

Upon mitochondrial dysfunction, CHOP acts as a rheostat that attenuates prolonged stress and delays the onset of cardiomyopathy.
Abstract
In response to disturbed mitochondrial gene expression and protein synthesis, an adaptive transcriptional response sharing a signature of the integrated stress response (ISR) is activated. We report an intricate interplay between three transcription factors regulating the mitochondrial stress response: CHOP, C/EBPβ, and ATF4. We show that CHOP acts as a rheostat that attenuates prolonged ISR, prevents unfavorable metabolic alterations, and postpones the onset of mitochondrial cardiomyopathy. Upon mitochondrial dysfunction, CHOP interaction with C/EBPβ is needed to adjust ATF4 levels, thus preventing overactivation of the ATF4-regulated transcriptional program. Failure of this interaction switches ISR from an acute to a chronic state, leading to early respiratory chain deficiency, energy crisis, and premature death. Therefore, contrary to its previously proposed role as a transcriptional activator of mitochondrial unfolded protein response, our results highlight a role of CHOP in the fine-tuning of mitochondrial ISR in mammals.
INTRODUCTION
Mitochondrial diseases are a heterogeneous group of devastating disorders characterized by respiratory chain dysfunction (1). Although mitochondrial disorders have distinct tissue and organ presentation, they seem to activate common stress responses evolved to mitigate the negative impact of respiratory deficiency on cellular and organismal metabolism (1). It appears that mitochondrial stress responses precede respiratory chain deficiency, thereby suggesting that they constitute an early event in pathogenesis of mitochondria-related diseases (2). This suggests that monitoring the activation and/or alteration of mitochondrial stress responses may provide early diagnostic markers in these conditions. Moreover, manipulation of mitochondrial stress responses may be beneficial for patients with mitochondrial disease and thus therapeutically exploited (3, 4).
Initially, the mitochondrial unfolded protein response (UPRmt) was postulated to be a common stress response to respiratory chain dysfunction (5). UPRmt constitutes a transcriptional program that up-regulates mitochondrial chaperones and proteases aimed to restore the loss of organelle proteostasis. Notwithstanding that UPRmt was first described to be triggered by the accumulation of misfolded proteins within the mitochondrial matrix in mammalian cells (5), most of the subsequent mechanistic studies were performed in Caenorhabditis elegans (6). In contrast, many aspects of the mammalian UPRmt signaling are less well understood. In mammalian cells, it is thought that mitochondrial proteotoxic stress leads to CHOP [CCAAT/enhancer binding protein (C/EBP) homology protein] up-regulation resulting in up-regulated transcription of UPRmt-responsive genes (5, 7). The CHOP-binding sites in the UPRmt gene promoters are presumably flanked by two conserved regions named the mitochondrial UPR elements 1 and 2 (MURE1 and MURE2) (7, 8). The role of CHOP in governing transcription of UPRmt genes is however controversial as the transcription factors that bind to MURE1 and MURE2 elements have not been identified (7, 9). Nevertheless, multiple studies confirmed up-regulation of the CHOP mRNA in cells derived from patients with various mitochondrial disorders, as well as mitochondrial disease models (2, 10–12). This illustrates that although CHOP plays a pivotal role in mammalian mitochondrial stress responses, the underpinning mechanisms of its actions in the context of mitochondrial dysfunction are still obscure.
Recently, it became clear that unlike in C. elegans, mammalian UPRmt may not be the primary response to mitochondrial dysfunction but rather function as a part of more complex mitochondrial stress response (11–14). Mammalian cells treated with mitochondrial toxins exhibit transcriptional reprogramming mimicking the integrated stress response (ISR) arm of the UPR, which is centered on the activating transcription factor 4 (ATF4) (13, 14). Consistent with this, studies carried out in models with defects in different steps of mitochondrial DNA (mtDNA) expression and protein synthesis revealed activation of ISR transcriptional signatures (11, 12). ISR hallmarks are increased eIF2α phosphorylation, reduction in ternary eIF2:tRNAiMet:guanosine 5′-triphosphate (GTP) complex levels, and subsequent inhibition of global protein synthesis that is paralleled by selectively induced translation of a subset of inhibitory upstream open reading frame (uORF) containing stress-responsive mRNAs, including ATF4, CHOP, and GADD34 (15). CHOP induction during ISR is thought to lead to cell death via induction of Growth Arrest and DNA Damage-Inducible Protein 34 (GADD34)–mediated eIF2a dephosphorylation and activation of Endoplasmic Reticulum Oxidoreductase 1 Alpha (ERO1A) endoplasmic reticulum (ER) oxidase (16).
CHOP is a multifunctional transcription factor that dimerizes with members of the C/EBP and ATF/cyclic adenosine 3′,5′-monophosphate response element binding protein families (17). Although up-regulated in response to a wide variety of stresses such as growth arrest and DNA damage, amino acid and glucose deprivation, hypoxia, and ER stress, the role of CHOP in cellular physiology is incompletely understood. CHOP is considered to induce apoptosis, but its transcriptional targets largely overlap with those of ATF4, including genes promoting cell survival and growth (16, 18). These findings highlight the intricate interaction partner–dependent roles of CHOP under different stresses and in various tissues. They also point out the importance of understanding the context-dependent role of CHOP under different physiological conditions. In the context of mitochondrial respiratory chain dysfunction, the role of CHOP is particularly important as CHOP was proposed to be the main transcription factor that conveys specificity of the mitochondrial stress response (5).
Here, we aimed to decipher the role of CHOP in the regulation of the mitochondrial stress response. As a model for the most common cause of mitochondrial diseases, namely, loss of mitochondrial translation, we used mice deficient in the mitochondrial aspartyl transfer RNA (tRNA) synthase DARS2 specifically in heart and skeletal muscle (DARS2 KO) (2). We demonstrate a beneficial role of CHOP in mitochondrial mutants as its loss leads to a marked shortening of life span in DARS2/CHOP double knockout (DKO) as compared to DARS2 KO animals. The beneficial effects of CHOP appear to be independent of UPRmt activation but rather mediated by attenuation of harmful overactivation of the ISR and a consequent metabolic imbalance. We also provide mechanistic evidence that these effects stem from the interplay between CHOP, ATF4, and C/EBPβ in regulation of mitochondrial ISR targets.
RESULTS
CHOP delays pathological changes caused by DARS2 deficiency in heart
To determine the in vivo function of CHOP in the context of mammalian mitochondrial dysfunction, we intercrossed whole-body Chop−/− mice (CHOP KO) with heart and skeletal muscle-specific DARS2-deficient mice (Dars2fl/fl; Ckmm-Cre+/tg; DARS2 KO) (fig. S1, A and B) (2). The resulting animals deficient in both CHOP (whole body) and DARS2 (heart and skeletal muscle) (Dars2fl/fl; Ckmm-Cre+/tg; Chop−/− and DKO) were born in Mendelian ratios (fig. S1C). We previously showed that DARS2 depletion mediated by Ckmm-Cre expression induces dilated cardiomyopathy preceding any pathological phenotypes in skeletal muscle (2). Hence, we monitored the effects of CHOP loss on pathologies caused by DARS2 abrogation in the heart.
Approximately from 2 weeks of age, a large number of DKO mice became increasingly susceptible to sudden death during a routine ear-clipping handling for genotyping. This procedure was tolerated well up to postnatal day 13 (P13) by mice of all four genotypes; hence P13 (±1) was defined as the early stage of heart dysfunction in DKO animals (DKOE). It appeared that the deterioration of the health status of DKO mice characterized by lower spontaneous cage activity, piloerection, unsteady gait, and overall droopiness is a very rapid process as the interval from the first apparent symptoms to death of the mice at around P17 (±2) was between 24 and 48 hours. This interval was defined as the late/terminal stage in DKO mice (DKOL). Consequently, the life expectancy of DKOs was severely reduced (>60%) compared to DARS2 KOs, signifying the essential role for CHOP in adaptation to impaired mitochondrial protein synthesis in heart (Fig. 1A). CHOP deficiency in the absence of DARS2 resulted in dilated cardiomyopathy (Fig. 1B and fig. S1, D to F) characterized by increased expression of mRNAs encoding cardiac hypertrophy markers Nppa and Nppb (Fig. 1C). Although no gross morphological changes were observed upon hematoxylin and eosin (H&E) staining, ultrastructural analyses suggested a disrupted myocardial organization, characterized by severely disorganized sarcomeric structures, expected to cause disturbances in contractile function of DKOL hearts (Fig. 1, D and E). Therefore, DKOL animals display very similar pathological changes, as compared to the terminal stage DARS2 KO mice (2), whereby the onset of these pathologies is markedly accelerated upon CHOP loss.
Fig. 1. Phenotypic characterization of DKO animals.
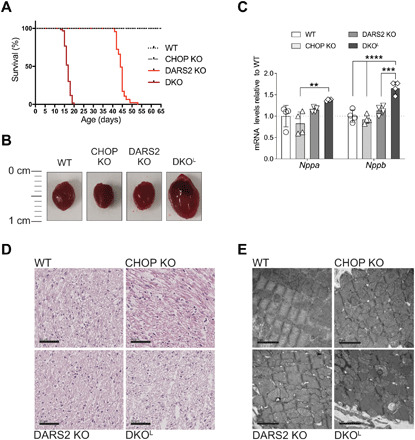
(A) Kaplan-Meyer survival curves for wild-type (WT; n = 36), CHOP KO (n = 35), DARS2 KO (n = 47), and DKO animals (n = 60). The life span of DKO in comparison to DARS2 KO mice is significantly decreased (P < 0.0001; log-rank test and Gehan-Breslow-Wilcoxon test). The viability of CHOP KO mice was WT-like in a 12-month follow-up. (B) Heart gross morphology. (C) Fold changes of the cardiac hypertrophy markers Nppa and Nppb obtained from the RNA sequencing dataset at P17 (±2) (n = 4). Bars represent means ± SD [multivariate analysis of variance (MANOVA) followed by one-way ANOVA and Tukey’s multiple comparisons test, **P < 0.05, ***P < 0.001, and ****P < 0.0001]. (D) H&E staining; (n = 3) at P17 (±2). Scale bars, 50 μm. (E) Transmission electron microscopy–based analyses of cardiac tissue biopsies; (n = 1) at P17 (±2). Scale bars, 2 μm.
Mitochondrial dysfunction is aggravated by the loss of CHOP
We next sought to identify pathways that are affected by the CHOP deficiency in the context of DARS2 KO. To this end, we compared global changes in mRNA levels to corresponding changes in the proteome in CHOP KO, DARS2 KO, and DKOL versus control hearts using the anota2seq algorithm (19). Scatter plots comparing mRNA and protein changes in DARS2 KO hearts revealed alterations in protein levels that were mainly independent of the mRNA levels, thus arguing for a prevalent impact of translational and/or protein stability changes on the proteome (Fig. 2A, fig. S2A, and table S1). In contrast, DKOL animals primarily showed congruent changes in mRNA and protein levels, which accounted for ~75% of detected alterations in protein levels (Fig. 2A, fig. S2A, and table S1).
Fig. 2. Loss of Chop causes strong respiratory defects that can only partially be explained by an exacerbated defect in mitochondrial translation.
