

Abstract

We have developed an efficient methodology for the synthesis of (2R,3S,4R)-2-hydroxymethyl-3,4-dihydroxy-6-aryl-7-aroylchromanes in which the chirality at the C-2, C-3, and C-4 positions is being drawn from C-glucopyranosyl aldehyde, which in turn can be efficiently synthesized from d-glucose. Thus, the synthesis starts with the transformation of sugar aldehyde into 1-(E-1-arylpropenon-3-yl)-3,4,6-tri-O-benzyl-d-glucals using Claisen–Schmidt type condensation reaction with different acetophenones and then to 1,2-disubstituted glucals via Pd(II)-catalyzed cross dehydrogenative coupling reaction, which in turn has been efficiently converted into (2R,3S,4R)-chromanes via 6π-electrocyclization and in situ dehydrogenative aromatization.
Introduction
Chromane scaffolds are important structural units often found in many natural products and bioactive compounds that exhibit anticancer,1 anti-HIV,2 antiplasmodial,3 antitubercular,4 antibacterial, and antifungal activities.5 The chromane core makes the structural framework of complex compounds, including constituents of vitamin E, catechin, mucroquinone, equol, hematoxylin, brazilin, and other pharmaceutical drugs, such as symakalin, ormeloxifene, cromakalim, sideroxylonal A, procyanidin B3, etc. (Figure 1).6
Figure 1.

Examples of natural products and bioactive compounds with the chiral chromane-core.
The biological importance of chiral chromanes accelerated the development of efficient methods for their synthesis. Among many reported strategies, the reaction of salicylaldehyde and enolates or their equivalents from acetophenones has gained prominent attention.7 In most of the reported methods, the oxygenated heterocyclic ring of chromane has been constructed on the prefunctionalized aromatic ring systems to get such structural motifs, which generated different levels of complexity in bringing defined chirality at the C-2, C-3, or C-4 position of chromanes.8 Due to these prefunctionalization issues, utility of Pd-catalyzed direct C–C bond formation via cross dehydrogenative coupling (CDC) reaction at the C-1 or C-2 position of glycals has emerged as an excellent tool with an advantage of higher atom economy and fewer steps than conventional synthesis.9
The arduous task of constructing an aromatic system onto the glycopyranose ring to synthesize chiral chromanes was first addressed by Werz and co-workers10 using functionalized 2-bromoglycals. Recently, we have reported the synthesis of tetrasubstituted 2R,3S-chromane from C-1-substituted glucal diene using Pd(II)-catalyzed oxidative cross coupling reaction of different alkenes followed by thermal oxidative electrocyclization.11 Herein, we report a better divergent route for the synthesis of 1-(E-1-arylpropenon-3-yl)-3,4,6-tri-O-benzyl-d-glucals and their transformation into pentasubstituted (2R,3S,4R)-chromanes via Pd(II)-catalyzed cross dehydrogenative coupling reaction with various alkenes followed by 6π-electrocyclization and in situ dehydrogenative aromatization.
Results and Discussion
The precursor C-glucopyranosyl aldehyde 1 for the synthesis of stereochemically defined chromanes was synthesized from d-glucose following a literature procedure.12 It was envisioned that Claisen–Schmidt condensation of C-glucosyl aldehyde with acetophenones in the presence of base should led to the formation of chalcone-type of compounds, i.e., 1-(E-1-arylpropenon-3-yl)-3,4,6-tri-O-benzyl-d-glucals. With this aim, condensation of sugar aldehyde 1 with 4-methyl acetophenone (2a) was carried out in the presence of DBU and NaOMe as a base in EtOH and MeOH, respectively, which led to the formation of a mixture of glucal propenone 3a and glucal aldehyde 4 in different ratios with later as the dominant product (Scheme 1, Table 1, entries 1 and 2).
Scheme 1. Synthesis of C-1-Functionalized Unsaturated Sugar Derivatives 3a–j.

Table 1. Condensation of 4-Methyl Acetophenone (2a) with Sugar Aldehyde 1 in Various Solvents at 25 °C in the Presence of Different Bases and Reaction Times Ranging from 2 to 24 ha.
| entry | solventb | base | reaction time (h) | glucalpropenone 3a (% yield)c | glucal aldehyde 4 (% yield)c |
|---|---|---|---|---|---|
| 1 | EtOH | DBU‡ | 6 | 25 | 50 |
| 2 | MeOH | NaOMe‡ | 3 | 30 | 50 |
| 3 | DCM | Pyrolidine‡ | 3 | 0 | 75 |
| 4 | EtOH | Piperidine‡ | 24 | 0 | 75 |
| 5 | MeOH | NEt3-proline‡ (1:1) | 24 | 0 | 78 |
| 6 | EtOH | Ba(OH)2¶ | 8 | 65 | 0 |
| 7 | EtOH | KOH¶ | 2 | 75 | 0 |
| 8 | EtOH | LiOH¶ | 3 | 70 | 0 |
| 9 | EtOH | NaOH¶ | 2 | 85 | 0 |
| 10 | EtOH/H2O (1:1) | NaOAc‡ | 24 | NR | NR |
| 11 | EtOH | NaOH¶ | 2 | 92d |
Reaction conditions: Compound 1 (0.181 mmol), 2a (0.181 mmol); ‡base (1 equiv.); ¶5% aq. base (1 mL); NR = No reaction.
Solvent used (1 mL).
Isolated yield.
Reaction performed without 2a.
Further, the use of organic bases such as pyrrolidine, piperidine, or NEt3-proline (1:1) in DCM, EtOH, or MeOH, respectively, for condensation of compounds 1 and 2a led to the exclusive formation of glucal aldehyde 4 in 75 to 78% yields (Table 1, entries 3–5).13 However, the use of Ba(OH)2, KOH, LiOH, and NaOH in EtOH led to the exclusive formation of glucal propenone 3a in 65, 75, 70, and 85% yields, respectively (Table 1, entries 6–9). The use of NaOAc in EtOH/H2O (1:1) for the condensation reaction did not yield any product (Table 1, entries 10). The reaction carried out under the conditions of entry 9, but in the absence of acetophenone, (2a) led to the formation of glucal aldehyde 4 only in 92% yield (Table 1, entry 11). The analysis of results of these experiments showed that 5% aq. NaOH in EtOH is the most suitable and highest yielding base for synthesizing compounds 3a and also glucal aldehyde 4 in the absence of 4-methyl acetophenone (2a) (Table 1, entries 9 and 11). The optimized conditions were used for the synthesis of C-1-functionalized unsaturated sugar derivatives 3a–j by condensation of sugar aldehyde 1 with aryl methyl ketones 2a–j in 68–88% yields (Scheme 1).
A representative mechanism for the synthesis of glucalpropenone 3a from the condensation of C-glucosyl aldehyde 1 and acetophenone (2a) is shown in Scheme 2. The formation of product 3a′ without a double bond in the sugar ring as shown in the bracket in Scheme 2 was not observed in any of the reaction conditions mentioned in Table 1.
Scheme 2. Proposed Mechanism for the Formation of Glucalpropenone 3a from Compounds 1 and 2a.

The formation of a mixture of glucalpropenone 3a and glucal aldehyde 4 in condensation reaction in the presence of DBU in EtOH and NaOMe in MeOH (Table 1, entries 1 and 2), the exclusive formation of glucal aldehyde 4 in reactions at entries 3–5 in Table 1, and exclusive formation of compound 3a in reactions at entries 6–9 in Table 1 have been observed. These results indicate that C-glucosyl aldehyde 1 first gets converted into glucal aldehyde 4, which then condenses with the enolate formed from acetophenone to yield compound 3a (Scheme 2). This is also supported by the HRMS (ESI) data analysis of the reaction mixture after time intervals of 10, 30, and 45 min of the addition of aq. NaOH to the mixture of compounds 1 and 2a, which showed peaks at m/z [M + Na]+ for aldehyde 1 (calcd 575.2404; found 575.2402), glucalpropenone 3a (calcd 583.2455; found 583.2461), and glucal aldehyde 4 (calcd 467.1829; found 467.1820), but the corresponding peak for saturated sugar propenone 3a′, which should appear at m/z 691.3030, was not observed. It was also demonstrated that glucalpropenone 3a can also be prepared from condensation of glucal aldehyde 4 and 4-methyl acetophenone (2a) in EtOH in the presence of 5% aq. NaOH in 90% yield.
Three C-1-substituted glucalpropenones 3a–c out of the 10 compounds 3a–j synthesized by Claisen–Schmidt type condensation of sugar aldehyde 1 and aryl methyl ketones 2a–c were converted into 1,2-disubstituted glucals 6a–i by their Pd(II)-catalyzed cross dehydrogenative coupling (CDC) reaction with different terminal alkenes 5a–g. Initially, the Pd(II)-catalyzed CDC reaction for the synthesis of 1,2-disubstituted glucals was optimized by carrying out the reaction in the presence of different alkenes in a solvent or in a mixture of solvents. Thus, the reaction of glucalpropenone 3a and 4-methylstyrene (5a) in the presence of Pd(OAc)2 (10 mol %) and Cu(OAc)2, AgOAc, or AgOTf (2 equiv.) as an oxidant in a mixture of DMF/DMSO (9:1, v/v) at 80 °C led to the formation of the desired 1,2-disubstituted glucal 6a in 20, 50, and 40% yields, respectively (Table 2, entries 1–3).
Table 2. Optimization of Condition for CDC Reaction of C-1-glucalpropenone 3a with 4-Methylstyrene (5a) to Synthesize 1,2-Disubstituted Glucal 6a at 80 °C for 12 ha.

| entry | catalyst (10 mol %) | oxidant (equiv.) | co-oxidant (equiv.) | solvent | yield of 6a (%)b |
|---|---|---|---|---|---|
| 1 | Pd(OAc)2 | Cu(OAc)2 (2) | - | DMF/DMSO (9:1) | 20 |
| 2 | Pd(OAc)2 | AgOAc (2) | - | DMF/DMSO (9:1) | 50 |
| 3 | Pd(OAc)2 | AgOTf(2) | - | DMF/DMSO (9:1) | 40 |
| 4 | Pd(OAc)2 | Cu(OAc)2(1) | AgOAc (2) | DMF/DMSO (9:1) | 80 |
| 5 | Pd(OAc)2 | CuI (1) | AgOTf (2) | DMF/DMSO (9:1) | 85 |
| 6 | Pd(OAc)2 | Cu(OAc)2(1) | AgOAc (2) | THF | 50 |
| 7 | Pd(OAc)2 | Cu(OAc)2(1) | AgOAc (2) | DMF | 65 |
| 8 | Pd(OAc)2 | Cu(OAc)2(1) | AgOAc (2) | AcCN | 55 |
| 9 | Pd(OAc)2 | Cu(OAc)2(1) | AgOAc (2) | acetone | 60 |
| 10 | Pd(OAc)2 | Cu(OAc)2(1) | AgOAc (2) | DCE | 70 |
| 11 | Pd(OAc)2 | Cu(OAc)2(1) | AgOAc (2) | dioxane | NR |
| 12 | Pd(OAc)2 | Cu(OAc)2 (1) | AgOAc (2) | toluene | NR |
| 13 | PdCl2 | CuI (1) | AgOTf (2) | DMF/DMSO (9:1) | 40 |
| 14 | Pd(PPh3)2Cl2 | CuI (1) | AgOTf (2) | DMF/DMSO (9:1) | 50 |
Reaction conditions: Compound 3a (0.18 mmol), 5a (0.2 mmol); solvent used (2 mL); NR = No reaction.
Isolated yield.
Usage of a mixture, rather than lone oxidants, i.e., Cu(OAc)2 (1 equiv.)/AgOAc (2 equiv.) or CuI (1 equiv.)/AgOTf (2 equiv.), in a mixture of DMF/DMSO (9:1, v/v) increased the yields of formation of 6a to 80 and 85%, respectively (Table 2, entries 4–5). However, a change of the solvent system from DMF/DMSO to pure THF, DMF, acetonitrile, acetone, and dichloroethane resulted in lower yields, while the use of dioxane and toluene did not yield the desired product 6a at all (Table 2, entries 6–12). The change of Pd-salt from Pd(OAc)2 to PdCl2 or Pd(PPh3)2Cl2 also resulted in the lowering of yields to 40 and 50% (Table 2, entries 13–14).
