

Abstract
Ketamine is an anesthetic, analgesic, and antidepressant whose secondary metabolite (2R,6R)-hydroxynorketamine (HNK) has N-methyl-d-aspartate-receptor-independent antidepressant activity in a rodent model. In humans, naltrexone attenuates its antidepressant effect, consistent with opioid pathway involvement. No detailed biophysical description is available of opioid receptor binding of ketamine or its metabolites. Using molecular dynamics simulations with free energy perturbation, we characterize the binding site and affinities of ketamine and metabolites in μ and κ opioid receptors, finding a profound effect of the protonation state. G-protein recruitment assays show that HNK is an inverse agonist, attenuated by naltrexone, in these receptors with IC50 values congruous with our simulations. Overall, our findings are consistent with opioid pathway involvement in ketamine function.
Keywords: Ketamine; norketamine; (2R,6R)-hydroxynorketamine; opioid receptors; molecular dynamics; free energy perturbation
Introduction
Part of the World Health Organization Model List of Essential Medicines, ketamine is administered primarily for its sedative and analgesic properties. One of its primary advantages in clinical medicine is hemodynamic stability compared to other common anesthetics such as propofol. It is also a rapidly acting antidepressant with striking efficacy particularly in severely depressed patients. In humans, pretreatment with naltrexone attenuates this antidepressant effect, suggesting the involvement of the opioid system.1 Opioid antagonists also attenuate the antidepressant effect in a rodent model.2
Ketamine is described as an N-methyl-d-aspartate (NMDA) receptor antagonist, binds pentameric ligand-gated ion channels,3,4 and binds and/or causes activation of various G-protein coupled receptors (GPCRs) including opioid and olfactory receptors.5−10 A functional role for opioid receptor binding is suggested by inhibition of ketamine’s effect on the mouse tail flick test by opioid antagonists.11 Further mouse studies showed that a greater antidepressant effect of R-ketamine than S-ketamine12 and the metabolite (2R,6R)-hydroxynorketamine (HNK) demonstrated antidepressant activity. The mGlu2 receptor and brain-derived neurotrophic factor are thought to underlie this effect,13,14 but a definitive molecular mechanism is not yet fully described.
Antidepressant effects might occur directly, through the binding of ketamine or its metabolites to GPCRs themselves, or indirectly, through modulation of endogenous opioid pathways. To evaluate the possibility of direct action on opioid receptors, using molecular dynamics (MD) simulations, we computationally characterized the binding of ketamine and its metabolites to μ and κ opioid receptors (MOR and KOR, respectively), determining both binding modes and affinities. Given the near-physiologic pKa of ketamine, protonation states of both the ligand and receptor were evaluated. We related these data to the results of G-protein recruitment assays. While neither ketamine nor norketamine had a significant effect on G-protein recruitment, HNK was an inverse agonist in both MOR and KOR, which is attenuated by naltrexone pretreatment. Consistent with and building upon previous work,1,6,7,13 our findings describe a biophysical underpinning for the opioid-receptor-dependent antidepressant sequelae of ketamine.
Results and Discussion
Ketamine Binds to Horse Spleen Apoferritin
As a validation of our methodology in a well-characterized protein, we first studied the interaction of S-ketamine with horse spleen apoferritin (HSAF). This 24-mer binds a variety of psychoactive molecules, such as propofol, isoflurane, sevoflurane, barbiturates, and the fluorescent general anesthetic 1-aminoanthracene (1-AMA), at an interfacial site between monomers similar to transmembrane binding sites in the GABAA receptor.15−19
In equilibrium MD simulations, both neutral and charged S-ketamine molecules placed in the interfacial binding site settled into a stable configuration in the site within 20 ns. Using free energy perturbation (FEP) MD, we calculated the binding affinities of these species.17 Including solvation, restraint, and charge correction energies, the dissociation constant KD calculated by FEP MD of neutral S-ketamine was 562 nM, and that of protonated S-ketamine was >1 M. Because the interfacial binding site is relatively hydrophobic, it was unsurprising that the positively charged species of S-ketamine had such low affinity. Residues within 5 Å of the neutral S-ketamine, at least 50% of the time, were L24, S27, Y28, L31, A55, and R59.
We determined the corresponding experimental binding affinity of S-ketamine by measuring the fluorescence change upon its displacement of 1-AMA. At pH 7.0, KD was 42 μM (95% CI = 22.4 to 78.8 μM, Hill slope = −0.8 ± 0.25; Supporting Figure 1).
As ligand charge states exist in equilibrium, in order to compare the computational predictions with experimental results, we determined the contribution of each charge state to the overall binding affinity. In the aqueous state at pH = 7.0, the proportion of protonated ketamine is 76%, because its pKa = 7.5.20 However, because in the bound state the effective pKa of functional groups can be shifted,21 we did not know a priori what proportion of bound ligands was protonated. We derived this from the binding affinity of each ligand species (eqs 4 and 5; full derivation of the method in the Supporting Information). Because the protonated form has a much lower affinity, the experimental KD of 42 μM must primarily reflect the neutral form. The fraction of protonated S-ketamine is 98.7%, calculated using eq 4 as well as the simplified eq 5. In this case, the ligand pKa would effectively be 8.87 in the bound state. These calculations for all ligands and receptors evaluated in this manuscript are shown in Supporting Table 3.
Predicted Opioid Receptor Binding Conformations of Ketamine and Metabolites
We used MD simulations to predict the conformations of bound ketamine as well as its metabolites norketamine and (2R,6R)-hydroxynorketamine (HNK) in MOR and KOR. We evaluated both neutral and +1 charged H6.52 in each receptor, as we anticipated that this positively charged residue in the binding pocket would affect the ligand affinity. There were therefore eight conditions for ketamine, eight for norketamine, and four for HNK (because we did not vary its enantiomeric state), for a total of 20 simulations.
As with HSAF, we placed a ligand molecule in the orthosteric binding pocket of each free receptor and refined each pose using equilibrium MD. We chose this binding pocket because it is well-described in crystal structures and includes residues that interact with opioid ligands (D3.32, Y3.33, M3.36, and H6.52), and alternative sites are not well-described. Docking of each ligand in the binding pocket revealed a large variety of possible binding modes with similar scores (see Supporting Figure 2), which suggested that a docked conformation would not provide a reliable starting point for simulation. We evaluated R and S enantiomers of ketamine and norketamine and varied the protonation state of all ligands (0 or +1 charge), as well as the protonation state of H6.52 in the binding pocket (H297 in MOR and H291 in KOR).
Each ligand arrived at a resting bound configuration within 30 ns (Figure 1b, Supporting Figure 3). This was expected given the relatively few internal degrees of freedom of these compact molecules. Supporting Table 2 lists residues within 6.5 Å of each ligand in 50% or more of trajectory frames; these are largely orthosteric binding pocket residues. Only protonated ligands are listed, because they had much higher binding affinities (described below).
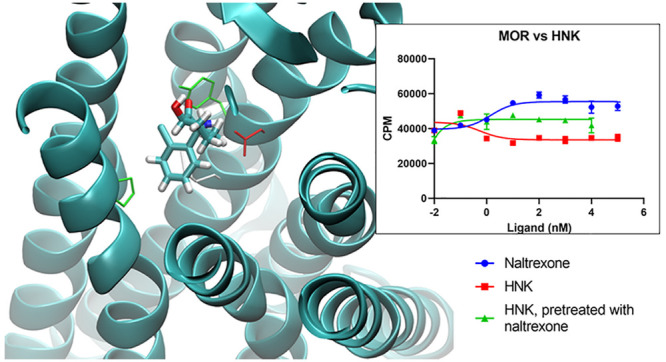
Figure 1.

