

Abstract
High levels of Aβ impair neuronal function at least in part by disrupting normal synaptic transmission and causing dysfunction of neural networks. This network dysfunction includes abnormal synchronization of neuronal activity resulting in epileptiform activity. Over time, this aberrant network activity can lead to the depletion of calcium-dependent proteins, such as calbindin, Fos, and Arc, and compensatory inhibitory remodeling of hippocampal circuits, including GABAergic sprouting and ectopic expression of the inhibitory neuropeptide Y (NPY) in dentate granule cells. Here we present detailed protocols for detecting and quantifying these alterations in mouse models of Alzheimer’s disease (AD) by immunohistochemistry. These methods are useful as surrogate measures for detecting chronic aberrant network activity in models of AD and epilepsy. In addition, since we have found that the severity of these changes relates to the degree of Aβ-dependent cognitive impairments, the protocols are useful for quantifying biomarkers of cognitive impairment in mouse models of AD.
Keywords: Hippocampus, Dentate gyrus, Granule cell, Mossy fiber, Hilus, Neuropeptide Y, NPY, Calbindin, Seizure, Epilepsy, Excitability, GABA, GABAergic sprouting, Ectopic expression, Excitatory, Inhibitory, Immunohistochemistry, Aβ, Alzheimer’s disease, Biomarkers, Immediate early gene, Fos, Arc, Neuronal activity
1. Introduction
Transgenic mice expressing Alzheimer’s disease (AD)-related genes, such as genes encoding mutant amyloid precursor protein (APP) and presenilin 1, have become a mainstay for studying the molecular pathogenesis of AD and for the preclinical development of new therapeutic strategies. These mouse models have proved useful because their brain architecture and circuitry are similar in many ways to those of humans, and because they display deficits in learning and memory that can be quantified by standard behavioral measures of hippocampal functions, including the Morris water maze, radial arm maze, and contextual fear conditioning. However, these tests have considerable downsides, including requirements for specialized equipment and dedicated space, expertise in the neurobehavioral analysis of mice, and high inter-animal variability in behavior, which increases the number of mice required to obtain conclusive results and makes the procedures very labor intensive (1, 2).
While behavior likely will never be supplanted as the ultimate measure of brain function, more convenient surrogate measures that provide reliable correlates of hippocampal dysfunction in AD mouse models would be very useful. Amyloid plaque load was initially considered a potential surrogate marker for the severity of AD-related changes in mouse models, but it is now clear that plaque density does not correlate with learning and memory deficits in AD mouse models (3–7), or indeed in patients with AD (8–10). Here, we present a protocol for other immunohistochemical measures that do correlate with cognitive deficits in mouse models of AD. In the dentate gyrus of different lines of hAPP transgenic mice, we have detected depletion of the calcium-binding protein calbindin D-28K (Fig. 1), aberrant expression of the neuromodulator neuropeptide Y (NPY) (Fig. 2), and reductions in the number of Fos- or Arc-positive granule cells (Fig. 3) (7, 11–15). Some of these changes have also been observed by other groups in other AD models (16), as well as in patients with AD (7). Importantly, the changes in calbindin, NPY, and Fos/Arc in individual mice and in transgenic lines correlate with the severity of learning and memory impairments (7, 11–14, 17), and with each other (Figs. 3 and 4).
Fig. 1.
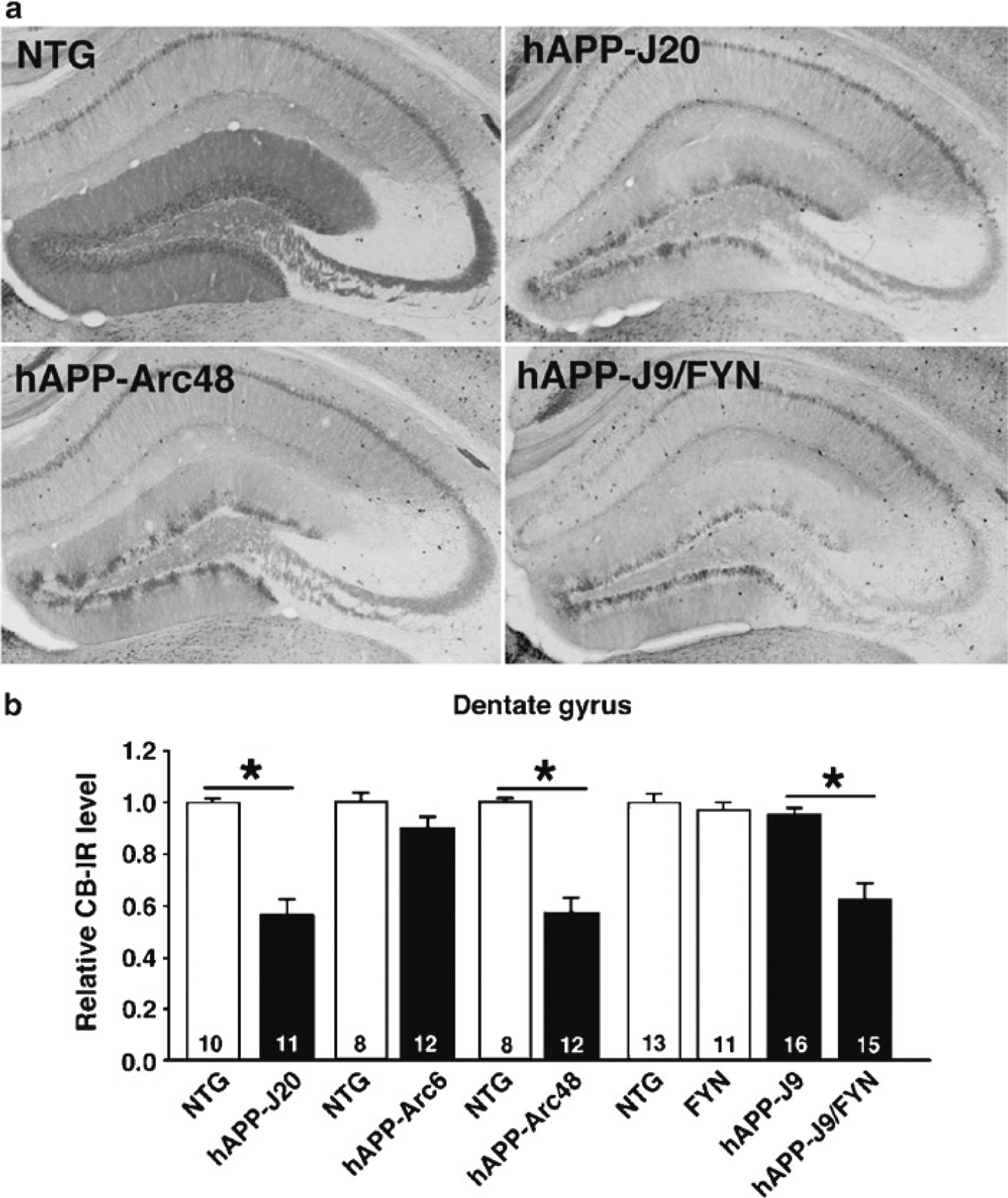
Calbindin depletion in multiple lines of cognitively impaired hAPP transgenic mice. Calbindin immunohistochemistry was performed and quantified as described here. (a) Calbindin depletion relative to levels in nontransgenic (NTG) mice was observed in transgenic mice from lines hAPP-J20 (expressing human APP [hAPP] with Swedish and Indiana mutations), hAPP-Arc48 (expressing hAPP with Swedish, Indiana, and Arctic mutations), and hAPP-J9/FYN (expressing both hAPP with Swedish and Indiana mutations and the Src-family tyrosine kinase Fyn). (b) Quantification of calbindin immunoreactivity (CB-IR) in the different lines. Calbindin was depleted in lines with Aβ-dependent cognitive impairments, including hAPP-J20 (originally demonstrated in ref. 7), hAPP-Arc48 (originally demonstrated in ref. 14), and hAPP-J9/FYN (originally demonstrated in ref. 12). Calbindin was not depleted or only slightly reduced in lines that do not display significant cognitive deficits, including hAPP-Arc6 (originally demonstrated in ref. 14) and hAPP-J9 (originally demonstrated in ref. 12). *P < 0.05 by Student’s t-test or Tukey–Kramer test.