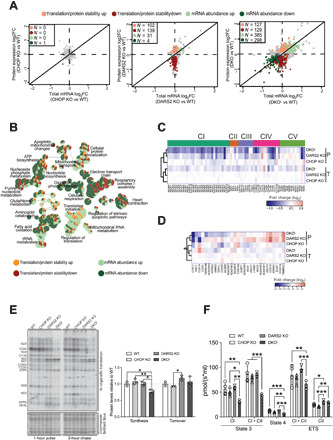
(A) Scatter plots of total mRNA and protein fold changes (FC) comparing CHOP KO, DARS2 KO, or DKOL to WT. The numbers of significantly regulated genes are indicated for translation/protein stability (red), and mRNA abundance (green). RNA sequencing and quantitative proteomics were performed on hearts of animals at P17 (±2) (n = 4). (B) A GO network of overrepresented terms among genes regulated via changes in translation/protein stability (up-regulated, light red; down-regulated, dark red) and mRNA abundance (up-regulated, light green; down-regulated, dark green) in DKO versus WT. Nodes represent identified GO terms, while the pie chart within each node indicates the proportion of genes regulated. (C and D) Heatmap of protein expression (P) and total mRNA (T) log2 fold changes of (C) the OXPHOS subunits grouped in respective complexes and (D) OXPHOS assembly factors (n = 4). (E) In organello translation assay (left) of cardiac mitochondria at P17 (±2). De novo protein synthesis was determined after 1 hour of 35S-methionine pulse labeling; protein turnover was assessed after 3 hours of the cold chase. Coomassie brilliant blue–stained gel was used as a loading control. Relative protein synthesis and turnover rates (right) (n = 3). (F) Oxygen consumption of intact cardiac mitochondria at P17 (±2). State 3: adenosine 5′-diphosphate (ADP)–stimulated respiration using CI or CI + CII substrates. State 4: Respiration upon addition of oligomycin. ETS, maximum respiration upon mitochondrial uncoupling (CI) and after addition of rotenone (CII) (n = 3 to 4). Bars represent means ± SD (MANOVA followed by one-way ANOVA and Tukey’s multiple comparisons test, *P < 0.05, **P < 0.01, and ***P < 0.001).
Gene Ontology (GO) analysis performed using ClueGO (20) and annotation from the GO consortium (21) on genes whose expression was reduced indicated that oxidative phosphorylation, electron transport, complex I assembly, adenosine 5′-triphosphate (ATP) biosynthesis, fatty acid oxidation, and heart contraction are predominantly disrupted in DKO hearts (Fig. 2B). This is consistent with the impairment of mitochondrial energy production and heart failure in DKO animals and similar to other models of mitochondrial cardiomyopathy (11). In contrast, translation, tRNA metabolism, mitochondrial RNA, and glutathione metabolism were primarily up-regulated pathways (Fig. 2B). We observed further perturbations in apoptotic pathways, amino acid catabolism, and purine nucleotide metabolism that contained a combination of up- and down-regulated gene expression changes (Fig. 2B).
A general down-regulation of steady-state levels of individual oxidative phosphorylation (OXPHOS) subunits detected in DARS2 KO hearts was further decreased in DKOL animals (Fig. 2C and fig. S2B). Intriguingly, while in DARS2 KO animals, most of the changes in the levels of OXPHOS subunits were not accompanied by alterations in mRNA abundance, numerous OXPHOS subunit-encoding genes exhibited congruent changes in mRNA and protein levels in DKOL animals (Fig. 2C). These include three of four subunits of succinate dehydrogenase (SDH; complex II), a complex fully encoded by nuclear DNA, usually up-regulated upon mitochondrial translation defects. This was further confirmed using an enzyme-histochemical assay, showing that substantial cyclooxygenase (COX) deficiency observed in DKOL animals is not accompanied by a compensatory SDH up-regulation (fig. S2C), as observed in DARS2 and other mitochondrial mutants (2, 22). Furthermore, while we detected a general compensatory up-regulation of OXPHOS assembly factors in DARS2 KO hearts, many were either unaltered or down-regulated in DKOL samples (Fig. 2D).
Although Dars2 deletion primarily interferes with mitochondrial protein synthesis, at P17, only a moderately dysbalanced mitochondrial translation was observed in DARS2 KO (Fig. 2E). In contrast, mitochondrial de novo protein synthesis in DKOL mice was significantly decreased and severely dysregulated, whereas the protein turnover remained unaffected (Fig. 2E). The exaggerated translation defect observed in DKOL animals was not caused by a decrease in mtDNA or mtRNA levels (fig. S2, D and E). Some mtRNAs were up-regulated (e.g., mt-COX3 and mt-ND1) in both DARS2 KO and DKO hearts, possibly as a compensatory response to defective protein synthesis (Fig. 2E and fig. S2E).
Severe dysregulation of mitochondrial translation in DKOL was accompanied with a strong decrease in the respiration capacity of all inducible states in mitochondria isolated from DKOL hearts (Fig. 2F). In contrast, no major defects in DARS2 KO heart mitochondria respiration were observed, thus suggesting compensation for the mitochondrial protein synthesis defect (Fig. 2F).
Unexpectedly, a comparable defect at the level of assembled respiratory chain complexes and supercomplexes was detected in DARS2 KO and DKOL mice despite higher levels of individual OXPHOS subunits in DARS2 KO (Fig. 2C and fig. S2F). These data suggest that, at early stages of DARS2 deficiency, nascent nuclear-encoded OXPHOS subunits are not efficiently incorporated in respiratory chain complexes in DARS2 KO hearts and are likely turned over at higher rates. Although DKOL and DARS2 KO mitochondria have comparable levels of respiratory chain supercomplexes (fig. S2F), DKOL mitochondria fail to sustain normal respiration (Fig. 2F). This suggests that the OXPHOS activity is further indirectly affected by CHOP deficiency that might lead to disruption of mitochondrial integrity or supply of critical metabolites.
CHOP deficiency causes a transcriptional reprogramming characterized by amplified ISR
CHOP deficiency in the context of mitochondrial dysfunction is expected to blunt the mitochondrial stress response (5). Therefore, by analyzing changes in the transcriptome, we compared pathways that are affected in DARS2-deficient hearts before and after CHOP depletion (table S2).
In DARS2 KO heart, relatively few mRNAs changed their expression, and most were up-regulated. Notably, using Cytoscape plug-in iRegulon, we demonstrated that two-thirds of these transcripts overlapped with an ISR signature activated by ATF4, which was also identified as the most prominent regulator of gene expression in DARS2 KO hearts (Fig. 3A and tables S2 and S3) (18, 23, 24). The most up-regulated transcripts in DARS2 KO hearts encoded enzymes involved in one-carbon metabolism, serine biosynthesis, and trans-sulfuration, as well as Gdf15 and Fgf21 (Fig. 3, A and B, and table S2), the two cytokines shown to be excreted from tissues upon OXPHOS deficiency (25, 26). Similar changes (Fig. 3A) were previously described in other cellular and in vivo models for mitochondrial OXPHOS defects, confirming that DARS2 deficiency causes a stress response relevant for many mitochondrial disease states (11–14).
Fig. 3. DKOL mice display strong overactivation of the ISR.
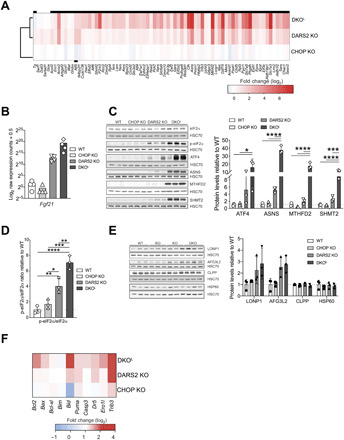
(A) Heatmap of total mRNA fold changes (log2) of significantly changed ATF4 target genes [as predicted by Cytoscape plug-in iRegulon (23, 24)], in DARS2 KO animals compared to WT controls. Black boxes above DKO and below CHOP KO rows indicate their respective significantly changed transcripts as compared to WT controls (n = 4). (B) Fgf21 log2 raw expression counts (sequenced reads, +0.5) as this gene was not detected in multiple WT and CHOP KO samples and hence was excluded during data filtering. Of note, these samples will obtain negative log2 values (n = 4). (C) Western blot analysis (left) and quantification of ISR markers (right). HSC70 was used as a loading control. Antibodies used were raised against proteins indicated in panels. Experiments were performed on cardiac lysates of mice at P17 (±2) (n = 3). (D) p-eIF2α/eIF2α ratio quantified from (C). (B to D) Bars represent means ± SD (MANOVA followed by one-way ANOVA and Tukey’s multiple comparisons test, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001). (E) Western blot analysis and quantification of UPRmt markers in WT, CHOP KO, DARS2 KO, and DKO animals at P17 (±2). Antibodies used were raised against proteins indicated in panels. HSC70 was used as a loading control. Bars represent means ± SD; no significant differences were detected (MANOVA: Wilk’s test, P = 0.176; Hotelling-Lawley’s test, P = 0.183; Pollai’s test, P = 0.232) (n = 3). (F) Heatmap of total mRNA fold changes (log2) for the selected alleged CHOP target genes involved in apoptosis (n = 4).
The ISR activation in DARS2 KO hearts was confirmed by increased eIF2α phosphorylation, accompanied by up-regulation of ATF4 (Fig. 3, C and D). These effects were further potentiated by CHOP loss, whereby induction of both eIF2α phosphorylation and ATF4 was more pronounced in DKOL relative to DARS2 KO hearts (Fig. 3, C and D). Transcript and protein levels of almost all ATF4 targets were highly up-regulated in DKOL as compared to DARS2 KO animals (Fig. 3, A to C). Consistently, further analysis of binding motifs in genes up-regulated in DKOL hearts established ATF4 as the most prominent signature (table S3) (23, 24). The most up-regulated transcripts in DARS2 KO and DKOL showed a notable overlap. To this end, of the top 11 most up-regulated transcripts, 8 overlapped, despite the 40-fold difference in the number of overall changes between the two models (table S2). The only difference was that these transcripts were, on average, more than 10-fold more up-regulated in DKOL than in DARS2 KO hearts (table S2). In contrast, UPRmt markers were not significantly changed in DARS2 KO or DKOL animals, adding evidence that UPRmt is neither an early nor prominent stress response in mammalian cells and tissues upon mitochondrial OXPHOS dysfunction (Fig. 3E). Instead, our data suggest a central role for ISR and ATF4-dependent regulation in the context of mitochondrial dysfunction in vivo and point to an unexpected role of CHOP in the suppression of the transcriptional overactivation of ATF4 targets.
CHOP is proposed to be involved in the regulation of apoptosis upon ER stress, although the exact mechanism remains controversial, as exogenously expressed CHOP has also been reported to positively regulate genes involved in protein synthesis and not apoptosis (16, 18). Henceforth, we analyzed changes in the expression levels of various apoptotic genes reported to be CHOP targets (27). Notably, proapoptotic members of the B-cell lymphoma 2 (BCL-2) family (Puma/Bbc3, Bid, Bax, and Bim/Bcl2l11) and genes encoding proteins involved in the activation or execution of apoptosis (Dr5/Tnfrsf10b, Casp3, and Ero1l) were not suppressed but often further up-regulated upon loss of CHOP in DARS2-deficient animal (Fig. 3F). Similarly, the steady-state level of proapoptotic protein BCL2-associated X protein (BAX) was up-regulated, and we observed a higher cleavage of caspase 3 in DKOL hearts as compared to control animals (fig. S3A). These results suggest that, unexpectedly, apoptosis may be up-regulated in DARS2-deficient hearts upon CHOP depletion and thus contribute to the detrimental phenotype observed in DKOL mice.
