

Abstract
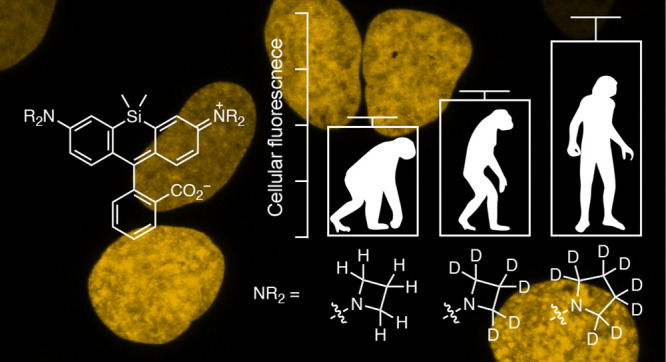
Fluorescence microscopy relies on dyes that absorb and then emit photons. In addition to fluorescence, fluorophores can undergo photochemical processes that decrease quantum yield or result in spectral shifts and irreversible photobleaching. Chemical strategies that suppress these undesirable pathways—thereby increasing the brightness and photostability of fluorophores—are crucial for advancing the frontier of bioimaging. Here, we describe a general method to improve small-molecule fluorophores by incorporating deuterium into the alkylamino auxochromes of rhodamines and other dyes. This strategy increases fluorescence quantum yield, inhibits photochemically induced spectral shifts, and slows irreparable photobleaching, yielding next-generation labels with improved performance in cellular imaging experiments.
Keywords: fluorescence, isotope effect, microscopy, organic chemistry, photochemistry, photobleaching, rhodamine, single-molecule imaging
Introduction
Fluorescence microscopy is a powerful tool to visualize the location and dynamics of biomolecules in living systems. The development of advanced microscopy techniques such as live-cell single-molecule imaging promises the visualization of proteins and other cellular components with high spatial and temporal precision. These new microscopy methods are photon-intensive, however, placing increased demands on the fluorescent labels. The unrelenting need for more photons is driving a renaissance in the field of small-molecule fluorophores, which exhibit the requisite brightness and photostability for advanced microscopy techniques and can be adapted to disparate imaging modalities and labeling strategies.1
There are several avenues for enhancing small-molecule fluorescent dyes.2 Increasing the fluorescence quantum yield (Φf) is an obvious way to improve imaging because brighter dyes translate more excitation light into emitted photons. Fluorophores can undergo various photochemical reactions that yield nonfluorescent products (i.e., photobleaching). Designing fluorophores with improved photostability enables longer duration imaging. Small-molecule dyes can undergo other photochemistry that elicits undesirable shifts in the absorption maximum (λabs) and fluorescence emission maximum (λem), which effectively broadens spectra and decreases excitation efficiency. Thus, improving fluorophore “chromostability” is also advantageous for imaging. Here, we describe a general method to improve small-molecule fluorophores through installation of deuterated auxochromes. This straightforward modification of dyes—the net addition of a few neutrons—enhances Φf, photostability, and chromostability, resulting in fluorophores with improved performance in cellular imaging experiments.
Results and Discussion
Rational optimization of small-molecule dyes requires an understanding of dye photophysics and the ability to easily modulate fluorophore structure using chemistry. We considered the rhodamine dyes, which persist in modern biological imaging due to their excellent brightness, superb photostability, and tunable spectral and chemical properties.3−8 The photophysics of rhodamines are well-understood due to their importance as laser dyes and biological probes.2 Rhodamines are also amenable to structural modification using a variety of synthetic organic chemistry strategies.4,9−11
The classic fluorophore tetramethylrhodamine (TMR, 1, Figure 1a) illustrates the intricate photophysics of rhodamine dyes. Absorption of a photon excites the dye from the ground state (1-S0) ultimately to the first excited state (1-S1). After excitation, the molecule can relax back to 1-S0 through different processes. Emission of a photon (fluorescence) competes with nonradiative decay pathways such as twisted internal charge transfer (TICT),12 where electron transfer from the aniline nitrogen to the xanthene system gives a charge-separated species with a twisted C–N bond (1-TICT); this decays back to 1-S0 without emitting a photon. TMR is susceptible to nonradiative decay via TICT, leading to a modest quantum yield (Φf = 0.41).13 Alternatively, the excited dye can undergo intersystem crossing (ISC) to the first triplet excited state (1-T1), from where it can sensitize singlet oxygen (1O2) and return to 1-S0. The 1O2 can oxidize the aniline nitrogen to the radical cation (1+•), which can undergo deprotonation to a carbon-centered radical (1•). Reaction with reactive oxygen species eventually results in loss of formaldehyde, giving the dealkylated trimethylrhodamine (2).2 This leads to a shift in λabs and λem. The distinct photobleaching reactions of rhodamines remain mysterious with multiple possible pathways (Figure 1a). Nevertheless, dealkylated rhodamines typically exhibit lower photostability, making this photochemical reaction a key initial step in photobleaching.