Fig. 2.
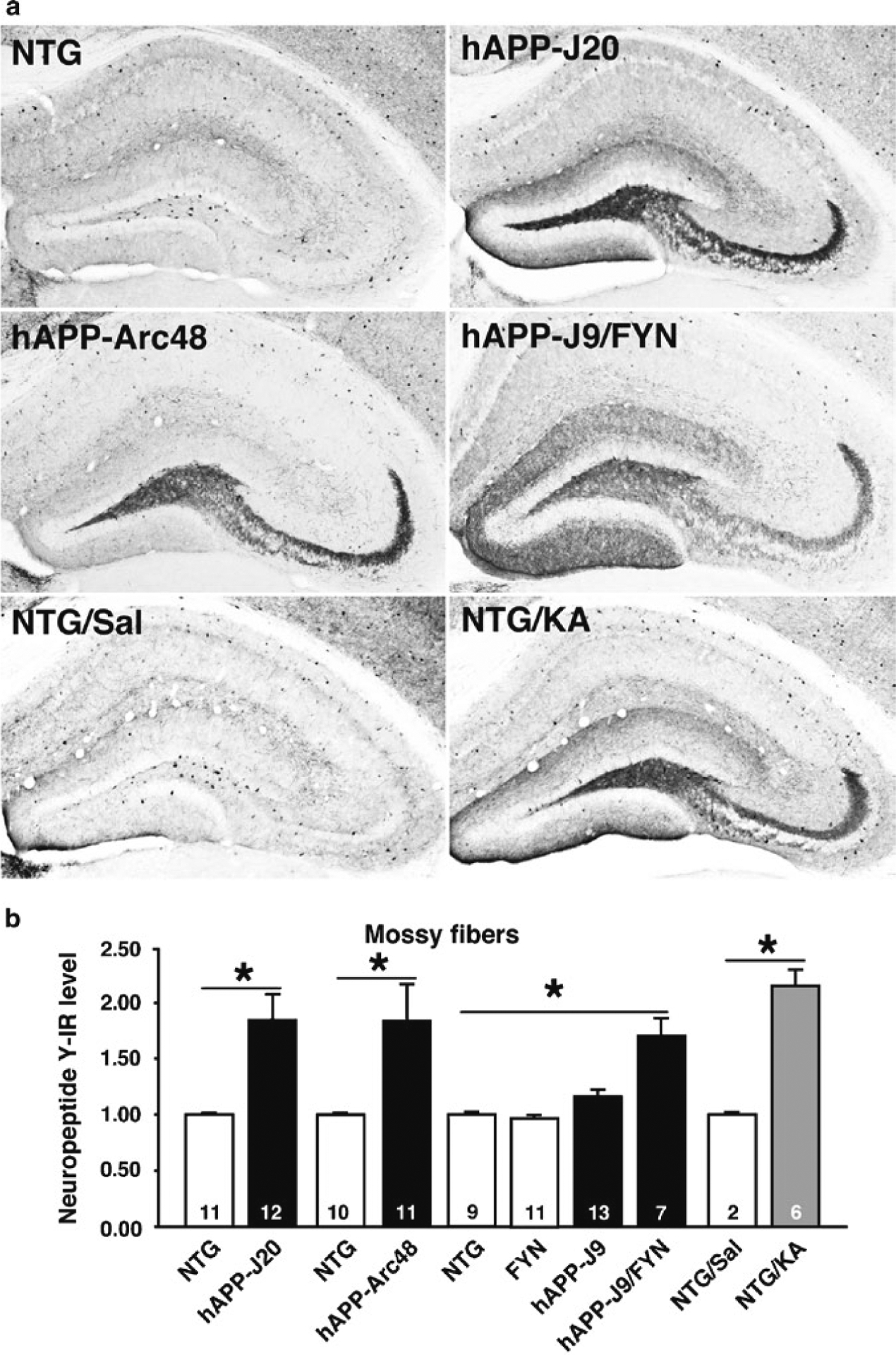
Aberrant NPY expression in multiple lines of hAPP transgenic mice. NPY immunohistochemistry was performed and quantified as described here. (a) Aberrant NPY expression was observed in transgenic mice from lines hAPP-J20, hAPP-Arc48, and hAPP-J9/FYN. Similar changes were seen 3 days after pharmacological induction of seizures by intraperitoneal injection of 25 mg/kg kainic acid in NTG mice (NTG/KA), but not in saline-treated NTG mice (NTG/Sal). (b) Quantification of NPY immunoreactivity (IR) in different groups of mice. NPY expression in mossy fibers was increased in lines with Aβ-dependent cognitive impairment, including hAPP-J20, hAPP-Arc48, and hAPP-J9/FYN, as well as after KA injection in NTG mice (originally demonstrated in ref. 13). *P < 0.05 by Student’s t-test or Tukey–Kramer test.
Fig. 3.