As we observed major changes in the abundance of proteins involved in amino acid metabolism, we next measured amino acid levels by liquid chromatography–tandem mass spectrometry. While only minor perturbations in amino acid levels were observed in DARS2 KO hearts, most amino acids were significantly up-regulated in DKO mice (fig. S3B). Of note, serine, glutamine, glutamate, and aspartate levels were not significantly changed in either DARS2 KO or DKOL relative to control hearts (fig. S3B). The unaltered serine levels, despite the increased levels of enzymes involved in serine synthesis [Phosphoglycerate dehydrogenase (PHGDH), Phosphoserine Aminotransferase 1 (PSAT1) , and Phosphoserine Phosphatase (PSPH)], suggest an increased flux of serine-derived one-carbon units for further methylation reactions into the one-carbon cycle. Similarly, glutamine and glutamate are likely used to replenish tricarboxylic acid cycle intermediates and aspartate production that is essential for nucleotide synthesis and cell proliferation (28, 29). Increased levels of citrate and isocitrate in DKOL, but not DARS2 KO, hearts suggest that glutamine primarily undergoes reductive metabolism (fig. S3C), as seen in the patient-derived cell lines harboring mtDNA mutations (30). Increased citrate levels can propagate intracellular acidosis, leading to hypocalcemia caused by reduced availability of Ca2+, further contributing to reduced contractility of the heart through a vicious circle of the excitation-contraction-metabolism impairment (31). Additional effects of elevated citrate levels on the regulation of metabolic enzyme and/or chromatin dynamics by acetylation may further contribute to accelerated pathological phenotypes observed in DKOL hearts.
ISR allows adaptation to mitochondrial mutants, while its overactivation affects cell proliferation
Next, we tested whether mitochondrial stress–induced ISR has a beneficial or detrimental role in conditions of mitochondrial dysfunction. For these analyses, we took advantage of two cell models for mitochondrial respiratory chain dysfunction: (i) mouse skin fibroblasts with severe mitochondrial dysfunction caused by the loss of COX10 (COX10 KO), an early assembly factor of the respiratory cytochrome c oxidase (32); and (ii) mouse embryonic fibroblasts (MEFs) treated with actinonin, an inhibitor of mitochondrial peptide deformylase causing impairment in mitochondrial translation (33).
In the COX10 KO cells, a robust activation of the ISR was detected as evidenced by increased levels of phosphorylated eIF2α, ATF4, and ATF4 targets (Fig. 4A and fig. S4A). To test whether increased ATF4 levels are a direct result of ISR activation, we incubated COX10 KO cells with the ISR inhibitor (ISRIB) (34). This treatment abrogated ATF4 induction and attenuated up-regulation of its downstream targets at both transcript and protein levels (Fig. 4A and fig. S4A). The phosphorylation of eIF2α remained unchanged (Fig. 4A), which was expected as ISRIB bolsters guanine-nucleoside exchange activity of eIF2B without affecting on phospho-eIF2α levels (34). Similarly, increased ATF4 levels induced by actinonin treatment were suppressed by ISRIB (Fig. 4B). Mirroring the results from DKOL mice, loss of CHOP combined with mitochondrial dysfunction induced by actinonin treatment greatly increased ATF4 protein and transcript levels, and expression of ATF4 targets Shmt2, Pycr1, and Mthfd2 (Fig. 4, B and C).
Fig. 4. Mitochondrial ISR has opposing effects depending on the levels of activation mediated by ATF4.
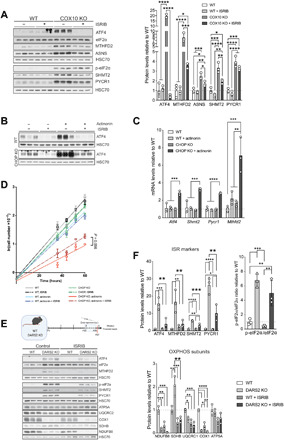
(A) Western blot analysis (left) and relative protein levels (right) of ISR markers and ATF4 downstream targets in immortalized COX10 KO and WT fibroblasts upon 48-hour treatment with DMSO (−) or ISRIB (+). (B) Western blot analysis of WT and CHOP KO MEFs treated for 48 hours with DMSO (−) or actinonin (+) in the presence (+) or absence (−) of ISRIB during the last 4 hours before protein isolation. (C) Relative transcript levels in WT and CHOP KO MEFs treated for 48 hours with DMSO (control) or actinonin. Tbp expression was used for normalization (n = 3). (D) Growth curves of respective exponential growth phases of WT and CHOP KO MEFs treated with DMSO (control), actinonin, and +/−ISRIB, respectively. Curves were determined using linear regression (n = 3). Bars represent means ± SD. (E) Western blot analysis of heart lysates from 4-week-old WT and DARS2 KO animals treated with control (DMSO) and ISRIB, according to the experimental setup presented in the schematic illustration (top). Animals are treated with daily injections of saline (control) or ISRIB solution for 7 days (blue boxes), starting at P19, and euthanized at P27 (red line) (n = 3). (F) Quantification of ISR markers (top), OXPHOS subunits (bottom), and p-eIF2α/eIF2α ratio (right) from the Western blot analysis at (E). (A, B, and F) Antibodies used were raised against proteins indicated in panels. HSC70 was used as a loading control. (A, C, and F) Bars represent means ± SD (MANOVA followed by one-way ANOVA and Tukey’s multiple comparisons test, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001).
Prevention of ISR overactivation in CHOP KO MEFs by ISRIB treatment resulted in a partial rescue of the proliferation defect induced by actinonin (Fig. 4D). In turn, wild-type (WT) cells treated with actinonin and CHOP KO cells grown under control conditions showed minor growth defects, which were not further affected by ISRIB (Fig. 4D). Therefore, CHOP deficiency, only in conditions of mitochondrial dysfunction, results in a detrimental ISR activation, which can be partially rescued by ISRIB treatment.
To assess the effect of ISRIB treatment in vivo, DKOL mice and respective controls were injected with ISRIB (5 μg/g) for up to 7 or 12 days, starting from 1 week of age (fig. S4B). Unfortunately, neither protocol resulted in the suppression of ATF4 levels or downstream targets in either DKO or DARS2 KO animals nor did it affect steady-state levels of OXPHOS subunits (fig. S4, C and D). However, this is not unexpected given the fact that ISRIB inhibits low-level ISR activity but does not affect strong ISR signaling (35), as observed in DKO mice. In contrast, a 7-day treatment of DARS2 KO animals with ISRIB, starting from P20, resulted in an apparent reduction of ISR markers (Fig. 4, E and F). Nevertheless, ISRIB-mediated suppression of ISR in DARS2 KO animals up to 4 weeks of age was not beneficial as it also prevented compensatory complex II (CII) up-regulation.
The transition from acute to prolonged ISR coincides with the rapid deterioration of the DKO phenotype
One of the hallmarks of the acute ISR is suppression of global protein synthesis, accompanied by translational activation of some uORF-containing mRNAs (15). To further understand the consequences of ISR activation in our model, we measured the global protein synthesis rate at P6, P13, and P17 in vivo in DKO and control hearts (36). At P6, cytoplasmic translation of all four genotypes was similar, in agreement with no phenotypes observed at this time point (fig. S5A). Coinciding with increased eIF2α phosphorylation, a 70% decrease in general protein synthesis was detected in mice at P13 (DKOE; Fig. 5A and fig. S5B). Within a few days, this effect seems to be reversed as we detected fully recovered protein synthesis rates in DKOL hearts at P17 (Fig. 5B and fig. S5C). This was despite unaltered eIF2α phosphorylation levels and activation of ATF4 and its targets that were comparable between DKOE and DKOL hearts (fig. S5D). These findings suggested a transition from acute to prolonged ISR, characterized by recovery of global protein synthesis and ongoing translation of ISR-sensitive mRNAs (37). These distinctions in global protein synthesis levels reflected different phenotypes of DKOE and DKOL mice. In the acute ISR, when global translation is strongly down-regulated, DKOE (P13 ± 1) animals cope better with the mitochondrial translation defect when compared to DARS2 KO animals (Fig. 5C). This is illustrated by the unaffected levels of OXPHOS complexes and supercomplexes in DKOE animals (Fig. 5D and fig. S5E). However, these effects are reversed when DKO animals reach the prolonged ISR stage, which is characterized by partial recovery of global mRNA translation and sustained ATF4-mediated transcriptional reprograming (fig. S5D). This reactivation of normal translation is likely to result in ER stress, and further energy crisis as protein synthesis is highly energy demanding (38). Consistently, we detected increased levels of the ER-chaperone binding immunoglobulin protein (BIP) in P17 DKOL hearts, which mirrored findings in DARS2-deficient hearts at the terminal state of 6 weeks of age (Fig. 5E). Levels of several ER Ca2+ transporter proteins were also profoundly disturbed [Ryanodine receptors (RyR), Sarco/endoplasmic reticulum Ca2+-ATPase 2 (SERCA2), and The inositol 1,4,5-trisphosphate receptor type 2 (IP3RII)], which may explain defects in the conductive system of the heart (Fig. 5F). Perturbed Ca2+ homeostasis due to the dysregulation of the ER Ca2+ transporters and increased Ca2+ release by ERO1α-stimulated IP3R activation may also contribute to ER stress leading to the development of fatal cardiomyopathy (Fig. 5, E and F). Therefore, although strong activation of ISR, as seen in DKOE animals, brings brief protection from the mitochondrial dysfunction, it cannot be sustained over prolonged period of time and results in a detrimental switch to a prolonged ISR program leading to additional ER stress, loss of Ca2+ homeostasis, and premature death.
Fig. 5. Strong suppression of protein synthesis by overactivated ISR is beneficial but cannot be sustained in vivo leading to devastating consequences.
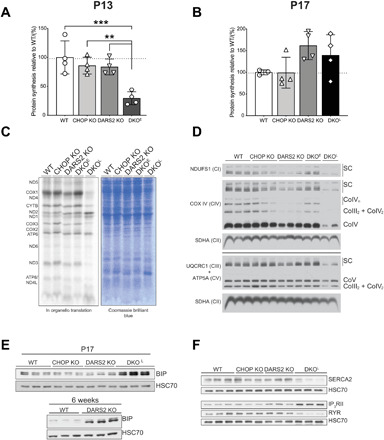
(A and B) The relative protein synthesis rate of animals injected with puromycin at (A) P13 and (B) P17. Bars represent means ± SD (one-way ANOVA and Tukey’s multiple comparisons test, **P < 0.01 and ***P < 0.001) (n = 4). (C) De novo synthesis in mitochondria isolated from WT, CHOP KO, DARS2 KO, and DKOE and DKOL animals after 1 hour of 35S-methionine pulse labeling followed by SDS-PAGE. Coomassie blue–stained gel was used as a loading control. (D) Blue native polyacrylamide gel electrophoresis (BN-PAGE) and subsequent Western blot analysis of OXPHOS complexes and supercomplexes in mitochondria isolated from WT, CHOP DO, DARS2 KO, and early (DKOE) and late-stage (DKOL) DKO animals. Subunit-specific antibodies (left) were used to detect respective complexes and supercomplexes (right) (n = 3). (E) Western blot analysis of BIP levels in WT, CHOP KO, DARS2 KO, and DKO at P17 (±2) (top) and WT and DARS2 KO at 6 weeks (bottom) (n = 3). (F) Western blot analysis proteins involved in the Ca2+ metabolism in WT, CHOP KO, DARS2 KO, and DKOL at P17 (±2) (n = 3). (E and F) HSC70 was used as a loading control (n = 3).
