

Abstract
Acid–base catalysis, which involves one or more proton transfer reactions, is a chemical mechanism commonly employed by many enzymes. The molecular basis for catalysis is often derived from structures determined at the optimal pH for enzyme activity. However, direct observation of protons from experimental structures is quite difficult; thus, a complete mechanistic description for most enzymes remains lacking. Dihydrofolate reductase (DHFR) exemplifies general acid–base catalysis, requiring hydride transfer and protonation of its substrate, DHF, to form the product, tetrahydrofolate (THF). Previous X-ray and neutron crystal structures coupled with theoretical calculations have proposed that solvent mediates the protonation step. However, visualization of a proton transfer has been elusive. Based on a 2.1 Å resolution neutron structure of a pseudo-Michaelis complex of E. coli DHFR determined at acidic pH, we report the direct observation of the catalytic proton and its parent solvent molecule. Comparison of X-ray and neutron structures elucidated at acidic and neutral pH reveals dampened dynamics at acidic pH, even for the regulatory Met20 loop. Guided by the structures and calculations, we propose a mechanism where dynamics are crucial for solvent entry and protonation of substrate. This mechanism invokes the release of a sole proton from a hydronium (H3O+) ion, its pathway through a narrow channel that sterically hinders the passage of water, and the ultimate protonation of DHF at the N5 atom.
Keywords: neutron diffraction, enzyme mechanism, deuteron, solvent, pH-dependent, dynamics
Introduction
Enzymes undergoing acid–base catalysis typically, where protons are transferred, show bell-shaped pH profiles of activity, with minimal activity observed at acidic and alkaline pH, flanking a pH optimum where the enzyme is most active. A simplistic model of the bell-shaped curve interprets the rising and falling inflection points to approximate two pKa values associated with catalytic residues, although in reality it can be more complex. The molecular basis for catalysis is often derived from crystal structures or NMR structures determined close to the optimal pH, sometimes augmented with atomistic simulations using both classical force fields and quantum chemistry methods. These structures provide useful information about the locations and motions of catalytic residues, solvent, ligands, and metal cofactors associated with catalysis. However, it is difficult to visualize catalytically important protons using X-ray crystallography because the single electron of hydrogen scatters X-rays weakly and protons do not scatter X-rays at all.1 Using neutrons instead of X-ray photons in the diffraction experiment helps to overcome this limitation, as neutrons scatter from atomic nuclei.2,3 Therefore, neutron crystallography is an ideal tool for studying the mechanisms of enzymes that employ acid–base catalysis.4−7
E. coli dihydrofolate reductase (ecDHFR) serves as a classic model of acid–base catalysis. DHFR catalyzes the NADPH-dependent reduction of 7,8-dihydrofolate (DHF) to 5,6,7,8-tetrahydrofolate (THF) for nucleotide and amino acid synthesis during cell proliferation.8−10 During the reaction, the N5 atom of DHF is protonated while its C6—C7 bond is reduced due to hydride transfer from NADPH. The source of the proton for reduction of N5 is unknown, and its position has never been visualized. As DHFR is an important drug target in certain cancers8,9 and rheumatoid arthritis11 as well as fungal12,13 and bacterial14 infections, a complete mechanistic understanding is a prerequisite for designing catalytically relevant drug molecules.10,12,13 The catalytic mechanism has been experimentally investigated through enzymology,15−18 biophysical studies,16,19 NMR,20 isotope labeling,18,21,22 electrical field measurements,23 and X-ray crystallography.9,10 Furthermore, numerous molecular dynamic (MD) studies have been integral to probing all aspects of the catalytic mechanism, including the role of coupled networks of residues and protein motions24,25 and the phenomenon of hydrogen tunneling for the hydride transfer step.26,27 Most importantly to this work are the computational studies linking Met20 loop conformations, solvent entry into the active site, and substrate protonation.28−30 However, the atomic details of DHF reduction, including the source and pathway of the proton linked to DHF reduction, have not been experimentally observed.
A conserved acidic residue (Asp27 in the E. coli enzyme) has been proposed to accept a proton from the N3 of DHF and transfer it to N5 through O4 tautomerization from the keto to the enol form.31,32 This mechanism appears unlikely due to the unfavorable proton transfer geometry between O4 and N5.28,33 The presence of both Asp27 and a bound catalytic mimic complex (DHF and NADP+) significantly raises the pKa value of the N5 atom of DHF from 2.6 to 6.5, the pH at which DHFR has maximal activity.19,29 This pKa value closely matches that of the hydride transfer step18 and likely describes the concentration of N5-protonated DHF substrate available. In an ecDHFR–folate–NADP+ ternary crystal structure (PDB entry 1RA2), a water molecule has been modeled within hydrogen-bonding distance to the N5 atom.10 The active site loop, known as the Met20 loop, is in an open conformation. Thus, a nearby solvent molecule could access the N5 atom as long as the active site loop containing Met20 adopts a conformation that is transiently at least partially open.9,28,29 When the Met20 loop is in the closed conformation, the pKa of the N5 atom is predicted to be elevated and favorable for protonation,18,29,30,34 and this catalytic pKa shift would also be relevant for hydride transfer. Therefore, loop dynamics is critical for catalysis: subtle loop opening could allow solvent entrance to the active site and subsequent closing accompanied by narrowing to necessitate direct protonation of N5. Foundational molecular dynamics simulations by Shrimpton and Allemann revealed that, in a Michaelis complex, water could access the N5 atom for protonation, and subsequently, the Met20 side chain would block the active site.28 Thus, high-resolution views of solvent structure and protonation states are needed to verify this as the potential mechanism for proton donation in DHFR.
Indeed, considerable effort has been made to visualize the protonated structures of the catalytic site of the inhibited complex of ecDHFR bound to methotrexate (MTX) (PDB 2INQ) and, more recently, the pseudo-Michaelis complex ecDHFR–folate–NADP+ (PDB 4PDJ) using neutron crystallography.35−37 Neither of these structures allowed us to visualize catalytic protons. Furthermore, deuterium isotope experiments conducted at extreme alkaline pH suggest pH-dependent alterations in the catalytic mechanism of ecDHFR.18 This prompted us to determine neutron diffraction structures spanning the kinetically characterized pH range of DHFR activity. We chose to study the ecDHFR–folate–NADP+ complex structure at lower pH so that protons relevant to the catalytic mechanism may be more easily observed as there may exist clear nuclear density for them, and they will have higher occupancies. Recently, this strategy was applied to a glycoside hydrolase and HIV-1 protease, where neutron structures across a range of pH enabled the observation of isolated protons in the active site and allowed the pKa determination of catalytic acidic residues.6,7
In this study, neutron diffraction experiments were conducted on the ecDHFR–folate–NADP+ complex at pH 4.5, which is ∼2 pH units below the pH optimum of the ecDHFR38 and well below the pH (7.0) of our previously determined pseudo-Michaelis structure. This acidic pH results in an ∼300-fold elevated concentration of free protons, increasing the probability of visualizing the elusive proton postulated to protonate the N5 atom of DHF. Indeed, by determining the neutron structure of the acidic pH pseudo-Michaelis complex (ecDHFR–folate–NADP+) at 2.1 Å resolution, the catalytic proton required for protonation of the substrate was observed for the first time. Moreover, a room temperature X-ray structure of the same ternary complex was determined at 1.65 Å resolution, allowing joint X-ray and neutron (XN) refinement. The combined X-ray and neutron diffraction data allow us to analyze the protonation states in the active site, compare pH-mediated changes between the acidic and neutral pH structures, model regulatory loop dynamics associated with ecDHFR activity, and propose a pathway for substrate protonation at N5, a critical step in the catalytic mechanism of this highly conserved enzyme. Additionally, quantum mechanics/molecular mechanics (QM/MM) calculations on a ternary complex model using the joint XN structure provide further support of this mechanism, indicating that progression from the bare proton state to protonation of N5 is barrierless and energetically favorable. Further, the calculations indicate a distortion in DHF to receive the proton that would be hindered in the DHF mimic and thus the basis, in part, for its capture of the bare proton state.
