

Abstract

C-C chemokine receptor type 5 (CCR5) is a member of the G protein-coupled receptor. CCR5 and its interaction with chemokine ligands have been crucial for understanding and tackling human immunodeficiency virus (HIV)-1 entry into target cells. In recent years, the change in CCR5 expression has been related to the progression of different cancer types. Patients treated with the CCR5 ligand, maraviroc (MVC), showed a deceleration in tumor development especially for metastatic colorectal cancer. Based on the crystal structure of CCR5, we herein describe a multistage virtual screening protocol including pharmacophore screening, molecular docking, and protein–ligand interaction fingerprint (PLIF) postdocking filtration for discovery of novel CCR5 ligands. The applied virtual screening protocol led to the identification of four hits with binding modes showing access to the major and minor pockets of the MVC binding site. Compounds 2–4 showed a decrease in cellular proliferation upon testing on the metastatic colorectal cancer cell line, SW620, displaying 12, 16, and 4 times higher potency compared to MVC, respectively. Compound 3 induced apoptosis by arresting cells in the G0/G1 phase of the cell cycle similar to MVC. Further in vitro assays showed compound 3 drastically decreasing the CCR5 expression and cellular migration 48 h post treatment, indicating its ability to inhibit metastatic activity in SW620 cells. The discovered hits represent potential leads for the development of novel classes of anticolorectal cancer agents targeting CCR5.
1. Introduction
C-C chemokine receptor type 5 (CCR5) is one of 19 human chemokine (CC) receptors belonging to family A of the G protein-coupled receptors (GPCRs).1 Like all members of the GPCR family, CCR5 shares the common molecular architecture of seven transmembrane (TM) helices linked by three extracellular loops (ECLs) and three intracellular loops (ICLs).2,3 The ECLs together with the N-terminus are involved in chemokine binding, whereas the ICLs as well as the C-terminus plays an important role in the G protein-mediated signal transduction. CC ligands bind to the CCR5 receptor, leading to activation of the signaling pathway mediated by heterotrimeric G proteins and causing cell motility.1−3 CCR5 is mainly expressed on the surface of white blood cells and plays an important role in human inflammatory responses to infection. CCR5 gained prominence as a coreceptor important for human immunodeficiency virus (HIV) host cell entry.4 Therefore, blocking the function of CCR5 by CCR5 inhibitors has been considered as an effective and relatively harmless HIV therapeutic strategy.4,5 Recent studies indicated that CCR5 is overexpressed in various types of cancer. CCR5 induces cancer cell homing to metastatic sites, augments the proinflammatory prometastatic immune phenotype, and enhances DNA repair, providing unusual cell survival and resistance to DNA-damaging agents.6,7 Consequently, CCR5 has been recognized as an exciting new therapeutic target for metastatic cancer, with clinical trials now targeting breast and colon cancers.8 A variety of small-molecule ligands have been identified that can modulate the activity of the CCR5 receptor.9,10
Several CCR5 ligands developed for HIV treatment are considered to be repurposed for cancer treatment.8 To date, maraviroc (MVC) is the only Food and Drug Administration (FDA)-approved CCR5 ligand for HIV treatment. MVC has been repositioned in clinical trials for cancer therapy. Indeed, patients treated with MVC showed a deceleration in tumor development.8 MVC has been discovered by high-throughput screening followed by a long optimization process.11 Later approaches to find CCR5 ligands used homology models of the CCR5 receptor12,13 and ligand-based fragment merging.14 In 2013, a crystal structure of CCR5 bound to MVC (Protein Data Bank (PDB): 4MBS)15 was published, providing a structural basis for the virtual discovery of CCR5 ligands of previously undescribed chemotypes. Maraviroc binds to an allosteric, and not orthosteric, binding site of the CCR5 receptor. Consequently, its pharmacological action should be described as that of a negative allosteric modulator, rather than of a competitive antagonist.16 However, the term “CCR5 antagonists” has been widely used for maraviroc and related compounds in the literature.8−10 Very recently, two pharmacophore-based virtual screening (VS) approaches for identification of novel CCR5 ligands have been reported. Mirza et al. discovered CCR5, CXCR4, and dual CCR5/CXCR4 inhibitors of partly novel chemotypes by screening the MolPort and Interbioscreen databases. However, the identified compounds were less active compared to the control ligands MVC and AMD300.17 Lin et al. screened the NCI database identifying potential CCR5 inhibitors with higher binding affinities than MVC as indicated by free energy calculations.18 However, the results were not supported by biological assays.18
In the present work, we describe the construction and validation of a virtual screening (VS) protocol that was used for mining the Specs database to discover novel CCR5 ligands as anticolorectal cancer agents.
2. Results
The X-ray crystallographic structure of CCR5 complexed with MVC (PDB code: 4MBS)15 was used for inferring chemical information on inhibitors’ binding to CCR5. MVC binds in an allosteric pocket located at the extracellular end of the TM bundle, occupying both the transmembrane site 1 (TMS1), or minor pocket, and transmembrane site 2 (TMS2), or major pocket. The minor pocket is delineated by residues from TM1, TM2, and TM7 and the major pocket between TM3-7.15,19 Structural information on previously reported CCR5 inhibitors was used to guide the discovery of novel CCR5 ligands. A highly selective ligand-based pharmacophore model was built and used along with docking and protein–ligand interaction fingerprints (PLIFs) postdocking filtration to screen the commercially available Specs database for novel CCR5 ligands. The four top-ranking hits were tested in vitro to evaluate their anticancer activities.
2.1. Pharmacophore Generation
Pharmacophore modeling is a powerful technique to identify ligands’ structural features important for biological activity. To build a pharmacophore model, a total of 2827 compounds with experimentally known CCR5 inhibitory activity were retrieved from the BindingDB (http://www.bindingdb.org).20 To guarantee maximal structural diversity, these compounds were grouped based on their chemical scaffolds and structural similarity, resulting in 39 different clusters (Table S1, Supporting Information). The most active compound from each cluster was selected, resulting in 39 training set compounds representing various chemical scaffolds.21−64 The selected compounds were then aligned on the coordinates of MVC obtained from its crystal structure bound to the CCR5 receptor (PDB code: 4MBS)15 using the align.svl script65 in MOE.66 For each compound, low energy conformations were generated and the conformer with the highest alignment score was selected. After visual examination, 16 compounds showing the highest structural alignment with MVC (F score ≤−200) were chosen as a basis for building the pharmacophore model using the Pharmacophore Elucidate module implemented in MOE66 (Figures 1 and S1). Eight different pharmacophore hypotheses (PH1–8) were postulated comprising features targeting the minor and major pockets of MVC’s binding site simultaneously (Figure S2). Six out of the eight hypotheses (PH1–6) had either a positive charge center or a hydrogen-bond donor feature representing a positively charged basic nitrogen atom. With the exception of PH7, all pharmacophore hypotheses had either a hydrophobic or an aromatic feature complementary to a deep hydrophobic pocket present in TMS2. Five models (PH1–4 and PH7) had a hydrophobic feature corresponding to the carbon linker between the quaternary nitrogen and the deep hydrophobic pocket binding feature. Finally, a hydrophobic spacer at the interface between TMS1 and TMS2 was present in six pharmacophore hypotheses (PH3–8).
Figure 1.

Two-dimensional (2D) chemical structures of CCR5 ligands used as training set compounds (T1–16) and their activity values (half-maximal inhibitory concentration (IC50)).
The plausibility of the resultant hypotheses was measured by the degree of active overlay expressed by the overlay score (Table 1). In addition, retrospective screening was conducted to evaluate the ability of hypotheses to separate the actives from inactives. To this end, a small-molecule test set was generated for model validation. Thus, 60 of the active CCR5 inhibitors from the literature that were not included in the training set were labeled as actives and a total of 2444 molecules, with similar physical properties as actives (molecular weight (MW), number of hydrogen-bond donors and hydrogen-bond acceptors, number of rotatable bonds, and octanol–water partition coefficient (log P)), were labeled as inactives. The inactive compounds comprised 91 experimentally confirmed inactive compounds, 201 CCR5 decoys from the GPCR Decoys Database (GDD),67 and 2152 inhibitors of other target proteins obtained from the GPCR Ligand Library (GLL)67 Drugbank.68 The multiconformation test set compounds were mapped to the pharmacophore hypotheses. As shown in Table 1, all models had an enrichment factor (EF) value larger than 1 (1 corresponds to random screening). Representing the basic nitrogen atom with a cationic feature decreased the number of mapped inactives as shown by the higher EF values of PH1, 3, and 5 compared to those of PH2, 4 and 6, respectively. PH1 had the highest overlay score of 12.3 and could retrieve 59 out of 60 known CCR5 inhibitors (sensitivity (Se) = 0.98), exclude 1844 out of a total of 2444 inactives (specificity (Sp) = 0.76), and showed a high mapping EF of 3.73. Introduction of additional spatial constraints in the form of ligand shape (LS) can prevent more inactives from mapping the pharmacophore model. Consequently, ligand shape defined by the cocrystallized structure of MVC was added as an additional constraint to PH1. The resultant hypothesis (PH9) was capable of excluding 2038 out of a total of 2444 inactives, showing superior specificity (0.84) and a higher mapping EF (5.29) compared to those of PH1. PH9 comprised four pharmacophore features: two hydrophobic, one aromatic, and a positively charged center (Figure 2A). Overlay of the crystallized coordinates of MVC on PH9 showed that the cationic feature represents the protonated tropane ring nitrogen that is engaged in a salt-bridge interaction with Glu2837.39. The aromatic feature corresponds to the phenyl group that reaches deep in the hydrophobic subpocket of TMS2 forming π–π interactions with Tyr1083.32. Finally, the two hydrophobic features are related to the isopropyl group of the triazole ring binding to TMS1 and to the aliphatic middle chain whose length was reported to be critical for maintaining the distance between the carboxamide nitrogen and the tropane nitrogen15 (Figure 2B). In conclusion, PH9 showed the capacity to recognize CCR5 inhibitors effectively and thus was selected for VS.
