

Iron-sulfur clusters enable cells to sense that they have the right amount of iron, and protect them from self-destruction.
Abstract
Intracellular iron levels are strictly regulated to support homeostasis and avoid iron-mediated ROS production. Loss of iron-sulfur cluster (ISC) synthesis can increase iron loading and promote cell death by ferroptosis. Iron-responsive element-binding proteins IRP1 and IRP2 posttranscriptionally regulate iron homeostasis. IRP1 binding to target mRNAs is competitively regulated by ISC occupancy. However, IRP2 is principally thought to be regulated at the protein level via E3 ubiquitin ligase FBXL5–mediated degradation. Here, we show that ISC synthesis suppression can activate IRP2 and promote ferroptosis sensitivity via a previously unidentified mechanism. At tissue-level O2 concentrations, ISC deficiency enhances IRP2 binding to target mRNAs independent of IRP1, FBXL5, and changes in IRP2 protein level. Deletion of both IRP1 and IRP2 abolishes the iron-starvation response, preventing its activation by ISC synthesis inhibition. These findings will inform strategies to manipulate ferroptosis sensitivity and help illuminate the mechanism underlying ISC biosynthesis disorders, such as Friedreich’s ataxia.
INTRODUCTION
The available cellular iron pool is strictly regulated to allow utilization of iron as an ion cofactor or in heme and iron-sulfur cluster (ISC) groups while avoiding iron-mediated reactive oxygen species (ROS) production (1). Excessive iron accumulation enhances oxidation of various macromolecules and can lead to cell death by mechanisms that include ferroptosis, a recently described iron-dependent form of cell death characterized by membrane lipid peroxidation (2, 3). Consequently, cellular iron regulation has a central role in modulating the sensitivity to ferroptosis, either facilitating it via iron accumulation or hindering it via iron sequestration (4).
The main regulators of the cellular iron pool, iron-responsive element (IRE)–binding proteins 1 and 2 (IRP1 and IRP2), are normally activated when cellular iron levels are low, initiating a rheostat-like iron-starvation response (5). When activated, the iron-starvation response increases the available iron pool by modulating iron uptake, storage, export, and use. Specifically, IRP1 is regulated through ISC occupancy and can function as an iron-responsive protein upon losing its ISC (6). Perturbations in ISC metabolism are thought to aberrantly activate the iron-starvation response via IRP1, overloading cellular iron pools and sensitizing cells to ferroptosis (7–9). Disorders of ISC biosynthesis, such as those observed in Friedreich’s ataxia and sideroblastic anemias, exhibit altered iron metabolism as part of their pathology (10–13). Therefore, a mechanistic investigation of how the iron-starvation response is regulated by ISC metabolism is important for understanding the cause of these human diseases and developing rational therapeutic strategies.
In prior work, we found that inhibition of ISC synthesis via suppression of the key biosynthetic protein NFS1 cysteine desulfurase (NFS1) sensitizes cancer cells to ferroptosis, a feature that may be responsible for positive selection of the NFS1 locus during lung tumorigenesis (9). We observed activation of the iron-starvation response upon inhibition of ISC synthesis and an increase in markers of lipid peroxidation and cell death upon treatment with ferroptosis-inducing drugs or compounds that induce oxidative stress (9). Cancer cells often exhibit enhanced production of ROS due to oncogene activation and dysregulation in antioxidant response pathways (14). Induction of ferroptosis has been proposed as a novel anticancer strategy, and knowing how to leverage this iron-catalyzed cell death pathway will be aided by mechanistic understanding of how cancer cells activate the iron-starvation response (15, 16). On the basis of our work and the canonical iron sensing pathway architecture, we hypothesized that activation of the iron-starvation response by IRP1 is the main facilitator of ferroptosis sensitivity in cells with perturbed ISC metabolism. Here, we unexpectedly find that IRP2, the major regulator of cellular iron homeostasis in vivo, is independently activated upon inhibition of ISC metabolism. IRP2 activation can occur independent of ubiquitin-mediated protein level regulation by SKP1-cullin-F-box ubiquitin ligase component FBXL5. Activation of either IRP is sufficient to activate the iron-starvation response and increase ferroptosis sensitivity, while blockade of both IRPs is necessary to induce ferroptosis resistance.
RESULTS
Suppression of ISC synthesis activates the iron-starvation response and sensitizes cells to ferroptosis
To identify determinants of ferroptosis sensitivity upon suppression of ISC biosynthesis, we transduced a panel of seven breast cancer cell lines with a short hairpin RNA (shRNA) targeting essential ISC biosynthesis protein NFS1 (shNFS1), whose effects on cell viability and the iron-starvation response can be completely rescued by an shRNA-resistant NFS1 complementary DNA (cDNA) (9). Of these cell lines, four exhibit increased sensitivity to the ferroptosis inducer erastin upon NFS1 suppression (ferroptosis-sensitive lines BT549, EFM19, Hs578t, and MDA-MB-231), while three do not (ferroptosis-resistant lines HCC1954, HCC70, and MCF7; Fig. 1A). ISC suppression canonically mediates activation of the iron-starvation response via activation of IRP1, which increases the stability of certain target mRNAs, such as those encoding the main iron uptake protein transferrin receptor (TFR1), while inhibiting the translation of mRNAs encoding other targets, such as iron storage protein ferritin (FTH1; Fig. 1B) (17–19). In the four ferroptosis-sensitive cell lines, we observe increased TFR1 and decreased FTH1 protein levels upon ISC synthesis inhibition, consistent with activation of the iron-starvation response. In contrast, ferroptosis-resistant lines display mild to no increase in TFR1 and/or low levels of basal FTH1 levels consistent with minimal engagement of the iron-starvation response (Fig. 1C). Upon treatment with excess iron or iron chelation, all nine cell lines alter TFR1 and FTH1 levels, indicating an intact response to direct iron modulation (fig. S1A). In prior work, we defined activation of NFS1 as important for lung tumor development (9). Therefore, we suppressed NFS1 or directly modulated iron levels in three lung cancer cell lines. We observed activation of the iron-starvation response and enhanced ferroptosis sensitivity upon ISC inhibition similar to that seen in breast cancer cell lines (fig. S1, B and C). These data suggest that engagement of the iron-starvation response is responsible for enhanced sensitivity to ferroptosis upon NFS1 suppression in cancer cells.
Fig. 1. Suppression of ISC synthesis activates the iron-starvation response and sensitizes cells to ferroptosis.
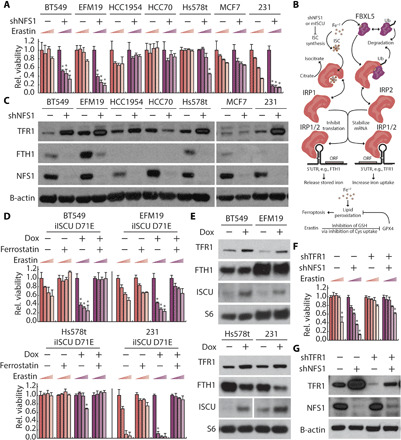
(A) Relative viability of indicated cell lines expressing an shRNA targeting NFS1 (shNFS1, +) relative to a nontargeting shRNA (shGFP, −) and treated with erastin (0, 2.5, 5, and 10 μM; 2 days), 3% O2. (B) Schematic of iron-starvation response and ferroptosis sensitivity. (C) Immunoblots from lysates derived from cell lines as in (A) for indicated proteins. ORF, open reading frame; GSH, glutathione. (D) Relative viability of indicated cell lines expressing doxycycline (dox)–inducible ISCU-DN treated with 100 nM dox (1 day) and then treated with 100 nM dox, 1 μM ferrostatin, and erastin (0, 2.5, 5, and 10 μM; 2 days) as indicated. (E) Immunoblots from lysates derived from indicated cell lines expressing dox-inducible ISCU-DN treated with 100 nM dox (1 day). (F) Relative viability of MDA-MB-231 cells expressing shGFP shNFS1 or an shRNA targeting TFR1 (shTFR1), treated with erastin (0, 2.5, 5, and 10 μM; 2 days). (G) Immunoblots of lysates derived from MDA-MB-231 cell line expressing indicated shRNAs for indicated proteins. *P < 0.01; error bars are SEM.
