

Abstract
The Chikungunya virus (CHIKV) is an arbovirus belonging to the genus Alphavirus of the Togaviridae family. CHIKV is transmitted by the mosquitoes and causes Chikungunya fever. CHIKV outbreaks have occurred in Africa, Asia, Europe, and the countries of Indian and Pacific Oceans. In 2013, CHIKV cases were registered for the first time in the Americas on the Caribbean islands. There is currently no vaccine to prevent or medicines to treat CHIKV infection. The CHIKV nonstructural protease (nsP2) is a promising potential target for the development of drugs against CHIKV infection because this protein is one of the key components of the viral replication complex and is involved in multiple steps of virus infection. In this work, novel analogues of the potential CHIKV nsP2 protease inhibitor, first reported by Das et al. in 2016, were identified using molecular modeling methods, synthesized, and evaluated in vitro. The optimization of the structure of the inhibitor allowed to increase the antiviral activity of the compound 2–10 times. The possible mechanism of action of the identified potential inhibitors of the CHIKV nsP2 protease was studied in detail using molecular dynamics (MD) simulations. According to the MD results, the most probable mechanism of action is the blocking of conformational changes in the nsP2 protease required for substrate recognition and binding.
Introduction
The Chikungunya virus (CHIKV) is an arbovirus of the genus Alphavirus of the Togaviridae family. CHIKV is spread by two mosquito species: Aedes aegypti and Aedes albopictus. Chikungunya fever, a disease caused by CHIKV infection in humans, usually lasts 5 to 7 days and is rarely life-threatening (the risk of death is around 1 case per 1000).1 However, it often results in severe long-lasting (months to years) joints’ dysfunction.2 There is no specific treatment for this disease, but nonsteroid anti-inflammatory drugs may be used to relieve pain and joint swelling. There is also no vaccine against CHIKV; therefore, the protective measures include the prevention of mosquito bites and destroying mosquito breeding sites. Global rates of CHIKV infection vary by the outbreak. When the virus was first discovered in West Africa in 1952,3 the spread of the virus was low. Since the 1960s, periodic outbreaks have been reported in Asia and Africa.4 However, since 2005, CHIKV has repeatedly re-emerged and caused major outbreaks in Africa, Asia, and America. In India, the virus reappeared after 32 years.5 Outbreaks have also been reported in previously unreported areas of CHIKV: Europe, the Caribbean, and South America. Local distribution has also been recorded in the United States and Australia.6 In 2005, the outbreak on the la Réunion island was the largest at that time: an estimated 266,000 cases were recorded on the island with a population of about 770,000.7 In 2006, 1.25 million cases were reported during the Indian outbreak.8 The sharp increase and expansion of the CHIKV incidence in recent years testify to our vulnerability to emerging infectious diseases transmitted by insects and emphasize the importance of finding effective drugs to treat such diseases.
CHIKV has small (∼70 nm in diameter) virions and a positive-sense RNA genome of approximately 11.6 kb. The genome contains two open reading frames encoding for nonstructural proteins (nsP1–nsP4) and structural proteins (C, E3, E2, E1, and 6K).9,10 Among CHIKV nonstructural proteins, which represent subunits of virus-encoded RNA replicase, nsP2 is the most attractive target for the development of new antiviral drugs. Aside its functions in virus RNA replication, it is also important for shutdown of the host cell transcription, inhibition of antiviral responses, and virion formation; most of these functions are associated with the C-terminal protease part of nsP2.11
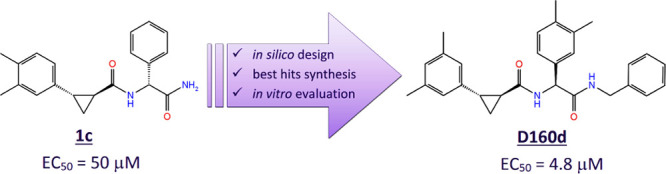
In recent years, many research groups have focused on identifying new inhibitors of CHIKV replication in order to develop a candidate drug for the treatment of CHIKV infection, as described in review articles by Subudhi et al.4 and Jadav et al.12 The use of computer-aided drug design (CADD) methods helps to accelerate the long process of developing new drug candidates and reduce their cost. In our previous work,13 a compound originating from the study by Bassetto et al.14 was selected as the lead structure for hit generation to identify novel inhibitors against the CHIKV nsP2 protease, combining CADD based on molecular docking, molecular dynamics (MD) simulations, and the pharmacophore approach with the use of cell-free and cell-culture experiments. The aim of our current study was to optimize the structure and increase the antiviral properties of the previously reported CHIKV nsP2 inhibitor13 (Scheme 1) using the structure–activity relationship for the newly designed and synthesized compounds.
Scheme 1. Most Active CHIKV Protease Inhibitor 1c from the Previous Study (Das et al. 2016), Its In Silico Modeled Analogues Designed for This Work and Chemical Synthesis of the Best Hits for Evaluation of Bioactivity.

Results and Discussion
Molecular Design and Molecular Docking
In this work, the CHIKV nonstructural protease nsP2 (PDB ID: 3TRK;15 see the Materials and Methods section) was used as a target for molecular design. Two potential binding sites of the CHIKV nsP2 protease were used for molecular docking (Figure 1). To search for more active analogues of the previously described inhibitor,13 158 compounds were designed modifying substituents and their positions in the phenyl rings (Scheme 1). It was shown in the previous work13 that only the relative configuration of cyclopropane substituents (trans-configuration) has an important role in the antiviral activity of trans-2-{[2-(3,4-dimethylphenyl)cyclopropyl]formamido}-2-phenylacetamide (1c), while the absolute configuration of the cyclopropane motif and the stereocenter of arylglycine do not impose any significant effect on the activity. According to these results, the S,S,S-stereoisomers of designed compounds were used for the molecular docking study, and during this procedure, only the cyclopropane substituents were fixed in the trans-configuration for all compounds, while the other bonds in the molecules were marked as flexible. The active site (hereinafter referred to as potential binding site 1; Figure 1) from the previous work,13 including the amino acid residues Cys1013 and His1083 (hereinafter, the numbers of amino acid residues correspond to their numbers in the nonstructural polyprotein of CHIKV) from the catalytic dyad and the conserved amino acid residue Trp1084, was chosen for the docking calculations. However, the number of selected compounds based on the calculated binding energy hits for the S,S,S-stereoisomers of designed compounds turned out to be too large (121 hits from 158 compounds; Table S1). An S,S,S-stereoisomer of the designed compound with a binding energy lower than or equal to −8.0 kcal/mol16 was considered as a binding energy hit. Thus, the binding energies of the selected hits for this active site were in the range of −9.4 to −8.0 kcal/mol. Therefore, another potential active site of CHIKV nsP2 at the C-terminal (hereinafter referred to as potential binding site 2; Figure 1) was used for further molecular docking, containing the following amino acids: Gln1039, Lys1045, Glu1157, Gly1176, His1222, Lys1239, Ser1293, Glu1296, and Met1297.17 The difference in the binding energy of small-molecule ligands with CHIKV nsP2 was not as large as that in the case of potential binding site 1. Thus, 28 hits from 158 compounds with the binding energies ranging from −8.7 to −8.0 kcal/mol were identified (Table S1). Analysis of the docking results at potential binding site 2 showed that the binding energy of the S,S,S-stereoisomers of designed compounds to the receptor is improved if the substituents R1 are located at the meta positions of the phenyl ring and R2 at the meta and para positions of the phenyl ring (see Scheme 1, Table S1).
