differential cytotoxicity response profiles using a one-way ANOVA statistical method, where yellow indicates significant toxicity, and the green represents the absence of toxicity. It is interesting that GOs also induced significant morphological alteration in KUP5 cells, which showed extensive cell swelling and the presence of large surface blebbing (Fig. S2). A time-lapsed video (Video 1) in the supplemental materials shows the dynamics of GO-L-induced morphological changes to KUP5 cells. This morphological change is in direct contrast to ZnO, a positive control for apoptotic cell death as demonstrated previously [29], which showed cell rounding and loss of filopodia (Fig. S2), suggesting a different cell death mechanism is involved for GOs. Additional data showed GOs failed to induce caspase 3/7 activation, while ZnO induced their activation, suggesting GOs induced non-apoptotic cell death (Fig. 2c–d). The morphological changes and caspase-3/7 data from GOs together were reminiscent of a previous report by Qu et al., which showed GO induced programmed necrosis in macrophages including J774A.1, RAW 264.7, and bone marrow-derived macrophages (BMDM) without triggering caspase-3 mediated apoptosis [28]. However, recent progress showed that programmed necrosis involves different mechanisms including necroptosis, pyroptosis, etc. [46,47]. The detailed mechanism involved in GO induced necrosis is still not clear. Thus we embarked on this study to elucidate the detailed mechanisms involved in GO-induced programmed necrosis.
Fig. 2.
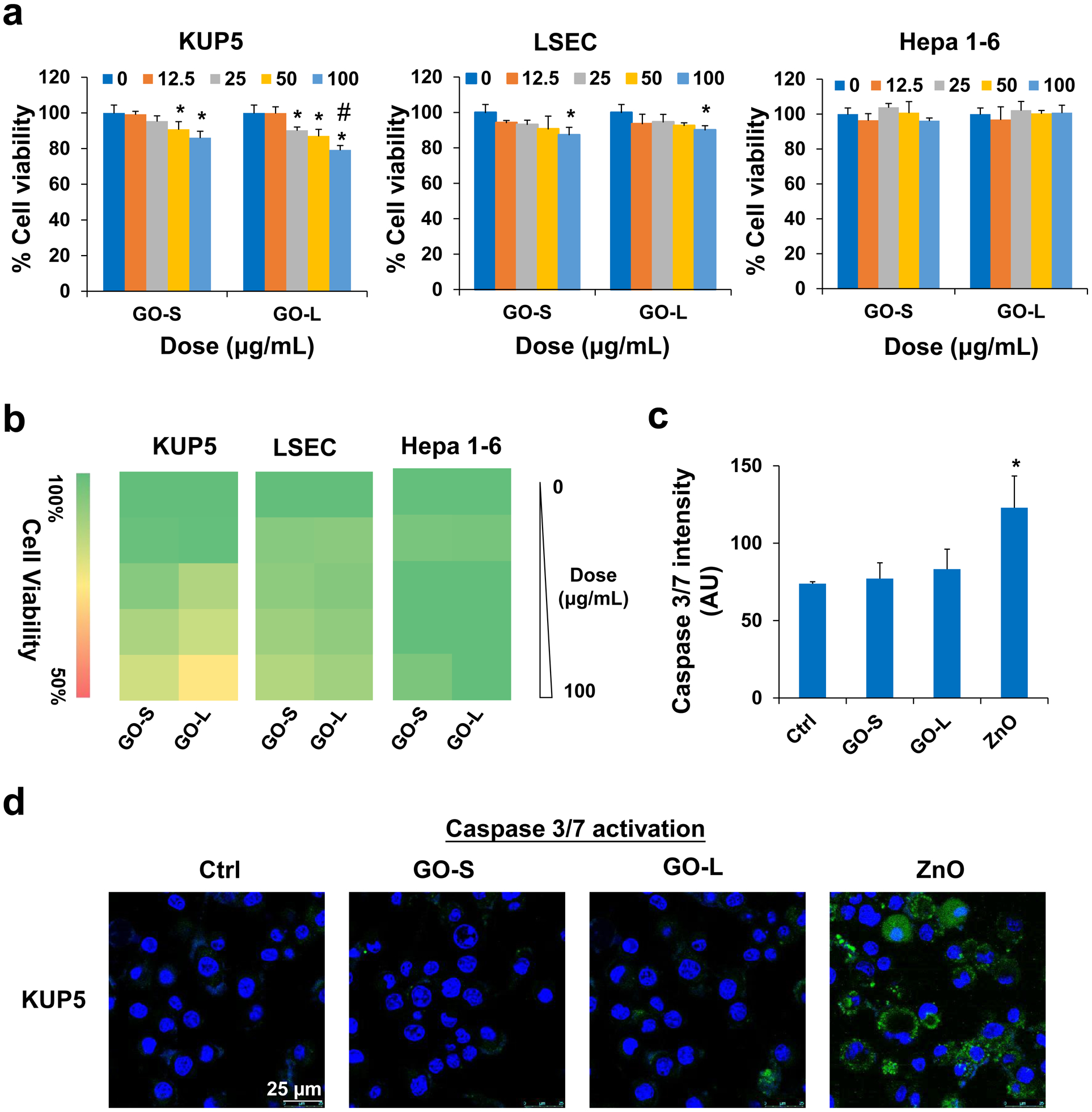
Cytotoxicity of GOs to KUP5, LSEC, and Hepa 1–6 cells. (a) Cell viability of KUP5, LSEC, and Hepa 1–6 cells on GO exposure for 24 h at a dose range of 0–100 μg/mL determined by MTS assay. The viability of untreated control cells was set to 100%. Asterisk (*) means P < 0.05, compared to the control, and #, P < 0.05, indicates significance between GO-S and GO-L treatments. (b) Heat maps display for KUP5, LSEC, and Hepa 1–6 cells according to the color scale in the sidebar on the left. (c) Quantification for caspase 3/7 activation in KUP5 cells under GO treatments. ZnO nanoparticle served as a positive control. The fluorescence intensity was monitored at excitation/emission wavelengths of 492/520 nm. * means P < 0.05, compared to the control. (d) Confocal images to demonstrate caspase 3/7 activation in KUP5 cells under GO treatments. The scale bar is 25 μm.
Lateral size-dependent cell association and uptake of GOs
The cytotoxicity induced by GO is dependent on its physical interactions with the cell membrane, following which there is the possibility of cellular uptake [48]. To shed light on the GO and cell interactions, we studied cell association and cellular uptake of GOs by KUP5, LSEC, and Hepa 1–6 cells. To visualize the GOs, we prepared FITC-BSA labeled GOs as described previously [25]. The liver cells were incubated with 12.5 μg/mL FITC-BSA-labeled GOs for 16 h and visualized under a confocal microscope. The cell membrane was counter-stained with Alexa Fluor 594-labeled WGA antibody (red) and the nucleus was stained with Hoechst 33342 dye (blue). As shown in Fig. 3a, fluorescent GO-S and GO-L were mostly present inside of KUP5 cells, which was confirmed by TEM images obtained from KUP5 cells treated by pristine GOs (Fig. 3b). For LSECs and Hepa 1–6 cells, while most GO-S could be visualized inside cells, most GO-L showed adsorption on the plasma membrane with limited cellular uptake. These confocal results were further confirmed by TEM images (Fig. S3–4).
Fig. 3.
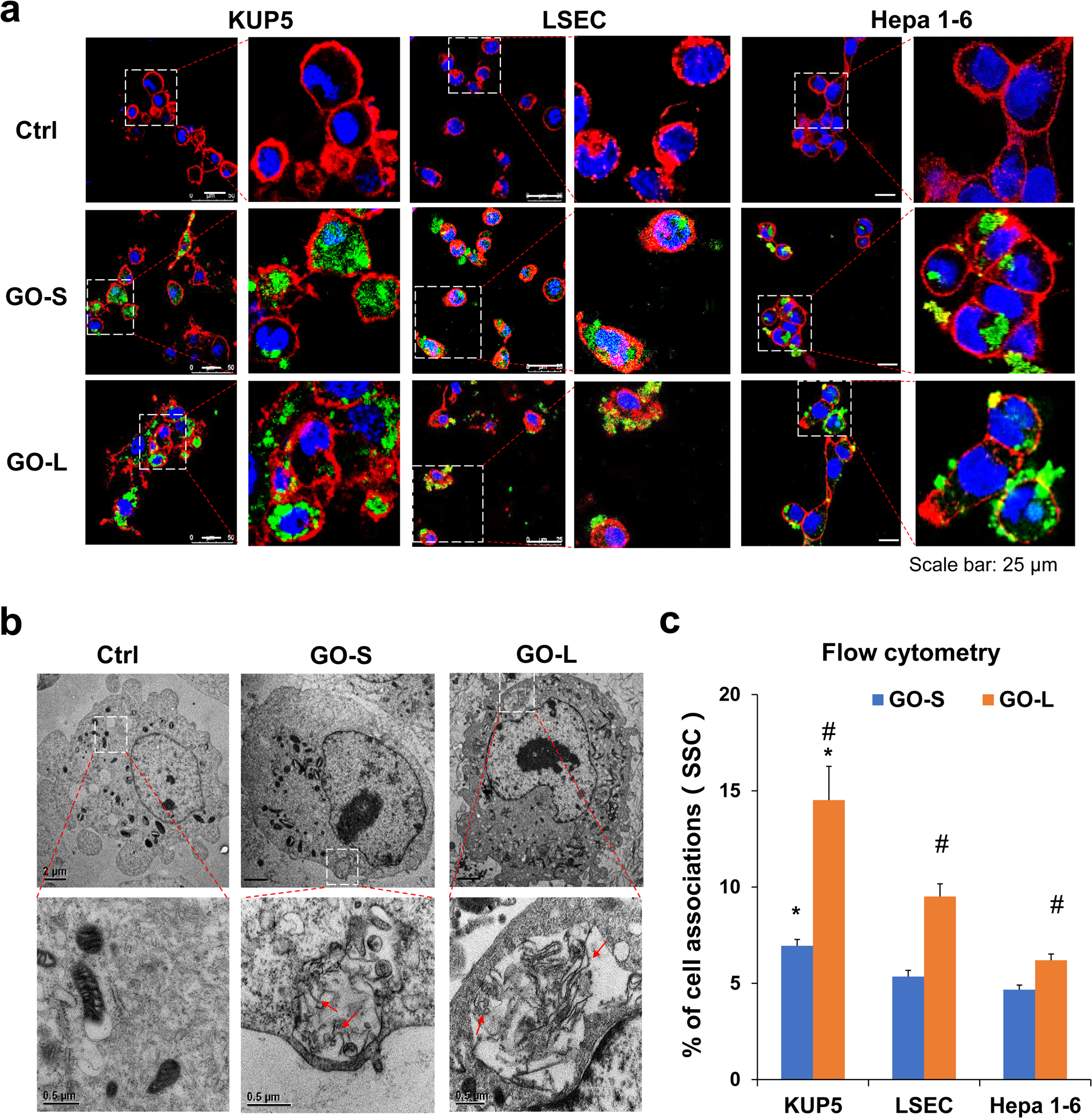
Determination of cellular association of GO in KUP5, LSEC, and Hepa 1–6 cells. (a) Confocal imaging of FITC-BSA-labeled GOs in KUP5, LSEC, and Hepa 1–6 cells. These cells were incubated with 12.5 μg/mL of FITC-labeled GOs (green) for 16 h, followed by staining with Hoechst 33342 dye (blue) and Alexa Fluor 594-labeled WGA antibody (red). The scale bar in the left panel image is 25 μm. (b) Visualizing the interactions of pristine GO with KUP5 cells by TEM. After exposure to 50 μg/mL of GO for 16 h, the cells were washed, fixed, and stained for TEM viewing under a JEOL 1200-EX microscope. Red arrows indicate GO sheets within the phagosomes. Scale bar in the upper panel is 2 μm; Scale bar in the lower panel is 0.5 μm. (c) Quantification for the percentage of SSC values by flow cytometry for GO-treated cells compared to untreated cells. *, P < 0.05, compares KUP5 cells to LSECs or Hepa 1–6 cells; #, P < 0.05, compares GO-L with GO-S.
