

Abstract
Most autoimmune diseases (AID) are linked to an imbalance between autoreactive effector T cells (Teffs) and regulatory T cells (Tregs). While blocking Teffs with immunosuppression has long been the only therapeutic option, activating/expanding Tregs may achieve the same objective without the toxicity of immunosuppression. We showed that low-dose interleukin-2 (ld-IL-2) safely expands/activates Tregs in patients with AID, such HCV-induced vasculitis and Type 1 Diabetes (T1D). Here we analyzed the kinetics and dose-relationship of IL-2 effects on immune responses in T1D patients. Ld-IL-2 therapy induced a dose-dependent increase in CD4+Foxp3+ and CD8+Foxp3+ Treg numbers and proportions, the duration of which was markedly dose-dependent. Tregs expressed enhanced levels of activation markers, including CD25, GITR, CTLA-4 and basal pSTAT5, and retained a 20-fold higher sensitivity to IL-2 than Teff and NK cells. Plasma levels of regulatory cytokines were increased in a dose-dependent manner, while cytokines linked to Teff and Th17 inflammatory cells were mostly unchanged. Global transcriptome analyses showed a dose-dependent decrease in immune response signatures. At the highest dose, Teff responses against beta-cell antigens were suppressed in all 4 patients tested. These results inform of broader changes induced by ld-IL-2 beyond direct effects on Tregs, and relevant for further development of ld-IL-2 for therapy and prevention of T1D, and other autoimmune and inflammatory diseases.
Keywords: Tolerance, Immunotherapy, Inflammation, Immunopathology, Pharmacokinetics
1. Introduction
Most self-reactive T lymphocytes are physically eliminated during thymic selection to avoid immune responses against self-antigens. Yet this process is imperfect, and potentially harmful self-specific effector T cells (Teffs) survive thymic deletion and populate the periphery. In the normal immune system, these autoreactive Teffs do not mediate autoimmunity as their expansion and function are inhibited by regulatory T cells (Tregs) [1]. In most autoimmune diseases (AID), there is an imbalance between Teffs that attack normal tissues and Tregs that normally control them. Tregs are essential players in the control of all immune responses, including responses to self, tumors, and infectious agents [1], and in the control of inflammatory disorders [2]. The discovery of Tregs revolutionized our understanding of AID pathophysiology and opened new avenues for the treatment of autoimmunity. Human beings and mice presenting a FOXP3 genetically-determined Treg defect develop multiple organ-specific AID [3–8]. Treg quantitative or qualitative defects have been described in common human AID and their mouse models [9–13]. Moreover, administration/restoration of Tregs improves symptoms in experimental animals and patients with AID [14–19]. Thus, while blocking Teffs with immunosuppressive drugs has long been the only therapeutic option for AID, activating/expanding Tregs is becoming a novel approach that may lead to improved outcomes with better safety [14–19].
Until recently, most Treg-based therapies have been based on the infusion of ex vivo expanded Tregs, as there were no drugs or molecules that could specifically expand/activate Tregs in vivo. We and others recently showed that IL-2, a molecule identified almost 30 years ago for its capacity to stimulate T cells in vitro, could be such a molecule [14–17]. We performed the first clinical trial aimed at inducing Tregs with IL-2 in patients with an AID [17], namely hepatitis C virus (HCV)-related vasculitis (NCT00574652) [20]. We treated these patients with low-dose (ld) IL-2 (1.5–3 million international units MIU/day; 52.5 MIU cumulative dose; Proleukin®). The treatment was very well tolerated. Importantly, we observed a major expansion of Tregs with subsequent clinical improvement in 8/10 patients [17]. Our results showed for the first time that ld-IL-2 could be exploited as an immunoregulatory drug for the treatment of AID, acting via Treg expansion/activation. Others have reported that low dose IL-2 is safe and beneficial for the treatment of alloimmunity, i.e. chronic graft versus host disease (GVHD) [15, 16].
Based on these results, we evaluated ld-IL-2 in patients with type 1 diabetes (T1D), a condition resulting from the chronic autoimmune destruction of pancreatic beta-cells, eventually leading to insulin deficiency. There are several reasons for using IL-2 in T1D patients: (i) several susceptibility genes are directly connected to the IL-2 pathway: IL-2, CD25 (IL-2RA) [21–23], IL-2RB [24], and PTPN2 [25, 26]; (ii) some studies reported Treg deficiency in peripheral blood [9, 10], and in pancreatic lymph nodes [27, 28]; (iii) at the time of clinical diagnosis, there is significant residual beta-cell function in most patients, so that immunotherapy could curtail inflammation, promote immune tolerance, and in turn preserve beta-cell mass and function [29]. In Non Obese Diabetic (NOD) mice, a model of spontaneous autoimmune diabetes with remarkable similarities to the human disease, IL-2 prevents T1D and we showed that a short course of IL-2 at diabetes onset led to disease reversal in one third of the mice [14, 30]. Finally, the use of immunosuppressants such as cyclosporine A (CsA), a calcineurin inhibitor that reduces T cell activation and expansion, provided proof of principle that newly-diagnosed T1D could be treated with immunotherapy [31–33]. CsA demonstrated clinical efficacy in prolonging endogenous insulin production, but remission from autoimmunity was limited to the duration of the treatment and CsA toxicity precluded its clinical use. Other immunosuppressive drugs or immunomodulators have been tested in clinical trials in T1D, both as short therapy courses or chronic regimens, only in some cases resulting in temporary preservation of insulin secretion with better results in subsets of responder patients, as reported recently for therapies with anti-CD3, anti-CD20 and CTLA4-Ig [34–38].
The identification of a dose of IL-2 capable of safely tipping the Treg/Teff balance towards Tregs is of major importance. In our vasculitis trial, we showed that IL-2 at the dose of 1.5 MIU induced Tregs in all 10 patients and was well tolerated. However, the dose to be used in T1D was not predictable as (i) some T1D patients may have defects in the IL-2/IL-2R activation pathway [39] (ii) HCV vasculitis is an antibody-mediated disease, while in T1D beta-cell destruction is mediated by pathogenic Teffs that could respond to IL-2 and thus exacerbate disease. Hence, we designed a dose finding trial to define safety and immunological responses; we previously reported that a 5-day course of single IL-2 injections, given at 0.33, 1 or 3 MIU, were well tolerated and stimulated Tregs [40].
Here, we report the results of detailed immunomonitoring of these patients, showing that ld-IL-2 induces a dose-dependent, regulatory milieu characterized by broad changes extending beyond the primary effect on Tregs. Thus, our study provides important information for further developing ld-IL2 therapies.
2. Materials and methods
2.1. Patient characteristics
Male and female patients aged from 18 to 55 years, with confirmed T1D were recruited at the Diabetology Department of the Pitié-Salpêtrière Hospital (Paris, France) and thereafter followed at the Clinical Investigation Centre-Paris Est. Written informed consent was obtained from all subjects before they were enrolled in the DF-IL2 trial (NCT01353833). Detailed patients description, safety analyses and peak effects of the treatment on Tregs have been reported [40]. This clinical trial was conducted according to Declaration of Helsinki principles. All human studies were approved by the appropriate institutional review boards.
2.2. Immunomonitoring
Blood samples were collected according to the planned protocol: absolute numbers of lymphocyte subsets and Tregs were monitored at each patient visit: day-1, before treatment; day 2–5, during IL-2 administration; day-6; 15, 22, and 60 end of the follow up.
Tregs phenotyping and cytokines assay were performed at baseline, day 6, 15, 22 and 60. Because of the limited amount of blood collected, other phenotypic markers where only investigated at baseline and day 15. When possible, cells were frozen for other investigations. Stat5 phosphorylation (pSTAT5) was examined on frozen cells from 2 controls and 3 patients per treated group. Beta-cell-specific Teff responses were studied in 1 control, 3 patients for the dose 0.33, 2 for the dose 1 and 4 for the dose 3.
2.3. Flow Cytometry and pStat5 analysis
Flow cytometry and basal pStat5 experiments were performed according to previously published methods [17, 41]. Monoclonal antibodies (mAbs) used were CD3-APCa750, CD4-ECD, CD8-APCa700, CD16-FITC, CD19-ECD, CD45RO-FITC, CD56-PCy7 and CD152-PE from Beckman Coulter; CD25-PE, CD45RA-APC and CD45RA-PCy7 from BD Biosciences; CD127-FITC from eBioscience; CD25-APC and LAP-PE from R&D Systems and GITR-PE from Miltenyi.
For pSTAT5 activation experiments, peripheral blood mononuclear cells (PBMCs) were cultured (500,000/well) for 30 min at 37° C and IL-2 was added for 15 min. Cells were then fixed with 1.6% paraformaldehyde, permeabilized with 100% methanol, stained for pSTAT5 and appropriate surface markers. Events were collected using a LSRFortessa flow cytometer (BD Biosciences) and data were analyzed using Diva software (BD Biosciences). To calculate EC50, after subtracting the value from the media control, binding data for each sample was normalized to 100% based on the maximal response and non-linear regression of these data was performed assuming a variable slope using Graph-Pad Prism 6.0.
