

Abstract
In order to improve the performance of well-established photocatalysts and to develop new potential photocatalyst materials, an understanding of the underlying mechanisms of photocatalytic reactions is of the utmost importance. An often neglected method for studying the mechanism is the investigation of isotope effects. Although just a few studies related to isotope effects exist, it has been shown to be a powerful tool for exploring mechanisms of photocatalytic processes. Most of the reports are focused on TiO2, which is the most studied photocatalyst, while there is a lack of data for other photocatalyst materials. This mini-review represents an overview of research utilizing isotope effects in the area of photocatalysis. The benefits and the importance of these studies will be highlighted, and the potential for these processes to be applied for the study of further photocatalytic reactions and different photocatalyst materials will be shown.
Introduction
Semiconductor photocatalysis is a versatile technology that has been applied to a broad range of applications from treatment of contaminated water and air to energy conversion and storage.1a−1e In designing and developing this process for practical commercial applications, it is critical to have a robust understanding of fundamental mechanistic processes that are occurring on the surface of the semiconductor material.1e−1j A broad range of physical and chemical methods have been used in developing our understanding of these surface processes over the past four decades.1j−1o One area that has not been applied to the same extent is the application of isotope effects to probe photocatalytic processes and mechanisms.
For this purpose, it is always necessary to perform two sets of experiments. In one experimental run one species (e.g., the photocatalyst) is labeled, and in a further run the same reaction is performed but with the same species unlabeled. This allows a comparison of both runs to make conclusions related to the mechanism of the investigated reaction.
The labeling of reactant molecules or the photocatalyst allows us to study the transfer of atoms between both species. For a correct interpretation of the obtained results, several points need to be considered. Besides a reaction during illumination, also a reaction in the dark needs to be performed since it might be possible that an exchange of atoms occurs spontaneously between the photocatalyst and the reactant molecules, which could lead to wrong conclusions for the reaction under illumination. Further, it is important to consider all possible reaction products that might be produced. For example, if CO2 is the reaction product and the incorporation of 18O is expected, it is necessary to detect C16O2, C16O18O, and C18O2.
In the case of the investigation of solvent isotope effects, the effect of the exchange of H2O by D2O on the rate constant is investigated. It can be recommended not only to perform experiments in pure H2O or D2O but also to consider mixtures of both solvents.
This paper explores the areas in which these techniques have been used successfully to date and also looks at the scope for more extensive application of such methods to studying the photocatalytic process promoted by semiconductor materials. In this mini-review, works from our groups as well as works from other groups are considered.
Isotopic Labeling of Semiconductor Photocatalyst Materials and Target Molecules for Reaction
Isotopic labeling is a method to investigate the incorporation of atoms from reactant molecules into the surface of the photocatalyst and vice versa during a reaction, which allows the mechanism of chemical processes on surfaces to be followed. In the case of TiO2 as a photocatalyst, oxygen labeling (exchange of 16O by 18O) is commonly used. For this, two different methods exist, since the reactant molecules can be either isotopic labeled or TiO2 itself.
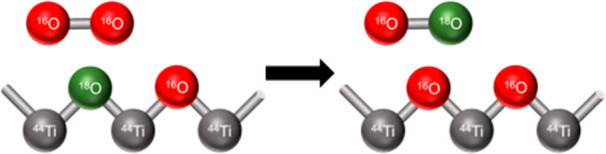
Courbon et al.2 showed that after 18O2 adsorption on Ti16O2 and following UV illumination 16O18O and 16O2 can be detected in the gas phase, while the 18O2 content decreases, which proves the incorporation of 18O2– into the surface of TiO2. Furthermore, they were able to confirm that only one surface oxygen anion is involved in the exchange at a time. The proposed mechanism of the oxygen isotopic exchange (OIE), as described by Pichat et al.,3 is depicted in Figure 1. After the excitation with light, with an energy equal to or higher than the band gap energy of TiO2, electron–hole pairs are formed. The conduction band electrons are able to reduce 18O2, which leads to the formation of superoxide radicals (18O2•–). Since the additional electron is in an antibonding orbital, the O–O bond is weakened in the superoxide radical. In the lattice of the oxide, an 16O2– anion transfers an electron to the conduction band, forming an 16O•– radical, which causes a weakened bond to the neighboring Ti4+ cations. Further, the generated species react with each other, leading to the incorporation of 18O2– in the surface and the release of 16O18O and 16O2 to the gas phase. However, Pichat et al.3 pointed out that there is, to the best of their knowledge, no proof for the existence of a complex between 18O2•– and 16O•–.
Figure 1.

Proposed mechanism of oxygen isotopic exchange by Pichat et al.3 Created in analogy to ref (3).
Tanaka4 suggested that the OIE reaction proceeds via an O3•– radical anion, while Murata et al.5 were able to confirm the formation of this species. Equation 1 describes the reaction mechanism based on the formation of an O3•– intermediate,6 which thus rules out a direct involvement of a superoxide species as proposed by Pichat et al.3 Courbon et al.2 found that the activity for the photooxidation of isobutane correlates with the activity for the OIE and thus concluded that an O•– species is involved in both reactions, which supports the mechanism.
| 1 |
The isotopic-labeled oxygen can be used also simultaneously with other unlabeled molecules to investigate the effect of the OIE. For example, Liao et al.7 reported that during UV illumination of a TiO2 surface no oxygen exchange between 18O2 and adsorbed CO occurs, while for adsorbed CO2, CO3, and HCOO species an oxygen exchange was observed.
A further approach is to use directly isotopic-labeled reactant molecules instead of 18O2. Isotopic-labeled water (H218O) was used by Nakamura et al.8 to support their proposed mechanism (Figure 2) for the oxygen evolution of rutile in contact with aqueous solutions during illumination, with Ti–O–O–OH and Ti–O–O–Ti as intermediates. Zhang et al.9 showed that 18O-enriched cyclohexanol and benzyl alcohol form in the presence of TiO2 and 16O2 during illumination in benzotrifluoride cyclohexanone and benzaldehyde that contain approximately 100% 16O. Further, they were able to exclude the possibility of oxygen transfer from the TiO2 to the molecule.
Figure 2.

Mechanism for the oxygen evolution of TiO2 (rutile) in aqueous solutions with a pH between 1 and 12 during illumination. Reprinted with permission from ref (8). Copyright 2004 American Chemical Society.