To quantify the cellular associations with GOs, flow cytometry was performed. When GOs are associated with the plasma membrane or taken up by the cells, an increase in cellular granularity can be measured by side scattering (SSC) [25]. As shown in Fig. 3c, the cellular SSC analysis demonstrated that the cellular association of GOs in KUP5 cells was significantly more than that in LSECs or Hepa 1–6 cells. Furthermore, cellularly associated GO-L was significantly higher than GO-S in KUP5, LSECs, and Hepa 1–6 cells. These results demonstrated that the cellular association of GOs with liver cells are lateral size- and cell type-dependent.
GO-induced plasma membrane lipid peroxidation in KUP5 only
Previously, we have shown GOs could induce plasma membrane lipid peroxidation to THP1 cells due to the presence of carbon radicals on GO surface [25]. The carbon radicals are typically more reactive than other surface functionalities and capable of oxidizing unsaturated lipids on the cell membrane by reacting with molecular dioxygen to generate superoxide radicals due to the presence of unpaired electrons. GO-S and GO-L have been shown to trigger abiotic ROS generation (Fig. S5) and may induce plasma membrane lipid peroxidation in liver cells. We used the BODIPY 581/591 C11 reagent, a lipid-soluble ratiometric fluorescent indicator of lipid oxidation, to evaluate the lipid peroxidation in KUP5, LSEC, and Hepa 1–6 cells. As shown in the confocal microscopy images (Fig. 4a), GO induced a substantial shift to green fluorescence at the expense of the red fluorescence in the plasma membrane of KUP5 cells, similar to the positive control of cumene hydroperoxide (CH). However, GOs failed to induce lipid peroxidation in LSECs or Hepa 1–6 cells despite the high association of GO-L with cell membranes. GO-induced lipid peroxidation was further quantified by flow cytometry. By calculating the percentage of cells exhibiting increased fluorescence intensity at 510 nm (Fig. 4b), we showed that the percentages of cells with lipid peroxidation were 7.9% and 12.6% in KUP5 cells for GO-S and GO-L, respectively. GO-L induced a significantly higher level of lipid peroxidation than GO-S. In contrast, GOs failed to induce lipid peroxidation in LSECs and Hepa 1–6 cells despite the uptake of GO-S inside of cells and GO-L adsorption on the cell membrane of these two cell types. These data suggest that membrane association alone is not sufficient to induce lipid peroxidation and thus other cellular mechanisms are involved.
Fig. 4.

Assessment of the plasma membrane lipid peroxidation production by GO in KUP5, LSEC, and Hepa 1–6 cells. (a) Confocal images to demonstrate the induction of lipid peroxidation by GO. KUP5, LSEC, and Hepa 1–6 cells were incubated with 12.5 μg/mL of GO for 16 h or 10 μM cumene hydroperoxide (CH, positive control) for 1 h. Cells were stained with 10 μM Image-iT lipid peroxidation sensor for 0.5 h and Hoechst 33342 for 15 min, respectively. The stained cells were visualized under a confocal microscope for red (reduced) and green (oxidized) fluorescence at excitation/emission wavelengths of 581/591 nm (Texas Red filter set) and 488/510 nm (traditional FITC filter), respectively. The scale bar is 25 μm. (b) Quantification for the percentage of cells with lipid peroxidation by flow cytometry. *, P < 0.05, compares to the control; #, P < 0.05, between GO-S and GO-L treatments.
GO-induced lipid peroxidation in KUP5 cells is dependent on phagocytosis
The differential effects on lipid peroxidation between KUP5 and LSEC or Hepa 1–6 cells may lie in the cellular uptake mechanisms among different cell types. KCs are residential macrophages in the liver, which are phagocytic cells that could uptake large particles (> 500 nm) through phagocytosis [21]. TEM images in Fig. 3b confirm that GOs were located in membrane-bound intracellular structures appearing to be the phagosomes. To determine the cellular uptake mechanisms, KUP5 cells were pre-treated with a phagocytosis inhibitor wortmannin (WM), a macropinocytosis inhibitor Cytochalasin D (Cyto D), and a clathrin-dependent endocytosis inhibitor Pitstop 2 under an incubation time and concentration without triggering cytotoxicity (data not shown). Confocal microscopy was used to visualize the cellular uptake of FITC-BSA-GO in KUP5 cells and 1 μm FITC-labeled polystyrene beads were used as a control particle for phagocytosis. As shown in Fig. 5a, WM significantly inhibited the uptake of FITC-BSA-GOs and 1 μm polystyrene beads, while Cyto D and Pitstop 2 could not inhibit the GO and bead uptake (Fig. S6), suggesting that KUP5 cells take up GOs predominantly through phagocytosis. For LSECs and Hepa 1–6 cells, only Pitstop 2 that blocks ligand access to the clathrin terminal domain could inhibit the GO uptake, suggesting GOs were taken up by clathrin-mediated endocytosis (Fig. S7–8). We then evaluated the effects of phagocytosis inhibition on GO-induced membrane lipid peroxidation in KUP5 cells. The flow data (Fig. 5b) and confocal images (Fig. S9), respectively, demonstrated that WM significantly inhibited the lipid peroxidation production by GOs in KUP5 cells.
Fig. 5.
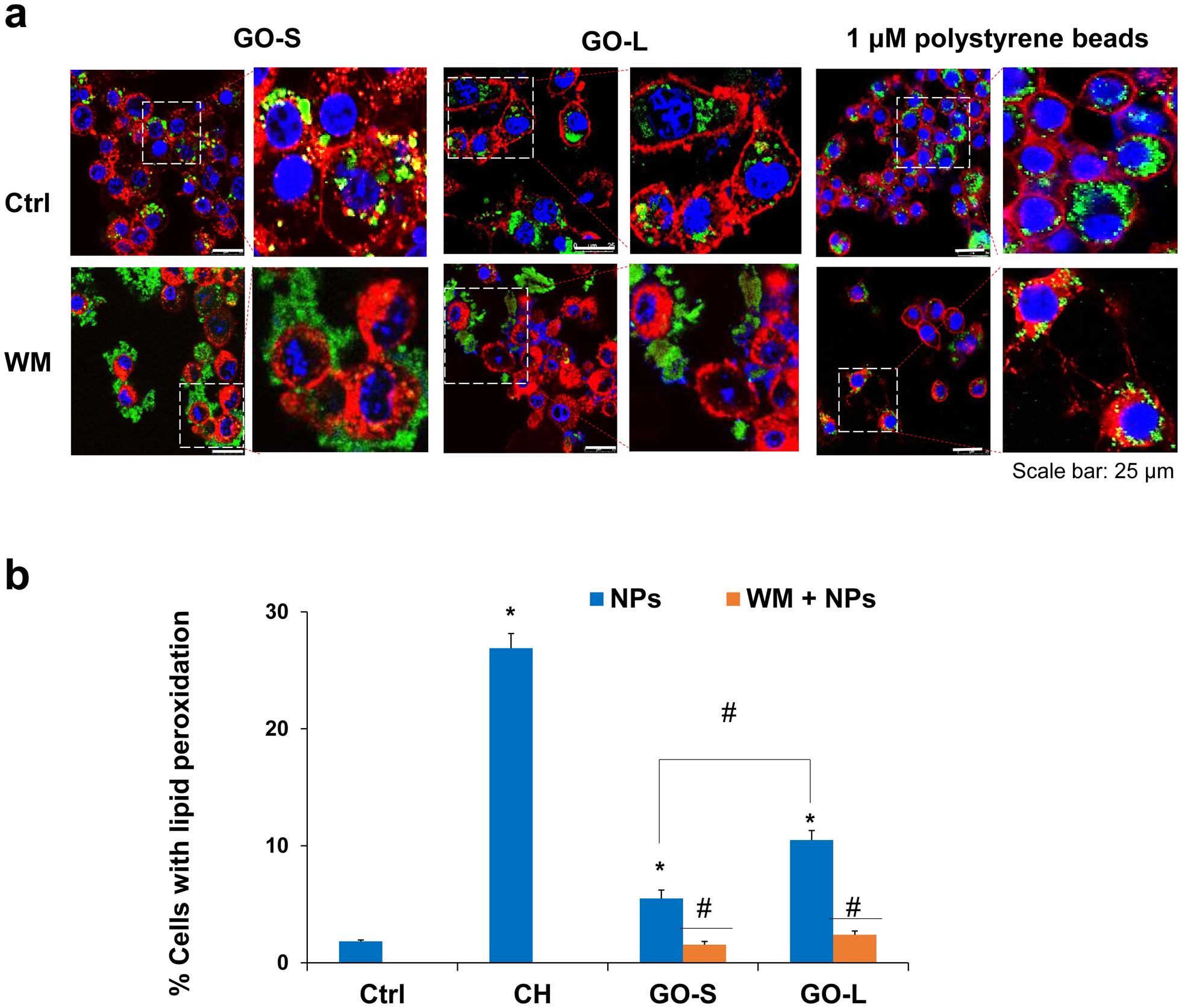
Determination of phagocytosis of GO by KUP5 cells. (a) Confocal images showing the cellular localization of FITC-GO in KUP5 cells under wortmannin (WM) treatment. Before exposure to GO, KUP5 cells were pretreated with 1 μM WM for 0.5 h. After staining with Hoechst 33342 dye and Alexa Fluor 594-labeled WGA antibody, the cells were visualized under a confocal microscope. The scale bar is 25 μm. (b) Quantification for the effects of WM on lipid peroxidation by flow cytometry. *, P < 0.05, compares to the control; #, P < 0.05, compares to GO treatment alone.