Figure 1.
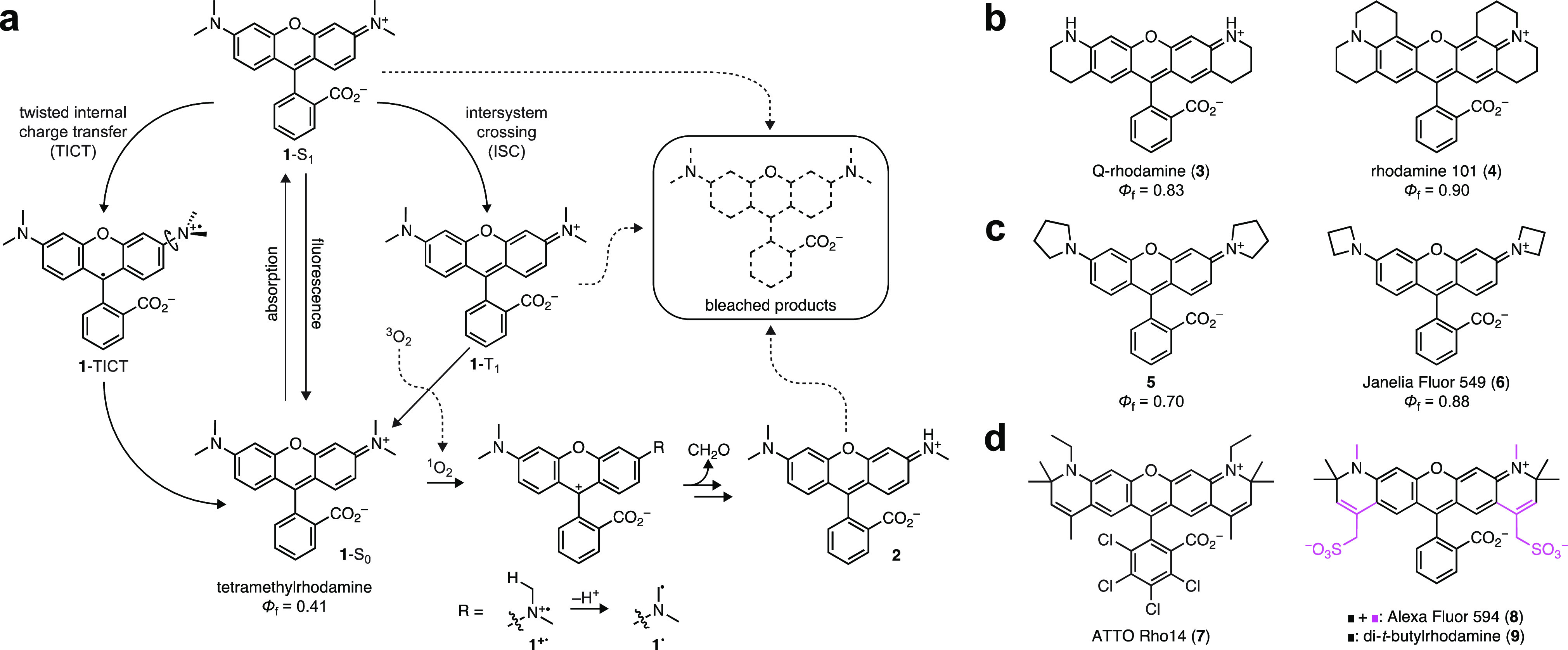
Photophysics of rhodamines and methods to improve rhodamine properties. (a) Photophysics of tetramethylrhodamine (TMR, 1). (b) Structures of rigidified rhodamines 3–4. (c) Structures of cyclic amine-containing rhodamines 5–6. (c) Structures of α-quaternary rhodamines 7–9.