(a) Key residue interactions of protonated HNK in MOR and KOR. In each case, the ligand is in the orthosteric binding pocket. Note the presence of D3.32 (with hydrogen bond) and H6.52. See Supporting Table 2 for comprehensive list of nearby residues. (b) Root mean square deviation (RMSD) of ligand over simulation time in equilibrium MD simulations prior to FEP, for HNK. Plots for all ligands as well as receptor pocket available in the Supporting Information. (c) ΔG vs lambda plots of ligand–protein decoupling FEP simulations for HNK, both neutral (left) and protonated (right), illustrating the smoothness of these curves, which are representative of all FEP simulations. Calculations for MOR and KOR as well as both binding pocket His protonation states are shown. Forward and backward legs of these interleaved double-wide sampled simulations are shown. More detailed plots, where each curve is labeled, are available in the Supporting Information.
Both enantiomers of ketamine interacted with similar subsets of residues in both MOR and KOR. In MOR, these included key interactions with D1473.32, Y1483.33, M1513.36, and H2976.52. D1473.32 and H2976.52 are key residues implicated in opioid binding. In KOR, both R- and S-ketamine interacted with D1383.32, Y1393.33, M1423.36, H2916.52, and W2876.48; the latter tryptophan is thought to interact with KOR agonists.22
R-Norketamine settled in the orthosteric binding pocket in MOR and KOR. MOR H297+ was the exception; the protonated R-norketamine settled into a separate binding pocket further from the lipid membrane, interacting primarily with transmembrane helices TM2 and TM7. In KOR, S-norketamine settled in a somewhat different orientation, interacting with L1012.46, A1042.49, D1052.50, and V1082.53 on TM2 but not D1383.32, Y1393.33, or H2916.52.
HNK bound the orthosteric binding pocket as well. Key interactions are included with D1473.32, Y1483.33, M1513.36, and H2976.52 in MOR and D1383.32, Y1393.33, M1423.36, and H2916.52 in KOR. In His+ receptors, HNK had more interactions with TM7 in both MOR and KOR (C7.38, A7.40, L7.41) than in the nonprotonated-His receptors.
The orthosteric binding pocket is water-exposed and can accommodate hydrophilic ligands (e.g., morphine at physiologic pH), though the vast majority of residues in the pocket are uncharged polar or hydrophobic; the charged residues are D2.50, D3.32, and H6.52. D3.32 interacted with the ligand in nearly all cases and would electrostatically stabilize a positive charge.
Predicted Binding Affinities of Ketamine and Metabolites to Opioid Receptors
Using double-decoupling FEP MD starting from the bound configurations determined above, we calculated the ligand binding affinity for each ligand–opioid receptor combination described above. This included decoupling charged species from the systems. Special attention was paid to mitigate the pitfalls of even state-of-the-art FEP MD for this difficult task. Electrostatic interactions of the charged ligand with charged residues in the binding pocket or with polar solvent have large interaction energy, and the final ΔG value will be the result of subtracting large numbers, so we used integrated double-wide sampling and multiple FEP runs to reduce this error. The interaction energy among charged periodic images resulting from decoupling of charged species causes overestimation of free energy change; we have used a standard box-size-dependent correction for this, described in the Methods section. Finally, for dynamics-based binding affinity calculations in general, protein conformational change upon binding may occur on too slow a time scale to be tractably sampled. Here, this would lead to systematic error in binding free energy predictions; to estimate convergence prior to FEP calculations, we quantified both ligand and binding pocket RMSD and bound-ligand rotational distributions (Supporting Figure 3).
We found, in general, that neutral ligands had low affinity (KD micromolar and above), but the protonated versions had high affinity (KD nanomolar and below) to both MOR and KOR (Figure 2 and Table 1). ΔG versus λ plots are shown in Figure 1 for HNK and in Supporting Figure 4 for all ligands. This result was reversed from the more hydrophobic binding site in HSAF and corresponded to the special case, discussed in the Methods section, of a rare but high-affinity ligand species.
Figure 2.

Bar plots of binding affinities of ketamine and metabolites with opioid receptors. These figures include the cost of desolvation from aqueous solution and binding to the receptor.
Table 1. Binding Free Energy (−ΔG) of Ketamine and Metabolites with GPCRs with Desolvation and Restraint Energies Includeda.
| |
neutral |
protonated |
|||
|---|---|---|---|---|---|
| –ΔG (kcal/mol) | KD (μM) | –ΔG (kcal/mol) | KD (nM) | ||
| S-Ketamine | |||||
| MOR | WT | 7.1 (6.5–7.9) | 7.9 (2.2–20) | 13.9 (12.6–15.2) | 0.096 (0.011–0.83) |
| H297+ | 7.6 (7.3–7.8) | 3.6 (2.4–5.3) | 10.5 (9.8–11.3) | 26 (7.5–89) | |
| KOR | WT | 2.6 (2.6–2.6) | 13 000 (13 000–13 000) | 14.1 (13.5–14.6) | 0.072 (0.029–0.18) |
| H291+ | 4.9 (4.3–5.4) | 310 (130–740) | 8.4 (7.7–9.0) | 910 (310–2700) | |
| R-Ketamine | |||||
| MOR | WT | 7.8 (7.4–8.2) | 2.4 (1.3–4.5) | 12.0 (11.4–12.6) | 2.1 (0.75–5.6) |
| H297+ | 4.6 (3.3–5.8) | 490 (60–4000) | 10.8 (9.9–11.7) | 16 (3.6–72) | |
| KOR | WT | 8.2 (7.9–8.6) | 1.1 (0.59–2.1) | 10.4 (9.2–11.5) | 32 (4.7–220) |
| H291+ | 2.9 (2.9–3.0) | 7500 (7100–8000) | 9.8 (9.1–10.5) | 88 (26–290) | |
| S-Norketamine | |||||
| MOR | WT | 2.7 (2.6–2.9) | 11 000 (8600–13 000) | 13.4 (12.3–14.5) | 0.22 (0.036–1.3) |
| H297+ | 3.8 (3.7–3.9) | 1900 (1600–2200) | 9.6 (8.6–10.6) | 120 (22–640) | |
| KOR | WT | 5.2 (4.8–5.7) | 160 (84–320) | 9.5 (9.1–10.0) | 130 (62–270) |
| H291+ | 6.0 (5.7–6.2) | 49 (32–76) | 6.5 (6.1–6.8) | 21 000 (12 000–37 000) | |
| R-Norketamine | |||||
| MOR | WT | 8.3 (8.1–8.4) | 1.1 (0.84–1.5) | 16.4 (16.1–16.8) | 0.0014 (0.000 81–0.0024) |
| H297+ | 9.2 (9.1–9.3) | 0.22 (0.19–0.25) | 4.4 (3.3–5.5) | 690 000 (110 000–4 100 000) | |
| KOR | WT | 2.0 (0.8–3.2) | 37 000 (5300–250 000) | 19.0 (18.8–19.2) | 0.000 020 (0.000 014–0.000 028) |
| H291+ | 4.9 (4.3–5.5) | 290 (110–740) | 16.5 (15.5–17.6) | 0.0012 (0.000 21–0.0062) | |
| HNK | |||||
| MOR | WT | 3.8 (1.9–5.7) | 1800 (75–45 000) | 11.4 (9.5–12.7) | 5.5 (0.71–140) |
| H297+ | 2.5 (1.7–3.2) | 16 000 (5300–64 000) | 8.3 (7.4–9.2) | 1000 (240–4600) | |
| KOR | WT | 7.4 (7.3–7.5) | 4.5 (4.0–5.1) | 12.8 (11.7–13.9) | 0.57 (0.10–3.6) |
| H291+ | 1.5 (0.6–2.4) | 86 000 (20 000–370 000) | 15.6 (13.9–16.7) | 0.0052 (0.000 84–0.090) | |
KD values are calculated as described in Methods from respective ΔG values by FEP MD and correspond to K0 and K1 in equation 2 and . All energies are favorable for binding. Ranges represent minimum and maximum values obtained in both directions of FEP calculation, using interleaved double-wide sampling. WT = without protonation of orthosteric histidine; H291+ and H297+ refer to protonated (+1 charge) histidine (Ballesteros-Weinstein 6.52).