Besides isotopic labeling of the reactant molecules, a further method is to label the photocatalyst. 18O-enriched surfaces can be prepared via different approaches. The surface of Ti16O2 can be 18O-enriched in contact with 18O2 at 750 K,10 under UV irradiation in H218O for 12 h,9 or by applying a potential of 1 V in an 18O-containing electrolyte during illumination.11 It needs to be taken into account that the 18O-enrichment is limited to the surface of TiO2.12 Kavan et al.13 reported the synthesis of isotopically pure Ti18O2 by the hydrolysis of TiCl4 in H218O, yielding anatase, which can be transformed to rutile by heating to 1000 °C in a vacuum.
Henderson10 investigated the formic acid decomposition at an 18O-enriched (100) TiO2 crystal without illumination. It was demonstrated that 18O-containing products are released during the decomposition (H2C18O, HC16O18OH, HC18O+, and H218O), which confirms the incorporation of lattice oxygen into the products. Henderson14 also investigated the same reaction at an 18O-enriched (110) TiO2 crystal in the dark. Similar as for the (100) crystal, the transfer of lattice oxygen to the product molecules was observed. Bogdanoff and Alonso-Vante11 studied the photoelectrooxidation of formic acid in the presence of 18O-enriched TiO2, but no incorporation of 18O was observed in the detected CO2 molecules. Civiš et al.15 reported for formic acid in contact with isotopically pure Ti18O2 during illumination that no oxygen exchange occurs between the oxygen atoms of Ti18O2 and the formic acid during adsorption and decomposition since strongly bonded formate species inhibit the exchange. Although no exchange takes place, C16O18O and C18O2 can be detected. These products are formed by the spontaneous exchange of oxygen between C16O2 molecules and Ti18O2. The comparison of the studies shows that there is no direct agreement, if lattice oxygen can be incorporated in the products of the formic acid decomposition or if products containing 18O can be detected at all. It needs to be noted that only Civiš et al.15 used isotopically pure Ti18O2, while in the other reports 18O-enriched surfaces were investigated.
Kavan et al.13 reported for the interface between isotopically pure anatase Ti18O2 and C16O2 that without illumination an OIE reaction occurs, while both C18O2 and C16O18O were detected in the gas phase. If the surface of anatase is covered with adsorbed HCl and water, no OIE was observed. Civiš et al.12 investigated the same interface in the dark and during illumination. As shown in Figure 3, an involvement of oxygen vacancies in the OIE reaction in the dark was found, while the whole process was found to be very fast. One oxygen atom of each C16O2 molecule coordinates to a vacancy, while the carbon atoms coordinate to lattice oxygen (Figure 3a). A CO3 bidentate species is formed with one oxygen atom from the C16O2 molecule being incorporated into the TiO2 lattice (Figure 3b). Afterward a C16O18O molecule is released from the surface, recreating an oxygen vacancy (Figure 3c). The major product of the OIE is C18O2 with a minor content of C16O18O. The adsorption of water on the surface did not suppress the OIE, and thus the water is not competing with CO2 for adsorption sites. By laser irradiation of the H216O-treated Ti18O2, it was possible to enhance the OIE reaction with C16O2. Further, as products of the photocatalytic reduction of C16O2, methane and C16O were detected.
Figure 3.
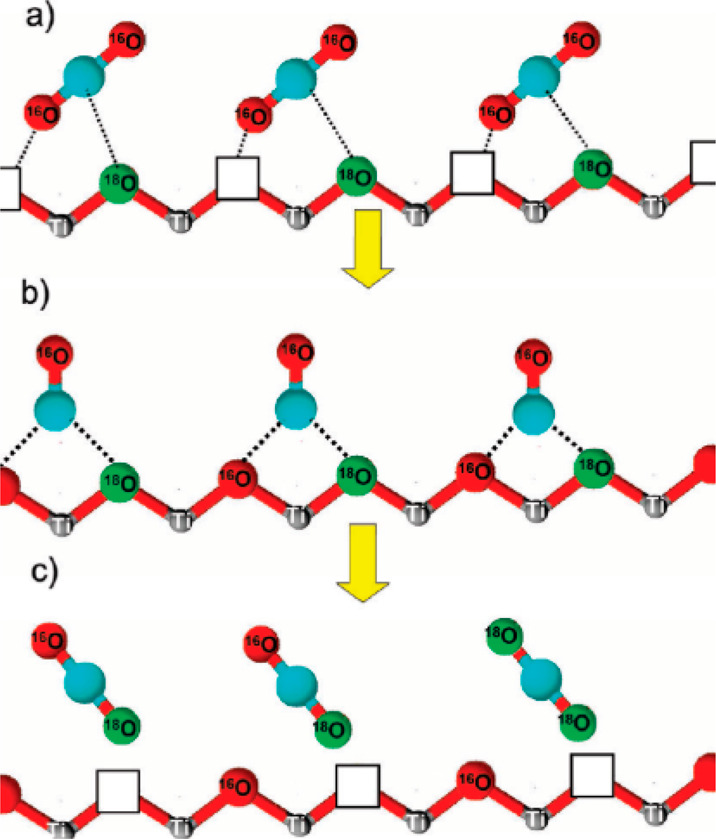
Mechanism of the spontaneous isotopic exchange between C16O2 and Ti18O2 with the involvement of oxygen vacancies. (a) Adsorption of C16O2 to the surface of Ti18O2. (b) Formation of a CO3 bidentate species. (c) Release of a C16O18O molecule. Reprinted with permission from ref (12). Copyright 2011 American Chemical Society.
