

Abstract
Activity prediction plays an essential role in drug discovery by directing search of drug candidates in the relevant chemical space. Despite being applied successfully to image recognition and semantic similarity, the Siamese neural network has rarely been explored in drug discovery where modelling faces challenges such as insufficient data and class imbalance. Here, we present a Siamese recurrent neural network model (SiameseCHEM) based on bidirectional long short-term memory architecture with a self-attention mechanism, which can automatically learn discriminative features from the SMILES representations of small molecules. Subsequently, it is used to categorize bioactivity of small molecules via N-shot learning. Trained on random SMILES strings, it proves robust across five different datasets for the task of binary or categorical classification of bioactivity. Benchmarking against two baseline machine learning models which use the chemistry-rich ECFP fingerprints as the input, the deep learning model outperforms on three datasets and achieves comparable performance on the other two. The failure of both baseline methods on SMILES strings highlights that the deep learning model may learn task-specific chemistry features encoded in SMILES strings.
Introduction
Given the virtually infinite chemical space, activity prediction plays an essential role in drug discovery by focusing exploration in a relevant space. Harnessing the growing number of high-resolution protein structures and the recent development in computer hardware such as GPUs, structure-enabled free-energy perturbation approaches could predict binding affinities to a satisfactory extent.1 However, such methods are not applicable in the absence of relevant protein structures, and they are most computationally expensive and require careful preparation of the simulation system, making their prospective application difficult in practice.2 In parallel, ligand-based approaches such as quantitative structure–activity relationship (QSAR)3 have long been established on the basis of the similar property principle of chemical informatics, stating that small molecules with similar structures are likely to exhibit similar biological activities.
Traditional QSAR approaches seek to establish a mathematical relationship between activity and computed molecular fingerprints or handcrafted descriptors. The advances in deep neural network have inspired novel approaches which learn task-specific representations using graph convolutions.4−7 A recent comprehensive evaluation demonstrates that a graph convolution model outperforms models using fixed molecular descriptors.8 A Siamese neural network consists of two identical subnetworks working in parallel to find the similarity between two different input vectors from the learned features. Unlike other modern deep learning models which rely on big data to perform well, it could learn from very little data and hence has become popular in the past few years. For example, it has recently been implemented to measure the transcriptional response similarity of two compounds using computed fingerprints as the input9 or to rank binding affinity of compounds within a congeneric series by applying convolutional neural network to their binding poses.10
Small-molecule drug discovery often starts with a lead compound, followed by a quick SAR exploration with analogues. With only a small amount of biological data available at the very beginning, the subsequent lead optimization presents a low-data problem.11 Lead optimization then results in one or several congeneric series where compounds differ in a few atoms around a unique scaffold. One-shot learning classification combined with graph convolutional neural networks has been shown to tackle this challenging issue of low data prevailing in real drug discovery projects.11 The imbalance between bioactive and inactive classes presents one additional challenge, as reflected by the low hit rate of high-throughput screening assays.12 The Siamese neural network has a competitive edge to cope with both low data and class imbalance.13
In this work, we build a Siamese recurrent neural network based on bidirectional long short-term memory (BiLSTM) architecture with a self-attention mechanism, operating on the SMILES representations of small molecules. Trained on random SMILES strings with the aim for the model to learn underlying chemical features, it performs robustly on five datasets for binary or categorical classification of bioactivities with N-shot learning. It consistently matches or outperforms the two popular baseline models, namely, random forest (RF) and support vector machine (SVM), which use chemistry-rich ECFP6 fingerprints. Taken together, the deep model called SiameseCHEM could build task-specific fingerprints and is applicable as an alternative tool for medicinal chemists in drug discovery.
Model Architecture
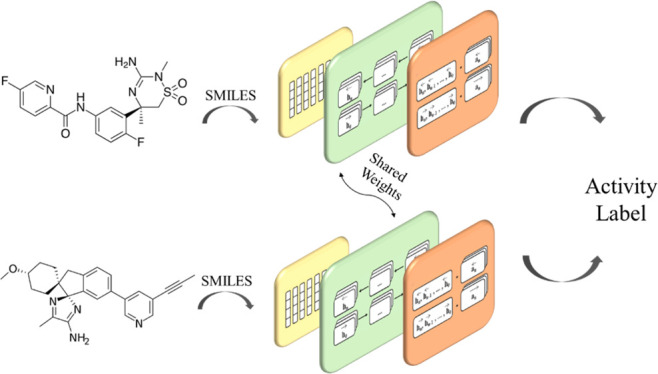
The SiameseCHEM deep learning model consists of a dual-branch network with shared weights and is implemented with PyTorch. The configuration consists of an embedding layer,14,15 three BiLSTM layers,16 an attention layer,17 and a final distance layer (Figure 1). The mathematical details are given in the Supporting Information.
Figure 1.

Siamese Recurrent Neural Network architecture.
Briefly, the embedding layer of 128 dimensions projects the discrete tokens into a continuous two-dimensional space, passed through three BiLSTM layers each of which has 128 hidden units. A dropout probability of 0.05 was used on the output weights of both the embedding layer and the BiLSTM layers. The self-attention mechanism17 was applied to the hidden states extracted from the last BiLSTM layer. Both the forward and backward hidden states from the last LSTM layer were concatenated to yield a hidden states matrix, which is operated by an attention matrix of 512 dimensions. The output is fed through a fully connected linear layer with the leaky ReLU activation function, resulting in an attentional vector of 256 dimensions. In the final stage, cosine similarity of the two attentional vectors of the input pair was computed and squeezed between 0 and 1 with a logistic sigmoid function.