For all protonated ligands, binding affinity was decreased when the orthosteric binding pocket histidine had +1 charge adjacent to the ligand (MOR H2976.52+ and KOR H2916.52+), as would be expected from electrostatic repulsion. There was no clear pattern to the effect for neutral ligands. Overall, the magnitude of the change was much lower than that due to ligand charge.
The protonated S-ketamine had higher affinity to both MOR and KOR than protonated R-ketamine unless H6.52 had +1 charge. Protonated R-norketamine had greater affinity for KOR than protonated S-norketamine. Protonated HNK had greater affinity for KOR than MOR, but when this histidine was protonated in MOR, HNK affinity decreased; in KOR, it increased. This was attributable to migration of the positively charged HNK away from the protonated histidine in KOR, such that it was able to settle into a favorable configuration (see Supporting Table 1).
In certain cases (S- and R-norketamine, HNK in MOR and S-ketamine in KOR), with a +1 charge, H6.52 the ligand interacted more with TM2 (Supporting Table 2), so the calculated affinities for these systems are not directly comparable. The protonated R-norketamine had a lower binding affinity than neutral R-norketamine to MOR H297+ because of migration to its alternative binding site near TM2 and TM7. Of note, the negatively charged D2.50 is in TM2, which would stabilize a positively charged ligand. HNK had stronger affinity in KOR but weaker affinity in MOR as a result of the protonated histidine, possibly related to increased interactions with TM2.
Existing experimental proxies for ketamine binding affinity to opioid receptors are limited to Ki values from radioligand inhibition experiments. Representative Ki values for S- and R-ketamine, respectively, are 11 and 28 μM in MOR and 24 and 100 μM in KOR.23 These are dependent on the hot ligand:7,9,10,24 in this case, [3H]DAMGO for MOR and [3H]U69593 for KOR. If we make the nontrivial approximation that Ki can be substituted for KD, our calculated affinities were in several cases higher than mathematically possible to reconcile with these experimental values (Supporting Table 3). Of note, the ketamine radioligand inhibition experiments were done using brain homogenates or CHO cells. This suggests the possibility that ketamine, as a nonspecific binder, would bind other sites in the experimental preparation and be relatively unavailable for binding to opioid receptors, leading to experimental underestimation of the binding affinity relative to our theoretical estimates.
Signal Transducer Recruitment Assays of Ketamine
We measured G-protein recruitment by MOR and KOR as a function of ligand concentration using [35S]GTPγS assays (see Methods section). No clear effect due to ketamine was observed at clinically relevant concentrations. There was a suggestion of inverse agonism by both enantiomers of norketamine, but this was unclear (Supporting Figure 5). R-Ketamine appeared to induce G-protein recruitment in MOR starting at roughly 10 μM ligand concentration. S-Ketamine appeared to induce G-protein recruitment starting at roughly 25 μM concentration, although we could not determine EC50 values; this is because plateau activation would have required well in excess of physiologically reasonable concentrations.
We tested whether ketamine may be a neutral antagonist for the G-protein pathway. In MOR, adding progressively increasing concentrations of R-ketamine to MOR pretreated with 60 or 200 nM methadone, a known MOR agonist, showed inhibition of G-protein recruitment at high micromolar concentrations. We did not increase the ketamine concentration enough to determine IC50, as these would be physiologically irrelevant concentrations. In KOR, R-ketamine trended toward inhibition of G-protein recruitment induced by either 50 or 250 nM nalbuphine, a known KOR agonist, again only at very high concentrations (Supporting Figure 6).
Given the predicted affinities and minimal activity in these assays, we would have expected to observe competitive neutral antagonism by ketamine at lower concentrations. We can again speculate that other binding partners for ketamine present in membrane preparations limited the ketamine available for competition. Also, opioid receptors may dimerize and distort the kinetics of competitive inhibition. Finally, there might be a high-affinity secondary bound configuration in the same orthosteric pocket, where ketamine binding does not displace methadone or nalbuphine. Some of us have predicted a similar situation with another small ligands in another class A GPCR, the α-2A adrenergic receptor.25 We concluded that ketamine might act as a neutral antagonist with respect to G-protein recruitment activity in both MOR and KOR; this activity is physiologically unimportant, or the experiment is confounded by the above factors.
Separately, we quantified β-arrestin recruitment as a function of S-ketamine concentration, for both MOR and KOR (see the Methods section). No significant change in β-arrestin recruitment was observed at micromolar or lower concentrations (Supporting Figure 7). We concluded that S-ketamine is an insignificant contributor to β-arrestin recruitment activity in both MOR and KOR.
As with radioligand inhibition assays, it is possible that [35S]GTPγS and β-arrestin assays may underestimate G-protein recruitment activity. These were conducted with cell-free membrane preparations, where the opioid receptors in question were amplified, but other membrane components are present in minimal quantities. Because ketamine and metabolites are not highly specific binders, the high-affinity charged ligand species may have relatively low availability at the binding pocket, resulting in less activation than would be expected from their affinities.
Inverse Agonism of HNK in MOR and KOR
In [35S]GTPγS assays with increasing ligand concentrations, HNK was an inverse agonist in both MOR and KOR (Figure 3). Fitting to a sigmoidal dose–response curve with a standard Hill slope of −1.0, the IC50 value for MOR was 0.56 nM (95% CI 0.41 to 4.7 nM), and for KOR, the IC50 was 2.1 × 10–5 nM. The latter extremely low IC50 value did not allow for a precise determination of confidence interval given the chosen ligand dilutions. For both MOR and KOR, the greatest degree of inhibition of G-protein recruitment was reached at 10 nM HNK; increased HNK concentrations did not increase inhibition.
Figure 3.
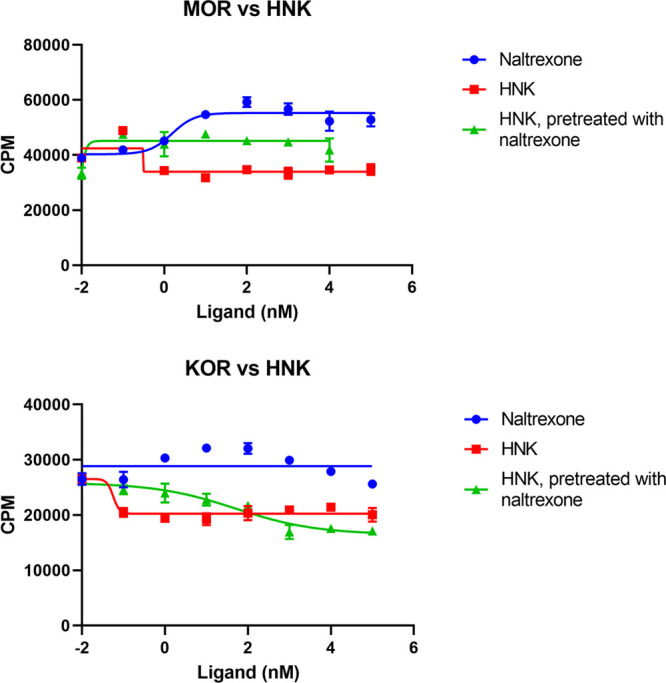
Sigmoidal dose–response inhibition curves from [35S]GTPγS assays, including standard deviations. Increasing concentrations of HNK depress activation in both MOR and KOR, suggesting an inverse agonist effect. This is attenuated by pretreatment with naltrexone in both MOR and KOR.
