

Abstract
Small molecules play a critical role in modulating biological systems. Knowledge of chemical–protein interactions helps address fundamental and practical questions in biology and medicine. However, with the rapid emergence of newly sequenced genes, the endogenous or surrogate ligands of a vast number of proteins remain unknown. Homology modeling and machine learning are two major methods for assigning new ligands to a protein but mostly fail when sequence homology between an unannotated protein and those with known functions or structures is low. In this study, we develop a new deep learning framework to predict chemical binding to evolutionary divergent unannotated proteins, whose ligand cannot be reliably predicted by existing methods. By incorporating evolutionary information into self-supervised learning of unlabeled protein sequences, we develop a novel method, distilled sequence alignment embedding (DISAE), for the protein sequence representation. DISAE can utilize all protein sequences and their multiple sequence alignment (MSA) to capture functional relationships between proteins without the knowledge of their structure and function. Followed by the DISAE pretraining, we devise a module-based fine-tuning strategy for the supervised learning of chemical–protein interactions. In the benchmark studies, DISAE significantly improves the generalizability of machine learning models and outperforms the state-of-the-art methods by a large margin. Comprehensive ablation studies suggest that the use of MSA, sequence distillation, and triplet pretraining critically contributes to the success of DISAE. The interpretability analysis of DISAE suggests that it learns biologically meaningful information. We further use DISAE to assign ligands to human orphan G-protein coupled receptors (GPCRs) and to cluster the human GPCRome by integrating their phylogenetic and ligand relationships. The promising results of DISAE open an avenue for exploring the chemical landscape of entire sequenced genomes.
Introduction
Small molecules like metabolites and drugs play an essential role in modulating physiological and pathological processes. The chemical modulation of a biological system results from its interaction with biomolecules, largely proteins. Thus, the genome-wide identification of chemical-protein interactions (CPI) will not only address many fundamental questions in biology (e.g., microbiome-host interaction mediated by the metabolite) but also provide new opportunities in drug discovery and precision medicine.1 Despite tremendous advances in genomics, the function of a vast number of proteins, particularly their ligands are to a great extent unknown. Among the approximately 3000 druggable genes that encode human proteins, only 5% to 10% have been targeted by an FDA-approved drug.2 The proteins that miss ligand information are orphan proteins in biology or considered as unlabeled data in terms of machine learning. It is a great challenge to assign ligands to orphan proteins, especially when they are significantly dissimilar from proteins with known structures or functions. Many experimental approaches have been developed to deorphanize the orphan proteins such as G-protein coupled receptors (GPCRs).3 However, they are both costly and time-consuming. Great effort has been devoted to the development of computational approaches, which may provide an efficient solution to generate testable hypotheses for elucidating the ligand of orphan proteins.
With the increasing availability of solved crystal structures of proteins, homology modeling and protein–ligand docking are major methods for the effort in deorphanization.4 However, the quality of homology models significantly deteriorates when the sequence identity between query and template is low. When a homology model with an acceptable quality is unavailable, structure-based methods, either physics-based or machine learning-based,5 could be unfruitful. Moreover, protein–ligand docking suffers from a high rate of false positives because it is incapable of accurately modeling conformational dynamics, solvation effect, crystallized water molecules, and other physical phenomena. Considering that around half of Pfam families do not have any structure information,6 it is necessary to develop a new sequence-based machine learning approach to deorphanization, especially for the proteins that are not close homologues of or even evolutionarily unrelated to the ones with solved crystal structures or known ligands.
Many machine learning methods have been developed to predict CPIs from protein sequences. Early works relied on feature engineering to create a representation of protein and ligand then use classifiers such as support vector machine7 and matrix factorization89 to make final prediction. End-to-end deep learning approaches have recently gained momentum.1011 Various neural network architectures, including Convolutional Neural Network (CNN),1213 seq2seq,1415 and Transformer,16 have been applied to represent protein sequences. These works mainly focused on filling in missing CPIs for the existing drug targets. When the sequence of orphan protein is significantly different from those used in the database or training data, the performance of many existing machine learning methods deteriorates significantly.8 As a result, existing machine learning methods are incapable of predicting genome-scale CPIs.
The failure of the machine learning method primarily comes from its generalization problem. The machine learning model is confined by its training data and cannot reliably predict new cases that are out of the domain of training data. Reducing data bias may lessen but not solve the problem. TransformerCPI16 addressed the data bias that comes from the chemicals, since the known pairs of interactions involve far more unique chemicals than unique proteins. But TransformerCPI did not confront the fundamental generalization problem on genome-wide CPI predictions, i.e., predicting ligand binding to remote orphan proteins. The fields of computer vision and natural language processing (NLP) have explored the generalization problem from several aspects. a major breakthrough is pretraining. Since the wide acknowledgment of the power of attention mechanism,52 a series of attention-based pretrained language models such as BERT,29 ALBERT,17 ROBERTA,53 etc. kept breaking records on most NLP benchmark tasks. The basic idea of pretraining is to train a model on a huge unlabeled corpus, which includes far more “vocabulary” than a downstream task data set. Due to the close analogue between protein sequence and human language, many laboratories have developed pretrained protein language models, such as TAPE,56 ProtTrans,54 and ESM.55 For example, TAPE uses Pfam,22 a database of thirty-one million protein domains, as the pretraining corpus. It is used on downstream tasks such as secondary structure prediction and contact prediction through fine-tuning and proved to improve downstream task performance. However, pretrained protein language models have not been applied to genome-wide CPI predictions. Moreover, biological information that is crucial to protein biochemical functions has not been fully incorporated into the protein sequence pretraining.
To extend the scope of state-of-the-art for the prediction of chemical binding to orphan proteins in entire sequenced genomes, we propose a new deep learning framework for improving protein representation learning so that the relationships between remote proteins or even evolutionary unrelated proteins can be detected. Inspired by the success in self-supervising learning of unlabeled data in NLP,17 and its application to biological sequences,18−20 we propose a new protein representation method, distilled sequence alignment embedding (DISAE), for the purpose of deorphanization of remote orphan proteins. Although the number of labeled proteins that have known ligands is limited, DISAE can utilize all available unlabeled protein sequences without the knowledge of their functions and structures. By incorporating biological knowledge into the sequence representation, DISAE can learn functionally important information about protein families that span a wide range of protein space. Furthermore, we devise a module-based pretraining-fine-tuning strategy using ALBERT.17 To our knowledge, it is the first time to apply a pretrained-fine-tuned protein sequence model for addressing the challenge of deorphanization of novel proteins on a genome-scale. Compared with state-of-the-art methods represented by TransformerCPI58 and TAPE,59 DISAE is innovative in many aspects: learning task, protein descriptor, chemical descriptor, model architecture, and training procedure.
In the benchmark study, DISAE significantly outperforms other state-of-the-art methods for the prediction of chemical binding to dissimilar orphan proteins by a large margin. The success of DISAE mainly comes from the use of multiple sequence alignment (MSA) and distilled sequence. Furthermore, the interpretability analysis of DISAE suggests that it learns biologically meaningful information. We apply DISAE to the deorphanization of G-protein coupled receptors (GPCRs). GPCRs play a pivotal role in numerous physiological and pathological processes. Due to their associations with many human diseases and high druggabilities, GPCRs are the most studied drug targets.21 Around one-third of FDA-approved drugs target GPCRs.21 Despite intensive studies in GPCRs, the endogenous and surrogate ligands of a large number of GPCRs remain unknown.3 Using DISAE, we can confidently assign 649 orphan GPCRs in Pfam with at least one ligand; 106 of the orphan GPCRs find at least one approved GPCR-targeted drugs as ligand with an estimated false positive rate lower than 0.05. These predictions merit further experimental validations. In addition, we cluster the human GPCRome by integrating their sequence and ligand relationships. The promising results of DISAE open an avenue for exploring the chemical landscape of all sequenced genomes. The code of DISAE is available on GitHub (https://github.com/XieResearchGroup/DISAE).
