

Abstract
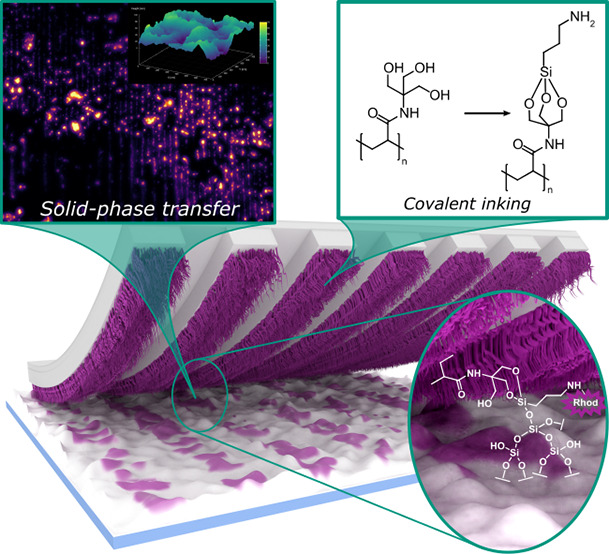
We present a microcontact printing (μCP) routine suitable to introduce defined (sub-) microscale patterns on surface substrates exhibiting a high capillary activity and receptive to a silane-based chemistry. This is achieved by transferring functional trivalent alkoxysilanes, such as (3-aminopropyl)-triethoxysilane (APTES) as a low-molecular weight ink via reversible covalent attachment to polymer brushes grafted from elastomeric polydimethylsiloxane (PDMS) stamps. The brushes consist of poly{N-[tris(hydroxymethyl)-methyl]acrylamide} (PTrisAAm) synthesized by reversible addition–fragmentation chain-transfer (RAFT)-polymerization and used for immobilization of the alkoxysilane-based ink by substituting the alkoxy moieties with polymer-bound hydroxyl groups. Upon physical contact of the silane-carrying polymers with surfaces, the conjugated silane transfers to the substrate, thus completely suppressing ink-flow and, in turn, maximizing printing accuracy even for otherwise not addressable substrate topographies. We provide a concisely conducted investigation on polymer brush formation using atomic force microscopy (AFM) and ellipsometry as well as ink immobilization utilizing two-dimensional proton nuclear Overhauser enhancement spectroscopy (1H–1H-NOESY-NMR). We analyze the μCP process by printing onto Si-wafers and show how even distinctively rough surfaces can be addressed, which otherwise represent particularly challenging substrates.
Keywords: microcontact printing, capillary-active substrates, silane chemistry, PDMS surface grafting, surface patterning, shuttled RAFT-polymerization
Introduction
Microstructured surfaces offer great potential in application fields such as microelectronics,1,2 information storage,3,4 (bio)sensing,5−7 or optoelectronics.8,9 In this context, microcontact printing (μCP) provides an inexpensive and straightforward access to surface patterning. As the most prominent subordinate technique among soft lithography,10 μCP represents a miniature variation of a classical stamping process and, thus, relies on the transfer of a chemical functionality, the ink, from a structured elastomeric stamp to a substrate.11 Ideally, the transfer of ink occurs exclusively at the area of contact between stamp and substrate surface imprinting the stamp negative onto the surface. Its ease of handling renders μCP suitable for upscaling to dimension of 4 in.,12 which represents a major advantage in comparison to alternative techniques for surface patterning, such as, dip pen,13 electron,14,15 ion beam16,17 or photolithography,18 etc., which often rely on tedious experimental protocols and require specialized and expensive instrumentation.
Numerous applications take advantage of μCP, such as microelectronic devices,19 biosensor technology,20,21 and tissue engineering.22,23 Subsequent applications of the surface-patterned substrates pose specified requirements on the chemical functionalities that may be introduced by μCP. In a first instance, this concerns the printing precision. As a parameter, which refers to the minimum size of details that can be transferred during the printing process, printing precision is determined by a complex interplay between stamp surface, ink rheology and substrate chemistry and topography.11 Here, the diffusive mobility of the ink may play a role as a limiting factor, as it may result in smearing, which drastically limits the resolvable details of the patterns. Another also very important parameter is the chemical composition of the imprinted area. Particularly important in sensing applications, the facile attachment of specific molecules to the patterns remains indispensable.24 In this regard, μCP enables a direct functionalization with the respective functionality or creation of binding sites for the attachment of said molecules in subsequent modification steps.25
Developed in the early 1990s by Kumar and Whitesides, μCP was first used to prepare patterned arrays of self-assembled monolayers (SAMs) of thiols using a structured stamp on a smooth gold substrate.26 Yet too, SAMs can be introduced on silver or copper surfaces,27 and smooth inorganic oxide substrates were employed using silane28 or carboxylic acids.29 These reports describe systems, which are straightforward to address via μCP due to the high affinity of the ink to the substrate, which counteracts diffusive ink smearing. Additionally, the smoothness and, thus, low capillarity of substrates also prevents uncontrolled topological ink flow, so that printing precisions even in the low micrometer range could be achieved.11,30,31 In spite of these high printing precisions, the method of printing SAMs on smooth metal surfaces demands very distinct requirements on the printing systems, and does not introduce chemical functionalities which could be utilized for a follow up-chemistry. In contrast, the microcontact chemistry method facilitates patterning with chemically well-defined groups.10,32 This technique relies on a precedent modification of both the stamp as well as the substrate, whereby each of the surfaces carries reaction partners that can be involved in a chemical process occurring under straightforward conditions (“click type-reactions”).33−38 Triggered upon their physical contact, either the cleavage39 or the formation of said chemical bond at the printing area results in substrate patterning with defined chemical groups with varying functionalities, from primary alcohols39 to, e.g., carbohydrates.34,37 Even printing precisions in the low nanometer range can be obtained (∼20 nm).33 However, this method is only compatible with very smooth surfaces and usually relies on a sophisticated pretreatment and, therefore, lacks universal applicability.
Within this study, we therefore aim at developing a universal approach, which can be used to address demanding substrates, such as porous silica, which can be applied for improving the efficiency of protein transfer.25 Given this porosity, this material represents a capillary-active substrate promoting ink smearing, which in turn limits its usual printing precision to the submillimeter range.25,40 In principle, these adverse conditions might be compensated by reducing the diffusive mobility of the ink.41 A convenient possibility hereto is to utilize macromolecular inks. As an example, poly(ethylene imine) has been employed to address silicon dioxide microspheres as substrates.42−44 Even though polymeric inks could offer binding sites allowing for additional functionalization, they tend to introduce three-dimensional alterations in the topography at the imprinted areas, which can represent a drawback for some applications.42
Addressing the limitations mentioned above, we therefore wanted to develop a feasible μCP routine capable to address difficult substrates with high capillarity. Furthermore, this routine should allow for a precise locally selective chemical surface modification without significantly altering the topography. Therefore, we aimed for the utilization of low molecular weight inks (LMWIs) while simultaneously minimizing ink smearing due to uncontrolled flow. In a recent study,41 we could show that introducing a surface-grafted polymer brush architecture to the stamp surface may serve to effectively limit the ink mobility and thereby enhancing printing precision. With this method, even inks possessing polar characteristics could be transferred, which are in general not straightforward to utilize in μCP.41,45 Inspired by this phenomenon, we now aimed for maximizing this limitation of ink mobility by intermediate covalent linkage between LMWI and stamp followed by bond transfer to the substrate therefore resembling a solid phase-like chemical reaction. In more detail, we wanted to create a suitable functionality onto the stamp surface tailored to host tri-alkoxysilane-based inks. These seemed appealing to us due to their commercial availability in a broad variety of molecular derivatives at usually bearable costs, which has rendered them ubiquitous in surface chemical modification.46 In particular we wanted to overcome typical issues that silanes are suffering from, like their considerably low affinity to stamps made from polydimethylsiloxane (PDMS) as well as a high tendency to uncontrollably hydrolyze and polymerize at ambient conditions.47,48 It might be mentioned here that, even though other materials also have been discussed, PDMS constitutes the most widely used stamp material.49 As a starting point for this endeavor, we envisioned polymer brush-modified PDMS-stamps utilizing a grafting-from process and the reversible addition–fragmentation chain transfer (RAFT) polymerization.50,51 Furthermore, we wanted to achieve a high control over the polymerization by employing the shuttled chain transfer agent (CTA) approach.52,53
Results and Discussion
Alkoxysilanes may undergo a transesterification reaction with surface hydroxy-moieties typically present on silicon oxide surfaces and other metal oxides, e.g., aluminum oxide or titania. For the case of silica, alkoxysilanes form highly stable −Si–O–Si– bonds via their reaction with surface silanol groups (Si–OH). If the employed silanes carry an organic substituent, this functionality is accordingly imparted onto the surface. An example of such a transfer of chemical functionality, utilizing the amino-terminated silane (3-aminopropyl)-trialkoxysilane (APTES), is illustrated in Figure 1A.54 Thereby, the alkoxy groups are usually trimethoxy or triethoxy substituents. Due to their brisk reactivity toward water, silanes are prone to intermolecular hydrolysis in the presence of moisture, leading to ill-defined oligomeric networks. This renders them rather unsuitable as a bare ink in μCP due to lack of controllability of their reaction during the printing process. By using a trivalent alcohol, however, it is possible to kinetically protect the tri-alkoxysilane due to an increased activation energy toward hydrolysis (Figure 1A).55,56 Notwithstanding this stabilization, the protected silane structure still remains reactive toward silanol groups, since Si–O–Si-bond formation is thermodynamically favored.56 As a suitable carrier for this type of silane-protecting units, we synthesized an acrylate-based polymer, grafted from the PDMS stamp surface containing a trivalent alcohol in its side chains. Consequently, the overall μCP routine comprises the three main steps summarized in Figure 1B. In step 1 we functionalize the surface of a PDMS stamp with a suitable polymer, as aforesaid via a grafting-from RAFT approach. In step 2, the ink is immobilized within the polymeric framework by forming the trisubstituted silane structure. With this ink-modified stamp, the printing process is performed in step 3, during which the ink is transferred from the grafted polymer brushes to the substrate. In the following sections, we will describe each step individually and analyze the respective outcome.