Given clinical evidence that pretreatment with naltrexone attenuates the antidepressant effect of ketamine,1 we hypothesized that naltrexone would attenuate HNK inverse agonism. In repeat [35S]GTPγS assays with naltrexone pretreatment (1 μM in MOR and 10 μM in KOR), the inverse agonist effect of HNK was obliterated in MOR, and in KOR, the IC50 was increased to 110 nM (Figure 3).
With the caveat that they reflect different biochemical processes, we could consider these IC50 values as rough proxies for HNK dissociation constants. The KD values calculated by FEP MD represented lower affinity than suggested by non-naltrexone IC50 values even for the high-affinity +1 charge HNK: KD values were 5.5 nM for MOR and 0.57 nM for KOR. This made the calculation of the proportion of protonated bound ligand mathematically impossible; the experimental value must be between the two FEP-calculated values. Despite this, the MOR KD and IC50 values are within an order of magnitude, and the KOR IC50 is low but imprecise in this range; therefore, it is a reasonable hypothesis that the experimental and theoretical values are compatible. Notably, FEP MD predicted a higher affinity of HNK to KOR than to MOR, which was borne out in the experiment.
Conclusions
In this study, we revealed molecular details of the bound configurations of ketamine and its metabolites to opioid receptors. We showed that the metabolite HNK has a high calculated affinity in the orthosteric binding site and is an inverse agonist for G-protein recruitment in both MOR and KOR in vitro and that this effect is inhibited by naltrexone pretreatment. This provides biochemical context to previous evidence suggesting that ketamine’s antidepressant effect is attenuated by naltrexone pretreatment. The IC50 values for HNK–MOR/KOR inverse agonism are in the low-nanomolar range, consistent with the relatively low doses of ketamine used in humans for depression: An example regimen is 0.1–0.75 mg/kg administered in an infusion over 40 min.26
Our data are consistent with the hypothesis that HNK is the primary ketamine-related compound with antidepressant activity and implicate G-protein recruitment by MOR and/or KOR as part of the molecular mechanism. Ostensibly, HNK binding allosterically modulates the conformation of TM5-7, in turn affecting G-protein recruitment, as proposed in other class A GPCRs.27−29 We speculate that these helices are stabilized, reducing constitutive G-protein recruitment and resulting in the inverse agonist effect. While ketamine and norketamine also have favorable binding affinities, we did not show that they have significant effects on G-protein recruitment.
This study cannot confirm whether HNK itself acts directly as an antidepressant or is part of a larger indirect mechanism involving the opioid system. There is controversy on this topic, with conflicting results in mouse models.13,34−36 We speculate that mouse models may not capture the same pathology as that underlying human depression. We propose that interaction of HNK with opioid receptors partly underlies the antidepressant activity, complementing previously described mechanisms involving mGlu2 and BDNF.
We did not address potential structural contributors to G-protein recruitment that could differentiate the neutral activity of ketamine from the inverse agonism of HNK. The patterns of ligand–residue interactions that confers agonist, antagonist, or inverse agonist activity in opioid receptors are as yet not fully described,32 though we envision future work attacking this problem for HNK. We do not consider here the possibilities of GPCR oligomerization or opioid receptor modulation indirectly by peptides. The mechanism of G-protein recruitment is an active area of investigation, with, for example, a recent description of the structure of an agonist bound KOR with Gi.33 We cannot yet exclude β-arrestin-dependent activity in MOR and KOR from ligands other than S-ketamine. In addition, there is evidence in rodent models that S-norketamine has antidepressant activity,30,31 which is intriguing given the suggestion of inverse agonism for this compound in the present study. Further investigation of norketamine could be considered.
Through the exploration of the relationship of affinity values with fractions of protonated ligand, we have provided a way to check how well experimental KD values reconcile with calculated affinities in this type of system. When the measured KD is correct and calculated affinities for each protonation state are both correct and reflect energetically plausible macrostates (e.g., the charge configuration of the binding pocket exists as simulated with ligand bound), a plausible distribution of ligand protonation states (and effective pKa) is yielded. This method gave a reasonable result in HSAF where we were able to measure KD.
Overall, our results support and build upon the finding by Williams et al. that implicates the opioid system in ketamine’s antidepressant activity by showing that pretreatment with naltrexone attenuates this effect.1 More work is required in this area. There may be benefit to further study in cell-based systems to address these questions, particularly in more clearly defining the cellular-level consequences of inverse agonism by HNK. We expect that continued study of both the molecular interactions of HNK as well as its potential use as an antidepressant will benefit human health.
Methods
We evaluated ketamine as well as its metabolites norketamine and (2R,6R)-hydroxynorketamine. We included R- and S-enantiomers for ketamine and norketamine and their protonated states.
Force Field Parameters
Molecular mechanics (MM) force field parameters for each ligand molecule, both neutral and charged versions, were required for MD simulations. No high-quality MM parameters with the CHARMM force field37,38 were available. We initially used the ParamChem web server to choose parameters for each ligand using the CGenFF force field,39,40 but this approach did not produce satisfactory parametrizations with sufficiently low ParamChem-reported penalty scores. Therefore, we manually refined those parameters. We employed QM methods, following the Force Field Took Kit workflow41 with modifications,42 including dihedral interaction corrections as necessary.
Quantum mechanics geometry optimizations and energy calculations were done with Gaussian09 Rev. E01 (Gaussian, Inc., Wallingford, CT, USA), at the B3LYP/6-31G** level of theory and basis set, for parity with parametrization approaches commonly used with the CHARMM force field. These were used to provide an initial minimized structure as well as “gold standard” energies with which the MM parameters could be fit.
Enthalpies of vaporization (sublimation) for each ligand were calculated as ΔHvap = ⟨Ugas⟩ – ⟨Ucondensed⟩/N + RT, where each U is the respective potential energy, angle brackets denote averages, N is the number of molecules, R is the gas constant, and T is the temperature. The potential energy averages were taken from simulations of a single ligand molecule in gas phase (2 ns) and of 300 ligand molecules in a periodic box (10 ns, NPT ensemble). Densities were calculated from the condensed phase simulations. We are unaware of experimental correlates of these parameters in the literature, but the heats of vaporization correlate with those calculated with the Joback method43 and adjusted to the simulation temperature. Desolvation energies (i.e., from aqueous to gas phase) were calculated by FEP MD as the energy of decoupling a single ligand molecule from a box of TIP3P water using the protocol described below. All these values are listed in Supporting Table 1.
Experimental Determination of Ketamine–HSAF Binding Affinity
In order to relate our approach to a model experimental system, we determined the binding affinity of S-ketamine to horse spleen apoferritin (HSAF). Fluorescence competition assays were performed as previously reported.19 The binding affinity of S-ketamine was determined by equilibrium binding of increasing amounts of S-ketamine with constant concentrations of 1-aminoanthracene (1-AMA) and HSAF. HSAF (0.3 μM) in 20 mM phosphate and 130 mM NaCl buffer at pH 7 was equilibrated with 11 μM 1-AMA. Each sample was made by adding a given concentration of S-ketamine (1 μM to 4 mM) in the same buffer to obtain 0.5 μL of total volume, mixing, and immediately measuring on a Shimadzu RF-5301PC spectrofluorophotometer (Shimadzu Corp., Kyoto, Japan). The fluorescence spectrum of 1-AMA was determined using light at 380 nm for excitation and recording emission at 510 nm. Excitation and emission slit widths were 5 and 10 nm, respectively. Fluorescence data were collected from 400 to 720 nm. All samples were run in triplicate. A fluorescence signal decrease from 1-AMA was observed as S-ketamine was titrated, indicating competition for binding. Data and error analysis were carried out using Prism 5.0 (GraphPad Software, La Jolla, CA, USA). Fluorescence was corrected by simple subtractions of baseline values for 1-AMA and HSAF. Fluorescence intensity versus concentration data were fit to variable slope Hill models. The Cheng-Prusoff equation was used to correct for the presence of the 1-AMA competitor.