Figure 1.

Potential binding sites 1 and 2 in the CHIKV nsP2 protease (PDB ID: 3TRK).
Compounds D27, D117, D120, D127, and D157 were selected for further study because of the best binding energies of their S,S,S-stereoisomers for both binding sites (Table S1), and compounds D27 and D127 were initially selected for synthesis. The calculated binding energies of the S,S,S-stereoisomers of selected compounds were in the range of −9.2 to −8.8 kcal/mol for potential binding site 1 and in the range of −8.7 to −8.0 kcal/mol for potential binding site 2. The choice of the compounds for further study and synthesis also depended on the type and positions of the substituents in the phenyl rings. The binding modes of all S,S,S-stereoisomers of selected compounds at potential binding site 1 of CHIKV nsP2 are the same (Figure 2). The S,S,S-stereoisomers of compounds D27, D117, D120, and D127 form three hydrogen bonds: one hydrogen bond by the NH2 group with the OH group of Tyr1079 and another hydrogen bond with the carbonyl oxygen of the side chain of Asn1082; the third hydrogen bond occurs between the NH group of the ligand and the OH group of Tyr1079 (Figure 2A–D). The substitution of the NH2 group with an OCH3 group in the S,S,S-stereoisomer of compound D157 results in the loss of hydrogen bonds with Tyr1079 and Asn1082, but the bond between the NH group of the ligand and the OH group of Tyr1079 is retained (Figure 2E). The loss of two hydrogen bonds causes a small displacement of the ligand at potential binding site 1, which in turn leads to the loss of a hydrophobic contact with the amino acid residue Cys1013 from the catalytic dyad, but at the same time, it promotes the binding of the ligand closer to the amino acid residue Trp1084, which is important for the CHIKV nsP2 activity.18
Figure 2.

Calculated binding modes of the S,S,S-stereoisomers of compounds D27 (A), D117 (B), D120 (C), D127 (D), and D157 (E) at potential binding site 1 of CHIKV nsP2 (PDB ID: 3TRK). Intermolecular hydrogen bonds are shown by dark-green dashed lines.
The binding modes of the S,S,S-stereoisomers of selected compounds also show the same tendency at potential binding site 2 (Figure 3). The S,S,S-stereoisomers of compounds D27, D117, D120, and D127 are hydrogen-bonded by the terminal NH2 group to the carboxyl group of Glu1204 (Figure 3A–D). As in the case of potential binding site 1, the substitution of the terminal NH2 group by the OCH3 group also leads to the loss of the hydrogen bond and to a small displacement of the S,S,S-stereoisomer of compound D157 at potential binding site 2 (Figure 3E) relative to the other selected compounds. As can be seen from the docking results for both potential binding sites of the CHIKV nsP2 protease (Figures 2 and 3), the NH2 group of the ligand can be the key to the antiviral activity of these compounds. Therefore, it was decided to study the biological activity of intermediates with the substituted terminal NH2 group (compounds D159–D161; Scheme 1).
Figure 3.

Calculated binding modes of the S,S,S-stereoisomers of compounds D27 (A), D117 (B), D120 (C), D127 (D), and D157 (E) at potential binding site 2 of CHIKV nsP2 (PDB ID: 3TRK). Intermolecular hydrogen bonds are shown by dark-green dashed lines.
Cytotoxicity of Compounds
First, all synthesized compounds, including intermediates with the substituted terminal NH2 group, were tested for their cytotoxicity effect in BHK-21 cells; concentrations ranging from 1 to 100 μM were used. To facilitate chemical synthesis and screening for a large array of potential inhibitors, all synthesized compounds were initially tested as equimolecular mixtures of four possible stereoisomers (R,R,R-, R,R,S-, S,S,R-, and S,S,S-stereoisomers). Based on the results of the previous work,13 we surmised that individual stereoisomers do not interfere with biological activities of each other. Among the intermediates with the substituted terminal NH2 group, D161 demonstrated a moderate cytotoxicity effect at a 100 μM concentration, and compounds D159 and D160 had no cytotoxic effect at concentrations of 1 up to 100 μM. Among the analogues of compound D160, compound D27 was toxic at all concentrations, compound D127 showed cytotoxicity at concentrations of more than 100 μM, and compound D163 showed cytotoxicity at concentrations of more than 10 μM; the other analogues were nontoxic (Table 1).
Table 1. Antiviral Activity and Cytotoxicity of Synthesized Compounds against CHIKV-NanoLuc in BHK-21 Cells.
| code | EC50 (μM) | CC50 (μM) |
|---|---|---|
| D27a | NAb | <0.1 |
| D119a | 32.0 | >100 |
| D127a | 29.6 | >100 |
| D159a | >100 | >100 |
| D160a | 10.5 | >100 |
| D160a | 26.8 | >100 |
| D160b | 70.7 | >100 |
| D160c | 8.7 | >100 |
| D160d | 4.8 | >100 |
| D161a | 13.9 | >100 |
| D162a | 95.9 | >100 |
| D163a | NAb | 10 |
| D164a | 50.5 | >100 |
| D165a | >100 | >100 |
| D166a | NAb | >100 |
Compounds were tested as equimolecular mixtures of four possible stereoisomers.
NA: inactive, that is, no activity at the maximum nontoxic concentration.