Both of these undesirable processes—TICT and dealkylation—can be mitigated through modifications in chemical structure. Rigidification of the rhodamine prevents rotation of the C–N bond and improves Φf,13 as evidenced by Q-rhodamine (3; Φf = 0.83)14 and rhodamine 101 (4; Φf = 0.90; Figure 1b).15 Decreasing the electron donor strength and minimizing homoallylic interactions can also decrease TICT and improve brightness. This was first demonstrated by Drexhage, a pioneer in fluorophore chemistry, who found that replacing the N,N-dimethylamino auxochromes in 1 with five-membered pyrrolidine rings afforded a brighter dye (5; Φf = 0.70; Figure 1c).13 We discovered that incorporation of smaller, four-membered azetidine rings further improved the brightness of rhodamines and other fluorophores, yielding the “Janelia Fluor” (JF) dyes.6−8 The azetidinyl-rhodamine 6 (JF549; Φf = 0.88) shows comparable Φf to the fully rigidified 4 (Figure 1b, c). The increased ionization potential of azetidine16 likely underlies the improved photostability of 6 because it suppresses formation of a radical cation (e.g., 1+•, Figure 1a). Finally, the dealkylation process can be inhibited by installing α-quaternary centers on the aniline nitrogens, thereby precluding deprotonation to form radicals such as 1• (Figure 1a). This structural motif was also introduced by Drexhage;17 it is found in a number of commercial fluorophores (e.g., 7–8),18 and this concept was revisited in the simplified di-t-butylrhodamine (9, Figure 1d).19
We envisioned an alternative strategy to increase brightness and photostability of small-molecule fluorophores such as 1 by replacing the hydrogen (H) atoms in the N-alkyl groups with deuterium (D). Oxidation of alkylamines can show remarkably large secondary isotope effects,20 suggesting that deuteration could reduce the electron donor strength of the auxochrome. This would decrease the efficiency of the TICT process and increase Φf. This effect would also slow 1O2-mediated oxidation (e.g., 1 → 1+•), and the stronger C–D bond could lower the rate of deprotonation (e.g., 1+• → 1•, Figure 1a). Together, these effects would likely decrease undesired dealkylation and improve both chromostability and photostability.
Deuterium substitution has been suggested as a strategy to improve fluorophores by altering vibrational modes.21 This idea is bolstered by the higher Φf and photostability observed for many fluorophores in deuterated solvents.22−24 Prior examples of deuterated dyes are limited, however, and are largely focused on direct attachment of D atoms to the aromatic system of the fluorophore. This substitution typically yields a negative or neutral effect on Φf as demonstrated for compounds 10–14 (Figure S1).25−27 The use of deuterated N-alkyl auxochromes to control electron and proton transfer represents a new hypothesis, which was initially tested with the classic fluorophore TMR (1). The deuterated analogue 1D was synthesized using a cross-coupling approach with fluorescein ditriflate (15) and dimethylamine-d6 (16; Figure 2a).9 Comparison of dyes 1 and 1D revealed quite similar λabs and λem, high extinction coefficients at λabs (ε; Table 1), and no change in the shape of the absorption or fluorescence emission spectra (Figure 1b). Deuteration did affect the brightness of the dye, however, with 1D showing a 22% increase in Φf compared to 1 (Table 1).28
Figure 2.

Deuterated tetramethylrhodamine. (a) Synthesis of 1D. (b) Normalized absorption (abs) and fluorescence emission (em) spectra of 1 and 1D. (c, d) LC–MS traces of 1 (c) and 1D (d) before and after photobleaching using 560 nm (1.02 W/cm2, 6 h). (e, f) Sequential absorption spectra of 1 (e) and 1D (f) during photobleaching using 560 nm (1.02 W/cm2). The magenta arrows highlight the shift in λabs and absorption intensity over time.
Table 1. Spectral Properties of Rhodaminesa.


All values are in 10 mM HEPES, pH 7.3 except for KL–Z, which was measured in 1:1 v/v dioxane:H2O.
The photostabilities of 1 and 1D were then compared in vitro. Solutions of both dyes were illuminated with 560 nm light and measured intermittently using tandem liquid chromatography–mass spectrometry (LC–MS) and absorption spectroscopy. For the parent TMR (1), LC–MS revealed clean demethylation to trimethylrhodamine (2; Figure 2c). This was also observed for the deuterated compound 1D but at a slower rate (Figure 2d, Figure S2a, b). The different degrees of demethylation were reflected in the absorption spectra, where 1 showed faster photobleaching and a more pronounced blue-shift in λabs compared to the deuterated 1D (Figure 2e, f, Figure S2c, d).
Based on this result with TMR (1), a series of matched pairs of rhodamine dyes with H- or D-substituted cyclic N-alkyl groups were synthesized (Table 1, Figure S3a). Like the TMR compounds 1 and 1D, deuteration did not significantly change λabs, λem, or spectral shape (Table 1, Figure S3b–e). The lactone–zwitterion equilibrium constant (KL–Z), a key determinant for rhodamine performance in biological environments,8 showed only minor changes with deuteration. The pyrrolidine dyes 5 and 5D exhibited the highest values with KL–Z > 4. The morpholino derivatives 18 and 18D showed a substantial shift to lower values (KL–Z < 0.2), due in part to the inductive electron-withdrawing properties of the oxygen atom.29 Like the TMR scaffold, where the deuterated 1D showed a higher Φf value compared to the parent dye 1 (Φf,D/Φf,H = 1.22), other deuterated rhodamines showed an increase in Φf, including dyes containing pyrrolidine (5/5D; Φf,D/Φf,H = 1.14), piperidine (17/17D; Φf,D/Φf,H = 1.50), and morpholine (18/18D; Φf,D/Φf,H = 1.18; Table 1). Notably, the azetidine-d6 compound (6D) showed no improvement in Φf over 6 (Table 1). This result is consistent with the hypothesis that the azetidine and deuterium substitutions both suppress TICT. Because the azetidine modification largely rescues quantum yield (cf. 4 and 6, Figure 1b, c), addition of deuterium does not offer further improvement to Φf.