Figure 1.

(A) Comparison of the reactivity of organosilanes (from left to right): the unprotected and highly reactive trialkoxysilane structure, the trialkoxysilane covalently attached on the appropriate polymer compound and kinetically protected forming a metastable adduct, trialkoxysilane covalently attached on the imprinted surface forming a stable structure. (B) General illustration of solid-phase transfer of reactive silanes by using μCP and polymer-grafted PDMS stamp including the steps: (Step 1) grafting polymer brushes from PDMS stamp, (Step 2) covalent attachment of APTES ink on the functionalities of the polymeric network, and (Step 3) transferring the APTES from the polymer to the imprinted substrate.
Preparation of Polymer-Brush PDMS Stamp
During this first step of the procedure, the polymer brush-modified PDMS stamps are prepared. For the preparation of the polymer brushes, we selected a controlled radical polymerization of hydroxy-bearing acrylic acid derivatives to enable the attachment of polymer chains to the PDMS stamp surface via a grafting from-method. A grafting from-approach was favored to a grafting to-strategy here, since it promises a reduced tendency for unspecific polymer adsorption and, therefore, a good control over the polymer architecture as well as an enhanced grafting density.53,57N-[Tris(hydroxymethyl)-methyl]acrylamide (TrisAAm), a commercially available acrylamide derivative, was selected as a monomer as it offers three hydroxy moieties simultaneously. This may facilitate the effective attachment of the trivalent alkoxysilane ink to a single monomeric unit within the final polymer.
The polymerization of TrisAAm was performed by RAFT polymerization mediated by the CTA 2-{[(Butylsulfanyl)carbothionyl]-sulfanyl}propanoic acid (PABTC), typically resulting in the formation of polymers with a narrow size distribution as well as a well-defined start- and end group fidelity.35,51 For the preparation of the polymer brush stamps, first, the CTA was attached covalently to the stamp surface by amide coupling to the surface amino groups that have been introduced to the PDMS by plasma activation and subsequent chemical vapor deposition (CVD) of APTES, followed by the RAFT polymerization using the CTA shuttle approach.52,53 During this process, the substrate to be grafted is subjected to the polymerization procedure under the presence of additional nonsurface-bound CTA. This allowed for the simultaneous formation of RAFT-induced polymers in solution and a shuttling of radicals between different sites on the surface, thus increasing control of the grafting-from process. Considering the controlled nature of this polymerization reaction, the characteristics of the simultaneously formed free polymers should be indicative of the characteristics of the formed brushes (Figure 2A). In addition, this procedure in principle allows for the customization of the degree of polymerization of the resulting polymer by a proper adjustment of the monomer-to-CTA ratio. Table S1 summarizes the results of different polymerization experiments. Size exclusion chromatography (SEC) results of polymers formed in solution provide Mn values for the different batches (14.90–43.69 kg mol–1 for polymerization experiments targeting a DP of 200) along with reasonably low dispersity values (Đ: 1.26–1.67) indicating a moderate control over the polymerization. A characteristic size exclusion chromatogram is given in Figure 2C showing the formation of a polymer with a monomodal size distribution. Increased Đ values for the surface-grafting method compared to a conventional RAFT-process (which are typically in the range of 1.1 < Đ < 1.3) could be explained by the presence of the PDMS sheet inside the reaction vessel, potentially limiting the overall chain transfer ability of the CTA by for instance diffusion processes into the PDMS polymeric framework. Nonetheless, the monomodal nature of the size distribution is highly indicative of a successful RAFT-mediated polymerization process.
Figure 2.

(A) Reaction scheme of the RAFT polymerization of PTrisAAm both in solution and grafted from the PDMS stamp surface; (B) Illustration of a PDMS stamp that was half covered with PTFE stripe to prevent polymerization in this area; (C) SEC curve of the shuttle-PTrisAAm (St3 in Table S1) from a grafting experiment (SEC measurements were performed in N-methyl-2-pyrollidone (NMP) with 0.5% LiBr (polystyrene standards); (D) AFM height profile of the grafted and non-grafted (PTFE stripe covered) part of the PDMS stamp (St 4) with a brush length of 70 ± 29 nm and its respective wetting behavior. Our approach leads to controlled polymer formation on the stamps with a narrow size distribution, whereby their presence on the surface could be proven via surface wetting and AFM measurements.
For the estimation of the polymer brush length on the stamp, we initially utilized atomic force microscopy (AFM). For this purpose, we covered a part of the stamp tightly with a poly(tetrafluoroethylene) (PTFE) stripe during the grafting procedure to block the formation of polymer brushes thereon (Figure S1A) and measured the height profile at the resulting boundary (Figure 2B). As a first indication of selective brush formation, the wetting behavior of treated PDMS stamps showed a contact angle of ∼30° for the grafted area of the PDMS. In contrast, the nongrafted part showed ∼90° (Figure 2D). This is well expected, since the high density of hydroxyl groups within the PTrisAAm should render the polymer-grafted surface hydrophilic, while PDMS represents a quite hydrophobic material. Furthermore, AFM performed in tapping mode at the boundary between grafted and nongrafted parts provided brush lengths in the range of 70 ± 29 nm (Figure 2D). The rather high deviation of the measured value could be explained chemically by the fact that the PDMS sheet interferes with the RAFT polymerization processes as the reaction partners as well as the solvent molecules can diffuse into the polymeric framework during the reaction. However, it is far more likely that the softness of PDMS and the adhesive nature interferes with the tip during AFM measurements, thus distorting the observed height profile. Accordingly, we were interested in the comparison with Si wafers, used as proxy for the stamps and receptive toward similar chemistry and surface grafted by the same routine. AFM analysis on the wafers as detected by scratching the polymer film with a needle and measuring the height profile of the resulting boundary revealed brush lengths of 11 ± 2 nm, which is significantly lower than the values obtained from PDMS stamps. In comparison, ellipsometry performed as an alternative technique for measuring the polymer layer thickness provided 20 ± 4 nm and was therefore approximately matching with the results detected by AFM. These results are much closer to realistic expectation for our polymer, and, thus, a more trustworthy result. Therefore, it is reasonable to assume these brush lengths as also being indicative for the brush lengths on PDMS stamps. Note that both techniques, although proven tools to precisely measure film thicknesses, cannot be applied directly to the surface of PDMS, since scratching the polymer film demands a high surface hardness and ellipsometry requires reflective surface characteristics thereof. Hence, we deem our measurement on Si-wafers the best possible approximation.