Synthesis of Compounds and Evaluation of Anti-CHIKV Activity
Compounds D27, D119, D127, and D159–D168 were synthesized, as described in the Supporting Information, by using Ugi multi-component reaction as the key step (Scheme 1).19,20 Since the Ugi reaction was not diastereoselective and racemic 2-arylcyclopropane-1-carboxylic acids were used as starting materials, equimolecular mixtures of four possible stereoisomers (two pairs of enantiomers) were obtained in these cases. Individual stereoisomers were isolated only for compound D160 with the highest antiviral activity and synthesized from (R,R)- and (S,S)-enantiomers (>94% ee) of trans-2-(3,5-dimethylphenyl)cyclopropane-1-carboxylic acid (see the Supporting Information). Compounds D159, D160, and D161 were tested as equimolecular mixtures of four possible stereoisomers (R,R,R-, R,R,S-, S,S,R-, and S,S,S-stereoisomers) for their ability to inhibit CHIKV infection in vitro at concentrations of 1 up to 100 μM in BHK-21 cells (Table 1). These compounds behaved differently against CHIKV infection. Compound D159 was inactive, the inhibitory effect of compounds D160 and D161 appeared at a 10 μM concentration, and the viral infection was almost completely inhibited at a 100 μM concentration. However, the inhibitory effect of compound D161 may be due to its moderate cytotoxic effect at a concentration of 100 μM.
For compounds D160 and D161, 50% effective concentration (EC50) was determined using concentrations from 0.1 up to 200 μM and cells infected at a multiplicity of infection (MOI) of 0.001 using CHIKV-NanoLuc, a marker-protein expressing variant of CHIKV from la Réunion island outbreak. At this low MOI, both compounds showed EC50 in the low-micromole range (10.5 and 13.9 μM, respectively). Among the potential inhibitors D159, D160, and D161, the antiviral activity of these compounds was found to depend on their substituents in the terminal NH2 group. For further biological evaluation of the detected dependence, three analogues of D160 were synthesized—compounds D27, D163, and D164, which have a different substituent in the terminal NH2 group. Based on the results of molecular design, compounds D119, D127, and D162 with different positions of the methyl and trifluoromethyl groups in the phenyl rings were selected for synthesis. To determine the dependence of the antiviral activity on the position of the methyl substituents R2 in the corresponding phenyl ring, an analogue of D159 having the methyl substituents at the meta and para positions (compound D165) was first synthesized, followed by its fluoro-substituted derivative D166. Among these, compounds D119, D127, and D164 were the most active—their EC50 were 32.0, 29.6, and 50.5 μM, respectively. Two compounds, D159 and D165, had EC50 greater than 100 μM. Other compounds showed no antiviral activity at their maximum nontoxic concentrations (Table 1).
The results of the measurement of the antiviral activity of these compounds confirmed that not only the substituent in the terminal NH2 group but also the positions of the substituents R2 in the phenyl ring of arylglycine significantly affect the anti-CHIKV activity. Attachment of a cyclopropyl substituent to the terminal NH2 group increased cytotoxicity; the 50% cytotoxic concentration (CC50) of compound D163 was approximately 10 μM, and the compound displayed no antiviral activity at the maximum noncytotoxic concentration (Table 1). In the absence of a substituent in the NH2 group (compound D27), the cytotoxic effect appeared already at a 0.1 μM concentration. However, insertion of a trifluoromethyl substituent R2 into the meta position of the phenyl ring (D119) did not increase cytotoxicity, but EC50 of this compound remained higher compared to that of D160. A similar effect was also achieved by introducing a cyclopentyl substituent into the NH2 group (D164), but such substitution had no significant effect on the antiviral activity of the compound. There was also no significant change in the activity when the position of the methyl groups R2 in the phenyl ring was varied, while the substituent in the NH2 group remained the same. Thus, for example, the activity of the D165 analogue remained at the same level as that of D159, but at the same time, the trifluoromethyl group R2 at the meta position of the phenyl in the arylglycine residue and the fluorine substituent R1 at the meta position of the phenyl in cyclopropane ring led to the complete loss of the activity (D166). Probably, the loss of the activity of compound D166 was due to a significant steric hindrance, which prevents the optimal binding of the ligand to the active center of CHIKV nsP2. It should be also noted that despite its structural similarity to D119, compound D162 showed three times less antiviral activity than compound D119 (EC50 = 95.9 and 32.0 μM, respectively; Table 1). Based on these results, it can be assumed that the position of the methyl groups (R1) in the phenyl ring of the cyclopropane moiety (Scheme 1) has also a significant effect on the antiviral activity.
Subsequently, to study the effect of the stereochemical configuration on the antiviral activity, four stereoisomers of compound D160, exhibiting the highest antiviral activity, were synthesized: D160a–d (Scheme 2; see the Supporting Information). All stereoisomers showed different activity against CHIKV. In both pairs of diastereoisomers, D160a and D160b (R,R,S- and R,R,R-stereoisomers, respectively) and D160c and D160d (S,S,R- and S,S,S-stereoisomers, respectively), the most active stereoisomers had the S-configuration of the stereocenter in arylglycine (D160a and D160d). Surprisingly, the antiviral activity was almost the same in the pair of S,S-cyclopropane diastereoisomers (D160c and D160d), while the change of the configuration of the stereocenter in arylglycine in the pair of R,R-stereoisomers significantly altered their antiviral activity (Table 1).
Scheme 2. Individual Stereoisomers for Compound D160 with the Denoted Configurations of the Stereocenters.

Letters R and S denote the absolute configurations of the stereocenters.
Determination of Inhibitory Activities Using the Cell-Free Protease Assay
Compounds with the highest antiviral activity were selected for the cell-free protease inhibition assay—D119, D127, D161, and the stereoisomers D160a–d. Compounds D119, D127, and D161 were tested as equimolecular mixtures of four possible stereoisomers. As is evident from increased amounts of the uncleaved substrate, all selected compounds inhibited the protease activity of nsP2 (Figure 4). However, in general, the effect on recombinant nsP2 was modest and only the stereoisomer D160a demonstrated strong inhibitory activity, as can be seen from the near absence of the cleavage product.
Figure 4.

Effects of selected compounds (1 mM) on the protease activity of CHIKV nsP2. The image combines two gels. The gels were transferred on the paper before scanning. Compounds D119, D127, and D161 represent equimolecular mixtures of four possible stereoisomers. The experiment was repeated three times, with very similar results.