The optimized conditions, i.e., the Pd(OAc)2-catalyst in the presence of a mixture of CuI (1 equiv.)/AgOTf (2 equiv.) as the oxidant in DMF/DMSO (9:1, v/v), were used for CDC reaction of glucalpropenone 3a with various styrenes and acrylates 5a–f, such as 4-methyl styrene (5a), styrene (5b), 4-nitrostyrene (5c), 2-chlorostyrene (5d), 2,2,2-trifluoroethylacrylate (5e), and benzyl acrylate (5f), to afford the corresponding 1,2-disubstituted glucals 6a–f with (E)-stereoselectivity in 78–88% yields. The broader substrate scope of the optimized reaction was further demonstrated by the successful CDC reaction of glucalpropenone 3b with styrene 5b and glucalpropenone 3c with 4-methyl styrene (5a) and 4-methoxy styrene (5g) to afford 1,2-disubstituted glucals 6g–i in 80 to 90% yields (Scheme 3).
Scheme 3. Synthesis of 1,2-Disubstituted Glucals 6a–i from Glucalpropenone 3a–c Using Pd(II)-Catalyzed CDC Reaction.

The CDC products 1,2-disubstituted glucals 6a–i containing an E,Z,E-triene system on 6π-electrocyclization in xylene in a sealed vessel at 160 °C followed by in situ dehydrogenative aromatization afforded (2R,3S,4R)-2-benzyloxymethyl-3,4-dibenzyloxy-6-aryl-7-aroyloxychromanes 7a–d and 7g–i in 66–73% yields. The electrocyclization of disubstituted glucals 6e and 6f bearing C-6 carbo(2,2,2-trifluoro)-ethoxy and carbo-benzyloxy substituents led to the decomposition of the starting material (Scheme 4). The electrocyclization reaction carried out in hexamethylphosphoramide (HMPA), ethylene glycol, or nitrobenzene at different temperatures either led to no reaction or decomposition of the starting material or formation of the product albeit in much lower yield. Further, an attempt to isolate the cyclohexadiene intermediate on electrocyclization of 1,2-disubstituted glucals by carrying out the reaction at a different temperature under a N2 atmosphere or by cooling the incomplete reaction mixture to −10 °C was unsuccessful.
Scheme 4. Synthesis of (2R,3S,4R)-Pentasubstituted Chromanes 8a–d and 8g–i from 1,2-Disubstituted Glucals 6a–i.
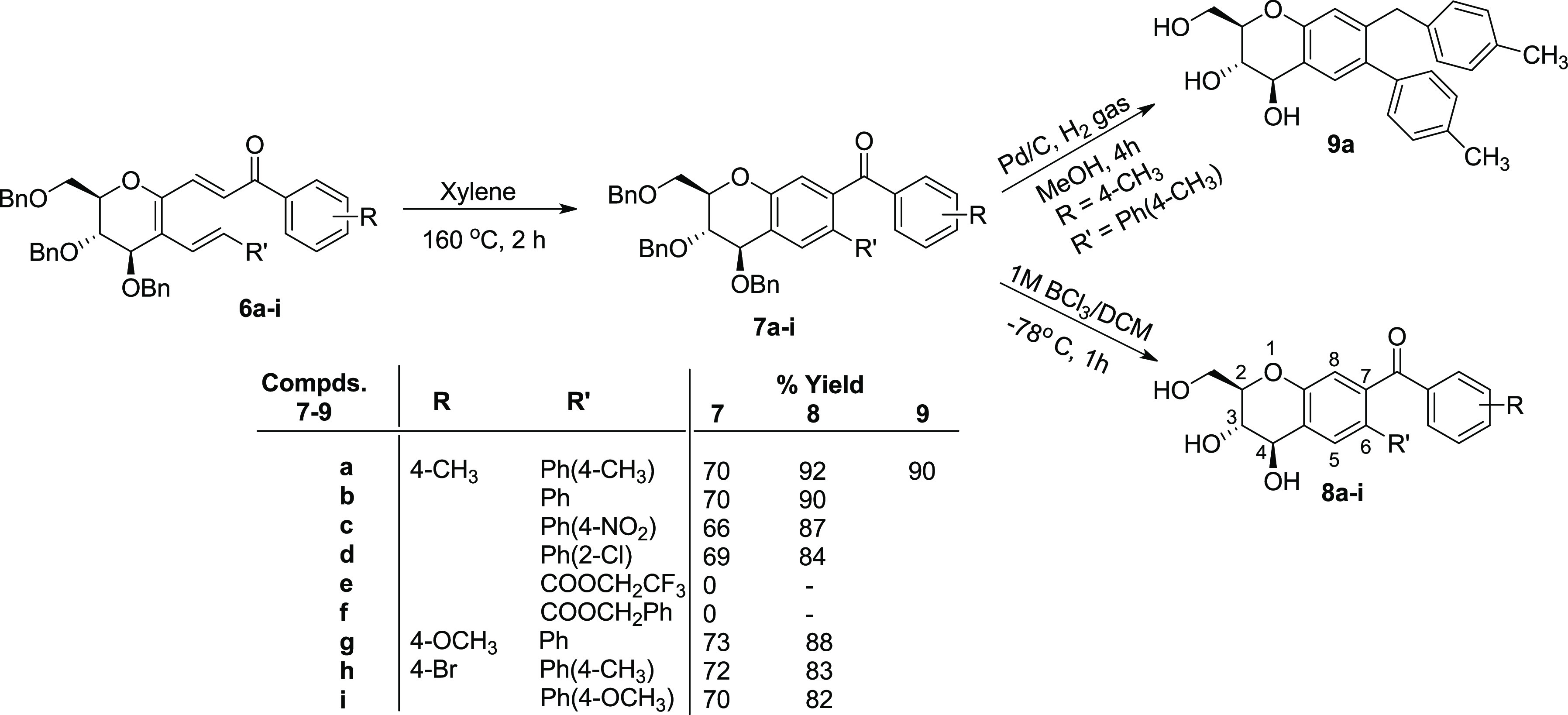
The debenzylation of compound 7a was initially tried by hydrogenation with 10% Pd/C-H2 in methanol at 25 °C. Although Pd/C-H2 in methanol efficiently affected complete debenzylation in compound 7a, it also led to the reduction of benzoyl to the benzyl group at the C-7 position of chromane to afford compound 9a.14 Finally, debenzylations of chromane 7a–d and 7g–i were affected with 1 M BCl3 in DCM at −78 °C to afford (2R,3S,4R)-2-hydroxymethyl-3,4-dihydroxy-6-aryl-7-aroyloxychromanes 8a–d and 8g–i in 82–92% yields (Scheme 4).15
A plausible mechanism for the formation of chromane 7via Pd-catalyzed CDC reaction of glucalpropenone 3 with styrene/acrylate 5 followed by 6π-electrocyclization reaction of the resulting 1,2-disubstituted glucal 6 starts with the heteroatom-directed electrophilic reaction of Pd(II) species at electron-rich C2-carbon of glucalpropenone 3. This follows hydrogen abstraction resulting in the formation of C2-palladized intermediate I, which on olefin coordination and carbopalladation afforded C2-alkyl-palladium intermediate II. Finally, β-hydride elimination from the second intermediate led to the formation of 1,2-disubstituted glucal 6. The Pd(0) generated after the reductive elimination step is regenerated to active Pd(II) species by CuI and AgOTf to maintain the continuity of the catalytic cycle. Further, 1,2-disubstituted glucal 6 on heating in xylene at 160 °C undergoes 6π-electrocyclization to an unstable cyclic diene intermediate III that spontaneously undergoes in situ dehydrogenative aromatization to afford chiral chromane 7 (Scheme 5).16
Scheme 5. Plausible Reaction Mechanism for Formation of 1,2-Disubstituted Glucal 6 and Its Conversion into (2R,3S,4R)-Pentasubstituted Chromane 7.

The structures of all synthesized compounds, i.e., 3a–j, 4, 6a–i, 7a–d, 7g–i, 8a–d, 8g–i, and 9a, were unambiguously established based on their IR, 1H-, 13C-, 19F NMR spectra and HRMS data analysis. The structure of known compound 4 was further confirmed by comparison of its spectral data with those reported in the literature.13a Further, the structure of compound 8a was unambiguously confirmed based on their X-ray data analysis (Figure 2, details in the Supporting Information).
Figure 2.

ORTEP diagram of compound 8a drawn in 50% thermal probability ellipsoids showing the atomic numbering scheme. Solvent molecules in the lattice are omitted for the sake of clarity. Only one molecule of the asymmetric unit has been shown.
Conclusions
We have described the efficient synthesis of C1-glucalpropenones in 68–88% yields and 1-formyl glucal in 92% yield. The glucalpropenones have been used as precursors for Pd(II)-catalyzed CDC reaction with styrenes and acrylates to synthesize 1,2-disubstituted glucals with (E)-stereoselectivity in 78–90% yields. Further, 1,2-disubstituted glucals have been subjected to 6π-electrocyclization on heating with xylene, which concomitantly affected in situ dehydrogenative aromatization to afford (2R,3S,4R)-2-benzyloxymethyl-3,4-dibenzyloxy-6-aryl-7-aroyloxychromanes in 66–73% yields. The debenzylation of synthesized chromanes has been achieved with boron trichloride in DCM to afford (2R,3S,4R)-trihydroxychromanes in 82–92% yields. C-7 Benzylchromane has also been synthesized by affecting debenzylation with Pd/C-H2 in methanol that led to the reduction of C-7 benzoyl to the benzyl group along with the removal of benzyl protection. The developed method is highly successful in generating a diversified library of chromanes with three inbuilt chiral centers derived from the precursor sugar. In addition, the developed methodology has the capability to generate diversity at both C6- and C7-positions of chromane. Although we have recently reported the synthesis of (2R,3S)-2-hydroxymethyl-3-hydroxychromanes from the sugar precursor, the advantage of the present synthesis is the import of three chiral centers from the sugar precursor into the chromane instead of only two and the possibility of generation of diversity at both C6- and C7-positions of chromane compared to only at the C6-position. Thus, the use of the present methodology for the synthesis of chromane shall generate a much larger library of structurally defined chromanes for drug discovery application and therefore is more useful.
Experimental Section
General
All commercially available reagents and absorbents were used without further purification. All solvents were distilled before use. The IR spectra were recorded on a PerkinElmer model 2000 FTIR spectrometer by making a KBr disk for solid samples. 1H-,13C-, and 19F- NMR spectra were recorded on JEOL Delta 400, 100.6, and 376 MHz spectrometers, respectively, using tetramethylsilane (TMS) as an internal standard. The chemical shift values are on the δ scale, and the coupling constant (J) are in hertz. HRMS recording was carried out using a Q-TOF mass spectrometer in ESI mode. The specific rotations of synthesized compounds were measured on a Rudolph autopol II automatic polarimeter using light of 589 nm wavelength. Analytical TLCs were performed on precoated fluorescent plates; visualization of the developed plates was performed under UV light or by charring with 5% alcoholic H2SO4 solution. Silica gel (100–200 mesh) was used for column chromatography.
General Procedure for Synthesis of 1-(E-1-Arylpropenon-3-yl)-3,4,6-tri-O-benzyl-d-glucals (3a–j)
To a solution of β-C-glucopyranosyl aldehyde 1 (600 mg, 1.09 mmol) and aryl methyl ketones 2a-j (1.09 mmol) in ethanol (12 mL), an aqueous solution of 5% NaOH (12 mL) was added dropwise with continuous stirring at 0 °C and further stirred for 2–6 h at 25 °C. After completion of the reaction as indicated by TLC examination, the reaction mixture was concentrated at reduced pressure keeping bath temperature below 40 °C and the thick liquid thus obtained was extracted with ethyl acetate (2 × 50 mL). The combined organic layer was dried over Na2SO4, filtered, and concentrated at reduced pressure to give the crude product, which was purified over a silica gel column with 5–10% ethyl acetate in petroleum ether as the eluent to afford pure products 3a–j in 68 to 88% yields.