Additional polymerization experiments were performed, varying the targeted DP of the resulting polymer by adjusting the CTA to monomer ratio during the polymerization method. The respective results are listed in Table S1. The SEC curves (Figure S2) as well as 1H NMR analysis of the resulting polymers indicate the formation of the different shuttle polymers with controllable chain lengths. AFM characterizations of the simultaneously formed polymer brushes as shown in Table S1 indicated that the most consistent characteristics could be observed for polymers with a degree of polymerization (DP) of 200, which motivated us to use this DP for all further experiments.
Incorporation of Reactive Silane ink
Equipped with a high density of hydroxy moieties in its side chain,58 it is expected that the three hydroxy functionalities of each TrisAAm-monomer unit within the polymer should form kinetically stable adducts with the trivalent 3-aminopropyl(triethoxy)silane (APTES). At best, this occurs in a way that APTES binds side on to single monomeric units of the polymer in analogy to the formation of a cage-like structure as it has been described for triethanolamine and APTES.56 To substantiate the formation of a polymer-APTES conjugate, proton nuclear magnetic resonance (1H NMR) spectroscopy was employed. Figure 3 provides a comparative overview over the respective 1H NMR spectra, i.e., the spectrum of the pristine polymer (A), the polymer-APTES conjugate (B), which was prepared from the polymer and APTES under NaOH catalysis at elevated temperatures, as well as a mixture of the polymer and APTES. The 1H NMR spectrum of the pristine PTrisAAm polymer (Figure 3A) reveals proton signals a–e corresponding to all protons present in the polymer structure. These signals can be found in the spectrum of the polymer-APTES conjugate as well (Figure 3B), along with additional peaks indicated as i, j, k, and m, which can be attributed to the protons emerging from APTES-bound hydrogen atoms. Similarly, the spectrum of a mixture of the polymer and APTES not subjected to the conditions of the conjugation reaction reveals these signals (Figure 3C). A comparison of both spectra B and C exposes that for Figure 3C, a coupling fine pattern of the signals i, j, k can be observed, which does not seem present in Figure 3B Being typical for compounds with a diversity of molecular architectures, this observation provides already a first indication that the APTES may be attached to the polymer structure in a covalent manner.
Figure 3.

1H NMR (400 MHz, DMSO-d6) spectra of (A) pristine PTrisAAm, (B) APTES conjugated PTrisAAm (structural proposal of the conjugates), (C) APTES mixed with PTrisAAm, (D) 1H–1H-NOESY spectra (400 MHz, DMSO-d6) of APTES conjugated PTrisAAm, and (E) APTES mixed PTrisAAm. The signal emphasized in purple in panel D shows a cross-correlation between the signals Hd-Hi, which is indicative for the formation of the APTES-polymer conjugate. In contrast, this signal is absent in panel E showing the mere mixture of the polymer and the ink, which further on solidifies the assumption that APTES binds to the polymer with proper inking conditions.
To further prove the formation of the suggested structure, two-dimensional nuclear Overhauser enhancement spectroscopy (1H–1H-NOESY-NMR) was performed, which monitors the scalar coupling of protons.60 The corresponding 1H–1H NOESY is shown in Figure 3D, and analysis is performed by studying the correlation between the characteristic peaks of the PTrisAAm-polymer and the conjugated APTES. For this purpose, the cross-correlation between the signal of a polymer proton, for instance d, and the signals of APTES protons is determined. Indeed, it is detected that d has a correlation with all protons of APTES, which is highlighted with the purple box in Figure 3D, particularly showing the correlation between d and i signals. Since the signal i represents a characteristic APTES proton not overlapping with other proton signals within the structure, its correlation Hi-Hd represents a clear evidence for the formation of said APTES-polymer conjugate. Moreover, the 1H–1H NOESY of the mixture of the polymer and APTES as shown in Figure 3E (Figure S3 and Figure S4 depict the NOESY of APTES and PTrisAAm, respectively, for comparison), well in contrast to the conjugate, does not show a correlation of the signals. In addition, the 1H–1H NOESY of the mixture of the compounds shown in Figure 3E lacks the cross-peak Hi-Hd as highlighted by the purple box therein, which would be expected for a mere mixture of the compounds. In conclusion, 1H NMR as well as 1H–1H NOESY data provide evidence for the side on-coordination of the APTES (note that the structure presented in Figure 3B is only a structural proposal, for a deeper discussion find an explanation under Figure S5 in the Supporting Information).
It should also be noted that the reaction was also performed with poly(2-hydroxyethyl) acrylate (PHEA) as carrier polymer in control experiments. During the reaction, however, a dense solid formed, which was insoluble in common solvents (Figure S6). We explain this observation by the formation of a gelatinous structure, in which the silane cross-links several polymer chains. This uncontrolled cross-linking leading to nonsoluble frameworks, however, could not be observed in the case of the PTrisAAm-polymer, which makes it a much more suitable pick for a polymer framework used for ink transfer via μCP.
Subsequently to the structural elucidation of the ink coordination to the polymer in solution, the immobilization of the ink molecules to polymer brushes is investigated. In order to verify that the polymer brushes remain on the grafted surface during the inking process, the brush thickness before and after the process was determined. For this purpose, ellipsometry analysis was again performed on brushes on Si-wafers, and it was proven that surface thicknesses of the brushes before and after inking the polymers are very similar to 20 ± 4 and 20 ± 1 nm, respectively (substrate W1 from Table S1, respective ellipsometry results are shown in Table S2). Along with the data obtained from 1H NMR spectroscopy, it can be concluded from this measurement that the conjugation procedures result in an attachment of the ink to the polymers tethered to a Si surface, which we expect also to occur on the PDMS stamp surfaces.
Printing of Reactive Silane
After having proven the formation of covalent bonds between the silane ink and the polymer brush stamps, the ink transfer to an activated inorganic oxide surface needs to be assessed. To ensure a profound analysis of the ink transfer, we employ a surface-oxidized silicon wafer as substrate for printing to gain further insight into the ink transfer on a nanometer level via AFM measurements. For this purpose, a line-grating PDMS stamp is used as depicted in Figure S1B to print a well-defined pattern onto said substrate. As a result of the stamp morphology, a regular stripe pattern is expected, whereby the stripe widths as well as their interspaces each should possess dimensions of 4 μm.
Figure 4 represents the printing results following the optimized experimental protocol as outlined in the Experimental Section. As indicated by AFM imaging (results shown in Figure 4A), it is apparent that a continuous stripe pattern with a dimension of 4 μm can be obtained precisely as an expected result from the stamp morphology. A closer look into the respective topography image reveals the formation of an ultrathin layer (height profile shown in the bottom row of Figure 4), indicating the formation of a mono- or oligomolecular APTES layer.
Figure 4.

Printing on a Si wafer with a structured PDMS stamp and its reuse. AFM-height images of the printing experiments of (A) 1st inking, (B) 2nd inking, and (C) 3rd inking of the stamp. In the top row, large scale AFM images of the respective printed areas are displayed. The scale bars represent 10 μm. The insets therein are small scale AFM micrographs of the stripe patterns (10 × 10 μm2). The bottom row depicts AFM height profiles corresponding to the above micrographs. As it can be seen, the stamp can be utilized at least twice without significant quality loss, while after the third time printing quality decreases due to an increasing occurrence of particulate structures. (D) shows a fluorescence widefield microscopy image (100 × 100 μm2) of a Si wafer subjected to μCP, which has been labeled covalently and selectively with fluorescein isothiocyanate (FITC) subsequently to the printing procedure. The printed stripe pattern of FITC fluorescence is indicative for the transfer of APTES during μCP. The scale bar in panel D is 10 μm.
We were, moreover, interested in recycling of the stamp after printing. The reutilization capacity of the PTrisAAm stamps should be an intrinsic feature of the method, since the trialkoxysilane binding to the polymer is reversible. To further on investigate the reversibility of ink binding, we exposed the polymer-ink-conjugate to the experimental conditions of the ink transfer. We could demonstrate that the conjugated APTES molecules detach from the PTrisAAm polymer (as indicated by NOESY measurements as illustrated in Figure S 7), thereby recovering the macromolecular carrier. This gives the incentive to evaluate the recycling potential of the stamps. Therefore, a single PTrisAAm polymer brush stamp was subjected to three consecutive printing processes with intermediate washing and reinking steps. As summarized in Figure 4, the stamp could be used at least twice (A and B) without significant loss of the printing quality. On the other hand, a third printing process (C) resulted in a visible decrease in quality. It is furthermore visible that an increasing number of printing steps is accompanied by the formation of particulate agglomerates at the printing area, which we consider condensation products from APTES, and whose formation becomes more likely with each reinking step.