Molecular Docking and MD Simulations of Synthesized Compounds
Molecular docking at both potential binding sites of the nsP2 protease was performed with the S,S,S-stereoisomers of all synthesized compounds and with four individual stereoisomers of the most active compound D160 (D160a–d). The molecular docking results are presented in Table 2. The calculated binding energies of the S,S,S-stereoisomers of compounds D27, D119, D127, and D159 and compounds D161–D166 were in the range of −9.1 to −7.1 kcal/mol for potential binding site 1 and in the range of −8.5 to −6.9 kcal/mol for potential binding site 2. The calculated binding energies of the individual stereoisomers D160a–d were in the range of −8.8 to −7.6 kcal/mol for potential binding site 1 and in the range of −9.2 to −8.8 kcal/mol for potential binding site 2. A comparison of the binding modes of the S,S,S-stereoisomer of the inactive compound D159 and the highest activity stereoisomer D160d at potential binding site 1 does not explain the absence of the antiviral activity of compound D159 as their binding modes are almost identical. Both compounds form hydrogen bonds with the OH group of Tyr1079 (Figure 5A,B). In addition, compound D160d forms a hydrogen bond with the carbonyl group of the side chain of Asn1082 (Figure 5B). At the same time, the analysis of the binding modes of these compounds shows a significant difference in the positions of the ligands at potential binding site 2. The S,S,S-stereoisomer of compound D159 forms only hydrophobic contacts with CHIKV nsP2 (Figure 5C), while compound D160d forms a hydrogen bond with Leu1203 (Figure 5D), which is part of the loop between the β7 strand and the α9 helix, which in turn plays a key role in the recognition and binding of the substrate.21
Table 2. Calculated Binding Energies, Ligand Efficiencies, and Binding Modes of Synthesized Compounds to CHIKV nsP2.
| binding site 1 |
binding site 2 |
||||||
|---|---|---|---|---|---|---|---|
| code | absolute configuration of stereocenters | ΔG (kcal/mol) | LEa | interactions (H-bonds) | ΔG (kcal/mol) | LEa | interactions (H-bonds) |
| D27 | S,S,S | –9.1 | 0.35 | Cys1013, Ala1046, Tyr1079 (HO···HN, HO···H2N), Asn1082 (O(carbonyl)···H2N), Trp1084, Leu1205 | –8.0 | 0.30 | Gln1039, Lys1045, Leu1203, Glu1204 (COO–···H2N), Pro1224, Asp1235, Lys1239 |
| D119 | S,S,S | –9.0 | 0.31 | Cys1013, Ala1046, Tyr1079, Asn1082, Trp1084, Leu1205 | –6.9 | 0.23 | Lys1045, Pro1191, Glu1204, Ile1221, His1222, Pro1224, Lys1239, Met1242, Leu1243 |
| D127 | S,S,S | –9.0 | 0.31 | Cys1013, Ala1046, Tyr1079 (HO···HN, HO···H2N), Asn1082 (O(carbonyl)···H2N), Trp1084, Leu1205 | –8.5 | 0.29 | Gln1039, Lys1045, Leu1203, Glu1204 (COO–···H2N), Ile1221, Pro1224, Asp1235, Lys1239, Leu1243 |
| D159 | S,S,S | –7.1 | 0.19 | Asn1011, Cys1013, Ser1048, Tyr1079 (HO···HN), Asn1082, Trp1084, Leu1205, Gln1241, Met1242, Asp1246 | –7.5 | 0.20 | Lys1045, Pro1191, Val1194, Leu1203, Glu1204, Lys1239, Leu1243 |
| D160a | R,R,S | –8.8 | 0.27 | Tyr1079, Asn1082, Trp1084, Lys1091, Met1242 | –9.0 | 0.27 | Pro1191, Leu1203, His1222, Lys1239 |
| D160b | R,R,R | –7.6 | 0.23 | Ser1048, Tyr1079 (HO···HN(substituted terminal group)), Trp1084, Lys1091, Met1242, Asp1246 | –8.8 | 0.27 | Lys1045, Pro1191, Leu1203, Ile1221, His1222, Met1238, Lys1239 |
| D160c | S,S,R | –8.0 | 0.24 | Asn1011, Cys1013, Ala1046, Tyr1047, Tyr1079, Asn1082, His1083, Trp1084, Asn1202, Glu1204, Leu1205 | –9.2 | 0.28 | Lys1045, Pro1191, Leu1192, Leu1203, Ile1221, His1222, Lys1239, Leu1243 |
| D160d | S,S,S | –8.8 | 0.27 | Cys1013, Ala1046, Tyr1079 (HO···HN, HO···HN(substituted terminal group)), Asn1082 (O(carbonyl)···HN(substituted terminal group)), Trp1084, Leu1205, Met1242 | –8.9 | 0.27 | Pro1191, Leu1192, Leu1203 (NH(peptide)···O(carbonyl)), Glu1204, Met1238, Lys1239, Met1242 |
| D161 | S,S,S | –7.6 | 0.25 | Asn1011, Cys1013, Ala1046, Tyr1079, Asn1082, Trp1084, Asn1202, Glu1204, Leu1205, Met1242 | –7.6 | 0.25 | Gly1176, Pro1191, Ile1221, His1222, Lys1239, Leu1243 |
| D162 | S,S,S | –8.2 | 0.29 | Asn1011, Cys1013, Ala1046, Tyr1047, Tyr1079, Asn1082 (O(peptide)···H2N), Trp1084, Asn1202, Glu1204, Met1242 | –7.6 | 0.27 | Lys1045, Leu1192, Leu1203, Glu1204 (COO–···H2N), Ile1221, Lys1239, Met1242, Leu1243 |
| D163 | S,S,S | –8.1 | 0.28 | Ala1046, Tyr1047, Ser1048, Tyr1079, Trp1084, Lys1091, Leu1205, Gln1241, Met1242 | –7.5 | 0.26 | Lys1045, Leu1203, Ile1221, His1222, Lys1239, Leu1243 |
| D164 | S,S,S | –8.1 | 0.26 | Ser1048, Tyr1079, Trp1084 | –8.1 | 0.26 | Pro1191, Leu1192, Leu1203 (NH···O(carbonyl)), Glu1204, Ile1221, Lys1239, Leu1243 |
| D165 | S,S,S | –8.0 | 0.21 | Cys1013, Ala1046, Tyr1047, Ser1048, Tyr1079, Asn1082, Trp1084, Gln1241, Met1242 | –8.0 | 0.21 | Lys1045, Pro1191, Val1194, Leu1203, Ile1221, Lys1239, Leu1243 |
| D166 | S,S,S | –8.4 | 0.21 | Ala1046, Tyr1079, Asn1082, Trp1084, Lys1091, Met1242 | –8.4 | 0.21 | Lys1045, Val1194, Leu1203, Ile1221, Lys1239, Leu1243 |
LE: ligand efficiency, that is, ΔG/N (heavy atoms).
Figure 5.

Calculated binding modes of (A) S,S,S-stereoisomer of D159 at potential binding site 1 of CHIKV nsP2, (B) S,S,S-stereoisomer D160d at potential binding site 1 of CHIKV nsP2, (C) S,S,S-stereoisomer of D159 at potential binding site 2 of CHIKV nsP2, and (D) S,S,S-stereoisomer D160d at potential binding site 2 of CHIKV nsP2 (PDB ID: 3TRK). Intermolecular hydrogen bonds are shown by dark-green dashed lines.
