

Abstract
Control over the placement and activity of biomolecules on solid surfaces is a key challenge in bionanotechnology. While covalent approaches excel in performance, physical attachment approaches excel in ease of processing, which is equally important in many applications. We show how the precision of recombinant protein engineering can be harnessed to design and produce protein-based diblock polymers with a silica-binding and highly hydrophilic elastin-like domain that self-assembles on silica surfaces and nanoparticles to form stable polypeptide brushes that can be used as a scaffold for later biofunctionalization. From atomic force microscopy-based single-molecule force spectroscopy, we find that individual silica-binding peptides have high unbinding rates. Nevertheless, from quartz crystal microbalance measurements, we find that the self-assembled polypeptide brushes cannot easily be rinsed off. From atomic force microscopy imaging and bulk dynamic light scattering, we find that the binding to silica induces fibrillar self-assembly of the peptides. Hence, we conclude that the unexpected stability of these self-assembled polypeptide brushes is at least in part due to peptide–peptide interactions of the silica-binding blocks at the silica surface.
Introduction
A key aspect of bionanotechnology is control of placement and activity of biomolecules on solid surfaces. For example, biofunctionalization of solid surfaces is crucial in biosensing and for implanted biomaterials.1 In the simplest approaches, biomolecules are either directly immobilized on surfaces through often nonspecific covalent bonds or physically adsorbed.2,3 Covalent coupling makes for stronger immobilization since bonds are typically irreversible. On the other hand, physical adsorption generates bonds to the surface that are typically reversible, but the process is much simpler.
In biosensing, an important example is the surface attachment of antibodies used to detect antigens. Direct nonspecific covalent immobilization of antibodies on surfaces may lead to significant loss of antibody activity, especially at high surface densities.4 This is supposedly caused by (permanent) misorientation of the antibodies leading to loss of accessibility of the antibody-binding region for the antigens and by (partial) denaturation of the antibodies caused by the direct contact with the surface (Figure 1a). The same mechanisms are operative when antibodies are directly physically adsorbed on surfaces (Figure 1b). One solution to avoid misorientation or denaturation is to indirectly attach antibodies (or other bioactive molecules) to the surface via polymer brushes (Figure 1c), which has the additional advantage that the performance of diagnostic devices can be improved using antifouling polymers.5 The technology for the synthesis and subsequent functionalization of antifouling polymer brushes on solid substrates has reached a sophisticated level.6,7 However, the required procedures for low-cost, bulk applications are still too complex and hence too expensive. Therefore, there is continued interest in the development of technologies for the generation of stable polymer brushes via simple adsorption.
Figure 1.

Biomolecule immobilization strategies illustrated for the case of antibodies. (a) Direct covalent attachment. (b) Direct physical adsorption. (c) Attachment to a covalently anchored polymer brush. (d) Attachment to a physically anchored PLL-g-PEG brush. (e) Physical immobilization through an SBP immobilization tag. (f) Physical immobilization through an SBP immobilization tag and antifouling polypeptide linker.
The most notable examples of noncovalently immobilized polymer brushes are PEG [poly(ethylene glycol)] copolymers such as PEG-g-PLL (poly l-lysine) and PEG-g-PEI (polyethylene imine).8 These have a so-called “bottle-brush” architecture, consisting of a polycationic main chain (PLL or PEI) to which short PEG side chains are grafted. The polycationic main chain adsorbs onto many relevant surfaces, the PEG side chains extend perpendicular from the polycationic main chain, thereby forming a brush. PEG is widely used as an antifouling material,6 while bioactive molecules can be readily attached to the PEG side chains using chemical procedures9−11 (Figure 1d).
A complementary approach to synthetic polymers is the use of recombinant or synthetic polypeptides. Polypeptides offer the advantage of precise control of polymer chemistry,12 including the conjugation of bioactive molecules such as antibodies, and the possibility to directly include other functional peptide or protein blocks in the design.13 Another advantage is the possibility to exploit many extensively studied solid-binding peptides (SBPs) as binding modules for specific surfaces. SBPs are short amino acid sequences that interact noncovalently with solid surfaces, with affinities in the micro- to nanomolar range14 and have been widely employed as immobilization tags for the direct immobilization of proteins on various surfaces (Figure 1e).15−17 However, as emphasized before, attachment to an antifouling polymer brush is preferred to prevent loss of activity of sensitive biomolecules such as antibodies.
There is some precedent for the use of SBPs as part of genetically engineered polypeptide designs. For example, sequences based on spider silk have been fused to sequences for silica-binding domains in order to synthesize biomimetic coatings for nanoparticles.18 The chimeric silk proteins form β-sheet-rich fibers on silica surfaces. In a similar strategy, Li et al. have used silica-binding domains to immobilize micelles of elastin-like polypeptides (ELPs) on silica nanoparticles and surfaces.19
Immobilization using a diblock polypeptide with an SBP and an inert hydrophilic domain such as a hydrophilic ELP (Figure 1f) is another promising option. This design has recently been explored by Li et al.20 for the immobilization of cells on gold surfaces. Here, we wish to explore a similar diblock design for coating another widely occurring type of surface, viz., silica surfaces.
Following Li et al.,20 for the inert and antifouling polypeptide blocks, we use ELPs. ELPs are based on sequence motifs from human tropoelastin,21 characterized by the pentapeptide motif EX = VPGXG, where X is any amino acid except proline, and have been well-characterized.22 Indeed, ELPs have found applications in biosensing,23 tissue engineering,24 nanoparticle coatings,25 drug-delivery systems,26 biomineralization studies,27 immunoassays,28 and molecular switches.29 To obtain inert and hydrophilic polypeptide brushes, we choose serine as a guest residue in the ELPs, X = S.
Some of us have previously developed a diblock polypeptide C–BK12 consisting of an oligolysine block BK12 = K12 and a random coil hydrophilic block with a collagen-like sequence, C = (GXaaYaa)132. The C–BK12 spontaneously assembles on DNA to form a dense brush.30 The same diblock polypeptide also assembles into dense hydrophilic brushes on the surface of nanodiamonds, providing the nanodiamonds with colloidal stability and promoting their uptake by cells.31
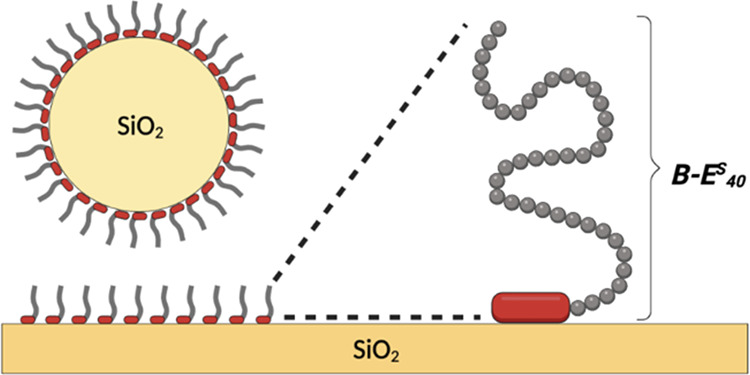
Building on our previous experience with the C–BK12 diblock, the basic design that we study here is again a simple diblock. We explore B–ES40 diblocks, where ES = (VPGSG) and B is a series of silica-binding peptide blocks. First, we characterized in detail the silica-binding properties of the B domains, using quartz crystal microbalance (QCM), atomic force microscopy (AFM), and single-molecule force spectroscopy (SMFS).32−35 Next, we demonstrate that the B–ES40 diblocks form dense polymer brushes on silica surfaces and show that the diblocks stabilize silica nanoparticles. We also explore interactions of coated silica nanoparticles with coated silica surfaces using a single-particle surface mobility assay. We find that even prolonged rinsing, except with very high ionic strength buffers, does not displace the diblocks from silica surfaces, which we attribute to strong peptide–peptide interactions of the SBPs on the silica surface, in addition to their inherent affinity to silica surfaces.
Materials and Methods
“Scan-Asyst air” AFM tips were purchased from Bruker, and the Maleimide-PEG-NHS molecule was purchased from Polypure. QCMD sensors were purchased from Biolin Scientific. A number of peptides were custom-ordered from PepScan (see Table 1), the reported purity is from HPLC analysis, reported mass is the experimental mass from mass spectrometry. Peptides were ion-exchanged to Na+ counterions by the manufacturer. Poly(l-lysine)-grafted poly(ethylene glycol) was purchased from SuSoS (Switzerland) with a grafting ratio of 3.5. The molecular weight of the PLL backbone and PEG side chains was 20 and 2 kDa, respectively. All other chemical reagents were purchased from Sigma-Aldrich.
Table 1. Purity and Molecular Weight of Synthetic Peptides.
| name | sequence | MW (Da) | purity (%) |
|---|---|---|---|
| BR14 | R14 | 2151.6 | >96 |
| BR13-Cys | R13C | 2204.7 | >99 |
| BRT | (RTHRK)4 | 2732.3 | >98 |
| BRT-Cys | (RTHRK)4C | 2835.5 | >92 |
| BRQ | (RQSSRGR)2 | 1672.9 | >92 |
| BRQ-Cys | (RQSSRGR)2C | 1776.1 | >98 |
| ES3 | (VPGSG)3 | 1210.3 | >99 |
| ES3-Cys | (VPGSG)3C | 1313.5 | >98 |
Buffer Preparation
10 mM phosphate buffer (PB) pH 7.4 with different amounts of added NaCl is used throughout. Buffer solutions were filtered and degassed before use.
Quartz Crystal Microbalance with Dissipation Monitoring (QCM-D)
A Q-Sense E4 (Biolin, Sweden) QCM-D instrument was used to quantify peptide and polypeptide binding to silica. QCM sensors coated with SiO2 were obtained from the instrument manufacturer and cleaned according to instructions of the manufacturer. Prior to measurements, the instrument was equilibrated with a starting buffer (PB with added NaCl as indicated) for at least 20 min at a flow rate of 50 μL/min. Analysis of QCMD data was performed using QSense software (Biolin). For some measurements, a linear filter was applied to correct for a small amount of baseline drift. Experimental observables are the changes in resonance frequency (Δf) and energy dissipation (D). If energy dissipation is small enough, the adsorbed mass Δm per unit area is calculated from the change in resonance frequency through the Sauerbrey equation
| 1 |
The oscillation frequency is an (odd) multiple (n = 1,3,5, ...) of the crystal resonance frequency (5 MHz). The numerical prefactor (for a 5 MHz crystal, per unit area of crystal surface) is C = 17.7 ng/(cm2·Hz). The active area of the crystal was 0.2 cm2. Adsorbed masses per unit area Γ obtained from the Sauerbrey equation versus the bulk concentration C were fitted using an effective Langmuir adsorption isotherm
| 2 |
where Γmax is the limiting adsorption, at high concentrations, and Kd,app is an apparent dissociation constant.