The photostability and chromostability of the brightest variants—those containing azetidine and pyrrolidine substituents—were then evaluated. Based on the photophysics of 1 (Figure 1a), dealkylation of pyrrolidinyl 5 should yield aldehyde 19 (Figure 3a). This was confirmed by LC–MS, which also revealed slower dealkylation of the deuterated 5D (Figure 3b, c, Figure S4a). Monitoring the bleaching of 5 by fluorescence emission gave a complicated set of spectra with an initial increase in intensity along with a hypsochromic spectral shift followed by rapid bleaching (Figure 3d). This result can be explained by the relatively low Φf of 5; dealkylation of this unoptimized dye yields the brighter trialkyl species 19, which then rapidly bleaches. Monitoring bleaching by absorption spectroscopy circumvents this Φf confound and shows a steady decrease in ε and blue-shift in λabs (Figure S4b). The bright JF549 (6) displayed a constant rate of bleaching with a concomitant shift in λem (Figure 3e). In comparison, the deuterated rhodamine congeners 5D and 6D bleached slower and exhibited higher resistance to undesirable dealkylation evidenced by the reduced shifts in λabs and λem (Figure 3d, e, Figure S4c, d). Based on these results, dyes 6D and 5D were given the monikers “JFX549” and “JFX554”, respectively, to denote the extra stability afforded by the deuterated auxochromes.
Figure 3.

Photostability and chromostability of 5, 5D, 6, and 6D. (a) Photochemical dealkylation of 5 to form aldehyde 19. (b, c) LC–MS traces of 5 (b) and 5D (c) before and after photobleaching. (d, e) Sequential fluorescence emission spectra of (d) 5 and 5D or (e) 6 and 6D during photobleaching using a 532 nm laser (0.96 W/cm2; 40 spectra taken over 40 min). The magenta arrows highlight the shift in λem and intensity over time.
The effect of deuteration on the properties of fluorophore:protein conjugates was then evaluated to determine if the improvements observed for the free dyes would translate to superior performance as fluorescent labels. The HaloTag30 ligand of 6 (JF549–HaloTag ligand, 20) was previously synthesized starting from a 6-carboxyfluorescein derivative.6 This approach was used to prepare the JFX549–HaloTag ligand (20D) and JFX554–HaloTag ligand (21D), along with the HaloTag ligand of 5 (21; Figure 4a, Scheme S1). Comparison of the HaloTag conjugates of 20 and 20Din vitro revealed a small but significant increase in Φf for the 20D:HaloTag conjugate (Φf = 0.89) compared to the nondeuterated 20-labeled protein (Φf = 0.87; Figure 4b). For the free dyes, 6D shows a slightly smaller Φf compared to 6 (Table 1), so this result suggests that deuteration suppresses a protein-bound-specific mode of nonradiative decay. Photobleaching experiments using the HaloTag protein labeled with ligands 20, 20D, 21, and 21D revealed that the HaloTag conjugates were more photostable and chromostable than the free dyes, demonstrating that the local environment around the fluorophore can substantially affect photophysics. Still, the HaloTag conjugates of deuterated dyes 20D and 21D exhibited slower bleaching compared to the HaloTag-bound 20 and 21 (Figure S5a, b).
Figure 4.

Performance of rhodamine ligands. (a) Structures of HaloTag ligands 20, 20D, 21, and 21D. (b) Φf of HaloTag protein conjugates of 20 and 20D. (c) Confocal microscopy images of live U2OS cells expressing HaloTag–histone H2B incubated with HaloTag ligands 20, 20D, 21, and 21D (200 nM, 2 h); ex/em = 561 nm/565–632 nm; scale bars: 21 μm. (d, e) SPT intensity (kilocounts per second, kcps) (d) or duration (s) (e) from cells labeled with 20, 20D, or 21D. All error bars: SEM.