The further study was focused on molecular docking and MD simulations of the stereoisomers D160a–d. During the molecular docking procedure, bonds in the ligands were fixed in their respective spatial configuration. A comparison of the calculated binding energies shows that the values of both binding sites are the same for the stereoisomers D160a and D160d (Table 2), while for the stereoisomers D160b and D160c, the best value is rather typical for potential binding site 2 (the difference with potential binding site 1 is 1.2 kcal/mol). The stereoisomers D160a and D160d were selected for further study using MD simulations. The MD simulations with a length of 50 nanoseconds (ns) were performed for both stereoisomers at both potential binding sites. In the case of the stereoisomer D160a, its complex with CHIKV nsP2 at potential binding site 2 was more stable compared to potential binding site 1 (Figure S1A,C). In the case of the stereoisomer D160d, the root-mean-square deviations (rmsds) of its complexes with the nsP2 protease at both potential binding sites were approximately similar (Figure S1B,D). Analysis of the MD trajectory at potential binding site 1 showed that both stereoisomers do not form any contacts with the amino acid residues from the catalytic dyad of the CHIKV nsP2 protease, but the stereoisomer D160a forms a long-term stacking (π–π) interaction with the side chain of the conserved amino acid residue Trp1084 (Figure 6A). The stereoisomer D160d, in turn, forms two long-term contacts—a hydrogen bond and a stacking (π–π) interaction with the side chain of Tyr1079 as well as a water bridge with the backbone of Glu1204 (Figure 6B). Thus, based on the MD results for potential binding site 1, it can be assumed that the stereoisomer D160a may have the highest antiviral activity due to a specific hydrophobic contact with the side chain of the conserved amino acid residue Trp1084, which is important for the activity of the nsP2 protease.18 However, experimental data show that the activity of the stereoisomer D160a in the cell-based assay is almost 6 times lower than the activity of D160d, 26.8 and 4.8 μM, respectively (Table 1). According to the MD results, the binding of both stereoisomers at potential binding site 2 is more specific than that at potential binding site 1. The stereoisomer D160a has two long-term contacts—a hydrogen bond with the backbone of His1222 and a hydrogen bond with the backbone of Lys1239, in addition to a short-term π-cation interaction with the side chain of Lys1239 as well as nonspecific hydrophobic interactions with Pro1191, Val1194, Leu1203, Ile1221, Leu1243, and Pro1224 (Figure 6C). The stereoisomer D160d forms several long-term contacts—a hydrogen bond and a water bridge with the backbone of Leu1203, two hydrogen bonds and several water bridges with Glu1204, and three π-cation interactions with Lys1045 and Lys1239 (Figure 6D). As can be seen from these data, the stereoisomer D160d remains bound to the amino acid residues of the loop between the β7 strand and the α9 helix (Leu1203, Glu1204, and Leu1205), which probably block the conformational changes of the CHIKV nsP2 protease required for substrate recognition and binding.21 It is worth noting that D160d binds to these amino acid residues at both potential binding sites, indicating that the most likely mechanism of its action is associated with blocking the binding of the substrate rather than the catalytic dyad.
Figure 6.

MD-calculated contacts of (A) R,R,S-stereoisomer D160a at potential binding site 1 of CHIKV nsP2, (B) S,S,S-stereoisomer D160d at potential binding site 1 of CHIKV nsP2, (C) R,R,S-stereoisomer D160a at potential binding site 2 of CHIKV nsP2, and (D) S,S,S-stereoisomer D160d at potential binding site 2 of CHIKV nsP2 (PDB ID: 3TRK). Interactions that occur more than 10% of the simulation time are shown.
Evaluation of Antiviral Activity at High MOI
The antiviral activity of the two stereoisomers, D160a and D160d, and compound D161 (an equimolecular mixture of four possible stereoisomers) was also analyzed using cells infected at MOI 10. Under these conditions, CHIKV infection is rapid and unlike low MOI infection, there is no spread of the virus from infected cells to noninfected cells. In this experiment, the inhibition of CHIKV infection was monitored by evaluation of the expression of CHIKV nsP1, nsP2, and capsid proteins (Figure 7). In this assay, compounds D160a and D161 exhibited the dose-dependent antiviral activity at a concentration greater than 50 μM, down-regulating production of viral nsP1 and nsP2 as well as capsid protein. The fact that inhibition required concentrations slightly above EC50 of the compounds (26.8 and 13.9 μM, respectively; Table 1) can be attributed to lower sensitivity of the assay performed at high MOI. Interestingly, in the case of the stereoisomer D160d, a slight decrease in the production of viral proteins was observed only at a concentration of 200 μM, a concentration much higher of EC50 (4.8 μM; Table 1). As the main difference of assays performed at low and high MOI is the presence of viral spread at the former case, it could be speculated that antiviral effects of the stereoisomer D160d may include inhibition of virion formation and/or release. Such an effect has been observed for deletion of residues 998–1003 of CHIKV ns-polyprotein; these residues are located close to the active site of the nsP2 protease.22 Additional studies are required to reveal whether the addition of the stereoisomer D160d mimics the effect of these mutations.
Figure 7.

Western blot analysis of proteins from CHIKV-infected cells that were treated using compounds D160a (R,R,S-stereoisomer) (A), D161 (an equimolecular mixture of four possible stereoisomers) (B), and D160d (S,S,S-stereoisomer) (C). BHK-21 cells infected with CHIKV-NanoLuc (MOI 10) were treated with increasing concentrations of the inhibitor. Cell lysates were collected 6 h post infection; proteins were separated using SDS-PAGE, transferred onto the PVDF membrane, and detected using indicated antibodies. β-Actin was detected as the loading control. Names of the proteins are indicated on the left; molecular masses of marker bands are indicated on the right. Neg—mock-infected BHK-21 cells treated with 1% DMSO; Pos—CHIKV-infected BHK-21 cells treated with 1% DMSO (no inhibitor).