Methods
Overview of Methodology
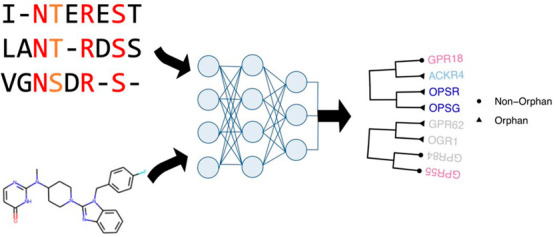
As illustrated in Figure 1A, the proposed method is designed to predict chemical binding to remote orphan proteins that do not have detectable relationships to annotated (i.e., labeled) proteins with known structures or functions. It is different from most of current works that focus on the assignment of functions to proteins that are homologous to annotated proteins. Our method mainly consists of two stages (Figure 1B,C). The first stage is for unsupervised learning of protein representations using only sequence data from all nonredundant sequences in Pfam-A families22 but without the need of any annotated structural or functional information. We develop a new algorithm, DISAE, for the self-supervised leaning (a special form of unsupervised learning) of protein representation. In contrast to existing sequence pretraining strategies that use original protein sequences as input,18−20 DISAE distills the original sequence into an ordered list of triplets by excluding evolutionarily unimportant positions from a MSA (Figure 1B). The purpose of sequence distillation is 2-fold: improving efficiency for the sequence pretraining, in which long sequences cannot be handled and reducing the noise in the input sequence. Then long-range residue–residue interactions are learned via the self-attention in the Transformer module of ALBERT. A self-supervised masked language modeling (MLM) approach is used at this stage. In the MLM, 15% triplets are randomly masked and assumed to be unknown. Then, the remaining triplets are used to predict what the masked triplets are. In the second stage, a supervised learning model is trained to predict if a chemical and a protein interact using the known CPIs that are collected from chemical genomics databases.23−26 The input of the supervised learning includes both the representation of chemical structures from neural fingerprint27 and the pretrained protein sequence embedding from the first stage. We develop a module-based fine-tuning strategy to balance the information learned from the unsupervised and the supervised stages, and apply an attention pooling28 to model the interaction between chemical substructures and protein residues. More details in the sequence representation learning, the architecture of neural network, benchmark data sets, training and evaluation procedure, and data analysis can be found in the section of Method.
Figure 1.

(A) Comparison of the scope of this study with that of current works. In existing methods, only annotated proteins (labeled data) are used in the model training. They work well when an orphan protein is homologous to the annotated protein but mostly fail when the orphan protein is dissimilar from the annotated protein. By contrast, this study uses both annotated and orphan proteins in a self-supervised-learning-fine-tuning framework and thus extends the scope to genome-wide remote orphan proteins that are out of the reach of state-of-the-art. (B) Illustration of protein sequence representation from distilled sequence alignment. The high and medium conserved positions are marked as red and orange, respectively. (C) Architecture of deep learning model for the whole-genome CPI prediction.
[Input of DISAE] Protein Sentence Representation from Distilled Sequence Alignment
A protein sequence is converted into an ordered list of amino acid fragments (words) with the following steps, as illustrated in Figure 1B.
-
1.
Given a sequence of interest S, an MSA is constructed by a group of similar sequences to S. In this paper, the precomputed alignments in Pfam22 are used.
-
2.
Amino acid conservation at each position is determined.
-
3.
Starting from the most conserved positions that are defined in the Pfam, a predefined number of positions in the alignment are selected by the ranking of conservation. In this study, the number of positions is set as 210. The length of the positions was not optimized.
-
4.
A word, either a single amino acid or a triplet of amino acids in the sequence, is selected at each position. The triplet may include gaps.
-
5.
Finally, all selected words from the sequence form an ordered list following the sequence order, i.e., the sentence representation of the protein.
The rationale for the distilled MSA representation is to only use functionally or evolutionarily important residues and ignore others. It can be considered a feature selection step. In addition, the use of MSA will allow us to correlate the functional information with the position encoding. Given a protein family, the residue positions will have a unified representation, thus facilitate the modeling of pairwise protein residue-ligand substructure interactions through the attention. The distilled sequence will not only reduce the noise but also increase the efficiency in the model training since the memory and time complexity of language model is O (n2). It is noted that we use the conservation to select residues in a sequence because it is relevant to protein function and ligand binding. Other criteria (e.g., coevolution) could be used depending on the downstream applications (e.g., protein structure prediction).
[Pretraining of DISAE] DISA) Using ALBERT
It has been shown that NLP algorithms can be successfully used to extract biochemically meaningful vectors by pretraining BERT29 on 86 billion amino acids (words) in 250 million protein sequences.39 A recently released light version of BERT, called ALBERT,17 boasts significantly lighter memory uses with better or equivalent performances compared to BERT.29 We extend the idea of unsupervised pretraining of proteins using ALBERT algorithm using distilled MSA representation. The distilled ordered list of triplets is used as the input for ALBERT pretraining. In this work, only the masked language model (MLM) is used for the pretraining.
[Fine-Tuning] Architecture of Deep Learning Model for Whole-Genome CPI Prediction
The deep learning model for the whole-genome CPI (chemical protein interaction) prediction is mainly composed of three components, as shown in Figure 1C, protein embedding by DISAE, chemical compound embedding, and attention pooling with multilayer perceptron (MLP) to model CPIs. DISAE is described in the previous section. Note that once processed through ALBERT, each protein is represented as a matrix of size 210 by 312, where each triplet is represented by a vector of length 312. During protein–ligand interaction prediction task, the protein embedding matrix is compressed using ResNet.40 Once processed through ResNet layers, each protein is represented as a vector of length 256, which contains compressed information for the whole 210 input triplets for the corresponding protein.
Neural molecular fingerprint41 is used for the chemical embedding. A small molecule is represented as a 2D graph, where vertices are atoms and edges are bonds. We use a popular graph convolutional neural network to process ligand molecules.27
The attentive pooling is similar to the design in ref (11), For each putative chemical–protein pair, the corresponding embedding vectors are fed to the attentive pooling layer, which in turn produces the interaction vector.
More details on the neural network model configurations can be found in Tables S2–S4 in the Supporting Information.
[Fine-Tuning] Module-Based Fine-Tuning Strategy
When applying ALBERT to a supervised learning task, fine-tuning17 is a critical step for the task-specific training following the pretraining. Pretrained ALBERT has already learned to generate protein representation in a meaningful way. However, it is also a design choice whether to allow ALBERT to be updated and trained together with the other components of the classification system during the fine-tuning.1742 Updating ALBERT during the fine-tuning will allow the pretrained protein encoder to better capture knowledge from the training data while minimizing the risk of significant loss of knowledge obtained from the pretraining. Hence, to find the right balance, we experiment with ALBERT models that are partially unfrozen.42 To be specific, major modules in the ALBERT model, embedding or transformer,43 are unfrozen separately as different variants of the model. The idea of “unfrozen” layers is widely used in NLP, e.g., ULMFiT42 where model consists of several layers. As training proceeds, layers are gradually unfrozen to learn the task-specific knowledge while safeguarding knowledge gained from pretraining. However, this is not straightforwardly applicable to ALBERT because ALBERT is not a linearly layered-up architecture. Hence, we apply a module-based unfrozen strategy.
Experiments Design
The purpose of this study is to build a model to predict chemical binding to novel orphan proteins. Therefore, we design experiments to examine the model generalization capability to the data not only unseen but also from significantly dissimilar proteins. We split training/validation/testing data sets to assess the performance of algorithms into three scenarios: 1. The proteins in the testing data set are significantly different from those in the training and validation data set based on the sequence similarity. 2. The ligands in the testing data are from a different gene family from that in the training/validation data. 3. The whole data set is randomly split like most of the existing work.
We use Long–Short-term Memory (LSTM),44 TransformerCPI, and TAPE as baselines, as shown in Table 1. Two variants of LSTM models are tested to compare with the above three groups of experiments: LSTM with distilled triplets and distilled singlets. For the LSTM and TAPE baselines, we only replace the pretrained ALBERT module with LSTM or TAPE, but other components of the whole architecture are the same, as in Figure 1C). For the TransformerCPI baseline, we test the original model of TransformerCPI on our data sets.