Lipid peroxidation in KUP5 cells mediated by NADPH oxidases
It is known that phagocytic uptake by macrophages is accompanied by the activation of a multi-subunit NADPH oxidase (NOX) complex in the phagosome, which is a major source of reactive oxygen species (ROS) generation [40,49]. We hypothesized that the lipid peroxidation production in response to phagocytosis could be mediated by the superoxide free radical generated by NOXs in KUP5 cells. NOX2, the only isoform of NADPH oxidase that KCs express [50], is the primary source of ROS generation in KUP5 cells. We used its inhibitor, dibenziodolium chloride (DPI), and siRNA knockdown of NOX2 subunits (p22phox and p47phox) to evaluate its role in GO-induced lipid peroxidation. As shown in the confocal images in Fig. 6a, DPI and siRNA knockdown of p22phox or p47phox in KUP5 cells significantly inhibited the lipid peroxidation induced by GOs. The quantification data by flow cytometry in Fig. 6b confirmed the NOX inhibitor or siRNA knockdown significantly reduced the percentages of cells with lipid peroxidation. We also used another inhibitor of NOX2, apocynin, which showed similar results (data not shown). These data suggest NOX activation is responsible for the GO-induced plasma membrane lipid peroxidation, which also explains the absence of lipid peroxidation in LSECs and Hepa 1–6 cells that could not trigger GO phagocytosis and NOX activation.
Fig. 6.
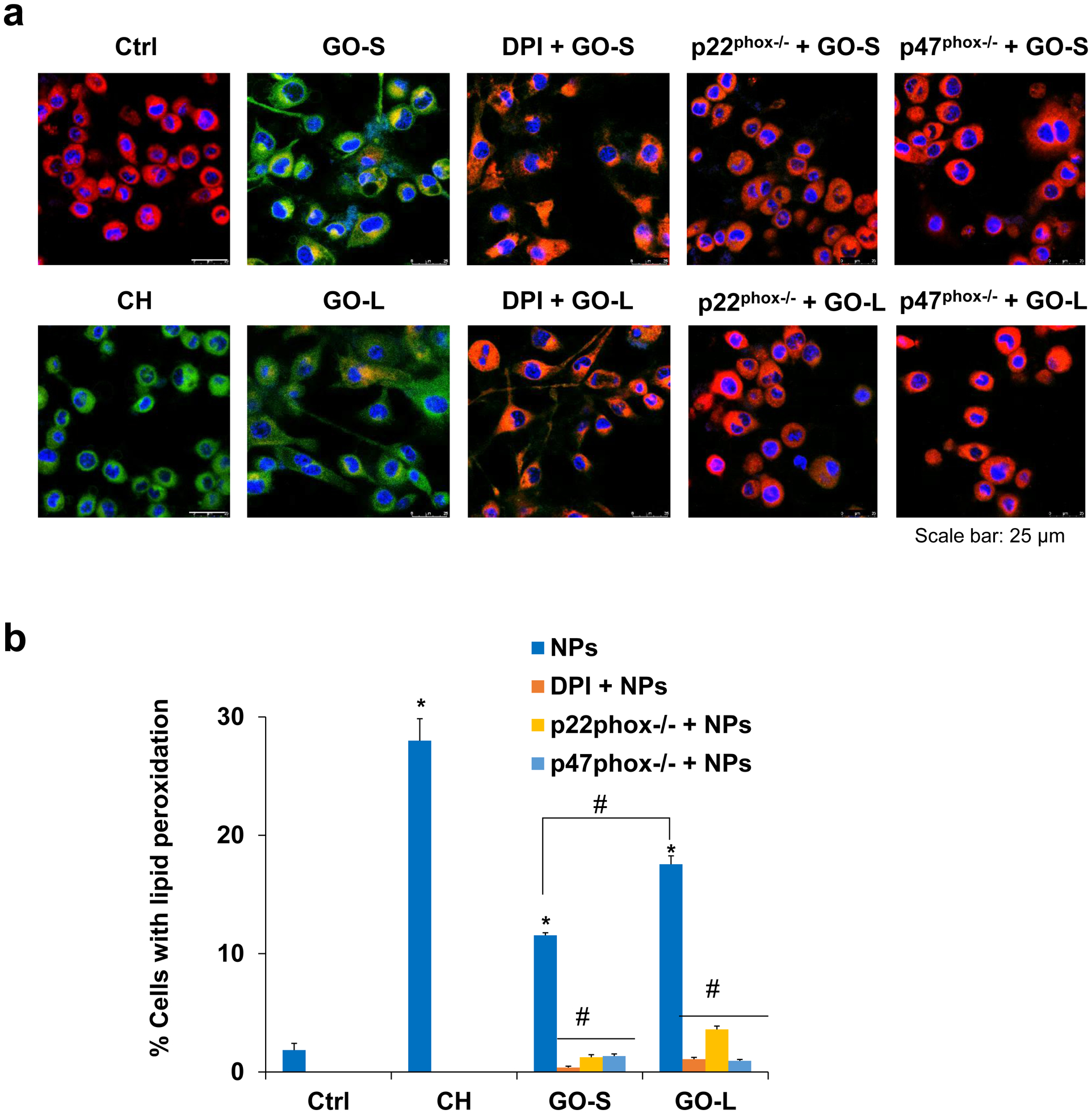
Assessment of the plasma membrane lipid peroxidation production by GO after inhibiting NADPH oxidase activity in KUP5 cells. (a) Confocal images to demonstrate the reduction of lipid peroxidation production in KUP5 under the treatment of NOX2 inhibitor, DPI, and siRNA knockdown of p22phox−/− or p47phox−/− in KUP5, respectively. The images were acquired to visualize the red (reduced) and green (oxidized) fluorescent dye at excitation/emission wavelengths of 581/591 nm and 488/510 nm. The scale bar is 25 μm. (b) Quantification for the percentage of cells with lipid peroxidation induced by GO under the various NADPH oxidase inhibitor treatments. *, P < 0.05, indicates significance compared with the control; #, P < 0.05, indicates significance compared to GO treatment alone.
Lipid peroxidation induced PLC activation, calcium flux, and mtROS generation
It is known that oxidized lipids as a result of lipid peroxidation is not only sufficient but also required for phospholipase C (PLC) activation, which then cleaves phospholipids on the cell membrane to diacylglycerol (DAG) and inositol triphosphate (IP3) [42,43]. IP3 serves as a second messenger to induce Ca2+ release from endoplasmic reticulum (ER) stores, one of the most powerful intracellular signals in a cell. Recent studies showed that lipid peroxidation-induced PLC activation and subsequent Ca2+ flux as well as mitochondrial ROS generation could drive a unique form of programmed necrosis, gasdermin D (GSDMD)-mediated pyroptosis [43,51,52]. To explore this possibility, we assessed the PLC activity using an EnzChek Direct Phospholipase C activity assay kit after GO exposure. For comparison purposes, we also included Gd2O3 nanoparticles, which has been demonstrated to be a pyroptosis inducer for KUP5 cells [28]. As shown in Fig. 7a, the PLC activity in KUP5 cells treated with GO-S and GO-L increased by 40.4% and 69.2% compared to the control, respectively. Furthermore, GO-L induced significantly stronger PLC activity than GO-S (P < 0.05). The role of PLC was confirmed by a PLC inhibitor U-73122. In addition, NADPH oxidase inhibitor DPI and phagocytosis inhibitor WM also inhibited the PLC activation (Fig. 7a), suggesting that phagocytosis and NOX2 activation are required for GO-induced PLC activation. Surprisingly, Gd2O3 also induced PLC activation, which has not been demonstrated before, suggesting a common pathway may be involved for GOs and Gd2O3.
Fig. 7.
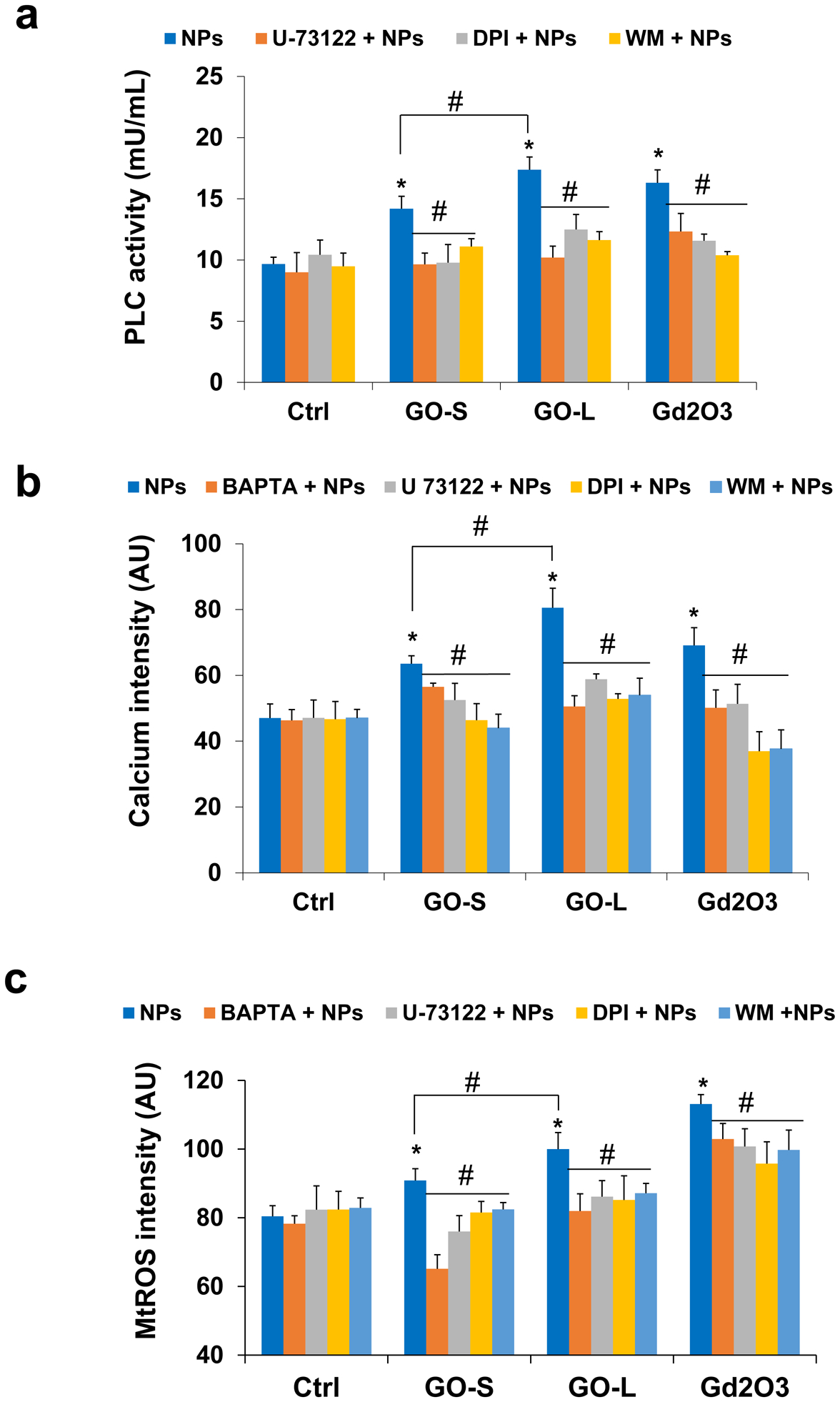
Determination of PLC activated intercellular calcium accumulation and mitochondrial ROS generation in KUP5 cells post-exposed to GO. (a) Assessment of PLC activity by EnzChek Direct Phospholipase C activity assay kit. KUP5 cells were pretreated with or without inhibitors (U-73122 as PLC inhibitor, DPI as NOX2 inhibitor, and WM as phagocytosis inhibitor) before treatment of 12.5 μg/mL of GO or Gd2O3 (positive control) for 16 h. (b) Quantification for intercellular calcium level in KUP5 cells under GO treatments with or without inhibitors. The fluorescence intensity was monitored at excitation/emission wavelengths of 494/516 nm. (c) Quantification for mtROS generation in KUP5 cells under GO treatments with or without inhibitors by a microplate reader. The fluorescence intensity was monitored at excitation/emission wavelengths of 510/580 nm. *, P < 0.05, indicates significance compared with the control; #, P < 0.05, indicates significance compared to NP treatment alone.