2.4. Plasma cytokine measurement
Plasma samples were collected, aliquoted and stored at −80°C until analyzed. Quantitative determination of 26 cytokines/chemokines (GM-CSF, IFN-α2, IFN-γ, IL-1RA, IL1a, IL1β, IL-2, IL-4, IL-5, IL-6, IL-7, IL-9, IL-10, IL-12p40, IL-12p70 , IL-13, IL-15, IL-17a, CXCL-10, CCL-2, CCL-3, CCL-4, CCL-2, TNF-α and IL-2RA) was performed using Human Milliplex HCYTOMAG-60 kits (Millipore) and TGF-beta using Human TGF-beta1 Platinium ELISA (eBioscience) in accordance with the manufacturer protocols.
2.5. Analysis of beta-cell antigen-specific T-cell responses
CD4+ and CD8+ T-cell responses were evaluated according to previously published assay [42].
2.6. Anti-IL-2 antibodies assay
The presence of plasma IL-2-binding antibodies was studied by ELISA, as previously described [43]. Data are the average of two replicates. Positive controls are from ApoE−/− mice that were injected three times a week with 25,000 IU of Proleukin® for 4 months. Mouse studies were approved by the appropriate institutional review boards.
IL-2 neutralising activity of plasma was assayed in a proliferation assay with Kit 225 cells [44]. 10,000 cells/well were cultured (37°C in 5% CO2) in 100 μL of RPMI (Gibco) and 10% of BSA. Plasma were preincubated for 1 hour with IL-2 10 IU/ml at 37°C and then added at the start of the cultures at a concentration of 20%. Cell proliferation was assayed by counting cells at day 3.
2.7. Transcriptomic analysis
Transcriptomic analysis was performed using the SurePrint G3 Human GE v2 8×60K Microarray (Agilent Technologies, ID: 039494) according to the manufacturer’s protocol and to previously published [17]. Cyanine-3 labeled cRNAs were prepared using the One-Color Low Input Quick Amp Labeling kit (Agilent Technologies).
The slides were scanned using a G2565CA Scanner System (Agilent Technologies), and using a scan protocol with a resolution of 3 μm and a dynamic range of 20 bit. The resulting .tiff images were analyzed with the Feature Extraction Software v10.7.3.1 (Agilent Technologies) and using the GE2_107_Sep09 protocol. Original dataset was normalized using quantile method implemented in Limma’s package. Probes with bad detection p-values (<0.01) in half of the samples of a biological group were discarded. Ranked gene lists were obtained using statistical score of paired eBayes implemented in Limma’s package. Gene set enrichment analyses were performed using the Blood Transcription Modules [45] added with a selection of molecular signatures from C2 database provided by GSEA software [46, 47]. Network analyses were performed using Cytoscape 2.8.3 and the plugin Enrichment Map dedicated to gene set enrichment analyses.
All datasets can be accessed in the ArrayExpress database (www.ebi.ac.uk/arrayexpress) under accession number E-MTAB-2858.
2.8. Statistical analysis
Data were compared between patients groups using a two-way analysis of variance (ANOVA) for repeated measures using GraphPad Prism version 5.0. When the p value of the time by treatment interaction was significant (p<0.05), each IL-2 group of patients was compared to placebo group.
Detailed statistical results of two-way ANOVA for repeated measures are shown in the Supplementary table 1.
Principal Component Analyses were then performed on log10-transformed data expressed as fold change compared to day 1. Multivariate analyses were performed using R platform (www.r-project.org).
3. Results
3.1. Clinical trial design, patients and safety
Results presented here are from a randomized, double-blind, placebo-controlled, dose-finding trial. Patients received a daily sub-cutaneous injection of placebo or 0.33, 1 or 3 MIU of IL-2 for 5 consecutive days. They were sampled every day just before the IL-2 injection for the first 5 days, then at day 6, 15, 22 and 60 (Supplementary Fig 1). Twenty-four patients (6 per dose) were included in the per-protocol analysis. No serious adverse events were reported during the trial [40].
3.2. Dose dependent effects on peripheral blood mononuclear cells
3.2.1. T, B and NK cells
There were no significant changes in CD4+ and CD8+ T cell, B cell and NK cell numbers in the two lower dose groups compared to placebo (Supplementary Fig 2). At the dose of 3 MIU/day, T, B and NK cell numbers were decreased approximately 2 fold during treatment, followed by an immediate return to baseline thereafter (Supplementary Fig 2). Regarding cell percentages (Supplementary Fig 3), a B cell reduction was observed at the two highest doses (p=0.03 at 1 MIU/day and 0.003 at 3 MIU/day) during treatment, followed by a return to baseline levels thereafter. No statistically significant variations of NK cell percentages were observed at the doses of 0.33 and 1 MIU/day while increase NK proportions were observed at the dose of 3 MIU/day (p=0.003). However, at this dose, individual variations of NK cell percentages remained in the range observed in the placebo group (see below). Finally, there was an increase in CD56bright regulatory NK cells [48] at the dose of 1 MIU/day (p=0.015) (Supplementary Fig 4).
3.2.2. CD4 and CD8 Tregs
Effects of treatment on the proportions of CD4+ Tregs are shown in Fig 1A. Comparisons between placebo and each of the three doses of IL-2 by repeated measures ANOVA showed that the day/treatment interaction was highly significant (p<0.0001). Tregs were expanded at all doses, and the duration of increase was dose-dependent. By day 15, Tregs returned to baseline at the dose of 0.33 MIU/day, but persisted elevated at the dose of 1 MIU/day; increased Treg levels were still detected at day 60 at the dose of 3 MIU/day. The increased percentage of CD4+ Tregs was associated with an increase in their absolute numbers (p<0.0001) (Fig1B), leading to an increase in the Tregs/Teff ratio (p<0.0001) (Fig 1C).
Fig 1: Dose-dependent increase of Treg in T1D patients treated by ld-IL-2.
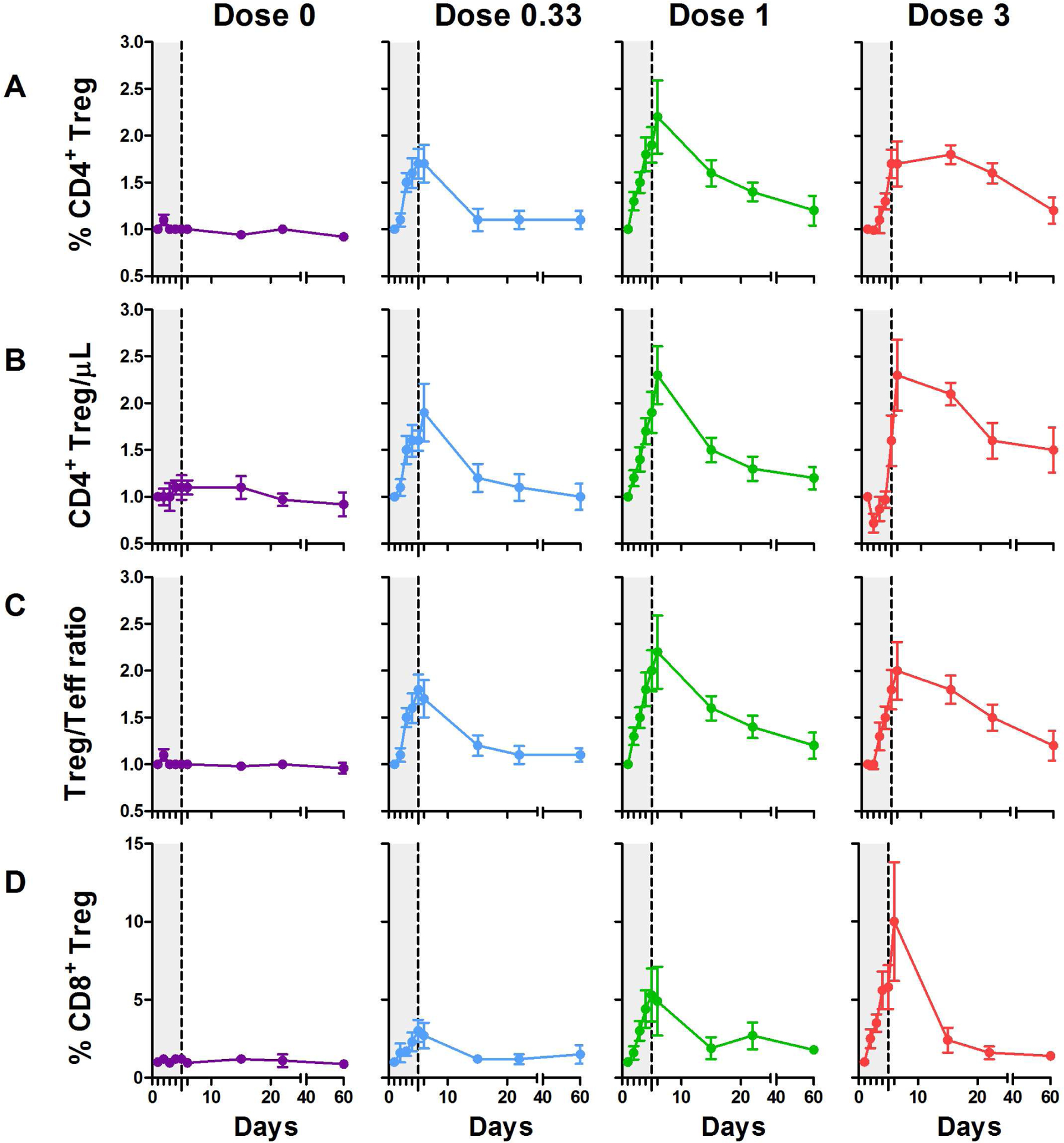
Each curve represents changes in CD25highCD127−Foxp3+ among CD4+ T cells (percentages in A and absolute numbers in B), in Treg/Teff ratio (C) and in percentages of CD8+CD25+Foxp3+ cells (D). Data are shown as mean ± SEM of individual results normalized by baseline values for each patient at different time points (the grey zone represents the 5 days IL-2 treatment).