Table 1. Statistical Parameters of Pharmacophore Hypotheses Built with CCR5 Training Set Compounds. Parameters of PH-1 and PH-9 are shown in bold.
| pharmacophore hypothesis | overlap | featuresb | TH | AH | TPR | FPR | EF |
|---|---|---|---|---|---|---|---|
| PH1 | 12.30 | 2Hyd | 600 | 59 | 0.98 | 0.24 | 3.73 |
| 1Aro | |||||||
| 1Cat | |||||||
| PH2 | 12.27 | 2Hyd | 901 | 59 | 0.98 | 0.36 | 2.56 |
| 1Aro | |||||||
| 1Don | |||||||
| PH3 | 11.49 | 2Hyd | 866 | 59 | 0.98 | 0.35 | 2.66 |
| 1Aro | |||||||
| 1Cat | |||||||
| PH4 | 11.49 | 2Hyd | 1138 | 59 | 0.98 | 0.46 | 2.05 |
| 1Aro | |||||||
| 1Don | |||||||
| PH5 | 11.45 | 2Hyd | 457 | 53 | 0.88 | 0.18 | 4.33 |
| 1Acc | |||||||
| 1Cat | |||||||
| PH6 | 11.44 | 2Hyd | 912 | 55 | 0.91 | 0.37 | 2.37 |
| 1Acc | |||||||
| 1Don | |||||||
| PH7 | 10.76 | 2Hyd | 1916 | 60 | 1.00 | 0.78 | 1.26 |
| 2Acc | |||||||
| PH8 | 10.46 | 2Hyd | 1440 | 59 | 0.98 | 0.59 | 1.64 |
| 2Acc | |||||||
| PH9 | 12.30 | 2Hyd | 406 | 59 | 0.98 | 0.16 | 5.29 |
| 1Aro | |||||||
| 1Cat | |||||||
| LS |
Total number of hits (TH), number of active hits (AH), true-positive rate (TPR), false-positive rate (FPR), enrichment factor (EF) of pharmacophore mapping.
Hydrophobic (Hyd), aromatic (Aro), cationic (Cat), H-bond donor (Don), H-bond acceptor (Acc), ligand shape (LS).
Figure 2.

Ligand-based pharmacophore model (PH9) for CCR5 inhibitors: (A) three-dimensional (3D) spatial arrangement and distance constraints between the chemical features of the pharmacophore model represented by blue (cationic center, Cat), orange (aromatic center, Aro), and green (hydrophobic, Hyd) spheres. (B) Overlay of the crystal coordinates of MVC on the pharmacophore model (PH9).
2.2. Molecular Docking
Molecular docking simulation studies were performed using GOLD 5.5 (Cambridge Crystallographic Data Centre, Cambridge, U.K.).69,70 The crystal structure of CCR5 cocrystallized with the FDA-approved inhibitor MVC (PDB code: 4MBS)15 was used in this study. To validate the docking protocol, MVC was first docked into its binding pocket of the CCR5 receptor. All of the resultant poses converged to a binding mode similar to that of the experimentally determined position of MVC, with the best ranking pose having a root-mean-square deviation (RMSD) value of 0.52 Å (Table S2). MVC binds to a deep pocket formed by residues in the extracellular part of the TM domain stabilizing the CCR5 receptor in an inactive state (Figure S3). This binding site is divided into minor (TMS1) and major (TMS2) pockets. TMS1 is defined by Tyr371.39, Trp862.60, Tyr892.63, Thr2847.40, and Met2877.43, while TMS2 is defined by Thr1955.39, Ile1985.42, Leu2556.55, Thr2596.59, and Met2797.35 in addition to a deep hydrophobic subpocket defined by Phe1093.33, Phe1123.36, Trp2486.48, and Tyr2516.51. Two residues, Glu2837.39 and Tyr1083.32, overlap at the interface of both pockets. The triazole ring of MVC occupies TMS1, forming aromatic interactions with Trp862.60. TMS2 is occupied by the difluorocyclohexyl group interacting with Thr1955.39 and Ile1985.42 in addition to a phenyl ring reaching deep in the hydrophobic subpocket and forming hydrophobic interactions with Tyr1083.32. Finally, the protonated nitrogen of the tropane group forms a salt bridge with Glu2837.39. The docking protocol was further assessed for the molecular recognition between ligands and their binding site in terms of enrichment performance. For that, docking of a test set comprising 20 active compounds and 537 inactive compounds was conducted to further check the docking ability to decrease the false-positive rate, and the validation test set was designed including any inactive compound that escaped the initial pharmacophore filtration. The enrichment of active compounds was assessed by the enrichment factor (EF) and area under the receiver operator characteristic (ROC) curve (AUROC) values. GoldScore showed good enrichment performance with an AUROC value of 0.79 and an EF at 1 and 5% of the ranked list of 4.64 and 3.97, respectively, as shown in Figure S4.
Protein–ligand interaction fingerprints (PLIFs) were further used to enhance the performance of the docking algorithm. PLIFs were previously reported to give better results than standard scoring functions in terms of identifying the correct binding modes of ligands and recovering active compounds in VS trials.71−73 Crystal structures and mutation studies74,75 identified Ile1985.42, Tyr1083.32, and Glu2837.39 as key residues that are essential for CCR5 receptor binding and thus can be used to filter ligands’ docked poses. Four PLIF models (PLIF-M1–PLIF-M4) based on one, two, or three essential residues (see Table 2 for details) were applied, and their performance was evaluated by screening the docked poses of the test set compounds (Figure S4). As shown in Table 2, filtering the docked poses based on PLIF-M1 resulted in excluding 26.82% of the FPs (specificity = 0.27). Additional filtration based on either Ile1985.42 (PLIF-M2) or Tyr1083.32 (PLIF-M3) besides Glu2837.39 resulted in excluding more FPs (specificity = 0.57 and 0.49, respectively). Filtration based on interactions with all three residues (PLIF-M4) resulted in a relatively similar reduction of the FP retrieval (specificity = 0.57). On the other hand, PLIF-M1 and M3 could successfully recognize all CCR5 inhibitors (sensitivity = 1), whereas for PLIF-M2 and M4, the retrieval rate was 95% (sensitivity = 0.95).
Table 2. Statistical Parameters of Postdocking Filtration of the Validation Database Using Various PLIF Models. Metrics of PLIF-M3 post-docking filtration are shown in bolda.
| PLIF model (PLIF-M) | key residue(s) | AH | TPR | FH | FPR |
|---|---|---|---|---|---|
| PLIF-M1 | Glu2837.39 | 20 | 1 | 393 | 0.73 |
| PLIF-M2 | Glu2837.39, Ile-985.42 | 19 | 0.95 | 232 | 0.43 |
| PLIF-M3 | Glu2837.39, Tyr1083.32 | 20 | 1 | 273 | 0.508 |
| PLIF-M4 | Glu2837.39, Ile-985.42, Tyr1083.32 | 19 | 0.95 | 231 | 0.43 |
Number of active hits (AH), true-positive rate (TPR), number of false hits (FH), false-positive rate (FPR).
In an attempt to improve the enrichment performance of the docking protocol, the filtered poses were rescored using three different scoring functions, DSXPDB,7676 ChemPLP, and ChemScore. As shown in Table 3, rescoring showed an improvement in the EF values compared to those of GoldScore with DSXPDB, showing the best performance based on the higher AUROC and EF values. Filtering the docked poses based on PLIF-M3 and rescoring the filtered poses using DSXPDB showed the highest AUROC value of 0.86 and an EF of the top 1 and 5% of the ranked list of 9.76 and 6.83, respectively (Figure S4D). Overall, postdocking processing using PLIF-M3 and rescoring the filtered poses using DSXPDB enhanced the enrichment performance of the docking algorithm and thus was used for prospective VS.
Table 3. Docking Enrichment Comparison Using Different Scoring Functions. Metrics of DSXPDB scores post PLIF-M3 filtration and Goldscore with no PLIF filtration are shown in bold for comparison.
| GoldScore |
ChemPLP |
ChemScore |
DSXPDB |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (re)scoring method/PLIF | AUROC | EF1% | EF5% | AUROC | EF1% | EF5% | AUROC | EF1% | EF5% | AUROC | EF1% | EF5% |
| none | 0.79 | 4.64 | 3.97 | 0.86 | 9.28 | 6.96 | 0.79 | 9.28 | 4.97 | 0.81 | 9.28 | 3.97 |
| PLIF-M1 | 0.74 | 8.26 | 4.91 | 0.83 | 8.26 | 5.90 | 0.78 | 8.26 | 5.90 | 0.84 | 8.26 | 7.86 |
| PLIF-M2 | 0.76 | 8.80 | 6.09 | 0.75 | 4.40 | 5.08 | 0.77 | 4.40 | 5.08 | 0.86 | 8.80 | 8.12 |
| PLIF-M3 | 0.77 | 4.88 | 4.88 | 0.80 | 4.88 | 4.88 | 0.79 | 4.88 | 6.83 | 0.86 | 9.76 | 6.83 |
| PLIF-M4 | 0.76 | 8.77 | 6.07 | 0.74 | 4.38 | 5.06 | 0.77 | 4.38 | 6.07 | 0.86 | 8.77 | 8.09 |
2.3. Virtual Screening
Our multistage virtual screening protocol (PH9–Docking–PLIF-M3–DSXPDB rescoring) was utilized to screen the Specs database77 comprising 213 504 structurally diverse compounds. As the database had undergone stringent druglike and desirable chemical group filters, we directly uploaded the database on Pharmit server78 to undergo a knowledge-based conformational search, resulting in a total of 2 819 976 conformers. The resultant conformers were screened using the pharmacophore model described earlier (PH9), narrowing down the database to 5847 compounds that fulfill the required chemical features and steric constraints. To examine their binding modes, these hit compounds were docked into the MVC binding site of the CCR5 receptor using GOLD (version 5.5). The generated poses were then filtered based on PLIF-M3, reducing the number of hits to 5374 compounds. The filtered poses were finally rescored using DSXPDB followed by visual examination of the top-scoring compounds. The structures of the top 1% scored compounds are shown in the Supporting Information (Figure S5). Although commercially available, the four highest-ranking compounds 1–4 (Figure 3A) have not been reported in the literature. All four hits showed a Tanimoto coefficient (Tc) less than 0.5 against the initial training set of 39 CCR5 inhibitors. The hits represent partly novel chemotypes distinct from known high-affinity CCR5 inhibitors.
Figure 3.

Chemical structures of hit compounds showing the basic nitrogen (blue) and functional groups fitting in TMS1 (green) and the hydrophobic subpocket of TMS2 (red). Interactions between the ligands and specific residues derived from the CCR5 X-ray structure (PDB code: 4MBS) are depicted by dotted lines. Structure of MVC is added for comparison.