1-[E-1-(4-Methylphenyl)propenon-3-yl]-3,4,6-tri-O-benzyl-d-glucal (3a)
It was obtained as a white solid (517 mg) in 85% yield; mp: 114–116 °C; IR (KBr, cm–1): 3030, 2922, 1665, 1626, 1607, 1297, 1098, 734, 697; [α]D27 = −67.0 (c 0.3, chloroform); 1H NMR (400 MHz, CDCl3): δ 2.42 (s, 3H), 3.83–3.96 (m, 3H), 4.19–4.23 (m, 1H), 4.31 (dd, 1H, J = 3.2 and 5.8 Hz), 4.58–4.71 (m, 5H), 4.84 (d, 1H, J = 11.3 Hz), 5.35 (d, 1H, J = 3.1 Hz), 7.07 (d, 1H, J = 15.1 Hz), 7.25–7.37 (m, 18H), 7.89 (d, 2H, J = 8.0 Hz); 13C{1H} NMR (100.6 MHz, CDCl3): δ 21.8, 68.4, 71.0, 73.5, 73.8, 74.1, 76.2, 77.3, 109.1, 123.0, 127.8, 127.9, 128.1, 128.6, 128.9, 129.4, 135.4, 137.6, 138.1, 138.2, 143.9, 150.7, 189.9; HRMS (ESI) m/z: [M + Na]+ calcd for C37H36NaO5 583.2455; found 583.2461.
1-[E-1-(4-Methoxyphenyl)propenon-3-yl]-3,4,6-tri-O-benzyl-d-glucal (3b)
It was obtained as a white solid (550 mg) in 88% yield; mp: 111–113 °C; IR (KBr, cm–1): 3031,1668, 1637, 1608, 1513, 1341, 1269, 1174, 1028, 733, 692; [α]D27 = −67.7 (c 0.3, chloroform); 1H NMR (400 MHz, CDCl3): δ 3.83–3.96 (m, 6H), 4.20–4.23 (m, 1H), 4.31 (dd, 1H, J = 3.1 and 5.6 Hz), 4.58–4.71 (m, 5H), 4.84 (d, 1H, J = 11.4 Hz), 5.35 (d, 1H, J = 3.0 Hz), 6.95 (d, 2H, J = 8.6 Hz), 7.07 (d, 1H, J = 15.2 Hz), 7.25–7.37 (m, 16H), 7.99 (d, 2H, J = 8.8 Hz); 13C{1H} (100.6 MHz, CDCl3): δ 55.6, 68.4, 71.0, 73.4, 73.8, 74.1, 76.2, 77.3, 108.9, 113.9, 122.9, 127.7, 127.9, 128.0, 128.5, 128.6, 130.9, 131.0, 137.3, 138.1, 138.2, 150.8, 163.6, 188.6; HRMS (ESI) m/z: [M + Na]+ calcd for C37H36NaO6 599.2404; found 599.2411.
1-[E-1-(4-Bromophenyl)propenon-3-yl]-3,4,6-tri-O-benzyl-d-glucal (3c)
It was obtained as a white solid (530 mg) in 78% yield; mp: 103–105 °C; IR (KBr, cm–1): 3028, 1627, 1599, 1541, 1395, 1299, 1069, 732, 694; [α]D27 = −59.6 (c 0.3, chloroform);1H NMR (400 MHz, CDCl3): δ 3.82–3.95 (m, 3H), 4.20–4.24 (m, 1H), 4.30 (dd, 1H, J = 3.2 and 5.4 Hz), 4.58–4.70 (m, 5H), 4.84 (d, 1H, J = 11.3 Hz), 5.38 (d, 1H, J = 3.3 Hz), 7.08 (d, 1H, J = 15.2 Hz), 7.29–7.37 (m, 16H), 7.61 (d, 2H, J = 8.4 Hz), 7.83 (d, 2H, J = 8.4 Hz); 13C{1H} NMR (100.6 MHz, CDCl3): δ 65.5, 68.4, 71.1, 73.5, 73.8, 74.0, 76.1, 77.3, 77.4, 109.9, 122.4, 127.1, 127.8, 127.9, 128.0, 128.1, 128.2, 128.5, 128.6, 128.7, 130.2, 132.0, 136.7, 138.0, 138.1, 138.6, 140.9, 150.5, 189.2; HRMS (ESI) m/z: [M + Na]+ calcd for C36H33BrNaO5 647.1404; found 647.1419.
1-[E-1-(2-Hydroxyphenyl)propenon-3-yl]-3,4,6-tri-O-benzyl-d-Glucal (3d)
It was obtained as a yellow solid (415 mg) in 68% yield; mp: 83–85 °C; IR (KBr, cm–1): 3416, 3030, 2360, 2341,1632, 1411, 617; [α]D27 = −73.8 (c 0.3, chloroform); 1H NMR (400 MHz, CDCl3): δ 3.83–3.96 (m, 3H), 4.23 (dt, 1H, J = 3.9 and 7.9 Hz), 4.31 (dd, 1H, J = 3.2 and 5.8 Hz), 4.59–4.71 (m, 5H), 4.84 (d, 1H, J = 11.3 Hz), 5.41 (d, 1H, J = 3.2 Hz), 6.88–6.92 (m, 1H), 7.00 (d, 1H, J = 8.4 Hz) 7.17 (d, 1H, J = 14.9 Hz), 7.27–7.51 (m, 17H), 7.81–7.83 (m, 1H) 12.74 (s, 1H); 13C{1H} NMR (100.6 MHz, CDCl3): δ 68.3, 71.1, 73.4, 73.8, 73.9, 76.0, 77.3, 110.2, 118.6, 118.9, 120.0, 121.4, 127.7, 127.8, 127.9, 128.0, 128.5, 128.6, 130.1, 136.6, 138.0, 138.1, 138.5, 150.4, 163.6, 193.9; HRMS (ESI) m/z: [M + Na]+ calcd for C36H34NaO6 585.2248; found 585.2251.
1-[E-1-(4-Nitrophenyl)propenon-3-yl]-3,4,6-tri-O-benzyl-d-glucal (3e)
It was obtained as a pale yellow solid (437 mg) in 68% yield; mp: 99–101 °C; IR (KBr, cm–1): 3028, 2866, 1673, 1629, 1601, 1592, 1530, 1304, 1046, 970, 728, 693; [α]D27 = −87.7(c 0.3, chloroform); 1H NMR (400 MHz, CDCl3): δ 3.83–3.96 (m, 3H), 4.23 (dd, 1H, J = 3.7 and 7.6 Hz), 4.31 (dd, 1H, J = 3.2 and 5.6 Hz), 4.64 (dt, 5H, J = 9.3 and 10.3 Hz), 4.84 (d, 1H, J = 11.3 Hz), 5.42 (d, 1H, J = 3.1 Hz), 7.11 (d, 1H, J = 15.1 Hz), 7.23–7.37 (m, 16H), 8.08 (d, 2H, J = 8.8 Hz) 8.30 (d, 2H, J = 8.7 Hz); 13C{1H} NMR (100.6 MHz, CDCl3): δ 68.3, 71.2, 73.5, 73.8, 73.9, 75.9, 77.3, 110.8, 122.1, 123.9, 127.7, 127.8, 127.9, 128.0, 128.5, 128.6, 129.5, 137.9, 138.1, 139.7, 142.6, 150.2, 150.3, 188.7; HRMS (ESI) m/z: [M + H]+ calcd for C36H34NO7592.2330; found 592.2313.
1-(E-1-Phenylpropenon-3-yl)-3,4,6-tri-O-benzyl-d-glucal (3f)
It was obtained as a white solid (475 mg) in 80% yield; mp: 87–89 °C; IR (KBr, cm–1): 3030, 2920, 1667, 1626, 1600, 1453, 1295, 1209, 1098, 736, 697; [α]D27 = −69.6 (c 0.3, chloroform); 1H NMR (400 MHz, CDCl3): δ 3.83–3.96 (m, 3H), 4.19–4.23 (m, 1H), 4.31 (dd, 1H, J = 3.3 and 5.6 Hz), 4.64 (ddd, 5H, J = 7.7, 15.8, and 16.8), 4.84 (d, 1H, J = 11.3 Hz), 5.36 (d, 1H, J = 3.0 Hz), 7.08 (d, 1H, J = 15.2 Hz), 7.25–7.37 (m, 16H), 7.47 (t, 2H, J = 7.6 Hz), 7.57 (t, 1H, J = 7.1 Hz), 7.97 (d, 2H, J = 7.7 Hz); 13C{1H} NMR (100.6 MHz, CDCl3): δ 68.3, 71.0, 73.5, 73.8,74.0, 76.2, 77.3, 109.6, 122.9, 127.7, 127.8, 127.9, 128.0, 128.5, 128.6, 128.7, 133.0, 137.9, 138.0, 138.1, 138.2, 150.5, 190.3; HRMS (ESI) m/z: [M + Na]+ calcd for C36H34NaO5 569.2298; found 569.2292.
1-[E-1-(4-Chlorophenyl)propenon-3-yl]-3,4,6-tri-O-benzyl-d-glucal (3g)
It was obtained as a white solid (473 mg) in 75% yield; mp: 97–99 °C; IR (KBr, cm–1): 3032, 2926, 1660, 1628, 1541, 1399, 1299, 1208, 1092, 732, 692; [α]D27 = −70.9 (c 0.3, chloroform); 1H NMR (400 MHz, CDCl3): δ 3.83–3.95 (m, 3H), 4.20–4.22 (m, 1H), 4.30 (dd, 1H, J = 3.2 and 5.4 Hz), 4.58–4.70 (m, 5H), 4.84 (d, 1H, J = 11.3 Hz), 5.38 (d, 1H, J = 2.9 Hz), 7.08 (d, 1H, J = 15.0 Hz), 7.25–7.36 (m, 16H), 7.44 (d, 2H, J = 8.4 Hz), 7.91 (d, 2H, J = 8.4 Hz); 13C{1H} NMR (100.6 MHz, CDCl3): δ 68.3, 71.1, 73.5, 73.8, 74.0, 76.0, 77.3, 109.7, 122.4, 127.7, 127.8, 127.9, 128.0, 128.5, 128.6, 129.0, 130.1, 136.2, 138.0, 138.1, 138.4, 139.4, 150.5, 189.0; HRMS (ESI) m/z: [M + Na]+ calcd for C36H33ClNaO5 603.1909; found 603.1875.
1-[E-1-(3-Bromophenyl)propenon-3-yl]-3,4,6-tri-O-benzyl-d-glucal (3h)
It was obtained as a white solid (543 mg) in 80% yield; mp: 110–112 °C; IR (KBr, cm–1): 3027, 2875, 1665, 1623, 1598, 1297, 1199, 1121, 1085, 961, 728, 692; [α]D27 = −63.4 (c 0.3, chloroform); 1H NMR (400 MHz, CDCl3): δ 3.83–3.96 (m, 3H), 4.20–4.22 (m, 1H), 4.30 (s, 1H), 4.58–4.71 (m, 5H), 4.84 (d, 1H, J = 11.3 Hz), 5.38 (d, 1H, J = 2.9 Hz), 7.08 (d, 1H, J = 15.1 Hz), 7.23–7.37 (m, 17H), 7.69 (d, 1H, J = 7.7 Hz), 7.87 (d, 1H, J = 7.7 Hz), 8.10 (s, 1H); 13C{1H} NMR (100.6 MHz, CDCl3): δ 68.3, 71.1, 73.5, 73.8, 74.0, 76.1, 77.3, 110.0, 122.3, 123.0, 127.1, 127.7, 127.8, 127.9, 128.0, 128.5, 128.6, 130.2, 131.6, 135.8, 138.1, 138.8, 139.7, 150.5, 188.9; HRMS (ESI) m/z: [M + H]+ calcd for C36H34BrO5625.1584; found 625.1599.