Table 1. Test Set Performance under Three Benchmark Settings Are Evaluated in ROC-AUC and PR-AUCa.
| ROC-AUC | ||||
|---|---|---|---|---|
| benchmark |
||||
| goal of comparison | model | dissimilar GPCR | kinase inhibitor | random |
| DISAE | ALBERT frozen transformer (distilled triplets) | 0.725 | 0.690 | 0.889 |
| the effect of pretraining | ALBERT frozen transformer (pretrained on GPCR) | 0.441 | 0.849 | |
| the effect of distilled sequence | ALBERT frozen transformer (distilled singlets) | 0.583 | 0.656 | |
| the effect of fine-tuning | ALBERT frozen embedding | 0.585 | 0.889 | |
| ALBERT all unfrozen | 0.680 | 0.891 | ||
| baseline against ALBERT | TransformerCPI (full sequence) | 0.570 | 0.680 | 0.896 |
| TransformerCPI (distilled singlets) | 0.645 | 0.682 | 0.897 | |
| TAPE (full sequence) | 0.610 | 0.640 | 0.825 | |
| TAPE (distilled singlets) | 0.680 | 0.619 | 0.829 | |
| LSTM (full sequence) | 0.524 | 0.662 | 0.911 | |
| LSTM (distilled singlets) | 0.652 | 0.642 | 0.907 | |
| LSTM (distilled triplets) | 0.476 | 0.667 | 0.908 | |
| PR-AUC | ||||
|---|---|---|---|---|
| benchmark |
||||
| goal of comparison | model | dissimilar GPCR | kinase inhibitor | random |
| DISAE | ALBERT frozen transformer (distilled triplets) | 0.589 | 0.673 | 0.783 |
| The effect of pretraining | ALBERT frozen transformer (pretrained on GPCR) | 0.215 | 0.728 | |
| the effect of distilled sequence | ALBERT frozen transformer (distilled singlets) | 0.370 | 0.477 | |
| the effect of fine-tuning | ALBERT frozen embedding | 0.278 | 0.783 | |
| ALBERT all unfrozen | 0.418 | 0.785 | ||
| baseline against ALBERT | TransformerCPI (full sequence) | 0.300 | 0.620 | 0.782 |
| TransformerCPI (distilled singlets) | 0.350 | 0.624 | 0.778 | |
| TAPE (full sequence) | 0.300 | 0.610 | 0.684 | |
| TAPE (distilled singlets) | 0.387 | 0.584 | 0.698 | |
| LSTM (full sequence) | 0.262 | 0.628 | 0.803 | |
| LSTM (distilled singlets) | 0.372 | 0.614 | 0.798 | |
| LSTM (distilled triplets) | 0.261 | 0.590 | 0.804 | |
ALBERT pretrained transformer-frozen model outperforms other models, and its performance is stable across all settings. Hence, it is recommended as the optimal configuration for the pretrained ALBERT model. Four variants of DISAE models are compared to the frozen transformer one. Unless specified in the parentheses, ALBERT is pretrained on whole Pfam proteins in the form of distilled triplets. The four DISAE variants are organized into three groups based on the goal of comparison. Three state-of-the-art models TAPE, TransformerCPI and LSTM are compared with the ALBERT pretrained models as baselines. Protein similarity based splitting uses a threshold of similarity score of 0.035 (Figure 2).
To examine the effect of different pretraining-fine-tuning algorithms, we organize experiments in three categories of comparisons, as shown in Table 1:
-
1.
The effect of vocabulary: Taking protein sequence as a sentence, its vocabulary could be built in many ways. We compare the use of the singlet with the triplet of vocabulary.
-
2.
The effect of pretraining: We assess how unlabeled protein sequences affect the performance of the classification. We compare ALBERT pretrained on whole Pfam alone against one pretrained on GPCRs alone and one without pretraining.
-
3.
The effect of fine-tuning: We compare three ALBERT models: ALBERT all unfrozen, ALBERT frozen embedding, and ALBERT frozen transformer.43 All these modes are pretrained on the whole Pfam.
Data Set
Our task is to learn from large-scale CPI data to predict unexplored interactions. The quality and quantity of the training samples are critical for biologically meaningful predictions. Despite continuous efforts in the community, a single data source typically curates an incomplete list of our knowledge of protein–ligand activities. Thus, we integrated multiple high-quality, large-scale, publicly available databases of known protein–ligand activities. We extracted, split, and represented protein–ligand activity samples for training and evaluation of machine learning-based predictions.
Sequence data for ALBERT pretraining
Proteins sequences are first collected from Pfam-A22 database. Then, sequences are clustered by 90% sequence identity, and a representative sequence is selected from each cluster. The 40 282 439 sequences (including 138 288 GPCR) are used for the ALBERT pretraining. To construct protein sentences from the distilled sequence alignment, the original alignment and conservation score from each Pfam-A family are used. As a comparison, the 35 181 GPCR sequences only from the GPCR family PF00001 are used to pretrain a separate ALBERT model.
We used Pfam alignments directly. From the consensus alignment sequence, we collected positions of high-confidence and low-confidence conserved amino acids together with conservatively substituted ones. We picked these positions from each of sequences. As a result, each sequence is represented by 210 amino acids, which may contain gaps. The 210 amino acids are then treated as a sentence of triplets. The triplets are used to train the ALBERT model with the following parameters: maximum sequence length = 256, maximum predictions per sentence = 40, word masking probability = 0.15, and duplication factor = 10. Note that the order of a pair of sequences may not be biologically meaningful. Thus, we did not apply the next sentence prediction task during the pretraining.
Binding assay data for supervised learning
We integrated protein–ligand activities involving any GPCRs from ChEMBL23 (ver. 25), BindingDB24 (downloaded Jan 9, 2019), GLASS25 (downloaded Nov 26, 2019), and DrugBank26 (ver. 5.1.4). Note that BindingDB also contains samples drawn from multiple sources, including PubChem, PDSP Ki, and U.S. patent. From ChEMBL, BindingDB, and GLASS databases, protein–ligand activity assays measured in three different unit types, pKd, pKi, and pIC50 are collected. Log-transformation was performed for activities reported in Kd, Ki, or IC50. For consistency, we did not convert it into different activity types. For instance, activities reported in IC50 are converted only to pIC50, but not any other activity types. The activities on a log-scale were then binarized based on the thresholds of activity values. Protein–ligand pairs were considered active if pIC50 > 5.3, or pKd > 7.3 or pKi > 7.3 and inactive if pIC50 < 5.0, pKd < 7.0 or pKi < 7.0 respectively.
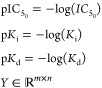 |
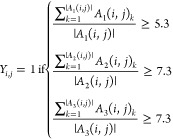 |
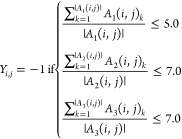 |
In the above equations, m and n are the total number of unique proteins and ligands, respectively. A1 (i, j), A2 (i, j), and A3 (i, j) is the list of all activity values for the ith protein and jth ligand in pIC50, pKd, and pKi, respectively. |A| denotes the cardinality of the set A. Note that there are gray areas in the activity thresholds. Protein–ligand pairs falling in the gray areas were considered undetermined and unused for training. If multiple activities were reported for a protein–ligand pair, their log-scaled activity values were averaged for each activity type and binarized accordingly. In addition, we collected active protein–ligand associations from DrugBank and integrated with the binarized activities mentioned above. Inconsistent activities (e.g., protein–ligand pairs that appear both active and inactive) were removed. There is a total of 9705 active and 25175 inactive pairs, respectively, in the benchmark set.
Benchmark
To test the model generalization capability, a protein similarity-based data splitting strategy is implemented. First, pairwise protein similarity based on bit-score is calculated using BLAST45 for all GPCRs in the data set. The similarity between proteini and proteinj is defined as
Then, according to the similarity distribution, a similarity threshold is set for splitting. The bit-score similarity threshold is 0.035. The sequences are clustered such that the sequences in the testing set are significantly dissimilar from those in the training/validation set, as shown in Figure 2. After splitting, there are 25 114, 6278, and 3488 samples for training, validation, and testing, respectively. Distribution of protein sequence similarity scores can be found in Figure S8.