Interplay of CHOP and C/EBPβ mediates the suppression of ATF4 overexpression
The prolonged activation of ISR in DKOL hearts may have adverse effects on cellular and organismal fate. GADD34, a regulatory subunit of the enzyme dephosphorylating eIF2α, is thought to function as ISR rheostat acting to restore protein synthesis and block excessive ATF4 activation (15). Unexpectedly, although CHOP was proposed to be a primary Gadd34 transcriptional activator (16), DKOL animals at P17 showed a significant up-regulation of Gadd34 transcripts to similar levels as those observed in terminal, 6-week-old DARS2 KO animals (Fig. 6A). This result suggests that CHOP may play a GADD34-independent role in the suppression of the overactivation of ATF4 induction and ATF4-mediated transcriptional reprogramming.
Fig. 6. CHOP interacts with C/EBPb protein to repress the ATF4 activation.
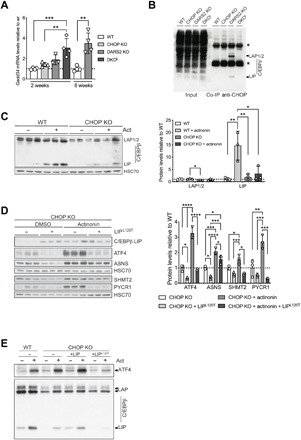
(A) Relative Gadd34 transcript levels at P17 (±2) WT, CHOP KO, DARS2 KO, and DKO animals, as well as in 6-week-old WT and DARS2 KO mice. Bars represent means ± SD, samples were normalized to WT mice of the respective age (P17: one-way ANOVA, *P < 0.05, **P < 0.01, and ***P < 0.001; 6 weeks: unpaired Student’s t test) (n = 4). (B) Coimmunoprecipitation (co-IP) of CHOP from WT, CHOP KO, DARS2 KO, and DKOL hearts. The CHOP and C/EBPβ interaction was monitored with Western blotting using an antibody against C/EBPβ. One percent of the input fractions was used as loading controls. Asterisks indicate the immunoglobulin G heavy and light chains. (C) Western blot analysis (left) and quantification (right) of the three CEBPβ isoforms LAP1, LAP2, and LIP in CHOP KO MEFs treated for 48 hours with actinonin along with the respective control (n = 3). (D) Western blot analysis (left) and quantification (right) of steady-state levels of ISR markers in actinonin-treated (48 hours) CHOP KO MEFs expressing the CEBPβ LIPL120T mutant variant along with the respective controls (n = 3). (E) Western blot analysis of the ATF4 and three CEBPβ isoforms in actinonin-treated (48 hours) CHOP KO MEFs expressing the CEBPβ LIP WT and CEBPβ LIPL120T mutant variant along with the WT cells and respective controls (n = 4). (C to E) Antibodies used were raised against proteins indicated in the panels. HSC70 was used as a loading control. (A, C, and D) Bars represent means ± SD (MANOVA followed by one-way ANOVA and Tukey’s multiple comparisons test, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001) (n = 3).
As a prerequisite for DNA binding, CHOP needs to heterodimerize with other transcription factors (17). To this end, to identify CHOP-interacting partners that may play a role in mitochondrial stress responses, we immunoprecipitated CHOP from DARS2 KO heart extracts, followed by mass spectrometry (table S4). Notably, besides CHOP, only six proteins were identified. Among those, the most enriched protein and the only transcription factor was C/EBPβ (table S4). These results were confirmed by Western blot analysis following coimmunoprecipitation (co-IP) against CHOP (Fig. 6B). Notably, CHOP and C/EBPβ appear to interact only upon mitochondrial dysfunction (i.e., DARS2 KO), despite similar levels of C/EBPβ in WT and DARS2 KO hearts (Fig. 6B and fig. S6A). The mass spectrometry analysis of C/EBPβ immunoprecipitates corroborated these results (table S5). In DKO hearts, C/EBPβ instead interacted with ATF4 and ATF3 (table S5). Previously, the induction of Atf3 was detected in the terminal stages mitochondrial stress responses along with UPRmt (12).
Further interplay of the three proteins is illustrated by the fact that mitochondrial dysfunction in C/EBPβ-deficient cells exacerbated the ISR stress and led to ATF4 activation similar to CHOP KO (fig. S6B). Interaction of CHOP with C/EBPβ was previously proposed in the context of mitochondrial dysfunction, wherein CHOP/C/EBPβ dimers are thought to bind and activate the promoters of UPRmt-responsive genes (5). Consistent with these results, we propose that C/EBPβ-CHOP heterodimers might act as suppressors of ATF4 overactivation upon mitochondrial dysfunction.
C/EBPβ is primarily regulated at the translational level and exists in three different isoforms, two activating [Liver-enriched activator protein (LAP1) and LAP2], and one inhibitory [Liver-enriched inhibitor protein (LIP)] (39). The C/EBPβ target genes are presumably positively regulated by LAP1/2 proteins, whereas LIP binding is thought to repress the transcription of respective promoter (39), although recently more complex functions have been proposed for C/EBPβ LIP in vivo (40). To further dissect the interplay between CHOP and C/EBPβ, we assessed the levels of all three C/EBPβ isoforms in different models of mitochondrial dysfunction. COX10 KO cells with strong chronic mitochondrial dysfunction presented an increase of all C/EBPβ isoforms (fig. S6C). Acute mitochondrial dysfunction caused by actinonin treatment in MEFs or DARS2 deficiency in heart had a milder effect on the levels of LAP isoforms (Fig. 6C and fig. S6D). Still, C/EBPβ LIP levels were strongly increased by actinonin treatment in WT cells (Fig. 6C). Notably, this effect was strongly blunted in CHOP-deficient cells and DKOL mice, indicating that an increase in C/EBPβ LIP levels is dependent on the CHOP presence (Fig. 6C and fig. S6D). In general, the CHOP presence seems to have a positive effect on the C/EBPβ levels in MEFs, indicating a regulation opposite to that of ATF4.
Under ER stress, CHOP and C/EBPβ LIP are shown to act in concert to exert their respective functions in the nucleus (41). According to the proposed model, CHOP depends on the interaction with C/EBPβ LIP to enter the nucleus, while the interaction with CHOP is thought to mask the nuclear export signal (NES) of C/EBPβ LIP, thereby reducing its exclusion from the nucleus and subsequent proteasomal degradation (41). To test whether C/EBPβ LIP plays a role in the direct regulation of the mitochondrial dysfunction–induced ISR, we expressed mutant LIPL120T, carrying a leucine-to-threonine substitution predicted to disrupt NES (42), in CHOP KO cells treated with actinonin (Fig. 6D). The expression of LIPL120T in CHOP KO cells resulted in intense ablation of basal and actinonin-induced ATF4 mRNA and protein levels and a marked decrease in the mRNA and protein levels of its downstream targets, even in the absence of mitochondrial insult (Fig. 6D and fig. S6E). Moreover, expression of LIPL120T mutant resulted in decreased expression of the endogenous C/ebpβ gene (fig. S6E). Intriguingly, moderate overexpression of WT C/EBPβ LIP in CHOP KO cells resulted in a mild further increase of ATF4 levels upon mitochondrial dysfunction (Fig. 6E). In contrast, C/EBPβ LIPL120T mutant suppresses ATF4 while also decreasing endogenous C/EBPβ levels (Fig. 6E). These results also suggest that mutant C/EBPβ LIPL120T does not require CHOP for its action.
It has been shown that ER stress leads to interdependent translocation and retention of C/EBPβ and CHOP inside the nucleus (41). Therefore, we next investigated the effects of mitochondrial stress on subcellular localization of C/EBPβ, CHOP, and ATF4. In both WT and CHOP KO cells, C/EBPβ and ATF4 were detected mainly in the nucleus (fig. S6F). The expression of either WT or mutant C/EBPβ LIP did not affect the subcellular localization of ATF4 in CHOP KO cells (Fig. 7A). Therefore, the ATF4 levels in CHOP-deficient cells appear not to be regulated through alterations in subcellular localization of LIP. Alternatively, in the absence of CHOP, C/EBPβ LIPL120T may bind ATF4 and prevent its translocation to the nucleus, thus promoting its degradation. To test this hypothesis, we incubated WT and CHOP KO cells in the presence or absence of the proteasome inhibitor MG132. In control conditions, both ATF4 and C/EBPβ were rapidly degraded, and only a modest fraction was retained and transported to the nucleus (Fig. 7B and fig. S6G). The rate of turnover, however, appeared not to be affected by mitochondrial function or CHOP deficiency (Fig. 7B and fig. S6G). In turn, mitochondrial dysfunction, induced by actinonin treatment, induced translocation of ATF4 to the nucleus and promoted activation of ISR. Of note, the expression of LIPL120T mutant resulted in lower levels of ATF4 in all fractions (Fig. 7B and fig. S6G). Overall, these results suggest that fine-tuning of mitochondrial stress responses is dependent on CHOP:C/EBPβ LIP interaction but not their subcellular localization nor their potential effects on the nuclear translocation of ATF4.
Fig. 7. Model of mitochondrial ISR regulation by CHOP-CEBPβ-ATF4 interplay.
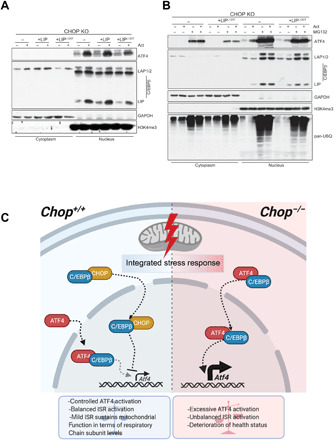
(A) Cell fractionation followed by the Western blot analysis of the ATF4 and three C/EBPβ isoforms in actinonin-treated (48 hours) WT or CHOP KO MEFs expressing WT C/EBPβ LIP or C/EBPβ LIPL120T mutant. (B) Cell fractionation followed by the Western blot analysis of the ATF4 and three C/EBPβ isoforms in actinonin-treated (48 hours) CHOP KO MEFs expressing WT C/EBPβ LIP and C/EBPβ LIPL120T mutant along with the WT cells and respective controls. The MG132 (15 μM) was applied in the last 6 hours of the actinonin treatment. Elevated protein ubiquitination reflects proteasome inhibition. (A and B) Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and H3K4me3 were used as loading controls and to determine quality of fractionation (n = 3). (C) CHOP levels increase early upon mitochondrial dysfunction leading to its association with C/EBPβ. The interaction with C/EBPβ likely promotes translocation of CHOP to the nucleus where it negatively regulates Atf4 levels and transcription of downstream ISR targets. Abrogation of CHOP results in increased ATF4:C/EBPβ association and transcription of ISR-regulated genes, created with BioRender.com.