The rhodamine-based HaloTag ligands 20, 20D, 21, and 21D were then evaluated in cellular experiments. All four ligands could label HaloTag–histone H2B fusions in living cells (Figure 4c) with no substantial differences in loading kinetics (Figure S5c). 20D gave a significantly longer fluorescence lifetime (τ) than 20 in live cells (Figure S5d), in line with the in vitro Φf measurements (Figure 4b). Photobleaching experiments in fixed cells demonstrated slightly slower bleaching for the deuterated 20D and 21D compared to cells labeled with 20 and 21 (Figure S5f, g). The utility of these dyes in live-cell single-particle tracking (SPT) was then assessed. In initial experiments, pyrrolidine compound 21 showed significantly poorer performance compared to 20 with substantially shorter average duration of individual molecules (Figure S5e). We therefore focused on the JF549–HaloTag ligand (20), JFX549–HaloTag ligand (20D), and JFX554–HaloTag ligand (21D) because the free fluorophores 5D, 6, and 6D exhibit the highest molecular brightness (ε × Φ; Table 1). Deuteration elicited modestly higher fluorescence intensity in cells for the azetidine 20D (8.91 ± 0.05 kcps; mean ± SEM) compared to 20 (8.60 ± 0.07 kcps) under equivalent imaging conditions (Figure 4d). Compound 20D showed no significant improvement in mean duration (11.2 ± 0.6 s; Figure 4e) over 20 (11.0 ± 0.4 s), indicating that in this case the higher photostability observed in vitro and in fixed cells does not translate to the live-cell environment. Remarkably, the deuterated pyrrolidine rhodamine ligand 21D exhibited significantly higher intensity (10.33 ± 0.07 kcps) and track length (13.6 ± 0.8 s) compared to azetidines 20 and 20D, making JFX554 an attractive new label for cellular imaging.31
This deuteration strategy was then applied to other fluorophores, including coumarins (22–22D), phenoxazines (23–23D), and fluorinated rhodamines (24–24D, 25–25D; Table 2, Scheme S2).6,11 Deuteration increased Φf for both the pyrrolidinyl coumarin (22/22D; Φf,D/Φf,H = 1.22) and pyrrolidinyl phenoxazine (23/23D; Φf,D/Φf,H = 1.38). The fluorinated rhodamines exhibited more nuanced behavior with the azetidinyl dyes 24 and 24D showing high Φf values of 0.83 and 0.88, respectively; 24D displayed similar properties to JF549 (6). The pyrrolidinyl dyes 25 and 25D exhibited considerably lower quantum yields, however, with Φf values of 0.30 and 0.37, respectively. This result is consistent with the TICT mechanism (Figure 1a). The higher electron donor strength of the pyrrolidine substituent, combined with the higher electron acceptor strength of the fluorinated xanthene system, promotes electron transfer and decreases Φf; the deuterated pyrrolidine in 25D partially rescues Φf.
Table 2. Spectral Properties of Other Deuterated Dyesa.

All values in 10 mM HEPES, pH 7.3.
Deuteration of red-shifted rhodamine variants was then explored through the synthesis of matched pairs of azetidinyl and pyrrolidinyl carborhodamines10 (26–26D, 27–27D) and Si–rhodamines11,32 (28–28D, 29–29D; Table 3, Scheme S2). For carborhodamines, deuteration increased Φf for both the azetidine-containing dyes (26/26D; Φf,D/Φf,H = 1.10) and the pyrrolidinyl fluorophores (27/27D; Φf,D/Φf,H = 1.30). The Si–rhodamine dyes were similar to the rhodamine series, however, with azetidines 28 (JF646) and 28D giving the same Φf. The deuterated pyrrolidine 29D exhibited a higher Φf than the parent dye (29/29D; Φf,D/Φf,H = 1.10). Like the rhodamine series (Table 1), deuteration had only minor effects on the lactone–zwitterion equilibrium, although the pyrrolidinyl dyes showed substantially higher KL–Z values compared to the azetidinyl dyes. The brightest new fluorophores, 28D and 29D, were named “JFX646” and “JFX650”, respectively.
Table 3. Spectral Properties of Red-Shifted Rhodamine Variantsa.


All values in 10 mM HEPES, pH 7.3 except for KL–Z, which was measured in 1:1 v/v dioxane:H2O.
ε > 150 000 M–1cm–1 in EtOH or TFE with 1% v/v TFA.