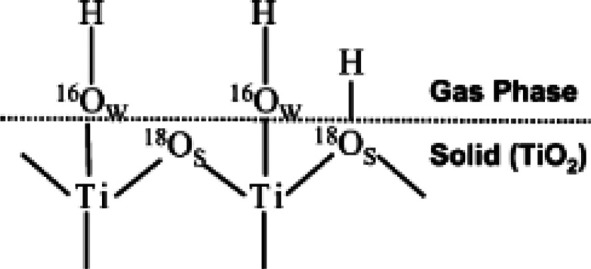
Montoya et al.16 investigated the H216O photooxidation in the presence of Ti18O2 and Ag+ ions as electron scavengers. During illumination, a higher 16O18O/16O2 quadrupole mass spectrometry (QMS) signal ratio as compared to the dark could be detected, which turned back to the initial value after switching off the light (Figure 4). In contrast, by applying Ti16O2 instead, independent from the illumination, no change in the QMS signal ratio appeared. Since in the case of Ti18O216O18O was evolved, it could be concluded that the photooxidation of water proceeds via a bridging oxygen from the lattice of TiO2, which is incorporated in the oxygen molecules. In the initial step, a 2-fold-coordinated (symbol: >) bridging oxygen (>Obr2–) (eq 2) or a 2-fold-coordinated protonated bridging oxygen (>OHbr) (eq 3) reacts with a photogenerated hole, leading to the formation of a 1-fold coordinated (symbol: −) bridging oxygen radical (−Obr•–) and a 1-fold coordinated bridging hydroxyl radical (−OHbr), respectively. The further steps yielding molecular oxygen according to the water redox photooxidation (WRP) mechanism are described elsewhere.17
| 2 |
| 3 |
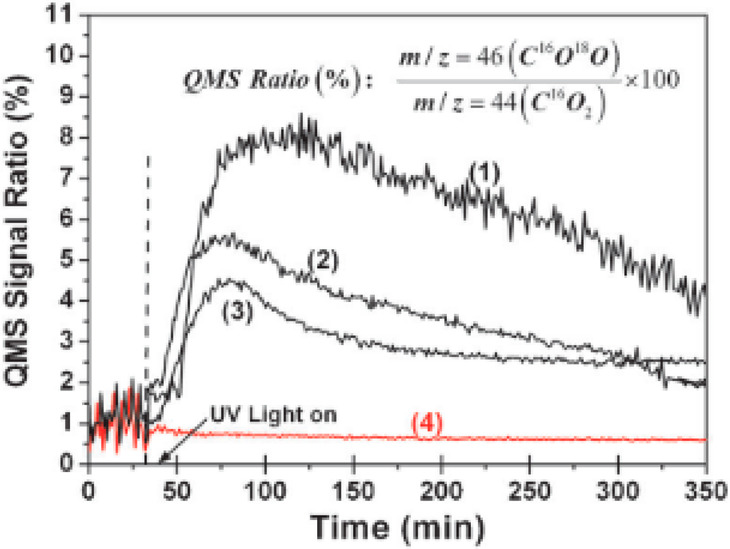
Melchers et al.18 employed Ti18O2 to analyze the mechanism of the anaerobic acetaldehyde degradation during illumination. In a previous study of the same authors, the incorporation of lattice oxygen into acetate after the adsorption of acetaldehyde was proposed, which resulted in the formation of CO2 and CH4.19 The comparison of the C16O18O/C16O2 QMS signal ratio of Ti16O2 and Ti18O2 showed that during illumination no change for Ti16O2 occurs, while for Ti18O2 the ratio increases (Figure 5). Consequently, the incorporation of lattice oxygen into the product molecules, and thus the proposed mechanism (Figure 6), could be proven.
Figure 4.
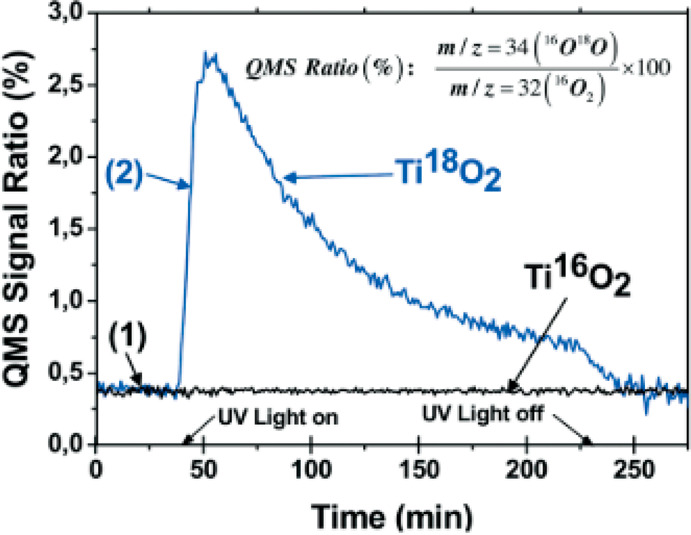
16O18O/16O2 QMS signal ratio from the photooxidation of H216O in the presence of Ag+ ions with Ti16O2 and Ti18O2. Reproduced from ref (16) with permission from The Royal Society of Chemistry.
Figure 5.

C16O18O/C16O2 QMS signal ratio from the aerobic degradation of acetaldehyde in the presence of Ti16O2 and Ti18O2 under illumination. Reprinted from ref (18). Copyright 2020, with permission from Elsevier.
Figure 6.
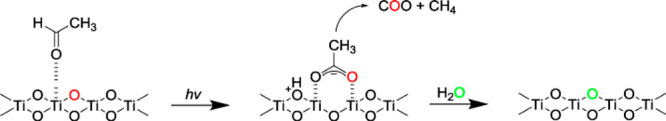
Reaction mechanism of the anaerobic degradation of acetaldehyde in the presence of TiO2 under illumination. Adapted from ref (18). Copyright 2020, with permission from Elsevier.
Montoya et al.20 studied the anaerobic oxidation of benzene in aqueous solutions in the presence of Ti18O2 and Ag+ as electron scavenger. Ti18O2 with an unlabeled hydrated surface (Figure 7), employing H216O, was used to distinguish between two possible reaction pathways. Either hydroxyl radicals are generated from adsorbed water species (16OHads•) or lattice oxygen is involved in the generation of radicals (−18Obr/–18OHbr•) and thus the oxidation of benzene. The C16O18O/C16O2 QMS signal ratio of Ti18O2 was during illumination higher compared to unlabeled Ti16O2, which indeed confirmed the involvement of bridging oxygens in the mineralization of benzene and the incorporation of these species into the product molecules.
Figure 7.
Ti18O2 surface with labeled bridging oxygens (18O) and unlabeled chemisorbed water species (16OH). Reprinted with permission from ref (20). Copyright 2013 American Chemical Society.