Figure 2.
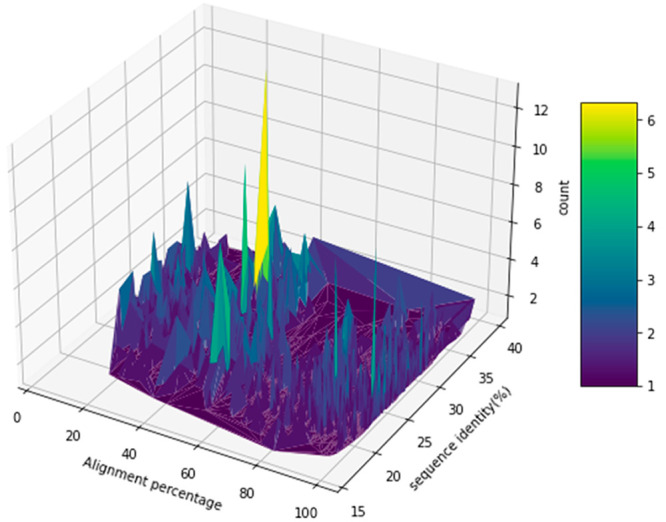
Distribution of sequence alignment percentage and percentage of sequence identity between proteins in the testing set (query) and those in the training/validation set (template) in the dissimilar protein benchmark.
Ensemble Model for the GPCR Deorphanization
All annotated GPCR-ligand binding pairs are used to build prediction models for the GPCR deorphanization. To reduce overfitting, an ensemble model is constructed using DISAE. Following the strategy of cross-validation,46 three DISAE models are trained. Like benchmark experiments, a hold-out set is selected based on protein similarity and is used for the early stopping at the preferred epoch for each individual model. Max-voting47 is invoked to make final prediction for the orphan human GPCRs.
To estimate the false positive rate of predictions on the orphan-chemical pairs, a prediction score distribution is collected for known positive pairs and negative pairs of testing data. If the prediction score of orphan pairs has the same distribution as that of the testing data, for each prediction score, a false positive rate can be estimated based on the score distribution of true positives and negatives.
SHAP Analysis
Kernel SHAP30 is used to calculate SHAP values for distilled protein sequences. It is a specially weighted local linear regression to estimate SHAP values for any model. The model used is DISAE that fine-tunes the ALBERT frozen transformer and is pretrained on whole Pfam in distilled triplets under the remote GPCR setting. The data used is the testing set generated under the same remote GRPC setting. Although the whole input features to the classifier consist of both distilled protein sequence and chemical neural fingerprint, only protein feature is of interest. Hence, when calculating the base value (a value that would be predicted if we do not know any features) required by SHAP analysis, all testing set protein sequences are masked with the same token, while chemical neural fingerprint remains untouched. Therefore, the base value is the average prediction score without protein feature and solely relying on chemical features. Since the distilled sequences are all set to be of length 210, the SHAP values are the feature importance for each of the 210 positions.
Statistical Test
To assess the statistical significance of the difference between the performance of DISAE and those of baselines, 200 test samples are randomly sampled with replacement from the dissimilar protein benchmark. For each set of 200 samples, the ROC-AUC and PR-AUC of DISAE, TAPE, and TransformerCPI models are calculated, respectively. Student’s t tests are carried out to compare the distribution of ROC-AUC and PR-AUC of the DISAE model with those of TAPE and TransformerCPI models, respectively.
Hierarchical Clustering of the Human GPCRome
The pairwise distance between two sequences is determined by the cosine similarity of the DISAE embedding vector after pretraining-fine-tuning, which incorporates both sequence and ligand information. The R package “ape” (Analyses of Phylogenetics and Evolution)48 and treeio49 are used to convert a distance matrix of proteins into a Newick tree format. The ggtree5051 package is used to plot the tree in a circular layout.
Results
Pretrained DISAE Triplet Vector Is Biochemically Meaningful
When pretraining the DISAE model with masked triplets of proteins, the masked word prediction accuracy reached 0.982 and 0.984 at the 80 000th and the 100 000th training step, respectively. For comparison, the pretraining accuracy when using BERT29 was 0.955 at 200 000th training step. This difference may be due to the larger batch sizes possible in ALBERT, in contrast to BERT. DISAE pretraining is subsequently used for the CPI prediction. To evaluate whether the pretrained DISAE vector is biochemically meaningful, we extracted the pretrained DISAE vector for all possible triplets. We then used t-SNE to project the triplet vector in a 2D space. As shown in Figure S1, the triplet vectors formed distinct clusters by the properties of amino acid side chains in the third amino acid of triplets, especially at levels 3 and 4. Triplets containing any ambiguous or uncommon amino acids, such as amino acid U for selenocysteine or X for any unresolved amino acids, formed a large cluster that did not form a smaller group (large group of black dots in each scatter plot), suggesting that the information regarding such rare amino acids are scarce in the pretraining data set. When there is no ambiguity in triplets, they form clearly separated clusters, implying that the pretraining process indeed allows the model to extract biochemically meaningful feature vectors from the proteins as sentences. The same clustering trends were also observed when triplets are grouped by individual amino acids, rather than their physicochemical properties of side chains (Figure S2).
DISAE Significantly Outperforms State-of-the-Art Models for Predicting Ligands of Remote Orphan Proteins
To evaluate the performance of DISAE for the prediction of ligand of remote orphan proteins, we perform experiments in a dissimilar protein benchmark. In this benchmark, the proteins in the testing data (i.e., query proteins) are significantly dissimilar from those in the training data (i.e., template proteins). As shown in Figure 2, the sequence identity of protein pairs between the query and the template are all less than 40%. The pairs with the percentage of alignment larger than 60% and the sequence identity larger than 30% only constitute about 8.83% of total pairs. Thus, homology-based methods cannot be applied to the majority of cases in benchmarking. We first evaluate the performance of baseline models LSTM, TransformerCPI, and TAPE in the dissimilar protein benchmark. In this conventional setting, the pretrained ALBERT model in Figure1C is replaced by LSTM or pretrained TAPE. In addition, there is no pretraining stage. Only labeled data are directly applied to supervised learning. As shown in Figure 3A and 3B, the performance of the conventional LSTM model with full length protein sequence is nearly random, with its ROC-AUC and PR-AUC being 0.524 and 0.262, respectively. TransformerCPI with full length protein sequence has ROC-AUC and PR-AUC 0.57 and 0.30, respectively; TAPE, 0.61, 0.30, respectively. It is clear that the attention mechanism and the pretraining can improve the performance of CPI predictions. As a comparison, DISAE has a ROC-AUC of 0.725 and a PR-AUC of 0.589, respectively, significantly outperforming the baselines. Furthermore, as shown in the inset of Figure 3A, DISAE far exceeds the baseline models at the range of low false positive rates. For example, at the false positive rate of 0.05, the number of true positives detected by DISAE is almost 6 times more than that correctly predicted by TransformerCPI or TAPE. When the train/test set is split at random, i.e., there are similar proteins in the testing set to those in the training set, the performance of LSTM models is relatively better than DISAE, TAPE, and TransformerCPI, as shown in Table 1. However, the performance of the LSTM model significantly deteriorates when the proteins in the testing set are different from those in the training set. The ROC-AUC and PR-AUC drop 28.1% and 53.4%, respectively, with distilled singlets as input. TransformerCPI also shows a similarly significant performance drop. The ROC-AUC and PR-AUC decrease 28.1% and 55.0%, respectively. On the contrary, DISAE could still maintain the ROC-AUC ∼ 0.7 (a drop of 18.4%) and the PR-AUC ∼ 0.5 (a drop of 24.8%). As expected, the performance drop of pretrained TAPE is less severe than LSTM and TransformerCPI but worse than DISAE. The ROC-AUC and PR-AUC reduce 17.8% and 44.6%, respectively, in the case of distilled singlet inputs. These results suggest that the supervised learning alone is prone to overfitting, as observed in the TransformerCPI and LSTM models, thus cannot be generalized to modeling remote orphan proteins. The pretraining of DISAE, which uses a large number of unlabeled distilled sequences from MSAs, improves the generalization power. The training curves in Figures S3–S5 further support the fact that DISAE is generalizable; thus, it can reliably maintain its high performance when used for the deorphanization of dissimilar proteins. When evaluated by the dissimilar protein benchmark, the accuracy of training keeps increasing with the increased epochs; and the performance of DISAE is slightly worse than most models. However, the PR-AUC of DISAE is relatively stable and significantly higher than other models of testing data. These observations further support the idea that DISAE can predict the ligand binding to novel proteins. Although the pretrained TAPE model can also improve generalizability, it is less powerful than DISAE because it incorporates less domain-specific information than DISAE.