The HaloTag ligands of the new Si–rhodamine compounds were then prepared (30D, 31–31D) based on the previous synthesis of JF646–HaloTag ligand6 (30; Figure 5a, Scheme S3). Like the analogous rhodamine ligands, the Φf of the deuterated azetidine JFX646–HaloTag ligand (30D; Φf = 0.73) was significantly higher than the parent ligand 30 (Φf = 0.65) when conjugated to the HaloTag protein (Figure 5b) despite the equivalent Φf values of the free fluorophores 28 and 28D (Table 3); 30D also showed longer τ in cells (Figure S6a). All four ligands could label HaloTag fusions in live cells (Figure 5c) with similar loading profiles (Figure S6b). Photobleaching experiments in fixed cells showed higher stability for 30D and 31D compared with the nondeuterated molecules 30 and 31 (Figure S6c, d). Mirroring the rhodamine series (Figure 4d,e), compounds 30, 30D, and 31D were evaluated in live-cell SPT experiments because the free dyes 28, 28D, and 29D exhibited the highest Φf values among the Si–rhodamines (Table 3). The deuterated Si–rhodamine HaloTag ligands showed superior performance with ligand 28D showing higher mean intensity (7.69 ± 0.05 kcps; Figure 5d) and longer average track length (16.4 ± 0.8 s; Figure 5e) compared to compound 28 (6.98 ± 0.04 kcps, 12.5 ± 0.8 s). The JFX650–HaloTag ligand (31D) exhibited the best overall performance (8.33 ± 0.04 kcps, 17.5 ± 0.3 s). Moving beyond SPT, these Si–rhodamine ligands were evaluated in confocal imaging experiments using a strain of S. cerevisiae expressing a Sec7-GFP–HaloTag fusion to visualize the late Golgi apparatus. The results were consistent with the SPT experiments, with the deuterated JFX ligands 30D and 31D exhibiting significantly higher brightness (Figure 5f) and photostability (Figure 5g, Figure S6e, f, Movie S1) compared to the JF646 ligand 30. The deuterated pyrrolidine JFX650–HaloTag ligand (31D) showed the best performance in the ensemble imaging in yeast.
Figure 5.

Performance of Si–rhodamine ligands. (a) Structures of HaloTag ligands 30, 30D, 31, and 31D. (b) Φ of HaloTag protein conjugates of 30 or 30D. (c) Confocal microscopy images of live U2OS cells expressing HaloTag–histone H2B incubated with HaloTag ligands 30, 30D, 31, and 31D (200 nM, 2 h); ex/em = 640 nm/656–700 nm; scale bars: 21 μm. (d, e) SPT intensity (kcps) (d) or duration (s) (e) from cells labeled with 30, 30D, or 31D. (f) Intensity from S. cerevisiae labeled with 30, 30D, or 31D (1 μM, 30 min). (g) Image montage of S. cerevisiae labeled with 30 or 31D; ex/em = 633 nm/650–795 nm; scale bar: 2 μm. All error bars: SEM.
Conclusion
Taking into account the known photophysical processes of rhodamines, we hypothesized that deuteration of the N-alkyl auxochromes of rhodamine dyes would improve brightness, chromostability, and photostability (Figure 1a). We based this idea on the isotope effects that deuterium exerts on electron and proton transfer processes and not the alteration of vibrational modes as previously proposed.21 This hypothesis was tested first using the classic fluorophore TMR (1; Figure 2) and then by synthesizing other deuterated rhodamine dyes. We discovered that deuteration maintained or improved Φf (Table 1) and enhanced chromo- and photostability (Figure 3). Synthesis of HaloTag ligands showed that deuterated rhodamines were superior labels for live-cell SPT experiments (Figure 4). This deuteration strategy was generalizable to other fluorophore classes (Table 2, Table 3), and the deuterated Si–rhodamines also showed improved performance in cellular imaging experiments (Figure 5).
Overall, this work establishes deuteration of N-alkyl auxochromes as a general strategy for improving the properties of small-molecule fluorophores. This work also demonstrates the predictive value and caveats of in vitro photobleaching experiments for the evaluation of probes intended for living cells. For the azetidine- and pyrrolidine-containing fluorophores, we observed variation in the relative performance of the free dyes 5, 5D, 6, and 6D in solution (Figure 3d, e), the corresponding ligands 20, 20D, 21, and 21D on the HaloTag protein (Figure S5a,b), the ligands in fixed cells (Figure S5f, g), and the ligands in living cells (Figure 4d, e). Although the general trend of deuteration improving brightness and photostability held throughout these experiments, evaluation in living cells was needed to reveal the superior performance of pyrrolidine-containing compounds JFX554 and JFX650. These new dyes can be used as direct replacements of the original Janelia Fluor 549 and Janelia Fluor 646 in bioimaging experiments.6 Future work will focus on utilizing these improved labels in other advanced imaging techniques, combining this deuteration strategy with complementary fluorophore tuning methods,7,8 and extending this straightforward isotope approach to improve other chromophores containing N-alkyl auxochromes.
Acknowledgments
We thank J.J. Macklin (Janelia) for advice on photobleaching experiments. This work was supported by the Howard Hughes Medical Institute (HHMI) and by the National Institutes of Health (NIH) through grants R01GM104010 and P30CA014599 (to B.S.G.) and T32GM007183 (to J.C.C.).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.1c00006.
The authors declare the following competing financial interest(s): Patents and patent applications describing azetidine- and deuterium-containing fluorophores (with inventors J.B.G. and L.D.L.) are assigned to HHMI.