We previously reported that the very small population of CD8+CD25+Foxp3+ T cells, which are suppressive and expanded in colorectal cancer [49], was expanded in the VASCU-IL-2 trial [17]. Here, we show a dose-dependent expansion of these CD8+ Tregs (p=0.0002), remarkably high at the highest IL-2 dose (p< 0.0001) (Fig 1D).
3.3. Treg immunophenotyping
Expanded Tregs displayed a memory phenotype (p=0.0051) [50] and phenotypic features characteristic of enhanced activation (Fig 2A). The expression of IL-2 receptor-alpha (CD25) was increased in Tregs (Fig 2B), but not in CD4+CD45RA− T effector/memory (TEM) cells (not shown), during IL-2 therapy, from day 2 or 3, for all doses (p<0.0001), and rapidly returned to baseline values thereafter. This was associated with increased expression of GITR (p=0.0058) and CTLA-4 (p<0.0001) (Fig 2C, 2D), whereas TGF-β latency-associated peptide (LAP) remained unchanged (data not shown).
Fig 2: Dose-dependent Treg activation in T1D patients treated by ld-IL-2.
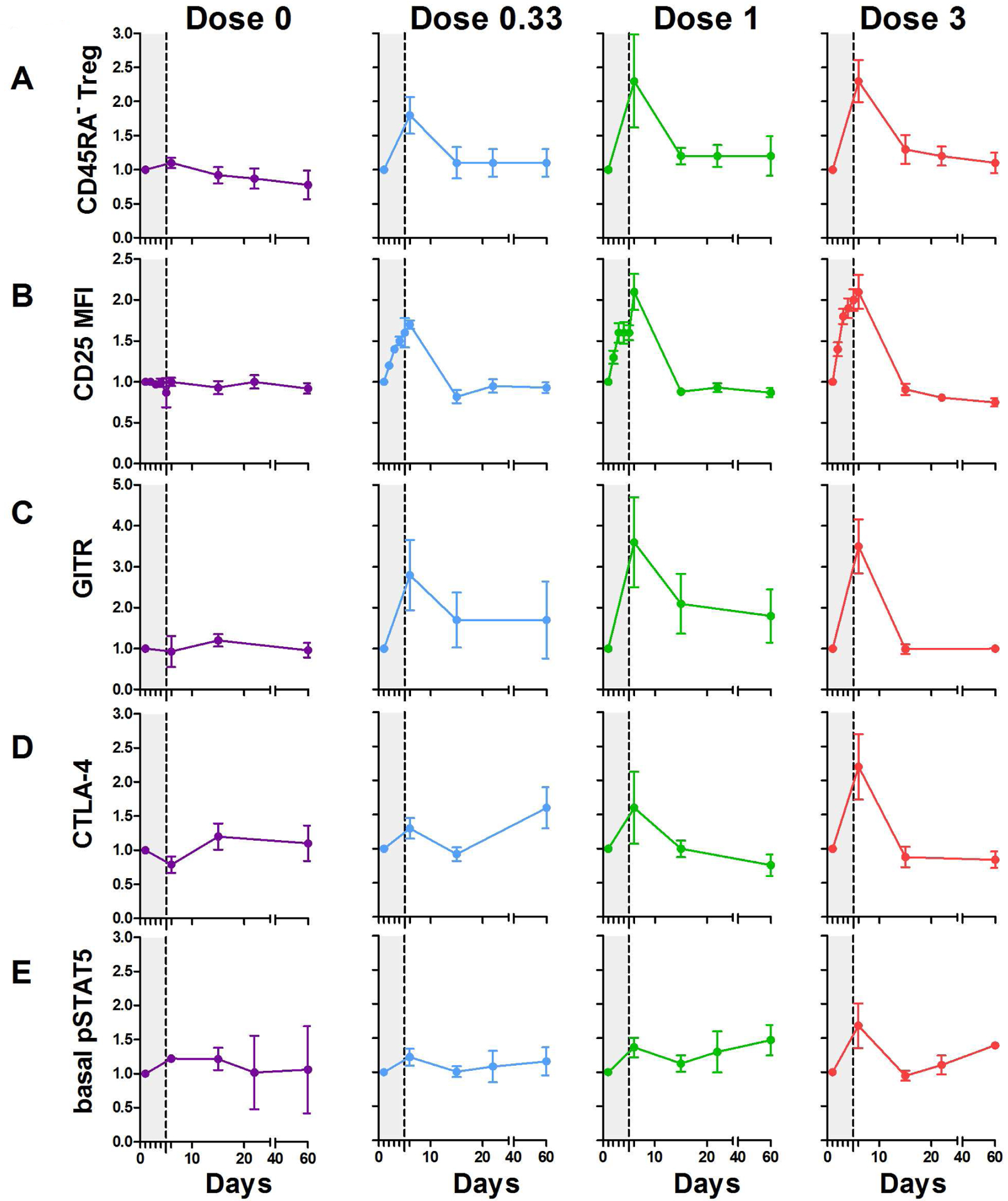
Curves represent changes in Treg activation markers. Percentage of CD25highCD127− CD45RA− Tregs among CD4+ cells (A). CD25 MFI (B), percentage of GITR+ (C), CTLA-4+ (D) and basal pSTAT5 expression (E) in CD4+CD25highCD127−Foxp3+Tregs. Data are shown as mean ± SEM of patients’ values normalized by individual baseline values at different time points (the grey zone represents the 5 days IL-2 treatment). Results are for all 6 patients per group, except for pSTAT5 measurement for which 2, 3, 3 and 3 patients were studied for the 0, .33, 1 and 3 MIU doses, respectively.
3.4. Tregs show stable, selective, and highly sensitive responses to IL-2.
An immediate consequence of IL-2 binding to the IL-2R is tyrosine phosphorylation of STAT5 (pSTAT5). Basal pSTAT5 levels in Tregs were transiently increased at the 3 MIU/day dose (p=0.0308) (Fig 2E), which may be related to the increased proportions of CD45RA− Tregs (Fig 2A). We also assessed the IL-2-dependent activation of pSTAT5 by CD4+ Treg and TEM cells by various amounts of IL-2, for patients’ samples that were obtained before and 4–6 days after the initiation of IL-2 therapy. For each patient’s sample examined, Tregs showed increased sensitivity to IL-2 when compared to TEM cells. These responses were largely comparable to cells from normal controls that did not receive IL-2 therapy (Fig 3A). This trend was similar regardless of the dose of IL-2 used to treat the patients. In addition, the responses during treatment were essentially identical to those detected before ld-IL-2 therapy. Thus, a short course of IL-2 therapy did not rapidly alter IL-2-dependent pSTAT5 activation. By calculating the EC50s of these responses, Tregs from T1D patients were approximately 15–20-fold more sensitive to IL-2 that TEM cells (Fig 3B). These analyses of frozen PBMCs resulted in IC50s in a similar range detected for fresh PBMCs, i.e 3–7 pM for Tregs and 30–35 pM for CD4+ TEM cells (A.Y, A.P. and T.R.M, unpublished data). Overall, these data are consistent with stable, highly sensitive, and selective responses by Tregs to IL-2 in these T1D patients.
Fig 3: Selective IL-2-dependent activation of STAT5 by Tregs does not vary after ld-IL- 2 therapy.