Materials and Methods
Neutron Crystallography
The crystal was initially screened for diffraction quality using a broad-bandpass Laue configuration using neutrons from 2.8 to 10 Å at the IMAGINE instrument at the High Flux Isotope Reactor (HFIR) at Oak Ridge National Laboratory.39−41 The 2.1 Å resolution neutron diffraction data were collected at 291 K on the monochromatic macromolecular neutron diffractometer BioDiff at the FRM II research reactor at the Heinz Maier-Leibnitz Zentrum (MLZ). This instrument is equipped with a cylindrical neutron image-plate detector.42 The neutron wavelength was set to 2.66 Å using a pyrolytic graphite monochromator (PG002). Two hundred eight frames of 0.3° oscillation and 50 min exposure time were recorded. The diffraction data were indexed and integrated using DENZO and then scaled with SCALEPACK.43,44 The 1.4 mm3 crystal of E. coli DHFR was in complex with folate and NADP+ at pH 4.5. This crystal was initially crystallized in the reservoir solution (18% PEG 400, 100 mM MnCl2, 20 mM imidazole at pH 7.0).36 The crystal quality was improved by the microseeding technique and reached the maximum size by the macroseeding technique with the decreased concentration of PEG 400 to 15%. After that, this crystal was sealed in a quartz capillary and vapor diffused for 1 year against the same reservoir solution made by 100% D2O. Before data collection, it was vapor diffused against the low pH solution (15% PEG 400, 100 mM MnCl2, 100 mM NaAc at pH 4.5) for 1 week. Hydrogen and its isotope deuterium possess neutron cross sections that allow these atoms to be located in nuclear density maps even at modest resolutions (∼2.5 Å).2,3 However, hydrogen has a negative neutron scattering length (−3.74 fm) while deuterium has a positive scattering length (+6.67 fm); thus, crystals that are to be used for neutron diffraction oftentimes undergo a period of hydrogen–deuterium exchange (HDX) prior to the experiment. Deuterium atoms stand out as strong positive peaks in difference nuclear density maps, yet negatively contoured nuclear density maps can be used to visualize hydrogen remaining in the model (nonexchanged hydrogens). A 1.65 Å resolution room temperature X-ray diffraction data set was collected using a smaller crystal on a Rigaku HighFlux HomeLab instrument equipped with a MicroMax-007 HF X-ray generator and Osmic VariMax optics. The diffraction images were obtained using an R-Axis IV++ image-plate detector. Diffraction data were integrated and scaled using the HKL3000 software suite indicating no appreciable radiation damage.43 Crystallographic data collection statistics are provided in Table S1.
Joint X-ray/Neutron Refinement
Because both data sets were collected at the same temperature with the same space group and almost identical unit cell dimensions, iterative joint X-ray and neutron (XN) refinement was performed using the program Phenix.refine,45 followed by model building. Atomic positions and individual atomic displacement parameters (B-factors) of all atoms were refined with stereochemical restraints, and occupancies of hydrogen/deuterium atoms at chemically exchangeable positions were refined and constrained to sum to unity. The refined Rwork and Rfree are 0.177 and 0.219, respectively (Table S1). To verify the identity of the deuteron (“D1”) modeled into the nuclear difference density peak in the active site near DOD47, we performed occupancy refinement against only the neutron diffraction data with a fixed B-factor for this atom at 30 Å2.
Due to poor nuclear density, we modeled the Met20 side chain in the neutron structure based on the position of one of the alternate conformers observed in the complementary electron density maps. This conformer was chosen as it does not form a close contact with the DOD47 and does not overlap with the position of the deuteron, as would be observed for the other Met20 conformer. Thus, the positioning of the side chain in the neutron structure is inferred, even though it is guided by the X-ray structure.
To compare the structures at pH 4.5 and pH 7.0 at higher resolution, we also refined their X-ray structures using Phenix.refine. After refinement, the models were validated using the PDB_REDO web server.46 The refined model without alternate conformations against the 1.65 Å resolution data at pH4.5 has the Rwork and Rfree values of 0.154 and 0.178, respectively. The model without alternate conformations refined against the 1.60 Å resolution data at pH7.0 has the Rwork and Rfree values of 0.165 and 0.187, respectively. We further performed ensemble refinement to compare the dynamics of both structures in different pH environments.47 All structural statistics are listed in Table S1.
Structural Analysis
Structural alignment was performed using the program Superpose incorporated in the CCP4 program suite.48 The distance difference matrix of Cα atoms generated from the X-ray structures at pH 4.5 and pH 7.0 was generated using the program DDMP.49 B factors of both X-ray structures were analyzed using the program Baverage in the CCP4 suite.48 All structural figures were created with Pymol (version 2.3.3).50
Molecular Dynamics Simulations and QM/MM Calculations with the Low-pH XN Structure
The starting point for the simulations was the pH 4.5 ecDHFR–folate–NADP+ XN crystal structure. The cocrystallized folate was modeled as dihydrofolate (DHF, N5 atom unprotonated), and NADP+ was modeled as NADPH by making the appropriate atom type reassignments and protonation changes. The unbound proton (technically a deuteron but referred to as proton from here) was modeled as a Li+ ion since CHARMM MM parameters are available for this ion, and it is of a similar van der Waals (vdW) radius, thus serving as a reasonable mimic. All water molecules identified in the XN structure were retained in their crystallographic coordinates. Bulk solvent surrounded the entire ternary complex in a cube with the size being the largest dimension of the protein plus 10 Å. Counterions (32 K+ and 22 Cl– ions) were added to the model to correspond to a 0.15 M concentration (physiological ionic strength) and an overall electrically neutral model. The final model contained 30 438 atoms. The all36 CHARMM force field51 was used for all moieties in the model.
MD simulations were primarily used to obtain an equilibrated bulk solvent phase surrounding the solute (enzyme, substrate, and cofactor) with minimal changes to the solute structure compared to the XN structure at low pH. Simulations were performed with the CHARMM/OpenMM interface51 in version c42b1 using periodic boundary conditions, particle mesh Ewald for long-range electrostatic interactions, and a Langevin thermostat (© = friction coefficient = 5) to maintain the temperature at 298 K. Harmonic restraints of 10.0 kcal mol–1 Å–2 were placed on all non-hydrogen atoms of the solute during the first 1 ns of the simulation, which used a 2 fs time step. The simulation was continued without restraints for another 5 ns with a 2 fs time step. The Li+ ion remained within 3.5 Å of the coordinates from the XN structure, and thus a snapshot from the end of the simulation was used for further investigation using QM/MM methods. The final snapshot was geometry optimized using the steepest descent algorithm for 1000 steps in order to remove the kinetic energy in the model.