The proposed binding modes of compounds 1–4 indicated similar orientations to that of MVC with a protonated amino group forming a salt bridge with Glu2837.39 and other groups occupying the major and minor pockets of the MVC binding site (Figure 4). The docked pose of compound 1 showed a 3,4 dimethoxyphenyl ring occupying TMS1 and a phenyl ring located in the hydrophobic subpocket of TMS2. The Glu2837.39 residue formed ionic interaction with the protonated isothiourea moiety. Interestingly, the 1-(3-ethoxy phenyl) pyrrolidinedione group projected toward ECL2 making several interactions with Ser180, a residue that is not involved in the binding of MVC.
Figure 4.

Graphical representation of the MVC binding pocket of CCR5 (PDB code: 4MBS) with the docked pose of compounds (A) 1, (B) 2, (C) 3, and (D) 4 (green sticks) showing residues from TMS1 and TMS2 (gray sticks). Only side-chain atoms are shown for clarity.
Docking of compound 2 showed its indole ring located in TMS1 and forming π–π interactions with Trp862.60, a 4H-pyran ring occupying TMS2 with a p-tolyl ring extending into the hydrophobic groove, making π–π interactions with Tyr1083.32 and a phenyl ring in a position topologically equivalent to the cyclohexyl ring of MVC projecting toward TM5.
The binding mode of compound 3 revealed the adamantane ring filling TMS1 and involved in hydrophobic interactions with Trp862.60. The basic amine formed a salt bridge with Glu2837.39 in addition to a cation−π interaction with Tyr1083.32. The TMS2 site accommodated a phenyl ring with an alkoxy substituent that could partially extend in the hydrophobic groove. Finally, a thiophene ring showed a similar binding orientation to that of the cyclohexyl ring of MVC extending toward TM5.
Lastly, docking of compound 4 revealed a benzyl ring fitting at the interface between TMS1 and TMS2 and a secondary amine slightly shifted toward TMS2 yet maintaining the salt-bridge interaction with Glu2837.39. The hydrophobic subpocket of TMS2 was occupied by one of the two phenyl rings, with the other phenyl ring oriented toward TM5. TMS1 was occupied by the benzyloxy group pointing toward TM1.
2.4. Cell Viability Assay
The effect of MVC and compounds 1–4 on cell proliferation was investigated in a 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) dye reduction assay on the SW620 CRC cell line as described previously.6 Surprisingly, for compound 1, a concentration-dependent (EC50 = 298.1 μM) increase in the SW620 CRC cell proliferation was observed after 48 h treatment, indicating a possible agonistic (positive allosteric) effect at CCR5 receptors (Figure 5). In contrast, compounds 2–4 behaved similar to MVC (CCR5 antagonist, negative allosteric modulator), causing a concentration-dependent decline in the survival rate of the SW620 cells. Interestingly, while MVC displayed IC50 = 401 μM, compounds 2–4 were significantly more potent with IC50 = 34.5, 25.0, and 96.2 μM, respectively (Figure 6). The most potent compound 3 was further investigated in western blot, cell cycle, and migration assays.
Figure 5.
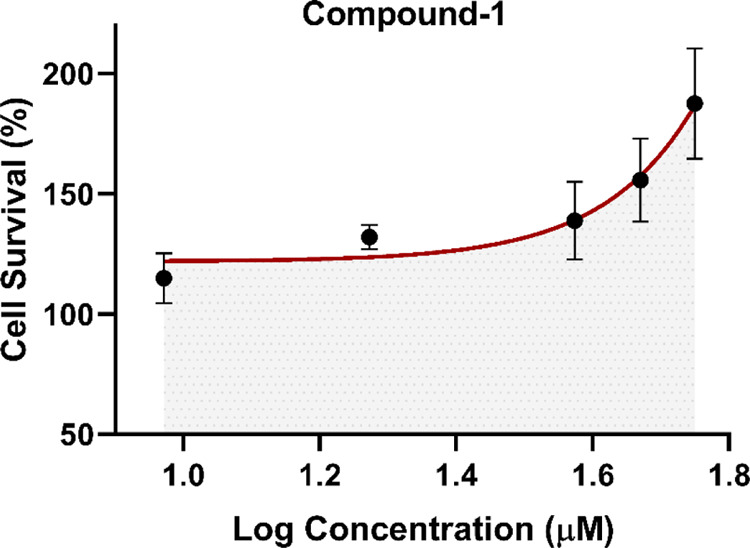
In vitro effect of compound 1 on the viability of SW620 CRC cells showing an approximate EC50 value of 300 μm.
Figure 6.

In vitro cytotoxic effects induced by MVC, compounds 2–4 in SW620 CRC cells 48 hrs post treatment. IC50 values with 95% confidence limits are given below the respective curves.
2.5. Cell-Cycle Assay
The effect of compound 3 on the cell cycle of SW620 CRC cells was examined as previously reported.6 As shown in Figure 7, the control samples showed a high distribution in the G0/G1 phase (46.5%), moderate distribution in the S phase (34.3–37.2%), and a significantly low distribution in the G2/M phase (8.5–12.5%). After 48 h treatment with compound 3, cells showed a high increase in the G0/G1 fraction by 8.6% and a nonsignificant change in the S phase relative to controls. This was followed by a subsequent decrease in the G2/M phase by 4.5%. The observed arrest of the cell cycle in the G0/G1 phase induced by compound 3 is similar to that previously reported by MVC.6
Figure 7.

Impact of compound 3 IC50 on the cell cycle of SW620 CRC cells at 48 h post treatment in comparison to the control samples C-1 (cells treated with compound 3 solvent) and C-2 (cells only).
2.6. Cell Migration Assay
The effect on migration and metastasis ability of SW620 CRC cells in response to compound 3 relative to MVC was investigated by the Boyden chamber assay.79 At 24 h post-treatment, a significant increase was observed in cell migration compared to controls treated with compound 3 (P-value = 0.0015). On the other hand, cells exposed to MVC showed a significant decrease in cell migration compared to controls (P-value = 0.0249). Compound 3 showed a significant decrease in cellular migration relative to controls 48 and 72 h post treatment with P-values of 0.0026 and 0.0014, respectively. On the other hand, cells treated with MVC showed no significance in cell migration compared to controls with P-values of 0.1413 and 0.9916, respectively (Figure 8). Overall, the cellular migration inhibitory effect of compound 3 was superior to that observed by MVC.
Figure 8.

Impact of compound 3 on migration of SW620 CRC cells at 24, 48, and 72 h post treatment in comparison to MVC, C-1 (cells treated with solvent of compound 3), C-2 (cells treated with MVC solvent), and C-3 (cells only).
2.7. Western Blot
To examine a possible involvement of CCR5 receptors in inhibiting SW620 CRC cells’ viability and migration, the expression level of the CCR5 receptor following exposure to compound 3 was examined using Western blot analysis.6 SW620 CRC cells were exposed to an IC50 concentration of compound 3. The expression levels of the CCR5 receptor were examined at the protein level after 12 and 48 h. After 12 h, a significant 8-fold increase in the CCR5 expression level compared to the control was observed. In contrast, after 48 h, the CCR5 expression level compared to the controls significantly decreased (Figure 9). These findings suggest a strong correlation between the observed cellular effects of compound 3 and the CCR5 receptor.
Figure 9.

Impact of compound 3 IC50 on the CCR5 protein expression level in SW620 CRC cells at 12 and 48 h post treatment in comparison to controls: C-1 (cells only), C-2 (cells treated with MVC solvent), and C-3 (cells treated with compound 3 solvent).
3. Discussion
The interest in the role of CCR5 in the onset and progression of tumorigenesis has led to the current focus on CCR5 as an exciting new therapeutic target for metastatic cancer with ongoing clinical trials targeting breast and colon cancers. The discovery of potent and selective CCR5 inhibitors with novel chemotypes appears promising for the development of antineoplastic agents. Herein, we utilized the 3D structure of CCR5 in complex with MVC15 and developed an integrated three-step protocol including pharmacophore modeling and molecular docking with PLIF postdocking filtration to identify novel CCR5 ligands. The pharmacophore model comprised four features covering the minor (TMS1) and major (TMS2) pockets of the MVC binding site in CCR5 as follows: (a) an aromatic moiety for interaction with Tyr1083.32 in the hydrophobic subpocket of TMS2; (b) a hydrophobic feature interacting with Trp862.60 in the TMS1 pocket, in addition to two features at the interface of the major and minor pockets; (c) a basic feature for interaction with Glu2837.39; and (d) a hydrophobic feature representing the carbon linker between the basic amine and the hydrophobic group located in TMS1 (Figure 2). The pharmacophore model revealed a symmetric distribution of hydrophobic/aromatic properties relative to the central cationic feature. Prospective VS using the multistage protocol resulted in discovery of four hit compounds 1–4 with binding modes mimicking that of MVC, occupying the major and minor pockets of the MVC binding site. Compounds 2–4 exhibited a remarkable reduction of cell proliferation of SW620 cells in a concentration-dependent manner displaying IC50 values 34.5, 25, and 96.2 μM and being 12, 16, and 4 times more potent than MVC, respectively. The most potent compound 3 induced apoptosis by arresting cells in the G0/G1 phase of the cell cycle similar to MVC. This appears to be in agreement with our previous work where targeting CCR5 by MVC induced a significant arrest in the G0/G1 phase of the cell cycle in CRC cells.6 Western blot and migrating assay showed compound 3 drastically decreasing the CCR5 expression and cellular migration 48 h post treatment, indicating its ability to inhibit metastatic activity in SW620 cells. The high CCR5 inhibitory activity of compound 3 might be explained by its high structural complementarity with the MVC binding site. It binds across both the major and minor pockets and forms the characteristic ionic interaction with Glu2837.39. However, its structure did not completely occupy the hydrophobic subpocket of the major pocket, suggesting possible future structural modification, introducing bulky hydrophobic substituents on the central phenyl ring to further increase the compound’s potency (Figure 4C).
Interestingly, compound 3 caused a significant increase in CCR5 expression in SW620 cells 12 h after exposure before a drastic decrease was observed after 48 h. In accordance, compound 3 affected cell migration in a similar manner with a significant increase in cell migration 24 h post treatment followed by complete abolishment after 48 h. These findings could suggest a positive feedback response as a result of a high CCR5 inhibitory effect of compound 3, where cells respond by initially increasing the CCR5 expression prior to complete removal of CCR5 receptors, indicating failure of the cells to restore its activity. This behavior is distinct from that observed for MVC, where no effect was observed on CCR5 expression levels at 48 h post treatment. Similar results were previously observed where MVC did not change the CCR5 expression, indicating MVC’s failure to induce CCR5 internalization.80
Unexpectedly, while MVC and compounds 2–4 inhibited proliferation of SW620 cells, compound 1 was found to increase SW620 cell proliferation, indicating its possible agonistic effect. This is remarkable considering that prospective VS campaigns often result in antagonists even when an agonist-bound VS model is used.81,82 The distinct pharmacological profiles of MVC, compound 1, and compound 3 need to be further assessed in functional assays.