1-[E-1-(3,4-Methylenedioxyphenyl)propenon-3-yl]-3,4,6-tri-O-benzyl-d-glucal (3i)
It was obtained as a white solid (519 mg) in 81% yield; mp:118–119 °C; IR (KBr, cm–1): 3029, 2891, 1662, 1630, 1593, 1450, 1303, 1244, 1088, 734, 692; [α]D27 = −72.1 (c 0.3, chloroform); 1H NMR (400 MHz, CDCl3): δ 3.82–3.95 (m, 3H), 4.18–4.22 (m, 1H), 4.30 (dd, 1H, J = 3.2 and 5.9 Hz), 4.58–4.70 (m, 5H), 4.84 (d, 1H, J = 11.3 Hz), 5.35 (d, 1H, J = 3.1 Hz), 6.05 (s, 2H), 6.85 (d, 1H, J = 8.2 Hz), 7.06 (d, 1H, J = 15.1 Hz), 7.25–7.37 (m, 16H), 7.49 (d, 1H, J = 1.2 Hz), 7.58 (dd, 1H, J = 1.2 and 8.3 Hz); 13C{1H} NMR (100.6 MHz, CDCl3): δ 68.3, 71.0, 73.4, 73.8, 74.0, 76.2, 77.2, 101.9, 107.9, 108.5, 109.0, 122.6, 125.0, 127.7, 127.8, 127.9, 128.0, 128.5, 128.6, 132.8, 137.9, 138.1, 138.2, 148.3, 150.7, 151.9, 188.1; HRMS (ESI) m/z: [M + Na]+ calcd for C37H34NaO7 613.2197; found 613.2181.
1-[E-1-(4-Hydroxyphenyl)propenon-3-yl]-3,4,6-tri-O-benzyl-d-glucal (3j)
It was obtained as an off white solid (464 mg) in 76% yield; mp: 136–138 °C; IR (KBr, cm–1): 3031, 2895, 1651, 1624, 1605, 1591, 1552, 1334, 1299, 1212, 1087, 964, 745, 696; [α]D27 = −71.3(c 0.3, chloroform); 1H NMR (400 MHz, CDCl3): δ 3.84–3.94 (m, 3H), 4.22 (d, 1H, J = 4.2 Hz), 4.29 (d, 1H, J = 3.3 Hz), 4.55–4.69 (m, 5H), 4.83 (d, 1H, J = 11.4 Hz), 5.35 (d, 1H, J = 3.0 Hz), 6.63 (bs, 1H) 6.85 (d, 2H, J = 8.8 Hz), 7.05 (d, 1H, J = 15.1 Hz), 7.25–7.36 (m, 16H), 7.89 (d, 2H, J = 8.4 Hz); 13C{1H} NMR (100.6 MHz, CDCl3): δ 68.4, 71.0, 73.5, 73.7, 74.0, 75.9, 109.0, 115.6, 122.8, 127.8, 127.9, 128.0, 128.1, 128.5, 128.6, 130.4, 131.4, 137.4, 137.9, 150.6, 160.9, 189.0; HRMS (ESI) m/z: [M + Na]+ calcd for C36H34NaO6 585.2248; found 585.2279.
Synthesis of 3,4,6-Tri-O-benzyl-1-formylglucal (4)
To a solution of β-C-glucopyranosyl aldehyde 1 (500 mg, 0.90 mmol) in ethanol (10 mL), an aqueous solution of 5% NaOH (10 mL) was added dropwise with continuous stirring at room temperature and further stirred for 1 h. After the completion of the reaction as indicated on TLC examination, the reaction mixture was concentrated at reduced pressure and the thick liquid thus obtained was extracted with ethyl acetate (2 × 50 mL). The combined organic layer was dried over Na2SO4, filtered, and concentrated at reduced pressure to give a crude product, which was purified over a silica gel column with 5% ethyl acetate in petroleum ether as an eluent to afford pure product 4 as light brown oil (370 mg) in 92% yield. It was characterized by comparing its 1H and 13C NMR data with those reported in the literature.13a
General Procedure for the Synthesis of 1,2-Disubstituted Glucals (6a–i)
Glucalpropenones 3a–c (400 mg, 1 equiv.), styrenes/acrylates 5a–g (1.1 equiv.), AgOTf (2 equiv.), CuI (1 equiv.), and Pd(OAc)2 (10 mol %) in a solvent mixture of DMF/DMSO (8 mL, v/v 9/1) were stirred at 80 °C in a 15 mL sealed tube for 12 h. After completion of the reaction as indicated on TLC examination, the resulting mixture was cooled to room temperature and extracted with ethyl acetate (2 × 50 mL). The combined organic phase was washed with brine (1 × 20 mL), dried over Na2SO4, and filtered. The organic phase was concentrated at reduced pressure, and the resulting residue was purified by silica gel column chromatography using ethyl acetate/petroleum ether (5–10%) as the eluent to furnish the desired products 6a–i in 78 to 90% yields.
1-[1-(4-Methylphenyl)propenon-3-yl]-2-(4-methyl)styryl-3,4,6-tri-O-benzyl-d-glucal (6a)
It was obtained as a yellow solid (410 mg) in 85% yield; mp: 182–185 °C; IR (KBr, cm–1): 3030, 2916 1724, 1656, 1604, 1573, 1502, 1452, 1367, 1288, 1209, 1180, 1093, 1022, 956, 846, 812, 738, 694; [α]D27 = −14.38 (c 0.1, dichloromethane);1H NMR (400 MHz, CDCl3): δ 2.35 (s, 3H), 2.43 (s, 3H), 3.82–3.89 (m, 2H), 4.18 (t, 1H, J = 4.0 Hz), 4.43–4.51 (dd, 3H, J = 10.7 and 20.4 Hz), 4.57 (s, 2H), 4.62 (d, 1H, J = 3.1 Hz), 4.74–4.81 (m, 2H), 6.60 (d, 1H, J = 15.9 Hz), 7.13 (d, 2H, J = 7.7 Hz), 7.24 (d, 3H, J = 7.0 Hz), 7.28–7.37 (m, 18H), 7.56 (d, 1H, J = 14.7 Hz), 7.93 (d, 2H, J = 7.1 Hz); 13C{1H} NMR (100.6 MHz, CDCl3): δ 21.4, 21.8, 68.5, 69.3, 71.1, 72.2, 73.3, 73.5, 76.0, 118.5, 121.8, 123.8, 126.5, 127.7, 127.8, 128.0, 128.1, 128.2, 128.4, 128.5, 128.6, 128.7, 128.9, 129.3, 129.4, 132.7, 134.9, 135.6, 137.6, 137.9, 138.1, 143.9, 148.9, 189.6; HRMS (ESI) m/z: [M + H]+ calcd for C46H45O5677.3262; found 677.3257.
1-[1-(4-Methylphenyl)propenon-3-yl]-2-styryl-3,4,6-tri-O-benzyl-d-glucal (6b)
It was obtained as a yellow solid (416 mg) in 88% yield; mp: 165–168 °C; IR (KBr, cm–1): 3032, 2877, 1726, 1654, 1606, 1573, 1494, 1452, 1367, 1292, 1211, 1182, 1091, 1020, 952, 846, 815, 1020, 738, 694; [α]D27 = −20.97 (c 0.1, dichloromethane); 1H NMR (400 MHz, CDCl3): δ 2.37 (s, 3H), 3.74–3.82 (m, 2H), 4.11 (t, 1H, J = 4.4 Hz), 4.36–4.44 (m, 3H), 4.50 (s, 2H), 4.54 (d, 1H, J = 3.5 Hz), 4.68–4.75 (m, 2H), 6.54 (d, 1H, J = 16.0 Hz), 7.15–7.17 (m, 3H), 7.19–7.24 (m, 11H), 7.27–7.33 (m, 9H), 7.50 (d, 1H, J = 14.7 Hz), 7.86–7.90 (m, 3H); 13C{1H} NMR (100.6 MHz, CDCl3): δ 21.4, 21.8, 68.5, 69.3, 71.1, 72.2, 73.3, 73.5, 76.0, 118.5, 121.8, 123.8, 126.5, 127.7, 127.8, 128.0, 128.1, 128.2, 128.4, 128.5, 128.6, 128.7, 128.9, 129.3, 129.4, 132.7, 134.9, 135.6, 137.6, 137.9, 138.1, 143.9, 148.9, 189.6; HRMS (ESI) m/z: [M + H]+ calcd for C45H43O5 663.3105; found 663.3106.
1-[1-(4-Methylphenyl)propenon-3-yl]-2-(4-nitro)styryl-3,4,6-tri-O-benzyl-d-glucal (6c)
It was obtained as a yellow solid (404 mg) in 80% yield; mp: 202–206 °C; IR (KBr, cm–1): 3061, 2908, 1654, 1583, 1556, 1512, 1452, 1371, 1332, 1292, 1209, 1180, 1083, 1016, 950, 910, 860, 819, 738, 696; [α]D27 = +175.30 (c 0.1, dichloromethane); 1H NMR (400 MHz, CDCl3): δ 2.38 (s, 3H), 3.73–3.83 (m, 2H), 4.14 (t, 1H, J = 3.9 Hz), 4.33–4.39 (m, 4H), 4.50 (s, 2H), 4.68–4.74 (m, 2H), 6.38 (d, 1H, J = 15.9 Hz), 7.13–7.15 (m, 2H), 7.19 (s, 6H), 7.23 (d, 5H, J = 8.2 Hz), 7.31–7.37 (m, 7H), 7.56 (d, 1H, J = 14.7 Hz), 7.83–7.89 (m, 3H), 8.09 (d, 2H, J = 8.7 Hz); 13C{1H} NMR (100.6 MHz, CDCl3): δ 21.9, 68.8, 69.7, 70.3, 72.2, 73.5, 73.9, 75.9, 116.9, 124.2, 125.2, 126.2, 126.8, 127.3, 127.7, 127.9, 128.2, 128.3, 128.5, 128.6, 128.7, 128.9, 129.5, 132.0, 135.3, 137.2, 137.7, 138.0, 144.2, 144.3, 146.6, 150.8, 189.3; HRMS (ESI) m/z: [M + H]+ calcd for C45H42NO7 708.2956; found 708.2939.
1-[1-(4-Methylphenyl)propenon-3-yl]-2-(2-chloro)styryl-3,4,6-tri-O-benzyl-d-glucal (6d)
It was obtained as a yellow solid (398 mg) in 80% yield; mp: 165–168 °C; IR (KBr, cm–1): 3034, 2875, 1651, 1606, 1575, 1496, 1369, 1323, 1290, 1211, 1182, 1095, 1031, 954, 910, 846, 815, 736, 694; [α]D27 = +76.33 (c 0.1, dichloromethane); 1H NMR (400 MHz, CDCl3): δ 2.43 (s, 3H), 3.76–3.86 (m, 2H), 4.11 (t, 1H, J = 3.7 Hz), 4.51–4.61 (m, 6H), 4.75 (s, 2H), 7.11 (d, 1H, J = 16.0 Hz), 7.18 (d, 1H, J = 7.4 Hz), 7.25 (d, 6H, J = 1.6 Hz), 7.29–7.34 (m, 10H), 7.37 (d, 4H, J = 4.3 Hz), 7.59 (d, 1H, J = 14.7 Hz), 7.68 (d, 1H, J = 7.8 Hz), 7.92–7.96 (m, 3H); 13C{1H} NMR (100.6 MHz, CDCl3): δ 21.8, 68.4, 70.7, 71.0, 72.1, 72.9, 73.5, 75.9, 117.8, 124.5, 125.5, 126.4, 127.1, 127.7, 127.8, 128.0, 128.1, 128.2, 128.3, 128.5, 128.7, 128.9, 129.5, 132.5, 133.3, 135.5, 135.6, 137.5, 137.7, 138.1, 144.1, 149.1, 189.5; HRMS (ESI) m/z: [M + H]+ calcd for C45H42ClO5 697.2715; found 697.2709.
1-[1-(4-Methylphenyl)propenon-3-yl]-2-(2,2,2-trifluoroethyl acrylat-3-yl)-3,4,6-tri-O-benzyl-d-glucal (6e)
It was obtained as a pale yellow gel (397 mg) in 78% yield; IR (KBr, cm–1): 3034, 2908, 1726, 1660, 1604, 1562, 1496, 1452, 1409, 1369, 1282, 1211, 1153, 1082, 960, 846, 817, 740, 694; [α]D27 = +37.15 (c 0.1, dichloromethane); 1H NMR (400 MHz, CDCl3): δ 2.43 (s, 3H), 3.78 (ddd, 2H, J = 5.9, 10.5 and 15.6 Hz), 4.13 (t, 1H, J = 3.4 Hz), 4.33 (s, 1H), 4.39–4.47 (m, 3H), 4.52 (s, 2H), 4.53–4.56 (m, 1H), 4.61–4.63 (m, 1H), 4.72 (q, 2H, J = 12.0 Hz), 5.78 (d, 1H, J = 15.5 Hz), 7.19 (dd, 2H, J = 2.8 and 6.6 Hz) 7.30–7.38 (m, 15H), 7.62 (d, 1H, J = 14.9 Hz), 7.81 (d, 1H, J = 14.9 Hz), 7.91 (d, 2H, J = 8.2 Hz), 7.96 (d, 1H, J = 15.5 Hz); 13C{1H} NMR (100.6 MHz, CDCl3): δ 21.8, 60.1, 68.1, 69.7, 70.3, 71.2, 72.0, 73.5, 76.3, 114.3, 114.4, 121.9, 127.3, 127.7, 127.9, 128.1, 128.3, 128.4, 128.6, 128.7, 128.8, 129.0, 129.5, 131.4, 135.1, 136.8, 137.5, 137.8, 141.1, 144.4, 154.1, 165.4, 189.3; 19F NMR (376 MHz, CDCl3): δ −73.6; HRMS (ESI) m/z: [M + H]+ calcd for C42H40F3O7 713.2721; found 713.2764.