He normal initialization18 was used for input-hidden weights, and hidden–hidden weights were initialized with a semi-orthogonal matrix.19 Biases were filled with zeros. The embedding layer weights have been initialized following a normal distribution with a mean of 0 and a standard deviation of 1. The hidden states were initialized with zeros. No batch normalization was applied as it did not improve model performance. The log-cosh loss function20 was chosen for its best performance among the contrastive loss,21 Huber loss,22 and L1 and L2 losses. Attempts of leveraging the triplet loss23 by feeding positive and negative examples to the anchor compound were unsuccessful. The model was trained using the Adam optimizer24 with an initial learning rate of 10–4, which was decayed with a factor of 0.1 after learning stagnated for 10 epochs. A gradient clipping of 1 was applied before updating the parameters of the optimizer in order to avoid gradient explosion.25 The model of each dataset was trained for 150 epochs with a batch size of 64. The training set was shuffled during each epoch. While the model performance was monitored on the validation set, no early-stopping was implemented.
Materials and Methods
Datasets
Five datasets against beta-secretase 1 (BACE1), C–C chemokine receptor type 5 (CCR5), dopamine receptor 2 (DRD2), epidermal growth factor receptor (EGFR), and nuclear receptor subfamily 1 group H member 2 (NR1H2) were collected from the publicly available database ExCAPE-DB26 and ChEMBL (version 25).27 The datasets were selected because they represent highly pursued drug target families. Since datasets extracted from the scientific literature often contain few inactive compounds, they were complemented by in-house inactive compounds.
Compounds with missing biological activity (pXC50) were discarded. Compounds having elements other than H, C, N, O, F, S, Cl, and Br were filtered out. Compounds with more than 50 heavy atoms or a SMILES string length greater than 150 were excluded. Compounds were then desalted and standardized, and chirality was unsigned using RDKit (v2020.03.1). Finally, data was collated and the median pXC50 value was taken as the biological activity if duplicates were present (Table S1). Compounds were divided into active and inactive by a threshold of 5 in pXC50, and the active class was further broken down into moderately active and strongly active with a threshold of 7 for the categorical classification. Each dataset was randomly split into a training set (50%), a validation set (40%), and a test set (10%).
Generation of Compound Pairs
There often exists class imbalance, for example, there are more active compounds in a dataset since inactive ones are seldomly reported. In this case, the inactive class was topped up by randomly adding inactive compounds from itself one at a time. Afterward, within each class, the first half of compounds were paired up with the second half, yielding pairs having similar biological activity. Compounds in the two classes were further paired up sequentially to yield pairs having dissimilar biological activity. Duplicate pairs were finally removed. This approach avoids the combinatorial explosion which would require a huge amount of computational resources for a big dataset. Meanwhile, it ensures that each compound will appear at least twice, one in the pairs having similar bioactivity and one in the pairs having dissimilar bioactivity. The resulting pairs are relatively balanced against each class.
Tokenization of SMILES Strings
A master dictionary was created for all five datasets. Each atom and explicit bond type in the SMILES strings was discretized into a token. Particular attention was paid to multilettered symbols (i.e., Br, Cl) and characters flanked by brackets (i.e., [nH], [N+], etc.), which were treated as special characters and represented by one token. Additional start-of-sequence and end-of-sequence characters were added to signal the start and end of the string. The final vocabulary consisted of 58 unique alphanumeric tokens. Each token is further mapped consecutively to an integer staring from 1. The SMILES string is then represented by a vector of integers corresponding to respective tokens, padded with zeros to reach the maximum length of 150.
Data Augmentation
Data augmentation is a strategy to increase the training data in order to enable invariance learning and, consequently, improve the model performance.28 In this context, two data augmentation strategies were studied. First, the training data was augmented by taking the unsampled pairs. A second approach is to generate multiple random SMILES strings per compound in the sampled pairs as previously reported.29−31 Randomized SMILES can be thought of as permutations of canonical SMILES strings.32 Given a molecular graph, different SMILES strings can be generated depending on the traversing route, and this can result in multiple random SMILES strings per compound in the sampled pairs. Training data was augmented with a 3-fold increase for both approaches.
Baseline Methods
Two classical machine learning methods, namely, RF and SVM, were chosen for comparison with the SiameseCHEM. Both RF and SVM prove to consistently perform well on a variety of tasks.8,33 For the RF, the number of trees was set to 100, and no maximum depth for the tree was specified. The Gini Index for information gain was used. For the SVM, the radial basis function was selected as the kernel function with a regularization parameter set to 1.0.
In addition, a Siamese multilayer perceptron (MLP) model was built with each branch consisting of two linear layers with 512 and 256 neurons, respectively, each followed by a dropout with a probability of 5% and a leaky ReLU activation function.
N-Shot Learning
A support set of N compounds with known bioactivity was constructed. The first half N compounds was taken without replacement from the pool of actives and the second from the inactive pool. The query compound was paired up with each compound in the support set to form N pairs. The resulting pairs were fed into a trained deep model which outputs a probability of having similar biological activity (similarity score). In comparison with the one-shot learning,13 the query compound was assigned the same activity class as the compound in the support set which has the highest similarity score. Notably, the process can be repeated k times each with a different support set. A consensus prediction out of the k times was designated as the final prediction.