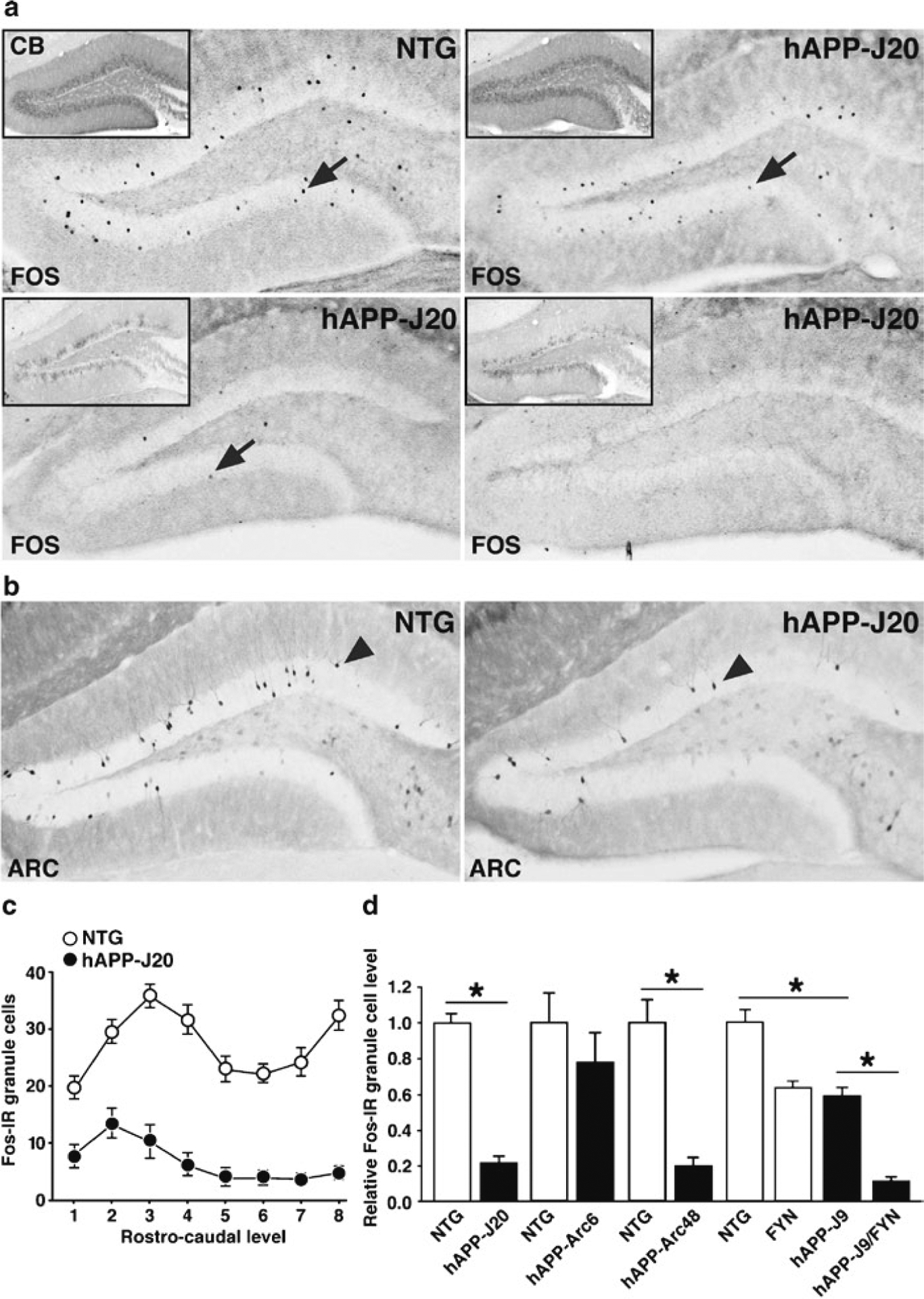
Reduced number of Fos- and Arc-positive dentate granule cells in hAPP mice. (a) Sections through the hippocampus of NTG or three different hAPP-J20 transgenic mice were immunostained for Fos. Inset shows calbindin immunoreactivity from the same animals. Some hAPP mice show normal levels of Fos and calbindin immunoreactivity (upper right), whereas most show varying degrees of reductions in both Fos and calbindin. (b) Arc-positive granule cells are also reduced in hAPP-J20 mice (originally demonstrated in ref. 11). (c) Plot of Fos-positive granule cells along the rostro–caudal axis of the dentate gyrus (1 = most rostral/dorsal; 8 = most caudal/ventral). The number of Fos-positive cells was reduced in hAPP-J20 mice along the entire axis. N =12–18 mice per genotype. (d) Quantification revealed reduced numbers of total Fos-positive granule cells in hAPP-J20 mice (originally demonstrated in ref. 7), hAPP-J9/FYN mice (originally demonstrated in ref. 12), and hAPP-Arc48 mice (originally demonstrated in ref. 14). *P < 0.05 by Student’s t-test or Tukey–Kramer test.
Fig. 4.
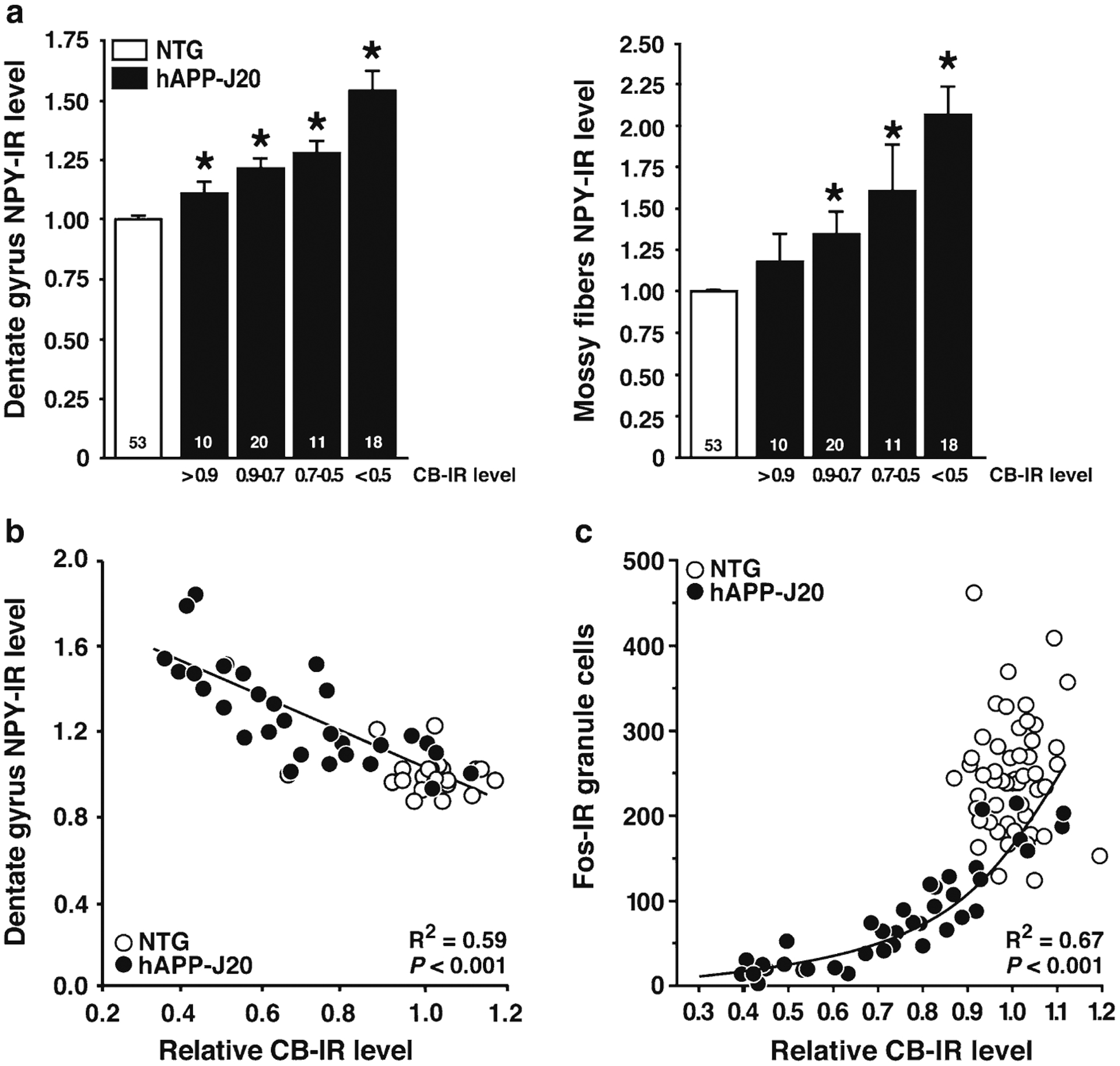
Correlations between calbindin, NPY, and Fos immunoreactivity in hAPP-J20 mice. (a) Compared with NTG controls, NPY expression was increased in the molecular layer of the dentate gyrus (left) and in the mossy fibers (right) in hAPP-J20 mice. Aberrant NPY expression was more prominent in hAPP-J20 mice with severe reductions in calbindin immunoreactivity. Numbers on bars indicate numbers of mice. * P < 0.05 versus NTG by Tukey–Kramer test. (b) hAPP-J20 transgenic mice showed interindividual variation in calbindin and NPY. Interestingly, these alterations were tightly correlated, suggesting a common underlying mechanism. The correlation and resulting R2 and P-value are for hAPP-J20 mice only. (c) Nonlinear correlation between calbindin immunoreactivity and the number of Fos-immunoreactive granule cells in hAPP-J20 mice. A few hAPP-J20 mice displayed normal levels of both calbindin- and Fos-immunoreactive cells. The number of Fos-immunoreactive cells is a highly sensitive measure and can be strongly reduced in hAPP-J20 mice with only mild calbindin depletion. See also refs. 7, 13. R2 and P-values are for hAPP-J20 mice.
Changes in calbindin, NPY, Fos, and Arc expression in dentate granule cells probably share a common neurobiological basis, representing compensatory responses to excessive neuronal activity (13, 18). Indeed, evidence has been accumulating that imbalances between excitation and inhibition may be a major mode by which Aβ disrupts normal cognitive functioning at the network level (13, 15, 16, 19–21). For example, hAPP mice have interictal epileptiform spikes and nonconvulsive seizures (13, 16, 22). This aberrant excitatory activity likely causes the depletion of calcium-dependent proteins and compensatory inhibitory remodeling in the hippocampus, including changes in calbindin, Fos, Arc, and NPY (13, 16). Here we describe how to detect and quantify changes in these functionally relevant biomarkers in AD mouse models by immunohistochemistry.
2. Materials
2.1. Perfusion and Fixation of Brain Tissue