1-[1-(4-Methylphenyl)propenon-3-yl]-2-(benzyl acrylate-3-yl)-3,4,6-tri-O-benzyl-d-glucal (6f)
It was obtained as a pale yellow gel (411 mg) in 80% yield; IR (KBr, cm–1): 3032, 2868, 1707, 1658, 1602, 1562, 1494, 1452, 1373, 1290, 1209, 1165, 1076, 1016, 966, 817, 734, 694; [α]D27 = −37.89 (c 0.1, dichloromethane); 1H NMR (400 MHz, CDCl3): δ 2.43 (s, 3H), 3.77 (ddd, 2H, J = 5.8, 10.5, and 15.4 Hz), 4.10 (t, 1H, J = 3.6 Hz), 4.36 (d, 1H, J = 2.0 Hz), 4.42 (s, 2H), 4.50 (s, 2H), 4.57 (dd, 1H, J = 5.1 and 9.2 Hz), 4.70 (d, 2H, J = 2.1 Hz), 5.22 (s, 2H), 5.90 (d, 1H, J = 15.5 Hz), 7.17 (dd, 2H, J = 2.9 and 6.6 Hz), 7.24 (d, 2H, J = 2.4 Hz), 7.29–7.39 (m, 18H), 7.60 (d, 1H, J = 14.8 Hz), 7.83 (d, 1H, J = 14.9 Hz), 7.89–7.94 (m, 3H); 13C{1H} NMR (100.6 MHz, CDCl3): δ 21.8, 66.2, 68.2, 70.2, 70.3, 71.9, 72.0, 73.5, 76.3, 114.9, 1/16.7, 126.9, 127.7, 127.9, 128.1, 128.2, 128.3, 128.6, 128.7, 129.0, 129.5, 131.7, 135.2, 136.4, 137.1, 137.6, 137.9, 139.2, 144.2, 153.3, 166.9, 189.4; HRMS (ESI) m/z: [M + H]+ calcd for C47H45O7 721.3160; found 721.3168.
1-[1-(4-Methoxylphenyl)propenon-3-yl]-2-styryl-3,4,6-tri-O-benzyl-d-glucal (6g)
It was obtained as a yellow solid (424 mg) in 90% yield; mp: 177–180 °C; IR (KBr, cm–1): 3030, 2905, 2865, 1710, 1651, 1595, 1502, 1453, 1299, 1257, 1213, 1171, 1088, 1023, 960, 834, 742, 695; [α]D27 = +133.49 (c 0.1, dichloromethane); 1H NMR (400 MHz, CDCl3): δ 3.84–3.88 (m, 2H), 3.89 (s, 3H), 4.18 (dd, 1H, J = 3.9 and 5.1 Hz), 4.43–4.46 (m, 3H), 4.58 (s, 2H), 4.61 (d, 1H, J = 3.5 Hz), 4.78 (dd, 2H, J = 3.8 and 12.0 Hz), 6.61 (d, 1H, J = 16.0 Hz), 6.95 (d, 2H, J = 8.9 Hz), 7.22–7.25 (m, 3H), 7.27–7.30 (m, 5H), 7.33–7.34 (m, 7H), 7.36–7.40 (m, 6H), 7.57 (d, 1H, J = 14.7 Hz), 7.94 (d, 1H, J = 14.7 Hz), 8.03 (d, 2H, J = 8.9 Hz); 13C{1H} NMR (100.6 MHz, CDCl3): δ 55.6, 68.6, 69.5, 71.2, 72.3, 73.3, 73.5, 76.1, 114.0, 118.1, 122.9, 124.1, 126.6, 127.7, 127.8, 127.9, 128.1, 128.2, 128.4, 128.6, 128.7, 128.8, 129.2, 131.1, 132.4, 137.7, 137.9, 138.2, 149.2, 163.7, 188.4; HRMS (ESI) m/z: [M + H]+ calcd for C45H43O6 679.3054; found 679.3045.
1-[1-(4-Bromophenyl)propenon-3-yl]-2-(4-methyl)styryl-3,4,6-tri-O-benzyl-d-glucal (6h)
It was obtained as a yellow solid (417 mg) in 88% yield; mp: 184–188 °C; IR (KBr, cm–1): 2922, 2856, 1739, 1655, 1581, 1458, 1370, 1294, 1213, 1180, 1105, 1036, 1011, 956, 824, 743, 697; [α]D27 = +12.60 (c 0.1, dichloromethane); 1H NMR (400 MHz, CDCl3): δ 2.35 (s, 3H), 3.81–3.88 (m, 2H), 4.16–4.18 (m, 1H), 4.45–4.51 (m, 3H), 4.56–4.62 (m, 3H), 4.77 (s, 2H), 6.59 (d, 1H, J = 15.9 Hz), 7.13 (d, 2H, J = 6.5 Hz), 7.23–7.36 (m, 18H), 7.49 (d, 1H, J = 14.7 Hz), 7.60–7.62 (m, 2H), 7.87–7.89 (m, 2H), 7.97 (d, 1H, J = 14.7 Hz); 13C{1H} NMR (100.6 MHz, CDCl3): δ 21.4, 68.5, 69.5, 71.0, 72.2, 73.1, 73.5, 76.0, 119.2, 121.6, 123.0, 126.5, 127.7, 127.8, 128.1, 128.4, 128.6, 129.5, 129.7, 130.2, 132.0, 133.6, 134.7, 136.8, 137.5, 137.8, 138.1, 148.5, 188.9; HRMS (ESI) m/z: [M + H]+ calcd for C45H42BrO5 741.2210; found 741.2208.
1-[1-(4-Bromophenyl)propenon-3-yl]-2-(4-methoxy)styryl-3,4,6-tri-O-benzyl-d-glucal (6i)
It was obtained as a yellow solid (388 mg) in 80% yield; mp: 182–187 °C; IR (KBr, cm–1): 2921, 2853, 1647, 1579, 1510, 1456, 1368, 1279, 1172, 1098, 1030, 1005, 951, 819, 736, 679; [α]D27 = +81.09 (c 0.1, dichloromethane); 1H NMR (400 MHz, CDCl3): δ 3.80–3.89 (m, 5H), 4.16–4.18 (m, 1H), 4.42–4.45 (m, 2H), 4.50 (d, 1H, J = 11.2 Hz), 4.57 (s, 2H), 4.60 (d, 1H, J = 3.4 Hz), 4.77 (dd, 2H, J = 3.3 and 12.0 Hz), 6.57 (d, 1H, J = 16.0 Hz), 6.87 (d, 2H, J = 8.7 Hz), 7.14 (d, 1H, J = 16.0 Hz), 7.22–7.24 (m, 2H), 7.28–7.37 (m, 15H), 7.48 (d, 1H, J = 14.7 Hz), 7.61 (d, 2H, J = 8.5 Hz), 7.88 (d, 2H, J = 8.5 Hz), 7.97 (d, 1H, J = 14.7 Hz); 13C{1H} NMR (100.6 MHz, CDCl3): δ 55.4, 68.5, 69.4, 71.1, 72.2, 73.2, 73.5, 76.0, 114.2, 119.2, 120.6, 122.8, 127.7, 127.8, 127.9, 128.0, 128.1, 128.4, 128.5, 128.6, 128.7, 129.4, 130.2, 130.4, 132.0, 133.6, 136.9, 137.6, 137.8, 138.1, 148.2, 159.5, 188.9; HRMS (ESI) m/z: [M + H]+ calcd for C45H42BrO6 757.2159; found 757.2163.
General Procedure for the Synthesis of Chiral Chromane Derivatives (7a–d and 7g–i)
In a 15 mL sealed tube, compound 6a–i (350 mg, 1 equiv.) was dissolved in xylene (5 mL) and the reaction mixture was stirred at 160 °C for 2 h (progress of reaction was monitored by thin layer chromatography). After completion, the solvent was removed over a rotary evaporator and the thick liquid thus obtained was extracted with ethyl acetate (2 × 15 mL). The organic phase was washed with saturated aqueous NaCl (1 × 10 mL), dried over Na2SO4, and filtered. Ethyl acetate was evaporated on a rotavapor, and the resulting residue was purified by silica gel column chromatography using 5–10% ethyl acetate in petroleum ether as an eluent to obtain the desired products 7a–d and 7g–i in 66–73% yields. The reaction with 1,2-disubstituted glucaltrienones 6e and 6f led to the decomposition of the precursor, and product formation was not observed.
(2R,3S,4R)-2-Benzyloxymethyl-3,4-dibenzyloxy-6-(4-methyl)phenyl-7-(4-methyl)benzoyl-chromane (7a)
It was obtained as a light brown oily compound (244 mg) in 70% yield; IR (cm–1, thin film): 3030, 2918, 2864, 1660, 1604, 1568, 1483, 1452, 1415, 1361, 1292, 1247, 1207, 1176, 1087, 1026, 995, 912, 819, 792, 734, 696; [α]D27 = +47.53 (c 0.1, dichloromethane); 1H NMR (400 MHz, CDCl3): δ 2.24 (s, 3H), 2.34 (s, 3H), 3.88 (ddd, 2H, J = 4.1, 10.9 and 14.0 Hz), 4.17 (t, 1H, J = 6.9 Hz), 4.38–4.41 (m, 1H), 4.58–4.64 (m, 2H), 4.73–4.85 (m, 5H), 6.97–7.00 (m, 3H), 7.08 (dd, 4H, J = 8.0 and 15.6 Hz), 7.29–7.36 (m, 16H), 7.61 (d, 2H, J = 8.1 Hz); 13C{1H} NMR (100.6 MHz, CDCl3): δ 21.2, 21.8, 68.8, 72.7, 73.6, 73.7, 74.0, 76.6, 77.5, 117.0, 121.8, 123.8, 127.8, 127.9, 128.0, 128.1, 128.2, 128.5, 128.7, 129.0, 129.1, 133.8, 134.6, 136.6, 137.0, 137.9, 138.0, 138.1, 140.2, 144.0, 152.4, 197.7; HRMS (ESI) m/z: [M + H]+ calcd for C46H43O5 675.3105; found 675.3097.
(2R,3S,4R)-2-Benzyloxymethyl-3,4-dibenzyloxy-6-phenyl-7-(4-methyl)benzoylchromane (7b)
It was obtained as a light brown oily compound (244 mg) in 70% yield; IR (cm–1, thin film): 3030, 2954, 2877, 1726, 1664, 1607, 1449, 1409, 1365, 1297, 1252, 1213, 1171, 1088, 1036, 917, 754, 701; [α]D27 = −19.60 (c 0.1, dichloromethane); 1H NMR (400 MHz, CDCl3): δ 2.32 (s, 3H), 3.88 (ddd, 2H, J = 4.1, 10.9, and 13.7 Hz), 4.18 (t, 1H, J = 7.0 Hz), 4.39–4.42 (m, 1H), 4.62 (d, 2H, J = 6.8 Hz), 4.74–4.85 (m, 5H), 7.00 (s, 1H), 7.07 (d, 2H, J = 7.9 Hz), 7.17 (d, 3H, J = 3.7 Hz), 7.29–7.36 (m, 18H), 7.58 (d, 2H, J = 8.1 Hz); 13C{1H} NMR (100.6 MHz, CDCl3): δ 21.7, 68.8, 72.7, 73.6, 73.7, 74.0, 76.6, 77.5, 117.1, 123.9, 126.9, 127.8, 127.9, 128.0, 128.1, 128.2, 128.5, 128.6, 128.9, 130.3, 130.9, 133.8, 134.6, 137.9, 138.0, 138.1, 139.9, 140.3, 143.9, 152.6, 197.6; HRMS (ESI) m/z: [M + H]+ calcd for C45H41O5 661.2949; found 661.2957.