To confirm these results with an orthogonal method of ISC synthesis suppression, we overexpressed a doxycycline (dox)–inducible mutant of the essential ISC scaffold protein ISCU (ISCU-DN), which traps nascent ISCs and acts as a dominant negative inhibitor of ISC synthesis (20). Similar to NFS1 suppression, dox-induced overexpression of ISCU-DN sensitizes cells to ferroptosis, a phenotype that can be rescued by the lipophilic antioxidant and ferroptosis suppressor ferrostatin (Fer-1; Fig. 1D), and not observed in response to dox addition alone (fig. S2A). Cells expressing ISCU-DN exhibit increased TFR1 and decreased FTH1 protein levels, further supporting the correlation between ferroptosis sensitivity, ISC biosynthesis suppression, and activation of the iron-starvation response (Fig. 1E).
On the basis of these results, we hypothesized that cellular iron accumulation is required for enhanced ferroptosis sensitivity independent of other effects of ISC synthesis inhibition. Therefore, we expressed an shRNA targeting TFR1 in combination with NFS1 suppression. Combined suppression of TFR1 and NFS1 in MDA-MB-231 and BT549 cell lines restores TFR1 expression to basal levels and is sufficient to reverse the enhanced ferroptosis sensitivity caused by NFS1 suppression (Fig. 1, F and G, and fig. S2B). These data indicate that increased iron import via TFR1 up-regulation can be sufficient to increase sensitivity to ferroptosis upon inhibition of ISC synthesis, independent of other effects of ISC inhibition. We conclude that activation of the iron-starvation response upon ISC synthesis inhibition is necessary for increased sensitivity to ferroptosis.
IRP1 knockout cells activate the iron-starvation response upon inhibition of ISC synthesis and become sensitive to ferroptosis
We next considered how ISC synthesis inhibition triggers activation of the iron-starvation response. While both IRP1 and IRP2 can activate the iron-starvation response, IRP1 is known to activate this response via direct sensing of ISC deficiency (5). Thus, we expected that cells lacking IRP1 would not activate the iron-starvation response upon ISC synthesis suppression and, consequently, would not exhibit enhanced sensitivity to ferroptosis. To test this hypothesis, we expressed spCas9 and single guide RNAs (sgRNAs) targeting either IRP1 or IRP2 in MDA-MB-231 cells and isolated clones derived from single cells that lacked IRP1 or IRP2 protein. Unexpectedly, upon inhibition of ISC synthesis, these cell lines (control, IRP1 knockout, and IRP2 knockout), all exhibit increased ferroptosis sensitivity upon erastin or RSL3 treatment, increased lipid peroxidation, and modulated TFR1 and FTH1 protein levels to similar degrees (Fig. 2, A to C, and fig. S2, C to F). Moreover, in both wild-type (WT) and IRP1 knockout cells, NFS1 suppression increases the level of functionally active and chelatable intracellular iron, termed as the labile iron pool (Fig. 2B). Suppressing ISC biosynthesis using orthogonal methods, including with shRNAs targeting ISCU or upon expression of ISCU-DN, also modulates TFR1 and FTH1 levels and sensitizes cells to ferroptosis in both WT and IRP1 knockout cells (Fig. 2, E and F, and fig. S3). These data suggest that a protein aside from IRP1 can sense ISC deficiency and activate the iron-starvation response, mediating increased sensitivity to ferroptosis.
Fig. 2. IRP1 knockout cells activate the iron-starvation response upon inhibition of ISC synthesis and become sensitive to ferroptosis.
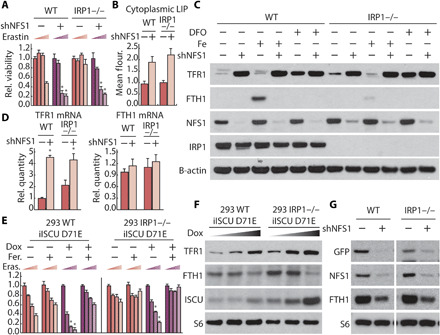
(A) Relative viability of parental (WT) and IRP1 knockout MDA-MB-231 cell line expressing a nontargeting shRNA (shGFP, −) or NFS1 (shNFS1, +), treated with erastin (0, 2.5, 5, and 10 μM; 2 days). (B) Labile iron pool of parental and IRP1 knockout MDA-MB-231 cell line expressing shGFP or shNFS1. (C) Immunoblots for indicated proteins from lysates derived from parental (WT) and IRP1 knockout MDA-MB-231 cell line expressing shGFP or shNFS1, treated with 50 μM Deferoxamine (DFO) and 35 μM iron nitrate as indicated (1 day). (D) Relative abundance of TFR1 and FTH1 mRNA in parental and IRP1 knockout MDA-MB-231 cell line expressing shGFP or shNFS1. (E) Relative viability of WT and IRP1 knockout 293 human embryonic kidney (HEK) cell line expressing dox-inducible ISCU-DN treated with 100 nM dox (1 day) and then treated with 100 nM dox, 1 μM ferrostatin (Fer.), and erastin (Eras.; 0, 2.5, 5, and 10 μM; 2 days) as indicated. (F) Immunoblots for indicated proteins of lysates derived from parental and IRP1 knockout HEK-293 cell line expressing dox-inducible ISCU-DN treated with dox (0, 10, and 100 μM; 1 day) as indicated. (G) Immunoblots for indicated proteins of lysates derived from parental and IRP1 knockout MDA-MB-231 cells expressing GFP reporter with 5′UTR IRE sequence from FTH1 mRNA and shRFP or shNFS1. *P < 0.01; error bars are SEM.
When activated, both IRP1 and IRP2 bind to hairpin-like structures called IREs in the 3′ untranslated region (3′UTR) or 5′UTR of target mRNAs. IRP binding to the 3′UTR stabilizes target mRNAs (e.g., TFR1), while binding to the 5′UTR inhibits translation (e.g., FTH1) (17–19). Hence, IRP binding increases both TFR1 protein and mRNA levels while decreasing FTH1 protein abundance, but not mRNA levels. Accordingly, TFR1, but not FTH1, mRNA levels increase in both WT, IRP1, and IRP2 knockout cells upon inhibition of ISC biosynthesis, further supporting activation of iron-starvation response upon ISC inhibition in these models (Fig. 2D and fig. S4, A to C).
We next sought to validate functional loss of IRP1 protein by orthogonal methods and determine whether the observed IRP1 knockout phenotypes would extend to other model systems. IRP1 functions as the cytosolic aconitase, converting citrate to isocitrate, when it is bound to an ISC (6). We therefore measured aconitase activity, IRE binding potential as demonstrated by electrophoretic mobility shift assay (EMSA) using a 32P-labeled IRE probe, and IRP1 mRNA levels. Consistent with complete deletion of IRP1, we observed no cytosolic aconitase activity, no IRP1 binding to a model IRE, and minimal IRP1 mRNA in MDA-MB-231 IRP1 knockout cells (fig. S5, A to C). We also generated three independent IRP1 knockout MDA-MB-231 clones. Upon suppression of NFS1 in all of these clones, we observed a similar increase in ferroptosis sensitivity and modulation of TFR1 and FTH1 levels, consistent with activation of the iron-starvation response (fig. S5, D and E). Last, we generated IRP1 and IRP2 knockout clones derived from two additional breast cancer cell lines, BT549 and EFM19. Upon inhibition of ISC biosynthesis in control, IRP1, and IRP2 knockout clones derived from these cell lines, we observed similar modulation of TFR1 and FTH1 levels, consistent with activation of the iron-starvation response in an IRP1-independent manner across several experimental systems (fig. S6, A and B).
TFR1 and FTH1 protein levels can be regulated by mechanisms other than the IRP-mediated iron-starvation response (21). We therefore created a reporter construct in which the green fluorescent protein (GFP) open reading frame is linked to the 5′UTR of the FTH1 mRNA (5′IRE-GFP). Hence, we anticipated that 5′IRE-GFP and FTH1 mRNAs would be regulated similarly via the iron-starvation response. NFS1 suppression reduced both GFP and FTH1 protein levels in MDA-MB-231 cells expressing 5′IRE-GFP, a phenotype observed in both control and IRP1 knockout cells (Fig. 2G). Together, these data demonstrate that the iron-starvation response can be activated by ISC deficiency even in the absence of IRP1.