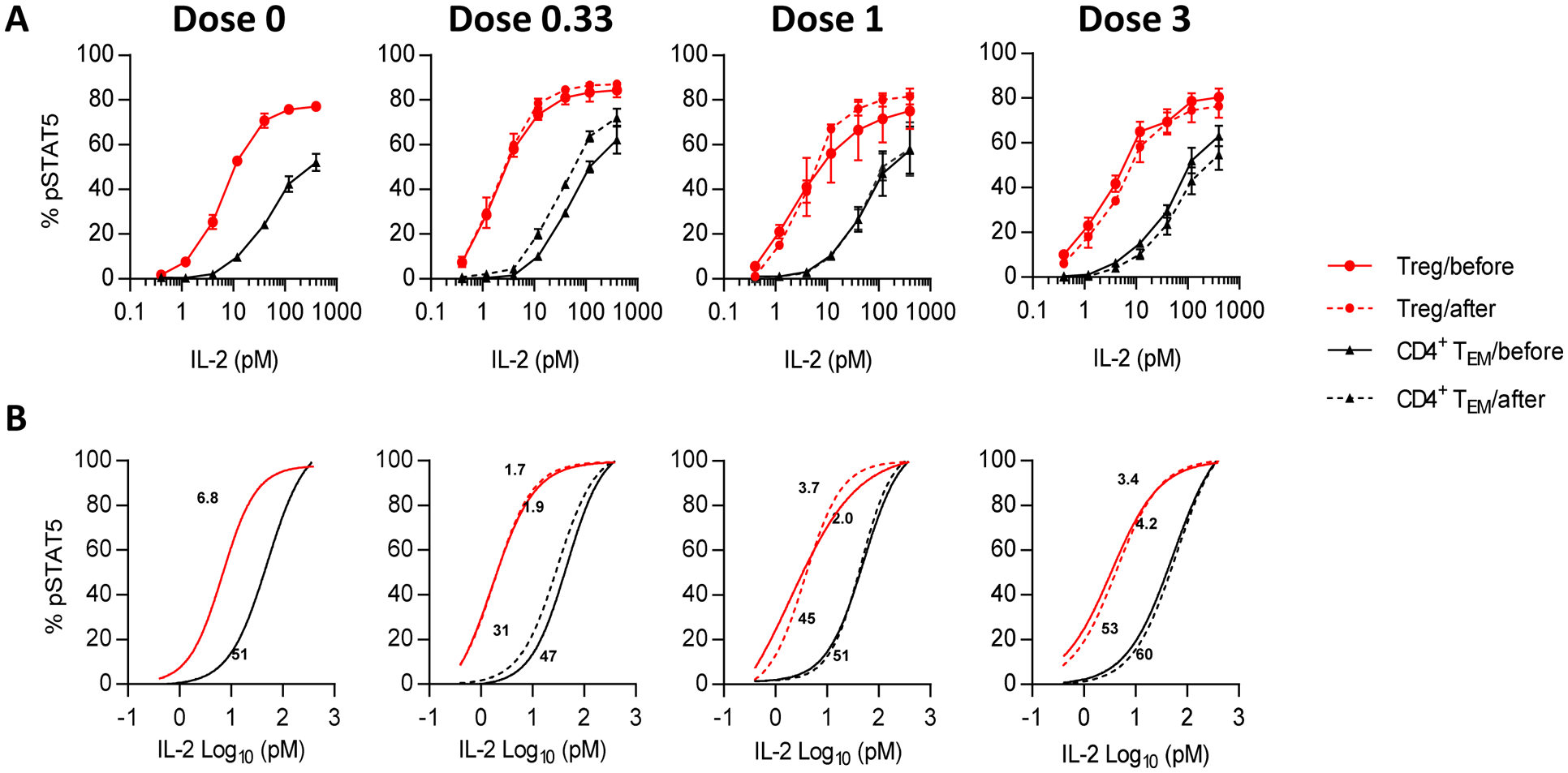
PBMC from controls (n=4) or patients treated with 0.33 MIU (n=3), 1 MIU (n=2), and 3 MIU (n=3) of IL-2 were stimulated with IL-2 for 15 minutes and the activation of pSTAT5 was determined for CD4+ Foxp3+ Tregs and CD4+ Foxp3− CD45RA− TEM cells. (A) Dose response curves and (B) non linear regression analyses of the binding data for the indicated cell populations. The numbers on the regression curves are the EC50s.
3.5. Dose dependent modulation of plasma cytokine profiles
A panel of 26 cytokines and chemokines were assayed to investigate whether IL-2 could influence the balance of Th1/Th2/Th17 cytokines during treatment. The entire cytokine profile was tested at day 6, according to dose and to the functional classification of cytokines. Dose dependent IL-2 levels could be detected in the plasma. A radar chart (Fig 4A) illustrates a clear trend for a dose-dependent increase of Treg-related cytokines, while the other cytokines are modestly affected at the 0.33 and 1 MIU/day doses. At 3 MIU/day, there are discrete and more pronounced cytokine increases, notably for CXCL10 and IL-5 whereas other cytokines tested remained unchanged (Fig 4A). Principal Component Analyses of these results illustrate this dose-dependent trend: the 0.33 MIU/day patients largely overlap with placebo-treated patients, while the 1 and 3 MIU/day patients show more distinct behaviors (Fig 4B).
Fig 4: Dose-dependent changes in cytokines and chemokines in the plasma of T1D patients treated by ld-IL-2.
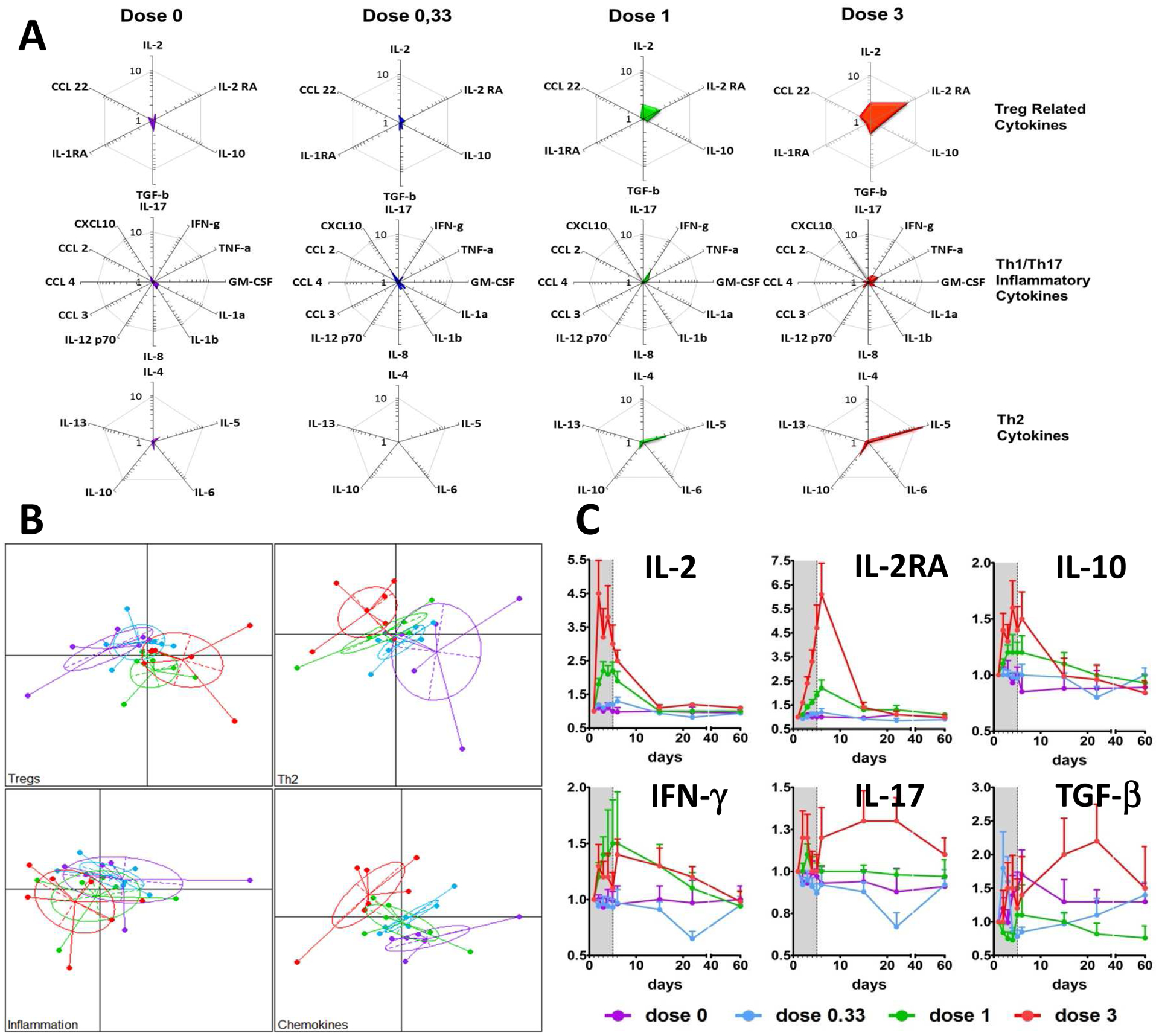
Plasma cytokines and chemokines were measured by luminex assay, except for TGF-beta measured by ELISA. Radar chart representation of Treg/anti-inflammatory, Th1/Th17/inflammatory and Th2 cytokines at day 6 of follow-up (A). For each cluster of cytokines, patient projections according to the first two PCA components (capturing more than 70% of the overall variability) (B). Time course changes of representative cytokines (C). Data are shown as mean ± SEM of patients’ values normalized by individual baseline values at different time points.
Repeated measures ANOVA for the day/treatment interaction were significant for IL-2, IL-2RA, IL-5, IL-10, IL-17, TNF-alpha, TGF-beta1, CCL22, and CXCL10 (p<0.0001 for IL-2, IL-2RA, IL-5, IL10, CXCL10 and TNF-alpha, p=0.0006 for TGF-beta1, p=0.0059 for IL-17 and p=0.0096 for CCL22) (Fig 4A and supplementary Table 1). The time-course changes for cytokines show a dose-dependent and rapid increase, with values already elevated at day 2 (Fig 4C). The increase of IL-17 was only observed at the 3 MIU/day dose (Fig 4C), but this was paralleled by an increase in the regulatory cytokine TGF-beta.