Figure 3.

Performance comparison of DISAE, TAPE, and TransformerCPI. (A and B) ROC- and PR-curves for the prediction of ligand binding to remote proteins. (C and D) ROC- and PR-curves for the classification on testing set in the cross-gene-family kinase inhibitor benchmark. All of the models are trained on distilled triplets.
A series of t tests is carried out to provide further contrast of the performance DISAE, TAPE and TransformerCPI. When comparing DISAE with TransformerCPI, the p-value for the t test of ROC-AUC and PR-AUC are p-value <1.29 × 10–64 and p-value <2.67 × 10–77, respectively; when comparing DISAE with TAPE, the p-value for the t test of ROC-AUC and PR-AUC are p-value <1.07 × 10–40 and p-value <4.88 × 10–67, respectively. Therefore, the advantage of DISAE over state-of-the-art is statistically significant. The sampled ROC-AUC and PR-AUC distributions are in Figure S9.
DISAE Outperforms State-of-the-Art Models for Predicting Ligand Binding Promiscuity Across Gene Families
We further evaluate the performance of DISAE and compare it with the baseline TransformerCPI, TAPE, and LSTM models on the prediction of ligand binding promiscuity across gene families. Specifically, we predict the binding of new kinase inhibitors to new GPCRs on the model that is trained using only GPCR sequences that are not known to bind to kinase inhibitors and chemicals that have not been annotated as kinase inhibitors. In other words, all GPCRs that bind to kinase inhibitors and all chemicals that are kinase inhibitors are excluded from the supervised training set. In this situation, there is no significant difference between the proteins in the testing set and those in the training set. The major difference between testing and training data comes from chemicals. Although the kinase and the GPCRs belong to two completely different gene families in terms of sequence, structure, and function, a few kinase inhibitors can bind to GPCRs as an off-target. We use these annotated GPCR-kinase inhibitor pairs as the test set. Interestingly, although DISAE is not extensively trained using a comprehensive chemical data set, DISAE outperforms the LSTM, TransformerCPI, and TAPE. As shown in Figure 3C,D, both the sensitivity and specificity of DISAE outperforms other models. The ROC-AUCs of DISAE, TransformerCPI, and TAPE model are 0.690, 0.68, and 0.64, respectively. Their PR-AUCs are 0.673, 0.620, and 0.61, respectively. This observation implies that the sequence pretraining captures certain ligand binding site information across gene families. It is noted that it is infeasible to apply a homology modeling-based method to infer ligand binding promiscuity across gene families.
Effect of Distilled Sequence Representation, Pretraining, and Fine-Tuning
To understand the contribution of each component in DISAE to the performance, we conduct a series of ablation studies as shown in Table 1 under the dissimilar protein and cross-gene-family benchmarks. For the three major pretraining-fine-tuning configurations of DISAE, distilled triplets sequence representation, pretrained on whole Pfam, and fine-tuned with frozen transformer (“ALBERT frozen transformer” in Table 1), are recommended as the best performed model for predicting ligands of remote orphan proteins.
-
1.
Triplet is preferred over singlet for predicting ligand binding to remote proteins
Under the same pretraining and fine-tuning settings, both ROC-AUC and PR-AUC of the ALBERT pretrained model that uses the distilled triplets are significantly higher than the those when the distilled singlet is used. However, for the LSTM model, the distilled singlet sometimes outperforms the distilled triplet. This can be because the triplet encodes more relational information on remote proteins than close homologues.
-
2.
Distilled sequence outperforms a full sequence for remote orphan proteins
Although the distilled sequence does not show clear advantage over the full sequences on the prediction performance in the kinase and randomly splitting benchmarks, it consistently outperforms the full sequence in the dissimilar GPCR benchmark for all four methods: DISAE, TAPE, TransformerCPI, and LSTM. Because the distilled sequence is derived from MSAs and functionally important residues, it could be more informative than the full sequence when predicting ligand binding. Even the distilled sequence alone could improve the baselines: when trained on distilled singlets, TransformerCPI, TAPE, and LSTM could improve ROC-AUC by 13%, 11%, and 24% to 0.645, 0.68, and 0.652 and improve PR-AUC by 17%, 29%, and 42% to 0.35, 0.387, and 0.372.
-
3.
Pretraining on a larger data set is preferred
With the same fine-tuning strategy, frozen transformer, and use of distilled triplets, the model that is pretrained overall Pfam performs better than that pretrained on the GPCR family alone in terms of both ROC-AUC and PR-AUC.
-
4.
Partial frozen transformer is preferred
With the same pretraining on whole Pfam and fine-tuning on the distilled triplets, the ALBERT pretrained transformer-frozen model outperforms all other models that have only embedding layers frozen or both transformer and embedding layers frozen.
DISAE Learns Biologically Meaningful Information
Interpretation of deep learning is critical for its real-world applications. To understand if the trained DISAE model is biologically meaningful, we perform the model explainability analysis using SHapley Additive exPlanation (SHAP).30 SHAP is a game-theoretic approach to explain the output of any machine learning model. Shapley values could be interpreted as feature importance. We utilize this tool to get a closer look into the internal decision making of DISAE’s prediction by calculating Shapley values of each triplet of a protein sequence. The average Shapley values of CPIs for a protein is used to highlight important positions for this protein.
Figure 4 shows the distribution of several residues of 5-hydroxytryptamine receptor 2B on its structure, which are among 21 (10%) residues with the highest SHAP values. Among them, 6 residues (T140, V208, M218, F341, L347, and Y370) are located in the binding pocket. L378 is centered in the functional conserved NPxxY motif that connects the transmembrane helix 7 and the cytoplasmic helix 8 and plays a critical role in the activation of GPCRs.3132 P160 and I161 are the part of intracellular loop 2, while I192, G194, I195, and E196 are located in the extracellular loop 2. The intracellular loop 2 interacts with the P-loop of G-protein.33 It is proposed that the extracellular loop 2 may play a role in the selective switch of ligand binding and determine ligand binding selectivity and efficacy3435.3637 The functional impact of other residues is unclear. Nevertheless, more than one-half of the top 21 residues ranked by SHAP values can explain the trained model. The enrichment of ligand binding site residues is statistically significant (p-value = 0.01). These results suggest that prediction from DISAE can provide biologically meaningful interpretations.
Figure 4.
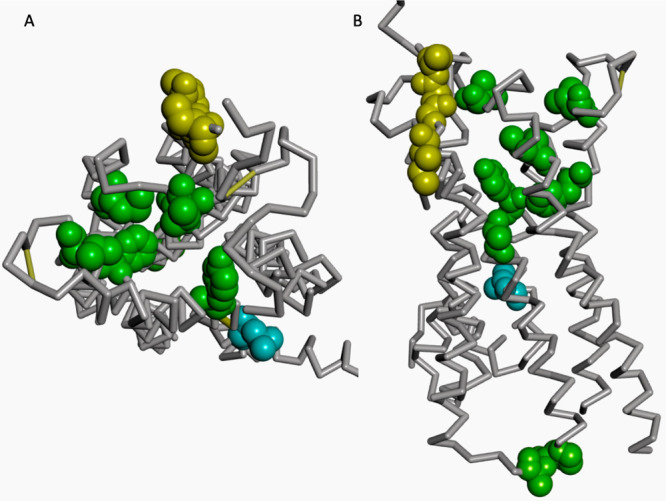
(A) Top and (B) side view of structure of 5-hydroxytryptamine receptor 2B (UNIPROT id: 5HTB2_HUMAN, PDB ID: 4IB4). The residues among those with the top 21 ranked SHAP values are shown in dark green, blue, yellow, and light green colored CPK mode for the amino acids in the binding pocket, NPxxY motif, extracellular loop, and intracellular loop, respectively.