Suppression of ISC synthesis increases IRP2 stability independent of IRP1 in atmospheric O2 levels
We next considered how the iron-starvation response could be activated by ISC synthesis inhibition if IRP1 is absent. Since IRP1 knockout cells are responsive to ISC deficiency, we hypothesized that ISC deficiency can activate IRP2. Levels of IRP2 are regulated posttranslationally by FBXL5-mediated ubiquitination and subsequent degradation (22, 23). This process can be mediated by direct binding of iron ions to FBXL5. Recently, a report indicated that FBXL5 also contains an ISC that is required for the IRP2:FBXL5 interaction and can thereby regulate IRP2 stability (24). We therefore considered whether suppression of ISC synthesis activates the iron-starvation response in IRP1 knockout cells by affecting IRP2 stability. In accordance with the prior studies (7, 25), inhibition of NFS1 increased IRP2 levels (Fig. 3A and fig. S2A). This effect of ISC synthesis inhibition on IRP2 levels was also observed to the same extent in IRP1 knockout cells, indicating that IRP2 protein level can be regulated independent of IRP1 (Fig. 3B and fig. S7A). To assess IRP2 stability in these conditions, we expressed an IRP2 cDNA under the control of a dox-repressible promoter, permitting CRISPR-Cas9–mediated deletion of both IRP1 and IRP2. Upon addition of dox, we observed that IRP2 levels decreased more slowly upon NFS1 suppression in 21% O2, indicating higher IRP2 protein stability (Fig. 3C). Together, these data demonstrate that inhibition of ISC synthesis results in increased IRP2 stability independent of IRP1 at 21% O2.
Fig. 3. Suppression of ISC synthesis increases IRP2 stability independent of IRP1 in atmospheric O2 levels.
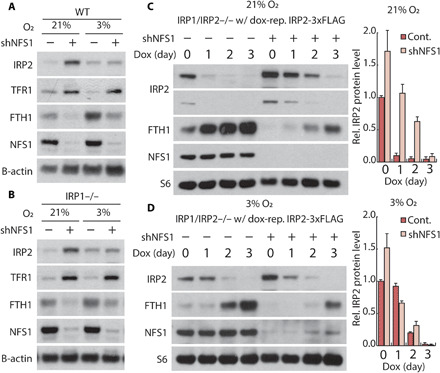
(A and B) Immunoblots for indicated proteins of lysates derived from parental (WT) (A) and IRP1 knockout (B) MDA-MB-231 cells expressing a nontargeting shRNA (shGFP, −) or shRNA targeting NFS1 (shNFS1, +) incubated in 3% or 21% O2. (C and D) Immunoblots for indicated proteins from lysates derived from IRP1/IRP2 double-knockout MDA-MB-231 expressing dox-repressible IRP2 and shGFP or shNFS1, treated with 500 nM dox for 0, 1, 2, or 3 days in 21% (C) or 3% O2 (D).
In contrast, cells cultured in tissue level O2 (3% O2) fail to up-regulate IRP2 protein levels upon ISC synthesis inhibition (Fig. 3A and fig. S7A) and do not exhibit a significant change in IRP2 stability (Fig. 3D). Prior studies demonstrated that the iron-starvation response is activated by either IRP2 stabilization or IRP1 activation. Because, under these conditions, IRP2 levels are unchanged and IRP1 is deleted, we expected to observe no change in the iron-starvation response upon ISC synthesis inhibition. However, suppression of ISC synthesis still resulted in modulation of TFR1 and FTH1 levels (Fig. 3, A and B). These data suggest that at 3% O2, ISC deficiency regulates IRP2 activity independent of FBXL5-mediated degradation. In contrast to the 21% O2 experienced by cells in standard culture conditions, cells in most tissues are exposed to oxygen levels near 3%. Thus, stability-independent regulation of IRP2 by ISC deficiency may be more physiologically relevant to disorders affecting ISC biosynthesis.
Changes in environmental O2 levels can affect ROS production. To analyze the effect of ROS on O2-dependent iron regulation, we treated cells with antioxidant and pro-oxidant compounds at 3 and 21% O2. We observed that the O2-dependent effects on IRP2 and iron-starvation response protein levels were not affected by these ROS-modulating compounds, indicating that O2-dependent changes in TFR1, FTH1, and IRP2 are not mediated by changes in ROS (fig. S7B). These results are in accordance with previously observed regulation of ISCs within resident proteins by environmental O2 levels, but not by physiological changes in ROS levels (9).
The transcription factor NRF2 is activated in a redox-dependent manner, resulting in increased expression of ferritin light chain (FTL) and heavy chain, as well up-regulation of glutathione synthesis (25). Hence, we asked whether NRF2 is involved in activation of IRP2 or the iron-starvation response upon ISC inhibition at different O2 levels. As expected, NRF2 activator KI696 reduced erastin sensitivity and increased FTH1 protein levels (fig. S7, C and D). NRF2 activation did not affect TFR1 levels or alter the activation of iron-starvation response upon NFS1 suppression (fig. S7C). Therefore, while activation of NRF2 can suppress ferroptosis, likely as a consequence of increased antioxidant defenses, NRF2 is not involved in regulation of IRP2 or the iron-starvation response downstream of ISC synthesis inhibition.
To better understand the impact of exposure to atmospheric O2 levels on IRP2 stability, we analyzed IRP2 levels and the iron-starvation response upon transferring IRP1 knockout cells from 3 to 21% O2. Both control and IRP1 knockout cells exhibit a decrease in basal IRP2 levels upon transfer to 21% O2 (fig. S8, A and B). Despite this decrease in IRP2 levels, TFR1 levels increase and FTH1 levels decrease, consistent with activation of the iron-starvation response. These changes in the expression of target proteins occur on the same time scale as O2-dependent degradation of ISCs, as assessed by aconitase activity (fig. S8C). Together, our results indicate that a reduction in ISC pools enhances IRP2 activity independent of changes in IRP2 protein levels and IRP1, revealing a previously unrecognized method of IRP2 regulation of the iron-starvation response.
FBXL5 binding is not required for regulation of IRP2 by ISCs
We next considered how IRP2 could be activated independent of FBXL5-mediated degradation in tissue-level O2. Consistent with a recent report of an ISC in FBXL5 mediating its interaction with IRP2 (25), we hypothesized that FBXL5 could compete with IREs for IRP2 binding and thus regulate IRP2 activity without any change in IRP2 levels. FBXL5-IRP2 binding was abrogated after suppression of ISC synthesis or upon mutation of the FBXL5 ISC binding site, while IRP2 protein levels remained unchanged (Fig. 4A). Therefore, ISCs mediate the IRP2-FBXL5 interaction. We then developed and tested an experimental system to assess IRP2 function independent of FBXL5 binding. We expressed an IRP2 R779E mutant incapable of binding FBXL5 (25) in IRP1/2 knockout cells expressing a dox-repressible flag-tagged IRP2 cDNA. Upon addition of dox, WT IRP2 is suppressed, and cells only express IRP2 R779E at near endogenous IRP2 protein levels, enabling us to characterize this IRP2 mutant in the absence of WT IRP1 or IRP2 (Fig. 4B). Since IRP2 R779E cannot bind FBLX5, we expected cells to be unable to regulate IRP2 levels in response to iron. Indeed, unlike WT IRP2, IRP2 R779E protein levels were unresponsive to addition of iron or an iron chelator (Fig. 4B). Compared to cells expressing WT IRP2, those expressing IRP2 R779E exhibited minimal changes in TFR1 and FTH1 levels upon iron addition or chelation. The residual regulation could be explained by mechanisms independent of the iron-starvation response, such as ferritinophagy, or by affecting ISC synthesis. Similar to WT IRP2, suppression of ISC synthesis did not affect IRP2 R779E levels in 3% O2. However, cells expressing IRP2 R779E retained activation of the iron-starvation response after suppression of NFS1 similar in magnitude to WT IRP2, as assessed by regulation of TFR1 and FTH1 protein and mRNA levels (Fig. 4C and fig. S9). Together, our data confirm prior reports that ISC binding can regulate the IRP2-FBXL5 interaction (24). We find that abrogation of IRP2-FBXL5 binding is not responsible for IRP2-mediated activation of iron-starvation response upon ISC synthesis inhibition at 3% O2.