3.6. Transcriptomic analysis
We compared the global transcriptome of PBMCs at baseline and day-6. For each IL-2 treatment dose (0.33, 1 and 3 MUI), Enrichment Map analysis was performed on significantly up- and down-regulated molecular signatures (q-value < 0.05). Network analyses of significantly enriched molecular signatures show a good consistency for all IL-2 doses (Fig 5). Whatever the IL-2 dose, there is a striking down regulation of B-cells related signatures, and an up regulation of molecular signatures related to cell cycle and transcription. Chemokines associated signatures are found down regulated with the 2 lowest IL-2 doses, but up-regulated at the dose of 3 MIU/day, a dose at which NK cells signatures also became up-regulated.
Fig 5: Clustering of significantly regulated molecular signatures from PBMCs.
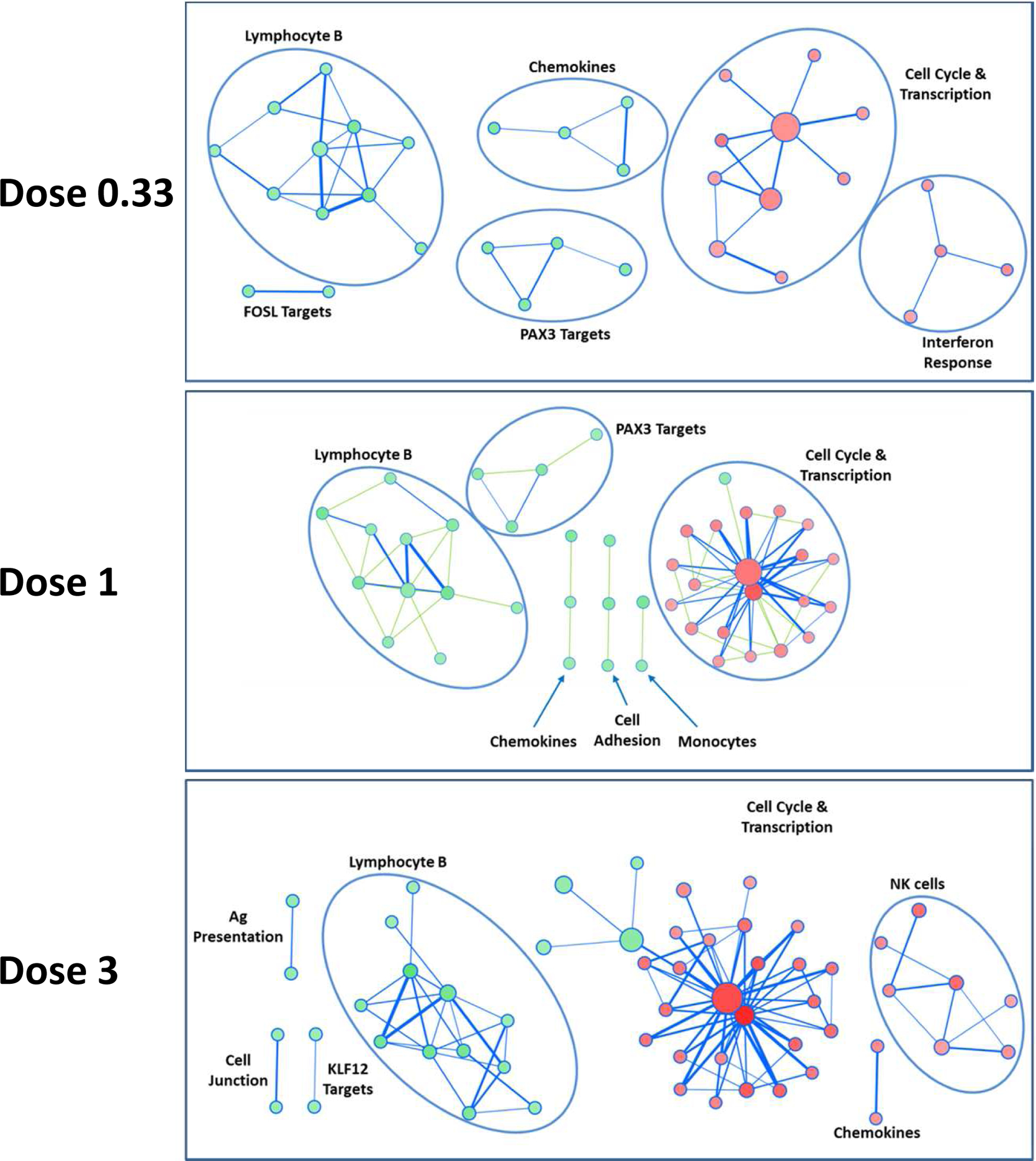
For each IL-2 treatment dose (0.33, 1 and 3 MIU), Enrichment Map analysis was performed on significantly up- and down-regulated molecular signatures, in red and green, respectively (q-value < 0.05). Clusters have been labelled according to their main biological feature. Size of nodes (circles) is proportional to the number of genes in the signature; width of bars linking nodes is related to the Jaccard coefficient between two signatures. Signatures without connection are not shown.
The molecular signature related to FOXP3 target genes [51] is non-significantly up-regulated signatures at the lowest dose (q-val=0.2), and significantly enriched and up regulated at the dose of 1 and 3 MIU/day.
3.7. Modulation of beta-cell-specific Teff responses
Preproinsulin (PPI) and glutamic acid decarboxylase (GAD) are beta-cell autoantigens commonly targeted by pathogenic Teffs in T1D patients. We therefore investigated T-cell responses directed to GAD, proinsulin (PI), C-peptide and the PPI signal peptide during ld-IL-2 treatment in 10 patients for whom samples from different time points (day 1, 6, 15 and 22) were available. Compared to pre-treatment values, many treated patients showed a reduction or stabilization of IFN-gamma responses to most antigens between day 6 and 22 (Fig 6). Specifically, we observed consistent suppression of all Teff responses tested in 1/3, 1/2 and 4/4 patients treated with 0.33, 1 and 3 MIU of IL-2, respectively. The suppressive effect on these responses was maintained after cessation of IL-2 therapy at the higher 3 MIU/day dose (Fig 6), up to day 22.
Fig 6: Modifications of β-cell-specific T-cell responses and Treg numbers in ld-IL-2-treated T1D patients.
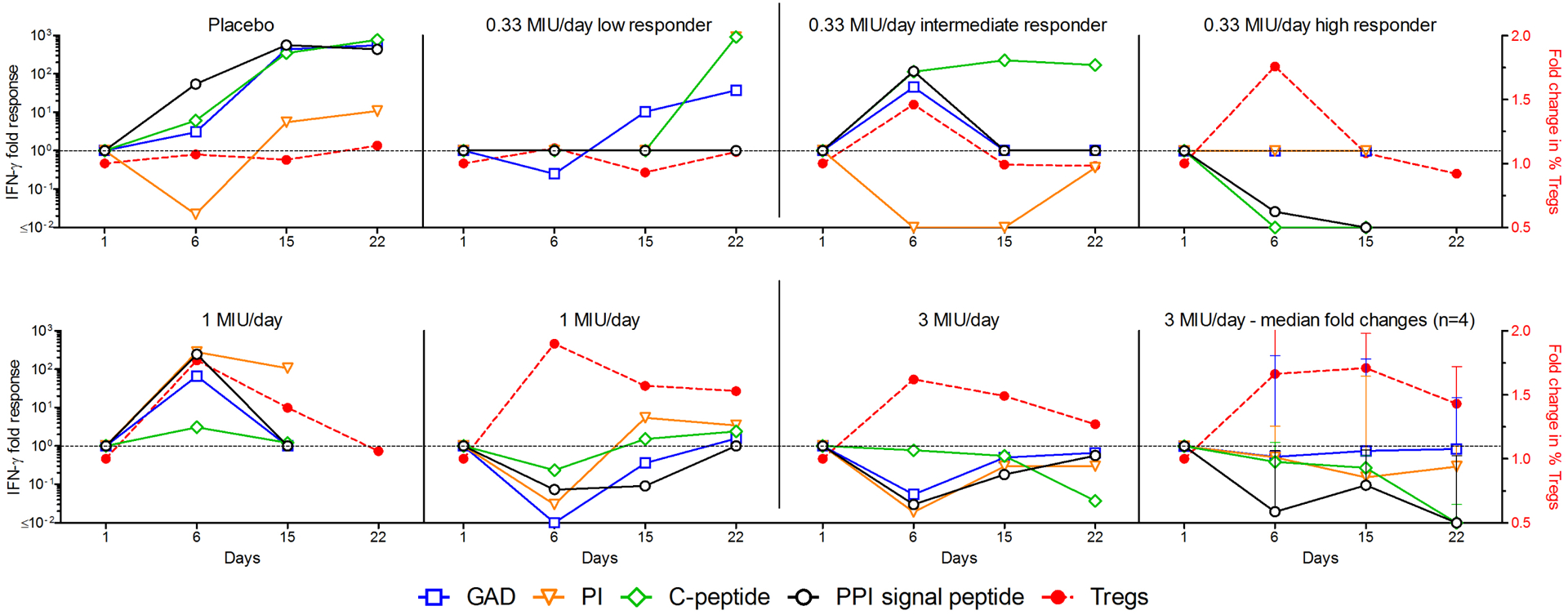
PBMCs from T1D patients were analyzed for IFN-PBMCs from T1D patients were analyzed for IFN-γ T-cell responses against β-cell protein antigens (left Y axis) at different time points. Percentages of Tregs are shown by the dashed red line (right Y axis). Results are expressed as fold changes normalized to day 1 (baseline). Each graph represents one patient except the bottom right graph which represents the median and range fold changes of 4 patients treated at 3 MIU /day.