(2R,3S,4R)-2-Benzyloxymethyl-3,4-dibenzyloxy-6-(4-nitro)phenyl-7-(4-methyl)benzoyl-chromane (7c)
It was obtained as a brown oily compound (230 mg) in 66% yield; IR (cm–1, thin film): 3062, 3030, 2921, 2858, 1710, 1662, 1601, 1564, 1516, 1454, 1344, 1291, 1254, 1212, 1177, 1094, 1028, 914, 854, 746, 699; [α]D27 = −9.81 (c 0.1, dichloromethane); 1H NMR (400 MHz, CDCl3): δ 2.28 (s, 3H), 3.81 (ddd, 2H, J = 4.0, 10.9 and 14.0 Hz), 4.11 (t, 1H, J = 7.0 Hz), 4.33–4.37 (m, 1H), 4.54 (d, 2H, J = 1.7 Hz), 4.66–4.80 (m, 5H), 6.96 (s, 1H), 7.05 (d, 2H, J = 8 Hz), 7.17–7.25 (m, 10H), 7.27 (s, 8H), 7.52 (d, 2H, J = 8.0 Hz), 7.97 (d, 2H, J = 8.5 Hz); 13C{1H} NMR (100.6 MHz, CDCl3): δ 21.8, 68.6, 73.1, 73.7, 73.8, 73.9, 76.5, 77.7, 117.7, 123.6, 124.4, 127.9, 128.1, 128.2, 128.5, 128.7, 129.3, 129.6, 130.3, 131.0, 131.5, 134.3, 137.7, 137.9, 140.2, 144.6, 146.7, 153.6, 196.7; HRMS (ESI) m/z: [M + H]+ calcd for C45H40NO7 706.2799; found 706.2809.
(2R,3S,4R)-2-Benzyloxymethyl-3,4-dibenzyloxy-6-(2-chloro)phenyl-7-(4-methyl)benzoyl-chromane (7d)
It was obtained as a pale yellow oily compound (241 mg) in 69% yield; IR (cm–1, thin film): 3032, 2918, 2867, 1663, 1607, 1566, 1458, 1406, 1364, 1297, 1212, 1170, 1087, 916, 831, 749, 699; [α]D27 = +37.53 (c 0.1, dichloromethane); 1H NMR (400 MHz, CDCl3): δ 2.25 (s, 3H), 3.78–3.85 (m, 2H), 4.12–4.15 (m, 1H), 4.33 (s, 1H), 4.47–4.62 (m, 3H), 4.64–4.76 (m, 4H), 6.98–7.07 (m, 5H), 7.14–7.27 (m, 18H), 7.53 (d, 2H, J = 8.1 Hz); 13C{1H} NMR (100.6 MHz, CDCl3): δ 21.7, 68.8, 71.4, 72.1, 73.6, 76.5, 77.2, 77.6, 117.6, 122.3, 126.4, 127.7, 127.8, 127.9, 128.0, 128.4, 128.5, 128.7, 129.4, 130.2, 132.0, 132.8, 134.4, 137.8, 138.0, 143.6, 152.9, 196.3; HRMS (ESI) m/z: [M + H]+ calcd for C45H40ClO5 695.2559; found 695.2555.
(2R,3S,4R)-2-Benzyloxymethyl-3,4-dibenzyloxy-6-phenyl-7-(4-methoxy)benzoylchromane (7g)
It was obtained as a pale yellow oily compound (255 mg) in 73% yield; IR (cm–1, thin film): 3060, 3027, 2922, 2858, 1657, 1598, 1505, 1452, 1412, 1362, 1295, 1254, 1212, 1168, 1085, 1023, 911, 842, 802, 741, 697; [α]D27 = +53.56 (c 0.1, dichloromethane); 1H NMR (400 MHz, CDCl3): δ 3.78 (s, 3H), 3.86 (ddd, 2H, J = 4.6, 10.8, and 14.2 Hz), 4.17 (t, 1H, J = 6.9 Hz), 4.39 (dt, 1H, J = 4.1 and 7.8 Hz), 4.60 (d, 2H, J = 1.9 Hz), 4.72–4.83 (m, 5H), 6.74 (d, 2H, J = 8.8 Hz), 6.98 (s, 1H), 7.16 (d, 4H, J = 4.4 Hz), 7.28–7.35 (m, 17H), 7.65 (d, 2H, J = 8.9 Hz); 13C NMR (100.6 MHz, CDCl3): δ 55.5, 68.8, 72.7, 73.6, 73.7, 74.0, 77.3, 113.5, 117.0, 123.7, 127.0, 127.9, 128.1, 128.3, 128.5, 128.6, 128.9, 130.1, 130.9, 132.5, 133.7, 137.8, 138.0, 139.9, 140.3, 152.6, 163.5, 196.6; HRMS (ESI) m/z: [M + H]+ calcd for C45H41O6 677.2898; found 677.2879.
(2R,3S,4R)-2-Benzyloxymethyl-3,4-dibenzyloxy-6-(4-methyl)phenyl-7-(4-bromo)benzoyl-chromane (7h)
It was obtained as a light brown oily compound (251 mg) in 72% yield; IR (cm–1, thin film): 3075, 3024, 2862, 1605, 1522, 1477, 1345, 1217, 1106, 1069, 1020, 1005, 933, 851, 792, 749, 700, 673; [α]D27 = −16.36 (c 0.1, dichloromethane); 1H NMR (400 MHz, CDCl3): δ 2.25 (s, 3H), 3.83–3.93 (m, 2H), 4.17 (t, 1H, J = 7.0 Hz), 4.39–4.42 (m, 1H), 4.61 (d, 2H, J = 2.1 Hz), 4.74–4.84 (m, 5H), 6.97–7.03 (m, 4H), 7.28–7.32 (m, 5H), 7.33–7.35 (m, 12H), 7.40 (d, 2H, J = 8.5 Hz), 7.51 (d, 2H, J = 8.6 Hz); 13C{1H} NMR (100.6 MHz, CDCl3): δ 21.1, 68.8, 72.8, 73.6, 73.7, 74.0, 76.5, 77.4, 117.1, 124.4, 127.8, 127.9, 128.0, 128.1, 128.5, 128.6, 128.7, 129.1, 130.8, 131.5, 133.8, 135.9, 136.7, 136.9, 137.8, 138.0, 139.3, 152.6, 197.0; HRMS (ESI) m/z: [M + H]+ calcd for C45H40BrO5 739.2054; found 739.2042.
(2R,3S,4R)-2-Benzyloxymethyl-3,4-dibenzyloxy-6-(4-methoxy)phenyl-7-(4-bromo)benzoyl-chromane (7i)
It was obtained as a pale yellow oily compound (244 mg) in 70% yield; IR (cm–1, thin film): 3059, 3029, 2922, 2858, 1660, 1612, 1561, 1450, 1363, 1220, 1092, 938, 855, 765, 740, 699; [α]D27 = +54.48 (c 0.1, dichloromethane); 1H NMR (400 MHz, CDCl3): δ 3.37 (s, 3H), 3.83–3.93 (m, 2H), 4.16–4.19 (m, 1H), 4.38–4.42 (m, 1H), 4.62 (d, 2H, J = 2.4 Hz), 4.74–4.85 (m, 5H), 6.71 (d, 2H, J = 8.8 Hz), 6.99 (s, 1H), 7.04 (d, 2H, J = 8.7 Hz), 7.31–7.35 (m, 14H), 7.37–7.38 (m, 3H), 7.40–7.41 (m, 1H), 7.49 (d, 2H, J = 8.6 Hz); 13C{1H} NMR (100.6 MHz, CDCl3): δ 55.3, 65.5, 68.7, 72.9, 73.7, 74.0, 76.6, 77.5, 113.8, 117.0, 124.4, 127.1, 127.8, 127.9, 128.1, 128.5, 128.7, 130.7, 131.4, 131.5, 132.1, 133.4, 135.9, 137.8, 138.0, 139.3, 152.2, 158.8, 197.2; HRMS (ESI) m/z: [M + H]+ calcd for C45H40BrO6 755.2003; found 755.1997.
General Procedure for the Synthesis of Chiral Trihydroxychromane Derivatives (8a–d and 8g–i)
In a 15 mL glass reaction tube, compound 7a–d and 7g–i (200 mg) was dissolved in dichloromethane (2 mL) and stirred at −78 °C. After 10 min, 1 M BCl3 in dichloromethane (1 mL) was added dropwise to the reaction mixture and stirring was continued for another 0.5 to 1 h (progress of the reaction was monitored by TLC). On completion, the temperature of the reaction mixture was raised to −40 °C, and methanol (1 mL) was added and stirred for another 15 min. Further, the reaction mixture was stirred at room temperature for half an hour, neutralized with AmberliteIRA-402(OH) ion exchange resin, and filtered. The filtrate was concentrated using a rotavapor, and the crude product thus obtained was purified by silica gel column chromatography using 0.5–2% methanol in chloroform as an eluent to obtain the desired products 8a–d and 8g–i in 82–92% yields.
(2R,3S,4R)-2-Hydroxymethyl-3,4-dihydroxy-6-(4-methyl)phenyl-7-(4-methyl)benzoylchromane (8a)
It was obtained as an off white solid compound (110 g) in 92% yield; mp: 112–115 °C; IR (KBr, cm–1): 3404, 3020, 2960, 2904, 1661, 1606, 1566, 1486, 1417, 1291, 1214, 1177, 1111, 1060, 919, 797, 758, 666; [α]D27 = +106.43 (c 0.1, dichloromethane); 1H NMR (400 MHz, DMSO-d6): δ 2.19 (s, 3H), 2.30 (s, 3H), 3.63–3.76 (m, 2H), 3.82–3.86 (m, 1H), 3.98 (ddd, 1H, J = 2.2, 5.0, and 9.2 Hz), 4.58 (t, 1H, J = 7.0 Hz), 4.81 (t, 1H, J = 5.7 Hz), 5.50 (d, 1H, J = 5.3 Hz), 5.81 (d, 1H, J = 6.3 Hz), 6.73 (s, 1H), 7.02–7.07 (m, 4H), 7.21 (d, 2H, J = 8.2 Hz), 7.49–7.54 (m, 3H); 13C{1H} NMR (100.6 MHz, DMSO-d6): δ 21.1, 21.7, 61.0, 68.2, 70.4, 80.7, 115.5, 128.4, 128.7, 129.4, 129.7, 130.2, 130.3, 132.5, 134.6, 136.5, 137.4, 139.1, 144.4, 152.7, 197.1; HRMS (ESI) m/z: [M + H]+ calcd for C25H25O5 405.1697; found 405.1712.
(2R,3S,4R)-2-Hydroxymethyl-3,4-dihydroxy-6-phenyl-7-(4-methyl)benzoylchromane (8b)
It was obtained as an off white solid compound (106 mg) in 90% yield; mp: 110–112 °C; IR (KBr, cm–1): 3371, 3025, 2921, 1656, 1604, 1564, 1478, 1444, 1409, 1290, 1247, 1212, 1177, 1113, 1054, 907, 836, 757, 700, 667; [α]D27 = +23.56 (c 0.1, dichloromethane); 1H NMR (400 MHz, DMSO-d6): δ 2.30 (s, 3H), 3.62–3.68 (m, 1H), 3.69–3.75 (m, 1H), 3.81–3.85 (m, 1H), 3.96–4.00 (m, 1H), 4.58 (t, 1H, J = 7.1 Hz), 4.79 (t, 1H, J = 5.8 Hz), 5.49 (d, 1H, J = 5.2 Hz), 5.80 (d, 1H, J = 6.4 Hz), 6.76 (s, 1H), 7.15–7.22 (m, 7H), 7.50–7.52 (m, 3H); 13C{1H} NMR (100.6 MHz, DMSO-d6): δ 21.7, 61.4, 68.7, 71.3, 78.7, 116.4, 126.3, 127.0, 128.2, 128.8, 129.0, 129.5, 130.3, 134.0, 134.3, 139.5, 144.2, 152.4, 198.2; HRMS (ESI) m/z: [M + H]+ calcd for C24H23O5 391.1540; found 391.1544.