Supplementary Material
References
- Lavis L. D. Chemistry is dead. Long live chemistry!. Biochemistry 2017, 56, 5165–5170. 10.1021/acs.biochem.7b00529. [DOI] [PubMed] [Google Scholar]
- Zheng Q.; Lavis L. D. Development of photostable fluorophores for molecular imaging. Curr. Opin. Chem. Biol. 2017, 39, 32–38. 10.1016/j.cbpa.2017.04.017. [DOI] [PubMed] [Google Scholar]
- Lavis L. D.; Raines R. T. Bright ideas for chemical biology. ACS Chem. Biol. 2008, 3, 142–155. 10.1021/cb700248m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beija M.; Afonso C. A. M.; Martinho J. M. G. Synthesis and applications of rhodamine derivatives as fluorescent probes. Chem. Soc. Rev. 2009, 38, 2410–2433. 10.1039/b901612k. [DOI] [PubMed] [Google Scholar]
- Lavis L. D.; Raines R. T. Bright building blocks for chemical biology. ACS Chem. Biol. 2014, 9, 855–866. 10.1021/cb500078u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm J. B.; English B. P.; Chen J.; Slaughter J. P.; Zhang Z.; Revyakin A.; Patel R.; Macklin J. J.; Normanno D.; Singer R. H.; Lionnet T.; Lavis L. D. A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat. Methods 2015, 12, 244–250. 10.1038/nmeth.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm J. B.; Muthusamy A. K.; Liang Y.; Brown T. A.; Lemon W. C.; Patel R.; Lu R.; Macklin J. J.; Keller P. J.; Ji N.; Lavis L. D. A general method to fine-tune fluorophores for live-cell and in vivo imaging. Nat. Methods 2017, 14, 987–994. 10.1038/nmeth.4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm J. B.; Tkachuk A. N.; Xie L.; Choi H.; Mohar B.; Falco N.; Schaefer K.; Patel R.; Zheng Q.; Liu Z.; Lippincott-Schwartz J.; Brown T. A.; Lavis L. D. A general method to optimize and functionalize red-shifted rhodamine dyes. Nat. Methods 2020, 17, 815–821. 10.1038/s41592-020-0909-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm J. B.; Lavis L. D. Synthesis of rhodamines from fluoresceins using Pd-catalyzed C-N cross-coupling. Org. Lett. 2011, 13, 6354–6357. 10.1021/ol202618t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm J. B.; Sung A. J.; Legant W. R.; Hulamm P.; Matlosz S. M.; Betzig E.; Lavis L. D. Carbofluoresceins and carborhodamines as scaffolds for high-contrast fluorogenic probes. ACS Chem. Biol. 2013, 8, 1303–1310. 10.1021/cb4000822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm J. B.; Brown T. A.; Tkachuk A. N.; Lavis L. D. General synthetic method for Si-fluoresceins and Si–rhodamines. ACS Cent. Sci. 2017, 3, 975–985. 10.1021/acscentsci.7b00247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettig W. Charge separation in excited states of decoupled systems—TICT compounds and implications regarding the development of new laser dyes and the primary process of vision and photosynthesis. Angew. Chem., Int. Ed. Engl. 1986, 25, 971–988. 10.1002/anie.198609711. [DOI] [Google Scholar]
- Vogel M.; Rettig W.; Sens R.; Drexhage K. H. Structural relaxation of rhodamine dyes with different N-substitution patterns: A study of fluorescence decay times and quantum yields. Chem. Phys. Lett. 1988, 147, 452–460. 10.1016/0009-2614(88)85007-3. [DOI] [Google Scholar]
- Grimm J. B.; Klein T.; Kopek B. G.; Shtengel G.; Hess H. F.; Sauer M.; Lavis L. D. Synthesis of a far-red photoactivatable silicon-containing rhodamine for super-resolution microscopy. Angew. Chem., Int. Ed. 2016, 55, 1723–1727. 10.1002/anie.201509649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Würth C.; Grabolle M.; Pauli J.; Spieles M.; Resch-Genger U. Comparison of methods and achievable uncertainties for the relative and absolute measurement of photoluminescence quantum yields. Anal. Chem. 2011, 83, 3431–3439. 10.1021/ac2000303. [DOI] [PubMed] [Google Scholar]
- Cauletti C.; Di Vona M. L.; Gargano P.; Grandinetti F.; Galli C.; Lillocci C. Ring-size effects on the ionization potentials of N-substituted azacycloalkanes. J. Chem. Soc., Perkin Trans. 2 1986, 667–670. 10.1039/p29860000667. [DOI] [Google Scholar]
- Herrmann R.; Josel H.-P.; Drexhage K.-H.; Arden-Jacob J.. Pentacyclic compounds and their use as absorption or fluorescent dyes. U.S. Patent US5,750,409A, 1998.