In order to facilitate QM/MM calculations using DFTB3, DFT, and wave function-based methods, a spherical boundary model was constructed by deleting all atoms 30.0 Å away from the unbound proton without breaking any covalent bonds. This construction step retained a solvent shell around the entire enzyme, substrate, and cofactor. Subsequently, all atoms further than 20.0 Å from the unbound proton were kept fixed, and all other atoms were unrestrained. The QM region included the side chains of Met20 and Asp27, the dihydrofolate molecule, the nicotinamide moiety of NADPH, the water molecule closest to the unbound proton plus the proton (the hydronium ion), and the water molecule in contact with the hydronium ion. Two hydrogen link atoms were placed along the Cα–Cα axis at a distance of 1.09 Å from the Cα atom for the amino acid side chains. The third link atom was placed along the second C–C bond from the nicotinamide moiety. A total of 109 atoms were represented by the DFTB3 method.52 This structure was geometry optimized with the DFTB3/MM hybrid potential until the average gradient was less than 0.0001 kcal mol–1 Å–1 using the adopted basis Newton–Raphson algorithm. The reaction coordinate for the proton transfer reaction is the mass-weighted distance difference between the oxygen atom of the adjacent water (the “hydronium”) to the unbound proton and the N5 of DHF to the unbound proton. Each point on the potential energy profile was geometry optimized for at least 200 steps using the ABNR algorithm with a restraining force on the reaction coordinate set to 400 kcal mol–1 Å–1. We also estimated the proton affinity to the N5 atom of both DHF and folate (the “mimic” in the XN structure). The proton affinities of both molecules were calculated by geometry optimizing both molecules at the spin-component-scaled SCS-MP2/def2-SVPD level and then reoptimizing them with a proton added to N5 (see Figure S2). The differences in electronic energy between the neutral molecule and the protonated one result in an estimate of the proton affinity of each molecule.
The QM region was also modeled with spin-component-scaled second-order Møller–Plesset (SCS-MP2)53 method and the range-separated wB97X-D DFT method.54 Both methods used the def2-SVPD basis set55 with the def2/J56 auxiliary basis set using ORCA57 through the pDynamo program.58 The models were all geometry optimized without any restraints.
For the MD simulations comparing dynamics and RMSF between the pH 4.5 and 7.0 XN structures, for the lower pH model, the bare proton binding near N5-DHF has not been included since inclusion would likely require ad hoc restraints to the position of the bare proton when using classical force fields. Inclusion of this proton and possibly others represented in the surrounding solvent of future studies may provide further insight into the differential dynamics.
Data Deposition
Coordinates and structure factor files for all DHFR ternary complex structures described in the paper have been deposited into the Protein Data Bank with the following accession codes: neutron (7D6G), joint-refined XN at pH 4.5 (7D3Z), joint-refined XN at pH 7.0 (7D4L), ensemble-refined X-ray structure at pH 4.5 (7D49), and ensemble-refined X-ray structure at pH 7.0 (7D4X).
Results
Neutron and X-ray Structures of ecDHFR–Folate–NADP+ at pH 4.5
Neutron diffraction data extending to 2.1 Å resolution were collected at 291 K from a crystal of an ecDHFR–folate–NADP+ ternary complex at pH 4.5 (pD 4.9) (Table S1). In this experiment, catalytic hydrogens and other exchangeable protons were visualized as D atoms due to H/D exchange (see Materials and Methods). A 1.65 Å resolution X-ray diffraction data set was also collected using a smaller crystal to conduct joint X-ray/neutron refinement. Both structures have been refined to acceptable stereochemistry. The final model was refined to an Rfree of 0.219 for the neutron data and 0.178 for the X-ray data, respectively (Table S1).
We had previously determined the X-ray structure of this complex at pH 7.0 to 1.6 Å resolution.37 This now allows us to compare DHFR ternary complex structures at two different pH values. The backbone structures at pH 4.5 and pH 7.0 are similar, with a Cα atom RMSD of 0.104 Å. After superposition, the most significantly different regions are the Met20 regulatory loop (residues 9–24) and the F–G loop (residues 116–132) (Figure 1A). The difference distance matrix of Cα atoms shows that the Met20 loop and the F–G loop are shifted slightly closer (∼0.5 Å) to the region ranging from P39 to V75 (Figure 1B). The coordinate error estimates for the structures are 0.12 Å (pH 4.5) and 0.16 Å (pH 7.0), respectively. Thus, this shift is significant, with the acidic environment resulting in the active site cleft to be narrower at pH 4.5 compared to pH 7.0 and concomitantly shifting the pKa of the bound ligand.29
Figure 1.

Structural comparison between the X-ray structures at pH 4.5 and pH 7.0. (A) The Cα difference after superposition shows the most significant changes occur in the Met20 and F–G loop regions. (B) The distance different matrix shows that, in the pH 4.5 structure, the cleft around the folate substrate is slightly closed due to the shifts by the Met20 and F–G loops.
Because the pH 4.5 and 7.0 X-ray structures were determined at room temperature and have the same space group, isomorphous unit cell dimensions, and comparable diffraction resolutions (Table S1), we can compare atomic mobility using B-factor analysis with minimal bias from crystal packing or lattice artifacts. Overall, the B-factors of the pH 4.5 structure are significantly lower than those of the pH 7.0 structure (Table 1 and Figure 2A–B). The pH 4.5 structure has more ordered water molecules than the pH 7.0 structure (Table S1). Ensemble refinement implemented in PHENIX also shows that the pH 4.5 structure is more ordered, particularly the Met20 loop and the F–G loop (Figure 2C). MD simulations of both the pH 4.5 and 7.0 structures also indicate that the Met20 loop is less mobile at pH 4.5 though the two simulations result in very similar global dynamics (Figure 2D). The basis for dampened Met20 loop dynamics in the simulation of the lower pH model is difficult to discern since the residues that are uniquely protonated are not in direct contact with the Met20 loop.
Table 1. B-Factor Analysis of the X-ray Structures at pH 4.5 and pH 7.0.
| Wilson B | Ball | B(protein) | B(main chain) | B(side chain) | Bsolvent | |
|---|---|---|---|---|---|---|
| pH 4.5 | 13.6 | 17.5 | 15.9 | 12.9 | 17.8 | 33.5 |
| pH 7.0 | 18.1 | 21.4 | 20.5 | 17.2 | 22.2 | 33.7 |
Figure 2.

DHFR has dampened dynamics at acidic pH. (A, B) B-factor analysis between the X-ray structures at pH 4.5 and pH 7.0. (C) The ensemble refined structures show that the Met20 loop at pH 4.5 is significantly less dynamic than at pH 7.0. (D) MD simulations of the pH 4.5 and pH 7.0 X-ray structures provide root-mean-square fluctuations (RMSF) and also reveal dampened dynamics for the Met20 loop at acidic pH. (E) The (FopH4.5–FopH7.0) X-ray isomorphous difference map (contour level: 3.5 σ) shows that the Met20 side chain and HOH14 are more ordered at pH 4.5.