DISCUSSION
Understanding of the mitochondrial stress response in mammals remains incomplete. In the present study, we uncovered an intricate interplay between three transcription factors regulating the mitochondrial stress response: CHOP, C/EBPβ, and ATF4. Contrary to its previously proposed role as a transcriptional activator of UPRmt, we present strong evidence that CHOP, through its interaction with C/EBPβ, attenuates prolonged ISR and mitochondrial cardiomyopathy through regulation of ATF4 levels (Fig. 7). Our results argue that upon mitochondrial dysfunction, the interaction of CHOP with C/EBPβ is needed for the adjustment of an ATF4-regulated transcriptional program. Very early upon DARS2 depletion, Chop is increasingly expressed (2) and forms a complex with C/EBPβ, which might facilitate the translocation of CHOP:C/EBPβ heterodimers to the nucleus. Regulation of ATF4 levels by C/EBPβ isoform LIP inhibition was proposed during ultraviolet (UV) stress, but CHOP was shown not to play a role in this context (43).
Similar to CHOP, C/EBPβ is a pleiotropic transcription factor that contributes to the regulation of homeostasis in several tissues, including bone, skin, and fat (40). We showed that in the context of mitochondrial dysfunction, the C/EBPβ accumulates in the cell (in particular, LIP isoform) and dimerizes with CHOP to presumably prevent overactivation of an ATF4-mediated response. In the absence of CHOP, C/EBPβ dimerizes with ATF4, which correlates with further induction of ISR. Our data suggest that C/EBPβ also dimerizes with ATF3 when CHOP is absent in DKO animals. ATF3 is shown to be activated during the second stage of ISR (12, 44). Once expressed, ATF3 binds promoters of ISR-responsive genes, leading to a subsequent suppression of transcription back toward the basal level (44). It is possible that also in the DKO animals, ATF3:C/EBPβ interaction is part of the feedback loop intended to suppress the ATF4 overactivation. In contrast, the interaction of ATF4 with C/EBPβ positively activates targeted genes under different conditions (45), which might have a deleterious outcome leading to, e.g., skeletal muscle atrophy (46). In contrast, we show that a dominant-negative C/EBPβ LIPL120T fully suppresses Atf4 and C/ebpβ overexpression upon mitochondrial dysfunction and down-regulates even basal levels of these transcription factors. Our findings thus suggest that C/EBPβ acts as a promiscuous transcription factor in the context of mitochondrial dysfunction, whereby differential transcriptional activity and associated functional outcomes are determined via interactions with CHOP and ATF4 (Fig. 7C). Further work is however required to dissect precise mechanisms of the observed interplay between CHOP, ATF4, and C/EBPβ.
CHOP is a transcription factor that is ubiquitously expressed at very low levels but quickly activated by a variety of insults such as ER stress, amino acid deprivation, glucose starvation, and UV irradiation (47). To date, CHOP was mostly studied in the context of ER stress, where it was proposed to regulate many pro- and anti-apoptotic genes in the late phase of ISR (47, 48). While numerous functions related to cell proliferation, differentiation, and development have been described for this transcription factor, in unstressed conditions, CHOP-deficient mice do not present any conspicuous phenotype (48, 49). Nevertheless, these mice seem to be protected from transient renal insufficiency caused by acute tubular necrosis (49). CHOP depletion seems to be beneficial in various other conditions, e.g., by delaying the onset of metabolic disease in several diabetic models (50), protecting livers from diet-induced hepatosteatosis (51), or delaying the onset of brain ischemia-induced neuronal cell death (52). Collectively, these studies suggest that loss of CHOP often leads to beneficial effects by delaying apoptosis in vivo. Unexpectedly, in mitochondrial mutants, CHOP depletion does not seem to decrease levels of proteins involved in the activation of apoptosis, as even the proposed bona fide CHOP targets BH3 interacting-domain death agonist (BID), Bcl-2-like protein 11 (BIM), ERO1A, and Tribbles homolog 3 (TRIB3) further increase their levels in DKO mutants.
We also provide evidence that CHOP loss is detrimental in mitochondrial mutants as it leads to early-onset fatal mitochondrial cardiomyopathy. This is, at least in part, mediated by the overactivation of ISR that is paralleled by inhibition of global protein synthesis and appears to be beneficial for a short time as DKOE animals maintain higher levels of OXPHOS complexes and balanced mitochondrial translation. However, loss of CHOP mitigates sustained suppression of protein synthesis in vivo that results in rapid loss of OXPHOS complexes and mitochondrial respiration. This is likely to affect mitochondrial import capacity leading to vicious cycle of damaging events. Simultaneously, mRNA translation rates are restored in DKOL around P17, coinciding with a detrimental phenotype. This is partly reminiscent of a transition from the acute to prolonged ISR in the cellular model of ER stress (37). During the acute ISR phase, global translation is reduced, and only a subset of stress-responsive mRNAs are translated, whereas the prolonged ISR is characterized by recovery of global translation while still allowing execution of acute ISR translational programs (37). While the prolonged ISR appears to have a beneficial effect in vitro by preventing cell death under conditions of ER stress (37), we show that in vivo, mitochondrial dysfunction in the heart impedes a sustained chronic ISR program. To this end, recovery of protein synthesis escalates ER stress possibly by increasing ER load. Recovery of global translation is also expected to significantly increase the energy demand and thereby result in energy depletion caused by massively reduced respiratory capacity due to DARS2 loss. According to the “energy starvation” hypothesis, suboptimal ATP supply predisposes for the contractile dysfunction observed during heart failure (53). It was shown that even very few cardiomyocytes with severe mitochondrial dysfunction are sufficient to promote ventricular arrhythmias, which lead to heart failure (54). Considering the severe impairment of electron transport chain (ETC) function in DKO mice, the occurrence of cardiac arrhythmias in those animals, contributing to the pathology, seems likely.
The pathology observed in DKOL animals is not a DARS2-specific phenomenon but a prevalent cardiac phenotype in mutants affecting mitochondrial gene expression and translation, as shown by a comparative study of five different models (11). At the molecular level, we demonstrated markedly increased serine synthesis and remodeling of the one-carbon cycle in hearts of DARS2 KO, DKOL mice, and cell culture models, attributable to OXPHOS deficiency and not to the loss of DARS2 in particular. Moreover, similar changes are described in other models and different tissues (11, 13, 14, 55). The vast majority of these alterations have been attributed to ATF4, which has been identified as a major regulator of amino acid metabolism feeding into the folate cycle during ISR induced by different stress signals including mitochondrial dysfunction (13, 14, 56). Although ATF4 may be activated by several different pathways, such as nuclear respiratory factor 2 (NRF2) stabilization or mechanistic (previously mammalian) target of rapamycin (mTOR) signaling (57, 58), we showed that ATF4 up-regulation caused by mitochondrial OXPHOS deficiency could be successfully prevented by suppression of the ISR.
In conclusion, we found a regulatory mechanism that fine-tunes the activation of the ISR upon mitochondrial dysfunction. We showed that CHOP is needed to prevent excessive activation of the ATF4-mediated stress response that results in cardiotoxic effects. This is mediated by CHOP interaction with C/EBPβ, which likely promotes CHOP:C/EBPβ heterodimer translocation to the nucleus. Our results also highlight an unforeseen opportunity of exploring a therapeutic intervention targeting ATF4 activity in various mitochondrial diseases.
MATERIALS AND METHODS
Generation and maintenance of mouse lines
DARS2 KO (Dars2fl/fl; Ckmm-Cre+/tg) mice were generated as previously described (2). WT control animals (Dars2fl/fl; Ckmm-Cre+/+ and Dars2+/fl; Ckmm-Cre+/+) were also obtained from this breeding. CHOP KO [B6.129S(Cg)-Ddit3tm2.1Dron/J] mice were obtained from the Jackson laboratory. Those mice are characterized by a Chop::LacZ KO allele, resulting in the whole-body KO of Chop (Chop−/−) (49).
Conditional Dars2-floxed mice (Dars2fl/fl) were crossed to CHOP KO mice (Chop−/−) to obtain CHOP-deficient animals with floxed Dars2 alleles (Dars2fl/fl; Chop−/−). Triple transgenic mice were generated by intercrossing of CHOP-deficient animals with floxed Dars2 alleles (Dars2fl/fl; Chop−/−), with transgenic mice harboring one copy of the Cre recombinase under control of the striated muscle creatine kinase (Ckmm) promoter (Ckmm-Cre+/tg). Resulting heterozygous triple transgenic mice (Dars2+/fl; Ckmm-Cre+/tg; Chop+/−) and CHOP-deficient animals with floxed Dars2 alleles (Dars2fl/fl; Chop−/−) were used to lastly generate CHOP KO (Dars2+/fl; Ckmm-Cre+/+; Chop−/− and Dars2fl/fl; Ckmm-Cre+/+; Chop−/−) and DKO (Dars2fl/fl; Ckmm-Cre+/tg; Chop−/−) mice. Genotyping for the Dars2 allele was performed as previously described (2). Genotyping for the Ckmm-Cre and Chop alleles was performed following the instructions of the Jackson laboratory using the protocol 22415 along with the primers oIMR3884, oIMR3885, and oIMR3886 for the Chop allele and the protocol Tg(Ckmm-Cre)5Khn along with the primers oIMR1085, oIMR6754, oIMR8744, and oIMR8745 for the Ckmm-Cre allele, respectively (www.jax.org). One- to 6-week-old animals were used in experiments approved and authorized by the Animal Ethics Committee of North-Rhein Westphalia (Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen) following the German and European Union regulations. Animal work was performed in conformity with the recommendations and guidelines of the Federation of European Laboratory Animal Science Associations.
Culture and maintenance of cells
Immortalized MEFs and fibroblasts were cultured in standard conditions, at 37°C and 5% CO2. The cell culture medium was composed of Dulbecco’s modified Eagle’s medium [glucose (4.5 g/liter), GlutaMAX, and sodium pyruvate; Gibco Life Technologies] supplemented with 10% “Fetal Bovine Serum Premium, South American Origin” (Biowest) and penicillin-streptomycin (Pen-Strep) (Gibco Life Technologies). In conditions of mitochondrial dysfunction (induced either genetically or by treatment), the medium was additionally supplemented with uridine (50 μg/ml). At 90% confluency, cells were split cell type–dependently in ratios ranging from 1:4 to 1:20.
Generation of immortalized MEF lines
Embryos from embryonic day 13.5 of intercrossed CHOP KO (Chop−/−) mice were used to isolate primary MEFs (59). Immortalization was achieved by transformation with the SV40 T antigen.
Drug treatments
For induction of mitochondrial dysfunction by actinonin treatment, 80% confluent cells were treated for 48 hours with 100 μM actinonin (Sigma-Aldrich). Proteasome was inhibited with 15 μM MG132 for the last 6 to 8 hours of treatment as indicated. Inhibition of the ISR was achieved by 4- or 48-hour 1 μM ISRIB (Sigma-Aldrich) treatments of 90% confluent cells. All compounds were solubilized in dimethyl sulfoxide (DMSO). Untreated cells were supplemented with corresponding amounts of the solvent. Treatments were renewed on a daily basis.
Transfection
Transfection of plasmids conferring hygromycin resistance (pTK-Hyg LIP, pTK-Hyg LIPwestern, pTK-Hyg LAP, and pTK-Hyg C/EBPβ) was performed with Lipofectamine 2000 or Lipofectamine LTX (Invitrogen) according to the manufacturer’s instructions using the forward transfection procedure. Seventy-two hours after transfection, the culture medium was replaced by hygromycin-supplemented (100 μg/ml) medium for negative selection of untransfected cells. Transfected cells were maintained in hygromycin-supplemented (100 μg/ml) medium.