To see if GO-induced PLC activation will lead to Ca2+ flux, we assessed intercellular calcium levels in KUP5 cells by staining the cells with a calcium dye, Fluo-4 AM, and then performed visualization under a confocal microscope. As shown in Fig. S10, the signal intensity of intercellular calcium in KUP5 cells substantially increased after GO exposure compared to the control, and Gd2O3 also increased the calcium signal. The Ca2+ flux in KUP5 cells was inhibited by BAPTA, a calcium-specific chelator, confirming the role of calcium. Furthermore, PLC inhibitor U-73122, NADPH oxidase inhibitor DPI, and phagocytosis inhibitor WM also attenuated the Ca2+ flux. The quantified data measured by a microplate reader in Fig. 7b confirmed GO-induced Ca2+ flux. GO-S and GO-L increased the intercellular calcium levels by 35.0% and 77.2% compared to the control, respectively. Furthermore, GO-L induced significantly stronger calcium elevations than GO-S (P < 0.05). Overall, these results indicated that GOs induced size-dependent PLC activation and Ca2+ flux in KUP5 cells.
The mitochondria take up calcium to regulate spatiotemporal patterns of calcium signaling that is critical for diverse cellular processes. However, excessive or sustained mitochondrial calcium uptake triggers mitochondrial damage, including increased mtROS production [53–55]. We assessed mtROS production in KUP5 cells treated with GOs using the MitoSOX Red fluorescent staining method. As shown in Fig. S11, the level of mtROS production in KUP5 cells after GO exposure significantly increased compared to the control, similar to Gd2O3. The quantified data measured by fluorescence intensity with a microplate reader in Fig. 7c demonstrated that the mtROS production in KUP5 cells treated with GO-S and GO-L increased by 8.8% and 20.4% compared to the control, respectively. GO-L induced a significantly stronger mtROS signal than GO-S (P < 0.05). The mtROS generation was also strongly inhibited by a calcium chelator BAPTA, a PLC inhibitor U-73122, an NADPH oxidase inhibitor DPI, and a phagocytosis inhibitor WM, respectively. These results indicate that GO was able to induce size-dependent mtROS generation in KUP5 cells and the mtROS generation.
GOs induced GSDMD-meditated pyroptosis in KUP5 cells through NLRP3 inflammasome activation
Calcium influx and mtROS generation have pivotal roles in NLRP3 inflammasome activation, as reflected in the processing of caspase-1 activation and secretion of IL-1β [52,54,55]. MtROS could lead to mitochondrial damage and release of oxidized mtDNA, which is a direct trigger of NLRP3 inflammasome activation [56]. Here, the caspase-1 activation by GOs in KUP5 cells was assessed by confocal microscopy to observe the cleavage of the substrate, FAM-YVAD-FMK. As demonstrated in Fig. 8a, the activation of caspase-1 by GOs significantly increased compared to the control, similar to Gd2O3. The quantified data measured by a microplate reader confirmed the caspase-1 activation in KUP5 cells, and GO-S and GO-L induced an increase in the caspase-1 signal by 12.7% and 26.1% compared to the control, respectively (Fig. 8b). GO-L induced significantly stronger caspase-1 activation than GO-S (P < 0.05). The caspase-1 activation was strongly inhibited by calcium chelator BAPTA, PLC inhibitor U-73122 (Fig. 8a–b), NADPH oxidase inhibitor DPI, and phagocytosis inhibitor WM (Fig. S12), respectively. These results indicate that GOs activate caspase-1 in KUP5 cells in a size-dependent manner.
Fig. 8.
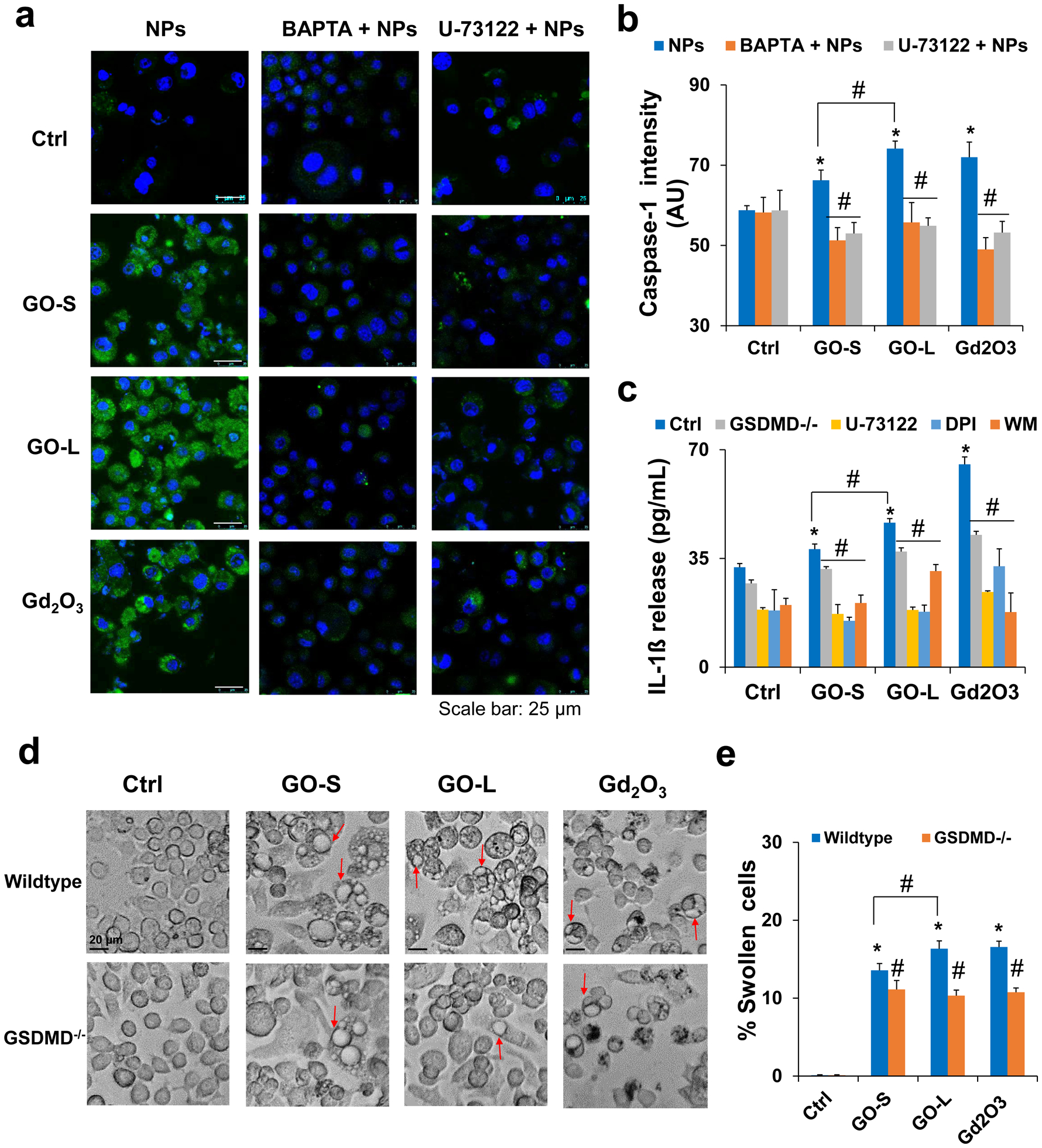
Induction of pyroptosis in KUP5 cells by GOs. (a) Assessment of caspase-1 activation in GO-treated KUP5 cells by a confocal microscope. The LPS-primed KUP5 cells were pretreated with or without inhibitors BAPTA and U-73122 before exposure to 12.5 μg/mL GO or Gd2O3 (positive control) for 16 h. Cells were stained with FAM-FLICA caspase substrate (green) for 1 h, stained with Hoechst 33342 (blue), and imaged using Leica Confocal SP8-SMD microscope. The scale bar is 25 μm. (b) Quantification for caspase-1 activation in KUP5 cells after GO treatments with or without inhibitors. The fluorescence intensity was monitored at excitation/emission wavelengths of 492/520 nm. (c) IL-1β release in KUP5 cells treated with GOs in the absence or presence of inhibitors or after siRNA GSDMD knockdown. Inhibitors included U-73122 as PLC inhibitor, DPI as NADPH oxidase inhibitor, and WM as phagocytosis inhibitor. LPS-primed (1 μg/mL, 4 h) KUP5 cells were exposed to GO or Gd2O3 for 24 h. Supernatants were collected to measure IL-1β release by ELISA. (d) Optical microscope images to compare the KUP5 cell morphological changes induced by 12.5 μg/mL of GO or Gd2O3 between wildtype cells and cells with GSDMD knockdown. Red arrows indicate the swollen cells. The scale bar is 20 μm. (e) The percentage of swollen cells was calculated based on the cells displaying at least two giant blebs in KUP5 cells. *, P < 0.05, indicates significance compared with the control; #, P < 0.05, indicates significance compared to NP treatment alone.