The docked poses showed compound 1 extending toward ECL2, which is not the case for compounds 2–4 (Figure 4A). ECL2 was reported to have a regulatory function in GPCR activation, with several residues in ECL2 playing a role in stabilizing the protein in the active state rather than the inactive one.83 One can only speculate that binding of compound 1 induces a conformational change in ECL2 distinct from that induced by compounds 2–4. However, it needs to be further investigated by, e.g., prolonged molecular dynamics (MD) simulations of the docked poses to examine for loop movements.
In conclusion, an integrated three-step protocol including pharmacophore modeling and molecular docking followed by PLIF postdocking filtration was applied for virtual screening of the Specs database to find CCR5 receptor ligands with novel chemical scaffolds. Our VS protocol led to the identification of four novel hit compounds. Three hits showed potency comparable to or higher than MVC in cellular assays on colorectal cancer cells. Although the discovered hits belong to novel chemical classes, their structures share a protonable basic group and several aromatic rings that, as indicated by molecular docking experiments, occupy the major and minor pockets of the MVC binding site. The discovered hits are potential leads for the development of novel classes of anticolorectal agents targeting CCR5.
4. Methodology
4.1. Compound Preparation
All molecules were prepared in Molecular Operating Environment (MOE)66 version 2016.10 by washing, partial charge calculation, and energy minimization using the MMFF94x forcefield and a gradient of 0.0001 kcal/(mol Å). Protonation and tautomeric states at pH 7.0 were generated by the Structure Protonation and Recognition System (SPORES).84 Multiconformations of compounds were generated by the low MD conformational search algorithm implemented in MOE using the following settings: energy window (7 kcal/mol), elimination of the duplicate conformer threshold (RMSD, 0.25 Å), total number of iterations (10 000 steps), rejection limit (100 steps), majorization–minimization (MM) iteration limit (500 steps), and maximum conformation limit (10 000 conformers).
4.2. Pharmacophore Model Generation
A total of 2827 previously known CCR5 inhibitors, characterized by their excellent experimental performances, were collected from the binding database (http://www.bindingdb.org).20 Compounds were clustered based on their chemical scaffolds using the publicly available sca.svl script85 in MOE, and structurally similar scaffolds were further clustered using the BIT_MACCS fingerprint86 and a Tanimoto coefficient (Tc) ≥85%. The most active compound from each cluster was selected for building the pharmacophore model by overlying it on the crystallized coordinates of MVC using the publicly available align.svl script65 in MOE.66 A stochastic conformational search with an energy window of 7 kcal/mol was conducted for each compound to compute a collection of alignments. The conformation with the highest similarity to MVC (most negative F score) was chosen as a basis to search for the common pharmacophoric features shared by all active compounds using the Pharmacophore Elucidate module implemented in MOE.66 The pharmacophore models were generated by the use of pharmacophore features and projected pharmacophore features based on the Unified annotation scheme. Models were automatically generated such that the resultant queries matched all training set compounds and had a maximum number of five features. The shortest allowed distance between features was set to 1.0 Å with clustering of features within 1.25 Å from each other. The generated pharmacophore hypotheses were validated by retrospective VS using a test set composed of 2504 compounds. The test set included 60 CCR5 inhibitors collected from the literature,20−53 which were labeled as actives. The remaining 2444 molecules were labeled as inactives, comprising 91 biologically confirmed inactive compounds, 201 CCR5 decoys obtained from the GPCR Decoys Database (GDD),67 and 2152 inhibitors of different targets including GPCRs [adenosine A2a receptor (AA2A), β-1 adrenergic receptor (ADRB1), β-2 adrenergic receptor (ADRB2), C-X-C chemokine receptor type 4 (CXCR4), and dopamine D3 receptor (DRD3)] and non-GPCRs [angiotensin converting enzyme inhibitors, carbonic anhydrase inhibitors, cyclooxygenase-1 (COX-1) inhibitors, and renin inhibitors], all obtained from the GPCR Ligand Library (GLL)67 and Drugbank.68 All molecules were prepared as described in the compound preparation Section 4.1. The test set compounds were mapped to the pharmacophore model using the pharmacophore search protocol available in MOE. The following metrics were used to evaluate the effectiveness of the models in the identification of active compounds: sensitivity (Se), specificity (Sp), and enrichment factor (EF). The best model was further refined by considering spatial information, where the crystallized coordinates of MVC were used as a template to derive a molecular shape constraint.
4.3. Molecular Docking
A genetic algorithm based on Cambridge Crystallographic Data Center (CCDC) Genetic Optimization for Ligand Docking (GOLD version 5.5)69,70 was employed for molecular docking using the crystal structure of CCR5 protein complexed with MVC (PDB: 4MBS).15 Binding site residues were defined by specifying the crystal structure ligand coordinates and using the default cutoff radius of 6 Å, with the “detect cavity” option enabled. The docking experiments were performed using the GoldScore scoring function. The search efficiency of the genetic algorithm was at 200% setting with the receptor kept rigid. Water molecules were kept in the pocket while allowing the ligand to displace them during the docking experiment. For each compound, 50 complexes were generated and clustered based on their RMSD with the threshold set at 0.75 Å using the complete linkage method. The quality of pose prediction was assessed by calculating the heavy atom RMSD between the docked poses and the original PDB coordinates of MVC. The docking protocol was further assessed in terms of enrichment performance by retrospective screening of a validation database of 557 compounds. A set of 20 previously reported CCR5 inhibitors along with 537 inactives, of which 91 biologically confirmed inactive compounds, 201 CCR5 decoys obtained from the GPCR Decoy Database (GDD),67 and 245 ligands for other GPCR and non-GPCR proteins were docked into the crystal structure of the CCR5 receptor. All of the docked poses were imported into the MOE database for calculating their protein–ligand interaction fingerprints (PLIF) rows, which were utilized for generating amino acid interaction fingerprints using eight types of interactions (side-chain hydrogen bonds (donor or acceptor), backbone hydrogen bonds (donor or acceptor), solvent hydrogen bonds (donor or acceptor), ionic interactions, and π interactions). The cavity used for the PLIF analysis consisted of the same set of residues used in the docking experiments. Finally, the resultant docked poses were filtered using a set of reference PLIFs (PLIF-M1-4) and rescored using three different scoring functions: ChemPLP, ChemScore in GOLD, and DrugScore (DSX) (version 0.9)76 utilizing the DrugScorePDB potential. The receiver operator characteristic (ROC) curves were plotted based on the true-positive ra tes (TPRs) and false-positive rates (FPRs) to evaluate the model optimization. The enrichment factor (EF) and area under the ROC curve (AUROC) values were calculated and used as model selection criteria for prospective VS runs. Figures were prepared using Pymol.87
4.4. Prospective Virtual Screening
The validated pharmacophore model (PH9) was utilized as a 3D query for screening the commercially available Specs database.77 Compounds were prepared as described in the compound preparation section, saved in the smi file format, and 3D conformations were generated using the knowledge-based conformational search method of the Pharmit server.78 The resultant conformations were mapped to the pharmacophore model such that hit molecules match all of the query features. Using GOLD, the identified hit compounds were docked into the MVC binding site of the CCR5 protein (PDB code: 4MBS)15 using the GoldScore scoring function. The search efficiency of the genetic algorithm was at 200% setting with the receptor kept rigid. Finally, the docked poses were filtered using PLIF-M3 and rescored using DSXPDB.76 The chemical similarity of selected compounds was calculated using the BIT_MACCS fingerprint86 and Tanimoto coefficient against the 39 initial training set compounds.
4.5. Cell Line
SW620, a metastatic human colon adenocarcinoma cell line, was obtained from American Type Culture Collection (ATCC). It was cultured using the Roswell Park Memorial Institute (RPMI)-1640 medium (Invitrogen, Darmstadt, Germany) supplemented with fetal bovine serum (10%) and l-glutamine (2 mM). The cell line was maintained under standard incubation conditions (37 °C and 5% CO2) with a humidified atmosphere. The free pathogenic contamination SW620 cell line was passaged routinely to keep the logarithmic growth phase of the cell population.
4.6. Compound Preparation
The compounds were purchased from SPECS,77 prepared using different solvents in a stock solution of 2 mg/mL, and stored at −20 °C. Compound 1 is dissolved in dimethyl sulfoxide (DMSO) (Merck, Germany), compounds 2 and 4 in ethanol (Merck, Germany), and compound 3 in methanol (Merck, Germany). MVC was prepared in a stock solution of 25 mg/mL in ethanol (Merck, Germany).
4.7. Statistical Analysis
The analysis was performed using GraphPad Prism 8.01 software in which data were expressed as the mean ± standard error of the mean (SEM). Statistical significance was calculated using one-way analysis of variance (ANOVA). A P-value of less than 0.05 was considered statistically significant (****P-value less than 0.0001, ***P-value less than 0.001, **P-value less than 0.01, *P less than 0.05).
4.8. Cell Viability Assay
The viability of SW620 cells was assessed by the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) dye reduction assay after treatment with MVC (Selzentry, Pfizer) and compounds 1–4. Briefly, 96-well plates were used to seed the SW620 cells at a preoptimized density (5 × 103 cells/well) and then treated with increasing concentrations of MVC (340–585 μM) and compounds 1–4 (4–123 μM) for 48 h. A 10 μL/well MTT solution (10 mg/mL in phosphate-buffered saline (PBS)) and dissolving newly formed formazan crystals with 100 μL of acidic 2-propanol (0.04 N HCl) were added to assess the surviving cell fractions from the controlled and treated groups (five replicates/sample). Using the enzyme-linked immunosorbent (ELISA) plate reader (Anthos Mikrosysteme GmbH, Krefeld, Germany), the optical density was measured at 540 nm wavelength with a 690 nm reference filter. The experiment was repeated twice with five replicates to validate the results. Cell survival rates were calculated as the percentages of untreated controls, and inhibitory concentrations (ICs) were determined by GraphPad Prism 8.01 software.