Montoya et al.21 investigated also the oxidation of benzene in acetonitrile with Ti18O2 in the presence of Ag+ as an electron scavenger to prove the incorporation of surface oxygen of TiO2 into the mineralization product CO2. The C16O18O/C16O2 QMS signal ratio of Ti18O2 is dependent on the water concentration, while a lower concentration yields higher ratios (Figure 8). For Ti16O2 the ratio under illumination does not increase, which confirms the incorporation of surface oxygen into the CO2 product molecules. Based additionally on their further findings (participation of TiO2 terminal oxygen atoms as hole traps and the dissociative adsorption of H2O into terminal oxygen vacancies), the authors were able to propose a terminal-oxygen indirect electron-transfer (TOIET) mechanism. The excitation of the labeled Ti18O2 leads to the formation of free photogenerated electrons (ef–) and holes (hf) (eq 4). A surface oxygen anion (18Os2–) is able to react with a free photogenerated hole, which causes the formation of a terminal radical (18Os) (eq 5). A physisorbed benzene molecule (C6H6) coordinates to a terminal radical, and an incipient covalent bond is formed (eq 6). The reaction with a further free photogenerated hole causes the formation of a phenol molecule (C6H618O), which contains an 18O atom that originates from the surface of Ti18O2 and an oxygen vacancy (V[18Os2–]) (eq 7). The vacancy can be healed by the dissociative adsorption of a H216O molecule, whereby an 16Os anion is incorporated into the surface of Ti18O2 (eq 8). As shown in eqs 9–11, the incorporated 16Os2– anion can also act as a hole scavenger, which finally results in the formation of a phenol molecule that contains an 16O atom (C6H616O). The resulting vacancy V[16Os] is healed by the dissociative adsorption of a further H216O molecule (eq 12). Ag+ ions are able to react with the free photogenerated electrons, which leads to the formation of metallic silver (eq 13). In eq 14, the complete process is summarized, which shows the exchange of an 18Os2– anion with an 16Os anion at the surface of Ti18O2.
| 4 |
| 5 |
| 6 |
| 7 |
| 8 |
| 9 |
| 10 |
| 11 |
| 12 |
| 13 |
| 14 |
Figure 8.
C16O18O/C16O2 QMS signal ratio from the anaerobic mineralization of benzene in acetonitrile in the presence of Ti18O2 (1–3) and Ti16O2 (4) under illumination. The following water concentrations were employed: (1) c(H2O) = 0.560 mmol L–1, (2) c(H2O) = 10 mmol L–1, (3) c(H2O) = 24 mmol L–1, and (4) c(H2O) = 24 mmol L–1. Copyright 2014 Wiley. Used with permission from ref (21).
Solvent Isotope Effects in Photocatalysis
Solvent isotope studies have been used extensively by organic chemists for decades. In this process, the relative rates of a reaction are compared, when they are carried out in normal water and deuterated water or “heavy” water. The solvent isotope effect (SIE) is the ratio of the rate constant in the “heavy” water (kD) to that observed in normal water (kH):
| 15 |
Typically the reactions in the heavy water solvent are significantly slower, and hence the rate constants are lower than those observed in normal water. The slower reaction rate in the deuterated solvent is due to the fact that the deuterated solvent has a lower vibrational zero-point energy, and hence a greater activation energy is required to dissociate the OD bond compared to OH bonds. Consequently, the rates are slower in deuterated solvents, which are involved in reactions.
Cunningham and Srijaranai were the first to report the use of this technique for a semiconductor photocatalytic process in 1988.22 In their investigation of the photocatalytic degradation of isopropanol (IPA) using TiO2, they observed a primary solvent isotope effect of 3. It was proposed that the reduced rate of IPA destruction in D2O was a result of the lower quantum efficiency for the formation of OD• radicals on the TiO2 surface in the heavy water solvent. As a result of this, there would be a lower number of OD• radicals on the TiO2 surface, which would be available to attack the isopropanol. On the basis of this proposal, they suggested that the photogeneration of hydroxyl radicals was the rate-determining step for the photocatalytic process.
Robertson et al. also observed a solvent isotope effect of 3 for the photocatalytic destruction of the cyanobacterial toxin microcystin-LR using a P25 TiO2 photocatalyst.23a The solvent isotope effect observed by Cunningham for the decomposition of IPA was the same as that reported by Robertson for the cyanotoxin, considering the substantial difference in structure and the molecular mass of the two substrates. Robertson suggested that this was also a confirmation of Cunningham and Srijaranai’s proposal that the hydroxyl radical generation on the photocatalyst surface was also the rate-determining step for the photocatalytic reaction.
In a subsequent study, Robertson and co-workers investigated the solvent isotope effect on the degradation of microcystin-LR (MC-LR) and another cyanobacterial chemical metabolite, geosmin (GSM), using a Hombikat K01/C TiO2 photocatalyst.23b In this case a solvent isotope effect of 1.5 was observed for microcystin and geosmin,23b which was approximately 50% lower than that found in the previous studies by Robertson et al. and Cunningham and Srijaranai (Table 1).
Table 1. Kinetic Isotope Effect Based on the Photocatalytic Destruction of GSM and MC-LR in Normal and Heavy Water with Hombikat K01/C TiO2 as a Photocatalysta.
| GSM |
MC-LR |
|||
|---|---|---|---|---|
| solvent | k (μM min–1) | relative rate | k (μM min–1) | relative rate |
| H2O | 1.56 | 1.0 | 8.55 | 1.0 |
| D2O | 0.97 | 0.62 | 5.44 | 0.64 |
Reprinted from ref (23b). Copyright 2011, with permission from Elsevier.
In this study, Robertson et al. proposed that the solvent isotope effect observed for both molecules was mediated via hydroxyl radicals, generated from the subsequent reduction of the superoxide radical anion, produced at the conduction band. After being generated, the superoxide would be hydrated or deuterated by the solvent to form a hydroperoxide ion (eq 16). The hydroperoxide ions may then interact to form hydrogen peroxide, which would then generate OH• (or OD•) radicals following an electron transfer from the conduction band again. This may be rate determining since O2 has to be generated at the conduction band prior to the interaction with the solvent and the subsequent formation of OD• or OH• species (eqs 17 and 18).
| 16 |
| 17 |
| 18 |
It was suggested that the observed solvent isotope effect could be a result of the rate of the reaction of the solvent with superoxide species rather than by the rate of reaction of OH• (or OD•) on the microcystin or geosmin. If the isotope reaction depended on this latter reaction, one would expect it to be the same no matter what photocatalyst or species being oxidized was utilized.
An interesting observation is the fact that the solvent isotope effect is approximately 3 for P-25 and approximately 1.5 for K01/C. They also suggested that since similar kinetic solvent isotope effects were observed for different substrate molecules on the same photocatalyst materials the interaction of the solvent with the photocatalyst and the rate of oxidation of the solvent were probably the rate-determining steps for the photocatalytic reaction, as opposed to conduction band reduction of oxygen as previously proposed by Gerischer and Heller.24 Furthermore, Robertson et al. proposed that the reason the kinetic solvent isotope effect observed in this subsequent work was smaller than that in their previous study and Cunningham’s work was due to the fact that different photocatalyst materials were employed,23b and hence the effect was likely to be dependent on the photocatalyst material.