Conclusions
In this work, the structure and antiviral properties of the previously found CHIKV nsP2 protease inhibitor13 were optimized using CADD methods, synthesis, and cell-based and cell-free experiments. Based on the structure of CHIKV nsP2 inhibitor 1c discovered in our previous study,13 158 novel analogues were designed, and 11 novel derivatives were synthesized and tested for their ability to inhibit both the enzymatic activity of the nsP2 protease and the replication of the virus as a whole. By optimizing the structure of the inhibitor, a series of compounds with antiviral activity 2–10 times higher than that of the initial compound 1c were obtained. The results of molecular modeling of the binding of the studied potential inhibitors showed that the most probable mechanism of inhibition of the CHIKV nsP2 protease activity is the blocking of conformational changes in nsP2 required for substrate recognition and binding. A study of the antiviral activity of the individual stereoisomers of the most active compound D160 showed that the antiviral activity significantly depends on the spatial configuration of all stereocenters of the compound, and the S,S,S-configuration of stereocenters is preferred. However, it is worth noting that despite the high antiviral activity in the cell-based assay at a low MOI, the stereoisomer D160d did not show significant inhibitory activity in the cell-free protease assay and the cell-based assay at a high MOI. The opposite situation was observed in the case of the stereoisomer D160a, showing the greatest inhibitory effect both in the cell-free protease assay and in the cell-based assay at a high MOI. This difference can probably be explained by the different bioavailability of these stereoisomers, but their mechanism of action needs to be further investigated for a more detailed explanation. Thus, both stereoisomers of compound D160 can be considered as new scaffolds for further optimization and development of new effective targeted inhibitors of CHIKV.
Materials and Methods
Molecular Modeling
Target
The crystal structure of the CHIKV nsP2 protease (PDB ID: 3TRK)15 was used for molecular docking. The structural model was measured by X-ray diffraction with a resolution of 2.40 Å. The raw crystal structure was corrected, and hydrogen atoms were automatically added to the protein using Schrödinger’s Protein Preparation Wizard of Maestro 10.7.23 All selenomethionines were converted to methionines. Water molecules were removed from the crystal structure.
Small-Molecule Library
The two-dimensional chemical structures of the S,S,S-stereoisomers of designed compounds were drawn using Schrödinger’s 2D Sketcher of Maestro. The ligand structures were prepared for molecular docking using LigPrep with the OPLS_2005 force field from the Schrödinger Suite.24 Generation of all possible states and ionization states was enumerated for each ligand using Epik at a pH of 7.0 ± 2. The stereoisomers were determined from the three-dimensional structures. PDB files for the molecular docking procedure were created from lowest-energy conformers for each ligand.
Molecular Docking
AutoDock Vina 1.1.225 was used for the docking studies to find out binding modes and binding energies of the S,S,S-stereoisomers of designed compounds to the receptor. In this study, two possible binding sites of the CHIKV nsP2 protease were selected for molecular docking. Potential binding site 1 of CHIKV nsP2 was specified with the residues Cys1013, His1083, and Trp1084 and was surrounded by a grid box sized 20 × 20 × 20 points with a spacing of 1.000 Å. Potential binding site 2 was specified with the residue His1222 and was surrounded by a grid box sized 24 × 24 × 24 points with a spacing of 1.000 Å. In the previously published work,13 it was reported that the configuration of the cyclopropane fragment of compound 1c was important for the antiviral activity; therefore, the cyclopropane bonds were fixed in trans-configuration for all compounds. The docking parameters were used in their default values, that is, one central processing unit to use, the number of output poses is 9, and the exhaustiveness is 8.
Molecular Dynamics
The MD simulations were carried out using the Desmond simulation package of Schrödinger LLC.26 In all runs, the NPT ensemble (isothermal–isobaric) was applied with the temperature of 300 K and a pressure of 1 bar. The simulation lengths were 50 ns with a relaxation time of 1 picosecond (ps). The OPLS_2005 force field parameters were used in all simulations.27 The long-range electrostatic interactions were calculated using the particle mesh Ewald method.28 The cutoff radius in Coulomb interactions was 9.0 Å. The water molecules were described using the simple point charge model.29 The Martyna–Tuckerman–Klein chain coupling scheme30 with a coupling constant of 2.0 ps was used for the pressure control, and the Nosé–Hoover chain coupling scheme30 was used for the temperature control. Nonbonded forces were calculated using an reversible reference system propagation algorithm integrator where the short-range forces were updated every step and the long-range forces were updated every three steps. The trajectories were saved at a 50.0 ps interval for analysis. The behavior and interactions between the ligands and protein were analyzed using the Simulation Interaction Diagram tool implemented in the Desmond MD package.
Cell Lines and Virus Strain
Baby hamster kidney (BHK-21) cells (ATCC CCL-10) were maintained in Glasgow’s minimal essential medium (GMEM; PAN Biotech) containing 7.5% fetal bovine serum, 2% tryptose phosphate broth, 20 mM N-2-hydroxyethylpiperazine-N-ethanesulfonic acid (HEPES), and 1% penicillin/streptomycin. The cell culture was maintained at 37 °C in an atmosphere of 5% CO2. The CHIKV-NanoLuc strain was obtained from the icDNA clone of pICRES1-NanoLuc, representing the LR2006OPY1 strain belonging to the East/Central/South African genotype.31 The virus stocks were stored at −80 °C. All virus experiments were conducted in accordance with the guidelines of the national authorities using appropriate biosafety laboratories under all necessary safety approvals.
Antiviral and Cytotoxicity Assays
Compounds
10 mM stocks of compounds were prepared by dissolving compounds in sterile dimethyl sulfoxide (DMSO) (Sigma, USA) and stored at −20 °C until further use.
Antiviral Activity Assay
BHK-21 cells were seeded on 24-well tissue culture plates (Thermo Fisher Scientific) at a density 2 × 105 cells/well in 400 μL of GMEM and were allowed to adhere overnight. BHK-21 cells were infected with the CHIKV-NanoLuc virus at an MOI of 0.001 plaque-forming unit/cell in an infection medium (200 μM/well) containing GMEM, 0.2% bovine serum albumin (BSA), 1% of the penicillin/streptomycin stock, and compounds at final concentrations ranging from 0.1 to 100 μM. After 1 h post infection, a complete growth medium (300 μL/well) containing compounds at final concentrations ranging from 0.1 to 100 μM was added. At 16 h post infection, the medium was discarded, cells were lysed, and the activity of nanoluciferase was measured using a Renilla luciferase assay system (Promega). The assay was carried out in three parallels. EC50 calculation was performed using GraphPad Prism version 8.0 for Windows (GraphPad Software, La Jolla, California, USA).32 The EC50 graphs are given in Figure S2.
Cytotoxicity Assay
BHK-21 cells were plated in 96-well plates containing the complete growth medium and were allowed to adhere overnight. The cells were then treated with compounds at the indicated concentrations and were incubated for 24 h. After 24 h of drug treatment, the cell viability was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay. Briefly, 10 μL of MTT solution (5 mg/mL) was added to each well and incubated at 37 °C for 4 h. After removing the medium, the crystals produced were dissolved in 100 μL of DMSO. The optical density of the solutions was measured at 540 nm. All experiments were performed in triplicate.