AFM Tip Functionalization
We followed previously published protocols for functionalization of AFM tips for single-molecule force measurements.36 AFM tips (Bruker) were cleaned with chloroform. Amino activation was performed by immersing the AFM tips in 30 μL of APTES and 10 μL of triethylamine, in a closed chamber under an argon atmosphere, for 2 h. Next, tips were cleaned again with chloroform and nitrogen gas and 1 mg of a maleimide-PEG-NHS linker was dissolved in 0.5 mL of chloroform and 30 μL of triethylamine. AFM cantilevers were then soaked in this solution (in a small Teflon beaker) for 3 h. Finally, tips were washed three times with chloroform. Peptides BR13Cys, BRTCys, BRQCys, and ES3Cys were covalently attached to maleimide groups of the PEG linkers attached to the AFM tips via thiol bonds. To this end, a solution was prepared of 100 μL of peptide (100 μM), 2 μL of EDTA (100 mM, pH7.5), 5 μL of HEPES (1 M pH 7.5), 2 μL of TCEP (100 mM), and 2 μL of HEPES (1 M, pH 9.6). Tips were immersed in the abovementioned solution for 4 h. Tips were washed with PB and stored at 4 °C in PB for not more than 2 weeks before use.
AFM Imaging in Air
Atomically flat silica surfaces were obtained using silicon wafers (Siltronic AG) with a 2–3 nm oxide layer due to natural oxidation with oxygen in air. Silica surfaces were cleaned with Milli-Q water and ethanol and plasma-cleaned for 5 min. For AFM imaging, peptide solutions, at concentrations as indicated, were filtered with a 10 kDa centrifugal filter and sonicated for 30 min to ensure that no peptide aggregates were present prior to imaging. Next, cleaned silica surfaces were immersed in 100 μL of peptide solution for 1 h. Silica samples were gently rinsed with Milli-Q water and carefully dried with nitrogen. Samples were imaged with a Multimode Bruker AFM (Bruker, California) using the automatic ScanAsyst imaging mode. ScanAsyst air tips were used with a nominal radius <10 nm.
Dynamic Light Scattering
For dynamic light scattering (DLS), a ZS-Nano (Malvern, UK) instrument was used, employing small-volume (20 μL) quartz cuvettes. Light scattering was measured at a scattering angle of 173°, at room temperature, T = 20 °C. Hydrodynamic sizes reported are the average of three measurements, obtained using the Zetasizer software version 7.13 (Malvern, UK). For DLS on peptide solutions, peptide solutions were filtered using a 10 kDa centrifugal filter and sonicated for 30 min to ensure that no peptide aggregates were present prior to the measurement. For each peptide sample, continuous (time-dependent) measurements were performed, and each reported hydrodynamic size is the average of three measurements (with duration of the measurement being controlled using the instrument). For measurements on silica nanoparticles coated with polypeptides, the polypeptide solution and the silica nanoparticles were sonicated for 15 min. The polypeptide solution was filtered using a 0.22 μm filter. Increasing concentrations of polypeptide were incubated with the silica nanoparticles for 10 min. For each sample, continuous measurements were performed, and each reported hydrodynamic size is the average of three measurements, with duration of the measurement being controlled using the instrument.
SMFS Measurements
Silica surfaces were prepared as described for AFM imaging in air. Functionalized tips prepared as described above were used, and experiments were performed in PB buffer pH 7.4 at the indicated concentration of NaCl. Single-molecule force-extension measurements, in a liquid flow cell, were performed using a ForceRobot 300 instrument (JPK, Berlin) which allowed to probe a grid of points (10 μm × 10 μm) on the silica surface for multiple times, to give a total of >2000 measurements per pulling rate. Pulling rates used here were 12, 10, 5, 2.5, 1.25, 0.625, 0.325, and 0.165 μm/s. A contact time between the tip and the surface of 0.2 s was set to let the peptides bind the surface. The spring constant and sensitivity of the cantilevers used here were calibrated with the contact-free thermal noise routine from the JPK software. The JPKSPM data analysis software, Excel (Redmond, WA), and Matlab (Natick, MA) were used to analyze the data and classify events. For each force-extension curve, a baseline was subtracted. Peptides studied here consist of no more than 21 amino acids. The PEG linker has an average number of 27 repeat units. Assuming a 0.4 nm contour length per monomer, we expect forces to vanish at extensions longer than about 20 nm. Data was therefore preclassified using the JPKSPM software, with force–distance curves for which there was still a significant force at extensions >20 nm not being analyzed. The loading rate for each curve was obtained as the slope of the force versus time curves immediately before rupture. To analyze the force extension curves, individual force extension curves were analyzed with a freely jointed chain (FJC) model.37 For M parallel chains
| 3 |
where F is the applied force, Ns is the total number of segments, kB is the Boltzmann constant, and T is the temperature. The contour length Lc determines the overall range of the force extension curve. The Kuhn length b = 0.7 nm determines the slope in the low-force regime and the curvature in the mid-force regime, while the segment elasticity M·Ks determines the slope in the high-force regime. Only events were analyzed with a single rupture event. A very small subset showed multiple smaller rupture events, and this subset of events was also not taken into account in the analysis.
The loading rate dependence of the rupture force data was fitted to the Friddle–de Yoreo model.38 This model accounts for the expected transition from a near-equilibrium regime with fast rebonding at low loading rates (Lr), to a regime of rapid irreversible nonequilibrium detachment at high loading rates, described using the Bell–Evans model. The near-equilibrium regime is characterized by an apparent equilibrium rupture force (feq) and the nonequilibrium regime is characterized by the unbinding rate koff and distance to the transition state xt
| 4 |
where
| 5 |
this is solved using the approximate relation ezE1(z) ≅ ln(1 + e–γ/z) where γ ≈ 0.577 is Euler’s constant. Furthermore, kB is the Boltzmann constant and T is the absolute temperature.
To calculate the free energy of binding per unit length ΔGb, we use the Manohar relationship39 for pulling polymers away from a surface, where ΔGb = γadhlmono and lmono is the length of the monomer (for which we take lmono = 0.35 nm). The tension γadh is
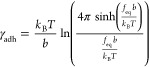 |
6 |
where b is the Kuhn length, for which we take b ≈ 0.7 nm. The free energy is expressed in thermal energies kBT, where T = 298 K is the temperature of the experiment.
Construction of Expression Plasmids for Polypeptides
To construct expression plasmids for the polypeptides, synthetic genes encoding BR9 = R9, BRT = (RTHRK)4, and BRQ = (RQSSRGR)2 were synthesized by Macrogen (Amsterdam, The Netherlands). A synthetic gene for ES40 was designed using the Codon scrambler tool developed by Tang and Chilkoti40 and synthesized by GenScript Inc (Piscataway, USA). All the synthetic fragments were designed in order to contain the features necessary for PRe-RDL cloning, as described by McDaniel et al.41 Sequences of the DNA fragments used are given in Table S1. The fragments were ligated into separate XbaI/EcorI-digested pET-24a(+) vectors, in order to construct a library of PRe-RDL compatible vectors. A total of 10 ng of plasmid DNA was transformed into Escherichia coli BL21 cells by means of electroporation. Colonies containing the correct DNA inserts were selected and confirmed by DNA sequencing. Next, plasmids for expression of the diblock polypeptides B–ES40 were constructed from the PRe-RDL plasmids for the elastin-like protein block ES40 and those for the binding blocks B, as described by McDaniel et al.41 Full amino acid sequences for the diblock polypeptides B–ES40 are given in Table S2.
Protein Expression
E. coli BL21 transformed with the expression plasmids for the polypeptides was cultured at 37 °C/200 rpm for 16 h in 10 mL of TB medium containing 50 μg/mL kanamycin. A starter culture was inoculated in 2 L of TB medium containing 50 μg/mL kanamycin and incubated for 24 h at 37 °C/200 rpm. After 8 h from the start of the incubation, IPTG was added to a final concentration of 1 mM and cells were grown overnight before harvesting.
Protein Isolation and Purification
Bacterial cells were centrifuged at 3300 rpm for 10 min at 4 °C and resuspended in 50 mL of cold phosphate-buffered saline (PBS). Cells were lysed using three cycles of a French press (SLM Aminco). Next, 4 mL of 10% (v/v) polyethyleneamine (PEI) was added to the cell lysates in order to precipitate DNA. The cell lysate was centrifuged for 15 min at 15,000 rpm at 4 °C in order to pellet the insoluble fraction. The ELP-containing polypeptides were first purified using inverse transition cycling. Ammonium sulphate was added to the supernatant to a final concentration of 0.5 M. The supernatant was heated to 37 °C and centrifuged for 10 min at 15,000 rpm at 37 °C. The pellet was resuspended in 10 mL of cold 20 mM HEPES pH 8.0 in order to solubilize the aggregated ELP. The ELP-enriched lysate was centrifuged for 10 min at 15,000 rpm at 4 °C in order to pellet the remaining insoluble matter. Next, polypeptides were further purified using ion-exchange chromatography. The low ionic strength buffer (buffer A) was 20 mM HEPES pH 8.0. The high ionic strength buffer was 20 mM HEPES pH 8.0 and 1 M NaCl (buffer B). Samples were injected in a cation-exchange column (UNO Q6, Bio-Rad Laboratories) and eluted with a linear gradient from buffer A to buffer B. The polypeptides of interest typically eluted at a salt concentration of 0.7 M, as found by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis. In some cases, SDS-PAGE indicated that a small amount of impurities was still present. These were then removed using a mild bake-out procedure: solutions were heated at 60 °C for 15 min, cooled in ice for 10 min, and then centrifuged for 15 min at 15,000 rpm at 4 °C in order to pellet the insoluble contaminants. The final purity of the polypeptides was assessed by SDS-PAGE.