An isomorphous difference map comparing the electron density between the pH 4.5 and pH 7.0 structures (FopH4.5–FopH7.0) reveals that the side chain of Met20 and a water molecule above the N10 atom of folate (HOH14 in the high-resolution X-ray structure at pH 4.5) are both more ordered at acidic pH (Figure 2E). These results suggest that ecDHFR is less dynamic in an acidic environment, particularly the Met20 loop. We note that the Met20 loops in both neutron structures are in a closed conformation, even though the Cα atom RMSD calculations described above show that their backbones do not perfectly align. This suggests that these are two distinct subconformations of the closed loop. Indeed, a surprising amount of conformational space is sampled by the Met20 loop in the ensemble refinement of the pH 7.0 model (Figure 2C). These motions and the alternative conformational states of the Met20 loop have been invoked as critical for catalysis.59 Although the dynamics of the Met20 loop in the pH 4.5 structure is reduced compared to the pH 7.0 structure, fluctuations are apparent. This mobility may play a role in solvent entry into the active site.
The neutron structure of the ecDHFR–folate–NADP+ complex at pH 4.5 is generally similar to our previously described pH 7.0 neutron structure.37 However, due to the acidic pH, deuterons are observed on many ionizable side chains, including all His residues (His45, His114, His124, His141, and His149), Glu48, Glu101, Glu139, and Asp132 (Figure S1). In contrast, His45, Glu101, and Glu139 are not protonated in the neutron structure determined at pH 7.0.37 These differences confirm that the newly obtained crystal is indeed in a lower pH environment. Additionally, our ability to model deuterons on chemically exchangeable side chains, especially those that titrate at acidic pH, verifies the quality of the maps.
Interactions in the Active Site and Loop Dynamics
To locate the deuterium atoms (the exchangeable protons) in the active site around the folate substrate, 2Fo–Fc and Fo–Fc nuclear density maps were generated and contoured at 1.5 and 3.5 σ, respectively. It is clear that the N3 atom of folate is protonated and forms a strong ionic interaction with the anionic Oδ2 atom of Asp27 at a distance of 1.8 Å (Figure 3A). The refined occupancy of the N3-associated deuterium is 0.99, compared to 0.83 in the pH 7.0 structure. The observation that Asp27 maintains its negative charge at pH 4.5 suggests that its carboxylate side chain forms a remarkably strong charge-assisted hydrogen bond with the protonated N3 atom of the substrate. The O4 atom of folate is in the keto form, the same as in the pH 7.0 structure. This confirms our previous proposal that keto-to-enol tautomerization is not necessary for ecDHFR catalysis and that the reaction proceeds through the stabilization of the keto isomer.37 These observations also suggest that important protein/substrate bonding interactions have not been adversely affected by the significant change in pH.
Figure 3.

N5 of substrate can be directly protonated by a solvent molecule. (A) 2Fo–Fc nuclear density map (contour level: 1.5 σ) and Fo–Fc nuclear density omit map (contour level: 3.5 σ) in the active site show a H-bond formed between the ionized Asp27 and the protonated N3 of folate at pH 4.5. The omit map was created without the deuterium contribution for neutron scattering. (B) 2Fo–Fc nuclear density map (contour level: 1.2 σ) and Fo–Fc nuclear density omit map (contour level: 3.8 σ) at pH 4.5. The omit map revealed a strong nuclear density peak near DOD47. We have modeled this as a deuteron (pink sphere), and it is H-bonded between DOD47 and the Sδ atom of the Met20 side chain. Importantly, this deuteron is also positioned near the substrate, ∼3.9 Å from the N5 atom. (C) 2Fo–Fc electron density map (contour level: 1.2 σ) shows the Met20 side chain has alternate conformations at pH 4.5 (D) 2Fo–Fc electron density map (contour level: 1.2 σ) shows the Met20 side chain is more dynamic at pH 7.0.
In ecDHFR, the dynamics of the Met20 loop has been implicated in catalytic turnover. Fluctuations in the loop are thought to enable the entrance of solvent molecules required for protonation of the N5 atom of DHF.9,28 Due to this flexibility, inherent crystallographic disorder, the relatively weak neutron scattering cross section of sulfur, as well as scattering cancellation effects from the hydrogenated side chain, the Met20 residue has poor nuclear density at 2.1 Å resolution (Figure 3B). However, it is visible in the 1.65 Å resolution electron density map (Figure 3C). In these maps, the side chain of Met20 could be modeled into two alternate conformations, with the minor one (∼40% occupancy) turning away from a nearby water (Figure 3C, labeled HOH14 in the X-ray structure and DOD47 in the neutron structure in Figure 3B) and its major one (∼60% occupancy) rotating 109° around its χ-3 angle and pointing toward this water molecule. In contrast, the pH 7 structure shows the occupancies of these side chain conformations to be switched, with the major conformation of the Met20 side chain (∼60%) rotating away from DOD47 and the minor one (∼40%) pointing toward the water (Figure 3D). Since the Met20 side chain alternate conformations can be observed at two different pH values, this suggests that the rotational dynamics of this residue has functional importance. In addition, the nature of this disorder changes as the pH is lowered, with the pH 7.0 structure being more extensively disordered at Met20 (Figure 2E) while the pH 4.5 structure shows two well-resolved conformations about the Cγ–Sδ torsion (Figure 3C–D).
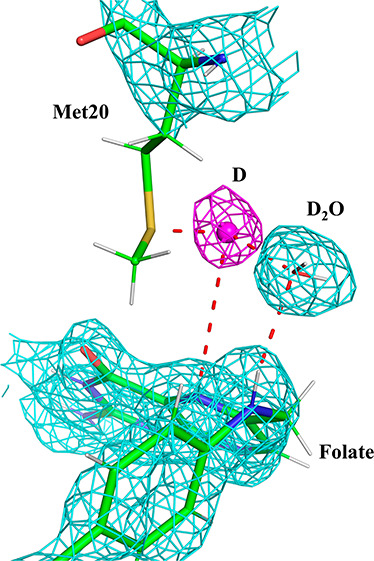
Due to the absence of nuclear density for the Met20 side chain as explained above, we modeled a singular conformation in the neutron structure based on electron density maps from the complementary X-ray structure (see Materials and Methods). In this model, DOD47 is within 3.4 Å of the Cε atom and 3.8 Å of the Sδ atom in the Met20 side chain (Figure 3B). DOD47 also forms a hydrogen bond with N10 of folate, an interaction that appears to be highly conserved in DHFR ternary structures (see Discussion below). Strikingly, in the pH 4.5 nuclear density maps, there is a strong nuclear density peak (at +3.8 σ contour level in the Fo–Fc omit map; Figure 3B) between the side chain sulfur atom (Sδ) of Met20 and the oxygen atom of DOD47. As this peak is not observed in the electron density maps and its shape is not consistent with a water molecule, we modeled a deuteron (labeled D1 in Figure 3B). Its occupancy refines to 0.78. As modeled and refined, the deuteron forms a typical hydrogen bond with the Sδ of Met20 (distance = 2.3 Å) and a low-barrier hydrogen bond with the oxygen (1.5 Å) of DOD47. We note that the deuteron is also 3.9 Å from the N5 atom of folate, the target of protonation in the DHFR mechanism. As it is bordered on either side by the Met20 side chain and the DOD47, the structural path from the deuteron to the N5 atom resembles a short narrow “channel” (Figure 3B), where steric constraints in the active site could serve as a type of conduit for substrate protonation.