Belhadj et al. used solvent isotope effects to study the adsorption of water and deuterium oxide on TiO2 surfaces in the dark and under UV(A) irradiation using in situ ATR-FTIR spectroscopy under aerobic and anaerobic conditions.25a Under dark conditions in a mixture of H2O and D2O solvents, an isotopic exchange was found to occur on the surface of the TiO2 material. Following irradiation with UV(A) light, the quantity of both OH and OD groups was found to be increasing in the presence of molecular oxygen. Additionally, hydroperoxide was generated through a photocatalytic process under aerobic conditions, which was believed to be produced as a result of the reduction of molecular oxygen adsorbed at the TiO2 surface by the photogenerated conduction band electrons, as opposed to being generated via water oxidation from valence band holes. It was also demonstrated from the spectroscopic studies that under conditions where the percentage of H2O was significantly less than that of D2O there was an exchange of solvent groups on the TiO2 surface with the OD– ions, having a stronger adsorption affinity to the photocatalyst compared to the OH– ions. Following illumination with UV light, both OH and OD groups were generated on the photocatalyst surface in the presence of oxygen. The generation of these groups also increased the hydrophilicity of the TiO2 surface. If the experiment was conducted under either a nitrogen or argon atmosphere, there was no evidence of the formation of OH and OD groups, and the hydrophilicity was inhibited (Figure 9). This result indicated that under UV irradiation oxygen played a critical role in both the photocatalytic response and the photoinduced hydrophilicity.
Figure 9.
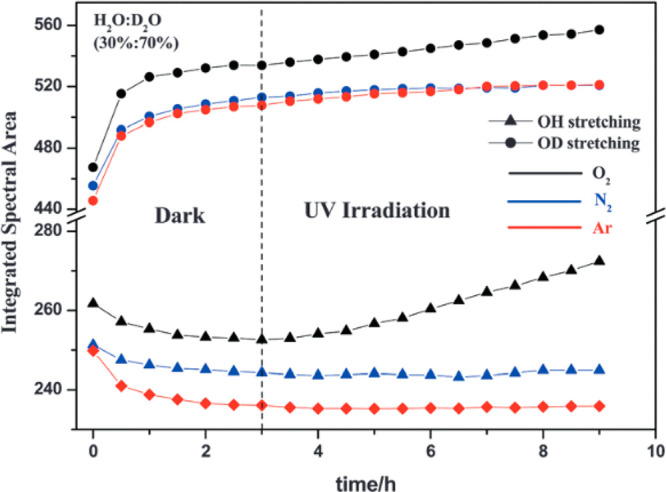
Evolution of the intensity of the integrated spectral areas of OH and OD stretching groups before and after UV irradiation: effect of dissolved O2, N2, and Ar on the adsorption of H2O–D2O on the TiO2 surface. Reprinted from ref (25a). Published by the PCCP Owner Societies.
In a subsequent study, the adsorption and photocatalytic degradation of acetate on TiO2 surfaces was investigated in H2O and D2O by both attenuated total reflection Fourier transformed infrared spectroscopy (ATR-FTIR) and EPR spectroscopy.25b Different interactions between the adsorbed acetate and OD groups resulted from the isotopic exchange on the TiO2 surface following adsorption of D2O. The interaction of the acetate with the TiO2 surface was found to be strongly influenced by the pH, and a range of surface complexes with the acetate were observed to form. Under acidic conditions, the formation of a bidentate structure involving two distinct Ti atoms appeared to be the preferred complex structure. At pH values close to the point of zero charge for the TiO2, the acetate favored a monodentate complex, formatted through adsorption to the positively charged TiO2 anatase material (Figure 10).
Figure 10.

Schematic representation for the adsorption of acetate on the anatase surface (UV100) in the dark at pH < pHzpc (A), pH ≈ pHzpc (B), pH > pHzpc (C). Reprinted from ref (25b). Copyright 2016, with permission from Elsevier.
Following irradiation with UV(A) light, hydroxyl radicals were observed under alkaline conditions, while methoxy radicals were generated under acidic conditions. Two different degradation pathways were proposed for the acetate under acidic and alkaline conditions (Figure 11), which were supported by the experimental studies performed using ATR-FTIR and EPR spectroscopy. Overall, the results of the EPR study suggested that under alkaline conditions acetate degradation was mainly promoted by attack by valence band generated hydroxyl radicals. Under acidic conditions, the degradation appeared to occur via direct oxidation via photogenerated valence band holes.
Figure 11.

Proposed mechanism for the photocatalytic reaction of acetate at pH 9 (A) and pH 3 (B). Reprinted from ref (25b). Copyright 2016, with permission from Elsevier.
Solvent isotope studies were also used for the investigation of the simultaneous photocatalytic degradation of formaldehyde and hydrogen evolution on a platinized TiO2 material under an oxygen-free atmosphere.25c Using QMS and ATR-FTIR spectroscopy for analysis, the main reaction products obtained from the photocatalytic degradation of 20% formaldehyde were hydrogen and carbon dioxide in a ratio of 2 to 1. From the solvent isotope study, it was found that the rate of mineralization of formaldehyde to CO2 is significantly reduced with increasing concentration of D2O. Following the investigation of the solvent isotope effect on the system using ATR-FTIR analysis, it was proposed that the formaldehyde oxidation was promoted by attack by OD• radicals, formed from the reaction with the photogenerated valence band hole. This reaction generated a surface-adsorbed deuterated formic acid (HCOOD), which subsequently underwent further oxidation by valence band holes in a photo-Kolbe-type reaction. The photogenerated conduction band electrons were proposed to simultaneously reduce H+ and D+, originating from both formaldehyde and D2O, to form molecular HD. The yield of the HD gas was found to be strongly influenced by the solvent and was maximized when the ratio of H2O:D2O was 20%:80%. The proposed mechanism for the simultaneous hydrogen production and formaldehyde oxidation in the presence of D2O is summarized in eqs 19–26 below:25c
| 19 |
| 20 |
| 21 |
| 22 |
| 23 |
| 24 |
| 25 |
| 26 |
Scope for Research Development and Focus
Each of the studies considered in this paper related to the labeling of photocatalyst materials and target molecules have utilized TiO2 as the photocatalyst. This is also the case for the studies dealing with solvent isotope effects. Although TiO2 is the most studied photocatalyst material, there has only been a relatively small number of reports related to isotope effects. As has been detailed above, the use of these isotope studies has enabled important insights to be gained for photocatalytic reactions on TiO2, but there is still significant scope for further studies using these techniques.