Mass Spectrometry
To confirm the identity of the polypeptides, their molecular weights were determined using matrix-assisted laser desorption/ionization (MALDI) mass spectrometry. Spectra were obtained using a Bruker UltraFlextreme machine. Samples were prepared for analysis following the instructions provided by the manufacturer.
N-Terminal Protein Sequencing
N-terminal sequencing was performed by Alphalyse (Odense, Denmark) on an ABI Procise 494 sequencer by Edman degradation chemistry.
Particle Mobility Assay
For glass substrate functionalization, glass slides (25 × 75 mm, #5, Menzel-Gläser) were precleaned by 30 min of sonication in isopropanol (VWR, absolute) and 10 min of sonication in Milli-Q. After the substrate was dried with a nitrogen stream, 1 min of oxygen plasma was applied to the slides to plasma-oxidize the surface. Custom-made fluid cell stickers (Grace Biolabs) with an approximate volume of 20 μL were then attached to the substrate and 0.5 mg/mL of PLL-g-PEG solution in Milli-Q, respectively. 0.5 mg/mL of BRT–ES40 solution in Milli-Q was immediately injected to the flow chamber and incubated for 5 h at room temperature.
For particle functionalization, vials with carboxyl-functionalized silica particles (Bangs Laboratories, 1 μm mean diameter) at a concentration of 1 mg/mL were incubated with, respectively, 0.9 mg/mL of BRT–ES40 in Milli-Q and 0.9 mg/mL of PLL-g-PEG in Milli-Q and placed on a rotating fin (VWR, The Netherlands) for 3 h at room temperature. Streptavidin-coated polystyrene superparamagnetic particles (10 mg/mL, Dynabeads MyOne Streptavidin C1, 65001, Thermo Scientific) at a concentration of 1 mg/mL in PBS (130 mM NaCl, pH 7.4) were incubated with 5 μL of 11 nt ssDNA (5′ TCACGGTACGA 3′ Biotin, Integrated DNA Technologies) at a concentration of 2 μM in PBS and mPEG-Biotin (PG1-BN-1k, Nanocs) at a concentration of 100 μM in PBS buffer for 70 min on a rotating fin. The particle mixtures were washed with 0.05 vol % Tween-20 (Sigma-Aldrich) and reconstituted in 1000 μL of PBS (130 mM NaCl, 7 mM Na2HPO4, and 3 mM NaH2PO4 at pH 7.4). Right before injecting particles to the flow chamber, the particle solution was sonicated with 10 pulses at 70% with 0.5 duty cycle (Hielscher, Ultrasound Technology).
For the particle mobility assay, particles with different modifications were added to the variously modified surfaces at a final concentration of 0.01 mg/mL in PBS. The trajectory of the particles was recorded for 1 and 5 min at a frame rate of 30 and 60 Hz with an integration time of 5 ms in a field of view of 883 × 552 μm2 with the Leica microscope (Dark field microscopy, Leica DMI5000M). By applying a Gaussian fitting over the intensity of pixels around the particle, the center of every particle can be determined to within approx. 20 nm and tracked over all frames.
Results and Discussion
At neutral pH, silica has a weak negative charge.42 Under these conditions and at low ionic strength, arginine has been found to adsorb most strongly to silica compared to the natural amino acids,43 at least in part due to the high pKa of its side chain. Indeed, in sequences of many SBPs, arginine is over-represented. While many sequences have been reported as being SBPs, a detailed characterization of their binding to silica has only been carried out for a few. Here, we choose to explore two of these, that is, BRT and BRQ, plus oligoarginine control peptides BRn = Rn, as binding domains B. The peptides were fused to ELP (repeat sequence of VPGSG) in order to generate brush-like diblocks B–ES40 (Figure 2).
Figure 2.

Schematic structure of B–ES40 diblocks for forming hydrophilic polypeptide brushes on silica surfaces.
The first SBP characterized is the highSP peptide44,45 which was engineered from a weaker binding precursor isolated from E. coli.46 The sequence of highSP will be referred to as BRT, after the first two residues, BRT = (RTHRK)4. BRT shows high affinity to silica, zinc, and other oxidized metals and has already been successfully employed as a binding tag.44,47 A second SBP that was evaluated is peptide SB7, which was isolated from the spore-coat protein CotB1 of Bacillus cereus.43 Its sequence will be referred to as BRQ, again after the first two residues, BRQ = (RQSSRGR)2. This peptide was designed as a short affinity purification tag, using silica-based materials.
Representative QCM-D data for the adsorption of the SBP BRT in PB to silica are shown in Figure 3a. The lowest concentration for which we observe significant binding is slightly below 1 μM. For this concentration, as well as for higher concentrations, adsorbed peptide layers form in less than 10 min. Flushing with PB buffer for 10 min does not lead to significant detachment of the adsorbed peptides. The out-of-phase response, or energy dissipation, is minimal and so was the dispersion between frequency shifts for different harmonics, indicating a rigidly adsorbed peptide layer that covibrates with the crystal oscillations.
Figure 3.
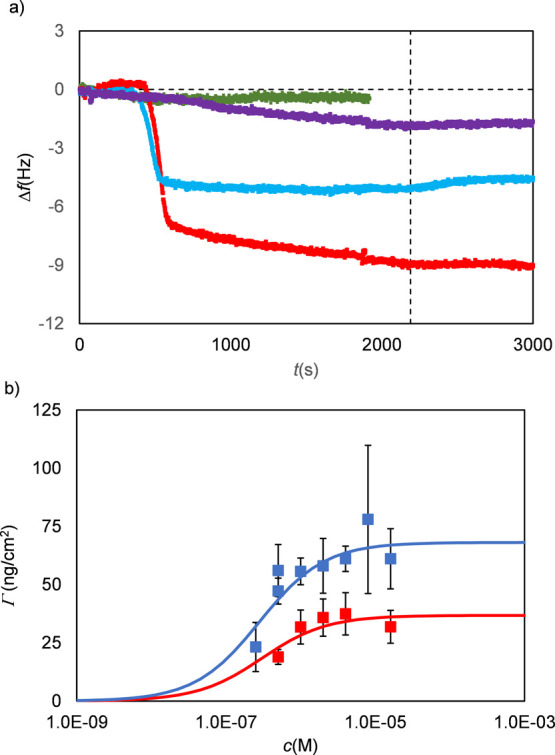
Representative QCM-D data for the adsorption of the SBP BRT and the short ELP ES3 to silica. (a) Frequency Δf (Hz) shift vs time t(s) for BRT at increasing peptide concentrations (purple, 0.175 μM; blue, 5 μM; and red, 10 μM) and for ES3 (green, 5 μM). The vertical dashed line indicates the start of washing with buffer (PB). (b) Representative curve of saturation adsorbed mass Γ (ng/cm2) vs peptide concentration c(M) for the SBP BRT. Blue filled squares: adsorption from PB buffer and red filled squares: adsorption from PBS buffer. Data were fitted using Langmuir isotherms (solid lines) to obtain apparent dissociation constants Kd,app (M–1) and maximum adsorbed masses Γmax (Table 2). Error bars are standard deviations for >3 independent measurements.
Eventually, we wish to construct diblock polypeptides B–ES40 with solid-binding domains B and long hydrophilic ELP domains ES40, where ES = VPGSG. For efficient brush formation, the much longer hydrophilic blocks should not compete with the binding blocks in binding to the silica. For example, it is well-known that PEG has a strong affinity for silica,48 which for PLL-g-PEG polymers may compete with the PLL backbone in adsorbing to silica surfaces. Therefore, we also performed QCM-D measurements on the adsorption of the short elastin-like peptide ES3 to silica. These results are included in Figure 3a. Within the accuracy of the instrument, we could not detect any adsorption of the elastin-like peptide, suggesting that this ELP is indeed a good choice as a hydrophilic block when aiming to construct self-assembling brushes on silica.
Since the out-of-phase response or dissipation D in the QCM-D experiments was shown to be negligible and there was hardly any dispersion between frequency shifts for different harmonics, the Sauerbrey equation can be applied to relate the frequency shift to adsorbed mass per unit area.14 Results for the saturation values of the adsorbed mass per unit area Γ as a function of peptide concentration c (in M) for the SBP BRT are shown in Figure 3b. Results for the other SBPs, BRQ and BR14, were very similar and are shown in Figure S1. A Langmuir equation was applied to fit the data to get apparent dissociation constants Kd,app and plateau values Γmax for the adsorbed mass per unit area.
Strictly speaking, the validity of the Langmuir equation requires that the peptides are noninteracting on the surface and that the adsorbed layer is in full equilibrium. For all three peptides, BRT, BRQ, and BR14, there are strong indications for irreversible rather than equilibrium adsorption and for an important role for peptide–peptide interactions at the solvent–silica interface. Hence, it is unlikely the peptides satisfy the requirements for the validity of the Langmuir equation. Therefore, we prefer to denote the dissociation constants obtained by the Langmuir fits of the QCM data as apparent dissociation constants Kd,app.
Values for dissociations constants Kd,app and plateau values Γmax are listed in Table 2. Within the rather large error of the measurements, there are no significant differences in the apparent dissociation constants of the peptides. Apparent dissociation constants Kd,app for all three peptides are on the order of μM. Saturation densities are on the order of 0.5 mg/m2. Both are fairly typical for adsorbed peptide layers.14,49,50 The saturation density corresponds to an approximate surface area per peptide on the order of 5 nm2.
Table 2. Parameters from Langmuir Fits of QCM-D Data.
| PB |
PBS |
|||
|---|---|---|---|---|
| peptide | Kd,app [106 M–1] | Γmax [ng/cm2] | Kd,app [106 M–1] | Γmax [ng/cm2] |
| BR14 | 1.4 | 90 | 0.7 | 70 |
| BRT | 3.6 | 70 | 3.3 | 40 |
| BRQ | 2.5 | 40 | 0.5 | 40 |
Next, we have attempted to image the ultrastructure of the adsorbed peptide layers by performing AFM imaging on dried peptide layers in air. Control measurements of clean silica slides did not show any feature with heights in excess of 200 pm. AFM images show surface features that are strongly dependent on the concentrations at which the layers are formed. For all peptides, at low concentrations, we observe a homogeneous background and small globular features, whereas at higher concentrations, a homogeneous background and surface-bound fibrils are observed. Selected results for the SBP BRT are shown in Figure 4a–c; an overview with results for all SBPs and concentrations tested is shown in Figure S2.