Cell growth estimation
To estimate differences in cell growth caused by CHOP deficiency and/or mitochondrial dysfunction, an equal number of cells were seeded and treated as indicated. The numbers of cells were determined at the indicated time points using the Countess Automatic Cell Counter (Invitrogen) combined with trypan blue staining.
Isolation of cardiac mitochondria
Freshly collected hearts were immediately transferred into 10 ml of prechilled mito-isolation buffer (MIB) [100 mM sucrose, 50 mM KCl, 1 mM EDTA, 20 mM N-tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid, and 0.2% bovine serum albumin (BSA) free from fatty acids (pH adjusted to 7.2)] supplemented with 1 μg of subtilisin (Sigma-Aldrich) per mg of tissue. Approximately 20 long strokes of a Potter S (Sartorius) homogenizer at 1000 rpm were required for homogenization. After centrifugation (800g, 5 min, 4°C), the mitochondria-containing supernatant was transferred into a fresh tube. Pelleted mitochondria (8500g, 5 min, 4°C) were resuspended in 30 ml of MIB and subjected to a third centrifugation step (700g, 5 min, 4°C). Last, mitochondria were pelleted (8500g, 5 min, 4°C) and resuspended in 100 μl of macrophage inflammatory protein without BSA. Protein concentration of mitochondria was determined using Bradford reagent (Sigma-Aldrich) according to the manufacturer’s instructions. Mitochondria were either immediately used (respirometry or in organello translation) or snap-frozen and stored at −80°C.
Oxygen consumption measurements
High-resolution respirometry using an Oxygraph-2k (OROBOROS Instruments) and a carbohydrate substrate-uncoupler-inhibitor titration protocol was conducted to determine mitochondrial oxygen consumption rates. First, the respiration medium (120 mM sucrose, 50 mM KCl, 20 mM tris-HCl, 1 mM EGTA, 4 mM KH2PO4, 2 mM MgCl2, and 0.1% BSA) was added to the Oxygraph chamber, and air equilibration was performed. Next, 25 μg of freshly isolated cardiac mitochondria was added. The respiration medium was supplemented with 2 mM pyruvate, 0.8 mM malate, 2 mM glutamate, and 2 mM adenosine 5′-diphosphate (ADP) to assess CI-dependent respiration. By providing additional 4 mM succinate, convergent CI- and CII-dependent respiration was determined. Inhibition of ATP-synthase-complex V (CV) by addition of oligomycin (1.5 μg/ml) allowed evaluating the coupling efficiency. The maximal capacity of the electron transfer system (ETS) was assessed by titration of carbonyl cyanide p-trifluoromethoxyphenylhydrazone (0.5 μM increments). Maximal capacity of the ETS of CII solely could be determined by inhibition of CI through addition of 0.5 μM rotenone. Last, inhibition of CIII by supplementation of 2.5 μM antimycin A allowed the determination of the residual oxygen consumption.
In organello translation
De novo mitochondrial translation was assessed by incubation (1 hour, 37°C, on rotating wheel) of 1.5 mg of freshly isolated mitochondria in 1 ml of 35S-translation buffer [100 mM mannitol, 10 mM Na-succinate, 80 mM KCl, 5 mM MgCl2, 1 mM KH2PO4, 25 mM Hepes (pH 7.4), 5 mM ATP, 200 μM GTP, 6 mM creatine phosphate, creatine kinase (60 μg/ml), cysteine (60 μg/ml), tyrosine (60 μg/ml), amino acids (60 μg/ml) (Ala, Arg, Asp, Asn, Glu, Gln, Gly, His, Ile, Leu, Lys, Phe, Pro, Ser, Thr, Trp, and Val), 35S-methionine (7 μl/ml)]. Subsequently, mitochondria were pelleted (12,000g, 2 min) and resuspended in 1 ml of nonradioactive translation buffer containing methionine instead of 35S-methionine. Half of the sample (“pulse fraction”) was pelleted again, resuspended in 100 μl of SDS–polyacrylamide gel electrophoresis (PAGE) loading buffer [50 mM tris-HCl (pH 6.8), 2% SDS (w/v), 10% glycerol (v/v), 1% β-mercaptoethanol, 12.5 mM EDTA, and 0.02% bromophenol blue], and lysed (30 min, room temperature) before transfer at −20°C. For the “cold chase” allowing to estimate the protein turnover, the remaining 500 μl of resuspended mitochondria was incubated for 3 hours at 37°C on a rotating wheel. Subsequently, the “chase fraction” was pelleted, resuspended in 100 μl of SDS-PAGE loading buffer, and lysed as the “pulse sample” before.
Separation of mitochondrial proteins was achieved by SDS-PAGE. Ten microliters per sample was loaded on a 15-cm-long, 15% polyacrylamide gel and run in a “SE600X Chroma Deluxe Dual Cooled Vertical Protein Electrophoresis Unit” (Hoefer) overnight at 80 V continuously. After fixing (50% methanol and 10% acetic acid) for 30 min, staining in Coomassie solution, and destaining (20% methanol and 10% acetic acid) of the polyacrylamide gel, the latter one was placed on Whatman paper (GE Healthcare) and dried (2 hours, 80°C) in a gel dryer. For detection of radioactive signals of de novo synthetized proteins, Amersham Hyperfilm MP (GE Healthcare) was exposed to the dried polyacrylamide gel.
SDS-PAGE Western blot analysis
Cellular protein lysates
Washed cell pellets were resuspended in cold radioimmunoprecipitation assay buffer [150 mM NaCl, 1% Triton X-100 (v/v), 0.5% Na-deoxycholate (w/v), 0.1% SDS (w/v), 50 mM tris-HCl (pH 7.4), 50 mM NaF, and 2 mM EDTA] supplemented with 1× protease inhibitor cocktail (Sigma-Aldrich) and 1× PhosSTOP phosphatase inhibitor cocktail (Roche). Next, cells were incubated 30 min on ice with brief vortexing every 10 min. Following 2× 45-s sonication, the lysates were cleared (10 min, 20,000g, 4°C) and transferred into fresh tubes.
Cardiac tissue protein lysates
Homogenization of 25 mg of cardiac tissue samples in 400 μl of cold organ lysis buffer [50 mM Hepes (pH 7.4), 50 mM NaCl, 1% Triton X-100 (v/v), 0.1 M NaF, 10 mM EDTA, 0.1% SDS (w/v), 10 mM Na-orthovanadate, 2 mM phenylmethylsulfonyl fluoride, 1× protease inhibitor cocktail (Sigma-Aldrich), and 1× PhosSTOP phosphatase inhibitor cocktail (Roche)] was performed with the Precellys CK 14 (Bertin Technologies) (5000 rpm, 30 s). Cleared protein lysates (45 min, 20,000g, 4°C) were transferred into fresh tubes. Determination of protein concentration was performed with Bradford reagent (Sigma-Aldrich) according to the manufacturer’s instructions. Protein lysates were stored at 80°C.
SDS–polyacrylamide gel electrophoresis
Protein samples were dissolved in SDS-PAGE loading buffer [50 mM tris-HCl (pH 6.8), 2% SDS (w/v), 10% glycerol (v/v), 1% β-mercaptoethanol, 12.5 mM EDTA, and 0.02% bromophenol blue] before denaturation. Depending on the required range of protein sizes, the proteins were separated on 8 to 15% acrylamide gels [stacking gel: 5% acrylamide-bisacrylamide (37.5:1), 12.5 mM tris-HCl, 0.1% SDS (w/v), 0.25% Ammonium persulfate (APS), and 0.25% Tetramethylethylenediamine (TEMED) (pH 6.8); separating gel: 8 to 15% acrylamide-bisacrylamide (37.5:1), 37.5 mM tris-HCl, 0.1% SDS (w/v), 0.1% APS, and 0.1% TEMED (pH 8.8)] in running buffer [25 mM tris-HCl, 250 mM glycine, and 0.1% SDS (w/v) (pH 8.3)].
Western blot
Transfer of proteins on a nitrocellulose membrane by Western blot was conducted in transfer buffer (30 mM tris-HCl, 240 mM glycine, 0.037% SDS, and 20% methanol) at 400 mA for 2 hours at 4°C. For a first evaluation of the transfer, shortly washed membranes (dH2O) were stained with Ponceau S solution (Sigma-Aldrich). Depending on the antibody requirements, destaining and blocking of membranes were performed for 1 hour either in 5% milk-PBST (Phosphate-Buffered Saline/Tween) or 3% BSA-TBST (Tris-Buffered Saline/Tween) on a gently shaking platform before subsequent immunodecoration with the indicated antibodies according to the manufacturer’s instructions. Secondary horseradish peroxidase–coupled antibodies (1:5000) were incubated for 1 hour before detection by Pierce ECL Western blotting substrate (Thermo Fisher Scientific). Densitometry-based quantification of Western blots was performed with ImageJ and Image Studio Lite Software.
Blue native polyacrylamide gel electrophoresis Western blot analysis
Blue native polyacrylamide gel electrophoresis (BN-PAGE) was performed on the basis of the “NativePAGE Novex Bis-Tris Gel System” (Invitrogen) according to the manufacturer’s instructions. For analysis of mitochondrial supercomplexes, 10 μg of mitochondria was lysed with 4% of digitonin. Analysis of individual mitochondrial complexes was conducted after lysis of 10 μg of mitochondria in 1% n-dodecyl-β-D-maltoside (DDM). After completion of lysis (15 min on ice), lysates were cleared (30 min, 20,000g, 4°C), and the resulting supernatant was loaded on a 4 to 16% bis-tris gradient gel. Subsequently, proteins were transferred to an Amersham Hybond polyvinylidene difluoride membrane (GE Healthcare) by Western blot and subsequently immunodecorated with indicated antibodies.
Analysis of label-free proteomics and RNA sequencing data
Independently normalized label-free proteomics and RNA sequencing data were scaled before analysis using the anota2seq algorithm (version 1.4.2) (19). Furthermore, datasets were reduced to genes identified on both platforms resulting in a total of 2556 mRNAs for analysis. Analysis of changes in protein levels and total mRNA was performed using the anota2seqAnalyze function to identify differences between CHOP KO, DARS2 KO, and DKO compared to WT. Changes were considered significant when passing the following parameters within the anota2seqSelSigGenes function: maxPAdj = 0.15, minSlopeTranslation = −1, maxSlopeTranslation = 2, selDeltaPT = log2(1.2), selDeltaP = 0, and selDeltaT = 0. Changes in translation or protein stability, as well as changes in mRNA abundance, were characterized using the anota2seqRegModes() function. GO analysis (60) was performed in Cytoscape (v 3.8.0) (23) using the ClueGO (v 2.5.7) app (20). Within ClueGO, four gene lists were provided corresponding to the identified modes for regulation of gene expression using anota2seq (i.e., translation/protein stability and mRNA abundance) divided into up- and down-regulated mRNAs. GO term inclusion parameter was set to a ≤ 5 gene overlap and <4% of total genes present in the GO term. For the resulting network, GO term grouping and fusion parameters were enabled, and only GO terms with a false discovery rate of <5% were displayed. Furthermore, anota2seq was applied on the full RNA sequencing dataset (14,174 protein coding transcripts) following the same approach as above. Master regulators among significantly up-regulated total mRNAs in the DARS2 KO versus WT comparison were detected using iRegulon (v1.3) with default settings (24).