In a previous study, it has been suggested that an open Met20 loop conformation enables solvent access to the N5 readily while the closed conformation prohibits solvent entry.10 Although it is likely for solvent to access N5 more readily in the open conformation, our study identifies the important catalytic water molecule DOD47 to be present in the closed conformation. Based on the proximity of the deuteron (D1) to DOD47, we propose the likelihood of DOD47 to be a short-lived hydronium (H3O+) species that could be the candidate for proton donation at N5. Previous neutron crystallographic studies of xylose isomerase and rubredoxin have identified H3O+ ions in their respective active sites.4,5
QM/MM Calculations on Proton Transfer from Hydronium to the N5 of DHF
In order to investigate the potential energy profile for the protonation of the N5 substrate atom from H3O+, QM/MM calculations were performed on a geometry-optimized ecDHFR–DHF–NADPH ternary complex based on the structure of the pH 4.5 XN model, with protons substituted for modeled deuterons. For these calculations, the catalytically optimal substrate dihydrofolate (DHF) was substituted for folate, and NADPH was substituted for the product cofactor NADP+. The QM region included the side chains of Met20 and Asp27, dihydrofolate, the nicotinamide moiety of NADPH, a hydronium ion (the water molecule closest to the unbound proton plus the proton), and the water molecule in contact with the hydronium ion.
A DFTB3/MM52 potential energy scan that transfers the unbound proton to the N5 atom of DHF is a barrierless process favorable by 58 kcal/mol (ΔE). Though this energy change is more significant than the predictions of past computational studies,34 this could be due to the subtle differences in the mechanism of protonation and the exclusion of a direct interaction between a water molecule and the N5 of DHF. Interestingly, while a minimum energy well is not observed for the process, a plateau region in the potential energy (PE) scan is seen when the proton is 2.37 Å from the oxygen atom of water, 2.44 Å from the sulfur atom of Met20, and 1.86 Å from the N5 of DHF (Figure 4A). Within this energetic plateau, the proton is positioned closer to N5 compared to the position of the potential deuteron in our neutron structure containing a mimic of DHF. The PE scan also indicates that there is no energy barrier that prevents protonation of N5 once the proton is unbound as modeled from our pH 4.5 structure. A local energy minimum well could trap the proton and prevent its transfer to the N5 atom or its reassociation with the water molecule to reform H3O+. However, the PE profile does not indicate that the unbound proton/deuteron resides in a potential energy well. The lack of a minimum for the configuration with the unbound proton could be due to crystal-packing effects and/or quantum dynamical effects to describe the delocalized nature of protons60 that are not included in the present computational model. In addition, our aim was to model the “unbound proton” configuration of the DHFR reactant state while, in contrast, the XN structure contains a mimic of DHF (folate), which has a weaker affinity (by 24 kcal/mol) for protons based on quantum chemistry calculations of DHF and the folate mimic (Figure S2 and Table S2).
Figure 4.
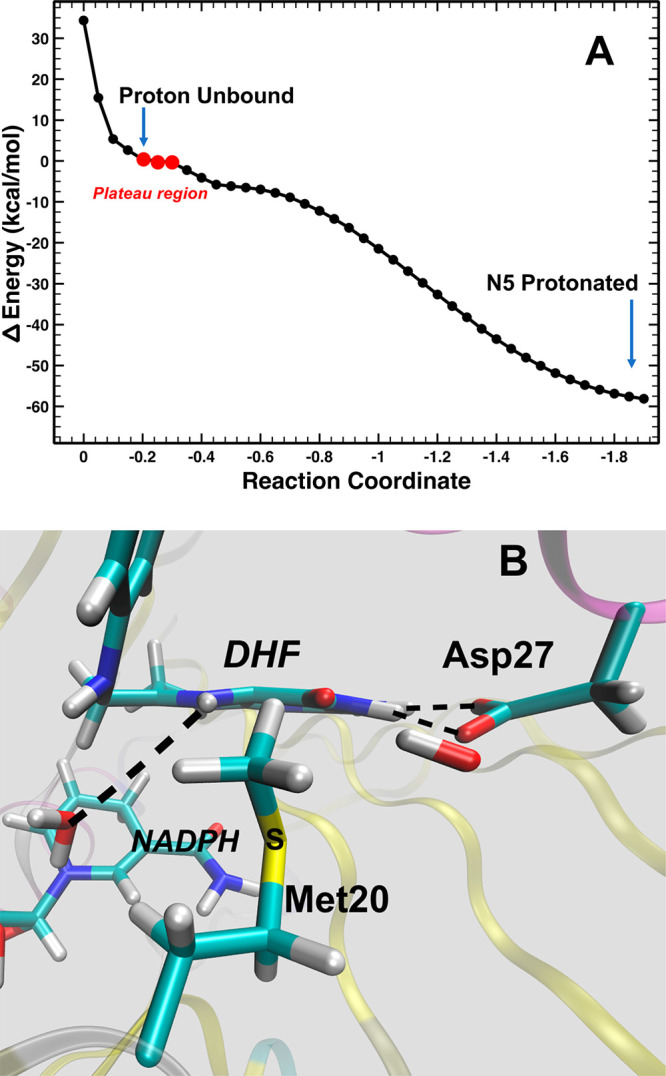
DFTB3/MM potential energy scan of transferring the position of the proton from the unbound position to the N5 of DHF. (A) The reaction coordinate is the mass-weighted distance difference between the oxygen atom of the adjacent water (the hydronium) to the unbound proton and the N5 of DHF to the unbound proton. (B) The approximate path and trajectory of the proton in the QM/MM calculations and MD simulations.
The QM region was also represented by the spin-component-scaled (SCS)-MP2 and wB97X-D DFT methods (see Materials and Methods for details of the models). The unbound proton is captured by the N5 atom of DHF in each case within a few optimization steps despite the closed conformation of the Met20 loop. Typically, a proton transfer originates from a hydrogen-bonded complex that resides within a local energy minimum with a short donor–acceptor distance (∼2.5 Å). Yet, in this closed conformation in which the N5 atom pKa is considerably increased,18,29,30 it appears that only a bare proton is able to traverse through the channel due to steric constraints in the active site directly adjacent to N5 (Figure 3B and Figure S3). The ring containing N5 assumes a “boatlike” conformation during the proton abstraction reaction and then, once the proton is captured, relaxes to a more planar structure (Figure 4B). Thus, this vibrational motion is likely to be a component of the coordinate that facilitates proton transfer to the N5 atom. Since the DHF mimic (folate) in the XN structure contains an additional double bond (between C7 and N8 in the pteridine ring), folate would be significantly less flexible, thus inhibiting the N5 capture of the unbound deuteron.