The categorical N-shot learning was lazily implemented as the proof of concept. Two deep models were trained separately on the two thresholds, namely, pXC50 of 5 and 7. The input pair was first fed to the model with the threshold of 7 and then fed to the second one only when the query compound was predicted to be inactive by the first one.
Model Evaluation
The Matthews correlation coefficient (MCC) ranges from −1 to 1 and is a preferred metric in bioinformatics to condense information in the confusion matrix.34
where TP, TN, FP, and FN are the number of true positives, true negatives, false positives, and false negatives, respectively. In addition, precision (or positive predictive value), recall (or sensitivity), and false positive rate are used to assess mispredictions.
Both Cohen’s weighted kappa (κ) in scikit-learn and Kendall’s coefficient (τ) in SciPy are adopted as the metric for the categorical classification.
Results and Discussion
Chemical Similarity of the Paired Compounds
The five datasets have rather diverse compounds, suggested by the high number of clusters giving rise to 5.8 compounds per cluster on average (Table 1). The clustering was performed using the Butina algorithm with a distance cutoff of 0.5 by the ECFP6 fingerprints.35 The chemical similarity of resulting pairs is centered around 0.15 across all five data sets, measured by the Tanimoto coefficient (TC) using the ECFP6 fingerprints (Table 1). Chemical similarity has been used for target predictions, and a TC value greater than 0.4 between a pair of compounds may suggest their similar biological effects.36 However, the average TC value for pairs having similar bioactivity is far low. Notably, the average TC for pairs of similar bioactivity is not higher than that for pairs of dissimilar activity. The indistinguishable distribution of TC values from pairs having similar and dissimilar activities (Figure 2) indicates that similarity in the biological activity cannot be discriminated based on the chemical similarity measured by the widely used ECFP6 fingerprints.
Table 1. Chemical Similarity of the Paired Compounds from the Five Datasets.
| datasets | Na | training pairs | validation pairs | TC of training setb | TC of validation setb |
|---|---|---|---|---|---|
| BACE1 | 20,450 (2490) | 14,788 | 2954 | 0.14 (0.13) | 0.14 (0.13) |
| CCR5 | 4998 (506) | 3104 | 616 | 0.20 (0.13) | 0.22 (0.17) |
| DRD2 | 106,341 (43,493) | 98,282 | 19,570 | 0.16 (0.14) | 0.16 (0.14) |
| EGFR | 11,364 (2134) | 5932 | 1184 | 0.21 (0.20) | 0.21 (0.20) |
| NR1H2 | 2712 (821) | 1956 | 382 | 0.19 (0.16) | 0.20 (0.16) |
Number of compounds and clusters (in brackets).
Mean Tanimoto coefficient of paired compounds in the subset having similar or dissimilar (in brackets) bioactivity.
Figure 2.
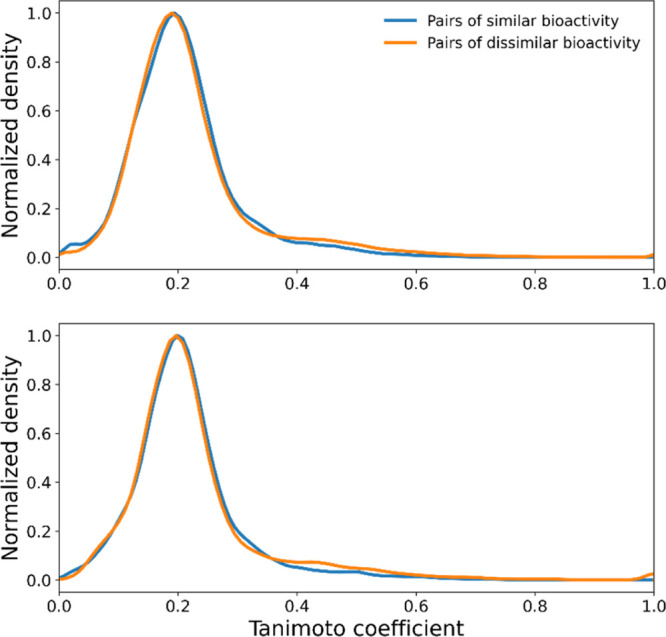
Distribution of Tanimoto coefficients of paired compounds from the dataset NR1H2. (Top) the training set and (bottom) the validation set.
Data Augmentation
With the aim to gain computational efficiency by training the deep model SiameseCHEM on a subset which is balanced against different activity classes, the impact of data augmentation was further evaluated. Both pair generation and randomizing SMILES data amplification strategies were pursued. Augmentation by additional unsampled pairs led to a slight decrease in performance for DRD2 and a marginal increase for both CCR5 and NR1H2 (Figure 3). Augmentation by random SMILES strings improved performance on four datasets, namely, CCR5, DRD2, EGFR, and NR1H2. The use of random SMILES strings presumably mitigates the risk for the model to capture casual correlations between the token-order and similarity labels. The combination of two approaches is comparable to the use of random SMILES strings alone. Following the finding here, subsequent models were trained with data augmentation by random SMILES strings.
Figure 3.