Application to the Hierarchical Classification and Deorphanization of Human GPCRs
With the established generalization power of DISAE, we use the ALBERT transformer-frozen model pretrained on whole Pfam in distilled triplets form to tackle the challenge of deorphanization of human GPCRs, due to its consistently excellent performance by all evaluation metrics in the benchmarks.
We define the orphan GPCRs as those which do not have known small molecule binders. 649 human GPCRs that are annotated in Pfam families PF00001, PF13853, and PF03402 are identified as the orphan receptors.
Studies have suggested that the classification of GPCRs should be inferred by combining sequence and ligand information.38 The protein embedding of the DISAE model after pretraining-fine-tuning satisfies this requirement. Therefore, we use the cosine similarity between the embedded vector of protein as a metric to cluster the human GPCRome, which includes both nonorphan and orphan GPCRs. The hierarchical clustering of GPCRs in the Pfam PF00001 based on embedding vector or full sequence similarity is shown in Figure 5. Figures S6 and S7 show the hierarchical clustering of PF13853 and PF03402, respectively. We also use family annotation from GPCRdb57 as color coding for GPCRs in Figure 5 to show the contrast between our embedding based clustering results and sequence based clustering results. While the sequence based clustering is generally clustering GPCRs from the same family together, our clustering tends to rearrange some of them. In general, DISAE embedding-based clustering results are more like sequence-based clustering at a finer resolution. For example, paralogs relaxin receptors RXFP1 and RXFP2 are the most similar in sequence, they are also the most similar in the DISAE embedding-based clustering. However, there are still inconstant examples, for instance, dopamine receptors DRD2 and DRD3 are very similar in sequence but they are not so close in the DISAE embedding tree. The divergence in the clustering of dopamine receptors may come from their binding site diversity.60
Figure 5.

Hierarchical clustering of human GPCRs in the Pfam PF00001 based on (A) cosine similarity between embedding vectors and (B) sequence similarity. Nonorphan and orphan GPCRs are labeled with circle and triangles, respectively. Names of GPCRs are colored coded by the family to which they belong.
Table 2 provides examples of predicted approved-drug bindings to the orphan GPCRs by DISAE with a high confidence (false positive rate <5.0 × 10–4). The complete list of orphans human GPCRs paired with 555 approved GPCR-targeted drugs is in Table S1. The predicted potential interactions between the approved drug and the orphan receptor will not only facilitate designing experiments to deorphanize GPCRs but also provide new insights into the mode of action of existing drugs for drug repurposing, polypharmacology, and side effect prediction.
Table 2. Example of Deorphanization Prediction from DISAE Ensemble Models.
| orphan receptor (uniprot ID) | Pfam | drug | drug target |
|---|---|---|---|
| A0A0C4DFX5 | PF13853 | Isoetharine | ADRB1, ADRB2 |
| A0A126GVR8 | PF13853 | Ganirelix | GNRHR |
| A0A1B0GTK7 | PF00001 | Levallorphan | OPRM1 |
| A0A1B0GVZ0 | PF00001 | Xamoterol | ADRB1, ADRB2 |
| A0A286YFH6 | PF13853 | Degarelix | GNRHR |
| A3KFT3 | PF13853 | Degarelix | GNRHR |
| C9J1J7 | PF00001 | Levallorphan | OPRM1 |
| C9JQD8 | PF00001 | Lixisenatide | GLP1R |
| E9PH76 | PF00001 | Ganirelix | GNRHR |
| E9PPJ8 | PF13853 | Xamoterol | ADRB1, ADRB2 |
Discussion
Our comprehensive benchmark studies demonstrate that DISAE can significantly improve the prediction of ligand binding to remote orphan receptors. The performance gain of DISAE comes from several major differences from state-of-the-art methods. Compared with pretrained TAPE from Pfam, which uses a single full protein sequence as the input, DISAE uses distilled triplets derived from the MSA. Our analysis suggests that all these components: MSA, distilled sequence, and triplet representation contribute to the excellent performance of DISAE. The MSA has two effects: capturing the evolutionary relationships between proteins and allowing consistent position encoding. The distilled sequence will not only reduce the memory complexity during the training, which allows the use of large batch size, but also improve the effectiveness of modeling long-range residue–residue interactions through the self-attention. Compared with the singlet residue representation, the triplet representation may better model ligand binding motifs. TransformerCPI is one of the best methods to date for the CPI predictions. The major difference between TransformerCPI and DISAE is that TransformerCPI does not use protein pretraining. Consequently, although TransformerCPI has an excellent performance when the test samples are similar to the training data, its generalization power when applied to dissimilar new samples is poor. Additionally, TransformerCPI uses SMILES and takes it as a 1D sequence for the chemical representation. In a high-level formulation, TransformerCPI is a sequence to sequence “translation” and uses a transformer to model CPIs. Instead DISAE uses a graph neural network to model chemical structures and an attention pooling to model protein residue-chemical substructure interactions. These differences may also contribute to performance discrepancy.
Conclusion
Our primary goal in this paper is to address the challenge of predicting ligands of orphan proteins that are significantly dissimilar from proteins that have known ligands or solved structures. To address this challenge, we introduce new techniques for the protein sequence representation by the pretraining of distilled sequence alignments, as well as module-based fine-tuning using labeled data. Our approach, DISAE, is inspired by the state-of-the-art algorithms in NLP. However, our results suggest that the direct adaption of NLP may be less fruitful. The successful application of NLP to biological problems requires the incorporation of domain knowledge in both the pretraining and fine-tuning stages. In this regard, DISAE significantly improves the state-of-the-art in the deorphanization of dissimilar orphan proteins. Nevertheless, DISAE can be further improved in several aspects. First, more biological knowledge can be incorporated into the pretraining and fine-tuning at both the molecular level (e.g., protein structure and ligand binding site information) and system level (e.g., protein–protein interaction network). Second, in the framework of self-supervised learning, a wide array of techniques can be adapted to address the problem of bias, sparsity, and noisiness in the training data. Put together, new machine learning algorithms that can predict endogenous or surrogate ligands of orphan proteins open a new avenue for deciphering biological systems, drug discovery, and precision medicine.
Acknowledgments
This project has been funded in whole or in part with federal funds from the National Institute of General Medical Sciences of National Institute of Health (R01GM122845), the National Institute on Aging of the National Institute of Health (R01AD057555), and the National Cancer Institute of National Institutes of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services nor does mention of trade names, commercial products or organizations imply endorsement by the U.S. Government. This Research was supported [in part] by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, and the Intramural Research Program of the NIH Clinical Center.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.0c01285.