Discussion
DHFR is an excellent model enzyme to study acid–base catalysis. The hydride transfer and the protonation steps are key steps in the catalytic mechanism that are experimentally difficult to visualize. In recent years, neutron diffraction has a proven track record for answering these types of mechanistic questions through the direct observation of catalytic protons.3,4,35,37,61,62 In this study, we directly visualize the catalytic proton originating from solvent, with access to the substrate possibly being mediated by side chain conformational dynamics.
The crucial steps in the DHFR catalytic mechanism involving hydride transfer and the substrate protonation have not been directly observed. Although the hydride transfer step is well established, the lack of identification of a catalytic residue has led to the proposal that protonation requires solvent mediation,10,63 and the regulatory Met20 loop could facilitate such solvent entry.28,29 In our pH 7.0 neutron structure of the ecDHFR ternary complex (4PDJ), we did observe and model a partially occupied water molecule within generous hydrogen-bonding distance of the N5 atom of folate.37 However, the modeling of this water was based solely on electron density, as we were unable to observe any nuclear density peak that could be modeled as a solvent molecule or a deuteron that could be the candidate responsible for the protonation of the N5 atom.37
In the present study, we conducted the neutron diffraction experiment with an ecDHFR ternary complex crystal at acidic pH (pH 4.5), 2 pH units below the enzyme’s optimum, to increase the chances of observing the elusive proton. For both pH values, two distinct conformations of the Met20 side chain could be modeled in the high-resolution X-ray structures that complemented the neutron structures (Figure 3C–D). The nuclear density for the Met20 side chain is poor at pH 4.5 (Figure 3B), possibly due to weak scattering, scattering cancellation effects, and crystallographic disorder. However, in the pH 4.5 nuclear density maps, a strong positive difference density peak is observed in the omit maps positioned between DOD47 and one of the conformations of the Met20 side chain. We ultimately modeled a deuteron (D1 in Figure 3B) within this peak. We based this identification on the following: (1) the peak is only observed in nuclear density, not electron density; (2) the shape of the density is almost perfectly spherical and not trapezoid or triangular as would be expected from a water molecule; (3) its unique position in the active site adjacent to Met20 and the folate (DHF) N5 as well as its very close distance to DOD47 of 1.5 Å (Figure 3B); and (4) its occupancy refines to nearly 0.8.35,64,65 We note that the neutron data reported here were collected near room temperature (291 K); thus, we cannot be certain that at growth temperatures optimal for most strains of E. coli (∼310 K) the deuteron would be observable. However, at room temperature, deuterons have been observed previously in a few neutron structures (such as in refs (4 and 66)). Indeed, we have previously used acidic pH and neutron crystallography to capture a deuteron in the active site of xylose isomerase, and it also formed low-barrier hydrogen bonds with surrounding oxygen atoms.4
In the conformation of Met20 as modeled in the neutron structure (Figure 3B), the Sδ atom may stabilize the deuteron as modeled in the nuclear density peak, with Met20, DOD47, and N5 triangulating the deuteron. We propose that this deuteron emanated from a H3O+ ion that originated from the DOD47 solvent molecule as observed in our neutron structure (Figure 3B). Indeed, the distance between the oxygen of DOD47 and the deuteron is very short, 1.5 Å, suggesting that this may be a short hydrogen bond67 such as a low-barrier hydrogen bond.4 DOD47 forms an H-bond with the N10 atom of folate (2.5 Å; Figure 3B; labeled as W47 in Figure 5). Inspection of crystal structures of the DHFR–folate–NADP+ complex from several different species shows that DOD47 is a conserved water molecule and is within H-bonding distance to the N10 atom in structures from H. sapiens (4M6K68), E. coli (1RX210), P. carinii (2CD269), and S. aureus (3FRD70).
Figure 5.

Interactions between folate and active site residues and solvent molecules. The H-bond distances are shown as dashed lines and are given in Å. Water molecules are numbered according to the water molecules modeled in the neutron structure (i.e., W47 = DOD47). Note the H+ atom triangulated between the Met20 side chain, W47, and the substrate N5 atom. This is based on the modeled deuteron in the pH 4.5 neutron structure.
Our studies have implications for the role of loop dynamics in DHFR catalysis. Previously, Sawaya and Kraut have implicated linkage between the open and closed Met20 loop conformations and regulation of catalysis, as supported by their analysis of a multitude of isomorphous crystal structures.10 In the open conformation, there is unrestricted solvent entry important for N5 protonation. They suggested that the solvent accessibility is prohibited in a closed conformation; however, it is in the closed conformation that catalysis occurs. In our structures, at both pH 7 and 4.5, we observe that the Met20 loop is in a closed conformation, yet a solvent molecule (DOD47) is seen in the active site and forms a hydrogen bond with the N10 atom of folate. In both structures, two distinct side chain conformations exist for the Met20 residue. These dynamics, for the global structure as well as specifically at Met20, are more pronounced at pH 7 (Figures 1–2). The Met20 side chain dynamics in the closed loop conformation and the proximity to DOD47 suggest a possible role for this residue in mediating solvent (proton) access to the substrate during catalysis. Our observations help to verify the previous important molecular dynamics simulations that were the first to reveal that solvent could access the active site of DHFR in a Michaelis complex;28 indeed, this insight in part laid the motivational groundwork for the current study.
We propose that DOD47 is the catalytic water that promotes protonation of the N5 atom in DHF. The deuteron (modeled as D1 in Figure 3B) possibly forms a low-barrier hydrogen bond with the oxygen atom of DOD47. Though its distance to N5 is beyond the hydrogen-bonding range (3.9 Å), the deuteron is positioned between the Met20 side chain and the DOD47 so as to define a pathway that could lead to protonation of N5 by the deuteron. Structural analysis of this “channel” reveals that only a hydrogen atom can fit within and travel along this pathway toward N5 to protonate it (Figure S3). For instance, modeling studies show that a water molecule (either as D2O or H2O) is sterically not tolerated due to the narrowing of this channel, strongly suggesting that the observed D1 is the catalytic proton.
Conclusions
Taken together, our observations are consistent with a mechanism in which a transient H3O+ intermediate (formerly DOD47) is stabilized at acidic pH. Molecular dynamics and QM/MM calculations show rapid and favorable transfer of the proton from the H3O+ to the N5 atom of DHF (Figure 4, Table S2). The computational studies also indicate that the protonation is accompanied by DHF ring distortion. Thus, we propose that, in the closed loop state of the Michaelis complex (DHF–NADPH), DOD47 obtains a proton from the solvent, thus becoming an H3O+ ion. This H3O+ ion is well positioned to protonate the N5 atom of DHF, access to which is facilitated by Met20 side chain dynamics (Figure 5). This specific active site environment aids in significantly increasing the pKa of N5 in DHF while still allowing a bare proton to traverse and ultimately reduce the N5 atom. Because of the conservation of DOD47 in ternary complex structures of DHFR, solvent-mediated protonation via a transient H3O+ may be a viable mechanism by which substrate protonation occurs in DHFR catalysis.