Fig. 4. FBXL5 binding is not required for regulation of IRP2 by ISCs.
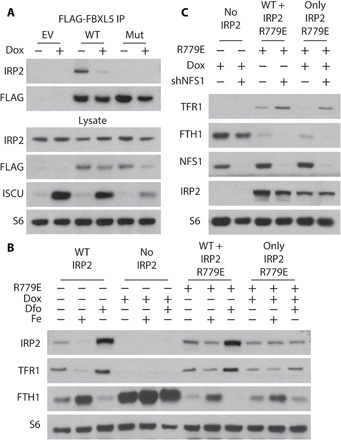
(A) Immunoprecipitation and immunoblots for indicated proteins of lysates derived from parental HEK-293 cell line expressing dox-inducible ISCU-DN transfected with empty vector (EV), FLAG-tagged FBXL5 (WT), or FBXL5 in which ISC-coordinating cysteine resides were mutated to alanine (ISCmut). Two days after transfection, cells are treated with 100 nM dox (1 day), and coimmunoprecipitation is performed using antibody against FLAG. (B) Immunoblots for indicated proteins of lysates derived from IRP1/2 double-knockout MDA-MB-231 cell line expressing dox-repressible WT IRP2 and ubiquitously expressed IRP2 R779E mutant, treated with 100 nM dox for 2 days and then treated with 100 nM dox, 50 μM DFO, or 35 μM iron nitrate (1 day) as indicated. (C) Immunoblots for indicated proteins of lysates derived from IRP1/2 double-knockout MDA-MB-231 cell line expressing dox-repressible WT IRP2 and ubiquitously expressed IRP2 R779E mutant, treated with 100 nM dox for 2 days and infected with shGFP or shNFS1.
Suppression of ISC synthesis increases IRP2-mRNA binding independent of altered IRP2 stability at tissue O2 levels
Because we observed that the iron-starvation response is activated by IRP2 upon ISC synthesis inhibition independent of FBXL5 binding, we hypothesized that ISC deficiency exerts this effect by enhancing IRP2-mRNA binding activity. We assessed the total IRE binding capacity of IRP2 by EMSA using a synthetic IRE substrate. At 21% O2, we observed enhanced IRP2:IRE binding upon ISC suppression in both control and IRP1 knockout cells, consistent with the observed increase in IRP2 levels under these conditions (Fig. 5, A and B, and fig. S10). In 3% O2, even though IRP2 protein levels were unchanged, ISC inhibition increased the IRP2:IRE binding potential (Fig. 5, C and D). Therefore, ISC deficiency regulates IRP2 at tissue-level O2 by a previously unrecognized FBXL5-independent mechanism.
Fig. 5. Suppression of ISC synthesis increases IRP2-mRNA binding independent of altered IRP2 stability at tissue O2 levels.
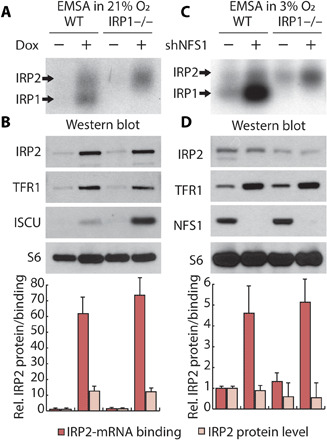
(A and B) EMSA using 32P-labeled IRE probe (A), or immunoblot for indicated proteins (B), of lysates derived from parental and IRP1 knockout HEK-293 cell line expressing dox-inducible ISCU-DN, treated with 100 nM dox (1 day), and incubated in 21% O2, lysis in 1% O2. (C and D) EMSA using 32P-labeled IRE probe (C) and immunoblot for indicated proteins (D) of lysates derived from parental and IRP1 knockout MDA-MB-231 cell line expressing shGFP or shNFS1, incubated in 3% O2, lysis in 1% O2.
IRP2 is sufficient to sense ISC deficiency and sensitize cells to ferroptosis
As IRP1 and IRP2 are the only proteins known to bind IRE elements and regulate the iron-starvation response (26), deleting both IRPs should eliminate any response to inhibition of ISC synthesis, including activation of the iron-starvation response and enhanced ferroptosis sensitivity. We could not obtain IRP1/2 double-knockout clones under standard culture conditions and hypothesized that these cells might experience iron starvation and loss of viability due to extremely low TFR1 and high FTH1 levels. We managed to obtain IRP1/2 knockout clones in iron- and antioxidant-supplemented media, and viability of these clones was dependent on continued iron supplementation (Fig. 6A and fig. S11A). These cells exhibited very low basal TFR1 levels and high basal FTH1 protein levels, which were not affected by iron, iron chelation, disruption of iron uptake, or NFS1 suppression (Fig. 6, B and C, and fig. S11, B and C). TFR1 mRNA levels were minimally affected by these perturbations (Fig. 6D), demonstrating that IRP1 and IRP2 are the only other major regulators of the iron-starvation response upon iron-level modulation or ISC deficiency. These phenotypes of IRP1/2 double-knockout cells were recapitulated in an orthogonal experimental system in which IRP1 was acutely suppressed by RNA interference (RNAi) in IRP2 knockout cells (fig. S12).
Fig. 6. IRP2 is sufficient to sense ISC deficiency and sensitize cells to ferroptosis.
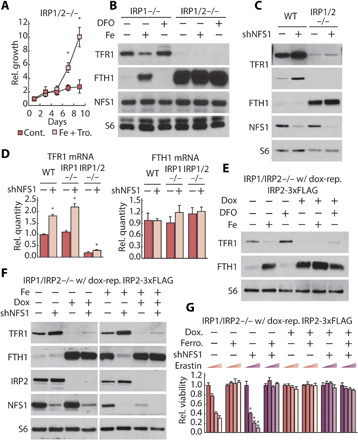
(A) Proliferation (relative cell number) of IRP1/2 double-knockout MDA-MB-231 cells grown in media supplemented with 35 μM iron nitrate and 50 μM Trolox or upon withdrawal of supplements at day 0. (B) Immunoblots for indicated proteins of lysates derived from IRP1 knockout and IRP1/2 double-knockout MDA-MB-231 cell line treated with 50 μM DFO and 35 μM iron nitrate (1 day) as indicated. (B and C) Immunoblots for indicated proteins of lysates derived from parental (WT) and IRP1/2 double-knockout MDA-MB-231 cell line expressing a nontargeting shRNA (shGFP, −) or shRNA targeting NFS1 (shNFS1, +). (D) Relative abundance of TFR1 and FTH1 mRNA in parental, IRP1 knockout, and IRP1/2 double-knockout MDA-MB-231 cell line expressing shGFP or shNFS1. (E and F) Immunoblots for indicated proteins of lysates derived from IRP1/IRP2 double-knockout MDA-MB-231 cells expressing dox-repressible IRP2 treated with 100 nM dox for 2 days, and then treated with 100 nM dox, 50 μM DFO, or 35 μM iron nitrate (1 day), or infected with shGFP or shNFS1. After drug selection for shRNA transduction, cells are treated with 100 nM dox and 35uM iron nitrate for 1 day. (G) Relative viability of cells as in (E) and (F). After drug selection for shRNA transduction, cells were treated with 100 nM dox, 1 μM ferrostatin, or erastin (0, 2.5, 5, and 10 μM) for 2 days as indicated.