3.8. Anti-IL-2 antibodies are not induced by ld-IL-2
We did not observe induction of anti-IL-2 antibodies and neutralizing antibodies in the plasma, at any time during the follow-up (Fig 7). While a very small reactivity against IL-2 could be detected in some patients, (i) it was not increased by IL-2 administration (Fig 7A and B) and (ii) it did not neutralize the effect of IL-2 on the proliferation of an IL-2-dependent cell line (Fig 7C).
Fig 7: Anti-IL-2 antibodies study.
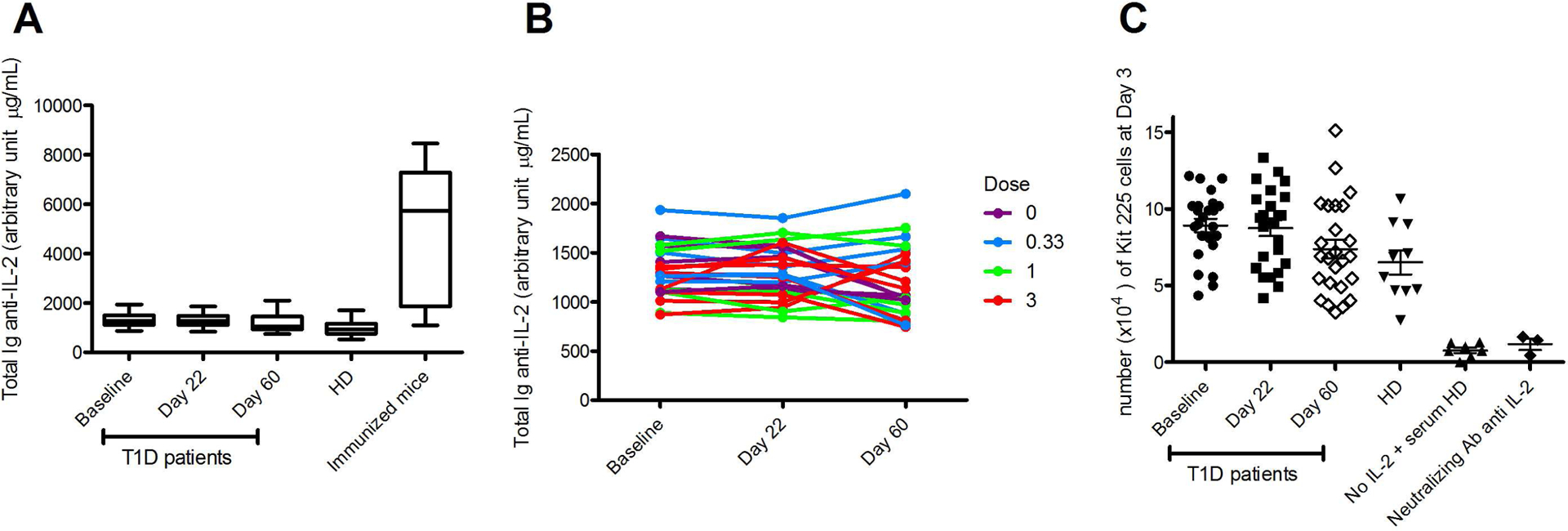
Patients’ plasma were tested for the presence of anti-IL-2 antibodies at different time points. (A) Mean values for anti-IL-2 Ig in patients and healthy donors (HD) plasma and for anti-human IL-2 Ig in plasma of mice treated for 4 months with human IL-2. (B) Individual time course of human Ig anti-IL-2 among normal and low responders group. (C) Proliferation of the IL-2 dependent Kit 225 cells in presence of patients and HD plasma, or an anti-IL-2 neutralizing antibody.
4. Discussion
4.1. On finding an optimal IL-2 dose for Treg specific expansion/activation
IL-2 has been initially described as a T cell growth factor [52] and developed for the treatment of conditions calling for the boosting of Teffs, i.e. cancer and infections [53, 54]. However, IL-2 is dispensable for the differentiation, survival and function of Teffs, as IL-2−/− mice develop T-cell-mediated autoimmunity [55]. In contrast, IL-2 is essential for the differentiation, survival and function of Tregs [56, 57]. IL-2 also has pleiotropic functions and can activate Teffs and notably memory CD8+ Teffs, and using IL-2 for treating T-cell mediated AID by promoting Treg function carries the risk of stimulating Teffs and could potentially worsen disease. Thus, therapeutic use of IL-2 to control autoimmunity requires identifying doses that selectively promote Treg function without activating effector populations.
The present study shows that the 0.3 to 3 MIU/day dose range is safe and effectively expanded/activated Tregs in T1D patients. The IL-2 dose-range of 0.3 to 1 MIU/day, given for 5 days effectively expands and activates Tregs without untoward effects on Teffs or NK cells. These results are in line with a recent study of low dose IL-2 (≈0.1 to 0.4 MIU/day for five days) in healthy adults [58]. At the dose of 3 MIU/day, the effect on Tregs is even more pronounced and importantly more durable; yet at this dose, we observed a trend toward NK cell expansion (Supplementary Fig 2), a trend for chemokine cytokine/chemokine increase (Fig 4), and more frequent mild to moderate side effects [40].
Under the assumption that chronic diseases will require maintaining increased proportion of Tregs over time, repeated treatment will likely be necessary. The appropriate dosing should consider the patient population, the duration and frequency of the treatment, and the cumulative dosing effects in relation to time. The duration of Tregs expansion, which is clearly dose-dependent, is a major parameter to take in consideration for designing regimen of administration. Using results from this trial, we modeled that at a dose of 1 MIU/day, a 5-day induction course followed by one injection every two weeks would allow to maintain a modest increase (< 30%) of Tregs throughout the treatment period, a dosing that we currently use in a trial investigating ld-IL-2 in 11 different AIDs (NCT01988506).
4.2. On the effect of IL-2 on the immune environment
At the dose of 1 and 3 MIU/day, we observed a significant and unexpected decrease in B cell percentages, correlating with increases in Treg but not NK cells (Supplementary Fig 2). Interpretation of this finding is not straightforward. A direct effect of IL-2 on mature B cells is unlikely because they do not express CD25 nor the signal transducing beta and gamma chains of the IL2 receptor [59]. The clinical significance of these findings remains to be clarified in future studies, particularly because a decrease in B cells could be advantageous for the treatment of B cell dependent AIDs.
IL-2 induces a clear shift of the peripheral blood immune environment towards a regulatory milieu, as reflected not only by changes in cell proportions, but also by plasma proteomics. This shift is dose-dependent, as well illustrated by the unsupervised statistical analyses (Fig 4B). These results suggest a broad anti-inflammatory effect of ld-IL-2 [17] and thus a broader therapeutic scope.
IL-10 and TGF-beta, two cytokines involved in suppressive functions, are also increased by IL-2 therapy, more pronouncedly at 3 MIU/day. At the same dose, there is an increase in IFN-gamma, which is usually classified as a Th1 cytokine. However, this is the only Th1 cytokine impacted and it was only modestly elevated, possibly coming from IFN-gamma secreting Tregs [60]. IL-17 was moderately increased only at the 3 MIU/day dose, a dose at which IL-2, IL-10 and TGF-beta were markedly increased; thus, the overall balance still appears tipped towards a regulatory milieu at this dose. Consistent with this interpretation, beta-cell-specific Teff responses were reduced in 6/9 patients tested during treatment, importantly in 4/4 treated with the 3 MIU/day dose (Fig 6), with no reductions in the placebo-treated patient tested. Thus, altogether, there is a clear IL-2 dose-dependent tuning of the immune response towards regulation of effector responses, including disease-specific, autoreactive Teff responses.
At the dose of 3 MIU/day, there was a marked increase of IL-5 levels (Fig 4A), a cytokine involved in eosinophil homeostasis. However, at the single and cumulative doses used, our available data show only a modest increase in eosinophil counts. This contrasts with the marked increase reported by others at doses of IL-2 that were higher and/or given over a longer time course [15, 61]. These results are compatible with IL-2 triggering IL-5 production by stimulating ILC2 cells, which are CD25+ [62]. However, ILC2 also produce IL-13, IL-9 and IL-4 [62], which were not increased in patients’ plasma. Of note, it has been reported that IL-5 promotes induction of antigen-specific Treg that suppress autoimmunity [63]. Importantly, at the 1 MIU/day dose, IL-5 was not induced, further pointing at this dose as safer choice for chronic administration.