These results demonstrate the power of comparative neutron and X-ray diffraction studies performed across a pH range, even when these pH values markedly differ from the pH optimum of the enzyme investigated. Structures solved with neutron diffraction at low pH can provide a “proton trap” as demonstrated here. The combination of X-ray and neutron diffraction with QM/MM calculations exemplifies a unique tool for visualizing dynamic systems where each technique provides valuable missing details complementing one another, painting a more complete picture of an enzyme mechanism. This is especially important for enzymes that have an acid–base catalytic mechanism, as it can provide not only the snapshot but also the potential trajectory of protons being donated or accepted along a reaction pathway.
Acknowledgments
Q.W. was supported by the National Natural Science Foundation of China (No. 32071264 and 31670790) and the Fundamental Research Funds for the Central Universities (No. KYXK202009). We thank the instrument scientists of the neutron diffractometer BioDiff at the FRM II, Drs. Andreas Ostermann and Tobias Schrader, for collecting neutron diffraction data. We thank the staff of the BL18U1 and BL19U1 beamlines at Shanghai Synchrotron Radiation Facility, Shanghai, P. R. China, for assistance during X-ray data collection. T.W. and C.L.B. III were supported by funding from the NIH, including RO1GM107233 and RO1GM130587. M.A.W. was also supported by the NIH, with funding through RO1GM139978. The Office of Biological and Environmental Research supported research at Oak Ridge National Laboratory’s Center for Structural Molecular Biology (CSMB), using facilities supported by the Scientific User Facilities Division, Office of Basic Energy Sciences, U.S. Department of Energy. A.K. and P.L. were supported by the U.S. Department of Energy’s (DOE) Office of Basic Energy Sciences. C.G.D. was supported by the NIH, with funding through RO1GM100887-01. We dedicate this to the memory of Dr. Elizabeth E. Howell, colleague, DHFR expert, artist, and friend, who passed away during the preparation of this manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.1c00417.
Figures S1–S3; Tables S1 and S2 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Fisher S. J.; Blakeley M. P.; Cianci M.; McSweeney S.; Helliwell J. R. Protonation-state determination in proteins using high-resolution X-ray crystallography: effects of resolution and completeness. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2012, 68, 800–809. 10.1107/S0907444912012589. [DOI] [PubMed] [Google Scholar]
- Blakeley M. P.; Hasnain S. S.; Antonyuk S. V. Sub-atomic resolution X-ray crystallography and neutron crystallography: promise, challenges and potential. IUCrJ 2015, 2, 464–474. 10.1107/S2052252515011239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Dell W. B.; Bodenheimer A. M.; Meilleur F. Neutron protein crystallography: A complementary tool for locating hydrogens in proteins. Arch. Biochem. Biophys. 2016, 602, 48–60. 10.1016/j.abb.2015.11.033. [DOI] [PubMed] [Google Scholar]
- Kovalevsky A. Y.; Hanson B. L.; Mason S. A.; Yoshida T.; Fisher S. Z.; Mustyakimov M.; Forsyth V. T.; Blakeley M. P.; Keen D. A.; Langan P. Identification of the elusive hydronium ion exchanging roles with a proton in an enzyme at lower pH values. Angew. Chem., Int. Ed. 2011, 50, 7520–7523. 10.1002/anie.201101753. [DOI] [PubMed] [Google Scholar]
- Cuypers M. G.; Mason S. A.; Blakeley M. P.; Mitchell E. P.; Haertlein M.; Forsyth V. T. Near-Atomic Resolution Neutron Crystallography on Perdeuterated Pyrococcus furiosus Rubredoxin: Implication of Hydronium Ions and Protonation State Equilibria in Redox Changes. Angew. Chem., Int. Ed. 2013, 52, 1022–1025. 10.1002/anie.201207071. [DOI] [PubMed] [Google Scholar]
- Gerlits O.; Wymore T.; Das A.; Shen C. H.; Parks J. M.; Smith J. C.; Weiss K. L.; Keen D. A.; Blakeley M. P.; Louis J. M.; Langan P.; Weber I. T.; Kovalevsky A. Long-Range Electrostatics-Induced Two-Proton Transfer Captured by Neutron Crystallography in an Enzyme Catalytic Site. Angew. Chem., Int. Ed. 2016, 55, 4924–4927. 10.1002/anie.201509989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Q.; Parks J. M.; Hanson B. L.; Fisher S. Z.; Ostermann A.; Schrader T. E.; Graham D. E.; Coates L.; Langan P.; Kovalevsky A. Direct determination of protonation states and visualization of hydrogen bonding in a glycoside hydrolase with neutron crystallography. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 12384–12389. 10.1073/pnas.1504986112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer B. I.; Dicker A. P.; Bertino J. R. Dihydrofolate reductase as a therapeutic target. FASEB J. 1990, 4, 2441–2452. 10.1096/fasebj.4.8.2185970. [DOI] [PubMed] [Google Scholar]
- Reyes V. M.; Sawaya M. R.; Brown K. A.; Kraut J. Isomorphous crystal structures of Escherichia coli dihydrofolate reductase complexed with folate, 5-deazafolate, and 5,10-dideazatetrahydrofolate: mechanistic implications. Biochemistry 1995, 34, 2710–2723. 10.1021/bi00008a039. [DOI] [PubMed] [Google Scholar]
- Sawaya M. R.; Kraut J. Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: crystallographic evidence. Biochemistry 1997, 36, 586–603. 10.1021/bi962337c. [DOI] [PubMed] [Google Scholar]
- Chaabane S.; Marzouk S.; Akrout R.; Ben Hamad M.; Achour Y.; Rebai A.; Keskes L.; Fourati H.; Bahloul Z.; Maalej A. Genetic Determinants of Methotrexate Toxicity in Tunisian Patients with Rheumatoid Arthritis: A Study of Polymorphisms Involved in the MTX Metabolic Pathway. Eur. J. Drug Metab. Pharmacokinet. 2016, 41, 385–393. 10.1007/s13318-015-0288-z. [DOI] [PubMed] [Google Scholar]
- Sharma M.; Chauhan P. M. Dihydrofolate reductase as a therapeutic target for infectious diseases: opportunities and challenges. Future Med. Chem. 2012, 4, 1335–1365. 10.4155/fmc.12.68. [DOI] [PubMed] [Google Scholar]
- Yuthavong Y.; Tarnchompoo B.; Vilaivan T.; Chitnumsub P.; Kamchonwongpaisan S.; Charman S. A.; McLennan D. N.; White K. L.; Vivas L.; Bongard E.; Thongphanchang C.; Taweechai S.; Vanichtanankul J.; Rattanajak R.; Arwon U.; Fantauzzi P.; Yuvaniyama J.; Charman W. N.; Matthews D. Malarial dihydrofolate reductase as a paradigm for drug development against a resistance-compromised target. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 16823–16828. 10.1073/pnas.1204556109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spina M.; Cuccioloni M.; Mozzicafreddo M.; Montecchia F.; Pucciarelli S.; Eleuteri A. M.; Fioretti E.; Angeletti M. Mechanism of inhibition of wt-dihydrofolate reductase from E. coli by tea epigallocatechin-gallate. Proteins: Struct., Funct., Genet. 2008, 72, 240–251. 10.1002/prot.21914. [DOI] [PubMed] [Google Scholar]