Figure 4.

Surface-induced fibril formation for SBP BRT on silica. (a–c) AFM images of BRT adsorbed onto silica from PB buffer and incubated for 1 h, imaged in air, for different concentrations and magnifications. Scale bars are 200 nm. (a) 3.66 μM, (b) zoom-in of region outlined in (a). (c) 36.6 μM. (d) Hydrodynamic diameter DH (nm) from DLS of 1 mg/mL SBP solutions in PB, as a function of time. Red circle: BRQ, green square: BRT, and blue triangle: BR14.
For the solid-binding peptides BRT and BRQ, surface-bound fibrils were observed when adsorption was performed at concentrations above about 5 μM, whereas for the BR14 control peptide, surface-bound fibrils were already observed at concentrations of around 0.5 μM, 1 order of magnitude lower. For each peptide, cross sections of multiple fibrils (n = 6) were analyzed and average fibril heights hfibril were calculated. Fibril heights for different peptides were similar, hfibril = 0.8 ± 0.2 nm for BRT, hfibril = 0.5 ± 0.2 nm for BRQ, and hfibril = 0.9 ± 0.4 nm for BR14. It should be noted that drying may have had some impact on the final morphology of the fibrils as imaged. This could possibly be checked by complementary AFM imaging experiments in liquid.
In order to establish whether fibril formation is induced by the silica surface or if fibrils form in solution and subsequently adsorb, we have monitored peptide solutions for long periods using DLS. Results are shown in Figure 4d. Peptide solutions of 1 mg/mL were carefully filtered and sonicated in order to ensure that no aggregates were present at the start of the measurement. The concentration of 1 mg/mL is well above the concentrations for which fibrils were observed in the AFM images. Hydrodynamic radii were measured for 1 h. During this time, the hydrodynamic diameter (DH) that was observed remained small, in the order of 2.5 nm, which is the size expected for monomeric peptides in solution. Samples measured again over 3 days and 1 week did not show any noticeable change in hydrodynamic size either (data not shown). Hence, fibril formation of the SBPs appears to be driven by interaction with silica surfaces and not start in the bulk solution.
Having established that the SBPs strongly interact with each other when adsorbed to silica surfaces, it is clear that a simple Langmuir adsorption isotherm cannot be used to describe the mechanism of peptide surface layer formation, even though the data of saturation adsorption Γmax versus concentration c can be fitted with a Langmuir isotherm.
To obtain information on the binding of individual SBPs to silica, in the absence of peptide–peptide interactions, we resort to the measurement of single-molecule force-extension curves using AFM. An AFM tip is functionalized with maleimide-terminated PEG linkers, to which cysteine-terminated SBPs are attached via thiol bonds at a low density, as sketched in Figure S3a. As shown in Figure S4d–f, with this procedure, there are typically a small number of peptides M <30 that interact simultaneously with the surface.38 The low density guarantees that during the experiments, peptides do not interact with each other.
The peptides studied here consist of no more than 21 amino acids, while the PEG linker has an average number of 27 repeat units; assuming an approximate size of 0.4 nm per monomer, a total contour length for the constructs of approximately 20 nm can be calculated. The absence of attraction at distances larger than this cutoff was used as a first selection criterion to identify relevant events. Histograms of rupture forces for the SBPs and the negative controls (respectively, the inert ELP peptide ES3 and the nonfunctionalized PEG linkers) are shown in Figure 5a,b. As expected, for the SBPs, we find a large number of rupture events and forces at rupture of order 100 pN. In contrast, for the negative controls, the number of events for which a rupture is detected is much lower, and typical rupture forces are lower, thus validating that in our experiment, we indeed measure adhesion forces of SBPs for the silica substrate. A typical force-extension curve for the SBP BR13Cys is shown in Figure 5c. All force-extension curves such as this one were fit with an FJC model for M parallel chains, to give the contour length Lc, breaking force Fbreak, and loading rate r at rupture, as well as an estimate of the number M of parallel chains in the rupture event. While most force-extension curves showed only a single rupture event and hence could be analyzed as described above, a small number of force-extension curves were also recorded that showed rupture in multiple steps.51 These rare events were excluded from the analysis. We find that most rupture events have a number of parallel bonds M < 10. To ensure that we measure binding and unbinding of individual peptides, we focus on the events corresponding to a low number of parallel bonds, M = 1, ..., 7. Histograms of the contour length Lc and the number of parallel bonds M are presented in Figure S4. For data with M = 1, ..., 7, rupture force versus loading rate was analyzed using the Friddle–de Yoreo (FdY) model for M parallel bonds that can detach and reattach.38,52 The FdY model has been used before to analyze the adhesion of proteins to substrates53,54 of polymer to minerals55 and of peptides to inorganic materials.56 At low loading rates, the rapid (re)binding and unbinding leads to a constant rupture force, but at higher loading rates, the model recovers the known logarithmic loading-rate dependence for the case in which rebinding is neglected.38 An example fit for the SBP BRT is shown in Figure 5d, fits for the other SBPs are given in Figure S3b,c. Fit parameters for all SBPs are listed in Table 3. An approximate binding free energy ΔG is calculated from the fitted Feq using a relation previously derived by Manohar,39 these values are also included in Table 3.
Figure 5.

Single-molecule AFM-based force spectroscopy. (a) Histogram of the number of events n vs breaking force Fbreak (pN) for SBPs BRQ-Cys, BRT-Cys, and BR13-Cys (as indicated). (b) Histogram of the number of events n vs breaking force Fbreak (pN) for negative controls (as indicated): just the PEG linker (no peptide conjugated) and the nonadsorbing elastin-like peptide ES3. (c) Example fit of single force curve for BR13-Cys with the FJC used to extract approximate numbers of parallel peptides M and contour lengths Lc. (d) Final result of data analysis, for the case of BRT-Cys: breaking force Fbreak (pN) versus loading rate r (pN/s), for selected event with M = 1, ..., 7. The red line is the fit with the Friddle–deYoreo model; fit parameters for all SBPs are given in Table 4.
Table 3. Fit Parameters for FdY Fits of Rupture Force Versus Loading Rate for SBP, Binding to Silica.
| SBP | koff (s–1) | xt (Å) | Feq (pN) | ΔG (kBT) |
|---|---|---|---|---|
| BR13 | 145.9 | 0.6 | 29.4 | 2.62 |
| BRQ | 246.4 | 0.3 | 46.1 | 3.81 |
| BRT | 287.4 | 0.4 | 40.0 | 3.36 |
Of special attention are the relatively high off-rates, for example, koff ≈ 2.9·102 s–1 for BRT, although still not as high as found previously (koff ≈ 2.6·103 s–1) for the adhesion of an even shorter and more weakly adhering peptide (GCRL) to silica.51 Hence, our data demonstrate that single SBPs detach rapidly, thereby supporting the hypothesis that the relative stability of SBP layers is caused by SBP–SBP interactions at the silica surface, which reinforce the surface layers.
Having established that all SBPs bind with similar affinities and have similar off-rates but form peptide layers that are not easily detached, B–ES40 diblock polypeptides were generated with binding blocks B = BRT, BRQ, and BR9. A shorter Rn block was used for the experiments with synthetic peptides (BR9 instead of BR14) since highly cationic polypeptides are often difficult to express in view of their possible toxicity to the production host, in our case E. coli.
Three fusion proteins were successfully produced and purified from E. coli. Representative SDS-PAGE and MALDI-TOF results for BRT–ES40 are shown in Figure 6. As expected, the overexpressed polypeptides are not found in the insoluble cell pellet (Figure 6a) but rather in the soluble extract obtained after cell disruption. The presence of positively charged silica-binding domains B in the fusion proteins was exploited for purification using cation-exchange chromatography. This allowed for nearly complete purification in one step. A final mild bake-out procedure was sufficient to further purify the desired proteins to an acceptable level (Figure 6a). As previously observed,57,58 the poor SDS-binding of ELPs leads to anomalously high molecular weights in SDS-PAGE when compared with globular protein molecular weight standards. To confirm the identity of the polypeptides, MALDI-TOF spectra were determined for the purified proteins (Figure 6b). The experimentally determined masses corresponded well with the theoretically expected values, as shown in Table 4. A further confirmation was obtained via N-terminal protein sequencing (data not shown).
Figure 6.