For future research, while also investigating the isotope effects of the photocatalytic decomposition of other substrates on TiO2 materials, it would be important to extend the studies to other photocatalyst materials as well. For example, Fe2O3, WO3, CdS, C3N4, and SrTiO3 are known as photocatalytically active materials, while the labeling of the catalyst or reactant molecules would allow us to get deeper insights into the corresponding mechanisms using these materials.
With respect to TiO2 itself, there are many more reaction mechanisms that should be investigated based on solvent isotope effects. In particular, the involvement of terminal hydroxyl or oxygen radicals in the photocatalytic mechanism, as one of the initial steps, might be either established or refuted.
The technique could also be used for kinetic studies, particularly in the case of rapidly decomposing intermediates, which may be more easily followed in the heavy water solvent.
Conclusion
The investigation of isotope effects represents a powerful tool in the area of photocatalysis to study the mechanisms of the reactions occurring on the surface of photocatalyst materials. Using this technique, it is possible to elucidate whether photocatalyst surface atoms are transferred into product molecules, while also the incorporation of atoms from reactant molecules into the surface of the photocatalyst can be observed. Furthermore, by exchanging H2O by D2O, the ratio of the rate constants between both solvents can be followed to investigate whether the generation of hydroxyl radicals is the rate-determining step of a reaction. Using D2O as a solvent has a further advantage since it allows the determination of whether hydrogen atoms in product molecules originate from reactant molecules or from the solvent. Consequently, it is important to perform such studies to allow the determination of the mechanistic pathway of the photocatalytic process. It should be noted that isotope effect studies are, however, not enough as the sole process to provide such detail, but in combination with other tools, they can provide important information on such processes.
In conclusion, it is clear that the application of isotope studies is a versatile and useful tool for studying photocatalytic reactions; however, the technique has been only applied in a relatively small number of investigations. There is therefore great scope for the further application of this technique in the field of semiconductor photocatalysis, and it is anticipated that this will be an area of growing interest within the photocatalytic research community over the next few years.
Acknowledgments
D.W.B. and C.G. gratefully acknowledge funding by the Federal Ministry of Education and Research (033RC029D). D.W.B. gladly acknowledges financial support by Saint-Petersburg State University (SPbU) within the Project ID: 73032813. The authors acknowledge St. Petersburg State University for the research grant (Pure ID 39054581). The authors also thank the Center for Computational Resources of St. Petersburg State University (Peterhof campus) for the provided CPU time.
Biographies

Carsten Günnemann received his M.Sc. degree in 2017 from the Gottfried Wilhelm Leibniz Universität Hannover under the supervision of Prof. Dr. Detlef W. Bahnemann, and he is currently a Ph.D. student in the same group. His research interests include transient absorption spectroscopy, (photo)electrochemistry, TiO2 films, and halide perovskites.

Prof. Dr. Detlef W. Bahnemann is Head of the Research Unit “Photocatalysis and Nanotechnology” at Leibniz University Hannover, Director of the Research Institute “Nanocomposite Materials for Photonic Applications” at Saint Petersburg State University, and Distinguished Professor at Shaanxi University of Science & Technology, Xi’an (Peoples Republic of China). His research topics including photocatalysis, photoelectrochemistry, solar chemistry, and photochemistry focused on the synthesis and physical-chemical properties of semiconductor and metal nanoparticles. His more than 500 publications have been cited over 61 000 times (h-index: 99). He is a Visiting Research Professor at Queens University Belfast, DeTao Master of Photocatalysis, Nanomaterials and Energy Applications, and holds a Guest Professorship at Tianjin University (China) and several visiting professorships in China, Malaysia, Saudi Arabia, and Britain.

Prof. Dr. Peter K.J. Robertson is Professor of Energy and Environmental Engineering at Queen’s University Belfast. His particular expertise is in the area of photocatalytic technology for both energy and water sustainability. This work has encompassed basic research on photocatalysis through to pilot process development for water treatment and for solar energy conversion and storage. He is also a member of the UK Engineering and Physical Science Research Council’s (EPSRC) Scientific Advisory Committee on Energy and the SUPERGEN High Level Group. He is also a member of sub Panel 8, Chemistry for REF2021 the UK National research assessment exercise. He is Chair of the Royal Irish Academy’s Physical, Chemical and Mathematical Sciences Committee. He holds visiting professor positions at the University of St Andrews, Ulster University, and Robert Gordon University. He is a Chartered Engineer, Chartered Scientist, and Chartered Chemist, a Fellow of the Royal Society of Chemistry, the Institute of Chemistry in Ireland, the Energy Institute, and an Associate Fellow of the Institution of Chemical Engineers.
The authors declare no competing financial interest.