(2R,3S,4R)-2-Hydroxymethyl-3,4-dihydroxy-6-(4-nitro)phenyl-7-(4-methyl)benzoyl-chromane (8c)
It was obtained as a brown gel (107 mg) in 87% yield; IR (KBr, cm–1): 3373, 2925, 1656, 1600, 1563, 1515, 1478, 1418, 1344, 1290, 1214, 1178, 1109, 1053, 1024, 912, 853, 757, 699, 665; [α]D27 = +17.66 (c 0.1, dichloromethane); 1H NMR (400 MHz, DMSO-d6): δ 2.32 (s, 3H), 3.65–3.68 (m, 1H), 3.70–3.77 (m, 1H), 3.82–3.85 (m, 1H), 4.01–4.04 (m, 1H), 4.59 (t, 1H, J = 7.1 Hz), 4.80 (t, 1H, J = 5.5 Hz), 5.52 (d, 1H, J = 5.4 Hz), 5.87 (dd, 1H, J = 1.1 and 6.2 Hz), 6.84 (d, 1H, J = 1.5 Hz), 7.25 (d, 2H, J = 7.6 Hz), 7.42–7.45 (m, 2H), 7.55–7.59 (m, 3H), 8.12 (dd, 2H, J = 1.6 and 8.7 Hz); 13C{1H} NMR (100.6 MHz,DMSO-d6): δ 21.7, 60.9, 68.0, 70.2, 80.9, 116.5, 124.0, 129.0, 129.8, 130.1, 130.3, 130.7, 130.8, 134.5, 139.2, 144.7, 146.6, 147.4, 153.8, 196.5; HRMS (ESI) m/z: [M + H]+ calcd for C24H22NO7 436.1391; found 436.1394.
(2R,3S,4R)-2-Hydroxymethyl-3,4-dihydroxy-6-(2-chloro)phenyl-7-(4-methyl)benzoyl-chromane (8d)
It was obtained as an off white solid compound (102 mg) in 84% yield; mp: 138–141 °C; IR (KBr, cm–1): 3370, 2923, 1659, 1607, 1564, 1456, 1409, 1286, 1214, 1174, 1033, 913, 828, 761; [α]D27 = +27.81 (c 0.1, dichloromethane); 1H NMR (400 MHz, DMSO-d6): δ 2.32 (s, 3H), 3.67–3.75 (m, 2H), 3.82–3.85 (m, 1H), 3.99–4.02 (m, 1H), 4.57 (t, 1H, J = 6.1 Hz), 4.78 (s, 1H), 5.50 (d, 1H, J = 5.3 Hz), 5.82 (d, 1H, J = 6.2 Hz), 6.82 (s, 1H), 7.23 (d, 5H, J = 7.9 Hz), 7.33 (d, 1H, J = 6.4 Hz), 7.40 (s, 1H), 7.54 (d, 2H, J = 8.1 Hz); 13C{1H} NMR (100.6 MHz, DMSO-d6): δ 21.7, 61.0, 68.1, 70.3, 80.8, 116.4, 125.8, 127.4, 128.7, 129.3, 129.4, 129.6, 130.3, 131.4, 132.3, 132.4, 134.5, 137.9, 139.2, 144.0, 153.0, 195.6; HRMS (ESI) m/z: [M + H]+ calcd for C24H22ClO5 425.1150; found 425.1121.
(2R,3S,4R)-2-Hydroxymethyl-3,4-dihydroxy-6-phenyl-7-(4-methoxy)benzoylchromane (8g)
It was obtained as a white solid compound (106 mg) in 88% yield; IR (KBr, cm–1): 3392, 3017, 2925, 2854, 1652, 1596, 1569, 1508, 1447, 1419, 1257, 1214, 1170, 1115, 1061, 1026, 908, 847, 759, 701, 667; [α]D27 = +70.35 (c 0.1, dichloromethane); 1H NMR (400 MHz, DMSO-d6): δ 3.63–3.76 (m, 2H), 3.78 (s, 3H), 3.81–3.86 (m, 1H), 3.96–4.00 (m, 1H), 4.57 (t, 1H, J = 7.0 Hz), 4.77 (t, 1H, J = 5.8 Hz), 5.47 (d, 1H, J = 5.3 Hz), 5.78 (d, 1H, J = 6.3 Hz), 6.74 (s, 1H), 6.92 (d, 2H, J = 8.9 Hz), 7.15–7.18 (m, 3H), 7.21–7.25 (m, 2H), 7.50 (s, 1H), 7.59 (d, 2H, J = 8.8 Hz); 13C{1H} NMR (100.6 MHz, DMSO-d6): δ 56.1, 61.0, 68.2, 70.4, 80.7, 114.3, 115.5, 127.3, 128.4, 128.8, 130.0, 130.3, 130.4, 132.4, 132.5, 139.3, 144.4, 152.8, 163.7, 195.9; HRMS (ESI) m/z: [M + H]+ calcd for C24H23O6 407.1489; found 407.1480.
(2R,3S,4R)-2-Hydroxymethyl-3,4-dihydroxy-6-(4-methyl)phenyl-7-(4-bromo)benzoyl-chromane (8h)
It was obtained as a white solid compound (105 mg) in 83% yield; mp: 140–144 °C; IR (KBr, cm–1): 3390, 2921, 1664, 1576, 1457, 1394, 1294, 1219, 1112, 1006, 918, 824, 779, 668; [α]D27 = +86.59 (c 0.1, dichloromethane); 1H NMR (400 MHz, DMSO-d6): δ 2.17 (s, 3H), 3.58–3.72 (m, 2H), 3.77–3.81 (m, 1H), 3.92–3.96 (m, 1H), 4.51–4.54 (m, 1H), 4.75 (t, 1H, J = 5.7 Hz), 5.46 (d, 1H, J = 5.2 Hz), 5.78 (d, 1H, J = 6.3 Hz), 6.77 (s, 1H), 6.98–7.02 (m, 4H), 7.45–7.47 (m, 3H), 7.56 (d, 2H, J = 8.6 Hz); 13C{1H} NMR (100.6 MHz, DMSO-d6): δ 21.1, 61.0, 68.1, 70.4, 80.7, 115.8, 128.0, 128.8, 129.0, 129.5, 130.3, 131.8, 132.2, 132.7, 136.1, 136.6, 137.2, 138.3, 152.9, 196.7; HRMS (ESI) m/z: [M + H]+ calcd for C24H22BrO5 469.0645; found 469.0641.
(2R,3S,4R)-2-Hydroxymethyl-3,4-dihydroxy-6-(4-methoxy)phenyl-7-(4-bromo)benzoyl-chromane (8i)
It was obtained as a white solid compound (106 mg) in 83% yield; mp: 150–154 °C; IR (KBr, cm–1): 3454, 2921, 1707, 1670, 1463, 1250, 1069, 831, 668; [α]D27 = +37.31 (c 0.1, dichloromethane); 1H NMR (400 MHz, DMSO-d6): δ 3.61 (m, 1H), 3.66 (s, 3H), 3.69–3.75 (m, 1H), 3.80–3.85 (m, 1H), 3.95–3.99 (m, 1H), 4.55 (t, 1H, J = 7.1 Hz) , 4.79 (t, 1H, J = 5.8 Hz), 5.50 (d, 1H, J = 5.3 Hz), 5.81 (d, 1H, J = 6.3 Hz), 6.79 (t, 3H, J = 4.3 Hz), 7.05 (d, 2H, J = 8.6 Hz), 7.48 (d, 3H, J = 8.3 Hz), 7.59 (d, 2H, J = 8.5 Hz); 13C{1H} NMR (100.6 MHz, DMSO-d6): δ 55.5, 61.0, 68.1, 70.4, 80.7, 114.3, 115.7, 127.9, 129.0, 130.1, 130.2, 131.8, 132.1, 132.4, 136.2, 138.2, 152.7, 158.8, 196.9; HRMS (ESI) m/z: [M + H]+ calcd for C24H22BrO5 469.0645; found 469.0641.
Synthesis of (2R,3S,4R)-2-Hydroxymethyl-3,4-dihydroxy-6-(4-methyl)phenyl-7-(4-methyl)benzylchromane (9a)
In a 10 mL round bottom flask, compound 7a (200 mg) in methanol (4 mL) was added followed by the addition of 10% Pd/C (20 mg) and the reaction mixture was stirred at 25 °C for 4 h under a H2 atmosphere. On completion (progress of the reaction was monitored by TLC), the reaction mixture was passed through celite to remove Pd/C and the filtrate was evaporated on a rotavapor to get the crude product. The crude product thus obtained was purified by silica gel column chromatography using 1% methanol in chloroform as an eluent to get the pure desired product as a white solid (104 mg) in 90% yield. mp: 140–143 °C; IR (KBr, cm–1): 3304, 3024, 2928, 1616, 1568, 1485, 1413, 1286, 1222, 1111, 1047, 997, 912, 879, 815, 767, 700; [α]D27 = +71.72 (c 0.1, dichloromethane); 1H NMR (400 MHz, DMSO-d6): δ 2.23 (s, 3H), 2.34 (s, 3H), 3.51–3.56 (m, 1H), 3.62–3.68 (m, 1H), 3.78 (s, 2H), 3.80–3.84 (m, 2H), 4.44 (t, 1H, J = 6.9 Hz), 4.70 (t, 1H, J = 5.5 Hz), 5.34 (d, 1H, J = 4.6 Hz), 5.54 (d, 1H, J = 6.2 Hz), 6.45 (s, 1H), 6.86 (d, 2H, J = 7.8 Hz), 7.03 (d, 2H, J = 7.7 Hz), 7.14 (d, 2H, J = 7.9 Hz), 7.19–7.22 (m, 3H); 13C{1H} NMR (100.6 MHz, DMSO-d6): δ 21.1, 21.3, 38.8, 61.2, 68.7, 70.6, 80.3, 116.9, 124.2, 129.1, 129.3, 129.4, 129.6, 129.9, 134.2, 135.3, 136.2, 138.5, 138.7, 139.0, 153.0; HRMS (ESI) m/z: [M + Na]+ calcd for C25H26O4Na 413.1723; found 413.1755.
Acknowledgments
We acknowledge the help of Mr. Adeel Feroze, a short time visitor in my laboratory from Siddhartha Public School, Delhi for the preparation of starting sugar aldehyde, and Dr. Kuldeep Mahiya, Department of Chemistry, Government College, Adampur, Haryana, India, for solving the crystal data. We are grateful to the Institute of Eminence, University of Delhi for providing financial support to strengthen research and development. We are thankful to CIF-USIC, University of Delhi, for providing the NMR spectral and HRMS recording facility. V.K.M. thanks CFEES-DRDO, Delhi for providing the Junior Research Fellowship. B.S. thanks University Grants Commission (UGC), New Delhi for providing the Teacher Research Fellowship under the Faculty Development Program.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c00103.
The authors declare no competing financial interest.