- Panchuk-Voloshina N.; Haugland R. P.; Bishop-Stewart J.; Bhalgat M. K.; Millard P. J.; Mao F.; Leung W. Y.; Haugland R. P. Alexa dyes, a series of new fluorescent dyes that yield exceptionally bright, photostable conjugates. J. Histochem. Cytochem. 1999, 47, 1179–1188. 10.1177/002215549904700910. [DOI] [PubMed] [Google Scholar]
- Butkevich A. N.; Bossi M. L.; Lukinavicius G.; Hell S. W. Triarylmethane fluorophores resistant to oxidative photobluing. J. Am. Chem. Soc. 2019, 141, 981–989. 10.1021/jacs.8b11036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull L. A.; Davis G. T.; Rosenblatt D. H.; Williams H. K. R.; Weglein R. C. Oxidations of amines. III. Duality of mechanism in the reaction of amines with chlorine dioxide. J. Am. Chem. Soc. 1967, 89, 1163–1170. 10.1021/ja00981a023. [DOI] [Google Scholar]
- Turro N. J.; Ramamurthy V.; Scaiano J. C.. Modern Molecular Photochemistry of Organic Molecules; University Science Books: 2010. [Google Scholar]
- Stryer L. Excited-state proton-transfer reactions. A deuterium isotope effect on fluorescence. J. Am. Chem. Soc. 1966, 88, 5708–5712. 10.1021/ja00976a004. [DOI] [Google Scholar]
- Magde D.; Wong R.; Seybold P. G. Fluorescence quantum yields and their relation to lifetimes of rhodamine 6G and fluorescein in nine solvents: Improved absolute standards for quantum yields. Photochem. Photobiol. 2002, 75, 327–334. . [DOI] [PubMed] [Google Scholar]
- Lee S. F.; Verolet Q.; Furstenberg A. Improved super-resolution microscopy with oxazine fluorophores in heavy water. Angew. Chem., Int. Ed. 2013, 52, 8948–8951. 10.1002/anie.201302341. [DOI] [PubMed] [Google Scholar]
- Dawson W. R.; Windsor M. W. Fluorescence yields of aromatic compounds. J. Phys. Chem. 1968, 72, 3251–3260. 10.1021/j100855a027. [DOI] [Google Scholar]
- Frampton M. J.; Accorsi G.; Armaroli N.; Rogers J. E.; Fleitz P. A.; McEwan K. J.; Anderson H. L. Synthesis and near-infrared luminescence of a deuterated conjugated porphyrin dimer for probing the mechanism of non-radiative deactivation. Org. Biomol. Chem. 2007, 5, 1056–1061. 10.1039/b700408g. [DOI] [PubMed] [Google Scholar]
- Kolmakov K.; Belov V. N.; Bierwagen J.; Ringemann C.; Muller V.; Eggeling C.; Hell S. W. Red-emitting rhodamine dyes for fluorescence microscopy and nanoscopy. Chem. - Eur. J. 2010, 16, 158–166. 10.1002/chem.200902309. [DOI] [PubMed] [Google Scholar]
- A preprint of our work appeared simultaneously with an independent report from Broichhagen and coworkers that details the improved properties and utility of deuterated TMR (1D) and a novel deuterated Si-tetramethylrhodamine; 10.1101/2020.08.17.253880. [DOI]
- Corrie J. E. T.; Munasinghe V. R. N. Substituent effects on the apparent pK of the reversible open-to-closed transition of sultams derived from sulforhodamine dyes. Dyes Pigm. 2008, 79, 76–82. 10.1016/j.dyepig.2008.01.008. [DOI] [Google Scholar]
- Los G. V.; Encell L. P.; McDougall M. G.; Hartzell D. D.; Karassina N.; Zimprich C.; Wood M. G.; Learish R.; Ohana R. F.; Urh M. HaloTag: A novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 2008, 3, 373–382. 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- Although the deuterated pyrrolidine-d8 is relatively expensive (∼$500/g), this low-molecular-weight reagent is used in late-stage, high-yielding Pd-catalyzed cross-coupling reactions, thereby increasing the overall cost of the multistep synthesis by only a marginal amount.
- Lukinavicius G.; Umezawa K.; Olivier N.; Honigmann A.; Yang G.; Plass T.; Mueller V.; Reymond L.; Correa I. R. Jr.; Luo Z. G.; Schultz C.; Lemke E. A.; Heppenstall P.; Eggeling C.; Manley S.; Johnsson K. A near-infrared fluorophore for live-cell super-resolution microscopy of cellular proteins. Nat. Chem. 2013, 5, 132–139. 10.1038/nchem.1546. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.