Anesthetic agent, such as pentobarbital.
Perfusion pump or large-capacity syringes and tubing.
Surgical tools.
Normal saline: 0.9% NaCl.
2× Phosphate buffer (2× PB): 160 mM Na2HPO4, 400 mM NaH2PO4, pH 7.4. For a 1-L stock solution, dissolve 22.72 g of Na2HPO4 and 5.52 g of NaH2PO4 × 1H2O in water and adjust pH to 7.4. Store at room temperature for weeks or at −20°C for months.
1 M NaOH: 40 g of NaOH in 1 L of water.
8% Paraformaldehyde (8% PFA, w/v): For 1 L, measure 80 g of PFA (see Note 1), add about 600 ml of water that has been heated to ~65°C in a microwave, place the mixture on a hot-plate in a fume hood, and stir with continued heating until the solution reaches, but does not exceed, 85°C (see Note 2). Turn off the heat and continue stirring. At this point, the solution should be whitish and cloudy. Add two to four drops of 1 M NaOH and wait for 2–4 min. The solution will become less cloudy and more transparent. Repeat the addition of NaOH until the PFA mixes fully in the solution. The goal is to obtain a completely transparent solution with minimal addition of NaOH, because high concentrations of NaOH increase the pH and affect the fixative properties of PFA. Add heated water to a volume of 1 L. Let the solution cool to room temperature while stirring. Aliquot and store at 4°C for use within 1–2 days or at −20°C for longer periods of storage.
4% PFA in 1× PB: Prepared as 1:1 mixture of 8% PFA and 2× PB. If the solutions are stored at −20°C, thaw in a 40–50°C water bath. Wait until both solutions are transparent. Do not use a microwave to thaw PFA.
2.2. Sectioning of Brain Tissue
Phosphate-buffered saline (PBS): Purchase 10× sterile stock solution (pH 7.4), dilute to 1×.
30% Sucrose in PBS (w/v).
Sliding microtome or vibratome (see Note 3).
10% Sucrose in PBS.
Cryoprotectant solution: 30% ethylene glycol, 30% glycerol, 40% 1× PBS (v/v/v). Store at −20°C.
#1 sable paintbrush.
2.3. Immunohistochemistry
Shaking platform, such as IKA Vibrax.
10-cm dishes.
24-well plates.
Transfer pipettes, pipette tips, and Parafilm (see Note 4).
PBS/Tx: 0.5% Triton X-100 in PBS.
Peroxidase quenching solution: 3% H2O2 and 10% methanol in PBS.
Blocking solution: 10% normal goat serum (Vector Laboratories), 1% blocking-grade dry milk (Bio-Rad), and 0.2% gelatin from porcine skin, type A (Sigma) in PBS/Tx.
Antibody incubation solution: 3% normal goat serum (Vector Laboratories) and 0.2% gelatin from porcine skin, type A (Sigma) in PBS.
Rabbit anti-calbindin D-28K antibody (Swant).
Rabbit anti-NPY antibody (Immunostar).
Rabbit anti-c-Fos (Ab-5, EMD Biosciences).
Rabbit anti-Arc (gift from Dr. Paul F. Worley).
Biotinylated goat anti-rabbit antibody (Vector Laboratories).
Vectastain Elite ABC Kit Standard (Vector Laboratories).
100 mM Tris-HCl, pH 7.4.
DAB solution: 0.25 mg/ml 3,3′-diaminobenzidine tetrahydrochloride (DAB) and 0.036% H2O2 in 100 mM Tris-HCl, pH 7.4. Dissolve one 10 mg tablet of DAB (Sigma) in 40 ml of 100 mM Tris (see Note 5). Filter through filter paper. Place 10 ml of filtered DAB solution in a glass scintillation vial and add 12 μl of 30% H2O2 (see Note 6). Use immediately.
Dissecting microscope (for visualizing progress of development reaction).