4.9. Cell-Cycle Assay
The effect of compound 3 on the cell cycle was determined by propidium iodide (PI) fluorescent staining and flow cytometry analysis (FACS). In brief, SW620 cells were seeded in 25 cm2 cell culture flasks at a preoptimized density of 350 000 cells and then treated with the compounds’ IC50. The cells were harvested after 48 h from the treatment and suspended in 0.1 mL of PBS followed by the addition of ice-cold ethanol (70%) for fixation. The cells were resuspended after an incubation period of 2 h at 4 °C using PBS containing RNase-A (1 mg/mL) to digest their RNA and then incubated again for 30 min at 37 °C. Afterward, the analysis was done immediately in less than 30 min using a FACS Canto (BD Biosciences, San Jose, CA) after the addition of PI (50 μg/mL). Ten thousand events (cells) were analyzed from each sample, using ModFit LT software. The cells’ distributions in G0/G1, S, and G2/M phases of the cell cycle were calculated. Experiments were repeated twice to validate the results.
4.10. Cell Migration Assay
The cell migratory potential of the SW620 cell line in response to compound 3 treatment was assessed using the Boyden Chamber assay. In brief, SW620 cells were seeded in 25 cm2 cell culture flasks at a preoptimized density of 350 000 cells and then treated with the compounds’ IC50. After 48 h from the treatment, the cells were harvested and suspended in Opti-MEM medium and seeded with an equal cell density of 50 000 cells in 8 μm pore-size hanging cell culture inserts (Millipore, Switzerland). Afterward, the inserts were transferred to 24-well plates in which each well contains 700 μL of RPMI medium supplemented with 10% FBS and incubated for 24, 48, and 72 h. The migrated cells were quantified after each incubation period using a fluorescence reader (Synergy 2, Biotek, Germany) with excitation (560/15) and emission (590/20) filters after the addition of 140 μL of Cell Titer Blue dye (Promega, Germany) for 4 h at 37 °C.
4.11. Western Blot
The protein expression level of CCR5 in response to compound 3 treatment was assessed using western blot. Briefly, SW620 cells were seeded in 25 cm2 cell culture flasks at a preoptimized density of 350 000 cells and then treated with the compounds’ IC50. Cells were harvested after the treatment and washed in PBS after being transferred to 1.5 mL microcentrifuge tubes. Radioimmunoprecipitation assay (RIPA) buffer (150 mM sodium chloride, 1.0% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 50 mM Tris, pH 8.0) supplemented with complete protease inhibitor cocktail tablets (Roche, Mannheim, Germany) was used to lyse the cell pellet. The produced lysate was agitated for 30 min at 4 °C, and after spinning at 14 000 rpm at 4 °C for 20 min, the supernatant was collected using the Pierce protein assay. The supernatant was quantified for protein concentration, and the total protein lysates (20 μg) were subjected to electrophoresis using 4–20% polyacrylamide gel electrophoresis (PAGE) gels (Nippon Genetics Europe). After the protein was transferred onto poly(vinylidene difluoride) (PVDF) membranes, the membranes were then probed for CCR5 protein using specific antibody CKR-5 (Santa Cruz Biotechnology, Heidelberg) as per the manufacturer’s instructions. Using antimouse horseradish peroxidase (HRP) conjugated antibody (Cell Signaling Technologies, Germany) and then visualization using the ECL System (Amersham, Germany), immunoblots were developed. The endogenous reference levels of GAPDH (Santa Cruz Biotechnology, Heidelberg) were used to normalize the protein expression. Experiments were repeated twice to validate the results. The relative concentrations were assessed using ImageJ software in which densitometric analysis of digitized autographic images was analyzed.
Acknowledgments
This work was partially funded by the Federal Ministry of Education and Research (BMBF) and Deutscher Akademischer Austauschdienst (DAAD). For their help in compound testing, lab members of the Toxicology and Chemotherapy Unit (G-401) at the German Cancer Research Centre (DKFZ), Heidelberg, Germany, especially Micha Sagini and Michael Zepp, are thanked. For his help in script-writing, Mohamed El-Zohairy, a master student in Informatics at Technical University of Munich (TUM), Munich, Germany, is thanked.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c00681.
Structural alignment of training set compounds; pharmacophore models (PH2–8) overlaid on the crystal coordinates of MVC; superimposed cocrystallized and redocked ligand; ROC curves of various PLIF models; list of the pharmacophore training set compounds with their structures, source, and scaffold information; pose-retrieval docking data of MVC; structures of top 1% scoring compounds (PDF)
Smiles of all compounds (XLSX)
The authors declare no competing financial interest.
Supplementary Material
References
- Bachelerie F.; Ben-Baruch A.; Burkhardt A. M.; Combadiere C.; Farber J. M.; Graham G. J.; Horuk R.; Sparre-Ulrich A. H.; Locati M.; Luster A. D.; Mantovani A.; Matsushima K.; Murphy P. M.; Nibbs R.; Nomiyama H.; Power C. A.; Proudfoot A. E. I.; Rosenkilde M. M.; Rot A.; Sozzani S.; Thelen M.; Yoshie O.; Zlotnik A. International Union of Pharmacology: LXXXIX: update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol. Rev. 2014, 66, 1–79. 10.1124/pr.113.007724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kufareva I.; Salanga C. L.; Handel T. M. Chemokine and chemokine receptor structure and interactions: implications for therapeutic strategies. Immunol. Cell Biol. 2015, 93, 372–383. 10.1038/icb.2015.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen S. J.; Crown S. E.; Handel T. M. Chemokine: receptor structure, interactions, and antagonism. Annu. Rev. Immunol. 2007, 25, 787–820. 10.1146/annurev.immunol.24.021605.090529. [DOI] [PubMed] [Google Scholar]
- Barmania F.; Pepper M. S. Cc chemokine receptor type five (CCR5): an emerging target for the control of HIV infection. Appl. Transl. Genomics 2013, 2, 3–16. 10.1016/j.atg.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westby M.; van der Ryst E. CCR5 antagonists: host-targeted antivirals for the treatment of HIV infection. Antiviral Chem. Chemother. 2005, 16, 339–354. 10.1177/095632020501600601. [DOI] [PubMed] [Google Scholar]
- Pervaiz A.; Ansari S.; Berger M. R.; Adwan H. CCR5 blockage by maraviroc induces cytotoxic and apoptotic effects in colorectal cancer cells. Med. Oncol. 2015, 32, 158 10.1007/s12032-015-0607-x. [DOI] [PubMed] [Google Scholar]
- Pervaiz A.; Zepp M.; Mahmood S.; Ali D. M.; Berger M. R.; Adwan H. CCR5 blockage by maraviroc: a potential therapeutic option for metastatic breast cancer. Cell. Oncol. 2019, 42, 93–106. 10.1007/s13402-018-0415-3. [DOI] [PubMed] [Google Scholar]
- Jiao X.; Nawab O.; Patel T.; Kossenkov A. V.; Halama N.; Jaeger D.; Pestell R. G. Recent Advances Targeting CCR5 for Cancer and Its Role in Immuno-Oncology. Cancer Res. 2019, 79, 4801–4807. 10.1158/0008-5472.CAN-19-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junker A.; Kokornaczyk A. K.; Strunz A. K.; Wünsch B.. Selective and Dual Targeting of CCR2 and CCR5 Receptors: A Current Overview. In Chemokines; Tschammer N., Ed.; Topics in Medicinal Chemistry; Springer: Cham, 2014; Vol. 14. [Google Scholar]
- Qi B.; Fang Q.; Liu S.; Hou W.; Li J.; Huang Y.; Shi J. Advances of CCR5 antagonists: From small molecules to macromolecules. Eur. J. Med. Chem. 2020, 208, 112819 10.1016/j.ejmech.2020.112819. [DOI] [PubMed] [Google Scholar]
- Dorr P.; Westby M.; Dobbs S.; Griffin P.; Irvine B.; Macartney M.; Mori J.; Rickett G.; Smith-Burchnell C.; Napier C.; Webster R.; Armour D.; Price D.; Stammen B.; Wood A.; Perros M. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob. Agents Chemother. 2005, 49, 4721–4732. 10.1128/AAC.49.11.4721-4732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellenberger E.; Springael J. Y.; Parmentier M.; Hachet-Haas M.; Galzi J. L.; Rognan D. Identification of nonpeptide CCR5 receptor agonists by structure-based virtual screening. J. Med. Chem. 2007, 50, 1294–1303. 10.1021/jm061389p. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Zhou E.; Yu K.; Zhu J.; Zhang Y.; Xie X.; Li J.; Jiang H. Discovery of a novel CCR5 antagonist lead compound through fragment assembly. Molecules 2008, 13, 2426–2441. 10.3390/molecules13102426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thum S.; Kokornaczyk A. K.; Seki T.; De Maria M.; Ortiz Zacarias N. V.; de Vries H.; Weiss C.; Koch M.; Schepmann D.; Kitamura M.; Tschammer N.; Heitman L. H.; Junker A.; Wünsch B. Synthesis and biological evaluation of chemokine receptor ligands with 2-benzazepine scaffold. Eur. J. Med. Chem. 2017, 135, 401–413. 10.1016/j.ejmech.2017.04.046. [DOI] [PubMed] [Google Scholar]