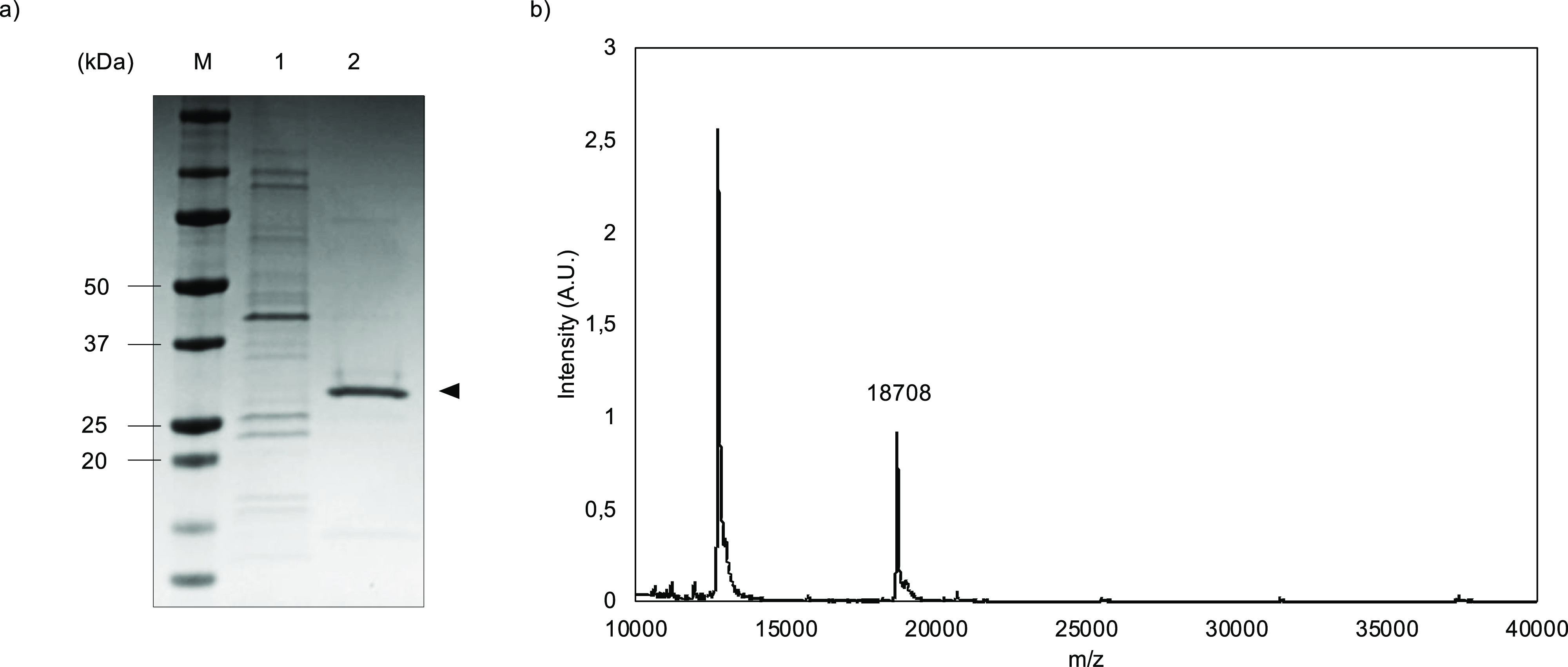
Purification and characterization of recombinant B–ES40 diblock polypeptides, example data for BRT–ES40. (a) SDS-PAGE analysis. Lane 1: insoluble pellet; lane 2: purified polypeptide; and lane M: molecular mass marker. (b) MALDI-TOF spectrum.
Table 4. Molecular Weights of Diblock Polypeptides B–ES40 as Computed from the Amino Acid Sequence and as Determined Using MALDI-TOF.
| diblock polypeptide | Mtheo (kg/mol) | Mexp (kg/mol) |
|---|---|---|
| BR9–ES40 | 17.452 | 17.458 |
| BRQ–ES40 | 17.645 | 17.652 |
| BRT–ES40 | 18.704 | 18.708 |
Next, layer formation on silica surfaces was studied using QCM-D for the B–ES40 diblocks and compared to that of the binding blocks B alone. A representative comparison for 1 μM BRT peptide versus the same molar concentration of BRT–ES40 diblock polypeptide is shown in Figure 7. At this concentration, saturated layers were formed for both the peptide and the diblock polypeptide. The curves of the frequency shift Δf and dissipation D versus time are substantially different for the two cases: for the diblock polypeptide, both the frequency shift and the dissipation are much higher. This indicates that due to the ES40 tails, not only the adsorbed mass per unit area is much higher but also that the highly hydrated nonadsorbed ES40 tails do not covibrate with the quartz crystal, in contrast to the strongly adsorbed BRT domains, leading to high dissipation.59 Similar results were found for the other diblock polypeptides BRQ–ES40 and BR9–ES40. This confirms that the diblock polypeptides do indeed form self-assembled ES40 brushes as intended.
Figure 7.
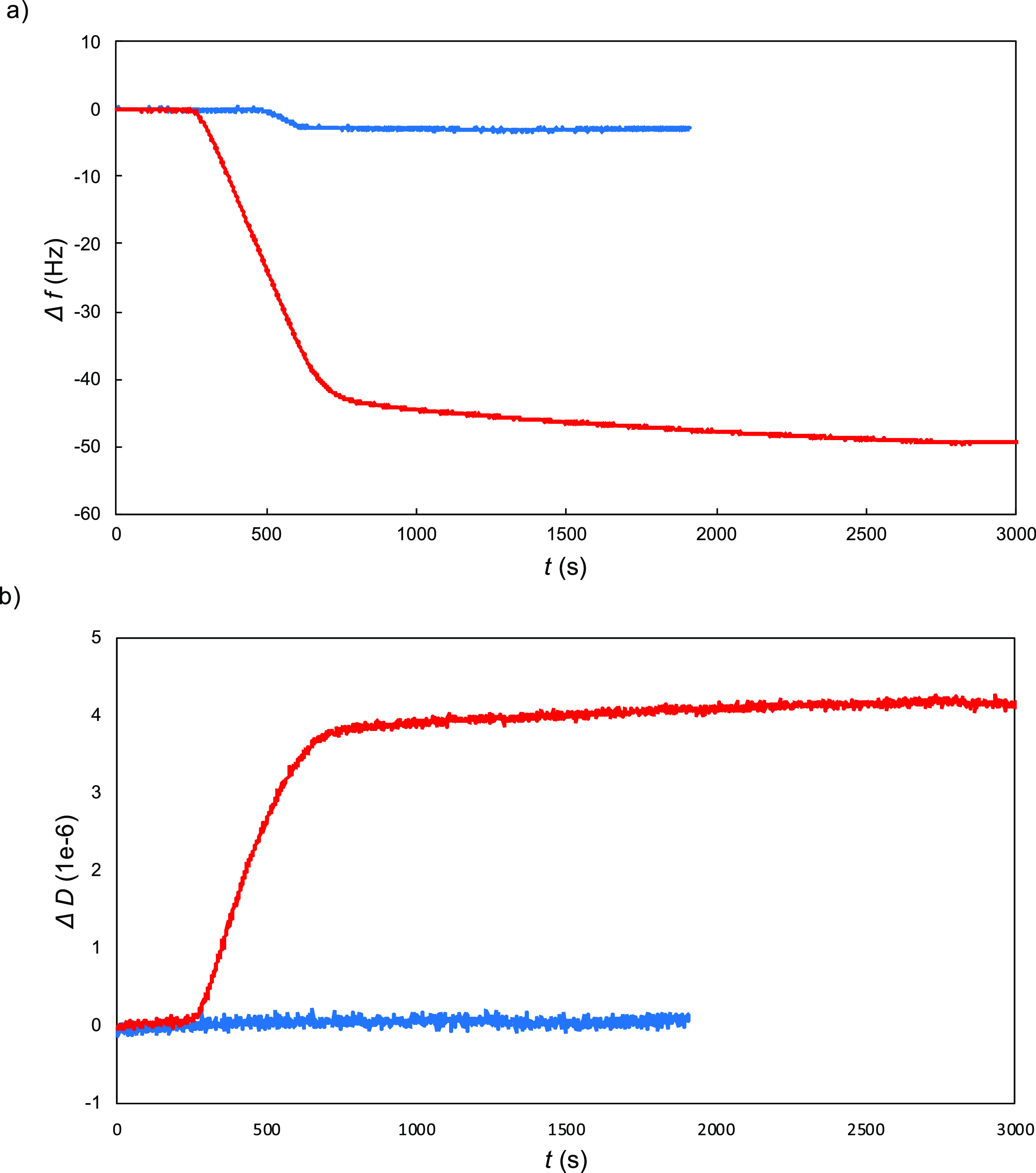
Representative comparison of QCMD data for adsorption to silica for 1 μM B–ES40 diblock polypeptides with the corresponding data for the SBP B. Red: diblock polypeptides BRT–ES40 and blue: SBP BRT. (a) Frequency shift vs time. (b) Dissipation vs time.
Given the complexities and uncertainties in precisely fitting QCM-D data for cases when significant dissipation is present,3 the frequency change Δf was used instead as a qualitative measure for the adsorbed amount. Limiting values of the frequency change Δf versus concentration are shown in Figure 8, whereas for the individual binding blocks, we found that saturated layers were formed at concentrations around approximately 1 μM, and for the diblock polypeptides, it seems that a saturation value for the frequency change is already reached at concentrations well below 1 μM. This could be caused by the steric repulsion of the ES40 tails limiting further adsorption as was also found for similar diblock polypeptides adhering to double-stranded DNA.60
Figure 8.

Limiting frequency shifts Δf for B–ES40 diblocks versus polypeptide concentration c (μM). Green: B = BRT, blue: B = BRQ, and red: B = BR9.
Some further aspects of the polypeptide brushes self-assembled from B–ES40 on silica were investigated for the specific case of B = BRT. First, we consider the dependence of brush formation on solution conditions. The adsorption of the cationic-binding peptides being largely electrostatically driven, we expect a significant dependence of the layer formation on ionic strength. Indeed, this is borne out by the QCM-D results shown in Figure 9.
Figure 9.

Salt-dependence of brush formation for 5 μM BRT–ES40. Frequency shift Δf vs time t. (a) Adsorption of BRT–ES40 from PB followed by washing with PBS (150 mM NaCl), followed by washing with a high salt buffer (PBS + 0.5 M NaCl). (b) Adsorption from PB followed by washing with PB (blue), compared with adsorption from PBS followed by washing with PBS (red).
As shown in Figure 9a, an adsorbed layer is rapidly formed when flowing a solution of 5 μM of BRT–ES40 in PB over a clean silica surface. Upon rinsing with PB buffer, there is negligible change in the frequency shift, indicating minimal desorption, as was also found for the peptide binding blocks B by themselves. Upon shifting to PBS (=PB + 150 mM NaCl), a significant decrease is observed for the magnitude of the frequency shift. Finally, upon flushing in PB + 500 mM NaCl, the frequency shift drops nearly to the baseline. A separate channel was used to correct for the effect of the buffer by itself on the crystal oscillation frequency. The experiments indicate that the salt stability of the self-assembly polypeptide brushes is at best moderate.
In another QCM-D experiment, brushes were assembled at different ionic strengths rather than exposed to a higher ionic strength after having been formed at low ionic strength. Results are shown in Figure 9b. A 5 μM solution of BRT–ES40 in either PB or PBS was flown over a clean silica sensor. After a saturated layer had formed, the sensor was rinsed with either PB or PBS for 4 h and the shift in frequency of the sensor was monitored. We find that the frequency shift for assembly in PBS is nearly but not exactly the same, as the shift when the layer is first formed in PB and subsequently exposed to PBS. Note that for this experiment, we performed prolonged rinsing with buffer and even after many hours of rinsing, there is hardly any shift in frequency. This indicates that for a given buffer composition, there is hardly any polypeptide desorption neither when rinsing with PB for saturated layers formed in PB nor for PBS for saturated layers formed in PBS.