4.3. On IL-2 and therapy and prevention of AIDs
AIDs often develop over a long period of time and are chronic, with a progressive aggravation. Early intervention with ld-IL2 could help reestablished a proper immune milieu and slow-down or even reverse the process. This is exemplified in T1D, for which at diagnosis most patients still maintain a functional mass of pancreatic β-cells and produce insulin upon stimulation. Halting beta-cell autoimmune destruction and preserving residual beta-cells soon after diagnosis could prevent further disease progression, thus improving clinical management and long-term outcomes for patients with T1D. In this perspective, our initial results showing the blunting of beta-cell-specific Teffs during ld-IL-2 are very encouraging. We caution that such analyses could be performed only in a subset of patients and thus further study and validation are needed, which we will pursue in our efficacy trial of ld-IL-2 in patients with T1D (NCT01862120).
We believe that there may be even greater potential for using ld-IL-2 to prevent AIDs. Providing a boost to Tregs in patients at risk for AIDs, such as autoantibody-positive relatives of T1D patients, could help delay or prevent the onset of the full-blown disease. Moreover, boosting Tregs with ld-IL2 could also prevent or decrease the frequency and severity of relapses in relapsing-remitting diseases like multiple sclerosis (MS).
Finally, ld-IL-2 has now been used successfully in the setting of chronic and acute GVHD [15, 64] and several AIDs, such as HCV-vasculitis [17], T1D [40], alopecia areata [65], and SLE [66–68]. A therapeutic benefit was observed in most patients treated in these trials. This changes the paradigm of a highly focused effect of ld-IL-2 on Tregs to one where IL-2 has a broader regulatory influence on the homeostasis of the immune system, and notably inflammation. As such, future studies should explore further the effects of IL-2 and define efficacious ld-IL-2 regimens that can benefit patients currently suffering from several forms of AID and inflammatory disorders.
Supplementary Material
Highlights:
We performed the 1st randomized double-blind placebo-controlled trial of ld-IL-2
We report the detailed kinetics and dose-relationship of ld-IL-2 effects
We report detailed immunomonitoring of the broad modification of immune responses
Our results are key to optimize the use of IL-2 for therapy of autoimmune diseases
Acknowledgments
We are indebted to Christophe Misse, director of the Département de la Recherche Clinique et du Développement (DRCD) of the AP-HP, for initiating the support for this study. We thank the patients and the members of the DF-IL2-study group (listed below), and more specially Gilbert Bensimon for the study design and Christine Payan for data management and statistical analyses. We also thank all the personnel of the Endocrinology department, the Clinical Investigation Center Paris-est (CIC-PE) and the Clinical Investigation Center for Biotherapies (CIC-BTi) for their participation and more especially Nathalie Fery and Cornélia Degbe DK, MR and TRM are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
MR and DK are inventors on a patent application related to the therapeutic use of ld-IL-2, which belongs to their academic institutions and has been licensed to ILTOO pharma. MR, GC and DK hold shares in ILTOO pharma. HPP is an employee of ILTOO pharma. No other potential conflicts of interest relevant to this article were reported.
This work was funded by the DRCD, the CIC-PE and CIC-BTi, the INSERM and French state funds managed by the ANR within the Investissements d’Avenir programme (ANR-11-IDEX-0004-02). RM was supported by the Aviesan/AstraZeneca program. JB and AL were supported by the Immune Tolerance Network (NIH Contract #N01 AI15416), an international clinical research consortium headquartered at the Benaroya Research Institute and supported by the National Institute of Allergy and Infectious Diseases and the Juvenile Diabetes Research Foundation. This work was also supported by the Diabetes Research Institute Foundation, Hollywood, FL, the Peacock Foundation, Inc., Miami, FL, and the Anton E. B. Schefer Foundation, which supported AY, AP and TRM.
MR supervised immunomonitoring, participated to the study design and analyzed results; GC, RM, AS, ND, WC, RL, AL, JB, GA, HPP, AY, AP and TRM performed some experiments and analyzed results; AH was the principal clinical investigator of the study, participated to the study design and analyzed results; DK was the principal investigator of the study, conceived the study, participated to the study design, analyzed results and wrote the manuscript. All authors edited the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol, 2010;10:490–500. [DOI] [PubMed] [Google Scholar]
- [2].Ait-Oufella H, Salomon BL, Potteaux S, Robertson AK, Gourdy P, Zoll J et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat Med, 2006;12:178–80. [DOI] [PubMed] [Google Scholar]
- [3].Campbell DJ, Koch MA. Phenotypical and functional specialization of FOXP3+ regulatory T cells. Nat Rev Immunol, 2011;11:119–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wing K, Sakaguchi S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat Immunol, 2010;11:7–13. [DOI] [PubMed] [Google Scholar]
- [5].Bacchetta R, Passerini L, Gambineri E, Dai M, Allan SE, Perroni L et al. Defective regulatory and effector T cell functions in patients with FOXP3 mutations. J Clin Invest, 2006;116:1713–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet, 2001;27:20–1. [DOI] [PubMed] [Google Scholar]
- [7].Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet, 2001;27:68–73. [DOI] [PubMed] [Google Scholar]
- [8].Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol, 2003;4:330–6. [DOI] [PubMed] [Google Scholar]
- [9].Brusko TM, Wasserfall CH, Clare-Salzler MJ, Schatz DA, Atkinson MA. Functional defects and the influence of age on the frequency of CD4+ CD25+ T-cells in type 1 diabetes. Diabetes, 2005;54:1407–14. [DOI] [PubMed] [Google Scholar]
- [10].Lindley S, Dayan CM, Bishop A, Roep BO, Peakman M, Tree TI. Defective suppressor function in CD4(+)CD25(+) T-cells from patients with type 1 diabetes. Diabetes, 2005;54:92–9. [DOI] [PubMed] [Google Scholar]
- [11].Long SA, Cerosaletti K, Bollyky PL, Tatum M, Shilling H, Zhang S et al. Defects in IL-2R signaling contribute to diminished maintenance of FOXP3 expression in CD4(+)CD25(+) regulatory T-cells of type 1 diabetic subjects. Diabetes, 2010;59:407–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Buckner JH. Mechanisms of impaired regulation by CD4(+)CD25(+)FOXP3(+) regulatory T cells in human autoimmune diseases. Nat Rev Immunol, 2010;10:849–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Long SA, Buckner JH. CD4+FOXP3+ T regulatory cells in human autoimmunity: more than a numbers game. J Immunol, 2011;187:2061–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Grinberg-Bleyer Y, Baeyens A, You S, Elhage R, Fourcade G, Gregoire S et al. IL-2 reverses established type 1 diabetes in NOD mice by a local effect on pancreatic regulatory T cells. J Exp Med, 2010;207:1871–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea EP 3rd et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med, 2011;365:2055–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Matsuoka K, Koreth J, Kim HT, Bascug G, McDonough S, Kawano Y et al. Low-dose interleukin-2 therapy restores regulatory T cell homeostasis in patients with chronic graft-versus-host disease. Sci Transl Med, 2013;5:179ra43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Saadoun D, Rosenzwajg M, Joly F, Six A, Carrat F, Thibault V et al. Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N Engl J Med, 2011;365:2067–77. [DOI] [PubMed] [Google Scholar]
- [18].Tang Q, Bluestone JA, Kang SM. CD4(+)Foxp3(+) regulatory T cell therapy in transplantation. Journal of molecular cell biology, 2012;4:11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wood KJ, Sakaguchi S. Regulatory T cells in transplantation tolerance. Nat Rev Immunol, 2003;3:199–210. [DOI] [PubMed] [Google Scholar]
- [20].Cacoub P, Poynard T, Ghillani P, Charlotte F, Olivi M, Piette JC et al. Extrahepatic manifestations of chronic hepatitis C. MULTIVIRC Group. Multidepartment Virus C. Arthritis Rheum, 1999;42:2204–12. [DOI] [PubMed] [Google Scholar]
- [21].Dendrou CA, Plagnol V, Fung E, Yang JH, Downes K, Cooper JD et al. Cell-specific protein phenotypes for the autoimmune locus IL2RA using a genotype-selectable human bioresource. Nat Genet, 2009;41:1011–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Qu HQ, Montpetit A, Ge B, Hudson TJ, Polychronakos C. Toward further mapping of the association between the IL2RA locus and type 1 diabetes. Diabetes, 2007;56:1174–6. [DOI] [PubMed] [Google Scholar]
- [23].Vella A, Cooper JD, Lowe CE, Walker N, Nutland S, Widmer B et al. Localization of a type 1 diabetes locus in the IL2RA/CD25 region by use of tag single-nucleotide polymorphisms. Am J Hum Genet, 2005;76:773–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Todd JA. Etiology of type 1 diabetes. Immunity, 2010;32:457–67. [DOI] [PubMed] [Google Scholar]