- Tan Q.; Zhu Y.; Li J.; Chen Z.; Han G. W.; Kufareva I.; Li T.; Ma L.; Fenalti G.; Li J.; Zhang W.; Xie X.; Yang H.; Jiang H.; Cherezov V.; Liu H.; Stevens R. C.; Zhao Q.; Wu B. Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science 2013, 341, 1387–1390. 10.1126/science.1241475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han B.; Salituro F. G.; Blanco M. J. Impact of Allosteric Modulation in Drug Discovery: Innovation in Emerging Chemical Modalities. ACS Med. Chem. Lett. 2020, 11, 1810–1819. 10.1021/acsmedchemlett.9b00655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirza M. U.; Saadabadi A.; Vanmeert M.; Salo-Ahen O. M. H.; Abdullah I.; Claes S.; De Jonghe S.; Schols D.; Ahmad S.; Froeyen M. Discovery of HIV entry inhibitors via a hybrid CXCR4 and CCR5 receptor pharmacophore-based virtual screening approach. Eur. J. Pharm. Sci. 2020, 155, 105537 10.1016/j.ejps.2020.105537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H-Y.; Ho Y.; Liu H-L. Structure-based Pharmacophore Modeling to Discover Novel CCR5 Inhibitors for HIV-1/Cancers Therapy. J. Biomed. Sci. Eng. 2019, 12, 10–30. 10.4236/jbise.2019.121002. [DOI] [Google Scholar]
- Arimont M.; Sun S.-L.; Leurs R.; Smit M.; de Esch I. J. P.; de Graaf C. Structural Analysis of Chemokine Receptor-Ligand Interactions. J. Med. Chem. 2017, 60, 4735–4779. 10.1021/acs.jmedchem.6b01309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilson M. K.; Liu T.; Baitaluk M.; Nicola G.; Hwang L.; Chong J. Bindingdb in 2015: a public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1053. 10.1093/nar/gkv1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armour D. R.; De Groot M. J.; Price D. A.; Stammen B. L. C.; Wood A.; Perros M.; Burt C. The discovery of tropane derived CCR5 receptor antagonists. Chem. Biol. Drug Des. 2006, 67, 305–308. 10.1111/j.1747-0285.2006.00376.x. [DOI] [PubMed] [Google Scholar]
- Barber C. G.; Blakemore D. C.; Chiva J. Y.; Eastwood R. L.; Middleton D. S.; Paradowski K. A. 1-Amido-1-phenyl-3- piperidinylbutanes CCR5 antagonists for the treatment of HIV. Part 1. Bioorg. Med. Chem. Lett. 2009, 19, 1075–1079. 10.1016/j.bmcl.2009.01.009. [DOI] [PubMed] [Google Scholar]
- Ernst J.; Dahl R.; Lum C.; Sebo L.; Urban J.; Miller S. G.; Lundström J. Anti-HIV-1 entry optimization of novel imidazopiperidine-tropane CCR5 antagonists. Bioorg. Med. Chem. Lett. 2008, 18, 1498–1501. 10.1016/j.bmcl.2007.12.058. [DOI] [PubMed] [Google Scholar]
- Fan X.; Zhang H.; Chen L.; Long Y. Efficient synthesis and identification of novel propane-1, 3-diamino bridged CCR5 antagonists with variation on the basic center carrier. Eur. J. Med. Chem. 2010, 45, 2827–2840. 10.1016/j.ejmech.2010.03.003. [DOI] [PubMed] [Google Scholar]
- Finke P. E.; Meurer L. C.; Oates B.; Shah S. K.; Loebach J. L.; Mills S. G.; MacCoss M.; Castonguay L.; Malkowitz L.; Springer M. S.; Gould S. L.; DeMartino J. A. Antagonists of the human CCR5 receptor as anti-HIV-1 agents. Part 3: a proposed pharmacophore model for 1-[n-(methyl)-n-(phenylsulfonyl) amino]-2-(phenyl)-4-[4-(substituted) piperidin-1-yl] butanes. Bioorg. Med. Chem. Lett. 2001, 11, 2469–2473. 10.1016/S0960-894X(01)00491-7. [DOI] [PubMed] [Google Scholar]
- Hale J. J.; Budhu R. J.; Mills S. G.; MacCoss M.; Gould S. L.; DeMartino J. A.; Springer M. S.; Siciliano S. J.; Malkowitz L.; Schleif W. A.; Hazuda; Miller M.; Kessler J.; Danzeisen R.; Holmes K.; Lineberger J.; Carella; Carver G.; Emini E. A. 1, 3, 4-trisubstituted pyrrolidine CCR5 receptor antagonists. Part 3: polar functionality and its effect on anti-HIV-1 activity. Bioorg. Med. Chem. Lett. 2002, 12, 2997–3000. 10.1016/S0960-894X(02)00605-4. [DOI] [PubMed] [Google Scholar]
- Hu S.; Wang Z.; Hou T.; Ma Xi.; Li J.; Liu T.; Xie X.; Hu Y. Design, synthesis, and biological evaluation of novel 2-methylpiperazine derivatives as potent CCR5 antagonists. Bioorg. Med. Chem. Lett. 2015, 23, 1157–1168. 10.1016/j.bmc.2014.12.052. [DOI] [PubMed] [Google Scholar]
- Imamura S.; Ichikawa T.; Nishikawa Y.; Kanzaki N.; Takashima K.; Niwa S.; Iizawa Y.; Baba M.; Sugihara Y. Discovery of a piperidine-4-carboxamide CCR5 antagonist (tak-220) with highly potent anti-HIV-1 activity. J. Med. Chem. 2006, 49, 2784–2793. 10.1021/jm051034q. [DOI] [PubMed] [Google Scholar]
- Kazmierski W. M.; Anderson D. L.; Aquino C.; Chauder B. A.; Duan M.; Ferris R.; Kenakin T.; Koble C. S.; Lang D. G.; McIntyre M. S.; Peckham J.; Watson C.; Wheelan P.; Spaltenstein A.; Wire M. B.; Svolto A.; Youngman M. Novel 4, 4-disubstituted piperidine-based C-C chemokine receptor-5 inhibitors with high potency against human immunodefficiency virus-1 and an improved human ether-a-go-go related gene (hERG) profile. J. Med. Chem. 2011, 54, 3756–3767. 10.1021/jm200279v. [DOI] [PubMed] [Google Scholar]
- Kazmierski W. M.; Aquino C.; Chauder B. A.; Deanda F.; Ferris R.; Jones-Hertzog D. K.; Kenakin T.; Koble C. S.; Watson C.; Wheelan P.; Yang H.; Youngman M. Discovery of bioavailable 4, 4-disubstituted piperidines as potent ligands of the chemokine receptor 5 and inhibitors of the human immunodefficiency virus-1. J. Med. Chem. 2008, 51, 6538–6546. 10.1021/jm800598a. [DOI] [PubMed] [Google Scholar]
- Kim D.; Wang L.; Hale J. J.; Lynch C. L.; Budhu R. J.; Maccoss M.; Mills S. G.; Malkowitz L.; Gould S. L.; DeMartino J. A.; Springer M. S.; Hazuda D.; Miller M.; Kessler J.; Hrin R. C.; Carver G.; Carella A.; Henry K.; Lineberger J.; Schleif W. A.; Emini E. A. Potent 1, 3, 4-trisubstituted pyrrolidine CCR5 receptor antagonists: effects of fused heterocycles on antiviral activity and pharmacokinetic properties. Bioorg. Med. Chem. Lett. 2005, 15, 2129–2134. 10.1016/j.bmcl.2005.02.030. [DOI] [PubMed] [Google Scholar]
- Lemoine R. C.; Petersen A. C.; Setti L.; Wanner J.; Jekle A.; Heilek G.; deRosier A.; Ji C.; Berry P.; Rotstein D. Evaluation of secondary amide replacements in a series of CCR5 antagonists as a means to increase intrinsic membrane permeability. Part 1: Optimization of gemdisubstituted azacycles. Bioorg. Med. Chem. Lett. 2010, 20, 704–708. 10.1016/j.bmcl.2009.11.072. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Zhou E.; Yu K.; Zhu J.; Zhang Y.; Xie X.; Li J.; Jiang H. Discovery of a novel CCR5 antagonist lead compound through fragment assembly. Molecules 2008, 13, 2426–2441. 10.3390/molecules13102426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S. F.; Chen B.; Davey D.; Dunning L.; Jaroch S.; May K.; Onuffer J.; Phillips G.; Subramanyam B.; Tseng J. L.; Wei R. G.; Wei M.; Ye B. CCR5 receptor antagonists: Discovery and SAR of novel 4-hydroxypiperidine derivatives. Bioorg. Med. Chem. Lett. 2007, 17, 1883–1887. 10.1016/j.bmcl.2007.01.050. [DOI] [PubMed] [Google Scholar]
- Nishizawa R.; Nishiyama T.; Hisaichi K.; Matsunaga N.; Minamoto C.; Habashita H.; Takaoka Y.; Toda M.; Shibayama S.; Tada H.; Sagawa K.; Fukushima D.; Maeda K.; Mitsuya H. Spirodiketopiperazine-based CCR5 antagonists: Lead optimization from biologically active metabolite. Bioorg. Med. Chem. Lett. 2007, 17, 727–731. 10.1016/j.bmcl.2006.10.084. [DOI] [PubMed] [Google Scholar]
- Palani A.; Shapiro S.; Clader J. W.; Greenlee W. J.; Vice S.; McCombie S.; Cox K.; Strizki J.; Baroudy B. M. Oximinopiperidino- piperidine-based CCR5 antagonists. Part 2: synthesis, SAR and biological evaluation of symmetrical heteroaryl carboxamides. Bioorg. Med. Chem. Lett. 2003, 13, 709–712. 10.1016/S0960-894X(02)01063-6. [DOI] [PubMed] [Google Scholar]
- Palani A.; Tagat J. R. Discovery and development of small molecule chemokine coreceptor CCR5 antagonists. J. Med. Chem. 2006, 49, 2851–2857. 10.1021/jm060009x. [DOI] [PubMed] [Google Scholar]