NLRP3 inflammasome and caspase-1 activation lead to cleavage of pro-IL-1β to mature IL-1β. We assessed IL-1β release from KUP5 cells after GO treatment by ELISA. As shown in Fig. 8c, the secretion of IL-1β in KUP5 cells increased by 12.9% and 44.6% for GO-S and GO-L, respectively, compared to the control. GO-L induced significantly stronger IL-1β release than GO-S (P < 0.05). Furthermore, the IL-1β secretion induced by GOs in KUP5 cells was significantly inhibited by PLC inhibitor U-73122, NADPH oxidase inhibitor DPI, and phagocytosis inhibitor WM, respectively. These results indicate that GOs induce size-dependent IL-1β release.
The induction of caspase-1 activation and IL-1β production in KUP5 cells as well as the morphological change including cell swelling and surface blebbing are suggestive of a unique form of cell death known as pyroptosis, which is mediated by caspase-1 activation and the formation of surface membrane pores by N-terminal of GSDMD upon its cleavage by caspase-1 [57–59]. We compared the KUP5 cell morphological changes by 12.5 μg/mL GO through optical microscopy to confirm the pyroptosis in KUP5 cells. As shown in Fig. 8d, the amount of swollen KUP5 cells significantly increased compared to the control, similar to the positive control of Gd2O3. The quantified data in Fig. 8e demonstrated that the amount of swollen KUP5 cells treated with GO-S and GO-L increased by 13.1% and 17.7% compared to the control, respectively. GO-L induced significantly more cellular swelling and surface blebbing than GO-S (P < 0.05) in KUP5 cells. To confirm the role of GSDMD in GO-induced pyroptosis, the siRNA knockdown of GSDMD was performed following the procedure in our previous reports [28,29]. The exposure to GO demonstrated a significant reduction in IL-1β release as well as cell swelling and surface blebbing (Fig. 8c–e) in GSDMD−/− KUP5 cells compared to the wildtype. The cellular swelling and surface blebbing were also significantly inhibited by a calcium chelator BAPTA, a PLC inhibitor U-73122, an NADPH oxidase inhibitor DPI, and a phagocytosis inhibitor WM, respectively (Fig. S13). Taken together, these results show that GO induces size-dependent pyroptosis in KUP5 through caspase-1 activation, IL-1β release, GSDMD cleavage, and the formation of membrane pores.
GO induced pyroptosis in KUP5 cells is a universal feature
It is well known that there are large variations among GO samples in terms of the physicochemical properties including the oxidation levels and surface chemical groups. To assess the sensitivity of our results to these other parameters, we prepared additional three GO samples (GO-1, −2, −3) that showed different physicochemical properties from GO-S and GO-L in terms of lateral size, thickness, and surface functional groups (Fig. S14, Tab. 3, and Tab. S1). These GOs induced similar response profiles in all cell types, and KUP5 cells were most sensitive to the cytotoxic effects of GOs compared to LSECs and Hepa 1–6 cells (Fig. S15). The confocal images in Fig. S16–17 confirmed the phagocytosis of GO-1, GO-2, and GO-3 by KUP5 cells, based on the reduction of intracellular GO after WM treatment. Similarly, these GOs also triggered plasma membrane lipid peroxidation in KUP5 cells (Fig. 9a), inhibiting NADPH oxidase activity or phagocytosis of GO-1, GO-2, and GO-3 significantly reduced the lipid peroxidation (Fig. S18). As shown in Fig. 9a, GO-induced lipid peroxidation also increased PLC activity, which triggered calcium accumulation, mtROS generation (Fig. S19a), caspase-1 activation (Fig. S19b) as well as IL-1β release and subsequent cell swelling and surface blebbing (Fig. 9b–c). Similarly, the siRNA knockdown of GSDMD, inhibition of GO phagocytosis, NADPH oxidase activity, and PLC activity protected the cells against pyroptosis in KUP5 cells (Fig. 9b–c and Fig. S20). These data suggest the pyroptosis in KUP5 cells is a universal feature for different GO preparations. However, the cytotoxic effects of GOs could not be directly compared due to differences in properties. The contribution from a specific property (e.g., lateral size in this case) can only be established when other properties are largely similar.
Tab. 3.
Physicochemical properties of additional GOs
| GO samples | GO-1 | GO-2 | GO-3 | |
|---|---|---|---|---|
| AFM | Lateral size (nm) | 140 ± 90 | 69 ± 58 | 1108 ± 1047 |
| Height (nm) | 2.5 ± 1.7 | 0.7 ± 0.2 | 1.1 ± 0.3 | |
| At.% from XPS | C-C/C=C | 46.1 ± 5.1 | 41.6 ± 3.7 | 30.3 ± 5.1 |
| C-OH | 15.5 ± 5.6 | 17.8 ± 1.6 | 9.7 ± 0.5 | |
| C-O | 24.9 ± 2.6 | 24.2 ± 5.0 | 24.6 ± 1.1 | |
| C=O | 7.0 ± 2.2 | 16.4 ± 0.3 | 30.5 ± 4.8 | |
| O=C-OH | 3.8 ± 1.6 | - | 4.9 ± 1.9 | |
| π → π* | 2.7 ± 0.7 | - | - |
Fig. 9.
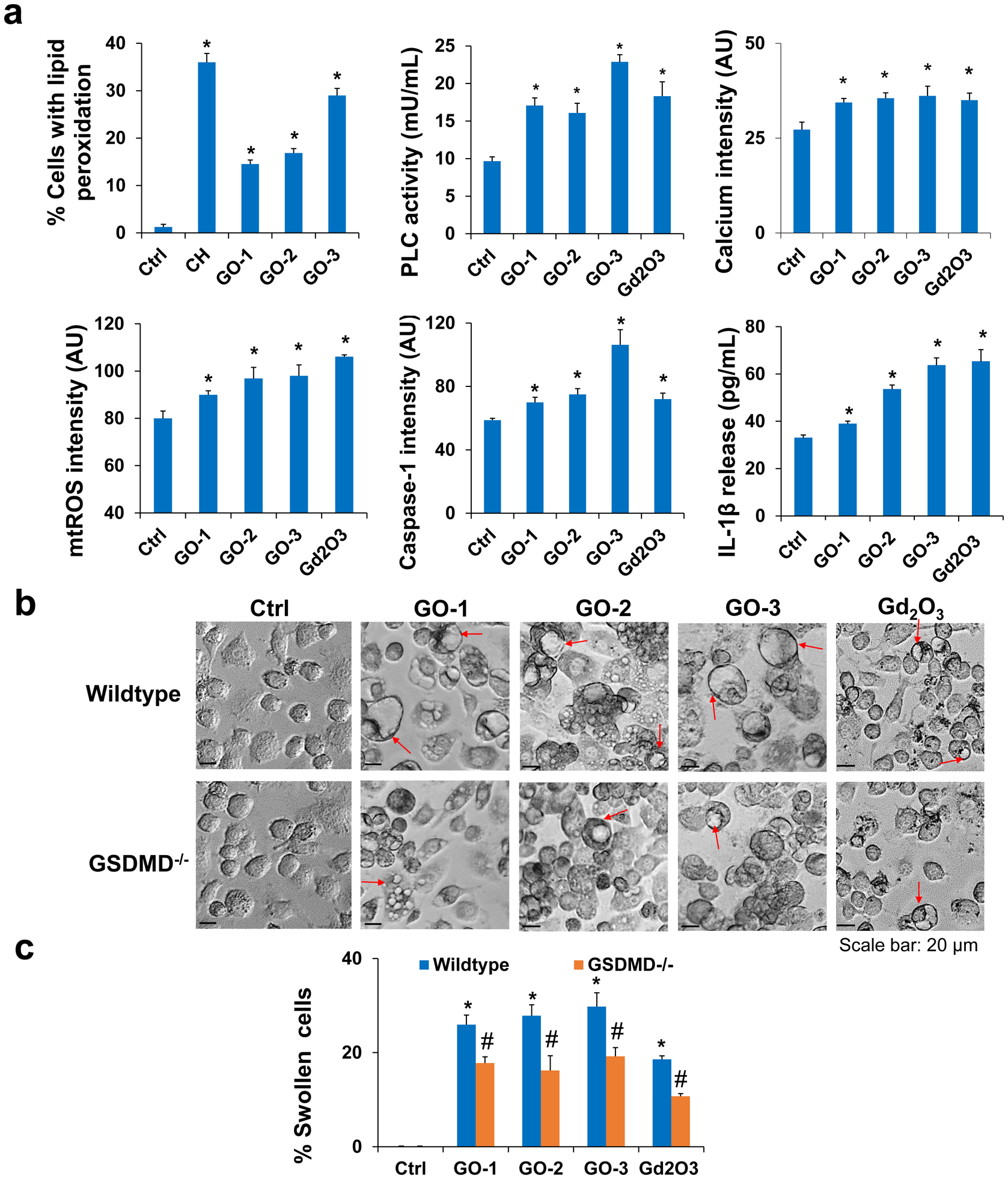
Additional GOs with different properties induced pyroptosis in KUP5 cells. (a) GO samples induced GSDMD-mediated pyroptosis in KUP5 cells including lipid peroxidation, PLC activation, calcium flux, mtROS generation, caspase-1 activation, and IL-1β release. (b) Optical microscope images to compare the cell morphological changes induced by 12.5 μg/mL of GOs in wild-type or GSDMD−/− KUP5 cells. Red arrows indicate the swollen cells. The scale bar is 20 μm. (c) The percentage of swollen cells was calculated in wild-type or GSDMD−/− KUP5 cells. *, P < 0.05, indicates significance compared with the control. #, P < 0.05, indicates significance between wild-type and GSDMD−/− KUP5 cells.
Pyroptosis signal pathway in KUP5 cells as a function GO lateral size
GO’s physicochemical properties were found to highly link with cellular responses to GO. To better understand the role of size in GO-induced pyroptosis, we performed correlation studies by looking at with cellular responses in KUP5 cells, e.g., cellular association, lipid peroxidation, IL-1β release, as a function GO physicochemical properties, including lateral size, hydrodynamic size, C-O, C=O, C-C/C=C, and C-OH groups. The results, represented by the heat map in Fig. 10a, showed a higher positive coefficient of determination (R2) between all cellular responses in the pyroptosis signal pathway and GO lateral and hydrodynamic sizes. In comparison to GO sizes, GO surface C=O groups were the only functional group contributed partly to the pyroptosis signal pathway, potentially due to chemical reactions. The correlation plots in Fig. 10b showed that GO hydrodynamic size, an important parameter in this study, was highly correlated with GO lateral size (R2=0.84) and also showed higher R2 with all cellular responses. Furthermore, GO lateral size showed excellent correlation coefficients with the pyroptosis signal pathway in KUP5 cells (Fig. S21). Taken together, GO lateral size played a vital role in the pyroptosis of KUP5 cells, no matter other properties can be largely similar or different.
Fig. 10.