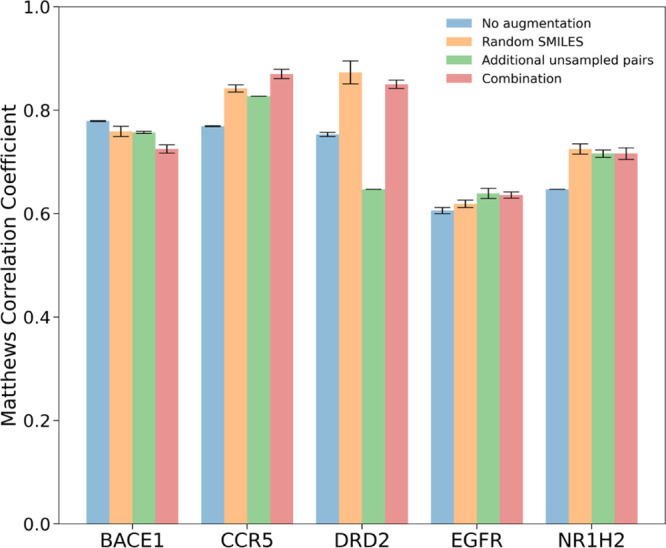
Performance of the deep model SiameseCHEM on the validation set with data augmentation. The coefficients are the mean value from the last five epochs, and the error bar depicts the standard deviation.
Effect of Thresholds
Table 2 shows the performance of the deep model on the validation set in terms of MCC, recall, and precision using a threshold of 5 or 7 in pXC50. The performance drops significantly with a threshold of 7. The reasons are two-fold. First, there are few active compounds with a threshold of 7, and the chemical diversity is underrepresented in the training set. Second, pairs are more biased toward inactive compounds which are predominant. However, both recall and precision are generally acceptable, showcasing the good discriminative power of the deep model. Notably, the decrease in performance with a threshold of 7 was more pronounced with the two baseline machine learning methods (Table 3).
Table 2. Performance on the Validation Set with a Threshold of 5 or 7 in pXC50a.
| dataset | threshold | MCC | recall | precision |
|---|---|---|---|---|
| BACE1 | 5 | 0.77 | 0.92 | 0.84 |
| 7 | 0.55 | 0.61 | 0.87 | |
| CCR5 | 5 | 0.83 | 0.93 | 0.91 |
| 7 | 0.61 | 0.83 | 0.75 | |
| DRD2 | 5 | 0.78 | 0.94 | 0.84 |
| 7 | 0.56 | 0.90 | 0.59 | |
| EGFR | 5 | 0.67 | 0.82 | 0.86 |
| 7 | 0.38 | 0.48 | 0.79 | |
| NR1H2 | 5 | 0.74 | 0.89 | 0.84 |
| 7 | 0.43 | 0.79 | 0.54 |
The numbers are the mean values from the last five epochs.
Table 3. Performance Comparison with Baseline Methods on the Validation Seta.
| dataset | threshold | SiameseCHEM | MLP (ECFP6) | RF (SMILES) | RF (ECFP6) | SVM (SMILES) | SVM (ECFP6) |
|---|---|---|---|---|---|---|---|
| BACE1 | 5 | 0.77 | 0.73 | 0.20 | 0.60 | –0.13 | 0.09 |
| 7 | 0.55 | 0.44 | –0.10 | 0.11 | –0..9 | –0.06 | |
| CCR5 | 5 | 0.83 | 0.83 | 0.51 | 0.80 | –0.13 | 0.49 |
| 7 | 0.61 | 0.52 | 0.07 | 0.34 | –0.03 | –0.01 | |
| DRD2 | 5 | 0.78 | 0.23 | –0.10 | –0.13 | –0.04 | –0.03 |
| 7 | 0.56 | –0.01 | –0.12 | –0.08 | –0.02 | –0.11 | |
| EGFR | 5 | 0.67 | 0.71 | 0.41 | 0.67 | 0.25 | 0.52 |
| 7 | 0.38 | 0.022 | –0.06 | 0.06 | –0.04 | –0.06 | |
| NR1H2 | 5 | 0.74 | 0.60 | 0.14 | 0.55 | –0.15 | –0.05 |
| 7 | 0.43 | 0.04 | 0.0 | –0.10 | 0.0 | 0.0 |
MCCs averaged from the 10-fold stratified cross-validation.
Comparison with Baseline Models
To benchmark the deep model, two popular machine learning methods,8,33 RF and SVM, were implemented with scikit-learn. The two methods were trained on the same training sets for the binary classification but taking a single compound as input using either the ECFP6 fingerprints or the tokenized SMILES strings. Each compound in a pair from the validation set was then fed into the baseline models separately. The similarity label of a pair is determined by the predicted activity class of each individual compound. To have a robust comparison on performance, the 10-fold stratified cross-validation was performed on all models, repartitioning the training and validation sets with a 80:20 split. The results are summarized in Table 3.