- Pryde D. C.; Corless M.; Fenwick D. R.; Mason H. J.; Stammen B. C.; Stephenson P. T.; Ellis D.; Bachelor D.; Gordon D.; Barber C. G.; Wood A.; Middleton D. S.; Blakemore D. C.; Parsons G. C.; Eastwood R.; Platts M. Y.; Statham K.; Paradowski K. A.; Burt C.; Klute W. The design and discovery of novel amide CCR5 antagonists. Bioorg. Med. Chem. Lett. 2009, 19, 1084–1088. 10.1016/j.bmcl.2009.01.012. [DOI] [PubMed] [Google Scholar]
- Rotstein D. M.; Gabriel S. D.; Manser N.; Filonova L.; Padilla F.; Sankuratri S.; Ji C.; deRosier A.; Dioszegi M.; Heilek G.; Jekle A.; Weller P.; Berry P. Synthesis, SAR and evaluation of [1, 4]-bipiperidinyl-4-yl-imidazolidin-2-one derivatives as novel CCR5 antagonists. Bioorg. Med. Chem. Lett. 2010, 20, 3219–3222. 10.1016/j.bmcl.2010.04.077. [DOI] [PubMed] [Google Scholar]
- Rotstein D. M.; Melville C. R.; Padilla F.; Cournoyer D.; Lee E. K.; Lemoine R.; Petersen A. C.; Setti L. Q.; Wanner J.; Chen L.; Filonova L.; Loughhead D. G.; Manka J.; Lin X. F.; Gleason S.; Sankuratri S.; Ji C.; Derosier A.; Dioszegi M.; Heilek G.; Jekle A.; Berry P.; Mau C. I.; Weller P. Novel hexahydropyrrolo [3, 4-c] pyrrole CCR5 antagonists. Bioorg. Med. Chem. Lett. 2010, 20, 3116–3119. 10.1016/j.bmcl.2010.03.095. [DOI] [PubMed] [Google Scholar]
- Seto M.; Aikawa K.; Miyamoto N.; Aramaki Y.; Kanzaki N.; Takashima K.; Kuze Y.; Iizawa Y.; Baba M.; Shiraishi M. Highly potent and orally active CCR5 antagonists as anti-HIV-1 agents: synthesis and biological activities of 1-benzazocine derivatives containing a sulfoxide moiety. J. Med. Chem. 2006, 49, 2037–2048. 10.1021/jm0509703. [DOI] [PubMed] [Google Scholar]
- Shah S. K.; Chen N.; Guthikonda R. N.; Mills S. G.; Malkowitz L.; Springer M. S.; Gould S. L.; Demartino J. A.; Carella A.; Carver G.; Holmes K.; Schleif W. A.; Danzeisen R.; Hazuda D.; Kessler J.; Lineberger J.; Miller M.; Emini E. A.; MacCoss M. Synthesis and evaluation of CCR5 antagonists containing modified 4-piperidinyl-2-phenyl-1-(phenylsulfonylamino)-butane. Bioorg. Med. Chem. Lett. 2005, 15, 977–982. 10.1016/j.bmcl.2004.12.044. [DOI] [PubMed] [Google Scholar]
- Shankaran K.; Donnelly K. L.; Shah S. K.; Caldwell C. G.; Chen P.; Finke P. E.; Oates B.; MacCoss M.; Mills S. G.; DeMartino J. A.; Gould S. L.; Malkowitz L.; Siciliano S. J.; Springer M. S.; Kwei G.; Carella A.; Carver G.; Danzeisen R.; Hazuda D.; Holmes K.; Kessler J.; Lineberger J.; Miller M. D.; Emini E. A.; Schleif W. A. Syntheses and biological evaluation of 5-(piperidin-1-yl)-3-phenylpentylsulfones as CCR5 antagonists. Bioorg. Med. Chem. Lett. 2004, 14, 3589–3593. 10.1016/j.bmcl.2004.03.112. [DOI] [PubMed] [Google Scholar]
- Skerlj R.; Bridger G.; Zhou Y.; Bourque E.; McEachern E.; Danthi S.; Langille J.; Harwig C.; Veale D.; Carpenter B.; Ba T.; Bey M.; Baird I.; Wilson T.; Metz M.; MacFarland R.; Mosi R.; Bodart V.; Wong R.; Fricker S.; Huskens D.; Schols D. Mitigating hERG inhibition: design of orally bioavailable CCR5 antagonists as potent inhibitors of R5 HIV-1 replication. ACS Med. Chem. Lett. 2012, 3, 216–221. 10.1021/ml2002604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skerlj R.; Bridger G.; Zhou Y.; Bourque E.; McEachern E.; Langille J.; Harwig C.; Veale D.; Yang W.; Li T.; Zhu Y.; Bey M.; Baird I.; Sartori M.; Metz M.; Mosi R.; Nelson K.; Bodart V.; Wong R.; Fricker S.; Mac Farland R.; Huskens D.; Schols D. Design and synthesis of pyridin-2-ylmethylaminopiperidin-1-yl-butyl amide CCR5 antagonists that are potent inhibitors of m-tropic (R5) HIV-1 replication. Bioorg. Med. Chem. Lett. 2011, 21, 6950–6954. 10.1016/j.bmcl.2011.09.133. [DOI] [PubMed] [Google Scholar]
- Skerlj R.; Bridger G.; Zhou Y.; Bourque E.; McEachern E.; Metz M.; Harwig C.; Li T. S.; Yang W.; Bogucki D.; Zhu Y.; Langille J.; Veale D.; Ba T.; Bey M.; Baird I.; Kaller A.; Krumpak M.; Leitch D.; Satori M.; Vocadlo K.; Guay D.; Nan S.; Yee H.; Crawford J.; Chen G.; Wilson T.; Carpenter B.; Gauthier D.; Macfarland R.; Mosi R.; Bodart V.; Wong R.; Fricker S.; Schols D. Design of substituted imidazolidinylpiperidinylbenzoic acids as chemokine receptor 5 antagonists: potent inhibitors of R5 HIV-1 replication. J. Med. Chem. 2013, 56, 8049–8065. 10.1021/jm401101p. [DOI] [PubMed] [Google Scholar]
- Strizki J. M.; Xu S.; Wagner N. E.; Wojcik L.; Liu J.; Hou Y.; Endres M.; Palani A.; Shapiro S.; Clader J. W.; Greenlee W. J.; Tagat J. R.; McCombie S.; Cox K.; Fawzi A. B.; Chou C. C.; Pugliese-Sivo C.; Davies L.; Moreno M. E.; Ho D. D.; Trkola A.; Stoddart C. A.; Moore J. P.; Reyes G. R.; Baroudy B. M. SCH-C (SCH 351125), an orally bioavailable, small molecule antagonist of the chemokine receptor CCR5, is a potent inhibitor of HIV-1 infection in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 12718–12723. 10.1073/pnas.221375398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupple P. A.; Batchelor D. V.; Corless M.; Dorr P. K.; Ellis D.; Fenwick D. R.; Galan S. R.; Jones R. M.; Mason H. J.; Middleton D. S.; Perros M.; Perruccio F.; Platts M. Y.; Pryde D. C.; Rodrigues D.; Smith N. N.; Stephenson P. T.; Webster R.; Westby M.; Wood A. of N-{(1S)-1-(3-fluorophenyl)-3-[(3-endo)-3-(5-isobutyryl-2-methyl-4,5,6,7-tetrahydro-1H-imidazo[4,5-c]pyridin-1-yl)-8-azabicyclo[3.2.1]oct-8-yl]propyl}acetamide (PF-232798). J. Med. Chem. 2011, 54, 67–77. 10.1021/jm100978n. [DOI] [PubMed] [Google Scholar]
- Tagat J. R.; McCombie S. W.; Steensma R. W.; Lin S.; Nazareno D. V.; Baroudy B.; Vantuno N.; Xu S.; Liu J. Piperazine-based CCR5 antagonists as HIV-1 inhibitors. I: 2 (S)-methyl piperazine as a key pharmacophore element. Bioorg. Med. Chem. Lett. 2001, 11, 2143–2146. 10.1016/S0960-894X(01)00381-X. [DOI] [PubMed] [Google Scholar]
- Thoma G.; Beerli C.; Bigaud M.; Bruns C.; Cooke N. G.; Streiff M. B.; Zerwes H. Reduced cardiac side-effect potential by introduction of polar groups: discovery of NIBR-1282, an orally bioavailable CCR5 antagonist which is active in vivo. Bioorg. Med. Chem. Lett. 2008, 18, 2000–2005. 10.1016/j.bmcl.2008.01.108. [DOI] [PubMed] [Google Scholar]
- Wei R. G.; Arnaiz D. O.; Chou Y.; Davey D.; Dunning L.; Lee W.; Lu S.; Onuer J.; Ye B.; Phillips G. CCR5 receptor antagonists: Discovery and SAR study of guanylhydrazone derivatives. Bioorg. Med. Chem. Lett. 2007, 17, 231–234. 10.1016/j.bmcl.2006.09.052. [DOI] [PubMed] [Google Scholar]
- Xue C. B.; Chen L.; Cao G.; Zhang K.; Wang A.; Meloni D.; Glenn J.; Anand R.; Xia M.; Kong L.; Huang T.; Feng H.; Zheng C.; Li M.; Galya L.; Zhou J.; Shin N.; Baribaud F.; Solomon K.; Scherle P.; Zhao B.; Diamond S.; Emm T.; Keller D.; Contel N.; Yeleswaram S.; Vaddi K.; Hollis G.; Newton R.; Friedman S.; Metcalf B. Discovery of INCB9471, a potent, selective, and orally bioavailable CCR5 antagonist with potent anti-HIV-1 activity. ACS Med. Chem. Lett. 2010, 1, 483–487. 10.1021/ml1001536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.; Lin X. F.; Padilla F.; Gabriel S. D.; Heilek G.; Ji C.; Sankuratri S.; deRosier A.; Berry P.; Rotstein D. M. Discovery of a potent, selective and orally bioavailable 3, 9-diazaspiro [5.5] undeca-2-one CCR5 antagonist. Bioorg. Med. Chem. Lett. 2009, 19, 209–213. 10.1016/j.bmcl.2008.10.115. [DOI] [PubMed] [Google Scholar]
- Zhang H. S.; Feng D. Z.; Chen L.; Long Y. Q. Discovery of novel-(S)-alpha-phenyl-gamma-amino butanamide containing CCR5 antagonists via functionality inversion approach. Bioorg. Med. Chem. Lett. 2010, 20, 2219–2223. 10.1016/j.bmcl.2010.02.023. [DOI] [PubMed] [Google Scholar]
- Duan M.; Peckham J.; Edelstein M.; Ferris R.; Kazmierski W. M.; Spaltenstein A.; Wheelan P.; Xiong Z. Discovery of N-benzyl-N′-(4-pipyridinyl) urea CCR5 antagonists as anti-HIV-1 agents (I): Optimization of the amine portion. Bioorg. Med. Chem. Lett. 2010, 20, 7397–7400. 10.1016/j.bmcl.2010.10.033. [DOI] [PubMed] [Google Scholar]
- Kim D.; Wang L.; Caldwell C. G.; Chen P.; Finke P. E.; Oates B.; MacCoss M.; Mills S. G.; Malkowitz L.; Gould S. L.; DeMartino J. A.; Springer M. S.; Hazuda D.; Miller M.; Kessler J.; Danzeisen R.; Carver G.; Carella A.; Holmes K.; Lineberger J.; Schleif W. A.; Emini E. A. Discovery of human CCR5 antagonists containing hydantoins for the treatment of HIV-1 infection. Bioorg. Med. Chem. Lett. 2001, 11, 3099–3102. 10.1016/S0960-894X(01)00654-0. [DOI] [PubMed] [Google Scholar]
- Lemoine R. C.; Petersen A. C.; Setti L.; Chen L.; Wanner J.; Jekle A.; Heilek G.; deRosier A.; Ji C.; Rotstein D. M. Evaluation of a 4-aminopiperidine replacement in several series of CCR5 antagonists. Bioorg. Med. Chem. Lett. 2010, 20, 1830–1833. 10.1016/j.bmcl.2010.02.004. [DOI] [PubMed] [Google Scholar]