Additional figures and tables mentioned in the manuscript (PDF)
Author Contributions
L.X. conceived and planned the experiments. T.C. and H.L. developed and implemented the algorithm. T.C., H.L., K.A.A., and Y.Q. carried out experiments. L.X., T.C., H.L., Y.Q., and K.A.A. contributed to the interpretation of the results. T.C., H.L., K.A.A., Y.Q., R.N., and L.X. wrote the manuscript. All authors provided critical feedback and helped shape the research, analysis, and manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Oprea T. Exploring the dark genome: implications for precision medicine. Mamm. Genome 2019, 30, 192. 10.1007/s00335-019-09809-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers G.; Austin C.; Anderson J.; Pawlyk A.; Colvis C.; Margolis R.; Baker J. Glimmers in illuminating the druggable genome. Nat. Rev. Drug Discovery 2018, 17, 301–302. 10.1038/nrd.2017.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laschet C.; Dupuis N.; Hanson J. The G Protein-Coupled Receptors deorphanization landscape. Biochem. Pharmacol. (Amsterdam, Neth.) 2018, 62. 10.1016/j.bcp.2018.02.016. [DOI] [PubMed] [Google Scholar]
- Ngo T.; Kufareva I.; Coleman J.; Graham R.; Abagyan R.; Smith N. Identifying ligands at orphan GPCRs: Current status using structure-based approaches. Br. J. Pharmacol. 2016, 173, 2934. 10.1111/bph.13452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S.; Li Y.; Chen S.; Xu J.; Yang Y. Predicting drug–protein interaction using a quasi–visual question answering system. Nature Machine Intelligence 2020, 2, 134–140. 10.1038/s42256-020-0152-y. [DOI] [Google Scholar]
- Lewis T.; Sillitoe I.; Andreeva A.; Blundell T.; Buchan D.; Chothia C.; Cozzetto D.; Dana J.; Filippis I.; Gough J.; Jones D.; Kelley L.; Kleywegt G.; Minneci F.; Mistry J.; Murzin A.; Ochoa-Montaño B.; Oates M.; Punta M.; Rackham O.; Stahlhacke J.; Sternberg M.; Velankar S.; Orengo C. Genome3D: exploiting structures to help users understand their sequences. Nucleic Acids Res. 2015, 43 (D1), D382–D386. 10.1093/nar/gku973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z.; Zhou S.; Wang Y.; Liu H.; Guan J.; Chen Y. P. Effectively identifying compound-protein interactions by learning from positive and unlabeled examples. IEEE/ACM Trans. Comput. Biol. Bioinf., PP 2016, 1832–1843. 10.1109/TCBB.2016.2570211. [DOI] [PubMed] [Google Scholar]
- Lim H.; Poleksic A.; Yao Y.; Tong H.; He D.; Zhuang L.; Meng P.; Xie L. Large-Scale Off-Target Identification Using Fast and Accurate Dual Regularized One-Class Collaborative Filtering and Its Application to Drug Repurposing. PLOS Comput. Biol. 2016, 12. 10.1371/journal.pcbi.1005135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim H.; Gray P.; Xie L.; Poleksic A. Improved genome-scale multi-target virtual screening via a novel collaborative filtering approach to cold-start problem. Sci. Rep. 2016, 6, 38860. 10.1038/srep38860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen M.; Zhang Z.; Niu S.; Sha H.; Yang R.; Yun Y.; Lu H. Deep-Learning-Based Drug-Target Interaction Prediction. J. Proteome Res. 2017, 16, 1401–1409. 10.1021/acs.jproteome.6b00618. [DOI] [PubMed] [Google Scholar]
- Gao K. Y.; Fokoue A.; Luo H.; Iyengar A.; Dey S.; Zhang P.. Interpretable Drug Target Prediction Using Deep Neural Representation. In Proceedings of the Twenty-Seventh International Joint Conference on Artificial Intelligence, IJCAI-18; International Joint Conferences on Artificial Intelligence Organization, 2018; pp 3371–3377. [Google Scholar]
- Nguyen T.; Le H.; Quinn T. P.; Nguyen T.; Le T. D.; Venkatesh S. GraphDTA: Predicting drug-target binding affinity with graph neural networks. Bioinformatics 2020, btaa921. 10.1093/bioinformatics/btaa921. [DOI] [PubMed] [Google Scholar]
- Lee I.; Keum J.; Nam H. DeepConv-DTI: Prediction of drug-target interactions via deep learning with convolution on protein sequences. PLoS Comput. Biol. 2019, 15, e1007129. 10.1371/journal.pcbi.1007129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutskever I.; Vinyals O.; Le Q.. Sequence to Sequence Learning with Neural Networks, 2014; pp 10; https://arxiv.org/abs/1409.3215v3.
- Karimi M.; Wu D.; Wang Z.; Shen Y. DeepAffinity: interpretable deep learning of compound-protein affinity through unified recurrent and convolutional neural networks. Bioinformatics 2019, 35 (18), 3329–3338. 10.1093/bioinformatics/btz111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.; Tan X.; Wang D.; Zhong F.; Liu X.; Yang T.; Luo X.; Chen K.; Jiang H.; Zheng M. TransformerCPI: Improving compound-protein interaction prediction by sequence-based deep learning with self-attention mechanism and label reversal experiments. Bioinformatics 2020, 36, 4406–4414. 10.1093/bioinformatics/btaa524. [DOI] [PubMed] [Google Scholar]
- Lan Z.; Chen M.; Goodman S.; Gimpel K.; Sharma P.; Soricut R.. ALBERT: A Lite BERT for Self-supervised Learning of Language Representations, 2019; https://arxiv.org/abs/1909.11942.
- Rao R.; Bhattacharya N.; Thomas N.; Duan Y.; Chen X.; Canny J. F.; Abbeel P.; Song Y. S.. Evaluating Protein Transfer Learning with TAPE. CoRR, abs/1906.08230, 2019. [PMC free article] [PubMed]
- Bepler T.; Berger B.. Learning protein sequence embeddings using information from structure CoRR, abs/1902.08661, 2019.
- Min S.; Park S.; Kim S.; Choi H. S.; Yoon S.. Pre-Training of Deep Bidirectional Protein Sequence Representations with Structural Information, 2019; https://arxiv.org/abs/1912.05625.
- Hauser A.; Attwood M.; Rask-Andersen M.; Schiöth H.; Gloriam D. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discovery 2017, 16, 829. 10.1038/nrd.2017.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Gebali S.; Mistry J.; Bateman A.; Eddy S.; Luciani A.; Potter S.; Qureshi M.; Richardson L.; Salazar G.; Smart A.; Sonnhammer E.; Hirsh L.; Paladin L.; Piovesan D.; Tosatto S.; Finn R. The Pfam protein families database in 2019. Nucleic Acids Res. 2018, D427. 10.1093/nar/gky995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaulton A.; Bellis L.; Bento A.; Chambers J.; Davies M.; Hersey A.; Light Y.; McGlinchey S.; Michalovich D.; Al-Lazikani B.; Overington J. ChEMBL: a Large-scale Bioactivity Database for Drug Discovery. Nucleic Acids Res. 2012, 40, D1100–7. 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Lin Y.; Liu M.; Gilson M. K. The Binding Database: data management and interface design. Bioinformatics 2002, 18, 130–9. 10.1093/bioinformatics/18.1.130. [DOI] [PubMed] [Google Scholar]
- Chan W. K. B.; Zhang H.; Yang J.; Brender J. R.; Hur J.; Ozgur A.; Zhang Y. GLASS: A comprehensive database for experimentally validated GPCR-ligand associations. Bioinformatics 2015, 31, 3035. 10.1093/bioinformatics/btv302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D.; Knox C.; Guo A.; Shrivastava S.; Hassanali M.; Stothard P.; Chang Z.; Woolsey J. DrugBank: A Comprehensive Resource for in Silico Drug Discovery and Exploration. Nucleic Acids Res. 2006, 34, D668–72. 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvenaud D.; Maclaurin D.; Iparraguirre J.; Bombarell R.; Hirzel T.; Aspuru-Guzik A.; Adams R.. Convolutional Networks on Graphs for Learning Molecular Fingerprints, 2015; pp 2224–2232https://arxiv.org/abs/1509.09292.
- dos Santos C. N.; Tan M.; Xiang B.; Zhou B.. Attentive Pooling Networks CoRR, abs/1602.03609, 2016.
- Devlin K.BERT: Pre-training of Deep Bidirectional Transformers for Language Understanding. Proceedings of the 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, Vol. 1 (Long and Short Papers); Association for Computational Linguistics, 2019; pp 4171–4186. [Google Scholar]
- Lundberg S.; Lee S.-I.. A Unified Approach to Interpreting Model Predictions, 2017; https://arxiv.org/abs/1705.07874.