Using different solvents, we could also show that the imprinted layer withstands a thorough washing procedure of the sample as indicated by the respective AFM image (Figure S8). This observation points toward a covalent and stable attachment of the ink to the substrate. To prove the transfer of reactive amino functionalities, we exposed a printed pattern to the amino-receptive dye fluorescein isothiocyanate (FITC). Fluorescence widefield microscopy revealed that the printing pattern could be labeled with a dye selectively binding to amino functionalities (Figure 4D), which strongly suggests that the amino-functional APTES ink was transferred. Since the labeling protocol involving several washing steps did not affect the printing pattern integrity, the results shown in Figure 4D suggest that the printed layer is attached covalently to the substrate.
As a further proof, we performed X-ray photoelectron spectroscopy (XPS) on a silicon wafer, which has been previously subjected to μCP using a flat stamp. This result clearly provides evidence for the transfer of amino functionalities during the printing process. All XPS spectra and a more detailed discussion of the results can be found in the Supporting Information of the manuscript (Figure S9).
Note that the transfer of oligomeric PDMS (oPDMS) represented a pivotal problem during initial experiments. Accordingly, printing experiments, during which this aspect has not been addressed properly, resulted in the formation of stripe patterns with an unexpectedly high topography of ∼280 nm as indicated via AFM (respective AFM images as well as height profile shown in Figure S10) that could be reduced to a few nanometers by washing with an organic solvent. Being a well-known problem in PDMS microcontact printing,61,62 finding a strategy to avoid said formation has proven crucial throughout this study and has been overcome by extending the PDMS stamp curing time to 16 h and performing an additional printing step by applying a certain force at elevated temperature to squeeze the remaining insides out of the stamp.
Having shown that our method is suitable for μCP of a trialkoxysilane to a silicon wafer, the next step was to demonstrate the applicability of our routine to topographically demanding oxide materials. Because of the capillarity of the surfaces having a rough topography, a pronounced diffusive motion of the ink on the substrate is highly likely. This would tremendously promote ink smearing. Accordingly, these substrates are in general not easy to address by conventional μCP, which motivated us to assess whether our method could retain a decent printing precision on these surfaces.
As a demanding substrate, we used a glass superficially modified with a silica hydrogel, which has been prepared via a sol–gel process leading to an artificial Lotus precursor.63 The resulting silica surface features a roughness of ∼61 nm as indicated by AFM measurements (Figure S11). Due to its topograpy, we, therefore, consider said substrates challenging for μCP using LMWI. Literature reports focusing on μCP on capillary-active silica surfaces almost exclusively utilize macromolecular compounds as ink.25
The samples’ treatment with an air plasma prior to the printing procedure renders it active for silane printing. In Figure 5(A-C), the printing results are shown, where all PDMS stamps utilized have been equipped with a line-grating profile (4 μm width × 4 μm distance). For comparison, μCP experiments were performed using (A) a bare PDMS stamp as well as utilizing a polymer brush-modified stamp (B) without and (C) with covalently attached ink molecules. We would like to mention here that the attachment of the trialkoxysilane ink requires basic conditions and heat, which was not applied for the case of Figure 5A and B. As ink, the fluorescent dye rhodamine 6G (Rh6G) was used in Figure 5A,B, which can only bind electrostatically to the negatively charged surface of silica. In contrast thereto, for Figure 5C, a conjugate of APTES and rhodamine B isothiocyanate (RhBITC) was used, which was attached onto the APTES-inked stamp prior to printing. For the low molecular weight compounds utilized as ink, an increased printing precision would be expected in the order from bare to polymer without attached ink toward covalently bound ink. This can mainly be explained by the polymer-associated decrease in ink mobility on the stamp surface.41
Figure 5.

Fluorescence microscopy images of the printing experiments performed on silica gel modified glass with rough topography (∼61 nm) and in their insets in the enlarged regions: (A) with bare PDMS stamp-without grafted polymer and without covalently attached ink (Rh6G), (B) printing with polymer grafted PDMS stamp-without covalently attached ink (Rh6G), and (C) printing with polymer grafted PDMS stamp-with covalently attached ink (APTES). The images were processed with similar brightness adjustment and contrast editing routines. The exclusiveness of ink transfer to the uneven surface features reveals a substantial increase in printing precision. The scale bars are 40 μm.
Indeed, the comparison of the micrographs in Figure 5 reveals an increase of printing accuracy from panels A–C, which we may identify by the increasingly pronounced interruptions of the imprinted stripes. Note, that the uneven topography of the rough silica-based substrate only allows the stamp to contact the very tips of the surface. Hence, if ink transfer is limited to the area of contact only, these tips would appear bright, while non-contacted, i. e. deeper, parts of the topography should remain dark. Accordingly, in Figure 5A, a continuous stripe pattern can be observed, while in Figure 5B, the stripe pattern appears more interrupted. We explain this increased printing precision by the active transfer of fluorophores from the stamp to elevated regions of the substrate, which are in direct contact with the stamp during the printing process, whereas the locations of the imprinted area which remain dark are spots, that are not in direct contact with the stamp and which do not experience diffusive ink smearing. In contrast to Figure 5A, where the bright fluorescence signals are distributed all over the stripe areas, the stripes are more interrupted in Figure 5B, which can be explained by a more controlled transfer of the ink to the direct contacting sites and the mediocre prevention of ink smearing to the noncontacting areas. In Figure 5C, the fluorescence signals possess a superior local selectivity, which indicates a transfer of the ink exclusively at the area of contact between surface features and stamp (Figure 5C). Therefore, here as we aimed for, we see fluorescence signals only on the very tips of the surface and not continuous stripes, indicating the precise contact with unevenly distributed rough topography of the printing substrate and the enhanced mobility of the ink. Subjecting these samples to a washing procedure (Figure S12) resulted in significant ink smearing for the samples shown in Figure 5A,B, whereas for Figure 5C, the pronounced signals remained. This can be considered as evidence for the covalent attachment of the ink to the substrate. It might be added here that for the visualization of the imprinted surface patterns we adjusted the brightness curves of the micrographs in order to decrease the oversaturation of the larger silica aggregates.
Conclusion
In summary, we developed a microcontact printing method based on a polymer-supported ink transfer. In detail, PDMS stamps were functionalized with poly{N-[tris(hydroxymethyl)-methyl]acrylamide} (PTrisAAm) polymer brushes, able to covalently immobilize organosilanes, such as (3-aminopropyl)triethoxysilane, and, in a second step, transfer them to oxide surfaces. As a feature of this method, uncontrolled ink flow of organosilanes as ink on the substrate surface was suppressed, which is reflected in the fact that high printing precisions were achieved even on capillary-active substrates that exhibit a considerable roughness. Owing to the versatility of organosilane chemistry regarding its plethora of functionalities, as well as the fact that it can be applied to a broad variety of oxide surfaces, we believe that the idea of involving this covalent microcontact printing routine holds potential for the functionalization of various substrates with relatively complex geometries and promise to achieve precise printing results. We expect that these advances might stimulate further interest in microcontact printing. Being capable of introducing different anchor groups with a high precision and a defined chemical composition paves the way for the application of a broad follow-up chemistry.
Experimental Section
Materials and Methods
Unless stated otherwise, all chemicals were used as purchased. PDMS preparation is performed by using the commercially available standard kit of SYLGARD 184 from Dow Corning. N-[Tris(hydroxymethyl)methyl]acrylamide (contains ≤7% KCl, 93%), 4,4′-azobis(4-cyanovaleric acid) (>98%), (3-aminopropyl)triethoxysilane (APTES, >99.8%), chorotrimethylsilane (98%), ammonium hydroxide solution (28.0–30.0% w/w NH3 basis), trimethylamine (99.5%), rhodamine B isothiocyanate (mixed isomers), and rhodamine 6G (95%) were purchased from Sigma-Aldrich. NaOH (>99.8%), ethanol (96%), diethyl ether (≥99.5%), and acetic acid (99.5%) were obtained from Chemsolute. 1,4-Dioxane (>99.5%) and dichloromethane (>99.8%) were purchased from Roth. N,N-Dimethylformamide (99.8%) was obtained from Merck. Hydrogen peroxide (30%), N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimid hydrochloride (99%), and calcium chloride (>98%) were used as it is purchased from Roth. Plasma treatment for the surface activation was performed using a PlasmaFlecto 10 oven.