The self-assembled polypeptide brushes could be used to stabilize and functionalize not only macroscopic flat silica surfaces but also silica nano- and microparticles. To investigate brush formation around silica nanoparticles, DLS was performed on nonporous silica particles with a reported hydrodynamic diameter of DH = 163 nm. A dilute suspension of these particles was incubated for 5 min with varying concentrations of the diblock polypeptide BRT–ES40. As shown in Figure 10, for the bare silica particles, a hydrodynamic diameter of 161 ± 0.8 nm was determined, in agreement with the manufacturer specifications. Upon increasing the BRT–ES40 diblock concentration, the particle diameter increases until it stabilizes at a final diameter of approximately 189 nm at 0.5 μM BRT–ES40. This implies an approximate hydrodynamic brush height of h ≈ 14 nm. In a separate DLS experiment, it was found that the hydrodynamic diameter of the free BRT–ES40 diblock, which is dominated by the coil size of the ES40 block, was DH = 9 ± 1 nm. This suggests that the ES40 chains are somewhat stretched. A similar result was found for polypeptide brushes self-assembled around DNA.60
Figure 10.

Formation of stable polymer brush on silica particles measured with DLS. Particle diameter vs concentration of BRT–ES40. Bare nonporous silica particles were incubated with increasing concentrations of BRT–ES40 and the particle size was measured by means of DLS. Each measurement was repeated three times, error bars are contained within the size of the dot. The gray-dashed line represents the measured hydrodynamic diameter of bare silica particles.
In searching for effective hydrophilic brush-forming polypeptides, we were motivated by a novel single-molecule biosensing platform in which the mobility of a functionalized micrometer-sized particle, tethered to a functionalized surface, is modulated by single-molecule interactions, as reported by some of us.61−63 This new biosensing platform allows for continuous monitoring of analytes and is based on affinity interactions, for example, an antibody sandwich or competition assay. For such a platform, it is essential to completely block interaction of the tethered particles with nonfunctionalized surfaces. As a first demonstration of the functionality of the self-assembled brushes in a biosensing application, we demonstrate the effectiveness of the BRT–ES40 diblocks in blocking nonspecific particle–surface interactions. Note that for full functionality of the polypeptides in biosensing, we will also need to show adequate antifouling against smaller biomolecules and long-term stability of the brushes. This will be the subject of later studies.
Blocking of nonspecific interactions by polypeptide brushes formed by BRT–ES40 diblocks on silica nanoparticles and glass slides was tested using a particle mobility assay, as illustrated in Figure 11. The interactions between particles and surface are directly reflected by the mobility.64 To record the mobility of micron-sized particles over time, we used dark-field microscopy. The position of every particle at each frame is localized with an accuracy of about 20 nm and then reconstructed into a trajectory over time. Silica particles and magnetic particles with specific modifications are sedimented onto the surface of coated glass slides and their trajectories are reconstructed. For each particle, a two-dimensional diffusion coefficient D2D is determined from the mean-squared displacement ⟨(r⇀(t + τ) – r⇀(t))2⟩ = 4D2Dτ. By analyzing the particle trajectories and setting a threshold for the diffusion coefficient, freely moving particles and stuck particles are distinguished and percentages are obtained for particles that ultimately become stuck. Results are given in Table 5, for various types of particles and for particle and surface coatings with BRT–ES40 diblock polypeptides and PLL-g-PEG copolymers, the current standard for self-assembled antifouling brushes. While both BRT–ES40 and PLL-g-PEG coatings are effective in preventing nonspecific interactions if both the particle and the surface are nonfunctionalized, various functionalized particles with biosensing applications such as ssDNA-, biotin-PEG-, and streptavidin-coated particles stick stronger to PLL-g-PEG-coated glass slides. In contrast, minimal nonspecific interactions are found between these functionalized particles and glass slides coated with BRT–ES40, demonstrating the usefulness of BRT–ES40 as a blocker of nonspecific particle–surface interactions.
Figure 11.
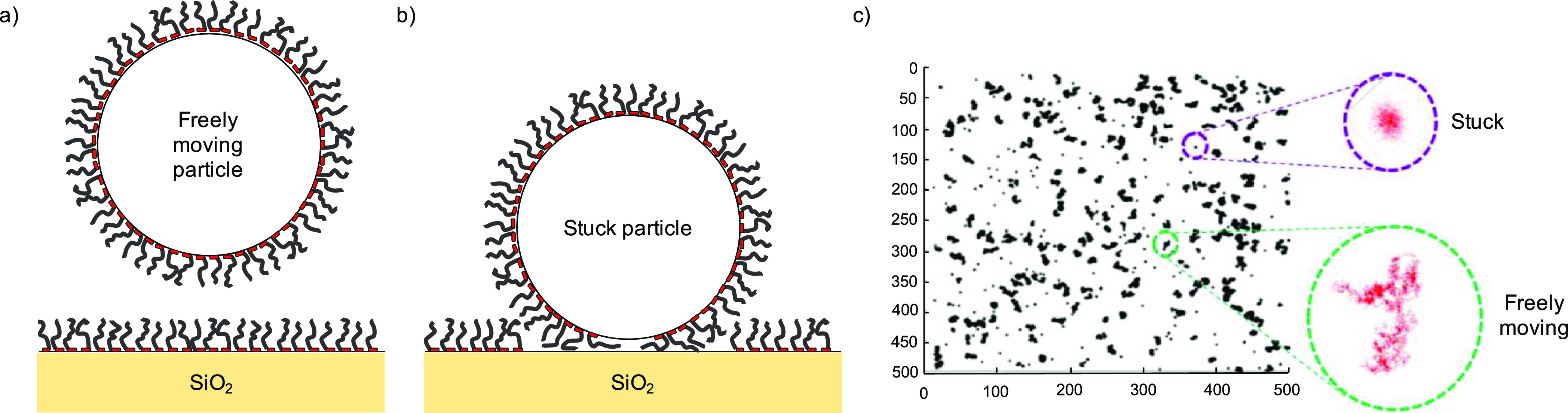
Schematic representation of the particle mobility assay. Particles coated with biomolecules such as ssDNA, streptavidin, and BRT–ES40 sediment to the surface of the substrate and show Brownian motion. The mobility and fraction of stuck particles is influenced by the interactions between particles and surfaces. (a,b) Schematic of a particle not interacting with the surface and a particle strongly interacting. (c) Example of particle position trajectories recorded over time for multiple particles. The insets show typical observed trajectories for a stuck and freely moving particle.
Table 5. Fraction of Differently Modified Particles Stuck to Differently Modified Silica Surfaces (%), as Obtained from a Particle Mobility Assay.
| glass
modification |
||
|---|---|---|
| particle type, modification | BRT–ES40 | PLL-g-PEG |
| silica, BRT–ES40 | 1.3 ± 0.6 | 3.8 ± 0.9 |
| silica, PLL-g-PEG | 1.9 ± 0.6 | 2.5 ± 0.2 |
| polystyrene, ssDNA | 1.0 ± 0.7 | 25.0 ± 14.6 |
| polystyrene, biotin-PEG | 1.7 ± 0.8 | 12.6 ± 6.0 |
| polystyrene, streptavidin | 3.2 ± 1.0 | 80.1 ± 13.0 |
Concluding Remarks
By comparing surface binding of different arginine-rich silica-binding peptides B, we have established that in-plane, peptide–peptide interactions play a crucial role in the formation of adsorbed silica-binding peptide layers. Presumably thanks to the cooperative nature of the layer formation by the SBPs B, the self-assembled brushes formed by the B–ES40 diblocks are excellent blockers of various nonspecific interactions.
Possible advantages of these polypeptides over other self-assembling brush systems such as PLL-g-PEG are that the former can directly be used as recombinant silica-immobilization tags for functional proteins. Also, we have found that at least for some cases, such as in blocking nonspecific interactions of functionalized particles with glass surfaces, PLL-g-PEG does not perform as effectively as BRT–ES40. Nonetheless, further testing is necessary to evaluate the antifouling properties of our polymer brush compared to PLL-g-PEG. Since binding peptides have been investigated for many types of surfaces, such as plastics, metals, and other oxides, the polypeptide approach also appears to be easier to generalize than the synthetic adsorbing bottle-brush systems.
These diblocks are also a good starting point for the design of polypeptides that form brushes with even better salt resistance, as may be required for some applications. Stronger attachment of the brushes to the silica could possibly be engineered by arranging for yet more binding cooperativity. For example, elaborating on a previous example of silica-binding ELP micelles,19 one could try designing triblock polypeptides featuring an additional “self-assembly” midblock, which could, for example, be a more hydrophobic ELP block. Alternatively, one could try to include multiple binding tags in a linear topology, although in this case, the gain in binding strength needs to be balanced against an inevitable reduction of the grafting density of the polymer brush.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biomac.1c00067.
Sequences of synthetic genes for diblock expression, amino acid sequence of diblock polypeptides, QCM-D data for SBPs BRQ and BR14, AFM images of SBPs BRT, BRQ, and BR14, schematic and data analysis of single-molecule AFM-based force spectroscopy for SBPs BRQ and BR13Cys, and event selection for measurement of force-extension curves using AFM (PDF)
Author Contributions
N.A. and F.A.G.-M. contributed equally. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
F.A.G.-M. was funded by the Industrial Partnership Programme “Designed hybrid networks using protein-like block copolymers” as a part of the agreement between Unilever Research and Development B.V. and The Netherlands Organisation for Scientific Research (NWO) with project number 15HSM10. N.A. and Y.-T.L. were funded by NWO-TTW-OTP project number 15481.
The authors declare no competing financial interest.