- Fritze O.; Filipek S.; Kuksa V.; Palczewski K.; Hofmann K.; Ernst O. Role of the conserved NPXXY(X)5,6F motif in the rhodopsin ground state and during activation. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 2290–5. 10.1073/pnas.0435715100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trzaskowski B.; Latek D.; Yuan S.; Ghoshdastider U.; Debiński A.; Filipek S. Action of Molecular Switches in GPCRs - Theoretical and Experimental Studies. Curr. Med. Chem. 2012, 19, 1090–109. 10.2174/092986712799320556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilger D.; Masureel M.; Kobilka B. K. Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol. 2018, 25, 4. 10.1038/s41594-017-0011-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolley M.; Conner A. Understanding the common themes and diverse roles of the second extracellular loop (ECL2) of the GPCR super-family. Mol. Cell. Endocrinol. 2016, 449. 10.1016/j.mce.2016.11.023. [DOI] [PubMed] [Google Scholar]
- Peeters M.; Van Westen G.; Li Q.; IJzerman A. Importance of the extracellular loops in G protein-coupled receptors for ligand recognition and receptor activation. Trends Pharmacol. Sci. 2011, 32, 35–42. 10.1016/j.tips.2010.10.001. [DOI] [PubMed] [Google Scholar]
- Seibt B.; Schiedel A.; Thimm D.; Hinz S.; Sherbiny F.; Mueller C. The second extracellular loop of GPCRs determines subtype-selectivity and controls efficacy as evidenced by loop exchange study at A2 adenosine receptors. Biochem. Pharmacol. (Amsterdam, Neth.) 2013, 1317. 10.1016/j.bcp.2013.03.005. [DOI] [PubMed] [Google Scholar]
- Perez-Aguilar J. M.; Shan J.; LeVine M. V.; Khelashvili G.; Weinstein H. A Functional Selectivity Mechanism at the Serotonin-2A GPCR Involves Ligand-Dependent Conformations of Intracellular Loop 2. J. Am. Chem. Soc. 2014, 136, 16044. 10.1021/ja508394x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz N.; Langenhan T.; Schöneberg T. Revisiting the classification of adhesion GPCRs. Ann. N. Y. Acad. Sci. 2019, 1456 (1), 80. 10.1111/nyas.14192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rives A.; Goyal S.; Meier J.; Guo D.; Ott M.; Zitnick C.; Ma J.; Fergus R.. Biological Structure and Function Emerge from Scaling Unsupervised Learning to 250 Million Protein Sequences. bioRxiv 622803, 2019. [DOI] [PMC free article] [PubMed]
- He K.; Zhang X.; Ren S.; Sun J.. Deep Residual Learning for Image Recognition CoRR, abs/1512.03385, 2015.
- Duvenaud D.; Maclaurin D.; Iparraguirre J.; Bombarell R.; Hirzel T.; Aspuru-Guzik A.; Adams R.. Convolutional Networks on Graphs for Learning Molecular Fingerprints, 2015, pp 2224–2232; https://arxiv.org/abs/1509.09292.
- Howard J.; Ruder S.. Universal Language Model Fine-tuning for Text Classification, 2018; pp 328–339; https://arxiv.org/abs/1801.06146.
- Vaswani A.; Shazeer N.; Parmar N.; Uszkoreit J.; Jones L.; Gomez A.; Kaiser L.; Polosukhin I.. Attention Is All You Need, 2017; https://arxiv.org/abs/1706.03762.
- Van Houdt G.; Mosquera C.; Napoles G. A Review on the Long Short-Term Memory Model. Artificial Intelligence Review 2020, 53, 5929. 10.1007/s10462-020-09838-1. [DOI] [Google Scholar]
- Altschul S.; Madden T.; Schaffer A.; Zhang J.; Zhang Z.; Miller W.; Lipman D. Gapped BLAST and PSI-BLAST: a new generation of protein databases search programs. Nucleic Acids Res. 1997, 25, 3389–402. 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohavi R.A Study of Cross-Validation and Bootstrap for Accuracy Estimation and Model Selection. Morgan Kaufmann, 1995; pp 1137–1143; https://dl.acm.org/doi/10.5555/1643031.1643047. [Google Scholar]
- Leon F.; Floria S.; Badica C.. Evaluating the effect of voting methods on ensemble-based classification. 2017 IEEE International Conference on INnovations in Intelligent SysTems and Applications (INISTA); Gdynia, 2017; pp 1–6. [Google Scholar]
- Paradis E.; Schliep K. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. 10.1093/bioinformatics/bty633. [DOI] [PubMed] [Google Scholar]
- Wang L.-G.; Lam T. T.-Y.; Xu S.; Dai Z.; Zhou L.; Feng T.; Guo P.; Dunn C. W.; Jones B. R.; Bradley T.; Zhu H.; Guan Yi; Jiang Y.; Yu G. treeio: an R package for phylogenetic tree input and output with richly annotated and associated data. Mol. Biol. Evol. 2019, 599. 10.1093/molbev/msz240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G.; Smith D. K.; Zhu H.; Guan Y.; Lam T. T.-Y. ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods in Ecology and Evolution 2017, 8 (1), 28–36. 10.1111/2041-210X.12628. [DOI] [Google Scholar]
- Yu G.; Lam T. T.-Y.; Zhu H.; Guan Yi Two methods for mapping and visualizing associated data on phylogeny using ggtree. Methods in Ecology and Evolution 2018, 35 (2), 3041–3043. 10.1093/molbev/msy194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahdanau D.; Cho K.; Bengio Y.. Neural Machine Translation by Jointly Learning to Align and Translate, 2014; https://arxiv.org/abs/1409.0473.
- Liu Y.; Ott M.; Goyal N.; Du J.; Joshi M.; Chen D.; Levy O.; Lewis M.; Zettlemoyer L.; Stoyanov V.. RoBERTa: A Robustly Optimized BERT Pretraining Approach, 2019; https://arxiv.org/abs/1907.11692.
- Elnaggar A.; Heinzinger M.; Dallago C.; Rihawi G.; Wang Y.; Jones L.; Gibbs T.; Feher T.; Angerer C.; Steinegger M.; Bhowmik D.; Rost B.. ProtTrans: Towards Cracking the Language of Life’s Code Through Self-Supervised Deep Learning and High Performance Computing, 2020; https://arxiv.org/abs/2007.06225. [DOI] [PubMed]
- Rives A.; Meier J.; Sercu T.; Goyal S.; Lin Z.; Liu J.; Guo D.; Ott M.; Zitnick C.; Ma J.; Fergus R.. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences, 2020; 10.1101/622803. [DOI] [PMC free article] [PubMed]
- Rao R.; Bhattacharya N.; Thomas N.; Duan Y.; Chen X.; Canny J.; Abbeel P.; Song Y. S.. Evaluating Protein Transfer Learning with TAPE, 2019; https://arxiv.org/abs/1906.08230. [PMC free article] [PubMed]
- Kooistra A. J.; Mordalski S.; Pándy-Szekeres G.; Esguerra M.; Mamyrbekov A.; Munk C.; Keseru G. M; Gloriam D. E., GPCRdb in 2021: integrating GPCR sequence, structure and function. Nucleic Acids Res., 1080. 10.1093/nar/gkaa1080. [DOI] [PMC free article] [PubMed]
- Chen L.; Tan X.; Wang D.; Zhong F.; Liu X.; Yang T.; Luo X.; Chen K.; Jiang H.; Zheng M. TransformerCPI: improving compound–protein interaction prediction by sequence-based deep learning with self-attention mechanism and label reversal experiments. Bioinformatics 2020, 36 (16), 4406–4414. 10.1093/bioinformatics/btaa524. [DOI] [PubMed] [Google Scholar]
- Rao R.; Bhattacharya N.; Thomas N.; Duan Y.; Chen X.; Canny J.; Abbeel P.; Song Y. S. bioRxiv 676825; Evaluating Protein Transfer Learning with TAPE NeurIPS Proc. 2019, 10.1101/676825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin J.; Fan T.; Guo P.; Wang J. Identification of functional divergence sites in dopamine receptors of vertebrates. Comput. Biol. Chem. 2019, 83, 107140. 10.1016/j.compbiolchem.2019.107140. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.