Cell-Free Protease Inhibition Assay
Full-length recombinant CHIKV nsP2 was used as the protease. The recombinant protein substrate contained the nsP2 cleavage site (residues P10 to P′5 from the nsP1/nsP2 cleavage site) placed between enhanced green fluorescent protein (EGFP) and thioredoxin. The recombinant proteins were expressed and purified, as described in detail earlier.33,34 The protease inhibition assay was carried out at 30 °C for 1.5 h in a 10 μL volume in the protease assay buffer [20 mM HEPES (pH 7.2), 2 mM dithiothreitol]. The CHIKV nsP2 final concentration was 1.4 μM, the substrate’s final concentration was 6 μM, and the inhibitor’s final concentration was 1 mM; 10% DMSO was used as a solvent control. Protease inhibition assay reaction products (5.5 μL) were analyzed by 10% sodium dodecyl sulfate (SDS)-PAGE and Coomassie blue staining.
Western Blot Analysis
90% confluent BHK-21 cells (1 × 106 cells/well in 35 mm diameter plates) were infected with CHIKV-NanoLuc at an MOI of 10 in the presence of the stereoisomers D160a, D160d, and compound D161 in the infection medium (GMEM, 0.2% BSA, 20 mM HEPES). The stereoisomer D160a was tested at concentrations of 5 up to 200 μM, the stereoisomer D160d at concentrations of 1 up to 200 μM, and compound D161 at concentrations of 10 up to 200 μM. Control cells were infected in the presence of 1% DMSO used as a solvent control. At 1 h post infection, the complete growth medium containing the same concentration of tested compounds or DMSO was added. Cells were incubated at 37 °C for 6 h, collected and lysed in 100 μL of the SDS sample buffer (50 mM Tris–HCl pH 6.8, 2% SDS, 10% glycerol, 0.2% bromophenol blue), and boiled at 100 °C for 8 min. Proteins were separated by SDS-PAGE in 10% gels and transferred onto hydrophilic polyvinylidene fluoride (PVDF) membranes. The CHIKV proteins were detected using the corresponding rabbit polyclonal antibodies (all generated in-house); β-actin (sc-47778; Santa Cruz Biotechnology) was used as a loading control. The membranes were incubated with the appropriate secondary antibodies conjugated to fluorescent infrared dyes (LI-COR), and the signals were visualized with the LI-COR Odyssey Fc Imaging System.
Acknowledgments
The current work was financially supported by the EU European Regional Development Fund through the Centre of Excellence in Molecular Cell Engineering (project no. 2014-2020.4.01.15-0013), Estonia. D.G.K. and M.O. acknowledge support from Tallinn University of Technology (grant no. B58).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c00625.
Calculated binding energies of small-molecule ligands to the receptor CHIKV nsP2; description of the chemical synthesis of new potential inhibitors and copies of their 1H and 13C NMR spectra; rmsd of the atomic positions for the stereoisomers D160a and D160d with CHIKV nsP2; and determination of EC50 of the selected compounds in BHK-21 cells (PDF)
Author Contributions
L.I. performed molecular docking and MD simulations; L.I., J.T.-T., and M.K. analyzed modeling results. L.I., K.R., and E.Ž. performed biological experiments; L.I., K.R., E.Ž., A.M., and M.K. analyzed experimental data. M.O., D.G.K., and M.L. designed and performed the chemical synthesis of the compounds. A.M. and M.K. coordinated the project. All authors participated in the preparation of the manuscript and approved the final manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Caglioti C.; Lalle E.; Castilletti C.; Carletti F.; Capobianchi M. R.; Bordi L. Chikungunya virus infection: an overview. New Microbiol. 2013, 36, 211–227. [PubMed] [Google Scholar]
- Silva L. A.; Dermody T. S. Chikungunya virus: epidemiology, replication, disease mechanisms, and prospective intervention strategies. J. Clin. Invest. 2017, 127, 737–749. 10.1172/jci84417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver S. C.; Osorio J. E.; Livengood J. A.; Chen R.; Stinchcomb D. T. Chikungunya virus and prospects for a vaccine. Expert Rev. Vaccines 2012, 11, 1087–1101. 10.1586/erv.12.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subudhi B.; Chattopadhyay S.; Mishra P.; Kumar A. Current Strategies for Inhibition of Chikungunya Infection. Viruses 2018, 10, 235. 10.3390/v10050235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahariya C.; Pradhan S. K. Emergence of chikungunya virus in Indian subcontinent after 32 years: A review. J. Vector Borne Dis. 2006, 43, 151–160. [PubMed] [Google Scholar]
- Burt F. J.; Rolph M. S.; Rulli N. E.; Mahalingam S.; Heise M. T. Chikungunya: a re-emerging virus. Lancet 2012, 379, 662–671. 10.1016/s0140-6736(11)60281-x. [DOI] [PubMed] [Google Scholar]
- Roth A.; Hoy D.; Horwood P. F.; Ropa B.; Hancock T.; Guillaumot L.; Rickart K.; Frison P.; Pavlin B.; Souares Y. Preparedness for Threat of Chikungunya in the Pacific. Emerg. Infect. Dis. 2014, 20, e130696 10.3201/eid2008.130696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muniaraj M. Fading chikungunya fever from India: beginning of the end of another episode?. Indian J. Med. Res. 2014, 139, 468–470. [PMC free article] [PubMed] [Google Scholar]
- Khan A. H.; Morita K.; Parquet M. D. C.; Hasebe F.; Mathenge E. G.; Igarashi A. Complete nucleotide sequence of chikungunya virus and evidence for an internal polyadenylation site. J. Gen. Virol. 2015, 83, 3075–3084. 10.1099/0022-1317-83-12-3075. [DOI] [PubMed] [Google Scholar]
- Singh S. K.; Unni S. K. Chikungunya virus: host pathogen interaction. Rev. Med. Virol. 2011, 21, 78–88. 10.1002/rmv.681. [DOI] [PubMed] [Google Scholar]
- Fros J. J.; van der Maten E.; Vlak J. M.; Pijlman G. P. The C-terminal Domain of Chikungunya Virus nsP2 Independently Governs Viral RNA Replication, Cytopathicity, and Inhibition of Interferon Signaling. J. Virol. 2013, 87, 10394–10400. 10.1128/jvi.00884-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadav S. S.; Sinha B. N.; Hilgenfeld R.; Jayaprakash V. Computer-Aided Structure Based Drug Design Approaches for the Discovery of new Anti-CHIKV Agents. Curr. Comput.-Aided Drug Des. 2017, 13, 346–361. 10.2174/1573409913666170309145308. [DOI] [PubMed] [Google Scholar]