References
- a Hoffmann M. R.; Martin S. T.; Choi W.; Bahnemann D. W. Environmental Applications of Semiconductor Photocatalysis. Chem. Rev. 1995, 95, 69–96. 10.1021/cr00033a004. [DOI] [Google Scholar]; b Robertson P. K. J.; Robertson J. M. C.; Bahnemann D. W. Removal of Microorganisms and Their Chemical Metabolites from Water Using Semiconductor Photocatalysis. J. Hazard. Mater. 2012, 211–212, 161–171. 10.1016/j.jhazmat.2011.11.058. [DOI] [PubMed] [Google Scholar]; c Pang X.; Skillen N.; Gunaratne N.; Rooney D. W.; Robertson P. K. J. Removal of Phthalates from Aqueous Solution by Semiconductor Photocatalysis: A Review. J. Hazard. Mater. 2021, 402, 123461. 10.1016/j.jhazmat.2020.123461. [DOI] [PubMed] [Google Scholar]; d Jiang C.; Moniz S. J. A.; Wang A.; Zhang T.; Tang J. Photoelectrochemical Devices for Solar Water Splitting – Materials and Challenges. Chem. Soc. Rev. 2017, 46, 4645–4660. 10.1039/C6CS00306K. [DOI] [PubMed] [Google Scholar]; e Loeb S. K.; Alvarez P. J. J.; Brame J. A.; Cates E. L.; Choi W.; Crittenden J.; Dionysiou D. D.; Li Q.; Li-Puma G.; Quan X.; et al. The Technology Horizon for Photocatalytic Water Treatment: Sunrise or Sunset?. Environ. Sci. Technol. 2019, 53, 2937–2947. 10.1021/acs.est.8b05041. [DOI] [PubMed] [Google Scholar]; f Schneider J.; Matsuoka M.; Takeuchi M.; Zhang J.; Horiuchi Y.; Anpo M.; Bahnemann D. W. Understanding TiO2 Photocatalysis: Mechanisms and Materials. Chem. Rev. 2014, 114, 9919–9986. 10.1021/cr5001892. [DOI] [PubMed] [Google Scholar]; g Kisch H. Semiconductor Photocatalysis-Mechanistic and Synthetic Aspects. Angew. Chem., Int. Ed. 2013, 52, 812–847. 10.1002/anie.201201200. [DOI] [PubMed] [Google Scholar]; h Zhu S.; Wang D. Photocatalysis: Basic Principles, Diverse Forms of Implementations and Emerging Scientific Opportunities. Adv. Energy Mater. 2017, 7, 1700841. 10.1002/aenm.201700841. [DOI] [Google Scholar]; i Ahmed A. Y.; Kandiel T. A.; Oekermann T.; Günnemann C.; Bahnemann D. Mechanistic Investigations of Photoelectrochemical Water and Methanol Oxidation on Well-Defined TiO2 Anatase (101) and Rutile (110) Surfaces. ACS Appl. Energy Mater. 2019, 2, 5308–5318. 10.1021/acsaem.9b01163. [DOI] [Google Scholar]; j Sieland F.; Schneider J.; Lippmann T.; Bahnemann D. W.; Dong C.-L. Understanding Charge Transfer Processes on Metal Oxides: A Laser-Flash-Photolysis Study. Proc. SPIE 2016, 9935, 99350G. 10.1117/12.2239261. [DOI] [Google Scholar]; k Rigby S. J.; Al-Obaidi A. H. R.; Lee S.-K.; McStay D.; Robertson P. K. J. The Application of Raman and Anti-Stokes Raman Spectroscopy for in Situ Monitoring of Structural Changes in Laser Irradiated Titanium Dioxide Materials. Appl. Surf. Sci. 2006, 252, 7948–7952. 10.1016/j.apsusc.2005.10.003. [DOI] [Google Scholar]; l Lee S.-K.; Mills A.; O’Rourke C. Action Spectra in Semiconductor Photocatalysis. Chem. Soc. Rev. 2017, 46, 4877–4894. 10.1039/C7CS00136C. [DOI] [PubMed] [Google Scholar]; m Li Y.; Lin B.; Ge L.; Guo H.; Chen X.; Lu M. Real-Time Spectroscopic Monitoring of Photocatalytic Activity Promoted by Graphene in a Microfluidic Reactor. Sci. Rep. 2016, 6, 28803. 10.1038/srep28803. [DOI] [PMC free article] [PubMed] [Google Scholar]; n Liqiang J.; Yichun Q.; Baiqi W.; Shudan L.; Baojiang J.; Libin Y.; Wei F.; Honggang F.; Jiazhong S. Review of Photoluminescence Performance of Nano-Sized Semiconductor Materials and Its Relationships with Photocatalytic Activity. Sol. Energy Mater. Sol. Cells 2006, 90, 1773–1787. 10.1016/j.solmat.2005.11.007. [DOI] [Google Scholar]; o Mendive C. B.; Bredow T.; Blesa M. A.; Bahnemann D. W. ATR-FTIR Measurements and Quantum Chemical Calculations Concerning the Adsorption and Photoreaction of Oxalic Acid on TiO2. Phys. Chem. Chem. Phys. 2006, 8, 3232. 10.1039/b518007b. [DOI] [PubMed] [Google Scholar]
- Courbon H.; Formenti M.; Pichat P. Study of Oxygen Isotopic Exchange over Ultraviolet Irradiated Anatase Samples and Comparison with the Photooxidation of Isobutane into Acetone. J. Phys. Chem. 1977, 81, 550–554. 10.1021/j100521a012. [DOI] [Google Scholar]
- Pichat P.; Courbon H.; Enriquez R.; Tan T. T. Y.; Amal R. Light-Induced Isotopic Exchange between O2 and Semiconductor Oxides, a Characterization Method That Deserves Not to Be Overlooked. Res. Chem. Intermed. 2007, 33, 239–250. 10.1163/156856707779238667. [DOI] [Google Scholar]
- Tanaka K. Intermediate of Oxygen Exchange Reaction over Illuminated Titanium Dioxide. J. Phys. Chem. 1974, 78, 555–556. 10.1021/j100598a019. [DOI] [Google Scholar]
- Murata C.; Hattori T.; Yoshida H. Electrophilic Property of O3– Photoformed on Isolated Ti Species in Silica Promoting Alkene Epoxidation. J. Catal. 2005, 231, 292–299. 10.1016/j.jcat.2005.01.012. [DOI] [Google Scholar]
- Mikhaylov R. V.; Lisachenko A. A.; Titov V. V. Investigation of Photostimulated Oxygen Isotope Exchange on TiO2 Degussa P25 Surface upon UV–Vis Irradiation. J. Phys. Chem. C 2012, 116, 23332–23341. 10.1021/jp305652p. [DOI] [Google Scholar]
- Liao L. F.; Lien C. F.; Shieh D. L.; Chen M. T.; Lin J. L. FTIR Study of Adsorption and Photoassisted Oxygen Isotopic Exchange of Carbon Monoxide, Carbon Dioxide, Carbonate, and Formate on TiO2. J. Phys. Chem. B 2002, 106, 11240–11245. 10.1021/jp0211988. [DOI] [Google Scholar]