The deep model SiameseCHEM shows a good performance with MCC greater than 0.65 across all five datasets for the threshold of 5. Notably, the deep model achieves better performances on BACE1, DRD2, and NR1H2 (p-value < 0.05) than the RF model which uses the ECFP6 fingerprints. For CCR5 and EGFR, it is not significantly different from the RF. The performance drops with the threshold of 7. However, the decrease in performance is even more prominent for the two baseline methods. They also have bigger variance across the datasets. The ECFP6 fingerprints are descriptors of atom-centric substructures and rich in chemistry information.37 Interestingly, the inferior performance of both baseline models which use the tokenized SMILES strings highlights that SMILES strings themselves are not discriminative of biological similarity. Hence, the deep model proves to be capable of learning relevant chemical features rather than making casual correlations. In addition, we compared to a Siamese MLP model which used the ECFP6 fingerprints as the input. The overall performance of MLP is comparable to that of SiameseCHEM, except on the DRD2 dataset (Table 3), which suggests the potential limitations of using the fixed molecular descriptors. The finding corroborates the competitive performance of deep learning-based feature representations.38−41 Alternative deep learning models, such as variational autoencoders,42 transformer,43 and seq2seq,44 therefore could be implemented within the Siamese framework to extract task-specific features.
N-Shot Learning
The SiameseCHEM deep model was retrained on a merged set consisting of both the training and the validation set to make full use of available compounds with known activity and then evaluated on the test set. A maximum of 5000 instances were sampled randomly without replacement per dataset from the respective original test set for evaluation of the model performance. The effect of the number of reference compounds (N) in a support set on inference of the activity label was first investigated (Figure 4). The predictive power of the deep model increases progressively with the increase in the number of reference compounds with which the query compound is paired. Concomitantly, the false positive rate declines gradually, enabling a more accurate discern of true active compounds. There is a big jump on performance (p-value < 0.05) from using two reference compounds which is essentially one-shot learning to four compounds. The performance reaches the plateau with 32 reference compounds for BACE1, DRD2, and EGFR, 16 for CCR5, and 8 for NR1H2. The number of reference compounds per dataset appears to correlate with the data size, suggesting that more diverse compound collections would require more reference compounds to be compared with. However, the number of reference compounds required is rather small in comparison with the number of clusters per dataset (Table 1), highlighting the unique merit of the Siamese neural network to cope with the scarce data and its strong discriminative power. Noteworthily, the one-shot learning (N = 2) achieves an acceptable performance on the three datasets of BACE1, DRD2, and NR1H2 with a false positive rate of 20% on average. In addition, the same procedure can be iterated k-times each with a different support set, and the improvement on performance is marginal with iterations (Table S2), presumably because the number of reference compounds has been optimized and big enough for reliable inference.
Figure 4.

Performance of binary classification via N-shot learning with regard to the number of reference compounds in the support set (N). The error bars indicate the 95% CI bounds, evaluated by the 10 repeated predictions each with a different support set.
The categorical classification was further explored using two deep models, each trained with a threshold of 5 or 7 in pXC50, respectively. The Cohen’s weighted kappa (κ) indicates a good agreement on the three datasets of BACE1, CCR5, and DRD2, as well as a modest agreement on the other two datasets of EGFR and NR1H2. The Kendall’s correlation coefficients (τ) are consistent with the Cohen’s weighted kappa, revealing a similar trend (Table 4).
Table 4. Performance of the Categorical Classification via N-Shot Learning.
| dataset | Na | κb | τc |
|---|---|---|---|
| BACE1 | 32 | 0.69 | 0.74 |
| CCR5 | 16 | 0.68 | 0.73 |
| DRD2 | 32 | 0.79 | 0.83 |
| EGFR | 32 | 0.56 | 0.61 |
| NR1H2 | 8 | 0.57 | 0.65 |
The number of reference compounds in the support set.
Cohen’s weighted kappa (κ) measures the degree of absolute agreement between the ground truth and predictions, with the value ranging from −1 to 1. It treats all misclassifications equally.
Kendall’s correlation coefficient (τ) measures the ordinal association between the ground truth and predictions, with the value ranging from −1 to 1. It penalizes ordinal misclassification more heavily than the kappa statistics.
Nonadditivity Analysis
Nonadditivity analysis studies whether the same transformations between related molecules have the same effect by assessing the experimental uncertainties, and strong nonadditivity is indicative of potential QSAR outliers.45 The results of nonadditivity analysis are summarized in Table 5 and illustrated by Figure 5. The estimated experimental uncertainty ranges from 0.10 for DRD2 to 0.58 for NR1H2. The three datasets of BACE1, EGFR, and NR1H2 have the estimated uncertainty around 0.5 log unit, slightly higher than 0.3 from the in-house homogeneous data. The percentage compounds outside the 95% confidence interval (CI) ranges from 0.17% for DRD2 to 6.71% for BACE1. The outliers from the nonadditivity analysis represent less than 5% the pairs which were wrongly labelled by the deep model. Overall, there is lack of an obvious correlation between the deep model performance and severity of nonadditivity. While nonadditivity may have a big impact on regressions, it could have little impact on classifications.
Table 5. Nonadditivity Analysis of the Five Datasets.
| nonadditivity metrics | BACE1 | CCR5 | DRD2 | EGFR | NR1H2 |
|---|---|---|---|---|---|
| estimated uncertainty | 0.55 | 0.28 | 0.10 | 0.48 | 0.58 |
| % Cpds outside 95% CI | 6.71 | 6.66 | 0.17 | 4.81 | 2.77 |
| mispredictionsa | 493 | 102 | 441 | 265 | 82 |
| mispredictions with an outlierb | 23 | 2 | 0 | 10 | 1 |
Number of total pairs whose similarity labels were wrongly predicted by the deep model SiameseCHEM.