Surface characterization via AFM was performed using Bruker′s FastScan with ScanAsyst instrument. Surface topography of the samples was measured by using standard tapping mode with a 0.2 Hz scanning rate for Figure 2D, 0.2 Hz for the AFM images in Figures 4 and S8, 1 Hz for Figure S10, and 0.4 Hz for Figure S11 AFM images. Data evaluation was done by using the program NanoScope Analysis version 1.9.
Measurements were performed with a HC PL FLUOTAR 40x/0.80 dry objective and an aperture of 0.8. Data evaluation was done with the program ImageJ 1.52n (available from https://imagej.nih.gov/ij/download.html). To achieve proper focus on the fluorescence microscopy images, measurements were performed as a z-stack which was transformed into a maximum projection image for the fluorescence (FL) channel. Contrast was enhanced with 0.5% saturated pixels. The brightness adaptation curve was applied with the following interpolated brightness values: 30–64, 65–130, 125–216, and 190–250 (from first value to second value) for Figure 4D and 0–0, 5–40, 30–140, 65–190, 130–225, 190–245, and 255–255 for Figure 5 and Figure S12. This was done in order to visualize the imprinted patterns alongside strongly oversaturated particulate agglomerates from silica. Background subtraction was done using ImageJ by the rolling ball algorithm with radius 100 px. Image brightness was adjusted by the “auto” option. The image contrast value was adjusted by “auto” for Figure 4D, and for Figure 5 as well as Figure S12, the brightness histogram was readjusted from 50 to 255.
SEC measurements were conducted by using a device from Agilent Technologies with GRAL and GRAM columns from PSS (column 1-precolumn: 10 μm 8 × 50 mm column, column 2:7 μm 8 × 300 mm long) at 25 °C. NMP with 0.5% LiBr was used as eluent with a flow rate of 0.500 mL min–1. NMR measurements were recorded on a Bruker 400 MHz spectrometer.
n
Ellipsometry measurements were performed on dry Si wafer samples by using a Multiscope from Optrel GbR (Kleinmachnow, Germany). The null ellipsometer configuration was used with an angle of incidence of 70°. The thicknesses of the polymer brush grafted surfaces were calculated by the software “Elli”, version 5.2 (Optrel GbR). The our-layer model was used with the parameters as follows: layer 1, air ( = 1.0000, k = 0); layer 2, organic layer (n = 1.5000, k = 0); layer 3, SiO2 (d = 1.0 nm, n = 1.4580, k = 0); and layer 4, silicon (n = 3.8858, k = −0.0200). The measurements were performed on three different spots of each sample and the average values were reported.
XPS measurements were performed on an AXIS Supra+ instrument (Kratos Analytical, U.K.). For this purpose, monochromatic Al Kα-radiation (300 W) was used for excitation. The instrument was operated in hybrid mode with electrostatic and magnetic lenses. For neutralization of the sample charges, thermal electrons from a filament were used. During the measurement, the instrument was operated with a takeoff angle of 90° with an analysis depth of ∼10 nm. Data analysis was performed using CASA-XPS software. For peak fitting, Gaussian functions were used.
Preparation of PDMS Stamp
The PDMS stamp was prepared by mixing the Sylgard 184 precursor elastomer and the curing agent (w/w 10:1) from Dow Corning Company. After degassing, the mixture is poured over a Si master with a template (dimensions of 4 μm width, 4 μm interspaces and 3.6 or 7 μm height of a stripe) or a flat glass that has serves 2 mm thickness and the poured mixture was cured at 80 °C for 16 h. The obtained elastomeric gel was washed in DCM (distilled) 3 times for 24 h. After vacuum drying the washed PDMS at 42 °C for 3 h, the obtained 5% weight loss is indicated as getting rid of the free oligomers produced. Prepared stamps were printed on bare glass with 1 N, 60 °C, 16 h, as sacrificial printing, before starting the surface functionalization.
Substrate Cleaning and Surface Activation
The RCA cleaning mixture was prepared by mixing Milli-Q water, ammonium hydroxide solution (28.0–30.0% NH3 basis), and hydrogen peroxide in a 5:1:1 ratio, respectively. A silicon wafer ((1,0,0), p-Type) was immersed in the mixture at 80 °C for 20 min. The cleaned substrate was rinsed with Milli-Q water and ethanol. Surface activation of both the Si wafer substrate and the PDMS surface was done by plasma treatment with 100 W, 60 s, 100 air.
Amino-Functionalization of PDMS Stamp and Si Wafer
In order to covalently attach the CTA, later controlling the RAFT polymerization, on to the PDMS surface and Si wafer, the cleaned and plasma activated surfaces were functionalized via chemical vapor deposition (CVD) using APTES. For this purpose, 200 μL of APTES and 200 μL of ammonium hydroxide solution (28.0–30.0% w/w NH3 basis) were placed in separate glass vials inside a 60 mL PFA-chamber (Carl-Roth) containing plasma treated PDMS stamp or Si wafer. The vessel was tightly closed subjected to CVD at 70 °C for 2 h.
Synthesis and Surface Attachment of the CTA
As a CTA of the RAFT polymerization of TrisAAm, 2-{[(butylsulfanyl)carbothioyl]sulfanyl}propanoic acid (PABTC) was used. The synthesis was performed according to the literature procedure.64 Upon completion of CVD of the substrate, a solution of PABTC (5 mg, 0.021 mol), N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimid hydrochloride (EDC, 5 mg, 0.026 mol), and triethylamine (5 μL, 0.0358 mmol) in dry DMF (1 mL) was prepared. The amide coupling on the amino functionalized substrate was done by dipping into the prepared CTA solution for overnight. The substrate was washed by dipping in ethanol and dried under soft air stream.
Surface Initiated RAFT Polymerization of N-[Tris(hydroxymethyl)methyl]acrylamide (TrisAAm) (Grafting from Approach)
RAFT polymerization of TrisAAm was performed using a ratio of initiator/CTA/monomer in a 1:15:3000 ratio. TrisAAm (350 mg, 2 mmol) was dissolved in 1,4-dioxane/Milli-Q water (1:1; 4 mL) solvent. The monomer solution of TrisAAm was first filtered through an Al2O3 column and then through a PVDF filter (0.2 μm pore size) to remove the inhibitor. From the filtered monomer solution (2 mL), 175 mg of 1 mmol TrisAAm was mixed with PABTC (1.2 mg, 0.005 mmol). The initiator ACVA (0.093 mg, 0.0003 mmol) was added to the reaction mixture. The final reaction mixture containing monomer, CTA and initiator dissolved in the reaction solution was purged using a nitrogen stream for 30 min to remove oxygen. The degassed reaction solution was transferred into reaction tubes containing a nitrogen atmosphere and PABTC modified substrate. The reaction tubes were closed with a rubber septa and placed into an oil bath at 70 °C for 14 h.
Inking: Covalent Attachment of APTES to the PTrisAAm in Solution
DMF was dried using CaCl2 and filtered through a PVDF filter with a pore size of 0.2 μm. PTrisAAm (50 mg) was dissolved in dried DMF (2.2 mL) and stirred in an Eppendorf tube until it dissolved completely. APTES (63 mg, 0.28 mmol) was added to the solution with the catalyst NaOH (0.11 mg, 0.0028 mmol). The reaction tube was placed in an oil bath at 100 °C for 3 h. The obtained product was precipitated in cold diethyl ether. The precipitate dissolved in DMSO and precipitated again in isopropanol three times as a cycle. The final product was dried under vacuum.