We then generated an inducible IRP1/2-deficient system that could be propagated in the short term without iron supplementation, enabling us to assess the impact of introducing IRP2 mutants expressed at endogenous levels. To accomplish this, we used IRP1/2 knockout cells expressing dox-repressible flag-tagged IRP2 cDNA at levels similar to endogenous IRP2 (dox-OFF IRP2 cells). In the absence of dox (IRP2 expressed), iron chelation and NFS1 suppression regulate the iron-starvation response, consistent with intact IRP2 function, and suppression of exogenous IRP2 via addition of dox reverts these IRP1/2 double-knockout cells to an iron and ISC pool-insensitive state (Fig. 6, E and F). Addition of dox was also sufficient to significantly dampen the increased TFR1 mRNA levels observed upon inhibition of ISC synthesis (fig. S13). Ferroptosis sensitivity correlates with the ability of cells to activate the iron-starvation response, as NFS1 suppression in Dox-treated cells (IRP1/2 suppressed) did not affect sensitivity to erastin, a phenotype that exogenous IRP2 was able to restore upon dox withdrawal (Fig. 6G). dox-treated cells were significantly more resistant to ferroptosis even after prolonged erastin treatment that induces ferroptosis in IRP2-expressing cells (fig. S14A), and these cells exhibited a significantly lower labile iron pool (fig. S14B) and retained sensitivity to other toxic compounds (fig. S14C). These data demonstrate that IRP1/2 is required to maintain sufficient intracellular iron levels for normal cellular function and to permit ferroptosis induction.
We next wanted to consider whether activation of IRPs by ISC synthesis inhibition is required to promote ferroptosis in cells that can maintain iron levels sufficient to enable normal cellular function. Therefore, we overexpressed in dox-OFF IRP2 cells a cDNA encoding the TFR1 open reading frame but lacking 3′UTR IRE elements that cause instability of the mRNA in the absence of IRPs (dox-OFF IRP2 + TFR1). Upon dox addition (IRP1/2 deficient), TFR1 protein levels were sustained, and long-term viability was maintained without iron supplementation (Fig. 7, A and B). These data demonstrate that maintaining TFR1 protein expression is sufficient to restore cell viability to IRP1/2 knockout cells. Significantly, maintaining TFR1 levels is also sufficient to restore ferroptosis sensitivity to IRP1/2-deficient cells, consistent with the notion that IRP1/2 deletion inhibits ferroptosis due to lack of intracellular iron (Fig. 7B). Next, we suppressed ISC synthesis and measured activation of the iron-starvation response and sensitivity to ferroptosis. In the presence of IRP2 (no dox), ISC suppression activated the iron-starvation response and sensitized cells to ferroptosis (Fig. 7, B and C). In contrast, cells deficient in both IRP1 and IRP2 (+ dox) exhibited sensitivity to ferroptosis, which was not modulated by ISC deficiency (Fig. 7C). Together, these data demonstrate that ISC deficiency can activate the iron-starvation response via either IRP1 or IRP2, and this activation is required for the increased ferroptosis sensitivity upon ISC suppression.
Fig. 7. IRP1/2 double-knockout cells expressing TFR1 can proliferate without iron supplementation and are sensitive to ferroptosis, but cannot sense ISC deficiency and enhance ferroptosis sensitivity.
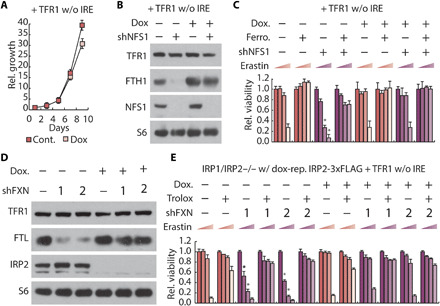
(A) Proliferation (relative cell number) of MDA-MB-231 cells expressing dox-repressible IRP2, TFR1 open reading frame (TFR1 without IRE) with 100 nM dox as indicated. (B) Immunoblots of IRP1/IRP2 double-knockout MDA-MB-231 cells expressing dox-repressible IRP2, TFR1 open reading frame, and nontargeting shRNA (shGFP, −) or shRNA targeting NFS1 (shNFS1, +), treated with 100 nM dox. (C) Relative viability of cells as in (B), treated with 100 nM dox, 1 μM ferrostatin, or erastin (0, 2.5, 5, and 10 μM) for 2 days as indicated. (D) Immunoblots of MDA-MB-231 cells expressing dox-repressible IRP2, TFR1 open reading frame, and shGFP or shRNAs targeting FXN (shFXN, 1 or 2), treated with 100 nM dox. (E) Relative viability of cells as in (D), treated with 100 nM dox, 50 μM Trolox, or erastin (0, 2.5, 5, and 10 μM) for 2 days as indicated. Asterisks indicate significance comparing shGFP to shNFS1 or shFXN for each group. P < 0.05, error bars are SEM.
Triplet-repeat expansion mutation of the gene encoding the protein frataxin (FXN), an allosteric activator of ISC synthesis, causes the disease Friedreich’s ataxia (27). Dysregulation of iron metabolism is observed in disease tissues, suggesting that inappropriate regulation of IRP proteins could be key to disease pathology. Therefore, we asked whether suppressing FXN sensitizes cells to ferroptosis through activation of the iron-starvation response. Similar to NFS1 suppression, expression of shRNAs targeting FXN (shFXN_1 and shFXN_2) in dox-OFF IRP2 + TFR1 cells activated the iron-starvation response and sensitized cells to ferroptosis (Fig. 7, D and E, and fig. S14D). Removal of both IRP1 and IRP2 (dox-OFF IRP2 cells +TFR1 + dox) abolished the enhanced ferroptosis sensitivity facilitated by ISC deficiency (Fig. 7E). These data indicate that dysregulation of IRP2 could result in altered iron metabolism in Friedreich’s ataxia via the FBXL5-independent mechanism described (Fig. 8).
Fig. 8. IRP2 regulation by ISC deficiency.
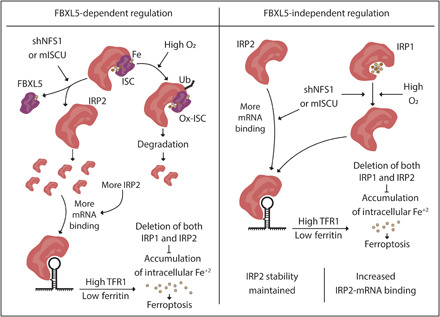
Model for IRP2 activation in ISC deficiency and subsequent ferroptosis sensitivity. At atmospheric O2 levels, the FBXL5 ISC is oxidized, and IRP2 is ubiquitinated and degraded. Partial inhibition of ISC synthesis by shNFS1 or a dominant negative ISCU (mISCU) abrogates the FBXL5:IRP2 interaction, leading to increased IRP2 stability and IRP2 binding to target mRNAs. At physiologic O2 levels, the FBXL5 ISC is reduced, and degradation of IRP2 is limited. Partial inhibition of ISC synthesis does not further alter IRP2 stability but still releases IRP2 from FBXL5. ISC inhibition enhances IRP2 binding to target mRNAs independent of FBXL5 binding. IRP2 activation increases intracellular Fe+2 pool and sensitize cells to ferroptosis. Deletion of both IRP1 and IRP2 is sufficient to inhibit the iron accumulation and subsequent ferroptosis sensitivity.
DISCUSSION
In prior work, we described how inhibition of ISC synthesis primes cells for ferroptosis induced by oxidative stress, which we proposed is due to IRP-mediated activation of the iron-starvation response (9). Here, we explore mechanistically how ISC synthesis inhibition elicits these effects. We show that while breast cancer cell lines can activate the iron-starvation response upon direct iron addition or iron chelation, only some cell lines effectively activate the iron-starvation response upon ISC inhibition. Those lines that activate the iron-starvation response become sensitive to ferroptosis induction, while those that do not maintain resistance. These data suggest that an inability to engage IRPs may be responsible for this difference, perhaps due to incomplete suppression of ISC synthesis. We found that suppression of TFR1 is sufficient to inhibit ISC-mediated ferroptosis sensitivity, indicating that the myriad other effects of ISC synthesis inhibition are not sufficient to sensitize cells to ferroptosis.