Number of wrongly predicted pairs having at least one compound outside the 95% CI from the nonadditivity analysis.
Figure 5.
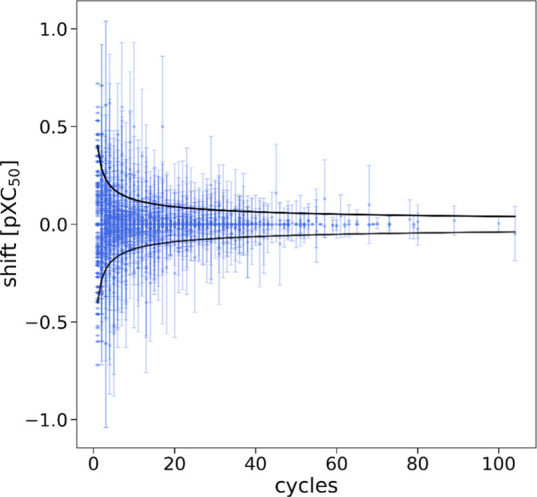
Additivity shift per compound for the DRD2 dataset for illustration of the results summarized in Table 5. Shown is the average additivity shift per compound and the standard deviation of the shift. Black lines indicate the 95% CI for a perfectly additive dataset with an experimental uncertainty of σ = 0.1 log unit.
Conclusions
We have developed a deep model SiameseCHEM, which is a Siamese recurrent neural network based on the BiLSTM architecture with a self-attention mechanism. Trained on random SMILES strings, it is capable of classifying the bioactivity of small molecules via N-shot learning. It outperforms the baseline methods of RF and SVMs which use the precomputed chemistry-rich ECFP6 fingerprints and demonstrates that it learns task-specific chemical features encoded by the SMILES strings. It is shown to have the advantage of coping with data paucity and class imbalance, two prevailing challenges for QSAR modelling in drug discovery. The current study constitutes a stepping stone to use the Siamese neural network for regression and may open a new avenue for further exploration in QSAR. The source code is publicly available at https://github.com/MolecularAI/Siamese-RNN-Self-Attention.
Acknowledgments
The authors would like to thank Panagiotis-Christos Kotsias, Huifang You, Jiazhen He, Dean Sumner, Atanas Patronov, Simon Johansson, and Josep Arús-Pous from the Molecular AI group and Vignesh Subramanian from the Computational Chemistry group at AstraZeneca for the fruitful discussions on coding and chemoinformatic topics.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c01266.
Mathematical details of the Siamese neural network; preparation of the datasets; MCC scores and false positive rates in N-shot learning strategies for inference repetition; and distribution of the cluster size, actives/inactives, and Tanimoto coefficients for each dataset (PDF)
The authors declare the following competing financial interest(s): DF, EN, HZ and CT are employees of AstraZeneca and own stock options.
Supplementary Material
References
- Wang L.; Wu Y.; Deng Y.; Kim B.; Pierce L.; Krilov G.; Lupyan D.; Robinson S.; Dahlgren M. K.; Greenwood J.; Romero D. L.; Masse C.; Knight J. L.; Steinbrecher T.; Beuming T.; Damm W.; Harder E.; Sherman W.; Brewer M.; Wester R.; Murcko M.; Frye L.; Farid R.; Lin T.; Mobley D. L.; Jorgensen W. L.; Berne B. J.; Friesner R. A.; Abel R. Accurate and Reliable Prediction of Relative Ligand Binding Potency in Prospective Drug Discovery by Way of a Modern Free-Energy Calculation Protocol and Force Field. J. Am. Chem. Soc. 2015, 137, 2695–2703. 10.1021/ja512751q. [DOI] [PubMed] [Google Scholar]
- Cournia Z.; Allen B.; Sherman W. Relative Binding Free Energy Calculations in Drug Discovery: Recent Advances and Practical Considerations. J. Chem. Inf. Model. 2017, 57, 2911–2937. 10.1021/acs.jcim.7b00564. [DOI] [PubMed] [Google Scholar]
- Nantasenamat C.; Isarankura-Na-Ayudhya C.; Prachayasittikul V. Advances in Computational Methods to Predict the Biological Activity of Compounds. Expet Opin. Drug Discov. 2010, 5, 633–654. 10.1517/17460441.2010.492827. [DOI] [PubMed] [Google Scholar]
- Kearnes S.; McCloskey K.; Berndl M.; Pande V.; Riley P. Molecular Graph Convolutions: Moving beyond Fingerprints. J. Comput. Aided Mol. Des. 2016, 30, 595–608. 10.1007/s10822-016-9938-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schütt K. T.; Arbabzadah F.; Chmiela K. R. M. S.; Tkatchenko A. Quantum-Chemical Insights from Deep Tensor Neural Networks. Nat. Commun. 2017, 8, 13890. 10.1038/ncomms13890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coley C. W.; Barzilay R.; Green W. H.; Jaakkola T. S.; Jensen K. F. Convolutional Embedding of Attributed Molecular Graphs for Physical Property Prediction. J. Chem. Inf. Model. 2017, 57, 1757. 10.1021/acs.jcim.6b00601. [DOI] [PubMed] [Google Scholar]