Q5 site-directed mutagenesis
The “Q5 Site-Directed Mutagenesis Kit” (New England Biolabs) was used to introduce a point mutation (L120T) in the pTK-Hyg LIP plasmid (41). For primer design, the New England Biolabs (NEB) online design software “NEBaseChanger” was used. All three steps described in the protocol [exponential amplification, Kinase, Ligase & DpnI treatment (KLD) reaction, and transformation] were performed as indicated in the manual.
Surface sensing of translation
Protein synthesis was determined using the nonradioactive technique called surface sensing of translation described in (61). This assay is based on the incorporation of the structural analogue of tyrosyl-tRNA puromycin in nascent polypeptide chains and subsequent detection of puromycylated proteins using an anti-puromycin–specific antibody.
Briefly, mice were injected at the indicated time points intraperitoneally with 0.04 μmol of puromycin dissolved in phosphate-buffered saline (PBS) per gram of body weight. Thirty minutes after injection, the animals were euthanized, and collected tissues were snap-frozen in liquid nitrogen. Subsequently, protein lysates of the collected tissues were prepared and processed by SDS-PAGE and Western blot. The relative signal intensity of the anti-puromycin–specific antibody is proportional to the relative protein synthesis rates at the time point of puromycin injection.
ISRIB injections
Briefly, mice were injected intraperitoneally with 5 μg of ISRIB (stock solution: 5 mg/ml in DMSO, dissolved in PBS up to the weight-dependent injection volume of 30 to 50 μl) per gram of body weight or the corresponding amount of PBS-dissolved solvent (DMSO) on a daily basis for the indicated time periods. One day after the last injection, the animals were euthanized, and collected tissues were snap-frozen in liquid nitrogen. Subsequently, protein lysates of the collected tissues were prepared and processed by SDS-PAGE and Western blot.
Statistical analysis
Numerical data are expressed as means ± SD. Statistical analysis was performed using the indicated statistical tests. If not indicated differently, statistical significance was considered for P < 0.05. With exception of multivariate analysis of variance (MANOVA) and omics analyses, all statistical tests were performed, and graphs were plotted using GraphPad Prism 8.0 software. MANOVA was performed with XLSTAT version 2020.3 software.
Acknowledgments
We wish to thank the CECAD Imaging and Proteomics Core Facilities for excellent support. Funding: The work was supported by Aleksandra Trifunovic’s grants of the Deutsche Forschungsgemeinschaft [DFG; German Research Foundation (SFB 1218)–Projektnummer 269925409 and TR 1018/8-1] and the Center for Molecular Medicine Cologne, University of Cologne. S.K. received scholarship from the Cologne Graduate School of Ageing Research (CGA). I.T. acknowledges Senior Scholar Award from Le Fonds de recherche du Québec–Santé (FRQS) and support from Canadian Institutes for Health Research (MOP-363027) and Joint Canada-Israel Health Research Program (JCIHRP) (108589-001) to I.T. and O.L. O.L.’s lab was supported by grants from the Swedish Research Council (2016-02891), the Swedish Cancer Society (19 0314), and the Wallenberg Academy Fellows program (2013.0181). M.H.’s laboratory is supported by NIH R01 DK060596 grant. Author contributions: Conceptualization: A.T., S.K., C.O., K.Sz., O.L., I.T., and M.H. Data curation: S.K., C.O., A.T., S.B., O.L., and K.Sz. Formal analysis: S.K., C.O., A.T., S.B., O.L., and K.Sz. Funding acquisition: A.T., S.K., O.L., I.T., and M.H. Investigation: S.K., C.O., A.K., K.Se., K.Sz., C.L., S.B., and O.L. Visualization: S.K., C.O., A.T., O.L., I.T., and M.H. Writing: A.T., S.K., C.O., O.L., and I.T. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Further information and requests for resources and reagents should be addressed to and will be fulfilled by A.T. Mouse and cell lines requests include signing of material transfer agreement.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/22/eabf0971/DC1
REFERENCES AND NOTES
- 1.Suomalainen A., Battersby B. J., Mitochondrial diseases: The contribution of organelle stress responses to pathology. Nat. Rev. Mol. Cell Biol. 19, 77–92 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Dogan S. A., Pujol C., Maiti P., Kukat A., Wang S., Hermans S., Senft K., Wibom R., Rugarli E. I., Trifunovic A., Tissue-specific loss of DARS2 activates stress responses independently of respiratory chain deficiency in the heart. Cell Metab. 19, 458–469 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Jain I. H., Zazzeron L., Goli R., Alexa K., Schatzman-Bone S., Dhillon H., Goldberger O., Peng J., Shalem O., Sanjana N. E., Zhang F., Goessling W., Zapol W. M., Mootha V. K., Hypoxia as a therapy for mitochondrial disease. Science 352, 54–61 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khan N. A., Nikkanen J., Yatsuga S., Jackson C., Wang L., Pradhan S., Kivela R., Pessia A., Velagapudi V., Suomalainen A., mTORC1 regulates mitochondrial integrated stress response and mitochondrial myopathy progression. Cell Metab. 26, 419–428.e5 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Zhao Q., Wang J., Levichkin I. V., Stasinopoulos S., Ryan M. T., Hoogenraad N. J., A mitochondrial specific stress response in mammalian cells. EMBO J. 21, 4411–4419 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shpilka T., Haynes C. M., The mitochondrial UPR: Mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Biol. 19, 109–120 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Aldridge J. E., Horibe T., Hoogenraad N. J., Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PLOS ONE 2, e874 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horibe T., Hoogenraad N. J., The chop gene contains an element for the positive regulation of the mitochondrial unfolded protein response. PLOS ONE 2, e835 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jadiya P., Tomar D., Mitochondrial protein quality control mechanisms. Genes (Basel) 11, 563 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Silva J. M., Wong A., Carelli V., Cortopassi G. A., Inhibition of mitochondrial function induces an integrated stress response in oligodendroglia. Neurobiol. Dis. 34, 357–365 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Kuhl I., Miranda M., Atanassov I., Kuznetsova I., Hinze Y., Mourier A., Filipovska A., Larsson N.-G., Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals. eLife 6, e30952 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Forsstrom S., Jackson C. B., Carroll C. J., Kuronen M., Pirinen E., Pradhan S., Marmyleva A., Auranen M., Kleine I.-M., Khan N. A., Roivainen A., Marjamaki P., Liljenback H., Wang L., Battersby B. J., Richter U., Velagapudi V., Nikkanen J., Euro L., Suomalainen A., Fibroblast growth factor 21 drives dynamics of local and systemic stress responses in mitochondrial myopathy with mtDNA deletions. Cell Metab. 30, 1040–1054.e7 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Quiros P. M., Prado M. A., Zamboni N., D’Amico D., Williams R. W., Finley D., Gygi S. P., Auwerx J., Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 216, 2027–2045 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bao X. R., Ong S. E., Goldberger O., Peng J., Sharma R., Thompson D. A., Vafai S. B., Cox A. G., Marutani E., Ichinose F., Goessling W., Regev A., Carr S. A., Clish C. B., Mootha V. K., Mitochondrial dysfunction remodels one-carbon metabolism in human cells. eLife 5, e10575 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Costa-Mattioli M., Walter P., The integrated stress response: From mechanism to disease. Science 368, eaat5314 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marciniak S. J., Yun C. Y., Oyadomari S., Novoa I., Zhang Y., Jungreis R., Nagata K., Harding H. P., Ron D., CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 18, 3066–3077 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ron D., Habener J. F., CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant-negative inhibitor of gene transcription. Genes Dev. 6, 439–453 (1992). [DOI] [PubMed] [Google Scholar]
- 18.Han J., Back S. H., Hur J., Lin Y.-H., Gildersleeve R., Shan J., Yuan C. L., Krokowski D., Wang S., Hatzoglou M., Kilberg M. S., Sartor M. A., Kaufman R. J., ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 15, 481–490 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oertlin C., Lorent J., Murie C., Furic L., Topisirovic I., Larsson O., Generally applicable transcriptome-wide analysis of translation using anota2seq. Nucleic Acids Res. 47, e70 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bindea G., Mlecnik B., Hackl H., Charoentong P., Tosolini M., Kirilovsky A., Fridman W. H., Pages F., Trajanoski Z., Galon J., ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25, 1091–1093 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.The Gene Ontology Consortium , The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 47, D330–D338 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuhl I., Kukat C., Ruzzenente B., Milenkovic D., Mourier A., Miranda M., Koolmeister C., Falkenberg M., Larsson N. G., POLRMT does not transcribe nuclear genes. Nature 514, E7–E11 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., Ideker T., Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Janky R., Verfaillie A., Imrichova H., Van de Sande B., Standaert L., Christiaens V., Hulselmans G., Herten K., Naval Sanchez M., Potier D., Svetlichnyy D., Kalender Atak Z., Fiers M., Marine J. C., Aerts S., iRegulon: From a gene list to a gene regulatory network using large motif and track collections. PLoS Comput. Biol. 10, e1003731 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tyynismaa H., Carroll C. J., Raimundo N., Ahola-Erkkila S., Wenz T., Ruhanen H., Guse K., Hemminki A., Peltola-Mjosund K. E., Tulkki V., Oresic M., Moraes C. T., Pietilainen K., Hovatta I., Suomalainen A., Mitochondrial myopathy induces a starvation-like response. Hum. Mol. Genet. 19, 3948–3958 (2010). [DOI] [PubMed] [Google Scholar]