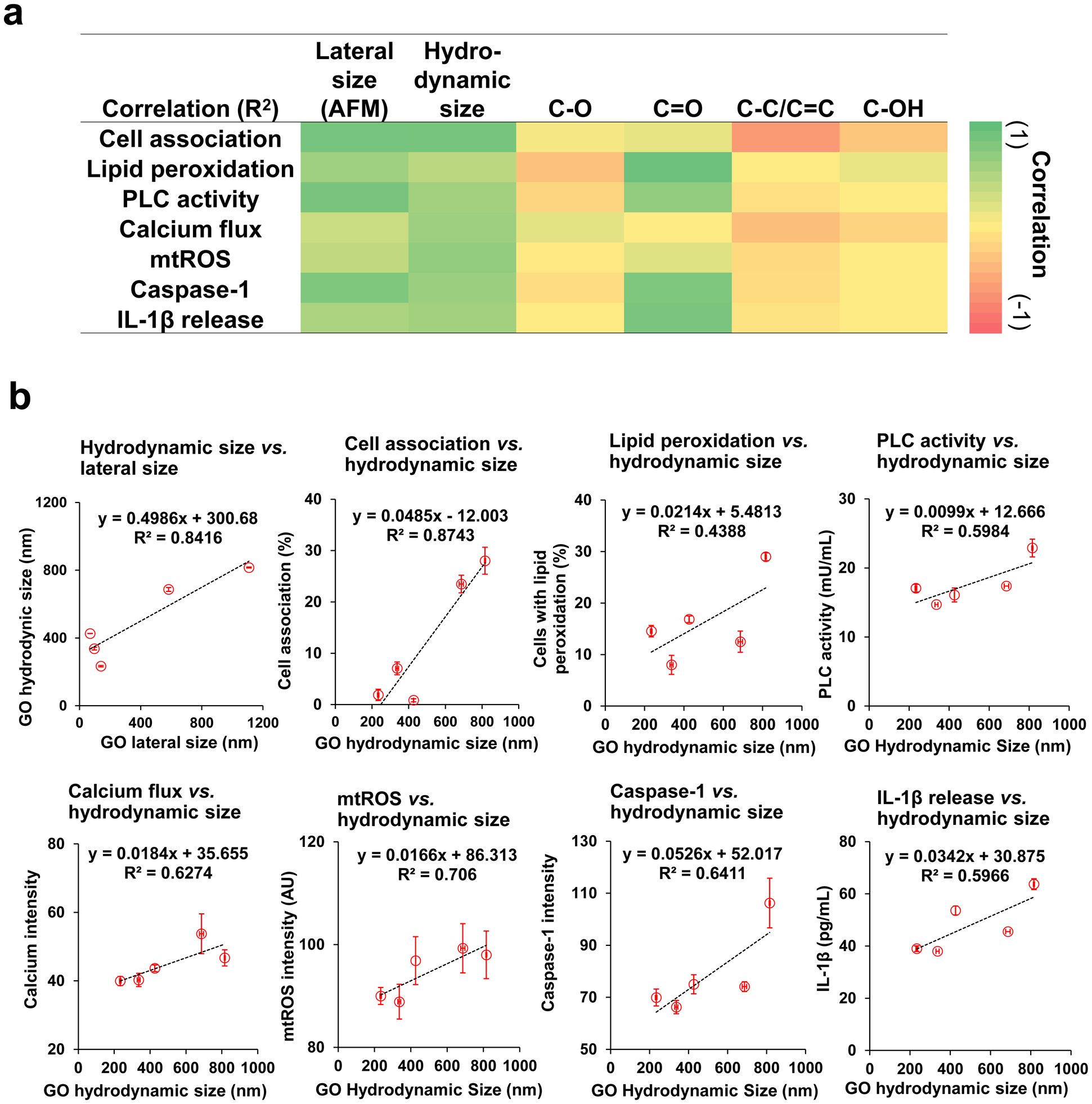
Determination correlations of GO physiochemical properties with the pyroptosis signal pathways in KUP5 cells. (a) Heat map to demonstrate the correlations of GO lateral size, hydrodynamic size, C-O, C=O, C-C/C=C, and C-OH groups, with cellular responses in KUP5 cells, including GO cell association, lipid peroxidation, PLC activation, calcium flux, mtROS generation, caspase-1 activation, and IL-1β release. (b) The correlation plots of GO lateral size against GO hydrodynamic size and GO hydrodynamic size against the cellular responses.
Additional signaling pathways and cell death mechanisms induced by GOs
It has been reported that GOs induce TLR-4 dependent programmed necrosis, possibly necroptosis, in macrophages, which involves the activation of TLR-4 and TNF-α production [27]. We compared the ability of GOs to induce TNF-α production in KUP5 cells. We found all GO samples including GO-S, GO-L, and GO-1, −2, −3 induced TNF-α production (Fig. S22a), consistent with previous reports [27]. In addition, we tested the GO-induced IL-1β release and cell swelling in the presence of CLI-095 (TLR-4 inhibitor), NEC-1 (necroptosis inhibitor). Both parameters were significantly reduced after the TLR4 inhibitor but not by NEC-1(Fig. S22b–d). This indicated the GO-induced programmed necrosis may not involve necroptosis. Furthermore, we know Gd2O3 could induce pyroptosis via lysosomal damage, cathepsin B release, which results in NLRP3 inflammasome activation [28]. To probe differences between Gd2O3 and GO-induced pyroptosis, we compared the IL-1β release and cell swelling in the presence of a cathepsin B inhibitor, CA-074-Me, and found that it did not reduce the IL-1β and cell swelling while it inhibited Gd2O3-induced effects (Fig. S22b–d). These results indicate that cathepsin B release-mediated NLRP3 inflammasome activation may not play a vital role in KUP5 cells after GO exposure.
Discussion
In this study, we determined the effects of GOs with two lateral sizes and similar physicochemical properties, GO-S and GO-L, on three major liver cell types, KCs, LSECs, and hepatocytes. We demonstrated the differential effects of GO on the cellular uptake, signaling pathway activation, and cytotoxicity in KUP5, LSEC, and Hepa 1–6 cells. We found GO induced lateral size-dependent toxicity to Kupffer cells but minimal toxicity to LSECs and hepatocytes. For Kupffer cells, GOs were taken up by phagocytosis, with higher GO-L uptake than GO-S. The phagocytosis induced plasma membrane lipid peroxidation mediated by NADPH oxidase, leading to PLC activation, calcium flux, and mtROS generation, which triggers NLRP3 inflammasome and caspase-1 activation, resulting in IL-1β release as well as GSDMD-mediated pyroptosis. GOs were less associated with LSECs and Hepa 1–6 cells and GOs failed to induce significant lipid peroxidation or cytotoxicity to LSECs and Hepa 1–6 cells. GO effects to these liver cells and pyroptotic cell death to KUP5 cells were further confirmed using additional GO samples (GO-1, GO-2, and GO-3), suggesting it is a universal feature.
An important finding of the current study is the delineation of the lateral size-dependent cellular uptake and toxicity of GOs in KCs, LSECs, and hepatocytes. Although previous work on GOs is available to show that differently sized GO induced differential cytotoxicity or inflammation responses to the lung or parenchymal hepatocytes in a mouse model [19,26,60], the interaction between different sizes of GO in major liver cell types has not been performed. The role of lateral size can be established because GO-S and GO-L had otherwise similar physicochemical properties, and those properties have profound impacts on GO-cell interactions. We found GOs induced significant toxicity to KUP5 cells, while the toxicity to LSEC and Hepa 1–6 was significantly lower. Furthermore, GO-L was significantly more toxic to KUP5 cells than GO-S. The reason for the differential toxicity and size-dependence is likely due to the quantity of cellular association or uptake by different cell types. Both GOs showed significantly higher cellular association or uptake to KUP5 cells than LSECs and Hepa 1–6 cells. In addition, we found GO-L showed greater cell association with the plasma membrane compared to GO-S in liver cells, which is similar to previous studies in lung cells [19,60]. This size-dependence may be attributed to the contact area or interactions between the flat 2D GO nanosheets and the cell membrane, the larger the contact area, the stronger the binding force and the higher cellular uptake in macrophages by phagocytosis [19,61–63]. For LSEC and Hepa 1–6 cells, although GO-S showed significant cellular uptake, GO-L was mostly on the cell surface, suggesting a different cellular uptake mechanism. Studies have shown that FITC-PEG-GOs can be readily taken up through clathrin-mediated endocytosis in HepG2 hepatocytes [38]. Similarly, LSECs with high clathrin-coated pits per membrane unit are more proficient in eliminating soluble macromolecules and smaller particles (in the 200 nm size range) by clathrin-mediated endocytosis [30,35]. Because clathrin-mediated endocytosis has a size limit at 200 nm, GO-L could not be taken up into the cells and they mostly adsorb on the membranes (Fig. 2a). Our result was supported by earlier work reports where it was found Kupffer cells actively swallowed the majority of GOs and only a small amount of GOs was found inside hepatocytes and the large-sized GO was located at the edges of hepatocytes in vivo [26]. Thus, it is reasonable to understand that GOs had more cell association and significant lipid peroxidation and toxicity in KUP5 cells than those in LSECs and Hepa 1–6 cells due to the phagocytosis of GOs by KCs. The differences in cellular uptake mechanisms have a profound impact on GO cytotoxicity.
We found that phagocytosis of GOs induced significant plasma membrane lipid peroxidation in KUP5 cells but not LSECs or Hepa 1–6 cells. This result can be attributed to the phagocytosis of GOs triggering NADPH oxidase, which generates copious superoxide that induces lipid peroxidation, while LSEC and hepatocyte did not. Although GOs have been shown to display carbon radicals on their surface that are capable of inducing lipid peroxidation when they interact, our current study showed that GOs by themselves could not induce lipid peroxidation (e.g., LSEC and Hepa 1–6) despite the presence of intrinsic oxidative stress generation and GO-membrane interactions. Furthermore, the higher lipid peroxidation produced by GO-L in KUP5 cells was consistent with the higher uptake of GO-L despite GO-L showed less abiotic ROS generation than GO-S (Fig. S5). GO-induced lipid peroxidation required phagocytosis and NADPH oxidase activation, which have been confirmed by phagocytosis and NOX2 inhibitors as well as NOX2 p22phox and p47phox subunit siRNA knockdown.