- Wu Z.; Ramsundar B.; Feinberg E. N.; Gomes J.; Geniesse C.; Pappu A. S.; Leswing K.; Pande V. MoleculeNet: A Benchmark for Molecular Machine Learning. Chem. Sci. 2018, 9, 513–530. 10.1039/c7sc02664a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K.; Swanson K.; Jin W.; Coley C.; Eiden P.; Gao H.; Guzman-Perez A.; Hopper T.; Kelley B.; Mathea M.; Palmer A.; Settels V.; Jaakkola T.; Jensen K.; Barzilay R. Analyzing Learned Molecular Representations for Property Prediction. J. Chem. Inf. Model. 2019, 59, 3370–3388. 10.1021/acs.jcim.9b00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon M.; Park D.; Lee J.; Jeon H.; Ko M.; Kim S.; Choi Y.; Tan A.-C.; Kang J. ReSimNet: Drug Response Similarity Prediction Using Siamese Neural Networks. Bioinformatics 2019, 35, 5249–5256. 10.1093/bioinformatics/btz411. [DOI] [PubMed] [Google Scholar]
- Jiménez-Luna J.; Pérez-Benito L.; Martínez-Rosell G.; Sciabola S.; Torella R.; Tresadern G.; De Fabritiis G. DeltaDelta Neural Networks for Lead Optimization of Small Molecule Potency. Chem. Sci. 2019, 10, 10911–10918. 10.1039/c9sc04606b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altae-Tran H.; Ramsundar B.; Pappu A. S.; Pande V. Low Data Drug Discovery with One-Shot Learning. ACS Cent. Sci. 2017, 3, 283–293. 10.1021/acscentsci.6b00367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallari R.; Swearingen E.; Liu W.; Ow A.; Young S. W.; Huang S.-G. A Generic High-Throughput Screening Assay for Kinases: Protein Kinase a as an Example. J. Biomol. Screen 2003, 8, 198–204. 10.1177/1087057103252306. [DOI] [PubMed] [Google Scholar]
- Koch G.; Zemel R.; Salakhutdinov R.. Siamese Neural Networks for One-Shot Image Recognition; University of Toronto, 2015. [Google Scholar]
- Pennington, Jeffrey Socher R.; Manning C.. GloVe: Global Vectors for Word Representation. Proceedings of the 2014 Conference on Empirical Methods in Natural Language Processing (EMNLP); Association for Computational Linguistics: Doha,Qatar, 2014; pp 1532–1543.
- Mikolov T.; Chen K.; Corrado G.; Dean J.. Efficient Estimation of Word Representations in Vector Space. International Conference on Intelligent Text Processing and Computational Linguistics; ICLR, 2013.
- Hochreiter S.; Schmidhuber J. Long Short-Term Memory. Neural Comput. 1997, 9, 1735–1780. 10.1162/neco.1997.9.8.1735. [DOI] [PubMed] [Google Scholar]
- Zheng S.; Yan X.; Yang Y.; Xu J. Identifying Structure-Property Relationships through SMILES Syntax Analysis with Self-Attention Mechanism. J. Chem. Inf. Model. 2019, 59, 914–923. 10.1021/acs.jcim.8b00803. [DOI] [PubMed] [Google Scholar]
- He K.; Zhang X.; Ren S.; Sun J.. Delving Deep into Rectifiers: Surpassing Human-Level Performance on ImageNet Classification. Proceedings of the IEEE International Conference on Computer Vision; ICCV, 2015.
- Saxe A. M.; McClelland J. L.; Ganguli S.. Exact Solutions to the Nonlinear Dynamics of Learning in Deep Linear Neural Networks. 2nd International Conference on Learning Representations, ICLR 2014—Conference Track Proceedings; ICLR, 2014; pp 1–15.
- Chen P.; Chen G.; Zhang S.. Log Hyperbolic Cosine Loss Improves Variational Auto-Encoder. ICLR 2019 Conference Blind Submission; ICLR, 2019; pp 1–15.
- Hadsell R.; Chopra S.; LeCun Y.. Dimensionality Reduction by Learning an Invariant Mapping. Proceedings of the IEEE Computer Society Conference on Computer Vision and Pattern Recognition; CVPR, 2006; Vol 2, pp 1735–1742.
- Huber P. J. Robust Estimation of a Location Parameter. Ann. Math. Stat. 1964, 35, 73–101. 10.1214/aoms/1177703732. [DOI] [Google Scholar]
- Schroff F.; Kalenichenko D.; Philbin J.. FaceNet: A Unified Embedding for Face Recognition and Clustering. Proceedings of the IEEE Computer Society Conference on Computer Vision and Pattern Recognition; CVPR, 2015; pp 815–823.
- Kingma D. P.; Ba J. L.. Adam: A Method for Stochastic Optimization. 3nd International Conference on Learning Representations, ICLR 2014—Conference Track Proceedings; ICLR, 2015; pp 1–15.
- Pascanu R.; Mikolov T.; Bengio Y.. On the Difficulty of Training Recurrent Neural Networks. 30th International Conference on Machine Learning, ICML; International Machine Learning Society (IMLS), 2013; pp 2347–2355.
- ExCAPE-DB . ExCAPE Chemogenomics Database. https://solr.ideaconsult.net/search/excape/ (accessed Feb 10, 2019).