1% Gelatin. Mix 1 g of gelatin from porcine skin, type A (Sigma) in 100 ml of warm water (see Note 7).
Superfrost Plus slides.
Slide racks and staining jars.
Xylene.
Cytoseal or Permount mounting medium.
2.4. Quantification of Results
Digital microscope and camera.
Image analysis software capable of densitometry.
Spreadsheet software.
Tally counter.
3. Methods
3.1. Perfusion and Fixation of Brain Tissue
Anesthetize animals. Levels of immediate-early genes such as Fos and Arc are tightly regulated by activity-dependent neuronal activity (11). Changes in levels of these proteins can be observed as soon as 45 min after environmental modification, such as moving the mice to the necropsy room, changing mouse cages, switching lights, etc. The day before perfusion, notify laboratory personnel and animal care staff to avoid any disturbance to the cages. When ready to perfuse, remove only a cage or two at a time from the home room, and proceed as quickly as possible to minimize the time to fixation. We perfuse mice from one or two cages every five or 10 min to ensure that all mice have a similar experience before the perfusion.
Flush-perfuse transcardially with normal saline for 45–60 s until the perfusate returns clear and no longer contains blood.
Remove the brain, bisect (see Note 8), and drop-fix one hemibrain in 4% PFA in 1× PB for 24–48 h at 4°C. Make sure that the fixative volume is at least ten times larger than the volume of the brain sample.
After the brains have been fixed at 4°C for 24–48 h, rinse twice for 2–5 h with PBS at 4°C. This reduces background during immunohistochemistry. Brains can be stored for 2–4 weeks in PBS at 4°C before sectioning.
3.2. Sectioning of Brain Tissue
Transfer the brains into 30% sucrose in PBS for 24–48 h at 4°C. Initially, the brains will float. They will sink when cryoprotected and ready for sectioning; do not section until the brains sink. Make sure that the volume of sucrose solution is at least ten times greater than the volume of the sample. Brains can be kept for 2 weeks at 4°C in 30% sucrose before sectioning. We recommend sectioning the brains within 1 month from the perfusion.
Place the hemibrains on the freezing stage of the microtome at room temperature and add a few drops of 10% sucrose around the hemibrains (see Note 9). Once the stage temperature is lowered, the 10% sucrose base will freeze and firmly attach the samples to the stage.
Set the temperature of the freezing stage at −20°C and wait for 5–10 min until the hemibrain and 10% sucrose base are completely frozen.
Collect ten subseries of floating sections (30 μm) per mouse hemibrain and place in 1.5-ml Eppendorf tubes containing 1.2 ml of ethylene glycol-based cryoprotectant medium (see Note 10). Each tube will contain 8–12 equidistant sections, 300 μm apart, throughout the rostro–caudal extent of the hemibrain. (These are standard parameters that could change depending on the experiment or animal model.)
Store the sections at −20°C until use.
3.3. Immunohistochemical Staining
The primary steps in the procedure are quenching endogenous peroxidase activity, blocking nonspecific binding, and incubating first with anti-calbindin, anti-NPY, anti-Fos, or anti-Arc primary antibody, then with a biotinylated secondary antibody, and finally with a horseradish peroxidase (HRP)-conjugated avidin–biotin complex. The HRP is detected by reaction with DAB, and the sections are mounted for quantification (see Note 11).
We routinely conduct the immunohistochemistry on floating microtome sections in a 24-well plate, with a 400-μl volume per well for most incubations and washes (the volume can be decreased to a minimum of ~250 μl for primary antibody reactions or other situations where reagents are limited). Unless indicated otherwise, all steps are conducted at room temperature (RT), and incubations/washes are done on a gently shaking platform.
Select one subseries for each animal.
Remove the sections from the cryoprotectant solution into a 10-cm dish containing PBS. Select the sections for immunohistochemical staining. For calbindin and NPY, the most important sections are those through the dorsal (rostral) hippocampus. Sections that are either anterior or posterior to the hippocampus can be discarded. For Fos and Arc, it is important to include every section through the dentate gyrus, from the most anterior where only the lower blade is visible, to the most posterior where the dentate takes on a circular shape. Transfer the desired sections to a 24-well plate, one well per animal, containing 400 μl of PBS per well.
Wash the sections 4 × 10 min at RT in PBS to remove cryoprotectant (see Note 12).
Wash the sections 2 × 15 min at RT in PBS/Tx.
Quench endogenous peroxidase activity by incubating the sections in PBS containing 3% H2O2 and 10% methanol for 15 min at RT. See Note 13.
Wash 4 × 15 min at RT in PBS/Tx.
Block nonspecific binding. Incubate the sections in PBS/Tx containing 10% normal goat serum (i.e., the normal serum from the species used to generate the secondary antibody), 1% dry milk, and 0.2% gelatin for 60 min at RT.
Wash 1 × 10 min in PBS/Tx at RT.
Primary antibody. Incubate the sections in PBS/Tx containing 3% normal goat serum, 0.2% gelatin, and primary antibody (1:30,000 rabbit anti-calbindin antibody, 1:8,000 rabbit anti-NPY antibody, 1:10,000 rabbit anti-Fos antibody, or 1:8,000 rabbit anti-Arc antibody) overnight at 4°C. Optimal primary antibody concentrations may differ between antibody batches. We recommend adjusting primary antibody concentration such that the development time is about 5 min.
Wash 5 × 15 min at RT in PBS/Tx.
Secondary antibody. Incubate the sections in PBS/Tx containing 3% normal goat serum, 0.2% gelatin, and 1:200 biotinylated goat anti-rabbit antibody for 60 min at RT.
Wash 5 × 15 min at RT in PBS/Tx. During these washes, prepare the ABC reagent for the next step. For a 24-well plate, add 45 μl of Reagent A (avidin) and 45 μl of Reagent B (biotinylated HRP) to 10 ml PBS/Tx and gently mix for 30 min at RT. The amount to prepare will depend on how many wells are being developed and can be scaled up or down, using 4.5 μl of each reagent per ml of PBS/Tx.
Avidin–biotin complex. Incubate the sections for 60 min at RT in ABC reagent that has been mixed as above.
Wash 1 × 15 in PBS/Tx at RT.
Wash 2 × 15 min in PBS at RT.
Wash 1 × 15 min in 100 mm Tris at RT (see Note 14).
Prepare the DAB solution as directed in Subheading 2.3.
Development reaction. We generally develop 24 wells at a time (one plate). Add DAB solution to each well at a constant interval. We typically use 10 s per well, which takes 4 min to add the DAB solution to a full 24-well plate. Monitor the progress of the development reaction under a dissecting microscope. The development time will vary depending on factors such as the batch and concentration of antibody and how the tissue was handled, but we generally aim for 5–6 min (see Note 15). Do not overdevelop and saturate the signal; light staining can be quantitated more reliably than dark staining. When the first well has reached the desired degree of development, stop the reaction by removing the DAB solution (see Note 5) and replacing it with 100 mM Tris. Continue to stop subsequent wells, again staggered 10 s apart (see Note 16).