S-Ketamine and (2R,6R)-hydroxynorketamine were obtained from Sigma-Aldrich (K1884, SML1873). R-Ketamine was obtained from Cayman Chemical (16519). The R- and S-norketamine were obtained from Tocris (5996, 6112).
G-Protein Activation ([35S]GTPγS) Assay
In this assay, levels of radioactive 35S-γ-GTP were measured after incorporation in the recruited G-protein complex.44 MOR membrane preparation was obtained from EMD Millipore (cat. number HTS101M) and PerkinElmer (cat. number 611055840UA), and KOR membrane preparation was obtained from PerkinElmer (cat. number ES-542-M400UA). The ligand was incubated with this membrane preparation. A G-protein preparation containing Gα, Gβ, Gγ, and radiolabeled GTP were added so that recruitment could occur. This reaction was then quenched, and free radiolabeled GTP was washed away; the remaining amount of radioactive 35S-γ-GTP bound to a Gα subunit was measured by its radioactivity in a scintillation counter as described previously.45 All experiments were run two to three times and, within each experiment, in duplicate at each data point. For each curve, one representative experiment is presented. Results in the form of scintillation counts were analyzed using GraphPad Prism 8.0 (GraphPad Software, San Diego, CA), and EC50/IC50 values were calculated by fit to a sigmoidal dose–response curve with a standard Hill slope of 1.0 or −1.0.
β-Arrestin Recruitment Assay
The previously described protocol was followed.46 In brief, HTLA cells, an HEK293 cell line stably expressing a tTA-dependent luciferase reporter and a β-arrestin2-TEV fusion gene, were maintained in DMEM with 10% FBS, 2 μg/mL puromycin, and 100 μg/mL hygromycin B in a humidified atmosphere at 37 °C in 5% CO2. For the transfection, cells were plated to 50–80% confluency and transfected with OPRM1 (for MOR) or OPRK1 (for KOR) using FuGENE HD (Promega Corp., Madison, WI, USA). The next day, transfected cells were trypsinized and transferred into poly-l-lysine coated and rinsed 96-well, white, clear-bottom cell culture plates in 50 μL of DMEM containing 1% dialyzed FBS and 1X penicillin/streptomycin at a density of 20 000 cells/well. After 7 h, 6X drug solutions were prepared in assay buffer (1X HBSS with 20 mM HEPES, pH 7.4), and 10 μL was added to each well. The following day, 60 μL of Bright Glo (Promega Corp.), diluted 4-fold, was added to each well and incubated 5 min at room temperature in the dark before luminescence was measured. Results in the form of luminescence units were analyzed using GraphPad Prism.
Simulations
We conducted equilibrium MD simulations of ligands bound to HSAF and μ and κ opioid receptors (MOR and KOR, respectively) derived from crystal structures47−49 (Protein Data Bank: 3F32, 5C1M, 4DJH). Missing residues were modeled with MODELER ModLoop.50 We used the inactive form of KOR available at the time of the study. The more recent nanobody-stabilized active form of KOR has a similar binding pocket, differing primarily in the displacement of TM6 and TM7 helices.48
We used CHARMM-GUI51 to embed each protein in a 70:30 ratio 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine:cholesterol lipid bilayer, with surrounding TIP3P water, 0.15 M NaCl, and excess ions for electroneutrality. The crystallographic cholesterol in MOR was preserved. We used the CHARMM36 force field with our ligand parameters. A single ligand molecule was placed in each orthosteric binding site, consistent with the location of the orthosteric binding pocket in crystal structures as well as docking calculations with AutoDock Vina,52 to provide the starting configuration for subsequent MD simulations. The docking calculations, using flexible side chains in the orthosteric pocket, yielded a wide variety of ligand configurations distributed throughout the pocket (see Supporting Figure 2). As side chain flexibility would be important to the resting configuration of these relatively small and simple ligands in a way that the simplified potential function of docking may not capture, we elected to use MD for refinement of the ligand coordinates. All simulations were conducted with NAMD 2.12 or 2.13 molecular dynamics software,53 using GPUs where possible. Minimization and equilibration with solute restraints preceded production simulation in the NPT ensemble. Each ligand was simulated in the binding site for at least 30 ns in equilibrium production MD simulation.
In order to assess whether these simulations arrived at a resting bound configuration, we calculated the root-mean-square deviation (RMSD) of the ligands in the binding pockets (Supporting Figure 3a) by first fitting the receptor with respect to the backbone of the first production simulation frame to remove global rotation and translation and then calculating the RMSD of the ligand heavy atoms with respect to the first production simulation frame. The RMSD of the binding pocket atoms was similarly calculated (Supporting Figure 3b). The distribution of the rotation angles of the ligands in the pocket was calculated as well (Supporting Figure 3c).
Using
FEP MD, we then calculated the standard binding affinity
of each ligand to the receptor as the negative of the energy of dissociation
from the receptor into bulk water. FEP MD has been successfully used
for calculating accurate ligand affinities in the class A GPCR orthosteric
pocket.54−56 There were 30–50 windows per run for each
leg, with 0.4 ns of equilibration plus 2 ns of production simulation
per window and λ schedules modified for more sampling in regions
with large-magnitude energy changes; each FEP run included a minimum
of 62 ns of production simulation, for a total of ∼3.2 μs
in FEP MD simulations including replicates. Interleaved double-wide
sampling was used, with sampling in both forward and backward directions
at each λ. To facilitate parallel execution, the FEP windows
were separated into groups of five, each starting from the same configuration.
The first window in each group used 1 ns rather than 0.4 ns of equilibration
time. Multiple runs were conducted for some systems; all results were
averaged. A flat-bottomed spherical volume restraint of radius 5 Å
was applied to the ligand center-of-mass to reduce the sampling of
very low probability states by the partly decoupled ligand.55,57 The thermodynamic process was therefore as follows: decoupling from
the receptor with the restraint, plus a correction for the restraint,
plus the energy of recoupling the ligand to water. The free energy
cost of imposing the restraint was calculated as 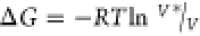 , where V* is the volume
of the sphere, V is the effective volume occupied
by a single ligand at 1 M concentration (∼1660 Å3), R is the gas constant, and T is the temperature. For the 5 Å sphere this penalty was 0.69
kcal/mol.
, where V* is the volume
of the sphere, V is the effective volume occupied
by a single ligand at 1 M concentration (∼1660 Å3), R is the gas constant, and T is the temperature. For the 5 Å sphere this penalty was 0.69
kcal/mol.
Correction for Particle Mesh Ewald (PME) Summation across Nonzero-Charge Systems During FEP MD
The FEP MD calculations of charged ligands by definition included non-neutral intermediate states, which shifts the electrostatic potential baseline due to the use of periodic boundary conditions and Ewald summation, and the resulting error in the computed free energy change scales as the inverse of the box length.58−60 This is a significant source of error, even in the setting of charged species having large-magnitude binding free energies when the binding pocket includes charged residues, as is the case in MOR and KOR. We calculated this relevant correction using the formula in Hummer et al.60 as −2.837297/L, where L is the box length and the equation is expressed in atomic units. For protein simulations, which were noncubic, L was chosen as the length of a cube of the same volume as the simulation. These corrections were included in the ΔG and KD values reported.
Binding Model for Two Ligand Species
Because each binding affinity calculation applies to a single ligand (and protein) protonation state, we must separately determine the proportions of these states to determine their contribution to the overall binding affinity. By the same token, given both an experimental dissociation constant KD as well as a computational state-specific KD, we can predict the proportions of the protonation states. A brief description follows; the full derivation is available in the Supporting Information.