Supplementary Material
References
- a Rawat P.; Verma S. M. Design and synthesis of chroman derivatives with dual anti-breast cancer and antiepileptic activities. Drug Des., Dev. Ther. 2016, 10, 2779–2788. 10.2147/DDDT.S111266. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ding J.; Yao J.; Xue J.; Li R.; Bao B.; Jiang L.; Zhu J.; He Z. Tumor-homing cell-penetrating peptide linked to colloidal mesoporous silica encapsulated (−)-epigallocatechin-3-gallate as drug delivery system for breast cancer therapy in vivo. ACS Appl. Mater. Interfaces 2015, 7, 18145–18155. 10.1021/acsami.5b05618. [DOI] [PubMed] [Google Scholar]
- Kashiwada Y.; Yamazaki K.; Ikeshiro Y.; Yamagishi T.; Fujioka T.; Mihashi K.; Mizuki K.; Cosentino L. M.; Fowke K.; Morris-Natschke S. L.; Lee K.-H. Isolation of rhododaurichromanic acid B and the anti-HIV principles rhododaurichromanic acid A and rhododaurichromenic acid from Rhododendron dauricum. Tetrahedron 2001, 57, 1559–1563. 10.1016/S0040-4020(00)01144-3. [DOI] [Google Scholar]
- a Presley C. C.; Valenciano A. L.; Fernández-Murga M. L.; Du Y.; Shanaiah N.; Cassera M. B.; Goetz M.; Clement J. A.; Kingston D. G. I. Antiplasmodial chromanes and chromenes from the monotypic plant species Koeberlinia spinose. J. Nat. Prod. 2018, 81, 475–483. 10.1021/acs.jnatprod.7b00579. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Roberts B. F.; Iyamu I. D.; Lee S.; Lee E.; Ayong L.; Kyle D. E.; Yuan Y.; Manetsch R.; Chakrabarti D. Spirocyclic chromanes exhibit antiplasmodial activities and inhibit all intraerythrocytic life cycle stages. Int. J. Parasitol.: Drugs Drug Resist. 2016, 6, 85–92. 10.1016/j.ijpddr.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pini E.; Poli G.; Tuccinardi T.; Chiarelli L. R.; Mori M.; Gelain A.; Costantino L.; Villa S.; Meneghetti F.; Barlocco D. New chromane-based derivatives as inhibitors of Mycobacterium tuberculosis salicylate synthase (mbtl): Preliminary biological evaluation and molecular modeling studies. Molecules 2018, 23, 1506. 10.3390/molecules23071506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Farhadi F.; Khameneh B.; Iranshahi M.; Iranshahy M. Antibacterial activity of flavonoids and their structure-activity relationship: An update review. Phytother. Res. 2019, 33, 13–40. 10.1002/ptr.6208. [DOI] [PubMed] [Google Scholar]; b Mellado M.; Espinoza L.; Madrid A.; Mella J.; Chávez-Weisser E.; Diaz K.; Cuellar M. Design, synthesis, antifungal activity, and structure-activity relationship studies of chalcones and hybrid dihydrochromane-chalcones. Mol. Diversity 2020, 24, 603–615. 10.1007/s11030-019-09967-y. [DOI] [PubMed] [Google Scholar]
- a Salamone S.; Colin C.; Grillier-Vuissoz I.; Kuntz S.; Mazerbourg S.; Flament S.; Martin H.; Richert L.; Chapleur Y.; Boisbrun M. Synthesis of new troglitazone derivatives: Anti-proliferative activity in breast cancer cell lines and preliminary toxicological study. Eur. J. Med. Chem. 2012, 51, 206–215. 10.1016/j.ejmech.2012.02.044. [DOI] [PubMed] [Google Scholar]; b Netscher T. Building up quarternary stereocenters of chromanes by asymmetric redox organocatalysis: A new entry to vitamin E. Angew. Chem., Int. Ed. 2014, 53, 14313–14315. 10.1002/anie.201409826. [DOI] [PubMed] [Google Scholar]; c Celebioglu A.; Uyar T. Antioxidant vitamin e/cyclodextrin inclusion complex electrospun nanofibers: Enhanced water solubility, prolonged shelf life, and photostability of vitamin E. J. Agric. Food Chem. 2017, 65, 5404–5412. 10.1021/acs.jafc.7b01562. [DOI] [PubMed] [Google Scholar]; d Coats A.; Jain S. Protective effects of nebivolol from oxidative stress to prevent hypertension-related target organ damage. J. Hum. Hypertens. 2017, 31, 376–381. 10.1038/jhh.2017.8. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Shen Y.-B.; Li S.-S.; Wang L.; An X.-D.; Liu Q.; Liu X.; Xiao J. Organocatalytic dearomative [4 + 2] cycloadditions of biomass-derived 2,5-dimethylfuran with ortho-quinone methides: Access to multisubstituted chromanes. Org. Lett. 2018, 20, 6069–6073. 10.1021/acs.orglett.8b02448. [DOI] [PubMed] [Google Scholar]; f Gogoi D.; Devi R.; Pahari P.; Sarma B.; Das S. K. cis-Diastereoselective synthesis of chromane-fused tetralins as B-ring-modified analogues of brazilin. Beilestein J. Org. Chem. 2016, 12, 2816–2822. 10.3762/bjoc.12.280. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Ville A.; Viault G.; Hélesbeux J.-J.; Guilet D.; Richomme P.; Séraphin D. Efficient semi-synthesis of natural δ-(R)-tocotrienols from a renewable vegetal source. J. Nat. Prod. 2019, 82, 51–58. 10.1021/acs.jnatprod.8b00517. [DOI] [PubMed] [Google Scholar]
- a Masesane I. B.; Desta Z. Y. Reactions of salicylaldehyde and enolates or their equivalents: Versatile synthetic routes to chromane derivatives. Beilstein J. Org. Chem. 2012, 8, 2166–2175. 10.3762/bjoc.8.244. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Tanaka K.; Kishimoto M.; Hoshino Y.; Honda K. Temperature controlled divergent synthesis of 4-alkoxy-or 4-alkenyl-chromanes via inverse electron demand cycloaddition with in situ generated ortho-quinone methides. Tetrahedron Lett. 2018, 59, 1841–1845. 10.1016/j.tetlet.2018.03.090. [DOI] [Google Scholar]; c Wang W.; Bai X.; Jin S.; Guo J.; Zhao Y.; Miao H.; Zhu Y.; Wang Q.; Bu Z. An unexpected FeCl3-catalyzed cascade reaction of indoles and o-hydroxychalcones for the assembly of chromane-bridged polycyclic indoles. Org. Lett. 2018, 20, 3451–3454. 10.1021/acs.orglett.8b01107. [DOI] [PubMed] [Google Scholar]; d Rouh H.; Liu Y.; Katakam N.; Pham L.; Zhu Y.-L.; Li G. Synthesis of functionalized chromene and chromane derivatives via cesium carbonate promoted formal [4 + 2] annulation of 2′-hydroxychalcones with allenoates. J. Org. Chem. 2018, 83, 15372–15379. 10.1021/acs.joc.8b02627. [DOI] [PubMed] [Google Scholar]
- a Carral-Menoyo A.; Misol A.; Gómez-Redondo M.; Sotomayor N.; Lete E. Palladium(II)-catalyzed intramolecular C-H alkenylation for the synthesis of chromanes. J. Org. Chem. 2019, 84, 2048–2060. 10.1021/acs.joc.8b03051. [DOI] [PubMed] [Google Scholar]; b Jakkampudi S.; Parella R.; Zhao J. C.-G. Stereoselective synthesis of chromane derivatives via a domino reaction catalyzed by modularly designed organocatalysts. Org. Biomol. Chem. 2019, 17, 151–155. 10.1039/C8OB02677G. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Li S.; Li F.; Gong J.; Yang Z. Palladium-catalyzed carbonylative cyclization of aryl alkenes/alkenols: A new reaction mode for the synthesis of electron-rich chromanes. Org. Lett. 2015, 17, 1240–1243. 10.1021/acs.orglett.5b00214. [DOI] [PubMed] [Google Scholar]; d Zhou L.; Li S.; Xu B.; Ji D.; Wu L.; Liu Y.; Zhang Z.-M.; Zhang J. Enantioselective Difunctionalization of Alkenes by a Palladium-Catalyzed Heck/Sonogashira Sequence. Angew. Chem. 2020, 132, 2791–2797. 10.1002/ange.201913367. [DOI] [PubMed] [Google Scholar]
- a Shang X.; Liu Z.-Q. Transition metal-catalyzed Cvinyl–Cvinyl bond formation via double Cvinyl–H bond activation. Chem. Soc. Rev. 2013, 42, 3253–3260. 10.1039/c2cs35445d. [DOI] [PubMed] [Google Scholar]; b Frihed T. G.; Bols M.; Pedersen C. M. C–H functionalization on carbohydrates. Eur. J. Org. Chem. 2016, 2740–2756. 10.1002/ejoc.201600121. [DOI] [Google Scholar]; c Bai Y.; Zeng J.; Cai S.; Liu X.-W. Palladium-Catalyzed Direct Cross-Coupling Reaction of Glycals with Activated Alkenes. Org. Lett. 2011, 13, 4394–4397. 10.1021/ol201734w. [DOI] [PubMed] [Google Scholar]; d Hussain N.; Tatina M. B.; Rasool F.; Mukherjee D. Diastereoselective sp2–sp3 coupling of sugar enol ethers with unactivated cycloalkenes: new entries to C-branched sugars. Org. Biomol. Chem. 2016, 14, 9989–9992. 10.1039/C6OB01949H. [DOI] [PubMed] [Google Scholar]; e Hussain N.; Tatina M. B.; Mukherjee D. Cross dehydrogenative coupling of sugar enol ethers with terminal alkenes in the synthesis of pseudo-disaccharides, chiral oxadecalins and a conjugated triene. Org. Biomol. Chem. 2018, 16, 2666–2677. 10.1039/C8OB00168E. [DOI] [PubMed] [Google Scholar]; f Begum Z.; Shankar G.; Sirisha K.; Reddy B. V. S. Carbohydr. Res. 2018, 461, 1–3. 10.1016/j.carres.2018.03.002. [DOI] [PubMed] [Google Scholar]; g de Robichon M.; Bordessa A.; Malinowski M.; Uziel J.; Lubin-Germain N.; Ferry A. Access to C-aryl/alkenylglycosides by directed Pd-catalyzed C–H functionalisation of the anomeric position in glycal-type substrates. Chem. Commun. 2019, 55, 11806–11808. 10.1039/C9CC05993H. [DOI] [PubMed] [Google Scholar]
- Leibeling M.; Koester D. C.; Pawliczek M.; Schild S. C.; Werz D. B. Domino access to highly substituted chromans and isochromans from carbohydrates. Nat. Chem. Biol. 2010, 6, 199–201. 10.1038/nchembio.302. [DOI] [PubMed] [Google Scholar]
- Maikhuri V. K.; Khatri V.; Kumar A.; Singh B.; Prasad A. K. Synthesis of Sugar Diene and Its Pd-Catalyzed Transformation into Chromanes. J. Org. Chem. 2020, 85, 7068–7076. 10.1021/acs.joc.0c00432. [DOI] [PubMed] [Google Scholar]
- Khatri V.; Kumar A.; Singh B.; Malhotra S.; Prasad A. K. Synthesis of β-C-glycopyranosyl aldehydes and 2,6-anhydro-heptitols. J. Org. Chem. 2015, 80, 11169–11174. 10.1021/acs.joc.5b01933. [DOI] [PubMed] [Google Scholar]
- a Sipos S.; Jablonkai I. Preparation of 1-C-glycosyl aldehydes by reductive hydrolysis. Carbohydr. Res. 2011, 346, 1503–1510. 10.1016/j.carres.2011.04.019. [DOI] [PubMed] [Google Scholar]; b Reddy M. R.; Hemaiswarya S.; Kommidi H.; Aidhen I. S.; Doble M. Acyl and benzyl-C-β-D-glucosides: Synthesis and biostudies for glucose-uptake-promoting activity in C2C12 mytotubes. Eur. J. Org. Chem. 2019, 6053–6070. 10.1002/ejoc.201901037. [DOI] [Google Scholar]
- Volkov A.; Gustafson K. P. J.; Tai C.-W.; Verho O.; Bäckvall J.-E.; Adolfsson H. Mild deoxygenation of aromatic ketones and aldehydes over Pd/C using polymethylhydrosiloxane as the reducing agent. Angew. Chem., Int. Ed. 2015, 54, 5122–5126. 10.1002/anie.201411059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend L. B.; Drach J. C.. Polysubstituted benzimidazoles as antiviral agents. U. S. Patent 5,360,7951994,.
- a Laha J. K.; Dayal N. A tandem approach to functionalized carbazoles from indoles via two successive regioselective oxidative heck reactions followed by thermal electrocyclization. Org. Lett. 2015, 17, 4742–4745. 10.1021/acs.orglett.5b02265. [DOI] [PubMed] [Google Scholar]; b Voigt K.; von Zezschwitz P.; Rosauer K.; Lansky A.; Adams A.; Reiser O.; de Meijere A. The twofold heck reaction on 1,2-dihalocycloalkenes and subsequent 6π-electrocyclization of the resulting (E, Z, E)-1,3,5-hexatrienes: A new formal {2+2+2}-assembly of six-membered rings. Eur. J. Org. Chem. 1998, 1521–1534. . [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.