Supplementary Material
References
- Sapsford K. E.; Algar W. R.; Berti L.; Gemmill K. B.; Casey B. J.; Oh E.; Stewart M. H.; Medintz I. L. Functionalizing Nanoparticles with Biological Molecules: Developing Chemistries That Facilitate Nanotechnology. Chem. Rev. 2013, 113, 1904–2074. 10.1021/cr300143v. [DOI] [PubMed] [Google Scholar]
- Vogler E. A. Protein Adsorption in Three Dimensions. Biomaterials 2012, 33, 1201–1237. 10.1016/j.biomaterials.2011.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabe M.; Verdes D.; Seeger S. Understanding Protein Adsorption Phenomena at Solid Surfaces. Adv. Colloid Interface Sci. 2011, 162, 87–106. 10.1016/j.cis.2010.12.007. [DOI] [PubMed] [Google Scholar]
- Saha B.; Songe P.; Evers T. H.; Prins M. W. J. The Influence of Covalent Immobilization Conditions on Antibody Accessibility on Nanoparticles. Analyst 2017, 142, 4247–4256. 10.1039/c7an01424d. [DOI] [PubMed] [Google Scholar]
- Yu Q.; Zhang Y.; Wang H.; Brash J.; Chen H. Anti-Fouling Bioactive Surfaces. Acta Biomater. 2011, 7, 1550–1557. 10.1016/j.actbio.2010.12.021. [DOI] [PubMed] [Google Scholar]
- Lowe S.; O’Brien-Simpson N. M.; Connal L. A. Antibiofouling Polymer Interfaces: Poly(Ethylene Glycol) and Other Promising Candidates. Polym. Chem. 2015, 6, 198–212. 10.1039/c4py01356e. [DOI] [Google Scholar]
- Baggerman J.; Smulders M. M. J.; Zuilhof H. Romantic Surfaces: A Systematic Overview of Stable, Biospecific, and Antifouling Zwitterionic Surfaces. Langmuir 2019, 35, 1072–1084. 10.1021/acs.langmuir.8b03360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gon S.; Fang B.; Santore M. M. Interaction of Cationic Proteins and Polypeptides with Biocompatible Cationically-Anchored PEG Brushes. Macromolecules 2011, 44, 8161–8168. 10.1021/ma201484h. [DOI] [Google Scholar]
- Zhang L.; Klein B. D.; Metcalf C. S.; Smith M. D.; McDougle D. R.; Lee H.-K.; White H. S.; Bulaj G. Incorporation of Monodisperse Oligoethyleneglycol Amino Acids into Anticonvulsant Analogues of Galanin and Neuropeptide y Provides Peripherally Acting Analgesics. Mol. Pharm. 2013, 10, 574–585. 10.1021/mp300236v. [DOI] [PubMed] [Google Scholar]
- Ding Y.; Zhang P.; Tang X.-Y.; Zhang C.; Ding S.; Ye H.; Ding Q.-L.; Shen W.-B.; Ping Q.-N. PEG Prodrug of Gambogic Acid: Amino Acid and Dipeptide Spacer Effects. Polymer 2012, 53, 1694–1702. 10.1016/j.polymer.2012.02.022. [DOI] [Google Scholar]
- Saxer S.; Portmann C.; Tosatti S.; Gademann K.; Zürcher S.; Textor M. Surface Assembly of Catechol-Functionalized Poly(L-Lysine)- Graftpoly(Ethylene Glycol) Copolymer on Titanium Exploiting Combined Electrostatically Driven Self-Organization and Biomimetic Strong Adhesion. Macromolecules 2010, 43, 1050–1060. 10.1021/ma9020664. [DOI] [Google Scholar]
- Urry D. W. Physical Chemistry of Biological Free Energy Transduction as Demonstrated by Elastic Protein-Based Polymers. J. Phys. Chem. B 1997, 101, 11007–11028. 10.1021/jp972167t. [DOI] [Google Scholar]
- Hassouneh W.; MacEwan S. R.; Chilkoti A. Fusions of Elastin-like Polypeptides to Pharmaceutical Proteins. Methods Enzymol. 2012, 502, 215–237. 10.1016/B978-0-12-416039-2.00024-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z.; Palafox-Hernandez J. P.; Law W.-C.; Hughes Z. E.; Swihart M. T.; Prasad P. N.; Knecht M. R.; Walsh T. R. Biomolecular Recognition Principles for Bionanocombinatorics: An Integrated Approach to Elucidate Enthalpic and Entropic Factors. ACS Nano 2013, 7, 9632–9646. 10.1021/nn404427y. [DOI] [PubMed] [Google Scholar]
- Sengupta A.; Thai C. K.; Sastry M. S. R.; Matthaei J. F.; Schwartz D. T.; Davis E. J.; Baneyx F. A Genetic Approach for Controlling the Binding and Orientation of Proteins on Nanoparticles. Langmuir 2008, 24, 2000–2008. 10.1021/la702079e. [DOI] [PubMed] [Google Scholar]
- Care A.; Bergquist P. L.; Sunna A. Solid-Binding Peptides: Smart Tools for Nanobiotechnology. Trends Biotechnol. 2015, 33, 259–268. 10.1016/j.tibtech.2015.02.005. [DOI] [PubMed] [Google Scholar]
- Care A.; Petroll K.; Gibson E. S. Y.; Bergquist P. L.; Sunna A. Solid-Binding Peptides for Immobilisation of Thermostable Enzymes to Hydrolyse Biomass Polysaccharides. Biotechnol. Biofuels 2017, 10, 1–16. 10.1186/s13068-017-0715-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong Po Foo C.; Patwardhan S. V.; Belton D. J.; Kitchel B.; Anastasiades D.; Huang J.; Naik R. R.; Perry C. C.; Kaplan D. L. Novel Nanocomposites from Spider Silk-Silica Fusion (Chimeric) Proteins. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 9428–9433. 10.1073/pnas.0601096103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Li N. K.; Tu Q.; Im O.; Mo C.-K.; Han W.; Fuss W. H.; Carroll N. J.; Chilkoti A.; Yingling Y. G.; et al. Functional Modification of Silica through Enhanced Adsorption of Elastin-Like Polypeptide Block Copolymers. Biomacromolecules 2018, 19, 298–306. 10.1021/acs.biomac.7b01307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Mo C.-K.; Chilkoti A.; Lopez G. P.; Carroll N. J. Creating Cellular Patterns Using Genetically Engineered, Gold- and Cell-Binding Polypeptides. Biointerphases 2016, 11, 021009. 10.1116/1.4952452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah M.; Hsueh P.-Y.; Sun G.; Chang H. Y.; Janib S. M.; MacKay J. A. Biodegradation of Elastin-like Polypeptide Nanoparticles. Protein Sci. 2012, 21, 743–750. 10.1002/pro.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Eldijk M. B.; McGann C. L.; Kiick K. L.; Van Hest J. C. M. Elastomeric Polypeptides. Top. Curr. Chem. 2012, 310, 71–116. 10.1007/128_2011_205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Shields C. W.; Huang J.; Zhang Y.; Ohiri K. A.; Yellen B. B.; Chilkoti A.; López G. P. Rapid Capture of Biomolecules from Blood: Via Stimuli-Responsive Elastomeric Particles for Acoustofluidic Separation. Analyst 2020, 145, 8087–8096. 10.1039/d0an01164a. [DOI] [PubMed] [Google Scholar]
- Girotti A.; Reguera J.; Rodríguez-Cabello J. C.; Arias F. J.; Alonso M.; Testera A. M. Design and Bioproduction of a Recombinant Multi(Bio)Functional Elastin-like Protein Polymer Containing Cell Adhesion Sequences for Tissue Engineering Purposes. J. Mater. Sci.: Mater. Med. 2004, 15, 479–484. 10.1023/B:JMSM.0000021124.58688.7a. [DOI] [PubMed] [Google Scholar]
- Zong J.; Cobb S. L.; Cameron N. R. Short Elastin-like Peptide-Functionalized Gold Nanoparticles That Are Temperature Responsive under near-Physiological Conditions. J. Mater. Chem. B 2018, 6, 6667–6674. 10.1039/c8tb01827h. [DOI] [PubMed] [Google Scholar]
- Shi P.; Aluri S.; Lin Y.-A.; Shah M.; Edman M.; Dhandhukia J.; Cui H.; MacKay J. A. Elastin-Based Protein Polymer Nanoparticles Carrying Drug at Both Corona and Core Suppress Tumor Growth in Vivo. J. Controlled Release 2013, 171, 330–338. 10.1016/j.jconrel.2013.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W.; MacEwan S. R.; Chilkoti A.; López G. P. Bio-Inspired Synthesis of Hybrid Silica Nanoparticles Templated from Elastin-like Polypeptide Micelles. Nanoscale 2015, 7, 12038–12044. 10.1039/c5nr01407g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Kim O.; Jung J.; Na K.; Heo P.; Hyun J. Simple Fabrication of a Smart Microarray of Polystyrene Microbeads for Immunoassay. Colloids Surf., B 2009, 72, 173–180. 10.1016/j.colsurfb.2009.03.031. [DOI] [PubMed] [Google Scholar]
- Lee J.; Jung J.; Na K.; Heo P.; Hyun J. Polypeptide-Mediated Switchable Microarray of Bacteria. ACS Appl. Mater. Interfaces 2009, 1, 1359–1363. 10.1021/am9002364. [DOI] [PubMed] [Google Scholar]
- Hernandez-Garcia A.; Werten M. W. T.; Stuart M. C.; De Wolf F. A.; De Vries R. Coating of Single DNA Molecules by Genetically Engineered Protein Diblock Copolymers. Small 2012, 8, 3491–3501. 10.1002/smll.201200939. [DOI] [PubMed] [Google Scholar]
- Zheng T.; Perona Martínez F.; Storm I. M.; Rombouts W.; Sprakel J.; Schirhagl R.; De Vries R. Recombinant Protein Polymers for Colloidal Stabilization and Improvement of Cellular Uptake of Diamond Nanosensors. Anal. Chem. 2017, 89, 12812–12820. 10.1021/acs.analchem.7b03236. [DOI] [PubMed] [Google Scholar]
- Thyparambil A. A.; Wei Y.; Latour R. A. Determination of Peptide - Surface Adsorption Free Energy for Material Surfaces Not Conducive to SPR or QCM Using AFM. Langmuir 2012, 28, 5687–5694. 10.1021/la300315r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.-O.; Jackman J. A.; Mochizuki M.; Yoon B. K.; Hayashi T.; Cho N.-J. Correlating Single-Molecule and Ensemble-Average Measurements of Peptide Adsorption onto Different Inorganic Materials. Phys. Chem. Chem. Phys. 2016, 18, 14454–14459. 10.1039/c6cp01168c. [DOI] [PubMed] [Google Scholar]