We take the probability of the occupancy of a protein binding site to be
| 1 |
where [L] is the concentration of the ligand (including all protonation states) and KD is the dissociation constant (note that when [L] = KD then P = 0.5). Including two protonation states, we define the occupancy of the site
| 2 |
where each fj is the proportion of systems in protonation state j, Kj is the ligand dissociation constant in protonation state j, and f0 + f1 = 1. The third term in eq 2 excludes the possibility of both ligands occupying the same site. Defining the fraction-weighted dissociation constants λ0 = K0/f0 and λ1 = K1/f1 yields
| 3 |
Let us call the dissociation constant across both protonation states K′D, such that when [L] = K′D, Peither = 1/2. Setting the left-hand side of eq 3 to 1/2 and solving for [L] yields
| 4 |
In the special case where the protonated form
has an extremely low affinity, so λ1 ≫ λ0, then 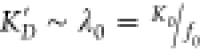 . Similarly, if the protonated form binds
much more strongly than the neutral form, λ1 ≫
λ0 and
. Similarly, if the protonated form binds
much more strongly than the neutral form, λ1 ≫
λ0 and 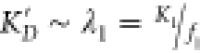 . So, to solve for f1 in these special
cases, we can use simplified expressions:
. So, to solve for f1 in these special
cases, we can use simplified expressions:
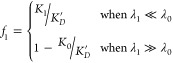 |
5 |
It is important to note that rare forms of the ligand may still determine the observed K′D if they have much higher affinities than those of the common form. In such a case, the dissociation constant observed from ligand titration would significantly overestimate the actual dissociation constant for the high-affinity species.
Acknowledgments
T.T.J. was supported by the National Institutes of Health postdoctoral training grant 5T32GM112596 and the Foundation for Anesthesia Education and Research (MRTG-BS-Joseph). High-performance computing resources and support were provided by the Office of Advanced Research Computing and RDI2 at Rutgers University and National Science Foundation XSEDE (award TG-MCB170017 to T.T.J.). R.L. was supported by 1R01GM111421, National Institutes of Health. A.Y. and L.Z. were supported by the intramural research program, National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.0c00741.
Detailed derivation of the binding model for two ligand species, densities and enthalpies of vaporization/sublimation, residues, ligand pKa, fluorescence curves, docked conformations, RMSDs, free energy perturbation molecular dynamics energy plots, G-protein activation assays, Competition [35S]GTPgS assays, and β-arrestin recruitment assays (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Williams N. R.; Heifets B. D.; Blasey C.; Sudheimer K.; Pannu J.; Pankow H.; Hawkins J.; Birnbaum J.; Lyons D. M.; Rodriguez C. I.; Schatzberg A. F. (2018) Attenuation of Antidepressant Effects of Ketamine by Opioid Receptor Antagonism. Am. J. Psychiatry 175, 1205. 10.1176/appi.ajp.2018.18020138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein M. E.; Chandra J.; Sheriff S.; Malinow R. (2020) Opioid System Is Necessary but Not Sufficient for Antidepressive Actions of Ketamine in Rodents. Proc. Natl. Acad. Sci. U. S. A. 117 (5), 2656–2662. 10.1073/pnas.1916570117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ion B. F.; Wells M. M.; Chen Q.; Xu Y.; Tang P. (2017) Ketamine Inhibition of the Pentameric Ligand-Gated Ion Channel GLIC. Biophys. J. 113 (3), 605–612. 10.1016/j.bpj.2017.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J.; Chen Q.; Willenbring D.; Mowrey D.; Kong X.-P.; Cohen A.; Divito C. B.; Xu Y.; Tang P. (2012) Structure of the Pentameric Ligand-Gated Ion Channel GLIC Bound with Anesthetic Ketamine. Struct. London Engl. 1993 20 (9), 1463–1469. 10.1016/j.str.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan R. K.; Rose M. A.; Duggan K. A. (1997) Evidence for Direct Interaction of Ketamine with Alpha 1- and Beta 2-Adrenoceptors. Clin. Exp. Pharmacol. Physiol. 24 (12), 923–926. 10.1111/j.1440-1681.1997.tb02720.x. [DOI] [PubMed] [Google Scholar]
- Gupta A.; Devi L. A.; Gomes I. (2011) Potentiation of μ-Opioid Receptor-Mediated Signaling by Ketamine: Opioid-Ketamine Interactions. J. Neurochem. 119 (2), 294–302. 10.1111/j.1471-4159.2011.07361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota K.; Okawa H.; Appadu B. L.; Grandy D. K.; Devi L. A.; Lambert D. G. (1999) Stereoselective Interaction of Ketamine with Recombinant Mu, Kappa, and Delta Opioid Receptors Expressed in Chinese Hamster Ovary Cells. Anesthesiology 90 (1), 174–182. 10.1097/00000542-199901000-00023. [DOI] [PubMed] [Google Scholar]
- Ho J.; Perez-Aguilar J. M.; Gao L.; Saven J. G.; Matsunami H.; Eckenhoff R. G. (2015) Molecular Recognition of Ketamine by a Subset of Olfactory G Protein-Coupled Receptors. Sci. Signal 8 (370), ra33-ra33 10.1126/scisignal.2005912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D. J.; Bouchal R. L.; deSanctis C. A.; Monroe P. J.; Amedro J. B.; Perrotti J. M.; Crisp T. (1987) Properties of the Interaction between Ketamine and Opiate Binding Sites in Vivo and in Vitro. Neuropharmacology 26 (9), 1253–1260. 10.1016/0028-3908(87)90084-0. [DOI] [PubMed] [Google Scholar]
- Smith D. J.; Pekoe G. M.; Martin L. L.; Coalgate B. (1980) The Interaction of Ketamine with the Opiate Receptor. Life Sci. 26 (10), 789–795. 10.1016/0024-3205(80)90285-4. [DOI] [PubMed] [Google Scholar]
- Pacheco D. d. F.; Romero T. R. L.; Duarte I. D. G. (2014) Central Antinociception Induced by Ketamine Is Mediated by Endogenous Opioids and μ- and δ-Opioid Receptors. Brain Res. 1562, 69–75. 10.1016/j.brainres.2014.03.026. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Li S.; Hashimoto K. (2014) R (−)-Ketamine Shows Greater Potency and Longer Lasting Antidepressant Effects than S (+)-Ketamine. Pharmacol., Biochem. Behav. 116, 137–141. 10.1016/j.pbb.2013.11.033. [DOI] [PubMed] [Google Scholar]
- Zanos P.; Highland J. N.; Stewart B. W.; Georgiou P.; Jenne C. E.; Lovett J.; Morris P. J.; Thomas C. J.; Moaddel R.; Zarate C. A.; Gould T. D. (2019) (2R,6R)-Hydroxynorketamine Exerts MGlu2 Receptor-Dependent Antidepressant Actions. Proc. Natl. Acad. Sci. U. S. A. 116 (13), 6441–6450. 10.1073/pnas.1819540116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto K.; Fogaça M. V.; Liu R.-J.; Duman C.; Kato T.; Li X.-Y.; Duman R. S. (2019) Activity-Dependent Brain-Derived Neurotrophic Factor Signaling Is Required for the Antidepressant Actions of (2R,6R)-Hydroxynorketamine. Proc. Natl. Acad. Sci. U. S. A. 116 (1), 297–302. 10.1073/pnas.1814709116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckenhoff R. G.; Xi J.; Shimaoka M.; Bhattacharji A.; Covarrubias M.; Dailey W. P. (2010) Azi-Isoflurane, a Photolabel Analog of the Commonly Used Inhaled General Anesthetic Isoflurane. ACS Chem. Neurosci. 1 (2), 139–145. 10.1021/cn900014m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woll K. A.; Peng W.; Liang Q.; Zhi L.; Jacobs J. A.; Maciunas L.; Bhanu N.; Garcia B. A.; Covarrubias M.; Loll P. J.; Dailey W. P.; Eckenhoff R. G. (2017) Photoaffinity Ligand for the Inhalational Anesthetic Sevoflurane Allows Mechanistic Insight into Potassium Channel Modulation. ACS Chem. Biol. 12 (5), 1353–1362. 10.1021/acschembio.7b00222. [DOI] [PubMed] [Google Scholar]