We then explore how ISC synthesis inhibition activates the iron-starvation response and hypothesize that IRP1 mediates this response based on canonical pathway architecture (5). However, deletion of IRP1 fails to block activation of the iron-starvation response or sensitivity to ferroptosis upon ISC synthesis inhibition. Instead, deletion of both IRPs is necessary to elicit this effect. We demonstrate that knockout of both IRP1 and IRP2 results in loss of cell viability accompanied by loss of TFR1 expression and depletion of the labile iron pool. These double-knockout cells can be maintained by providing sufficient non-transferrin bound iron or by expressing the TFR1 open reading frame, which is incapable of regulation by IRPs. These genetic manipulations enable us to generate a stable cellular model that lacks IRP-mediated iron sensing. Using these systems, we demonstrate that IRP-mediated sensing is necessary to sensitize cells to ferroptosis upon ISC synthesis inhibition. Moreover, we show that IRP2 can independently activate the iron-starvation response downstream of ISC synthesis inhibition, an unexpected finding, as IRP1-mediated activation is the anticipated model.
IRP2 stability can be controlled by FBXL5-mediated ubiquitination and proteasomal degradation (22, 23). Recently, biochemical studies have identified an ISC cofactor in FBXL5 (24). In this model, ISC occupancy enables a stable FBXL5:IRP2 interaction at low O2, but FBXL5 ubiquitin ligase activity is only activated once the ISC is oxidized at high O2. These data are consistent with our findings that IRP2 stability is affected by ISC synthesis inhibition at atmospheric O2 (21%), but not at tissue-level O2 (3%). We find that abrogation of this interaction, while preventing regulation of IRP2 levels by iron, fails to eliminate regulation of the iron-starvation response upon ISC synthesis inhibition. Unexpectedly, we find that despite ISC inhibition failing to affect IRP2 stability at 3% O2, IRP2:IRE binding is increased and activation of the iron-starvation response is enhanced. These data support a model in which IRP2 or another unknown IRP2-interacting protein contains an ISC or is affected by ISC levels, thereby modulating IRP2:IRE binding. As most cells in the body experience O2 levels near 3% (28), this mechanism of IRP2 activation is likely to be more physiologically relevant.
Prior studies comparing the function of IRP1 and IRP2 have concluded that the IRP2 arm is the major cellular iron sensor in most tissues in vivo, but its activation by ISC synthesis inhibition was assumed to be mediated by IRP1 (29). Demonstrating a direct link between ISC synthesis and IRP2 activation will be informative for designing therapeutic strategies where modulation of ferroptosis is warranted, such as in potentiating cancer cell death (15, 16), and for disorders caused by ISC synthesis inhibition. For example, myelodysplastic syndrome frequently exhibits mutation of the splicing factor 3b subunit 1 (SF3B1), and these patients can develop sideroblastic anemia characterized by the accumulation of iron-loaded erythroid precursors (30). Mutant SF3B1 is known to mis-splice the mitochondrial transporter ABCB7 (ATP binding cassette subfamily B member 7), affecting ISC pools via a poorly understood mechanism, and individuals harboring germline mutations in ABCB7 also exhibit sideroblastic anemia (31). In Friedreich’s ataxia, the most common form of inherited ataxia, a triple-repeat expansion mutation in the intron of the gene causes mis-splicing and low levels of FXN, an allosteric activator of ISC synthesis (13, 27). Patients with Friedreich’s ataxia develop tissue iron loading that is proposed to cause neuronal cell death and cardiac hypertrophy, leading to reduced life span. On the basis of our findings, we hypothesize that defective ISC synthesis and hyperactivation of the iron-starvation response can contribute to the pathology of sideroblastic anemia and Friedreich’s ataxia. Fibroblasts from patients with Friedreich’s ataxia exhibit increased ferroptosis sensitivity (32, 33). Hence, intervening in IRP2-mediated activation of the iron-starvation response may be useful in restoring iron homeostasis and ameliorating disease phenotypes.
METHODS
Reagents
Reagents were obtained from the following sources. Antibodies: b-actin (8457), FTH1 (3998), IRP1 (20272), IRP2 (37135), S6 (2217), and TFR1 (CD71, 13208) were from Cell Signaling Technologies; NFS1 (sc-365308), ISCU (sc-271468), and FECH (sc-377377) were from Santa Cruz Biotechnology; and FXN (ab113691) and FTL (ab218400) were from Abcam. Custom-made NRF2 antibody was provided by E. Schmidt, Montana State University. Cells: BT549, HCC1954, HCC70, HEK-293, Hs578t, MCF7, and MDA-MB-231 were from American Type Culture Collection; EFM19 was from DSMZ. Chemicals: RPMI (MT10040CV) was from Corning; PrimeStar HS DNA Polymerase Premix (R040A) was from Takara; RNeasy Plus Mini Kit (74136), Qiaprep Spin Miniprep Kit (27106), and QIAquick Gel Extraction Kit (28706) were from Qiagen. Erastin (S7242) was from Selleckchem. 6-Hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (Trolox, 238813), deferoxamine mesylate (D9533), ferrostatin-1 (SML0583), iron(III) nitrate (F8508) were from Sigma-Aldrich; KI696 (HY-101140) from MedChemExpress shRNAs were obtained from the The RNAi Consortium (TRC) or identical sequences cloned into pLKO.1p: shRFP, TRCN0000072203; shGFP, TRCN0000072186; shNFS1, TRCN0000229753; shTFRC, TRCN0000057659; shIRP1 TRCN0000056553; shFXN, TRCN0000006138 and TRCN0000006137; and shISCU, TRCN0000290051 and TRCN0000289988.
Cell culture
Cells were tested for the presence of mycoplasma by polymerase chain reaction (PCR), and the authenticity of cell lines was verified by short tandem repeat (STR) profiling. Cells were cultured in RPMI supplemented with 10% fetal bovine serum (Peak Serum) and 1% penicillin and streptomycin. When stated, O2 concentration was controlled by placing cells in an incubator maintaining 3% O2. Unless stated otherwise, cells are incubated in 3% O2 at least 3 days before experimentation. Single-cell clones are obtained by infecting parental cells with Cas9 system (pLentiCRISPR V2, Addgene), specific sgRNA for knockout, and cDNA vectors for exogenous expression for indicated genes. After selection, cells are plated in 96 wells in serial dilution and single clones are screened with Western blot. Target sequences for sgRNAs are used in the study: three different target sites for different IRP1 clones, (i) TGGCTCAGGAATCATCCACC, (ii) CCGCCAGGTTGGGGTAGTGG, and (iii) GCTCGCTACTTAACTAACAG; two different target sites for IRP2, (i) AATGCACCAAATCCTGGAGG and (ii) CCT GGAACTGGAATGGCTCA.
Cell proliferation and viability assays
Cell viability assay: After indicated treatment or infection, cells are counted using Beckman Z2 Coulter Counter, and 2000 cells per well are plated to 96 wells in triplicates. Further treatments are done as indicated, and cells are incubated in 3 or 21% O2. Cell viability is measured using the CellTiter-Glo Luminescent Cell Viability Assay (Promega G7573) in plate reader, and empty wells are used as baseline to correct background. Cell proliferation: After indicated treatment, cells are counted using Beckman Z2 Coulter Counter, and 25,000 cells per well are plated to 6 wells in triplicate. For 10-day proliferation assay, media are changed at day 6, and cells are counted at day 10.
Immunoblotting
Cells are washed with cold phosphate-buffered saline (PBS) and lysed by addition of lysis buffer [50 mM tris (pH 7.4), 150 mM NaCl, 1% NP-40, 0.1% sodium deoxycholate, 0.1% SDS, and 2 mM EDTA] with a protease inhibitor cocktail (Sigma-Aldrich, 5892791001). After 10-min incubation on ice, lysates are collected and centrifuged at 4°C at 21,000g for 10 min. Supernatant is collected, and protein concentration is quantified using a bicinchoninic acid (BCA) protein assay kit (Pierce 23225). Eight micrograms of protein was loaded into Bolt 4 to 12% bis-tris polyacrylamide gels (Thermo Fisher Scientific, NW04125BOX) with protein ladder (Thermo Fisher Scientific, LC5925), electrophoresed in running buffer (Thermo Fisher Scientific, B0002) at 110 V for 1.5 hours, and transferred in transfer buffer [N-cyclohexyl-3-aminopropanesulfonic acid (2.2 g liter−1), NaOH (0.45 g liter−1), and 10% ethanol] to a polyvinylidene difluoride membrane (Millipore, IPVH00010) at 40 V for 3 hours. Membranes are blocked with 5% bovine serum albumin (BSA), incubated with indicated antibody overnight at 4°C, washed three times with TBS-T (tris buffered saline, 0.1% Tween-20), incubated with secondary antibody for 1 hour at room temperature, washed three times with TBS-T, and developed using ECL substrate (Thermo Fisher Scientific, PI-32106) with autoradiography film (WorldWide, 41101001) in dark room.