Wash another 2 × 5 min in 100 mM Tris at RT.
Wash 2 × 15 min in PBS at RT (see Note 17).
Wash for 5 min with warmed 1% gelatin in water.
Move the sections to a 10-cm dish containing warm 1% gelatin and arrange in anatomical order for mounting.
Rest a clean SuperFrost Plus slide against the back of the 10-cm dish, wet the slide with gelatin, and use a paintbrush to gently slide the sections up onto the slide.
Blot excess gelatin solution from the edge of the slide and air dry overnight in a location protected from dust.
Clear the slides 2 × 5 min in xylene.
Coverslip with mounting medium and dry overnight.
3.4. Quantification of Results
Quantification of the results is an important aspect of the protocol and is performed on a digital microscope with image-analysis software that allows for densitometric measurement. All quantification should be performed by an experimenter blinded to the genotypes of the animals.
3.4.1. Quantification of Calbindin Immunoreactivity
Calbindin depletion occurs in dentate granule cells and is visible in both the cell bodies (granule cell layer) and their dendrites (molecular layer) (Fig. 1). We quantify calbindin immunoreactivity in the molecular layer. Calbindin immunoreactivity in area CA1 was not altered in the hAPP transgenic lines we have analyzed so far (Fig. 5); we therefore use this region as an internal control to normalize for nonspecific variations in signal intensity (see Note 18).
Fig. 5.
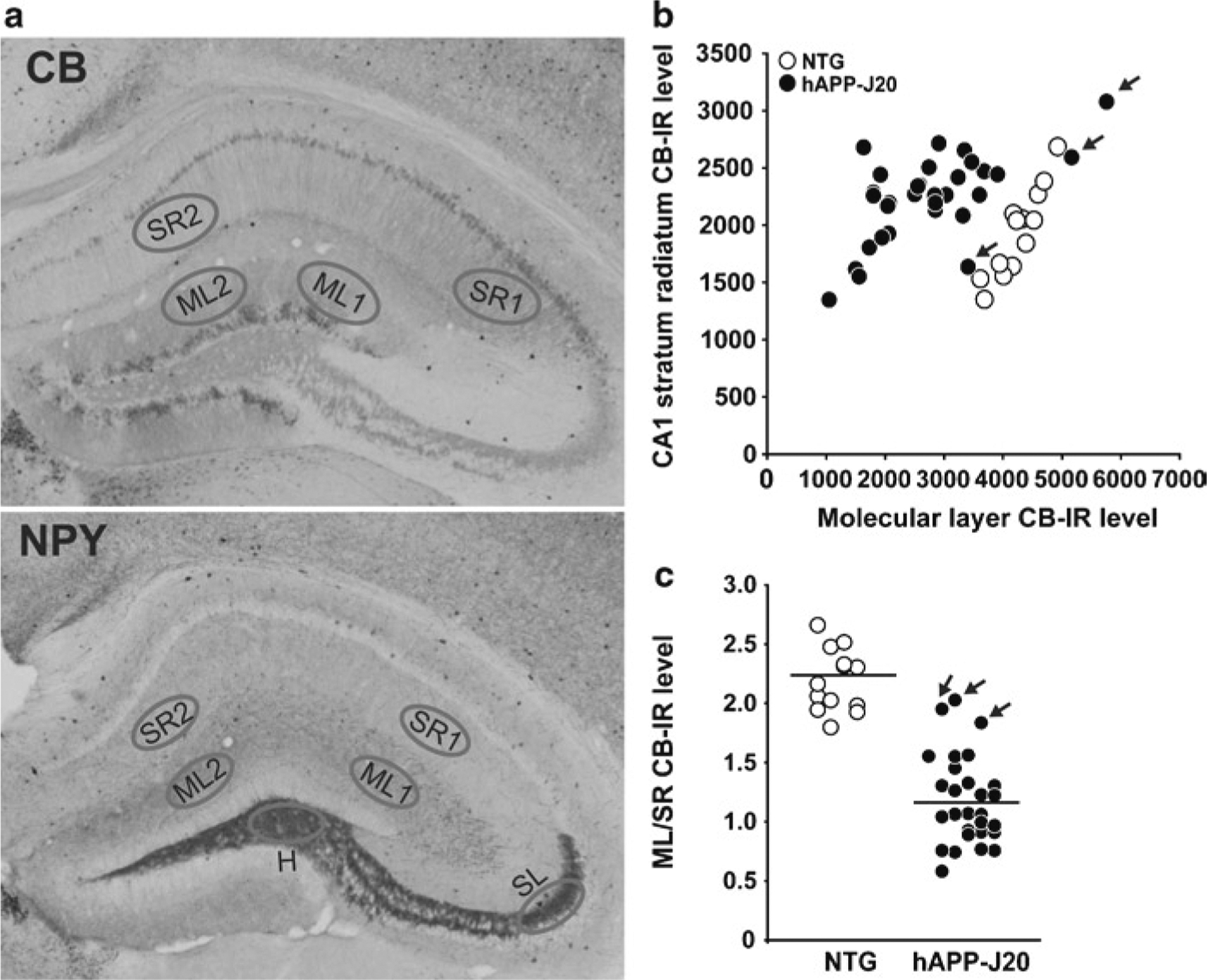
Quantification of calbindin and NPY immunoreactivity. (a) Images demonstrate calbindin depletion (top) and aberrant NPY expression (bottom) in hAPP-J20 mice. Regions of interest for densitometric quantification are indicated in the dentate molecular layer (ML) and hilus (H), CA1 stratum radiatum (SR), and CA3 stratum lucidum (SL). (b) Calbindin immunoreactivity in the dentate ML and CA1 SR. Most hAPP-J20 mice have reduced calbindin in the dentate ML relative to NTG mice, with three exceptions indicated by arrows. In contrast, there are no differences between the groups in CA1 SR. Plotting dentate ML versus CA1 SR, as shown here, reveals a correlation between calbindin IR in the ML and in SR, indicating nonspecific variation in immunostaining. Because there are no differences between NTG and hAPP-J20 mice in calbindin levels in CA1 SR, this measurement can be used to normalize these non-biological differences in staining and reduce the variability in the measurement of calbindin in dentate ML (see Subheading 3.4.1, Eq. 1). (c) The ratio of ML/SR calbindin immunoreactivity reveals calbindin levels in the dentate ML to be lower in hAPP-J20 mice than in NTG controls, with reduced interindividual variability compared to raw ML data. Arrows indicate the three hAPP mice with normal ML/SR calbindin levels.
Prepare the microscope, camera, and computer for image analysis. Adjust white balance.
Image the section at 10×.
Create an elliptical region of interest (ROI) roughly the width of the dentate gyrus molecular layer.