- Kim D.; Wang L.; Caldwell C. G.; Chen P.; Finke P. E.; Oates B.; MacCoss M.; Mills S. G.; Malkowitz L.; Gould S. L.; DeMartino J. A.; Springer M. S.; Hazuda D.; Miller M.; Kessler J.; Danzeisen R.; Carver G.; Carella A.; Holmes K.; Lineberger J.; Schleif W. A.; Emini E. A. Design, synthesis, and SAR of heterocycle-containing antagonists of the human CCR5 receptor for the treatment of HIV-1 infection. Bioorg. Med. Chem. Lett. 2001, 11, 3103–3106. 10.1016/S0960-894X(01)00655-2. [DOI] [PubMed] [Google Scholar]
- Bhalay G.; Abrecht B.; Akhlaq M.; Baettig U.; Beer D.; Brown Z.; Charlton S.; Dunstan A.; Bradley M.; Gedeck P.; Glen A.; Howe T.; Keller T.; Leighton-Davies J.; Li A.; McCarthy C.; Mocquet C.; Owen C.; Nicklin P.; Rosethorne E. Design and synthesis of a library of chemokine antagonists. Bioorg. Med. Chem. Lett. 2011, 21, 6249–6252. 10.1016/j.bmcl.2011.09.013. [DOI] [PubMed] [Google Scholar]
- Cumming J. G.; Brown S. J.; Cooper A. E.; Faull A. W.; Flynn A. P.; Grime K.; Oldfield J.; Shaw J. S.; Shepherd E.; Tucker H.; Whittaker D. Modulators of the human CCR5 receptor. Part 3: SAR of substituted 1-[3-(4-methanesulfonylphenyl)-3-phenylpropyl]-piperidinyl phenylacetamides. Bioorg. Med. Chem. Lett. 2006, 16, 3533–3536. 10.1016/j.bmcl.2006.03.089. [DOI] [PubMed] [Google Scholar]
- Hale J. J.; Budhu R. J.; Holson E. B.; Finke P. E.; Oates B.; Mills S. G.; MacCoss M.; Gould S. L.; DeMartino J. A.; Springer M. S.; Siciliano S.; Malkowitz L.; Schleif W. A.; Hazuda D.; Miller M.; Kessler J.; Danzeisen R.; Holmes K.; Lineberger J.; Carella A.; Carver G.; Emini E. 1, 3, 4-Trisubstituted pyrrolidine CCR5 receptor antagonists. Part 2: lead optimization affording selective, orally bioavailable compounds with potent anti-HIV activity. Bioorg. Med. Chem. Lett. 2001, 11, 2741–2745. 10.1016/S0960-894X(01)00545-5. [DOI] [PubMed] [Google Scholar]
- Finke P. E.; Oates B.; Mills S. G.; MacCoss M.; Malkowitz L.; Springer M. S.; Gould S. L.; DeMartino J. A.; Carella A.; Carver G.; Holmes K.; Danzeisen R.; Hazuda D.; Kessler J.; Lineberger J.; Miller M.; Schleif W. A.; Emini E. A. Antagonists of the human CCR5 receptor as anti-HIV-1 agents. Part 4: synthesis and structure–Activity relationships for 1-[N-(Methyl)-N-(phenylsulfonyl) amino]-2-(phenyl)-4-(4-(N-(alkyl)-N-(benzyloxycarbonyl) amino) piperidin-1-yl) butanes. Bioorg. Med. Chem. Lett. 2001, 11, 2475–2479. 10.1016/S0960-894X(01)00492-9. [DOI] [PubMed] [Google Scholar]
- Lemoine R. C.; Petersen A. C.; Setti L.; Baldinger T.; Wanner J.; Jekle A.; Heilek G.; deRosier A.; Ji C.; Rotstein D. M. Evaluation of a 3-amino-8-azabicyclo [3.2. 1] octane replacement in the CCR5 antagonist maraviroc. Bioorg. Med. Chem. Lett. 2010, 20, 1674–1676. 10.1016/j.bmcl.2010.01.080. [DOI] [PubMed] [Google Scholar]
- Finke P. E.; Meurer L. C.; Oates B.; Mills S. G.; MacCoss M.; Malkowitz L.; Springer M. S.; Daugherty B. L.; Gould S. L.; DeMartino J. A.; Siciliano S. J.; Carella A.; Carver G.; Holmes K.; Danzeisen R.; Hazuda D.; Kessler J.; Lineberger J.; Miller M.; Schleif W. A.; Emini E. A. Antagonists of the human CCR5 receptor as anti-HIV-1 agents. Part 2: structure–activity relationships for substituted 2-aryl-1-[N-(methyl)-N-(phenylsulfonyl) amino]-4-(piperidin-1-yl) butanes. Bioorg. Med. Chem. Lett. 2001, 11, 265–270. 10.1016/S0960-894X(00)00639-9. [DOI] [PubMed] [Google Scholar]
- Bondarev D.Align_all.svl, CCG SVL Exchange, 2017. https://svl.chemcomp.com.
- Molecular Operating Environment (MOE), version 2019.01; Chemical Computing Group ULC: Montreal, QC, 2019.
- Gatica E. A.; Cavasotto C. N. Ligand and decoy sets for docking to G protein-coupled receptors. J. Chem. Inf. Model. 2012, 52, 1–6. 10.1021/ci200412p. [DOI] [PubMed] [Google Scholar]
- Wishart D. S.; Knox C.; Guo A. C.; Cheng D.; Shrivastava S.; Tzur D.; Gautam B.; Hassanali M. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008, 36, D901–D906. 10.1093/nar/gkm958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones G.; Willett P.; Glen R. C. Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J. Mol. Biol. 1995, 245, 43–53. 10.1016/S0022-2836(95)80037-9. [DOI] [PubMed] [Google Scholar]
- Jones G.; Willett P.; Glen R. C.; Leach A. R.; Taylor R. Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Biol. 1997, 267, 727–748. 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- Da C.; Kireev D. Structural protein–ligand interaction fingerprints (SPLIF) for structure-based virtual screening: method and benchmark study. J. Chem. Inf. Model. 2014, 54, 2555–2561. 10.1021/ci500319f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anighoro A.; Bajorath J. Three-dimensional similarity in molecular docking: prioritizing ligand poses on the basis of experimental binding modes. J. Chem. Inf. Model. 2016, 56, 580–587. 10.1021/acs.jcim.5b00745. [DOI] [PubMed] [Google Scholar]
- Anighoro A.; Bajorath J. Binding mode similarity measures for ranking of docking poses: a case study on the adenosine A2A receptor. J. Comput.-Aided Mol. Des. 2016, 30, 447–456. 10.1007/s10822-016-9918-z. [DOI] [PubMed] [Google Scholar]
- Lau G.; Labrecque J.; Metz M.; Vaz R.; Fricker S. P. Specificity for a CCR5 inhibitor is conferred by a single amino acid residue role of ILE198. J. Biol. Chem. 2015, 290, 11041–11105. 10.1074/jbc.M115.640169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondru R.; Zhang J.; Ji C.; Mirzadegan T.; Rotstein D.; Sankuratri S.; Dioszegi M. Molecular interactions of CCR5 with major classes of small-molecule anti-HIV CCR5 antagonists. Mol. Pharmacol. 2008, 73, 789–800. 10.1124/mol.107.042101. [DOI] [PubMed] [Google Scholar]
- Neudert G.; Klebe G. DSX: a knowledge-based scoring function for the assessment of protein-ligand complexes. J. Chem. Inf. Model. 2011, 51, 2731–2745. 10.1021/ci200274q. [DOI] [PubMed] [Google Scholar]
- Specs. Available online: http://www.specs.net (accessed July 30, 2019).
- Sunseri J.; Koes D. R. Pharmit: interactive exploration of chemical space. Nucleic Acids Res. 2016, 44, W442–W448. 10.1093/nar/gkw287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grotendorst G. R. Spectrophotometric assay for the quantitation of cell migration in the Boyden chamber chemotaxis assay. Methods Enzymol. 1987, 147, 144–152. 10.1016/0076-6879(87)47105-X. [DOI] [PubMed] [Google Scholar]
- López-Huertas M. R.; Jiménez-Tormo L.; Madrid-Elena N.; Gutiérrez C.; Rodríguez-Mora S.; Coiras M.; Alcamí J.; Moreno S. The CCR5-antagonist Maraviroc reverses HIV-1 latency in vitro alone or in combination with the PKC-agonist Bryostatin-1. Sci. Rep. 2017, 7, 2385 10.1038/s41598-017-02634-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu J.; Wang S.; Balius T. E.; Singh I.; Levit A.; Moroz Y. S.; O’Meara M. J.; Che T.; Algaa E.; Tolmachova K.; Tolmachev A. A.; Shoichet B. K.; Roth B. L.; Irwin J. J. Ultra-large library docking for discovering new chemotypes. Nature 2019, 566, 224–229. 10.1038/s41586-019-0917-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth B. L. Molecular pharmacology of metabotropic receptors targeted by neuropsychiatric drugs. Nat. Struct. Mol. Biol. 2019, 26, 535–544. 10.1038/s41594-019-0252-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlshøj S.; Amarandi R. M.; Larsen O.; Daugvilaite V.; Steen A.; Brvar M.; Pui A.; Frimurer T. M.; Ulven T.; Rosenkilde M. M. Molecular mechanism of action for allosteric modulators and agonists in CC-chemokine Receptor 5 (CCR5). J. Biol. Chem. 2016, 291, 26860–26874. 10.1074/jbc.M116.740183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ten Brink T.; Exner T. E. pka based protonation states and microspecies for protein-ligand docking. J. Comput.-Aided Mol. Des. 2010, 24, 935–942. 10.1007/s10822-010-9385-x. [DOI] [PubMed] [Google Scholar]
- Kirsten G.Sca.svl, CCG SVL Exchange, 2012. https://svl.chemcomp.com.
- Durant J. L.; Leland B. A.; Henry D. R.; Nourse J. G. Reoptimization of MDL keys for use in drug discovery. J. Chem. Inf. Comput. Sci. 2002, 42, 1273–1280. 10.1021/ci010132r. [DOI] [PubMed] [Google Scholar]
- Lilkova E.et al. The PyMOL Molecular Graphics System, version 2.0; Schrödinger, LLC, 2015.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.