Electrophoretic mobility shift assay
Protocol was adapted from (7). Preparing probe: Following oligonucleotides are annealed by boiling and slowly cooling down in annealing buffer [10 mM tris-HCl (pH 7.5) and 10 mM MgCl2]; oligos: TAATACGACTCACTATA and GGAGTTCCGTCCAAGCACTGTTGAAGCAGGAACTCCTATAGTGAGTCGTATTA. After annealing, radiolabeled probes are synthesized using T7 RNA polymerase (New England Biolabs, M0251S) with uridine 5′-triphosphate [α-32P] (PelkinElmer, BLU007H250UC). Probe is purified using ethanol, acetate with tRNA as carrier for precipitation, and radioactivity of purified probe is measured. Preparing lysate: Lysis buffer [10 mM HEPES (pH 7.2), 3 mM MgCl2, 40 mM KCl, 5% glycerol, 5% glycerol, 0.2% octylphenoxypolyethoxyethanol (IGEPAL CA-630), 50 μM deferoxamine, and 0.5 mM fresh dithiothreitol (DTT)] is prepared with protease inhibitors (Sigma-Aldrich, 5892791001) and equilibrated in 1% O2. Cells are lysed in 1% O2 and incubated 10 min on ice, and lysates are collected and centrifuged at 4°C at 21,000g for 10 min. Supernatant is collected in 1% O2 and protein concentration is quantified using a BCA protein assay kit (Pierce 23225). EMSA: Radiolabed probe is heated to denature and cooled down for 10 min. A 50000-CPM radiolabeled probe per reaction is mixed with reaction buffer [40 mM KCl, 25 mM tris-Cl (pH 7.5), 50% glycerol, tRNA (5 mg/ml), and 0.5 M fresh DTT] with ribonuclease (RNase) inhibitors (Thermo Fisher Scientific 10777019). Twelve micrograms of protein in lysate buffer is mixed with reaction buffer 1:1 ratio, and samples are incubated in 1% O2 at room temperature for 10 min. After adding loading buffer, entire reaction is loaded to 8% polyacrylamide/(tris/borate/EDTA) gel, prerun for 30 min, and run for 4 hours at 200 V. Gel is exposed to film (Sigma-Aldrich, Z350370) and developed in dark room.
qPCR
RNA was isolated by column purification (RNeasy Kit, Qiagen). One microgram of total RNA is reverse transcribed to cDNA using reverse transcriptase (Thermo Fisher Scientific, 18090050) with 1 μl of RNase OUT (10777019, Invitrogen). Quantitative PCR (qPCR) was performed on cDNA using SYBR green quantification (Thermo Fisher Scientific, K0223). Expressions are quantified relative to ACTB and RPL13A. Primers used are as follows: actin beta (ACTB), AAGGGACTTCCTGTAACAATGCA (forward) and CTGGAACGGTGAAGGTGACA (reverse); FTH1, AGGTGCGCCAGAACTACCA (forward) and TCGCGGTCAAAGTAGTAAGACATGG (reverse); IRP1, CATTACCAAGCACCTCCGCC (forward) and GCTCGGTCAGCAATGGACAA (reverse); RPL13A, CATAGGAAGCTGGGAGCAAG (forward) and GCCCTCCAATCAGTCTTCTG (reverse); TFR1, AGGCCAATGTCACAAAACCAA (forward) and AGCCAATCATAAATCCAATCAAGAA (reverse); and FXN, CTTGCAGACAAGCCATACACG (forward) and ACACCCAGTTTTTCCCAGTCC (reverse).
In gel aconitase assay
Protocol was adapted from (7). Cells are washed with cold PBS and lysed by addition of lysis buffer [40 mM KCl, 25 mM tris-Cl (pH 7.5), 1% Triton X-100, 0.1 M fresh DTT, 1 M Na citrate, and 1 M MnCl2] with a protease inhibitor cocktail (Sigma-Aldrich, 5892791001). After 10-min incubation on ice, lysates are collected and centrifuged at 4°C at 21,000g for 10 min. Supernatant is collected, and protein concentration is quantified using a BCA protein assay kit (Pierce, 23225). Twelve micrograms of protein was loaded into 8% polyacrylamide/TBE gel supplemented with 1 M Na citrate and run at 175 V for 4 hours in running buffer [25 mM tris base, 192 mM glycine, and 3.6 mM citrate (pH 8)]. Gel was washed with distilled water and incubated at 37°C for 5 to 45 min in reaction solution [100 mM tris (pH 8.0), 1 mM nicotinamide adenine dinucleotide phosphate (Sigma-Aldrich, 10128031001), 2.5 mM cis-aconitate (Sigma-Aldrich, A3412), 5 mM MgCl2, 1.2 mM MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (Thermo Fisher Scientific, M6494), 0.3 mM phenazine methosulfate (Sigma-Aldrich, P9625), and isocitrate dehydrogenase (5 U/ml; Sigma-Aldrich, I2002)]. Gel is washed three times with distilled water for 5 min to remove background and gel is imaged.
Flow cytometry for labile iron pool
Cells are infected and treated as indicated. After washing with PBS, cells are incubated in 10 nM calcein AM (Thermo Fisher Scientific, C3099) in 1% BSA PBS for 10 min and washed with PBS two times. Cells are trypsinized and resuspended in 1% BSA PBS. Pyridoxal isonicotinoyl hydrazone (PIH) (Sigma-Aldrich, 528110) is added to one set of samples to chelate iron, and stained cells are analyzed with flow cytometry. Median of PIH-treated samples are subtracted from median of non–PIH-treated samples to assess the labile iron pool of cell.
Flow cytometry for lipid peroxidation
Cells are infected and treated as indicated. After washing with PBS, cells are incubated in 10 μM BODIPY 581/591 C11 (diluted in PBS) for 15 min in an incubator at 37°C and 21% O2 and washed with PBS. Cells are resuspended in 1% BSA PBS and analyzed with flow cytometry.
Statistical analysis
Cell viability, proliferation assays, and mRNA quantification are performed in at least biological triplicate, the entire experiment performed at least three times, and the results of these three experiments combined and reported as n = 3. Immunoblots and EMSAs are repeated from at least three independently generated cell lysates, and representative immunoblots and autoradiographs are shown. Student’s t distribution is assumed, which is not contradicted by data, and P values in the figures are the result of heteroscedastic Student’s t tests. No samples were excluded from analysis, and no sample size estimates were used.