For each animal, select two sections (a and b) through the dorsal hippocampus (roughly around −1.6 mm and −1.9 mm from bregma). Quantify the density of immunoreactivity in four areas of each section using the ROI tool: two segments of the dentate molecular layer (ML1a and ML2a) and two corresponding segments of the CA1 stratum radiatum (SR1a and SR2a) (see Fig. 5). Repeat for the second section (ML1b, ML2b, SR1b, and SR2b).
Repeat step 4 for each animal in the experiment.
- Calculate the calbindin immunoreactivity for each animal as follows:
This equation sums the ML density in two segments of each histological section, normalizes to the SR density for each section, and then averages the two sections to obtain the calbindin immunoreactivity for that animal. The resulting value should be ~2 for NTG mice. A lower value might indicate that the sections have been overdeveloped.(1) To enable comparison of data between experiments, determine the average calbindin level for all of the NTG animals in the experiment, and normalize every animal’s calbindin level to this value. The average value of the NTG animals will thus be 1.0 after normalization.
3.4.2. Quantification of NPY Immunoreactivity
In the normal mouse hippocampus, NPY is expressed in a subset of inhibitory interneurons. Dark staining is visible in the soma of these cells, and faint staining from the axons of these inhibitory interneurons is visible in the molecular layer, where they synapse onto the dendrites of granule cells (Fig. 2). In AD mouse models, NPY is aberrantly expressed by granule cells themselves, most prominently in their axons (the mossy fibers), which is visible as dark staining along the course of the mossy fibers through the hilus to their termination in stratum lucidum of area CA3 (Fig. 2). hAPP transgenic mice also exhibit axonal sprouting of NPY-expressing hilar interneurons, resulting in increased immunoreactivity in the dentate molecular layer (Fig. 2). We quantify both processes, measuring NPY immunoreactivity in the mossy fibers and in the dentate molecular layer. Because NPY immunoreactivity in area CA1 was not altered in the hAPP lines we have analyzed so far, we use this region as an internal control to normalize the signal (Fig. 5).
Prepare the microscope, camera, and computer for image analysis. Adjust the white balance.
Image the section at 10×.
Create an elliptical ROI roughly the width of the dentate gyrus molecular layer.
For each subject animal, select two sections (a and b) through the dorsal hippocampus with good dentate anatomy (roughly around −1.6 mm and −1.9 mm from the bregma). Quantify the density of immunoreactivity in six areas of each section using the ROI tool: two segments of the dentate molecular layer (ML1a and ML2a), two corresponding segments of the CA1 stratum radiatum (SR1a and SR2a), the dentate hilus (Ha), and CA3 stratum lucidum (SLa) (see Fig. 5). Repeat for the second section (ML1b, ML2b, SR1b, SR2b, Hb, and SLb).
Repeat step 4 for each animal in the experiment.
- Calculate the NPY immunoreactivity in the molecular layer for each animal as follows:
This equation sums the ML density in two segments of each histological section, normalizes to the SR density for each section, and then averages the two sections to obtain the NPY immunoreactivity in the molecular layer for that animal. This measure reflects axonal sprouting of NPY-positive hilar interneurons onto granule cell dendrites in the molecular layer.(2) - Calculate the NPY immunoreactivity in the mossy fibers for each subject animal as follows:
This equation calculates the hilus density, normalizes to SR density, for each histological section, then averages the two sections. Similar normalization and averaging are performed for the CA3 stratum lucidum signals from the two sections. Then the hilar and CA3 stratum lucidum signals are averaged to obtain the NPY immunoreactivity in the mossy fibers for each animal. This measure reflects the aberrant expression of NPY in granule cell axons.(3) To enable comparison of data between experiments, determine the average NPY value for all of the NTG animals in the experiment, and normalize every animal’s NPY level to this value. The average value of the NTG animals will be 1.0.
3.4.3. Quantification of Fos and Arc Immunoreactivity
In the normal mouse hippocampus, Fos and Arc are expressed in a small proportion of dentate granule cells. Fos-positive granule cells are easily visualized by their dark, round, nuclear staining (Fig. 3a), whereas Arc-positive granule cells show intense labeling in the soma and dendrites (Fig. 3b). The number of Fos- and Arc-positive granule cells are markedly reduced in hAPP transgenic mice with high levels of Aβ in the hippocampus (Fig. 3).
Starting at the most anterior section through the dentate gyrus, count the number of Fos- or Arc-positive cells in the granule cell layer in each section with a tally counter. Most positive cells exhibit strong and unequivocal staining, but there will be a few cells with less intense immunoreactivity. The experimenter must develop a consistent threshold for what will be considered a positive cell. All animals in a given cohort should be counted by the same experimenter at the same time. See Note 19.
Data can be plotted either as the number of Fos- or Arc-positive cells at a given rostro–caudal level (Fig. 3c) or as the total number of positive cells counted (Fig. 3d).
4. Notes
PFA is a toxic and volatile compound and must be handled in a fume hood.
PFA does not go into solution well at RT. However, avoid temperatures above 85°C, which will compromise the fixative properties of PFA.
We prefer the microtome over the vibratome, as it produces more uniform sections.
For washing, we use a transfer pipette with a fine (10 μl) Pipetman tip attached to the end with Parafilm. Remove as much liquid from the well as possible without damaging the sections. Tipping the plate back and forth can help collect the sections on one side of the well while removing the liquid from the other side.
DAB is a suspected carcinogen. When working with DAB and DAB-containing solutions, wear gloves, lab coat, and eye protection. Anything that comes into contact with DAB, including pipette tips, glassware and plasticware, and filter paper, should be treated with bleach or a solution of 3% potassium permanganate (KMnO4) and 2% sodium carbonate (Na2CO3).
Hydrogen peroxide is a substrate for peroxidase, and DAB is an electron donor in this reaction. The resulting oxidation of DAB produces a brown precipitate that is insoluble in xylenes and thus serves as a good immunohistochemical detection label.
It is important not to dissolve the gelatin in saline buffers, as the salts will crystallize on the sections as they dry.
We typically flash freeze one hemisphere on dry ice for future biochemical analyses, and drop-fix the other for immunohistochemistry. If desired, the entire brain can be fixed for immunohistochemistry without bisecting into separate hemibrains.
We routinely make small amounts of 10% sucrose by a 1:3 dilution of the 30% sucrose solution used for cryoprotection.
If the sections are intended for immunohistochemistry within a few days, the sections can be placed into PBS rather than cryoprotectant and stored at 4°C. This saves some time later, as the washes to remove cryoprotectant solution can be omitted.
A similar protocol can be performed for fluorescent labeling of calbindin or NPY. The following steps in Subheading 3.3 are modified. (a) Steps 5 and 6: quenching endogenous peroxidase and subsequent washing can be omitted. (b) Step 9: primary antibody concentrations are usually increased by 2–3× for fluorescence microscopy. (c) Step 11: use a fluorescent-labeled secondary antibody and incubate for 2 h. (d) Steps 13–26 (avidin–biotin–HRP complex, DAB development, and dry mounting in gelatin) are omitted. Instead, mount the PBS-washed sections with a fluorescence mounting medium and keep slides at 4°C.