- Krysiak S.; Liese S.; Netz R. R.; Hugel T. Peptide Desorption Kinetics from Single Molecule Force Spectroscopy Studies. J. Am. Chem. Soc. 2014, 136, 688–697. 10.1021/ja410278r. [DOI] [PubMed] [Google Scholar]
- Tamerler C.; Oren E. E.; Duman M.; Venkatasubramanian E.; Sarikaya M. Adsorption Kinetics of an Engineered Gold Binding Peptide by Surface Plasmon Resonance Spectroscopy and a Quartz Crystal Microbalance. Langmuir 2006, 22, 7712–7718. 10.1021/la0606897. [DOI] [PubMed] [Google Scholar]
- Ebner A.; Wildling L.; Gruber H. J.. Functionalization of AFM Tips and Supports for Molecular Recognition Force Spectroscopy and Recognition Imaging. Methods in Molecular Biology; Humana Press: New York, NY, 2019; Vol. 1886, pp 117–151. [DOI] [PubMed] [Google Scholar]
- Oesterhelt F.; Rief M.; Gaub H. E. Single Molecule Force Spectroscopy by AFM Indicates Helical Structure of Poly(Ethylene-Glycol) in Water. New J. Phys. 1999, 1, 6. 10.1088/1367-2630/1/1/006. [DOI] [Google Scholar]
- Friddle R. W.; Noy A.; De Yoreo J. J. Interpreting the Widespread Nonlinear Force Spectra of Intermolecular Bonds. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 13573–13578. 10.1073/pnas.1202946109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manohar S.; Jagota A. Sequence-Dependent Force Response during Peeling of Single-Stranded DNA from Graphite. Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys. 2010, 81, 021805. 10.1103/PhysRevE.81.021805. [DOI] [PubMed] [Google Scholar]
- Tang N. C.; Chilkoti A. Combinatorial Codon Scrambling Enables Scalable Gene Synthesis and Amplification of Repetitive Proteins. Nat. Mater. 2016, 15, 419–424. 10.1038/nmat4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel J. R.; MacKay J. A.; Quiroz F. G.; Chilkoti A. Recursive Directional Ligation by Plasmid Reconstruction Allows Rapid and Seamless Cloning of Oligomeric Genes. Biomacromolecules 2010, 11, 944–952. 10.1021/bm901387t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iler R. K.The Chemistry of Silica: Solubility, Polymerization, Colloid and Surface Properties and Biochemistry of Silica; Wiley, 1979. [Google Scholar]
- Abdelhamid M. A. A.; Ikeda T.; Motomura K.; Tanaka T.; Ishida T.; Hirota R.; Kuroda A. Application of Volcanic Ash Particles for Protein Affinity Purification with a Minimized Silica-Binding Tag. J. Biosci. Bioeng. 2016, 122, 633–638. 10.1016/j.jbiosc.2016.04.011. [DOI] [PubMed] [Google Scholar]
- Hassert R.; Pagel M.; Ming Z.; Häupl T.; Abel B.; Braun K.; Wiessler M.; Beck-Sickinger A. G. Biocompatible Silicon Surfaces through Orthogonal Click Chemistries and a High Affinity Silicon Oxide Binding Peptide. Bioconjugate Chem. 2012, 23, 2129–2137. 10.1021/bc3003875. [DOI] [PubMed] [Google Scholar]
- Gladytz A.; John T.; Gladytz T.; Hassert R.; Pagel M.; Risselada H. J.; Naumov S.; Beck-Sickinger A. G.; Abel B.; Friesner R. A.; et al. Peptides@mica: From Affinity to Adhesion Mechanism. Phys. Chem. Chem. Phys. 2016, 18, 23516–23527. 10.1039/C6CP03325C. [DOI] [PubMed] [Google Scholar]
- Thai C. K.; Dai H.; Sastry M. S. R.; Sarikaya M.; Schwartz D. T.; Baneyx F. o. Identification and Characterization of Cu2O- and ZnO-Binding Polypeptides by Escherichia Coli Cell Surface Display: Toward an Understanding of Metal Oxide Binding. Biotechnol. Bioeng. 2004, 87, 129–137. 10.1002/bit.20149. [DOI] [PubMed] [Google Scholar]
- Zernia S.; Frank R.; Weiße R. H.-J.; Jahnke H.-G.; Bellmann-Sickert K.; Prager A.; Abel B.; Sträter N.; Robitzki A.; Beck-Sickinger A. G. Surface-Binding Peptide Facilitates Electricity-Driven NADPH-Free Cytochrome P450 Catalysis. ChemCatChem 2018, 10, 525–530. 10.1002/cctc.201701810. [DOI] [Google Scholar]
- Malmsten M.; Linse P.; Cosgrove T. Adsorption of PEO-PPO-PEO Block Copolymers at Silica. Macromolecules 1992, 25, 2474–2481. 10.1021/ma00035a028. [DOI] [Google Scholar]
- Hnilova M.; So C. R.; Oren E. E.; Wilson B. R.; Kacar T.; Tamerler C.; Sarikaya M. Peptide-Directed Co-Assembly of Nanoprobes on Multimaterial Patterned Solid Surfaces. Soft Matter 2012, 8, 4327–4334. 10.1039/c2sm06426j. [DOI] [Google Scholar]
- Rimola A.; Sodupe M.; Ugliengo P. Affinity Scale for the Interaction of Amino Acids with Silica Surfaces. J. Phys. Chem. C 2009, 113, 5741–5750. 10.1021/jp811193f. [DOI] [Google Scholar]
- Meißner R. H.; Wei G.; Ciacchi L. C. Estimation of the Free Energy of Adsorption of a Polypeptide on Amorphous SiO2 from Molecular Dynamics Simulations and Force Spectroscopy Experiments. Soft Matter 2015, 11, 6254–6265. 10.1039/c5sm01444a. [DOI] [PubMed] [Google Scholar]
- Friddle R. W.; Battle K.; Trubetskoy V.; Tao J.; Salter E. A.; Moradian-Oldak J.; De Yoreo J. J.; Wierzbicki A. Single-Molecule Determination of the Face-Specific Adsorption of Amelogenin’s C-Terminus on Hydroxyapatite. Angew. Chem., Int. Ed. 2011, 50, 7541–7545. 10.1002/anie.201100181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter-Scherer V.; Cuellar-Camacho J. L.; Bhatia S.; Haag R.; Herrmann A.; Lauster D.; Rabe J. P. Force Spectroscopy Shows Dynamic Binding of Influenza Hemagglutinin and Neuraminidase to Sialic Acid. Biophys. J. 2019, 116, 1037–1048. 10.1016/j.bpj.2019.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfreundschuh M.; Alsteens D.; Wieneke R.; Zhang C.; Coughlin S. R.; Tampé R.; Kobilka B. K.; Müller D. J. Identifying and Quantifying Two Ligand-Binding Sites While Imaging Native Human Membrane Receptors by AFM. Nat. Commun. 2015, 6, 8857. 10.1038/ncomms9857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sand K. K.; Friddle R. W.; Deyoreo J. J. Quantifying the Free Energy Landscape between Polymers and Minerals. Sci. Rep. 2017, 7, 8663. 10.1038/s41598-017-09041-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J.; Battle K. C.; Pan H.; Salter E. A.; Chien Y.-C.; Wierzbicki A.; De Yoreo J. J. Energetic Basis for the Molecular-Scale Organization of Bone. Proc. Natl. Acad. Sci. U.S.A. 2015, 112, 326–331. 10.1073/pnas.1404481112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems L.; Van Westerveld L.; Roberts S.; Weitzhandler I.; Calcines Cruz C.; Hernandez-Garcia A.; Chilkoti A.; Mastrobattista E.; Van Der Oost J.; De Vries R. Nature of Amorphous Hydrophilic Block Affects Self-Assembly of an Artificial Viral Coat Polypeptide. Biomacromolecules 2019, 20, 3641–3647. 10.1021/acs.biomac.9b00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems L.; Roberts S.; Weitzhandler I.; Chilkoti A.; Mastrobattista E.; Van Der Oost J.; De Vries R. Inducible Fibril Formation of Silk-Elastin Diblocks. ACS Omega 2019, 4, 9135–9143. 10.1021/acsomega.9b01025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reviakine I.; Johannsmann D.; Richter R. P. Hearing What You Cannot See and Visualizing What You Hear: Interpreting Quartz Crystal Microbalance Data from Solvated Interfaces. Anal. Chem. 2011, 83, 8838–8848. 10.1021/ac201778h. [DOI] [PubMed] [Google Scholar]
- Storm I. M.; Kornreich M.; Hernandez-Garcia A.; Voets I. K.; Beck R.; Cohen Stuart M. A.; Leermakers F. A. M.; De Vries R. Liquid Crystals of Self-Assembled DNA Bottlebrushes. J. Phys. Chem. B 2015, 119, 4084–4092. 10.1021/jp511412t. [DOI] [PubMed] [Google Scholar]
- Visser E. W. A.; Yan J.; Van IJzendoorn L. J.; Prins M. W. J. Continuous Biomarker Monitoring by Particle Mobility Sensing with Single Molecule Resolution. Nat. Commun. 2018, 9, 1–10. 10.1038/s41467-018-04802-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubken R. M.; De Jong A. M.; Prins M. W. J. Multiplexed Continuous Biosensing by Single-Molecule Encoded Nanoswitches. Nano Lett. 2020, 20, 2296–2302. 10.1021/acs.nanolett.9b04561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J.; Van Smeden L.; Merkx M.; Zijlstra P.; Prins M. W. J. Continuous Small-Molecule Monitoring with a Digital Single-Particle Switch. ACS Sens. 2020, 5, 1168–1176. 10.1021/acssensors.0c00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagishima T.; Di Michele L.; Kotar J.; Eiser E. Diffusive Behaviour of PLL-PEG Coated Colloids on λ-DNA Brushes - Tuning Hydrophobicity. Soft Matter 2012, 8, 6792–6798. 10.1039/c2sm25296a. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.