- 26.Yatsuga S., Fujita Y., Ishii A., Fukumoto Y., Arahata H., Kakuma T., Kojima T., Ito M., Tanaka M., Saiki R., Koga Y., Growth differentiation factor 15 as a useful biomarker for mitochondrial disorders. Ann. Neurol. 78, 814–823 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu H., Tian M., Ding C., Yu S., The C/EBP homologous protein (CHOP) transcription factor functions in endoplasmic reticulum stress-induced apoptosis and microbial infection. Front. Immunol. 9, 3083 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sullivan L. B., Gui D. Y., Hosios A. M., Bush L. N., Freinkman E., Vander Heiden M. G., Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell 162, 552–563 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Birsoy K., Wang T., Chen W. W., Freinkman E., Abu-Remaileh M., Sabatini D. M., An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell 162, 540–551 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Q., Kirk K., Shurubor Y. I., Zhao D., Arreguin A. J., Shahi I., Valsecchi F., Primiano G., Calder E. L., Carelli V., Denton T. T., Beal M. F., Gross S. S., Manfredi G., D’Aurelio M., Rewiring of glutamine metabolism is a bioenergetic adaptation of human cells with mitochondrial DNA mutations. Cell Metab. 27, 1007–1025.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Porter G. A. Jr., Hom J., Hoffman D., Quintanilla R., de Mesy Bentley K., Sheu S. S., Bioenergetics, mitochondria, and cardiac myocyte differentiation. Prog. Pediatr. Cardiol. 31, 75–81 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Diaz F., Thomas C. K., Garcia S., Hernandez D., Moraes C. T., Mice lacking COX10 in skeletal muscle recapitulate the phenotype of progressive mitochondrial myopathies associated with cytochrome c oxidase deficiency. Hum. Mol. Genet. 14, 2737–2748 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richter U., Lahtinen T., Marttinen P., Myohanen M., Greco D., Cannino G., Jacobs H. T., Lietzen N., Nyman T. A., Battersby B. J., A mitochondrial ribosomal and RNA decay pathway blocks cell proliferation. Curr. Biol. 23, 535–541 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Sidrauski C., McGeachy A. M., Ingolia N. T., Walter P., The small molecule ISRIB reverses the effects of eIF2α phosphorylation on translation and stress granule assembly. eLife 4, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rabouw H. H., Langereis M. A., Anand A. A., Visser L. J., de Groot R. J., Walter P., van Kuppeveld F. J. M., Small molecule ISRIB suppresses the integrated stress response within a defined window of activation. Proc. Natl. Acad. Sci. U.S.A. 116, 2097–2102 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ravi V., Jain A., Ahamed F., Fathma N., Desingu P. A., Sundaresan N. R., Systematic evaluation of the adaptability of the non-radioactive SUnSET assay to measure cardiac protein synthesis. Sci. Rep. 8, 4587 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guan B. J., van Hoef V., Jobava R., Elroy-Stein O., Valasek L. S., Cargnello M., Gao X. H., Krokowski D., Merrick W. C., Kimball S. R., Komar A. A., Koromilas A. E., Wynshaw-Boris A., Topisirovic I., Larsson O., Hatzoglou M., A unique ISR program determines cellular responses to chronic stress. Mol. Cell 68, 885–900.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buttgereit F., Brand M. D., A hierarchy of ATP-consuming processes in mammalian cells. Biochem. J. 312 (Pt 1), 163–167 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Descombes P., Schibler U., A liver-enriched transcriptional activator protein, LAP, and a transcriptional inhibitory protein, LIP, are translated from the same mRNA. Cell 67, 569–579 (1991). [DOI] [PubMed] [Google Scholar]
- 40.Begay V., Baumeier C., Zimmermann K., Heuser A., Leutz A., The C/EBPβ LIP isoform rescues loss of C/EBPβ function in the mouse. Sci. Rep. 8, 8417 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chiribau C. B., Gaccioli F., Huang C. C., Yuan C. L., Hatzoglou M., Molecular symbiosis of CHOP and C/EBP beta isoform LIP contributes to endoplasmic reticulum stress-induced apoptosis. Mol. Cell. Biol. 30, 3722–3731 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.la Cour T., Kiemer L., Molgaard A., Gupta R., Skriver K., Brunak S., Analysis and prediction of leucine-rich nuclear export signals. Protein Eng. Des. Sel. 17, 527–536 (2004). [DOI] [PubMed] [Google Scholar]
- 43.Dey S., Savant S., Teske B. F., Hatzoglou M., Calkhoven C. F., Wek R. C., Transcriptional repression of ATF4 gene by CCAAT/enhancer-binding protein β (C/EBPβ) differentially regulates integrated stress response. J. Biol. Chem. 287, 21936–21949 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lopez A. B., Wang C., Huang C. C., Yaman I., Li Y., Chakravarty K., Johnson P. F., Chiang C. M., Snider M. D., Wek R. C., Hatzoglou M., A feedback transcriptional mechanism controls the level of the arginine/lysine transporter cat-1 during amino acid starvation. Biochem. J. 402, 163–173 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shan J., Zhang F., Sharkey J., Tang T. A., Ord T., Kilberg M. S., The C/ebp-Atf response element (CARE) location reveals two distinct Atf4-dependent, elongation-mediated mechanisms for transcriptional induction of aminoacyl-tRNA synthetase genes in response to amino acid limitation. Nucleic Acids Res. 44, 9719–9732 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ebert S. M., Bullard S. A., Basisty N., Marcotte G. R., Skopec Z. P., Dierdorff J. M., Al-Zougbi A., Tomcheck K. C., DeLau A. D., Rathmacher J. A., Bodine S. C., Schilling B., Adams C. M., Activating transcription factor 4 (ATF4) promotes skeletal muscle atrophy by forming a heterodimer with the transcriptional regulator C/EBPβ. J. Biol. Chem. 295, 2787–2803 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oyadomari S., Mori M., Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 11, 381–389 (2004). [DOI] [PubMed] [Google Scholar]
- 48.Ron D., Translational control in the endoplasmic reticulum stress response. J. Clin. Invest. 110, 1383–1388 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zinszner H., Kuroda M., Wang X., Batchvarova N., Lightfoot R. T., Remotti H., Stevens J. L., Ron D., CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 12, 982–995 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oyadomari S., Koizumi A., Takeda K., Gotoh T., Akira S., Araki E., Mori M., Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J. Clin. Invest. 109, 525–532 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rinella M. E., Siddiqui M. S., Gardikiotes K., Gottstein J., Elias M., Green R. M., Dysregulation of the unfolded protein response in db/db mice with diet-induced steatohepatitis. Hepatology 54, 1600–1609 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tajiri S., Oyadomari S., Yano S., Morioka M., Gotoh T., Hamada J. I., Ushio Y., Mori M., Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ. 11, 403–415 (2004). [DOI] [PubMed] [Google Scholar]
- 53.Katz A. M., Is the failing heart energy depleted? Cardiol. Clin. 16, 633–644 (1998). [DOI] [PubMed] [Google Scholar]
- 54.Baris O. R., Ederer S., Neuhaus J. F., von Kleist-Retzow J. C., Wunderlich C. M., Pal M., Wunderlich F. T., Peeva V., Zsurka G., Kunz W. S., Hickethier T., Bunck A. C., Stockigt F., Schrickel J. W., Wiesner R. J., Mosaic deficiency in mitochondrial oxidative metabolism promotes cardiac arrhythmia during Aging. Cell Metab. 21, 667–677 (2015). [DOI] [PubMed] [Google Scholar]
- 55.Nikkanen J., Forsstrom S., Euro L., Paetau I., Kohnz R. A., Wang L., Chilov D., Viinamaki J., Roivainen A., Marjamaki P., Liljenback H., Ahola S., Buzkova J., Terzioglu M., Khan N. A., Pirnes-Karhu S., Paetau A., Lonnqvist T., Sajantila A., Isohanni P., Tyynismaa H., Nomura D. K., Battersby B. J., Velagapudi V., Carroll C. J., Suomalainen A., Mitochondrial DNA replication defects disturb cellular dNTP pools and remodel one-carbon metabolism. Cell Metab. 23, 635–648 (2016). [DOI] [PubMed] [Google Scholar]
- 56.Harding H. P., Zhang Y., Zeng H., Novoa I., Lu P. D., Calfon M., Sadri N., Yun C., Popko B., Paules R., Stojdl D. F., Bell J. C., Hettmann T., Leiden J. M., Ron D., An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619–633 (2003). [DOI] [PubMed] [Google Scholar]
- 57.Ben-Sahra I., Hoxhaj G., Ricoult S. J. H., Asara J. M., Manning B. D., mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 351, 728–733 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.DeNicola G. M., Chen P. H., Mullarky E., Sudderth J. A., Hu Z., Wu D., Tang H., Xie Y., Asara J. M., Huffman K. E., Wistuba I. I., Minna J. D., DeBerardinis R. J., Cantley L. C., NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat. Genet. 47, 1475–1481 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Trifunovic A., Hansson A., Wredenberg A., Rovio A. T., Dufour E., Khvorostov I., Spelbrink J. N., Wibom R., Jacobs H. T., Larsson N. G., Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc. Natl. Acad. Sci. U.S.A. 102, 17993–17998 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ashburner M., Ball C. A., Blake J. A., Botstein D., Butler H., Cherry J. M., Davis A. P., Dolinski K., Dwight S. S., Eppig J. T., Harris M. A., Hill D. P., Issel-Tarver L., Kasarskis A., Lewis S., Matese J. C., Richardson J. E., Ringwald M., Rubin G. M., Sherlock G., Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nat. Genet. 25, 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goodman C. A., Hornberger T. A., Measuring protein synthesis with SUnSET: A valid alternative to traditional techniques? Exerc. Sport Sci. Rev. 41, 107–115 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Matic S., Jiang M., Nicholls T. J., Uhler J. P., Dirksen-Schwanenland C., Polosa P. L., Simard M. L., Li X., Atanassov I., Rackham O., Filipovska A., Stewart J. B., Falkenberg M., Larsson N. G., Milenkovic D., Mice lacking the mitochondrial exonuclease MGME1 accumulate mtDNA deletions without developing progeria. Nat. Commun. 9, 1202 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Albert M. C., Brinkmann K., Pokrzywa W., Gunther S. D., Kronke M., Hoppe T., Kashkar H., CHIP ubiquitylates NOXA and induces its lysosomal degradation in response to DNA damage. Cell Death Dis. 11, 740 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ewels P. A., Peltzer A., Fillinger S., Patel H., Alneberg J., Wilm A., Garcia M. U., Di Tommaso P., Nahnsen S., The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 38, 276–278 (2020). [DOI] [PubMed] [Google Scholar]
- 65.Kim D., Paggi J. M., Park C., Bennett C., Salzberg S. L., Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liao Y., Smyth G. K., Shi W., featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 67.Yates A. D., Achuthan P., Akanni W., Allen J., Allen J., Alvarez-Jarreta J., Amode M. R., Armean I. M., Azov A. G., Bennett R., Bhai J., Billis K., Boddu S., Marugan J. C., Cummins C., Davidson C., Dodiya K., Fatima R., Gall A., Giron C. G., Gil L., Grego T., Haggerty L., Haskell E., Hourlier T., Izuogu O. G., Janacek S. H., Juettemann T., Kay M., Lavidas I., Le T., Lemos D., Martinez J. G., Maurel T., McDowall M., McMahon A., Mohanan S., Moore B., Nuhn M., Oheh D. N., Parker A., Parton A., Patricio M., Sakthivel M. P., Abdul Salam A. I., Schmitt B. M., Schuilenburg H., Sheppard D., Sycheva M., Szuba M., Taylor K., Thormann A., Threadgold G., Vullo A., Walts B., Winterbottom A., Zadissa A., Chakiachvili M., Flint B., Frankish A., Hunt S. E., G I. I., Kostadima M., Langridge N., Loveland J. E., Martin F. J., Morales J., Mudge J. M., Muffato M., Perry E., Ruffier M., Trevanion S. J., Cunningham F., Howe K. L., Zerbino D. R., Flicek P., Ensembl 2020. Nucleic Acids Res. 48, D682–D688 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Robinson M. D., Oshlack A., A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Murru S., Hess S., Barth E., Almajan E. R., Schatton D., Hermans S., Brodesser S., Langer T., Kloppenburg P., Rugarli E. I., Astrocyte-specific deletion of the mitochondrial m-AAA protease reveals glial contribution to neurodegeneration. Glia 67, 1526–1541 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Aravamudhan S., Turk C., Bock T., Keufgens L., Nolte H., Lang F., Krishnan R. K., Konig T., Hammerschmidt P., Schindler N., Brodesser S., Rozsivalova D. H., Rugarli E., Trifunovic A., Bruning J., Langer T., Braun T., Kruger M., Phosphoproteomics of the developing heart identifies PERM1 - An outer mitochondrial membrane protein. J. Mol. Cell. Cardiol. 154, 41–59 (2021). [DOI] [PubMed] [Google Scholar]
- 71.Schwaiger M., Rampler E., Hermann G., Miklos W., Berger W., Koellensperger G., Anion-exchange chromatography coupled to high-resolution mass spectrometry: A powerful tool for merging targeted and non-targeted metabolomics. Anal. Chem. 89, 7667–7674 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/22/eabf0971/DC1