Inking: Covalent Attachment of APTES to the PTrisAAm Grafted from the Surface Initiated PDMS Stamp and Si Wafer
Inking solution (1 mg mL–1) was prepared by dissolving APTES (1.3 mg, 0.00587 mmol) in 1300 μL of dried DMF together with NaOH as catalyst (0.00234 mg, 5.87 × 10–5 mmol) per stamp and wafer in a closed Eppendorf tube, and the reaction took place in a 100 °C oil bath for 3 h. The inked stamp was washed in DCM for 3 days refreshing the solvent daily and dried in a vacuum oven at 42 °C for 3 h. The stamp that was reused for the second and third printing was washed prior to each reinking in 10% acetic acid aqueous solution for 3 days by refreshing the solvent and dried in a vacuum oven at 42 °C for 3 h, and it was continued with the same procedure as in the first inking. Note that, for the printing shown in Figure 4D, the stamp with grafted polymer on it was washed again in DCM for 3 days and dried at 42 °C for 3 h under vacuum prior to inking.
Transfer of APTES on to Substrates
To prepare the inked and washed stamp for printing, a sacrificial printing was done beforehand for each stamp. For this purpose, stamp was at first printed on chlorotrimethylsilane functionalized glass with 1 N, 60 °C, 16 h.
Stamp was spin coated (3000 rpm, 60 s) with a 1% acetic acid aqueous solution as the catalyst and dried with air stream. Printing on Si wafer was performed in a closed vessel with no weight on it just by laying the PDMS (with 4 μm stripe pattern) on to the surface activated substrate for 20 min at room temperature. The vessel also contained a separate of vial 20 μL of acetic acid and saturated NaCl aqueous solution. Washing of the printed substrate was done by filtered ethanol and Milli-Q water.
Fluorescence labeling of the printed Si wafer was done by 10 μg of fluorescein isothiocyanate per 1 mL of EtOH per substrate by dipping it in to the solution for 1 h and then washed with ethanol and carbonate buffer solution with pH 9.5.
For the stamp (St 6, Table S1) that is used for XPS measurements, a flat PDMS stamp that has no stripe pattern on it was printed under the same printing conditions as mentioned above, for printing on the Si wafer.
For the stamp used for printing on silica gel modified glass, labeling was done by using 10 μg of rhodamine B isothiocyanate per 1 mL of DMF (dried) or 1 mL of EtOH per stamp by dipping it in to the solution for 1 h and washing with ethanol. Printing was done with the rhodamine B isothiocyanate labeled stamp (with 4 μm stripe pattern) at the same conditions with an additional 200 g of weight on it. Washing of the printed substrate was done by filtered EtOH.
Transfer of Rhodamine 6G on to Substrate
The washed stamp was used after a sacrificial printing was done as mentioned in the previous subsection. Note that here there was no covalent attachment of ink into to polymer brushes. Inking was performed by drop casting 10 μg of rhodamine 6G dye per 1 mL of Milli-Q water for 1 h and printed at room temperature for 10 s with 200 g of weight on the silica gel modified glass substrate.
Preparation of the Printing Substrates
The RCA cleaning mixture was prepared by mixing Milli-Q water, ammonium hydroxide solution (28.0–30.0% NH3 basis), and hydrogen peroxide in a 5:1:1 ratio, respectively. The Si wafer was immersed in the mixture at 80 °C for 20 min. The cleaned substrate was rinsed with Milli-Q water and ethanol, respectively. Surface activation of the substrate was done by plasma treatment with 100 W, 60 s, and 100 air.
Silica gel modified glass was prepared using a trimethylsilyl [-Si(CH3)3] modified SiO2 sol. according to the literature procedure.63 The obtained sol was prepared as 1% w/w in n-propanol and used for dip coating the glass by withdrawing 400 mm min–1. Prior to dip coating, the glass surface was activated by plasma treatment of (100 W, 60 s, and 100 air), and chlorotrimethylsilane functionalization was performed by CVD at room temperature. It was dried for 1 h at 100 °C and treated with plasma (100 W, 60 s, and 100 air) right before printing.
Acknowledgments
The authors gratefully acknowledge the financial support of European Research Council (ERC) in the framework of the project REPLICOLL (Grant No. 648365). The authors thank the NMR core facility of the Institute of Chemistry (University of Potsdam) of Prof. Heiko Möller, Dr. Matthias Heydenreich, and Angela Krtitschka. Dr. Erik Wischerhoff is acknowledged for measuring the ellipsometry data. We also thank Sophie Laroque for performing SEC measurements. Dr. Andreas Holländer is acknowledged for providing useful advices in XPS data interpretation. M.H. gratefully acknowledges funding by the DFG (Emmy-Noether-Program, HA 7725/2-1) as well as the Open-Topic Postdoc program of the University of Potsdam.
Glossary
Abbreviations
- μCP
microcontact printing
- APTES
(3-aminopropyl)-triethoxysilane
- PDMS
polydimethylsiloxane
- TrisAAm
N-[tris(hydroxymethyl)-methyl]acrylamide
- RAFT
reversible addition–fragmentation chain-transfer
- AFM
atomic force microscopy
- NOESY
nuclear Overhauser enhancement spectroscopy
- SAM
self-assembled monolayers
- LMWI
low molecular weight inks
- CTA
chain transfer agent
- CVD
chemical vapor deposition
- DP
degree of polymerization
- PTFE
polytetrafluoroethylene
- PHEA
poly(2-hydroxyethyl) acrylate
- XPS
X-ray photoelectron spectroscopy
- RhBITC
rhodamine B isothiocyanate
- EDC
N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimid hydrochloride
- ACVA
4,4′-azobis(4-cyanovaleric acid)
- PABTC
2-{[(butylsulfanyl)carbothionyl]-sulfanyl}propanoic acid
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsapm.1c00024.
Supplemental 1H–1H NOESY data including further discussion; SEC data; AFM data, XPS data, and discussion; and fluorescence microscopy data (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Kamyshny A.; Magdassi S. Conductive Nanomaterials for Printed Electronics. Small 2014, 10 (17), 3515–3535. 10.1002/smll.201303000. [DOI] [PubMed] [Google Scholar]
- Hyun W. J.; Secor E. B.; Hersam M. C.; Frisbie C. D.; Francis L. F. High-resolution Patterning of Graphene by Screen Printing with a Silicon Stencil for Highly Flexible Printed Electronics. Adv. Mater. 2015, 27 (1), 109–115. 10.1002/adma.201404133. [DOI] [PubMed] [Google Scholar]
- Ditlbacher H.; Krenn J.; Lamprecht B.; Leitner A.; Aussenegg F. Spectrally Coded Optical Data Storage by Metal Nanoparticles. Opt. Lett. 2000, 25 (8), 563–565. 10.1364/OL.25.000563. [DOI] [PubMed] [Google Scholar]
- Lee J.; Yoo B.; Lee H.; Cha G. D.; Lee H.-S.; Cho Y.; Kim S. Y.; Seo H.; Lee W.; Son D.; Kang M.; Kim H. M.; Park Y. I.; Hyeon T.; Kim D.-H. Ultra-Wideband Multi-Dye-Sensitized Upconverting Nanoparticles for Information Security Application. Adv. Mater. 2017, 29 (1), 1603169. 10.1002/adma.201603169. [DOI] [PubMed] [Google Scholar]