Another significant finding of this work is the identification and elucidation of the detailed mechanism of GO-induced programmed necrosis, namely, GSDMD-mediated pyroptosis in KCs. Although reports have shown that GO can elicit caspase-1 dependent IL-1β production, and the IL-1 β production requires NADPH oxidase-generated reactive oxygen species and cellular uptake of GO [64], there are major gaps in the chain of evidence. Here, we demonstrated that all GOs could trigger GSDMD-mediated pyroptosis based on the following events: i) GO-induced NOX2 dependent lipid peroxidation in response to phagocytosis induces the activation of PLC in KUP5 cells, which cleaves PIP2 into DAG and IP3; 2) IP3, in turn, results in the mobilization of intracellular calcium stores, which induces mtROS generation when mitochondria take up excessive or sustained calcium; iii) Calcium flux and mtROS generation activate NLRP3 inflammasome, as reflected in the activation of caspase-1 and IL-1β release; iv) Caspase-1 is also responsible for cleavage of GSDMD, resulting in the release of N-terminal peptide fragments that integrated into the cell surface membrane, where peptide oligomerization leads to pore formation, cellular swelling, and giant cell blebs formation (Fig. 8c, S2 and Video 1). The hallmarks of pyroptosis including IL-1β release and cell swelling could be significantly inhibited by GSDMD siRNA knockdown for all GO samples, suggesting that pyroptosis is the cell death mechanism of KCs treated with GOs. This result is consistent with a recent study showing that in lethal polymicrobial sepsis animal model, lipid peroxidation drives the activation of PLCG1 in BMDMs, which leads to calcium flux and GSDMD-mediated pyroptosis. Inhibition of PLC activation by U-73122 offered protection against multi-organ failure and animal lethality in sepsis animal models [43]. Furthermore, we found the pyroptosis signal pathway in KUP5 cells was as a function GO lateral size (Fig. 10), even though many significant differences were present on surface function groups among GO-1, −2, −3. The pathway may be partly affected by GO surface functional groups, such as C=O groups. However, the GO samples in this study had various surface functional groups, compositions, and lateral size, and could not form the basis for comparisons on the effect of C=O groups. Further studies to detail the carbonyl group effects of GO with comparable physicochemical properties to cells are needed.
It is worth noting that GOs could engage other cell death mechanisms depending on the cell types and GO properties. For example, nano-sized GOs have been shown to induce apoptotic cell death in endothelial cells through activating c-Jun N-terminal kinase (JNK) pathways, autophagy, and elevated intracellular calcium levels, but not for micron-sized GO [34]. GOs have also been shown to induce apoptosis in HepG2 hepatocytes, A549 lung epithelial cells, T lymphocytes, U251 glioma cells, and A2780 ovarian cancer cells [47]. In addition, GOs are known to induce TLR-4 dependent necrosis in macrophages including J774A.1 and RAW 264.7 cells, in which GO induced activation of TLR-4 and subsequent autocrine TNF-α production, triggering the ligation of TNF receptor (TNFR) and formation of RIP1-RIP3 complex, namely the necrosome [27]. This process leads to necroptosis, another type of programmed necrosis, which can be inhibited by RIP inhibitor NEC-1. However, NEC-1 could not fully inhibit the necroptosis [27], suggesting that additional necrotic pathways independent of RIP1/RIP3 may be involved. We tested the possibility of inducing necroptosis in GO-treated KUP5 cells. We confirmed that GOs could activate TLR-4 pathways, inducing TNF-α production, and CLI-095, the TLR-4 inhibitor, being present in a significantly reduced percentage of swollen cells (Fig. S22). CLI-095 also reduced GO-induced IL-1β production, which is also reasonable because TLR-4 activation by LPS serves signal 1 for NLRP3 inflammasome activation involving the expression of pro-IL-1β and NLRP3 proteins. However, NEC-1 did not significantly inhibit the percentage of swollen cells as well as the IL-1β production, suggesting that for KUP5 cells, pyroptosis rather than necroptosis was involved. Further studies are needed to clarify the involvement of other programmed necrosis pathways. Interestingly, our results demonstrated the similarities and differences between GOs and Gd2O3, which served as the positive control of pyroptosis [28,29]. Gd2O3 has been reported previously to be taken up into lysosomes by KUP5 cells, which induced lysosomal damage and cathepsin B release, leading to NLRP3 inflammasome activation and GSDMD-mediated pyroptosis. Consistent with our previous report, a cathepsin B inhibitor, CA-074-Me [28], significantly inhibited Gd2O3 induced IL-1β production and reduced the number of swollen cells (Fig. S22), confirming that lysosomal damage plays a major role in Gd2O3 induced pyroptosis. However, GO-L induced IL-1β production and cell swelling could not be inhibited by CA-074-Me, highlighting the different signaling pathways engaged by GOs and Gd2O3, although Gd2O3 could also induce PLC activation, calcium flux, and mtROS generation. Currently, there is a lack of direct evidence of GO-induced pyroptosis in the liver in vivo. However, a recent report showed that there was more GO uptake by KCs than hepatocytes in mouse liver sections. In addition, GOs induced IL-1β production in primary KCs separated from the BALB/c mice. Furthermore, IL-1β was detected in liver homogenates of GO treated mice. Large GO (500 to 2000 nm) induced higher IL-1β production by primary KCs and in the liver homogenates than small GO (50 to 200 nm) [26], consistent with our results. Further studies to demonstrate GO induced pyroptosis in KCs in vivo are needed.
Conclusions
In this study, we showed that GOs induced lateral size-dependent toxicity to Kupffer cells but significantly lower toxicity to LSECs and hepatocytes. For KCs, GOs underwent cellular uptake through phagocytosis, with higher GO-L uptake than GO-S. Phagocytosis of GOs triggered NADPH oxidase-mediated plasma membrane lipid peroxidation, leading to PLC activation, calcium influx, and mtROS generation, which triggered NLRP3 inflammasome and caspase-1 activation, IL-1β release and GSDMD-mediated pyroptosis. For LSECs and hepatocytes, GOs showed reduced cell association compared to KCs, and GO-S was largely taken up while GO-L mostly adsorbed on the cell membrane. GOs did not induce significant lipid peroxidation or cytotoxicity in both cell lines. Using three additional GO samples with different properties, we confirmed that the differential cytotoxic responses and toxicity mechanism were similar to GO-S and GO-L, suggesting that it is a universal feature of GO. Among the GO properties including lateral sizes and surface functionalities, lateral size correlated well with the cellular response in KUP5 cells, indicating the key role of lateral size in pyroptosis. Overall this study provides a detailed mechanistic understanding of GO-induced toxicity in liver cells and establishes the role of lateral size in GO induced liver toxicity.
Supplementary Material
Highlights:
Graphene oxide induces differential toxicity in Kupffer cells, LSECs, and hepatocytes.
GOs induce lipid peroxidation that is dependent on phagocytosis and NADPH oxidase activation in Kupffer cells.
GO-induced lipid peroxidation triggers PLC, calcium flux, mtROS, caspase-1 activation, and pyroptosis.
GOs with different properties also trigger pyroptosis, suggesting it is a universal feature.
Lateral size plays a role in GO-induced pyroptosis and large GO shows stronger effects.
Acknowledgments
The research reported in this publication was supported by the Nanotechnology Health Implications Research (NHIR) Consortium of the National Institute of Environmental Health Sciences of the National Institutes of Health under Award Number (U01ES027237). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The engineered nanomaterials used in the research presented in this publication were procured/developed, characterized, and provided by the Engineered Nanomaterials Resource and Coordination Core established at Harvard T. H. Chan School of Public Health (NIH grant # U24ES026946) as part of the Nanotechnology Health Implications Research Consortium. The authors thank the CNSI Advanced Light Microscopy/Spectroscopy and Electron Imaging Center for NanoMachines Core Facilities, the Flow Cytometry Core Facility of Jonsson Comprehensive Cancer Center, the Microscopic Techniques and Electron Microscope Core Facility of Brain Research Institute, and the Integrated Molecular Technologies Core (CURE/P30 DK041301) at UCLA. This work made use of the Keck-II facility of Northwestern University’s NUANCE Center, which has received support from the SHyNE Resource (NSF ECCS-1542205), the IIN, and the Northwestern University MRSEC program (NSF DMR-1720139).
Footnotes
Declaration of competing interest
The authors declare no conflict of interest.