- [25].Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet, 2007;39:857–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang WY, Barratt BJ, Clayton DG, Todd JA. Genome-wide association studies: theoretical and practical concerns. Nat Rev Genet, 2005;6:109–18. [DOI] [PubMed] [Google Scholar]
- [27].Ferraro A, Socci C, Stabilini A, Valle A, Monti P, Piemonti L et al. Expansion of Th17 cells and functional defects in T regulatory cells are key features of the pancreatic lymph nodes in patients with type 1 diabetes. Diabetes, 2011;60:2903–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature, 2010;464:1293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Battaglia M, Roncarolo MG. Immune intervention with T regulatory cells: past lessons and future perspectives for type 1 diabetes. Semin Immunol, 2011;23:182–94. [DOI] [PubMed] [Google Scholar]
- [30].Tang Q, Adams JY, Penaranda C, Melli K, Piaggio E, Sgouroudis E et al. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity, 2008;28:687–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bougneres PF, Carel JC, Castano L, Boitard C, Gardin JP, Landais P et al. Factors associated with early remission of type I diabetes in children treated with cyclosporine. N Engl J Med, 1988;318:663–70. [DOI] [PubMed] [Google Scholar]
- [32].Feutren G, Papoz L, Assan R, Vialettes B, Karsenty G, Vexiau P et al. Cyclosporin increases the rate and length of remissions in insulin-dependent diabetes of recent onset. Results of a multicentre double-blind trial. Lancet, 1986;2:119–24. [DOI] [PubMed] [Google Scholar]
- [33].Stiller CR, Dupre J, Gent M, Jenner MR, Keown PA, Laupacis A et al. Effects of cyclosporine immunosuppression in insulin-dependent diabetes mellitus of recent onset. Science, 1984;223:1362–7. [DOI] [PubMed] [Google Scholar]
- [34].Skyler JS. Immune intervention for type 1 diabetes, 2012–2013. Diabetes technology & therapeutics, 2014;16 Suppl 1:S85–91. [DOI] [PubMed] [Google Scholar]
- [35].Orban T, Bundy B, Becker DJ, Dimeglio LA, Gitelman SE, Goland R et al. Costimulation modulation with abatacept in patients with recent-onset type 1 diabetes: follow-up 1 year after cessation of treatment. Diabetes Care, 2014;37:1069–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Herold KC, Gitelman SE, Ehlers MR, Gottlieb PA, Greenbaum CJ, Hagopian W et al. Teplizumab (anti-CD3 mAb) treatment preserves C-peptide responses in patients with new-onset type 1 diabetes in a randomized controlled trial: metabolic and immunologic features at baseline identify a subgroup of responders. Diabetes, 2013;62:3766–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hagopian W, Ferry RJ Jr., Sherry N, Carlin D, Bonvini E, Johnson S et al. Teplizumab preserves C-peptide in recent-onset type 1 diabetes: two-year results from the randomized, placebo-controlled Protege trial. Diabetes, 2013;62:3901–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Orban T, Beam CA, Xu P, Moore K, Jiang Q, Deng J et al. Reduction in CD4 Central Memory T-cell subset in Co-Stimulation Modulator Abatacept-Treated Patients with Recent-Onset Type 1 Diabetes is Associated with Slower C-Peptide Decline. Diabetes, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Garg G, Tyler JR, Yang JH, Cutler AJ, Downes K, Pekalski M et al. Type 1 diabetes-associated IL2RA variation lowers IL-2 signaling and contributes to diminished CD4+CD25+ regulatory T cell function. J Immunol, 2012;188:4644–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hartemann A, Bensimon G, Payan C, Jacqueminet S, Bourron O, Nicolas N et al. Low-dose Interleukin-2 induces regulatory T cells and is well tolerated in patients with type-1 diabetes: results of a phase I/II randomized, double-blind, placebo-controlled trial Lancet Diabetes & Endocrinology, 2013;1:295–305. [DOI] [PubMed] [Google Scholar]
- [41].Long SA, Rieck M, Tatum M, Bollyky PL, Wu RP, Muller I et al. Low-dose antigen promotes induction of FOXP3 in human CD4+ T cells. J Immunol, 2011;187:3511–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Martinuzzi E, Afonso G, Gagnerault MC, Naselli G, Mittag D, Combadiere B et al. acDCs enhance human antigen-specific T-cell responses. Blood, 2011;118:2128–37. [DOI] [PubMed] [Google Scholar]
- [43].Scharenberg JG, Stam AG, von Blomberg BM, Roest GJ, Palmer PA, Franks CR et al. The development of anti-interleukin-2 (IL-2) antibodies in cancer patients treated with recombinant IL-2. Eur J Cancer, 1994;30A:1804–9. [DOI] [PubMed] [Google Scholar]
- [44].Hori T, Uchiyama T, Tsudo M, Umadome H, Ohno H, Fukuhara S et al. Establishment of an interleukin 2-dependent human T cell line from a patient with T cell chronic lymphocytic leukemia who is not infected with human T cell leukemia/lymphoma virus. Blood, 1987;70:1069–72. [PubMed] [Google Scholar]
- [45].Li S, Rouphael N, Duraisingham S, Romero-Steiner S, Presnell S, Davis C et al. Molecular signatures of antibody responses derived from a systems biology study of five human vaccines. Nat Immunol, 2014;15:195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet, 2003;34:267–73. [DOI] [PubMed] [Google Scholar]
- [47].Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A, 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL et al. Innate or adaptive immunity? The example of natural killer cells. Science, 2011;331:44–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Chaput N, Louafi S, Bardier A, Charlotte F, Vaillant JC, Menegaux F et al. Identification of CD8+CD25+Foxp3+ suppressive T cells in colorectal cancer tissue. Gut, 2009;58:520–9. [DOI] [PubMed] [Google Scholar]
- [50].Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity, 2009;30:899–911. [DOI] [PubMed] [Google Scholar]
- [51].Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA et al. Foxp3-dependent programme of regulatory T-cell differentiation. Nature, 2007;445:771–5. [DOI] [PubMed] [Google Scholar]
- [52].Morgan DA, Ruscetti FW, Gallo R. Selective in vitro growth of T lymphocytes from normal human bone marrows. Science, 1976;193:1007–8. [DOI] [PubMed] [Google Scholar]
- [53].Chang AE, Rosenberg SA. Overview of interleukin-2 as an immunotherapeutic agent. Semin Surg Oncol, 1989;5:385–90. [DOI] [PubMed] [Google Scholar]
- [54].Giedlin MA, Zimmerman RJ. The use of recombinant human interleukin-2 in treating infectious diseases. Curr Opin Biotechnol, 1993;4:722–6. [DOI] [PubMed] [Google Scholar]
- [55].Sadlack B, Lohler J, Schorle H, Klebb G, Haber H, Sickel E et al. Generalized autoimmune disease in interleukin-2-deficient mice is triggered by an uncontrolled activation and proliferation of CD4+ T cells. Eur J Immunol, 1995;25:3053–9. [DOI] [PubMed] [Google Scholar]
- [56].Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H, Litsa D et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood, 2006;108:1571–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Malek TR, Castro I. Interleukin-2 receptor signaling: at the interface between tolerance and immunity. Immunity, 2010;33:153–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ito S, Bollard CM, Carlsten M, Melenhorst JJ, Biancotto A, Wang E et al. Ultra-low dose interleukin-2 promotes immune-modulating function of regulatory T cells and natural killer cells in healthy volunteers. Mol Ther, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol, 2012;12:180–90. [DOI] [PubMed] [Google Scholar]
- [60].Oldenhove G, Bouladoux N, Wohlfert EA, Hall JA, Chou D, Dos Santos L et al. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity, 2009;31:772–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Long SA, Rieck M, Sanda S, Bollyky JB, Samuels PL, Goland R et al. Rapamycin/IL-2 combination therapy in patients with type 1 diabetes augments Tregs yet transiently impairs beta-cell function. Diabetes, 2012;61:2340–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Walker JA, Barlow JL, McKenzie AN. Innate lymphoid cells--how did we miss them? Nat Rev Immunol, 2013;13:75–87. [DOI] [PubMed] [Google Scholar]
- [63].Tran GT, Hodgkinson SJ, Carter NM, Verma ND, Plain KM, Boyd R et al. IL-5 promotes induction of antigen-specific CD4+CD25+ T regulatory cells that suppress autoimmunity. Blood;119:4441–50. [DOI] [PubMed] [Google Scholar]
- [64].Kennedy-Nasser AA, Ku S, Castillo-Caro P, Hazrat Y, Wu MF, Liu H et al. Ultra low-dose IL-2 for GVHD prophylaxis after allogeneic hematopoietic stem cell transplantation mediates expansion of regulatory T cells without diminishing antiviral and antileukemic activity. Clinical cancer research : an official journal of the American Association for Cancer Research, 2014;20:2215–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Castela E, Le Duff F, Butori C, Ticchioni M, Hofman P, Bahadoran P et al. Effects of Low-Dose Recombinant Interleukin 2 to Promote T-Regulatory Cells in Alopecia Areata. JAMA dermatology, 2014. [DOI] [PubMed] [Google Scholar]
- [66].Klatzmann D Immunoregulation Without Immunosuppression: The Promise of Low Dose IL-2. FOCIS meeting Chicago 2014. [Google Scholar]
- [67].Von Spee-Mayer CSE, Rose A, Humrich J, Riemekasten G. Low-dose interleukin-2 therapy caused selective expansion of Tregs together with rapid reduction of disease activity in a patient with severe refractory SLE. EULAR meeting Paris, 2014. [Google Scholar]
- [68].Yu D Low-dose interleukin-2 in active systemic lupus erythematosus. FOCIS meeting Chicago 2014, 2014. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.