- Das P. K.; Puusepp L.; Varghese F. S.; Utt A.; Ahola T.; Kananovich D. G.; Lopp M.; Merits A.; Karelson M. Design and Validation of Novel Chikungunya Virus Protease Inhibitors. Antimicrob. Agents Chemother. 2016, 60, 7382–7395. 10.1128/AAC.01421-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassetto M.; De Burghgraeve T.; Delang L.; Massarotti A.; Coluccia A.; Zonta N.; Gatti V.; Colombano G.; Sorba G.; Silvestri R.; Tron G. C.; Neyts J.; Leyssen P.; Brancale A. Computer-aided identification, design and synthesis of a novel series of compounds with selective antiviral activity against chikungunya virus. Antiviral Res. 2013, 98, 12–18. 10.1016/j.antiviral.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 3TRK: Structure of the Chikungunya virus nsP2 protease. Research Collaboratory for Structural Bioinformatics Protein Data Bank, Spetember 28, 2011. https://www.rcsb.org/structure/3TRK (accessed 2021-01-27).; b Cheung J.; Franklin M.; Mancia F.; Rudolph M.; Cassidy M.; Gary E.; Burshteyn F.; Love J.. Structure of the Chikungunya virus nsP2 protease. 2011. Unpublished work. [Google Scholar]
- Shityakov S.; Foerster C. In silico predictive model to determine vector-mediated transport properties for the blood–brain barrier choline transporter. Adv. Appl. Bioinf. Chem. 2014, 7, 23–36. 10.2147/aabc.s63749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashad A. A.; Mahalingam S.; Keller P. A. Chikungunya Virus: Emerging Targets and New Opportunities for Medicinal Chemistry. J. Med. Chem. 2014, 57, 1147–1166. 10.1021/jm400460d. [DOI] [PubMed] [Google Scholar]
- Rausalu K.; Utt A.; Quirin T.; Varghese F. S.; Žusinaite E.; Das P. K.; Ahola T.; Merits A. Chikungunya virus infectivity, RNA replication and non-structural polyprotein processing depend on the nsP2 protease’s active site cysteine residue. Sci. Rep. 2016, 6, 37124. 10.1038/srep37124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moni L.; Denißen M.; Valentini G.; Müller T. J. J.; Riva R. Diversity-Oriented Synthesis of Intensively Blue Emissive 3-Hydroxyisoquinolines by Sequential Ugi Four-Component Reaction/Reductive Heck Cyclization. Chem.—Eur. J. 2015, 21, 753–762. 10.1002/chem.201404209. [DOI] [PubMed] [Google Scholar]
- Thompson M. J.; Chen B. Ugi Reactions with Ammonia Offer Rapid Access to a Wide Range of 5-Aminothiazole and Oxazole Derivatives. J. Org. Chem. 2009, 74, 7084–7093. 10.1021/jo9014529. [DOI] [PubMed] [Google Scholar]
- Narwal M.; Singh H.; Pratap S.; Malik A.; Kuhn R. J.; Kumar P.; Tomar S. Crystal structure of chikungunya virus nsP2 cysteine protease reveals a putative flexible loop blocking its active site. Int. J. Biol. Macromol. 2018, 116, 451–462. 10.1016/j.ijbiomac.2018.05.007. [DOI] [PubMed] [Google Scholar]
- Law Y.-S.; Wang S.; Tan Y. B.; Shih O.; Utt A.; Goh W. Y.; Lian B.-J.; Chen M. W.; Jeng U.-S.; Merits A.; Luo D. Interdomain Flexibility of Chikungunya Virus nsP2 Helicase-Protease Differentially Influences Viral RNA Replication and Infectivity. J. Virol. 2021, 95, e01470–20. 10.1128/jvi.01470-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Sastry G. M.; Adzhigirey M.; Day T.; Annabhimoju R.; Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]; b Schrödinger Suite 2020-4, Protein Preparation Wizard: Epik, Impact, and Prime; Schrödinger, LLC: New York, NY, 2020.
- Schrödinger Release 2020-4: LigPrep; Schrödinger, LLC: New York, NY, 2020.
- Trott O.; Olson A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bowers K. J.; Chow D. E.; Xu H.; Dror R. O.; Eastwood M. P.; Gregersen B. A.; Klepeis J. L.; Kolossvary I.; Moraes M. A.; Sacerdoti F. D.; Salmon J. K.; Shan Y.; Shaw D. E.. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. Proceedings of the 2006 ACM/IEEE Conference on Supercomputing; IEEE, 2006; p 43.; b Schrödinger Release 2020-4: Desmond Molecular Dynamics System; D. E. Shaw Research: New York, NY, 2020;; Schrödinger Release 2020-4: Maestro–Desmond Interoperability Tools; Schrödinger: New York, NY, 2020.
- Banks J. L.; Beard H. S.; Cao Y.; Cho A. E.; Damm W.; Farid R.; Felts A. K.; Halgren T. A.; Mainz D. T.; Maple J. R.; Murphy R.; Philipp D. M.; Repasky M. P.; Zhang L. Y.; Berne B. J.; Friesner R. A.; Gallicchio E.; Levy R. M. Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. 10.1002/jcc.20292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toukmaji A. Y.; Board J. A. Jr. Ewald summation techniques in perspective: a survey. Comput. Phys. Commun. 1996, 95, 73–92. 10.1016/0010-4655(96)00016-1. [DOI] [Google Scholar]
- Zielkiewicz J. Structural properties of water: Comparison of the SPC, SPCE, TIP4P, and TIP5P models of water. J. Chem. Phys.2005, 123, 104501. J. Chem. Phys. 2006, 124, 109901. 10.1063/1.2018637. [DOI] [PubMed] [Google Scholar]
- Martyna G. J.; Klein M. L.; Tuckerman M. Nosé-Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 1992, 97, 2635–2643. 10.1063/1.463940. [DOI] [Google Scholar]
- Utt A.; Quirin T.; Saul S.; Hellström K.; Ahola T.; Merits A. Versatile Trans-Replication Systems for Chikungunya Virus Allow Functional Analysis and Tagging of Every Replicase Protein. PloS One 2016, 11, e0151616 10.1371/journal.pone.0151616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GraphPad Prism for Windows, ver. 8.0; GraphPad Software: La Jolla, CA, 2020; www.graphpad.com (accessed 2020-09-19).
- Das P. K.; Merits A.; Lulla A. Functional cross-talk between distant domains of chikungunya virus non-structural protein 2 is decisive for its RNA-modulating activity. J. Biol. Chem. 2014, 289, 5635–5653. 10.1074/jbc.m113.503433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utt A.; Das P. K.; Varjak M.; Lulla V.; Lulla A.; Merits A. Mutations Conferring a Noncytotoxic Phenotype on Chikungunya Virus Replicons Compromise Enzymatic Properties of Nonstructural Protein 2. J. Virol. 2015, 89, 3145–3162. 10.1128/jvi.03213-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.