Appendix A. Supplementary data
Supplementary material related to this article can be found, in the online version, at DOI: https://doi.org/10.1016/j.nantod.2020.101061
References
- [1].Lan G, Quan Y, Wang M, Nash GT, You E, Song Y, Veroneau SS, Jiang X, Lin W, J. Am. Chem. Soc, 141 (2019) 15767–15772. 10.1021/jacs.9b08956 [DOI] [PubMed] [Google Scholar]
- [2].Yong Wang, Jun Mao, Xianguang Meng, Liang Yu, Dehui Deng, Chem. Rev, 119 (2018) 1806–1854. 10.1021/acs.chemrev.8b00501 [DOI] [PubMed] [Google Scholar]
- [3].Qiu L, He Z, Li D, Adv. Mater, 30 (2018) 1704850, 10.1002/adma.201704850. [DOI] [PubMed] [Google Scholar]
- [4].Georgakilas V, Tiwari JN, Kemp KC, Perman JA, Bourlinos AB, Kim KS, Zboril R, Chem. Rev, 116 (2016) 5464–5519. 10.1021/acs.chemrev.5b00620 [DOI] [PubMed] [Google Scholar]
- [5].Geim AK, Science, 324 (2009) 1530–1534. 10.1126/science.1158877 [DOI] [PubMed] [Google Scholar]
- [6].Bottari G, Herranz MÁ, Wibmer L, Volland M, Rodríguez–Pérez L, Guldi DM, Hirsch A, Martín N, D’Souza F, Torres T, Chem. Soc. Rev, 46 (2017) 4464–4500. 10.1039/c7cs00229g [DOI] [PubMed] [Google Scholar]
- [7].Sun M, Li J, Nano Today, 20 (2018) 121–137. 10.1016/j.nantod.2018.04.007 [DOI] [Google Scholar]
- [8].Nie C, Ma L, Li S, Fan X, Yang Y, Cheng C, Zhao W, Zhao C, Nano Today, 26 (2019) 57–97. 10.1016/j.nantod.2019.03.003 [DOI] [Google Scholar]
- [9].Lakshmanan R, Maulik N, Can. J. Physiol. Pharmacol, 96 (2018) 869–878. 10.1139/cjpp-2018-02252018-0225 [DOI] [PubMed] [Google Scholar]
- [10].Akhavan O, Ghaderi E, Shirazian SA, Rahighi R, Carbon, 97 (2016) 71–77. 10.1016/j.carbon.2015.06.079 [DOI] [Google Scholar]
- [11].Li R, Mansukhani ND, Guiney LM, Ji Z, Zhao Y, Chang CH, French CT, Miller JF, Hersam MC, Nel AE, Xia T, ACS Nano, 10 (2016) 10966–10980. 10.1021/acsnano.6b05692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zheng XT, Ananthanarayanan A, Luo KQ, Chen P, Small, 11 (2015) 1620–1636. 10.1002/smll.201402648 [DOI] [PubMed] [Google Scholar]
- [13].Palmieri V, Papi M, Nano Today, 33 (2020) 100883, 10.1016/j.nantod.2020.100883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Liu J, Dong J, Zhang T, Peng Q, Control J. Release, 286 (2018) 64–73. 10.1016/j.jconrel.2018.07.034 [DOI] [PubMed] [Google Scholar]
- [15].Reina G, González-Domínguez JM, Criado A, Vázquez E, Prato M, Chem. Soc. Rev, 46 (2017) 4400–4416. 10.1039/C7CS00363C [DOI] [PubMed] [Google Scholar]
- [16].Zhang Yi-Nan, Poon Wilson, Tavares Anthony, J., McGilvray Ian, D., J. Control. Release, 240 (2016) 332–348. 10.1016/j.jconrel.2016.01.020 [DOI] [PubMed] [Google Scholar]
- [17].Wen K, Chen Y, Chuang C, Chang H, Lee C, Tai N, J. Appl. Toxicol, 35 (2015) 1211–1218. 10.1002/jat.3187 [DOI] [PubMed] [Google Scholar]
- [18].Wu Y, Feng W, Liu R, Xia T, Liu S, ACS Nano, 14 (2020) 877–890. 10.1021/acsnano.9b08127 [DOI] [PubMed] [Google Scholar]
- [19].Ma J, Liu R, Wang X, Liu Q, Chen Y, Valle RP, Zuo YY, Xia T, Liu S, ACS Nano, 9 (2015) 10498–10515. 10.1021/acsnano.5b04751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mu Q, Su G, Li L, Gilbertson BO, Yu LH, Zhang Q, Sun Y, Yan B, ACS Appl. Mater. Inter, 4 (2012) 2259–2266. 10.1021/am300253c [DOI] [PubMed] [Google Scholar]
- [21].Dixon LJ, Barnes M, Tang H, Pritchard MT, Nagy LE, Compr. Physiol, 3 (2013) 785–797. 10.1002/cphy.c120026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].David A, Hume, Curr. Opin. Immunol, 18 (2006) 49–53. 10.1016/j.coi.2005.11.008 [DOI] [PubMed] [Google Scholar]
- [23].Boey A, Ho HK, Small, 16 (2020) e2000153, 10.1002/smll.202000153. [DOI] [PubMed] [Google Scholar]
- [24].Tsoi KM, MacParland SA, Ma X, Spetzler VN, Echeverri J, Ouyang B, Fadel SM, Sykes EA, Goldaracena N, Kaths JM, Conneely JB, Alman BA, Selzner M, Ostrowski MA, Adeyi OA, Zilman A, McGilvray ID, Chan WCW, Nat. Mater, 15 (2016) 1212–1221. 10.1038/nmat4718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Li R, Guiney LM, Chang CH, Mansukhani ND, Ji Z, Wang X, Liao Y, Jiang W, Sun B, Hersam MC, Nel AE, Xia T, ACS Nano, 12 (2018) 1390–1402. 10.1021/acsnano.7b07737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhang Y, Ma C, Wang Z, Zhou Q, Sun S, Ma P, Lv L, Jiang X, Wang X, Zhan L, Nanoscale, 12 (2020) 8147–8158. 10.1039/C9NR10713D [DOI] [PubMed] [Google Scholar]
- [27].Qu G, Liu S, Zhang S, Wang L, Wang X, Sun B, Yin N, Gao X, Xia T, Chen JJ, Jiang GB, ACS Nano, 7 (2013) 5732–5745. 10.1021/nn402330b [DOI] [PubMed] [Google Scholar]
- [28].Mirshafiee V, Sun B, Chang CH, Liao Y, Jiang W, Jiang J, Liu X, Wang X, Xia T, Nel AE, ACS Nano, 12 (2018) 3836–3852. 10.1021/acsnano.8b01086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang X, Chang CH, Jiang J, Liu X, Li J, Liu Q, Liao YP, Li L, Nel AE, Xia T, Small,16 (2020) e2000528, 10.1002/smll.202000528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sørensen KK, Simonsantamaria J, Mccuskey RS, Smedsrød B, Compr. Physiol, 5 (2015) 1751–1774. 10.1002/cphy.c140078 [DOI] [PubMed] [Google Scholar]
- [31].Kjeken R, Mousavi SA, Brech A, Gj En T, Berg T, Cell Tissue Res, 304 (2001) 221–230. 10.1007/s004410100348 [DOI] [PubMed] [Google Scholar]
- [32].Gissen P, Arias IM, J. Hepatol, 63 (2015) 1023–1037. 10.1016/j.jhep.2015.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Poisson J, Lemoinne S, Boulanger C, Durand FO, Moreau R, Valla D, Rautou PE, J. Hepatol, 66 (2016) 212–227. 10.1016/j.jhep.2016.07.009 [DOI] [PubMed] [Google Scholar]
- [34].Lim M, Jeung IC, Jeong J, Yoon S, Lee S, Park J, Kang Y, Lee H, Park Y, Lee HG, Lee S, Han BS, Song NW, Lee SC, Kim J, Bae K, Min J, Acta Biomater, 46 (2016) 191–203. 10.1016/j.actbio.2016.09.018 [DOI] [PubMed] [Google Scholar]
- [35].Liu Q, Wang X, Liu X, Kumar S, Gochman G, Ji Y, Liao Y, Chang CH, Situ W, Lu J, Jiang J, Mei K, Meng H, Xia T, Nel AE, ACS Nano, 13 (2019) 4778–4794. 10.1021/acsnano.9b01444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Chatterjee N, Eom HJ, Choi J, Biomaterials, 35 (2014) 1109–1127. 10.1016/j.biomaterials.2013.09.108 [DOI] [PubMed] [Google Scholar]
- [37].Duch MC, Budinger GRS, Liang YT, Soberanes S, Urich D, Chiarella SE, Campochiaro LA, Gonzalez A, Chandel NS, Hersam MC, Nano Lett, 11 (2011) 5201–5207. 10.1021/nl202515a [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Linares J, Matesanz MC, Vila M, Feito MJ, Gonçalves G, Vallet-Regí M, Marques PAAP, Portolés MT, ACS Appl. Mater. Inter, 6 (2014) 13697–13706. 10.1021/am5031598 [DOI] [PubMed] [Google Scholar]
- [39].von Kleist L, Stahlschmidt W, Bulut H, Gromova K, Puchkov D, Robertson MJ, MacGregor KA, Tomilin N, Pechstein A, Chau N, Chircop M, Sakoff J, von Kries JP, Saenger W, Krausslich HG, Shupliakov O, Robinson PJ, McCluskey A, Haucke V, Cell, 146 (2011) 471–484. 10.1016/j.cell.2011.06.025 [DOI] [PubMed] [Google Scholar]
- [40].Sun B, Wang X, Ji Z, Wang M, Xia T, Small, 11 (2015) 2087–2097. 10.1002/smll.201402859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Osaki T, Uchida Y, Hirayama J, Nishina H, Biol. Pharm. Bull, 34 (2011) 1343–1347. 10.1248/bpb.34.1343 [DOI] [PubMed] [Google Scholar]
- [42].Domijan AM, Kovac S, Abramov AY, J. Cell Sci, 127 (2013) 21–26. 10.1242/jcs.138370 [DOI] [PubMed] [Google Scholar]
- [43].Kang R, Zeng L, Zhu S, Xie Y, Liu J, Wen Q, Cao L, Xie M, Ran Q, Kroemer G, Wang H, Billiar TR, Jiang J, Tang D, Cell Host Microbe, 24 (2018) 97–108. 10.1016/j.chom.2018.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hu W, Peng C, Lv M, Li X, Zhang Y, Chen N, Fan C, Huang Q, ACS Nano, 5 (2011) 3693–3700. 10.1021/nn200021j [DOI] [PubMed] [Google Scholar]
- [45].Duan G, Kang SG, Tian X, Garate JA, Zhao L, Ge C, Zhou R, Nanoscale, 7 (2015) 15214–15224. 10.1039/c5nr01839k [DOI] [PubMed] [Google Scholar]
- [46].Frank D, Vince JE, Cell Death Differ, 26 (2019) 99–114. 10.1038/s41418-018-0212-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ou L, Lin S, Song B, Liu J, Lai R, Shao L, Int. J. Nanomed, 12 (2017) 6633–6646. 10.2147/IJN.S140526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zhang B, Wei P, Zhou Z, Wei T, Adv. Drug Deliver. Rev, 105 (2016) 145–162. 10.1016/j.addr.2016.08.009 [DOI] [PubMed] [Google Scholar]
- [49].Liang S, Kisseleva T, Brenner DA, Front. Physiol, 7 (2016), 10.3389/fphys.2016.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].De Minicis S, Bataller R, Brenner DA, Gastroenterology, 131 (2006) 272–275. 10.1053/j.gastro.2006.05.048 [DOI] [PubMed] [Google Scholar]
- [51].Russo AJ, Rathinam VAK, Cell Host Microbe, 24 (2018) 8–9. 10.1016/j.chom.2018.06.010 [DOI] [PubMed] [Google Scholar]
- [52].Rimessi A, Bezzerri V, Patergnani S, Marchi S, Cabrini G, Pinton P, Nat. Commun, 6 (2015) 6201, 10.1038/ncomms7201. [DOI] [PubMed] [Google Scholar]
- [53].Hoth M, Fanger CM, Lewis RS, J. Cell Biol, 137 (1997) 633–648. 10.1083/jcb.137.3.633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, Horng T, Proc. Natl. Acad. Sci. USA, 109 (2012) 11282–11287. 10.1073/pnas.1117765109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Horng T, Trends Immunol, 35 (2014) 253–261. 10.1016/j.it.2014.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, Wong J, Ding S, Seki E, Schnabl B, Nature, 560 (2018) 198–203. 10.1038/s41586-018-0372-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Bergsbaken T, Fink SL, Cookson BT, Nat. Rev. Microbiol, 7 (2009) 99–109. 10.1038/nrmicro2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F, Nature, 526 (2015) 660–665. 10.1038/nature15514 [DOI] [PubMed] [Google Scholar]
- [59].Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, Lieberman J, Nature, 535 (2016) 153–158. 10.1038/nature18629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Vranic S, Rodrigues AF, Buggio M, Newman L, White MRH, Spiller DG, Bussy C, Kostarelos K, ACS Nano, 12 (2017) 1373–1389. 10.1021/acsnano.7b07734 [DOI] [PubMed] [Google Scholar]
- [61].Li Y, Yuan H, von Dem Bussche A, Creighton M, Hurt RH, Kane AB, Gao H, Proc. Natl. Acad. Sci. USA, 110 (2013) 12295–12300. 10.1073/pnas.1222276110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Champion JA, Mitragotri S, Proc. Natl. Acad. Sci. USA, 103 (2006) 4930–4934. 10.1073/pnas.0600997103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Nel A, Xia T, Madler L, Li N, Science, 311 (2006) 622–627. 10.1126/science.1114397 [DOI] [PubMed] [Google Scholar]
- [64].Mukherjee SP, Kostarelos K, Fadeel B, Adv. Healthc. Mater, 7 (2018) 1700815, 10.1002/adhm.201700815. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.