- Yu Q.; Guan P.; Qin D.; Golden G.; Wallace P. M. Inverted Size-Dependence of Surface-Enhanced Raman Scattering on Gold Nanohole and Nanodisk Arrays. Nano Lett. 2008, 8 (7), 1923–1928. 10.1021/nl0806163. [DOI] [PubMed] [Google Scholar]
- Sun F.; Hung H.-C.; Sinclair A.; Zhang P.; Bai T.; Galvan D. D.; Jain P.; Li B.; Jiang S.; Yu Q. Hierarchical Zwitterionic Modification of a SERS Substrate Enables Real-Time Drug Monitoring in Blood Plasma. Nat. Commun. 2016, 7 (1), 1–9. 10.1038/ncomms13437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juste-Dolz A.; Avella-Oliver M.; Puchades R.; Maquieira A. Indirect Microcontact Printing to Create Functional Patterns of Physisorbed Antibodies. Sensors 2018, 18 (9), 3163. 10.3390/s18093163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogele K.; List J.; Pardatscher G.; Holland N. B.; Simmel F. C.; Pirzer T. Self-Assembled Active Plasmonic Waveguide with a Peptide-Based Thermomechanical Switch. ACS Nano 2016, 10 (12), 11377–11384. 10.1021/acsnano.6b06635. [DOI] [PubMed] [Google Scholar]
- Atwater H. A.; Polman A. Plasmonics for Improved Photovoltaic Devices. Nat. Mater. 2010, 9 (3), 205–213. 10.1038/nmat2629. [DOI] [PubMed] [Google Scholar]
- Lamping S.; Buten C.; Ravoo B. J. Functionalization and Patterning of Self-Assembled Monolayers and Polymer Brushes Using Microcontact Chemistry. Acc. Chem. Res. 2019, 52 (5), 1336–1346. 10.1021/acs.accounts.9b00041. [DOI] [PubMed] [Google Scholar]
- Kaufmann T.; Ravoo B. J. Stamps, Inks and Substrates: Polymers in Microcontact Printing. Polym. Chem. 2010, 1 (4), 371–387. 10.1039/B9PY00281B. [DOI] [Google Scholar]
- Zhou X.; Xu H.; Cheng J.; Zhao N.; Chen S.-C. Flexure-Based Roll-to-Roll Platform: A Practical Solution for Realizing Large-Area Microcontact Printing. Sci. Rep. 2015, 5, 10402. 10.1038/srep10402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G.; Hirtz M.; Fuchs H.; Zheng Z. Development of Dip-Pen Nanolithography (DPN) and Its Derivatives. Small 2019, 15 (21), 1900564. 10.1002/smll.201900564. [DOI] [PubMed] [Google Scholar]
- Vieu C.; Carcenac F.; Pepin A.; Chen Y.; Mejias M.; Lebib A.; Manin-Ferlazzo L.; Couraud L.; Launois H. Electron Beam Lithography: Resolution Limits and Applications. Appl. Surf. Sci. 2000, 164, 111–117. 10.1016/S0169-4332(00)00352-4. [DOI] [Google Scholar]
- Chen Y. Nanofabrication by Electron Beam Lithography and Its Applications: A Review. Microelectron. Eng. 2015, 135, 57–72. 10.1016/j.mee.2015.02.042. [DOI] [Google Scholar]
- Haufe W.Production of Microstructures by Ion Beam Sputtering. In Sputtering by Particle Bombardment III: Characteristics of Sputtered Particles, Technical Applications; Behrisch R., Wittmaack K., Eds.; Springer Berlin Heidelberg: Berlin, Heidelberg, 1991; pp 305–338. [Google Scholar]
- Qian H. X.; Zhou W.; Miao J.; Lim L. E. N.; Zeng X. R. Fabrication of Si Microstructures Using Focused Ion Beam Implantation and Reactive Ion Etching. J. Micromech. Microeng. 2008, 18 (3), 035003. 10.1088/0960-1317/18/3/035003. [DOI] [Google Scholar]
- Buschow K. J.; Cahn R. W.; Flemings M. C.; Ilschner B.; Kramer E. J.; Mahajan S. Encyclopedia of Materials. Sci. Technol. 2001, 1, 11. [Google Scholar]
- Stolz Roman L.; Inganäs O.; Granlund T.; Nyberg T.; Svensson M.; Andersson M. R.; Hummelen J. C. Trapping Light in Polymer Photodiodes with Soft Embossed Gratings. Adv. Mater. 2000, 12 (3), 189–195. . [DOI] [Google Scholar]
- Mrksich M.; Whitesides G. M. Patterning Self-Assembled Monolayers Using Microcontact Printing: A New Technology for Biosensors?. Trends Biotechnol. 1995, 13 (6), 228–235. 10.1016/S0167-7799(00)88950-7. [DOI] [Google Scholar]
- Dias A. D.; Kingsley D. M.; Corr D. T. Recent Advances in Bioprinting and Applications for Biosensing. Biosensors 2014, 4 (2), 111–136. 10.3390/bios4020111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. J.; Huie P.; Leng T.; Peterman M. C.; Marmor M. F.; Blumenkranz M. S.; Bent S. F.; Fishman H. A. Microcontact Printing on Human Tissue for Retinal Cell Transplantation. Arch. Ophthalmol. 2002, 120 (12), 1714–1718. 10.1001/archopht.120.12.1714. [DOI] [PubMed] [Google Scholar]
- Yu H.; Xiong S.; Tay C. Y.; Leong W. S.; Tan L. P. A Novel and Simple Microcontact Printing Technique for Tacky, Soft Substrates and/or Complex Surfaces in Soft Tissue Engineering. Acta Biomater. 2012, 8 (3), 1267–1272. 10.1016/j.actbio.2011.09.006. [DOI] [PubMed] [Google Scholar]
- Sathish S.; Ricoult S. G.; Toda-Peters K.; Shen A. Q. Microcontact Printing with Aminosilanes: Creating Biomolecule Micro-and Nanoarrays for Multiplexed Microfluidic Bioassays. Analyst 2017, 142 (10), 1772–1781. 10.1039/C7AN00273D. [DOI] [PubMed] [Google Scholar]
- Blinka E.; Loeffler K.; Hu Y.; Gopal A.; Hoshino K.; Lin K.; Liu X.; Ferrari M.; Zhang J. X. J. Enhanced Microcontact Printing of Proteins on Nanoporous Silica Surface. Nanotechnology 2010, 21 (41), 415302. 10.1088/0957-4484/21/41/415302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A.; Whitesides G. M. Features of Gold Having Micrometer to Centimeter Dimensions Can Be Formed through a Combination of Stamping with an Elastomeric Stamp and an Alkanethiol Ink Followed by Chemical Etching. Appl. Phys. Lett. 1993, 63, 2002–2004. 10.1063/1.110628Plea. [DOI] [Google Scholar]
- Colorado R. Jr.; Lee T.. Thiol-Based Self-Assembled Monolayers: Formation and Organization, 2001.
- Pujari S. P.; Scheres L.; Marcelis A. T. M.; Zuilhof H. Covalent Surface Modification of Oxide Surfaces. Angew. Chem., Int. Ed. 2014, 53 (25), 6322–6356. 10.1002/anie.201306709. [DOI] [PubMed] [Google Scholar]
- Tao Y. T. Structural Comparison of Self-Assembled Monolayers of n-Alkanoic Acids on the Surfaces of Silver, Copper, and Aluminum. J. Am. Chem. Soc. 1993, 115 (10), 4350–4358. 10.1021/ja00063a062. [DOI] [Google Scholar]
- Perl A.; Reinhoudt D. N.; Huskens J. Microcontact Printing: Limitations and Achievements. Adv. Mater. 2009, 21 (22), 2257–2268. 10.1002/adma.200801864. [DOI] [Google Scholar]
- Maksud M. I.; Yusof M. S.; Mahadi M.; Jamil A.. A Study on Printed Multiple Solid Line by Combining Microcontact and Flexographic Printing Process for Microelectronic and Biomedical Applications. Int. J. Integr. Eng. 2014, 5 ( (3), ). [Google Scholar]
- Wendeln C.; Ravoo B. J. Surface Patterning by Microcontact Chemistry. Langmuir 2012, 28 (13), 5527–5538. 10.1021/la204721x. [DOI] [PubMed] [Google Scholar]
- Mizuno H.; Buriak J. M. Nanoscale Patterning of Organic Monolayers by Catalytic Stamp Lithography: Scope and Limitations. ACS Appl. Mater. Interfaces 2009, 1 (12), 2711–2720. 10.1021/am900602m. [DOI] [PubMed] [Google Scholar]
- Wendeln C.; Roling O.; Schulz C.; Hentschel C.; Ravoo B. J. Modification of Surfaces by Chemical Transfer Printing Using Chemically Patterned Stamps. Langmuir 2013, 29 (8), 2692–2699. 10.1021/la305024a. [DOI] [PubMed] [Google Scholar]