- Vedula L. S.; Brannigan G.; Economou N. J.; Xi J.; Hall M. A.; Liu R.; Rossi M. J.; Dailey W. P.; Grasty K. C.; Klein M. L.; Eckenhoff R. G.; Loll P. J. (2009) A Unitary Anesthetic Binding Site at High Resolution. J. Biol. Chem. 284 (36), 24176–24184. 10.1074/jbc.M109.017814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley S.; Vedula L. S.; Bu W.; Meng Q. C.; Xi J.; Liu R.; Eckenhoff R. G.; Loll P. J. (2012) Recognition of Anesthetic Barbiturates by a Protein Binding Site: A High Resolution Structural Analysis. PLoS One 7 (2), e32070 10.1371/journal.pone.0032070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butts C. A.; Xi J.; Brannigan G.; Saad A. A.; Venkatachalan S. P.; Pearce R. A.; Klein M. L.; Eckenhoff R. G.; Dmochowski I. J. (2009) Identification of a Fluorescent General Anesthetic, 1-Aminoanthracene. Proc. Natl. Acad. Sci. U. S. A. 106 (16), 6501–6506. 10.1073/pnas.0810590106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen M. L.; Trevor A. J. (1974) On the Cerebral Accumulation of Ketamine and the Relationship Between Metabolism of the Drug and Its Pharmacological Effects. J. Pharmacol. Exp. Ther. 189 (2), 351–358. [PubMed] [Google Scholar]
- Onufriev A. V.; Alexov E. (2013) Protonation and PK Changes in Protein-Ligand Binding. Q. Rev. Biophys. 46 (2), 181–209. 10.1017/S0033583513000024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardy E.; Mosier P. D.; Frankowski K. J.; Wu H.; Katritch V.; Westkaemper R. B.; Aubé J.; Stevens R. C.; Roth B. L. (2013) Chemotype-Selective Modes of Action of κ-Opioid Receptor Agonists. J. Biol. Chem. 288 (48), 34470–34483. 10.1074/jbc.M113.515668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hustveit O.; Maurset A.; Oye I. (1995) Interaction of the Chiral Forms of Ketamine with Opioid, Phencyclidine, Sigma and Muscarinic Receptors. Pharmacol. Toxicol. 77 (6), 355–359. 10.1111/j.1600-0773.1995.tb01041.x. [DOI] [PubMed] [Google Scholar]
- La Regina A.; Petrillo P.; Sbacchi M.; Tavani A. (1988) Interaction of U-69,593 with Mu-, Alpha- and Kappa-Opioid Binding Sites and Its Analgesic and Intestinal Effects in Rats. Life Sci. 42 (3), 293–301. 10.1016/0024-3205(88)90638-8. [DOI] [PubMed] [Google Scholar]
- McKinstry-Wu A. R.; Woll K. A.; Joseph T. T.; Bu W.; White E. R.; Bhanu N. V.; Garcia B. A.; Brannigan G.; Dailey W. P.; Eckenhoff R. G. (2019) Azi-Medetomidine: Synthesis and Characterization of a Novel A2 Adrenergic Photoaffinity Ligand. ACS Chem. Neurosci. 10 (11), 4716–4728. 10.1021/acschemneuro.9b00484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade C. (2017) Ketamine for Depression, 4: In What Dose, at What Rate, by What Route, for How Long, and at What Frequency?. J. Clin. Psychiatry 78 (7), e852 10.4088/JCP.17f11738. [DOI] [PubMed] [Google Scholar]
- Rasmussen S. G. F.; DeVree B. T.; Zou Y.; Kruse A. C.; Chung K. Y.; Kobilka T. S.; Thian F. S.; Chae P. S.; Pardon E.; Calinski D.; Mathiesen J. M.; Shah S. T. A.; Lyons J. A.; Caffrey M.; Gellman S. H.; Steyaert J.; Skiniotis G.; Weis W. I.; Sunahara R. K.; Kobilka B. K. (2011) Crystal Structure of the B2 Adrenergic Receptor-Gs Protein Complex. Nature 477 (7366), 549–555. 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehl A.; Hu H.; Maeda S.; Zhang Y.; Qu Q.; Paggi J. M.; Latorraca N. R.; Hilger D.; Dawson R.; Matile H.; Schertler G. F. X.; Granier S.; Weis W. I.; Dror R. O.; Manglik A.; Skiniotis G.; Kobilka B. K. (2018) Structure of the M-Opioid Receptor-G i Protein Complex. Nature 558 (7711), 547. 10.1038/s41586-018-0219-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suomivuori C.-M.; Latorraca N. R.; Wingler L. M.; Eismann S.; King M. C.; Kleinhenz A. L. W.; Skiba M. A.; Staus D. P.; Kruse A. C.; Lefkowitz R. J.; Dror R. O. (2020) Molecular Mechanism of Biased Signaling in a Prototypical G Protein-Coupled Receptor. Science 367 (6480), 881–887. 10.1126/science.aaz0326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C.; Kobayashi S.; Nakao K.; Dong C.; Han M.; Qu Y.; Ren Q.; Zhang J.; Ma M.; Toki H.; Yamaguchi J.; Chaki S.; Shirayama Y.; Nakazawa K.; Manabe T.; Hashimoto K. (2018) AMPA Receptor Activation-Independent Antidepressant Actions of Ketamine Metabolite (S)-Norketamine. Biol. Psychiatry 84 (8), 591–600. 10.1016/j.biopsych.2018.05.007. [DOI] [PubMed] [Google Scholar]
- Yokoyama R.; Higuchi M.; Tanabe W.; Tsukada S.; Naito M.; Yamaguchi T.; Chen L.; Kasai A.; Seiriki K.; Nakazawa T.; Nakagawa S.; Hashimoto K.; Hashimoto H.; Ago Y. (2020) (S)-Norketamine and (2S,6S)-Hydroxynorketamine Exert Potent Antidepressant-like Effects in a Chronic Corticosterone-Induced Mouse Model of Depression. Pharmacol., Biochem. Behav. 191, 172876. 10.1016/j.pbb.2020.172876. [DOI] [PubMed] [Google Scholar]
- Cui X.; Yeliseev A.; Liu R. (2013) Ligand Interaction, Binding Site and G Protein Activation of the Mu Opioid Receptor. Eur. J. Pharmacol. 702 (1–3), 309–315. 10.1016/j.ejphar.2013.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mafi A.; Kim S.-K.; Goddard W. A. (2020) The Atomistic Level Structure for the Activated Human κ-Opioid Receptor Bound to the Full Gi Protein and the MP1104 Agonist. Proc. Natl. Acad. Sci. U. S. A. 117 (11), 5836–5843. 10.1073/pnas.1910006117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi J.; Toki H.; Qu Y.; Yang C.; Koike H.; Hashimoto K.; Mizuno-Yasuhira A.; Chaki S. (2018) (2 R,6 R)-Hydroxynorketamine Is Not Essential for the Antidepressant Actions of (R)-Ketamine in Mice. Neuropsychopharmacology 43 (9), 1900–1907. 10.1038/s41386-018-0084-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanos P.; Moaddel R.; Morris P. J.; Georgiou P.; Fischell J.; Elmer G. I.; Alkondon M.; Yuan P.; Pribut H. J.; Singh N. S.; Dossou K. S. S.; Fang Y.; Huang X.-P.; Mayo C. L.; Wainer I. W.; Albuquerque E. X.; Thompson S. M.; Thomas C. J.; Zarate C. A.; Gould T. D. (2016) NMDAR Inhibition-Independent Antidepressant Actions of Ketamine Metabolites. Nature 533 (7604), 481–486. 10.1038/nature17998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumsden E. W.; Troppoli T. A.; Myers S. J.; Zanos P.; Aracava Y.; Kehr J.; Lovett J.; Kim S.; Wang F.