Acknowledgments
We thank Possemato lab members for the helpful comments, and members of the Rouault lab for the assistance with EMSA and aconitase assays. Funding: Research was supported by the NIH (T32GM007308, T32GM115313, CA228202, CA168940, GM132491, CA198543, CA168940, and GM121994) and the Friedreich’s Ataxia Research Alliance. R.P. is a scholar of the Pew Charitable Trusts and the Alexander and Margaret Stewart Trust and an American Cancer Society Research Scholar. plentiCRISPR v2 was a gift from F. Zhang. FBXL5 plasmids were a gift from N. Zhang. KI696 compound and NRF2 antibody were a gift from T. Papagiannakopoulos. Author contributions: Conceptualization: R.P. and E.M.T. Methodology: R.P. and E.M.T. Investigation: E.M.T., S.W.A., V.O.S., and G.C.W.; Writing (original draft): E.M.T. and R.P. Writing (review and editing): E.M.T. and R.P. Funding acquisition, resources, and supervision: R.P. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/22/eabg4302/DC1
REFERENCES AND NOTES
- 1.Wang J., Pantopoulos K., Regulation of cellular iron metabolism. Biochem. J. 434, 365–381 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dixon S. J., Lemberg K. M., Lamprecht M. R., Skouta R., Zaitsev E. M., Gleason C. E., Patel D. N., Bauer A. J., Cantley A. M., Yang W. S., Morrison B. III, Stockwell B. R., Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cao J. Y., Dixon S. J., Mechanisms of ferroptosis. Cell. Mol. Life Sci. 73, 2195–2209 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.V. O. Sviderskiy, E. M. Terzi, R. Possemato, Iron–sulfur cluster metabolism impacts iron homeostasis, ferroptosis sensitivity, and human disease, in Ferroptosis in Health and Disease, D. Tang, Ed. (Springer International Publishing: Cham, 2019), pp. 215–237. [Google Scholar]
- 5.Hentze M. W., Muckenthaler M. U., Galy B., Camaschella C., Two to tango: Regulation of mammalian iron metabolism. Cell 142, 24–38 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Hirling H., Henderson B. R., Kuhn L. C., Mutational analysis of the [4fe-4s]-cluster converting iron regulatory factor from its RNA-binding form to cytoplasmic aconitase. EMBO J. 13, 453–461 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tong W. H., Rouault T. A., Functions of mitochondrial ISCU and cytosolic ISCU in mammalian iron-sulfur cluster biogenesis and iron homeostasis. Cell Metab. 3, 199–210 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Doll S., Conrad M., Iron and ferroptosis: A still ill-defined liaison. IUBMB Life 69, 423–434 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Alvarez S. W., Sviderskiy V. O., Terzi E. M., Papagiannakopoulos T., Moreira A. L., Adams S., Sabatini D. M., Birsoy K., Possemato R., NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 551, 639–643 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wingert R. A., Galloway J. L., Barut B., Foott H., Fraenkel P., Axe J. L., Weber G. J., Dooley K., Davidson A. J., Schmid B., Paw B. H., Shaw G. C., Kingsley P., Palis J., Schubert H., Chen O., Kaplan J., Zon L. I.; Tübingen 2000 Screen Consortium , Deficiency of glutaredoxin 5 reveals Fe-S clusters are required for vertebrate haem synthesis. Nature 436, 1035–1039 (2005). [DOI] [PubMed] [Google Scholar]
- 11.Ducamp S., Fleming M. D., The molecular genetics of sideroblastic anemia. Blood 133, 59–69 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gakh O., Bedekovics T., Duncan S. F., Smith D. Y. IV, Berkholz D. S., Isaya G., Normal and Friedreich ataxia cells express different isoforms of frataxin with complementary roles in iron-sulfur cluster assembly. J. Biol. Chem. 285, 38486–38501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vaubel R. A., Isaya G., Iron-sulfur cluster synthesis, iron homeostasis and oxidative stress in Friedreich ataxia. Mol. Cell. Neurosci. 55, 50–61 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sullivan L. B., Chandel N. S., Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2, 17 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hassannia B., Vandenabeele P., Vanden Berghe T., Targeting ferroptosis to iron out cancer. Cancer Cell 35, 830–849 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Dixon S. J., Stockwell B. R., The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 10, 9–17 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Aziz N., Munro H. N., Iron regulates ferritin mRNA translation through a segment of its 5’ untranslated region. Proc. Natl. Acad. Sci. U.S.A. 84, 8478–8482 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hentze M. W., Caughman S. W., Rouault T. A., Barriocanal J. G., Dancis A., Harford J. B., Klausner R. D., Identification of the iron-responsive element for the translational regulation of human ferritin mRNA. Science 238, 1570–1573 (1987). [DOI] [PubMed] [Google Scholar]
- 19.Koeller D. M., Casey J. L., Hentze M. W., Gerhardt E. M., Chan L. N., Klausner R. D., Harford J. B., A cytosolic protein binds to structural elements within the iron regulatory region of the transferrin receptor mRNA. Proc. Natl. Acad. Sci. U.S.A. 86, 3574–3578 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crooks D. R., Maio N., Lane A. N., Jarnik M., Higashi R. M., Haller R. G., Yang Y., Fan T. W. M., Linehan W. M., Rouault T. A., Acute loss of iron-sulfur clusters results in metabolic reprogramming and generation of lipid droplets in mammalian cells. J. Biol. Chem. 293, 8297–8311 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pietsch E. C., Chan J. Y., Torti F. M., Torti S. V., Nrf2 mediates the induction of ferritin H in response to xenobiotics and cancer chemopreventive dithiolethiones. J. Biol. Chem. 278, 2361–2369 (2003). [DOI] [PubMed] [Google Scholar]
- 22.Salahudeen A. A., Thompson J. W., Ruiz J. C., Ma H. W., Kinch L. N., Li Q., Grishin N. V., Bruick R. K., An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis. Science 326, 722–726 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vashisht A. A., Zumbrennen K. B., Huang X., Powers D. N., Durazo A., Sun D., Bhaskaran N., Persson A., Uhlen M., Sangfelt O., Spruck C., Leibold E. A., Wohlschlegel J. A., Control of iron homeostasis by an iron-regulated ubiquitin ligase. Science 326, 718–721 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang H., Shi H., Rajan M., Canarie E. R., Hong S., Simoneschi D., Pagano M., Bush M. F., Stoll S., Leibold E. A., Zheng N., FBXL5 regulates IRP2 stability in iron homeostasis via an oxygen-responsive [2Fe2S] cluster. Mol. Cell 78, 31–41.e5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dodson M., Castro-Portuguez R., Zhang D. D., NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 23, 101107 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo B., Yu Y., Leibold E. A., Iron regulates cytoplasmic levels of a novel iron-responsive element-binding protein without aconitase activity. J. Biol. Chem. 269, 24252–24260 (1994). [PubMed] [Google Scholar]
- 27.Tsai C. L., Barondeau D. P., Human frataxin is an allosteric switch that activates the Fe-S cluster biosynthetic complex. Biochemistry 49, 9132–9139 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Vaupel P., Kallinowski F., Okunieff P., Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: A review. Cancer Res. 49, 6449–6465 (1989). [PubMed] [Google Scholar]
- 29.Meyron-Holtz E. G., Ghosh M. C., Rouault T. A., Mammalian tissue oxygen levels modulate iron-regulatory protein activities in vivo. Science 306, 2087–2090 (2004). [DOI] [PubMed] [Google Scholar]
- 30.Malcovati L., Karimi M., Papaemmanuil E., Ambaglio I., Jädersten M., Jansson M., Elena C., Gallì A., Walldin G., Della Porta M. G., Raaschou-Jensen K., Travaglino E., Kallenbach K., Pietra D., Ljungström V., Conte S., Boveri E., Invernizzi R., Rosenquist R., Campbell P. J., Cazzola M., Hellström Lindberg E., SF3B1 mutation identifies a distinct subset of myelodysplastic syndrome with ring sideroblasts. Blood 126, 233–241 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dolatshad H., Pellagatti A., Liberante F. G., Llorian M., Repapi E., Steeples V., Roy S., Scifo L., Armstrong R. N., Shaw J., Yip B. H., Killick S., Kušec R., Taylor S., Mills K. I., Savage K. I., Smith C. W. J., Boultwood J., Cryptic splicing events in the iron transporter ABCB7 and other key target genes in SF3B1-mutant myelodysplastic syndromes. Leukemia 30, 2322–2331 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cotticelli M. G., Xia S., Lin D., Lee T., Terrab L., Wipf P., Huryn D. M., Wilson R. B., Ferroptosis as a novel therapeutic target for Friedreich’s ataxia. J. Pharmacol. Exp. Ther. 369, 47–54 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Du J., Zhou Y., Li Y., Xia J., Chen Y., Chen S., Wang X., Sun W., Wang T., Ren X., Wang X., An Y., Lu K., Hu W., Huang S., Li J., Tong X., Wang Y., Identification of Frataxin as a regulator of ferroptosis. Redox Biol. 32, 101483 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/22/eabg4302/DC1