- Bhairamadgi N. S.; Gangarapu S.; Caipa Campos M. A.; Paulusse J. M. J.; van Rijn C. J. M.; Zuilhof H. Efficient Functionalization of Oxide-Free Silicon(111) Surfaces: Thiol–Yne versus Thiol–Ene Click Chemistry. Langmuir 2013, 29 (14), 4535–4542. 10.1021/la400007y. [DOI] [PubMed] [Google Scholar]
- Rozkiewicz D. I.; Ravoo B. J.; Reinhoudt D. N. Reversible Covalent Patterning of Self-Assembled Monolayers on Gold and Silicon Oxide Surfaces. Langmuir 2005, 21 (14), 6337–6343. 10.1021/la050438i. [DOI] [PubMed] [Google Scholar]
- Wendeln C.; Heile A.; Arlinghaus H. F.; Ravoo B. J. Carbohydrate Microarrays by Microcontact Printing. Langmuir 2010, 26 (7), 4933–4940. 10.1021/la903569v. [DOI] [PubMed] [Google Scholar]
- Kaufmann T.; Gokmen M. T.; Wendeln C.; Schneiders M.; Rinnen S.; Arlinghaus H. F.; Bon S. A. F.; Du Prez F. E.; Ravoo B. J. Sandwich” Microcontact Printing as a Mild Route Towards Monodisperse Janus Particles with Tailored Bifunctionality. Adv. Mater. 2011, 23 (1), 79–83. 10.1002/adma.201003564. [DOI] [PubMed] [Google Scholar]
- Sharpe R. B. A.; Burdinski D.; Huskens J.; Zandvliet H. J. W.; Reinhoudt D. N.; Poelsema B. Chemically Patterned Flat Stamps for Microcontact Printing. J. Am. Chem. Soc. 2005, 127 (29), 10344–10349. 10.1021/ja052139l. [DOI] [PubMed] [Google Scholar]
- Ng E.; Gopal A.; Hoshino K.; Zhang X. Multicolor Microcontact Printing of Proteins on Nanoporous Surface for Patterned Immunoassay. Appl. Nanosci. 2011, 1 (2), 79–85. 10.1007/s13204-011-0009-0. [DOI] [Google Scholar]
- Sperling M.; Reifarth M.; Grobe R.; Böker A. Tailoring Patches on Particles: A Modified Microcontact Printing Routine Using Polymer-Functionalised Stamps. Chem. Commun. 2019, 55 (68), 10104–10107. 10.1039/C9CC03903A. [DOI] [PubMed] [Google Scholar]
- Zimmermann M.; John D.; Grigoriev D.; Puretskiy N.; Böker A. From 2D to 3D Patches on Multifunctional Particles: How Microcontact Printing Creates a New Dimension of Functionality. Soft Matter 2018, 14 (12), 2301–2309. 10.1039/C8SM00163D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John D.; Zimmermann M.; Böker A. Generation of 3-Dimensional Multi-Patches on Silica Particles via Printing with Wrinkled Stamps. Soft Matter 2018, 14 (16), 3057–3062. 10.1039/C8SM00224J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M.; Grigoriev D.; Puretskiy N.; Böker A. Characteristics of Microcontact Printing with Polyelectrolyte Ink for the Precise Preparation of Patches on Silica Particles. RSC Adv. 2018, 8 (69), 39241–39247. 10.1039/C8RA07955B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadhu V. B.; Perl A.; Péter M.; Rozkiewicz D. I.; Engbers G.; Ravoo B. J.; Reinhoudt D. N.; Huskens J. Surface Modification of Elastomeric Stamps for Microcontact Printing of Polar Inks. Langmuir 2007, 23 (12), 6850–6855. 10.1021/la063657s. [DOI] [PubMed] [Google Scholar]
- Haensch C.; Hoeppener S.; Schubert U. S. Chemical Modification of Self-Assembled Silane Based Monolayers by Surface Reactions. Chem. Soc. Rev. 2010, 39 (6), 2323–2334. 10.1039/b920491a. [DOI] [PubMed] [Google Scholar]
- Li H.; Zhang J.; Zhou X.; Lu G.; Yin Z.; Li G.; Wu T.; Boey F.; Venkatraman S. S.; Zhang H. Aminosilane Micropatterns on Hydroxyl-Terminated Substrates: Fabrication and Applications. Langmuir 2010, 26 (8), 5603–5609. 10.1021/la9039144. [DOI] [PubMed] [Google Scholar]
- Geissler M.; Kind H.; Schmidt-Winkel P.; Michel B.; Delamarche E. Direct Patterning of NiB on Glass Substrates Using Microcontact Printing and Electroless Deposition. Langmuir 2003, 19 (15), 6283–6296. 10.1021/la034317z. [DOI] [Google Scholar]
- Mukhopadhyay R. When PDMS Isn’t the Best. Anal. Chem. 2007, 79 (9), 3248–3253. 10.1021/ac071903e. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Zheng Z.; Huang Z.; Ling J. A CTA-Shuttled R-Group Approach: A Versatile Synthetic Tool towards Well-Defined Functional Cylindrical Polymer Brushes via RAFT Polymerization. Polym. Chem. 2017, 8 (17), 2659–2665. 10.1039/C7PY00167C. [DOI] [Google Scholar]
- Chapman R.; Gormley A. J.; Herpoldt K.-L.; Stevens M. M. Highly Controlled Open Vessel RAFT Polymerizations by Enzyme Degassing. Macromolecules 2014, 47 (24), 8541–8547. 10.1021/ma5021209. [DOI] [Google Scholar]
- Zheng Z.; Ling J.; Müller A. H. E. Revival of the R-Group Approach: A “CTA-Shuttled” Grafting from Approach for Well-Defined Cylindrical Polymer Brushes via RAFT Polymerization. Macromol. Rapid Commun. 2014, 35 (2), 234–241. 10.1002/marc.201300578. [DOI] [PubMed] [Google Scholar]
- Kerr A.; Hartlieb M.; Sanchis J.; Smith T.; Perrier S. Complex Multiblock Bottle-Brush Architectures by RAFT Polymerization. Chem. Commun. 2017, 53 (87), 11901–11904. 10.1039/C7CC07241D. [DOI] [PubMed] [Google Scholar]
- Arkles B. Tailoring Surfaces with Silanes. CHEMTECH 1977, 7, 766–778. [Google Scholar]
- Puri J. K.; Singh R.; Chahal V. K. Silatranes: A Review on Their Synthesis, Structure, Reactivity and Applications. Chem. Soc. Rev. 2011, 40 (3), 1791–1840. 10.1039/B925899J. [DOI] [PubMed] [Google Scholar]
- Shlyakhtenko L. S.; Gall A. A.; Lyubchenko Y. L. Mica Functionalization for Imaging of DNA and Protein-DNA Complexes with Atomic Force Microscopy. Methods Mol. Biol. 2012, 931, 295–312. 10.1007/978-1-62703-056-4_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conzatti G.; Cavalie S.; Combes C.; Torrisani J.; Carrere N.; Tourrette A. PNIPAM Grafted Surfaces through ATRP and RAFT Polymerization: Chemistry and Bioadhesion. Colloids Surf., B 2017, 151, 143–155. 10.1016/j.colsurfb.2016.12.007. [DOI] [PubMed] [Google Scholar]
- Saito N.; Sugawara T.; Matsuda T. Synthesis and Hydrophilicity of Multifunctionally Hydroxylated Poly(Acrylamides). Macromolecules 1996, 29 (1), 313–319. 10.1021/ma950419n. [DOI] [Google Scholar]
- Bax A. Two-Dimensional NMR and Protein Structure. Annu. Rev. Biochem. 1989, 58, 223–256. 10.1146/annurev.bi.58.070189.001255. [DOI] [PubMed] [Google Scholar]
- Wang X.; Sperling M.; Reifarth M.; Böker A. Shaping Metallic Nanolattices: Design by Microcontact Printing from Wrinkled Stamps. Small 2020, 16, 1906721. 10.1002/smll.201906721. [DOI] [PubMed] [Google Scholar]
- Yunus S.; de Looringhe C. d. C.; Poleunis C.; Delcorte A. Diffusion of Oligomers from Polydimethylsiloxane Stamps in Microcontact Printing: Surface Analysis and Possible Application. Surf. Interface Anal. 2007, 39 (12–13), 922–925. 10.1002/sia.2623. [DOI] [Google Scholar]
- Goswami D.; Medda S. K.; De G. Superhydrophobic Films on Glass Surface Derived from Trimethylsilanized Silica Gel Nanoparticles. ACS Appl. Mater. Interfaces 2011, 3 (9), 3440–3447. 10.1021/am200666m. [DOI] [PubMed] [Google Scholar]
- Larnaudie S. C.; Brendel J. C.; Jolliffe K. A.; Perrier S. Cyclic Peptide–Polymer Conjugates: Grafting-to vs Grafting-From. J. Polym. Sci., Part A: Polym. Chem. 2016, 54 (7), 1003–1011. 10.1002/pola.27937. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.