If the sections were in PBS rather than cryoprotectant, this step can be omitted.
Some tissues contain endogenous peroxidase activity that could react with DAB to induce deposition of stain during development, even in the absence of antigen/antibody/HRP. This step quenches endogenous peroxidase to avoid this problem. In brain sections, the main source of endogenous peroxidase is erythrocytes, and failure to block endogenous peroxide will result in staining of the vasculature.
Developing in Tris-based solution instead of PBS produces lower background.
If the reaction proceeds much faster (e.g., 1 min), it is difficult to ensure that each well is developed for equal times, and antibody concentrations or DAB concentrations should be reduced. If the reaction takes too long (e.g., >10 min), then concentrations can be increased.
We routinely have two people involved in stopping the development reaction. One person removes the DAB from the well, and the other person immediately adds the Tris. This allows us to move from well to well in 10-s intervals. If it takes longer than this to stop the reaction in each well, use a longer interval between wells when adding the DAB, so that each well has the same duration of development in DAB.
Sections can be stored at 4°C in PBS for up to a week before sectioning. If the sections will be mounted immediately, it is not necessary to change from Tris to PBS, and this step can be omitted.
Different sections may have different staining intensity due to different times of development or immunoreactivity of the tissue. An internal control effectively cancels out this non-biological variation. It is important to verify in pilot experiments that the signal of the control area (e.g., radiatum of CA1) is not significantly changed in the experimental groups.
Around 5% of hAPP-J20 mice have abnormally increased Fos and Arc levels due to recent epileptiform activity (13). We do not typically count the number of positive granule cells in these mice.
Acknowledgments
We thank members of the Mucke laboratory, particularly Nga Bien-Ly and Alice Thwin, who helped develop these protocols; Jeannie Chin, Irene Cheng, and Julie Harris for helpful comments on the manuscript; Paul Worley for Arc antibody; and Stephen Ordway and Gary Howard for editorial review. This work was supported by Stephen D. Bechtel Jr. Young Investigator Awards (J.J.P and E.D.R), and NIH grants NS054811 (E.D.R.), AG023501 (L.M.) and AG022074 (L.M.).
References
- 1.Crabbe JC, Wahlsten D, and Dudek BC (1999) Genetics of mouse behavior: interactions with laboratory environment. Science 284, 1670–72. [DOI] [PubMed] [Google Scholar]
- 2.Richter SH, Garner JP, and Würbel H (2009) Environmental standardization: cure or cause of poor reproducibility in animal experiments? Nat. Methods 6, 257–61. [DOI] [PubMed] [Google Scholar]
- 3.Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O’Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, and Duff K (1998) Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat. Med 4, 97–100. [DOI] [PubMed] [Google Scholar]
- 4.Westerman MA, Cooper-Blacketer D, Mariash A, Kotilinek L, Kawarabayashi T, Younkin LH, Carlson GA, Younkin SG, and Ashe KH (2002) The relationship between Aβ and memory in the Tg2576 mouse model of Alzheimer’s disease. J. Neurosci 22, 1858–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kobayashi DT, and Chen KS (2005) Behavioral phenotypes of amyloid-based genetically modified mouse models of Alzheimer’s disease. Genes Brain Behav 4, 173–96. [DOI] [PubMed] [Google Scholar]
- 6.Lesné S, Ming TK, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, and Ashe KH (2006) A specific amyloid-β protein assembly in the brain impairs memory. Nature 440, 352–57. [DOI] [PubMed] [Google Scholar]
- 7.Palop JJ, Jones B, Kekonius L, Chin J, Yu G-Q, Raber J, Masliah E, and Mucke L (2003) Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer’s disease-related cognitive deficits. Proc. Natl. Acad. Sci. U S A 100, 9572–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arriagada PV, Growdon JH, Hedley-Whyte ET, and Hyman BT (1992) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42, 631–39. [DOI] [PubMed] [Google Scholar]
- 9.Ingelsson M, Fukumoto H, Newell KL, Growdon JH, Hedley-Whyte ET, Frosch MP, Albert MS, Hyman BT, and Irizarry MC (2004) Early Aβ accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology 62, 925–31. [DOI] [PubMed] [Google Scholar]
- 10.Giannakopoulos P, Gold G, Kövari E, von Gunten A, Imhof A, Bouras C, and Hof PR (2007) Assessing the cognitive impact of Alzheimer disease pathology and vascular burden in the aging brain: the Geneva experience. Acta Neuropathol 113, 1–12. [DOI] [PubMed] [Google Scholar]
- 11.Palop JJ, Chin J, Bien-Ly N, Massaro C, Yeung BZ, Yu G-Q, and Mucke L (2005) Vulnerability of dentate granule cells to disruption of Arc expression in human amyloid precursor protein transgenic mice. J. Neurosci 25, 9686–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chin J, Palop JJ, Puoliväli J, Massaro C, Bien-Ly N, Gerstein H, Scearce-Levie K, Masliah E, and Mucke L (2005) Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer’s disease. J. Neurosci 25, 9694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu G-Q, Kreitzer A, Finkbeiner S, Noebels JL, and Mucke L (2007) Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 55, 697–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng I, Scearce-Levie K, Legleiter J, Palop J, Gerstein H, Bien-Ly N, Puoliväli J, Lesné S, Ashe K, Muchowski P, and Mucke L (2007) Accelerating amyloid-β fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J. Biol. Chem 282, 23818–28. [DOI] [PubMed] [Google Scholar]
- 15.Palop JJ, and Mucke L (2009) Epilepsy and cognitive impairments in Alzheimer disease. Arch. Neurol 66, 435–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Minkeviciene R, Rheims S, Dobszay MB, Zilberter M, Hartikainen J, Fülöp L, Penke B, Zilberter Y, Harkany T, Pitkänen A, and Tanila H (2009) Amyloid β-induced neuronal hyperexcitability triggers progressive epilepsy. J. Neurosci 29, 3453–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu G-Q, and Mucke L (2007) Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer’s disease mouse model. Science 316, 750–54. [DOI] [PubMed] [Google Scholar]
- 18.Vezzani A, Sperk G, and Colmers WF (1999) Neuropeptide Y: emerging evidence for a functional role in seizure modulation. Trends Neurosci 22, 25–30. [DOI] [PubMed] [Google Scholar]
- 19.Palop JJ, Chin J, and Mucke L (2006) A network dysfunction perspective on neurode-generative diseases. Nature 443, 768–73. [DOI] [PubMed] [Google Scholar]
- 20.Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, and Garaschuk O (2008) Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science 321, 1686–89. [DOI] [PubMed] [Google Scholar]
- 21.Palop JJ, and Mucke L (2010) Amyloid-β-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat. Neurosci 13:812–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vogt DL, Thomas D, Galvan V, Bredesen DE, Lamb BT, and Pimplikar SW (2009) Abnormal neuronal networks and seizure susceptibility in mice overex-pressing the APP intracellular domain. Neurobiol. Aging doi: 10.1016/j.neurobiolaging.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]