- Gaulton A.; Hersey A.; Nowotka M.; Bento A. P.; Chambers J.; Mendez D.; Mutowo P.; Atkinson F.; Bellis L. J.; Cibrián-Uhalte E.; Davies M.; Dedman N.; Karlsson A.; Magariños M. P.; Overington J. P.; Papadatos G.; Smit I.; Leach A. R. The ChEMBL Database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. 10.1093/nar/gkw1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard P.; Victorri B.; LeCun Y.; Denker J.. Tangent Prop - A Formalism for Specifying Selected Invariances in an Adaptive Network. Advances in Neural Information Processing Systems; Moody J., Hanson S., Lippmann R. P., Eds.; Morgan-Kaufmann, 1992; Vol. 4. [Google Scholar]
- Arús-Pous J.; Johansson S. V.; Prykhodko O.; Bjerrum E. J.; Tyrchan C.; Reymond J. L.; Chen H.; Engkvist O. Randomized SMILES Strings Improve the Quality of Molecular Generative Models. J. Cheminf. 2019, 11, 1–13. 10.1186/s13321-019-0393-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjerrum E. J.SMILES Enumeration as Data Augmentation for Neural Network Modeling of Molecules. 2017, arXiv preprint arXiv:1703.07076. [Google Scholar]
- Tetko I. V.; Karpov P.; Bruno E.; Kimber T. B.; Godin G.. Augmentation Is What You Need!. Artificial Neural Networks and Machine Learning—ICANN 2019: Workshop and Special Sessions; Springer International Publishing: Cham, 2019; pp 831–835.
- O’Boyle N. M. Towards a Universal SMILES Representation - A Standard Method to Generate Canonical SMILES Based on the InChI. J. Cheminf. 2012, 4, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D.; Wu Z.; Hsieh C.-Y.; Chen G.; Liao B.; Wang Z.; Shen C.; Cao D.; Wu J.; Hou T. Could Graph Neural Networks Learn Better Molecular Representation for Drug Discovery? A Comparison Study of Descriptor-Based and Graph-Based Models. J. Cheminf. 2021, 13, 12. 10.1186/s13321-020-00479-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chicco D.; Jurman G. The Advantages of the Matthews Correlation Coefficient (MCC) over F1 Score and Accuracy in Binary Classification Evaluation. BMC Genom. 2020, 21, 1–13. 10.1186/s12864-019-6413-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butina D. Unsupervised Data Base Clustering Based on Daylight’s Fingerprint and Tanimoto Similarity: A Fast and Automated Way to Cluster Small and Large Data Sets. J. Chem. Inf. Comput. Sci. 1999, 39, 747–750. 10.1021/ci9803381. [DOI] [Google Scholar]
- Lounkine E.; Keiser M. J.; Whitebread S.; Mikhailov D.; Hamon J.; Jenkins J. L.; Lavan P.; Weber E.; Doak A. K.; Côté S.; Shoichet B. K.; Urban L. Large-Scale Prediction and Testing of Drug Activity on Side-Effect Targets. Nature 2012, 486, 361–367. 10.1038/nature11159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers D.; Hahn M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- He J.; You H.; Sandström E.; Nittinger E.; Bjerrum E. J.; Tyrchan C.; Czechtizky W.; Engkvist O. Molecular Optimization by Capturing Chemist’s Intuition Using Deep Neural Networks. ChemRxiv 2021, 13, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao K.; Nguyen D. D.; Tu M.; Wei G.-W. Generative Network Complex for the Automated Generation of Drug-like Molecules. J. Chem. Inf. Model. 2020, 60, 5682–5698. 10.1021/acs.jcim.0c00599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter R.; Montanari F.; Noé F.; Clevert D.-A. Learning Continuous and Data-Driven Molecular Descriptors by Translating Equivalent Chemical Representations. Chem. Sci. 2019, 10, 1692–1701. 10.1039/c8sc04175j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Bombarelli R.; Wei J. N.; Duvenaud D.; Hernández-Lobato J. M.; Sánchez-Lengeling B.; Sheberla D.; Aguilera-Iparraguirre J.; Hirzel T. D.; Adams R. P.; Aspuru-Guzik A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci. 2018, 4, 268–276. 10.1021/acscentsci.7b00572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaswani A.; Shazeer N.; Parmar N.; Uszkoreit J.; Jones L.; Gomez A. N.; Kaiser Ł.; Polosukhin I.. Attention Is All You Need. Advances in Neural Information Processing Systems; Guyon I., Luxburg U. V., Bengio S., Wallach H., Fergus R., Vishwanathan S., Garnett R., Eds.; Curran Associates, Inc., 2017; Vol. 30. [Google Scholar]
- Vaswani A.; Shazeer N.; Parmar N.; Uszkoreit J.; Jones L.; Gomez A. N.; Kaiser L.; Polosukhin I.. Attention Is All You Need. Advances in Neural Information Processing Systems; Curran Associates, Inc., 2017; pp 6000–6010. [Google Scholar]
- Sutskever I.; Vinyals O.; Le Q. V. Sequence to Sequence Learning with Neural Networks. Adv. Neural Inf. Process. Syst. 2014, 4, 3104–3112. [Google Scholar]
- Kramer C. Nonadditivity Analysis. J. Chem. Inf. Model. 2019, 59, 4034–4042. 10.1021/acs.jcim.9b00631. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.