- Nakamura R.; Nakato Y. Primary Intermediates of Oxygen Photoevolution Reaction on TiO2 (Rutile) Particles, Revealed by in Situ FTIR Absorption and Photoluminescence Measurements. J. Am. Chem. Soc. 2004, 126, 1290–1298. 10.1021/ja0388764. [DOI] [PubMed] [Google Scholar]
- Zhang M.; Wang Q.; Chen C.; Zang L.; Ma W.; Zhao J. Oxygen Atom Transfer in the Photocatalytic Oxidation of Alcohols by TiO2: Oxygen Isotope Studies. Angew. Chem. 2009, 121, 6197–6200. 10.1002/ange.200900322. [DOI] [PubMed] [Google Scholar]
- Henderson M. A. Formic Acid Decomposition on the {110}-Microfaceted Surface of TiO2(100): Insights Derived from 18O-Labeling Studies. J. Phys. Chem. 1995, 99, 15253–15261. 10.1021/j100041a048. [DOI] [Google Scholar]
- Bogdanoff P.; Alonso-Vante N. A Kinetic Approach of Competitive Photoelectrooxidation of HCOOH and H2O on TiO2 Anatase Thin Layers via on-Line Mass Detection. J. Electroanal. Chem. 1994, 379, 415–421. 10.1016/0022-0728(94)87165-5. [DOI] [Google Scholar]
- Civiš S.; Ferus M.; Kubát P.; Zukalová M.; Kavan L. Oxygen-Isotope Exchange between CO2 and Solid Ti18O2. J. Phys. Chem. C 2011, 115, 11156–11162. 10.1021/jp201935e. [DOI] [Google Scholar]
- Kavan L.; Zukalova M.; Ferus M.; Kürti J.; Koltai J.; Civiš S. Oxygen-Isotope Labeled Titania: Ti18O2. Phys. Chem. Chem. Phys. 2011, 13, 11583–11586. 10.1039/c1cp20775j. [DOI] [PubMed] [Google Scholar]
- Henderson M. A. Complexity in the Decomposition of Formic Acid on the TiO2(110) Surface. J. Phys. Chem. B 1997, 101, 221–229. 10.1021/jp961494i. [DOI] [Google Scholar]
- Civiš S.; Ferus M.; Zukalová M.; Kubát P.; Kavan L. Photochemistry and Gas-Phase FTIR Spectroscopy of Formic Acid Interaction with Anatase Ti18O2 Nanopartciles. J. Phys. Chem. C 2012, 116, 11200–11205. 10.1021/jp303011a. [DOI] [Google Scholar]
- Montoya J. F.; Bahnemann D. W.; Salvador P.; Peral J. Catalytic Role of Bridging Oxygens in TiO2 Liquid Phase Photocatalytic Reactions: Analysis of H216O Photooxidation on Labeled Ti18O2. Catal. Sci. Technol. 2017, 7, 902–910. 10.1039/C6CY02457B. [DOI] [Google Scholar]
- Salvador P. Mechanisms of Water Photooxidation at N-TiO2 Rutile Single Crystal Oriented Electrodes under UV Illumination in Competition with Photocorrosion. Prog. Surf. Sci. 2011, 86, 41–58. 10.1016/j.progsurf.2010.10.002. [DOI] [Google Scholar]
- Melchers S.; Schneider J.; Bahnemann D. W. Isotopic Studies on the Degradation of Acetaldehyde on Anatase Surfaces. Catal. Today 2020, 340, 318–322. 10.1016/j.cattod.2018.10.016. [DOI] [Google Scholar]
- Melchers S.; Schneider J.; Emeline A. V.; Bahnemann D. W. Effect of H2O and O2 on the Adsorption and Degradation of Acetaldehyde on Anatase Surfaces—An In Situ ATR-FTIR Study. Catalysts 2018, 8, 417. 10.3390/catal8100417. [DOI] [Google Scholar]
- Montoya J. F.; Ivanova I.; Dillert R.; Bahnemann D. W.; Salvador P.; Peral J. Catalytic Role of Surface Oxygens in TiO2 Photooxidation Reactions: Aqueous Benzene Photooxidation with Ti18O2 under Anaerobic Conditions. J. Phys. Chem. Lett. 2013, 4, 1415–1422. 10.1021/jz400580b. [DOI] [PubMed] [Google Scholar]
- Montoya J. F.; Bahnemann D. W.; Peral J.; Salvador P. Catalytic Role of TiO2 Terminal Oxygen Atoms in Liquid-Phase Photocatalytic Reactions: Oxidation of Aromatic Compounds in Anhydrous Acetonitrile. ChemPhysChem 2014, 15, 2311–2320. 10.1002/cphc.201402043. [DOI] [PubMed] [Google Scholar]
- Cunningham J.; Srijaranai S. Isotope-Effect Evidence for Hydroxyl Radical Involvement in Alcohol Photo-Oxidation Sensitized by TiO2 in Aqueous Suspension. J. Photochem. Photobiol., A 1988, 43, 329–335. 10.1016/1010-6030(88)80029-7. [DOI] [Google Scholar]
- a Robertson P. K. J.; Lawton L. A.; Cornish B. J. P. A.; Jaspars M. Processes Influencing the Destruction of Microcystin-LR by TiO2 Photocatalysis. J. Photochem. Photobiol., A 1998, 116, 215–219. 10.1016/S1010-6030(98)00312-8. [DOI] [Google Scholar]; b Robertson P. K. J.; Bahnemann D. W.; Lawton L. A.; Bellu E. A Study of the Kinetic Solvent Isotope Effect on the Destruction of Microcystin-LR and Geosmin Using TiO2 Photocatalysis. Appl. Catal., B 2011, 108–109, 1–5. 10.1016/j.apcatb.2011.07.019. [DOI] [Google Scholar]
- Gerischer H.; Heller A. The Role of Oxygen in Photooxidation of Organic Molecules on Semiconductor Particles. J. Phys. Chem. 1991, 95, 5261–5267. 10.1021/j100166a063. [DOI] [Google Scholar]
- a Belhadj H.; Hakki A.; Robertson P. K. J.; Bahnemann D. W. In Situ ATR-FTIR Study of H2O and D2O Adsorption on TiO2 under UV Irradiation. Phys. Chem. Chem. Phys. 2015, 17, 22940–22946. 10.1039/C5CP03947A. [DOI] [PubMed] [Google Scholar]; b Belhadj H.; Melchers S.; Robertson P. K. J.; Bahnemann D. W. Pathways of the Photocatalytic Reaction of Acetate in H2O and D2O: A Combined EPR and ATR-FTIR Study. J. Catal. 2016, 344, 831–840. 10.1016/j.jcat.2016.08.006. [DOI] [Google Scholar]; c Belhadj H.; Hamid S.; Robertson P. K. J.; Bahnemann D. W. Mechanisms of Simultaneous Hydrogen Production and Formaldehyde Oxidation in H2O and D2O over Platinized TiO2. ACS Catal. 2017, 7, 4753–4758. 10.1021/acscatal.7b01312. [DOI] [Google Scholar]