-H.; Schmidt S.; Jenne C. E.; Yuan P.; Morris P. J.; Thomas C. J.; Zarate C. A.; Moaddel R.; Traynelis S. F.; Pereira E. F. R.; Thompson S. M.; Albuquerque E. X.; Gould T. D. (2019) Antidepressant-Relevant Concentrations of the Ketamine Metabolite (2R,6R)-Hydroxynorketamine Do Not Block NMDA Receptor Function. Proc. Natl. Acad. Sci. U. S. A. 116 (11), 5160–5169. 10.1073/pnas.1816071116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best R. B.; Zhu X.; Shim J.; Lopes P. E. M.; Mittal J.; Feig M.; MacKerell A. D. (2012) Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain X1 and X2 Dihedral Angles. J. Chem. Theory Comput. 8 (9), 3257–3273. 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauda J. B.; Venable R. M.; Freites J. A.; O’Connor J. W.; Tobias D. J.; Mondragon-Ramirez C.; Vorobyov I.; MacKerell A. D.; Pastor R. W. (2010) Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 114 (23), 7830–7843. 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; MacKerell A. D. (2012) Automation of the CHARMM General Force Field (CGenFF) I: Bond Perception and Atom Typing. J. Chem. Inf. Model. 52 (12), 3144–3154. 10.1021/ci300363c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; Raman E. P.; MacKerell A. D. (2012) Automation of the CHARMM General Force Field (CGenFF) II: Assignment of Bonded Parameters and Partial Atomic Charges. J. Chem. Inf. Model. 52 (12), 3155–3168. 10.1021/ci3003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayne C. G.; Saam J.; Schulten K.; Tajkhorshid E.; Gumbart J. C. (2013) Rapid Parameterization of Small Molecules Using the Force Field Toolkit. J. Comput. Chem. 34 (32), 2757–2770. 10.1002/jcc.23422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph T. T.; Hénin J.; Eckenhoff R. G.; Dmochowski I. J. (2018) Molecular Mechanics Parameterization of Anesthetic Molecules. Methods in Enzymology 602, 61–76. 10.1016/bs.mie.2018.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joback K. G.; Reid R. C. (1987) Estimation of Pure-Component Properties from Group-Contributions. Chem. Eng. Commun. 57 (1–6), 233–243. 10.1080/00986448708960487. [DOI] [Google Scholar]
- Harrison C.; Traynor J. R. (2003) The [35S]GTPgammaS Binding Assay: Approaches and Applications in Pharmacology. Life Sci. 74 (4), 489–508. 10.1016/j.lfs.2003.07.005. [DOI] [PubMed] [Google Scholar]
- Liu R.; Huang X.-P.; Yeliseev A.; Xi J.; Roth B. L. (2014) Novel Molecular Targets of Dezocine and Their Clinical Implications. Anesthesiol. J. Am. Soc. Anesthesiol. 120 (3), 714–723. 10.1097/ALN.0000000000000076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroeze W. K.; Sassano M. F.; Huang X.-P.; Lansu K.; McCorvy J. D.; Giguère P. M.; Sciaky N.; Roth B. L. (2015) PRESTO-Tango as an Open-Source Resource for Interrogation of the Druggable Human GPCRome. Nat. Struct. Mol. Biol. 22 (5), 362–369. 10.1038/nsmb.3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W.; Manglik A.; Venkatakrishnan A. J.; Laeremans T.; Feinberg E. N.; Sanborn A. L.; Kato H. E.; Livingston K. E.; Thorsen T. S.; Kling R. C.; Granier S.; Gmeiner P.; Husbands S. M.; Traynor J. R.; Weis W. I.; Steyaert J.; Dror R. O.; Kobilka B. K. (2015) Structural Insights into M-Opioid Receptor Activation. Nature 524 (7565), 315–321. 10.1038/nature14886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che T.; Majumdar S.; Zaidi S. A.; Ondachi P.; McCorvy J. D.; Wang S.; Mosier P. D.; Uprety R.; Vardy E.; Krumm B. E.; Han G. W.; Lee M.-Y.; Pardon E.; Steyaert J.; Huang X.-P.; Strachan R. T.; Tribo A. R.; Pasternak G. W.; Carroll F. I.; Stevens R. C.; Cherezov V.; Katritch V.; Wacker D.; Roth B. L. (2018) Structure of the Nanobody-Stabilized Active State of the Kappa Opioid Receptor. Cell 172 (1), 55–67. e15. 10.1016/j.cell.2017.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D.; Wang S.; McCorvy J. D.; Betz R. M.; Venkatakrishnan A. J.; Levit A.; Lansu K.; Schools Z. L.; Che T.; Nichols D. E.; Shoichet B. K.; Dror R. O.; Roth B. L. (2017) Crystal Structure of an LSD-Bound Human Serotonin Receptor. Cell 168 (3), 377–389. e12. 10.1016/j.cell.2016.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiser A.; Sali A. (2003) ModLoop: Automated Modeling of Loops in Protein Structures. Bioinformatics 19 (18), 2500–2501. 10.1093/bioinformatics/btg362. [DOI] [PubMed] [Google Scholar]
- Jo S.; Kim T.; Iyer V. G.; Im W. (2008) CHARMM-GUI: A Web-Based Graphical User Interface for CHARMM. J. Comput. Chem. 29 (11), 1859–1865. 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. (2010) AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 31 (2), 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kalé L.; Schulten K. (2005) Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 26 (16), 1781–1802. 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenselink E. B.; Louvel J.; Forti A. F.; van Veldhoven J. P. D.; de Vries H.; Mulder-Krieger T.; McRobb F. M.; Negri A.; Goose J.; Abel R.; van Vlijmen H. W. T.; Wang L.; Harder E.; Sherman W.; Ijzerman A. P.; Beuming T. (2016) Predicting Binding Affinities for GPCR Ligands Using Free-Energy Perturbation. ACS Omega 1 (2), 293–304. 10.1021/acsomega.6b00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Deng Y.; Roux B. (2006) Absolute Binding Free Energy Calculations Using Molecular Dynamics Simulations with Restraining Potentials. Biophys. J. 91 (8), 2798–814. 10.1529/biophysj.106.084301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deflorian F.; Perez-Benito L.; Lenselink E. B.; Congreve M.; van Vlijmen H. W. T.; Mason J. S.; Graaf de C.; Tresadern G. (2020) Accurate Prediction of GPCR Ligand Binding Affinity with Free Energy Perturbation. J. Chem. Inf. Model. 60 (11), 5563–5579. 10.1021/acs.jcim.0c00449. [DOI] [PubMed] [Google Scholar]
- Salari R.; Joseph T.; Lohia R.; Hénin J.; Brannigan G. (2018) A Streamlined, General Approach for Computing Ligand Binding Free Energies and Its Application to GPCR-Bound Cholesterol. J. Chem. Theory Comput. 14, 6560. 10.1021/acs.jctc.8b00447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.-L.; Aleksandrov A.; Simonson T.; Roux B. (2014) An Overview of Electrostatic Free Energy Computations for Solutions and Proteins. J. Chem. Theory Comput. 10 (7), 2690–2709. 10.1021/ct500195p. [DOI] [PubMed] [Google Scholar]
- Simonson T.; Roux B. (2016) Concepts and Protocols for Electrostatic Free Energies. Mol. Simul. 42 (13), 1090–1101. 10.1080/08927022.2015.1121544. [DOI] [Google Scholar]
- Hummer G.; Pratt L. R.; García A. E. (1996) Free Energy of Ionic Hydration. J. Phys. Chem. 100 (4), 1206–1215. 10.1021/jp951011v. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.