- Fierke C. A.; Johnson K. A.; Benkovic S. J. Construction and evaluation of the kinetic scheme associated with dihydrofolate reductase from Escherichia coli. Biochemistry 1987, 26, 4085–4092. 10.1021/bi00387a052. [DOI] [PubMed] [Google Scholar]
- Maharaj G.; Selinsky B. S.; Appleman J. R.; Perlman M.; London R. E.; Blakley R. L. Dissociation constants for dihydrofolic acid and dihydrobiopterin and implications for mechanistic models for dihydrofolate reductase. Biochemistry 1990, 29, 4554–4560. 10.1021/bi00471a008. [DOI] [PubMed] [Google Scholar]
- Bhabha G.; Lee J.; Ekiert D. C.; Gam J.; Wilson I. A.; Dyson H. J.; Benkovic S. J.; Wright P. E. A dynamic knockout reveals that conformational fluctuations influence the chemical step of enzyme catalysis. Science 2011, 332, 234–238. 10.1126/science.1198542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. T.; Francis K.; Layfield J. P.; Huang X.; Hammes-Schiffer S.; Kohen A.; Benkovic S. J. Escherichia coli dihydrofolate reductase catalyzed proton and hydride transfers: temporal order and the roles of Asp27 and Tyr100. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 18231–18236. 10.1073/pnas.1415940111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. Q.; Kraut J.; Blakley R. L.; Callender R. Determination by Raman spectroscopy of the pKa of N5 of dihydrofolate bound to dihydrofolate reductase: mechanistic implications. Biochemistry 1994, 33, 7021–7026. 10.1021/bi00189a001. [DOI] [PubMed] [Google Scholar]
- Casarotto M. G.; Basran J.; Badii R.; Sze K. H.; Roberts G. C. Direct measurement of the pKa of aspartic acid 26 in Lactobacillus casei dihydrofolate reductase: implications for the catalytic mechanism. Biochemistry 1999, 38, 8038–8044. 10.1021/bi990301p. [DOI] [PubMed] [Google Scholar]
- Luk L.; Ruiz-Pernia J. J.; Dawson W.; Roca M.; Loveridge E. J.; Glowacki D.; Harvey J.; Mulholland A.; Tunon I.; Moliner V.; Allemann R. Unraveling the role of protein dynamics in dihydrofolate reductase catalysis. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 16344–16349. 10.1073/pnas.1312437110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk L.; Ruiz-Pernia J.; Adesina A.; Loveridge E. J.; Tunon I.; Moliner V.; Allemann R. Chemical Ligation and Isotope Labeling to Locate Dynamic Effects during Catalysis by Dihydrofolate Reductase. Angew. Chem., Int. Ed. 2015, 54, 9016–9020. 10.1002/anie.201503968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adesina A.; Swiderek K.; Luk L.; Moliner V.; Allemann R. Electric Field Measurements Reveal the Pivotal Role of Cofactor-Substrate Interaction in Dihydrofolate Reductase Catalysis. ACS Catal. 2020, 10, 7907–7914. 10.1021/acscatal.0c01856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal P. K.; Billeter S. R.; Rajagopalan P. T.; Benkovic S. J.; Hammes-Schiffer S. Network of coupled promoting motions in enzyme catalysis. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 2794–2799. 10.1073/pnas.052005999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Fortunato G.; Lin J.; Agarwal P. K.; Kohen A.; Singh P.; Cheatum C. Evolution Conserves the Network of Coupled Residues in Dihydrofolate Reductase. Biochemistry 2019, 58, 3861–3868. 10.1021/acs.biochem.9b00460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Viloca M.; Truhlar D.; Gao J. Reaction-path energetics and kinetics of the hydride transfer reaction catalyzed by dihydrofolate reductase. Biochemistry 2003, 42, 13558–13575. 10.1021/bi034824f. [DOI] [PubMed] [Google Scholar]
- Stojkovic V.; Perissinotti L.; Willmer D.; Benkovic S. J.; Kohen A. Effects of the donor-acceptor distance and dynamics on hydride tunneling in the dihydrofolate reductase catalyzed reaction. J. Am. Chem. Soc. 2012, 134, 1738–1745. 10.1021/ja209425w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrimpton P.; Allemann R. K. Role of water in the catalytic cycle of E. coli dihydrofolate reductase. Protein Sci. 2002, 11, 1442–1451. 10.1110/ps.5060102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khavrutskii I. V.; Price D. J.; Lee J.; Brooks C. L. 3rd Conformational change of the methionine 20 loop of Escherichia coli dihydrofolate reductase modulates pKa of the bound dihydrofolate. Protein Sci. 2007, 16, 1087–1100. 10.1110/ps.062724307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mhashal A. R.; Vardi-Kilshtain A.; Kohen A.; Major D. T. The role of the Met(20) loop in the hydride transfer in Escherichia coli dihydrofolate reductase. J. Biol. Chem. 2017, 292, 14229–14239. 10.1074/jbc.M117.777136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller G. P.; Benkovic S. J. Stretching exercises--flexibility in dihydrofolate reductase catalysis. Chem. Biol. 1998, 5, R105–113. 10.1016/S1074-5521(98)90616-0. [DOI] [PubMed] [Google Scholar]
- Cummins P. L.; Gready J. E. Energetically most likely substrate and active-site protonation sites and pathways in the catalytic mechanism of dihydrofolate reductase. J. Am. Chem. Soc. 2001, 123, 3418–3428. 10.1021/ja0038474. [DOI] [PubMed] [Google Scholar]
- Gandour R. D. On the Importance of Orientation in General Base Catalysis by Carboxylate. Bioorg. Chem. 1981, 10, 169–176. 10.1016/0045-2068(81)90020-1. [DOI] [Google Scholar]
- Mhashal A.; Pshetitsky Y.; Cheatum C.; Kohen A.; Major D. Evolutionary Effects on Bound Substrate p K a in Dihydrofolate Reductase. J. Am. Chem. Soc. 2018, 140, 16650–16660. 10.1021/jacs.8b09089. [DOI] [PubMed] [Google Scholar]
- Bennett B.; Langan P.; Coates L.; Mustyakimov M.; Schoenborn B.; Howell E. E.; Dealwis C. Neutron diffraction studies of Escherichia coli dihydrofolate reductase complexed with methotrexate. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 18493–18498. 10.1073/pnas.0604977103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Q.; Kovalevsky A. Y.; Wilson M. A.; Bennett B. C.; Langan P.; Dealwis C. Preliminary joint X-ray and neutron protein crystallographic studies of ecDHFR complexed with folate and NADP+. Acta Crystallogr., Sect. F: Struct. Biol. Commun. 2014, F70, 814–818. 10.1107/S2053230X1400942X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Q.; Bennett B. C.; Wilson M. A.; Kovalevsky A.; Langan P.; Howell E. E.; Dealwis C. Toward resolving the catalytic mechanism of dihydrofolate reductase using neutron and ultrahigh-resolution X-ray crystallography. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 18225–18230. 10.1073/pnas.1415856111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshiro T.; Kuge Y.; Igarashi A.; Mochida K.; Iwakura M.; Uwajima T. Production of dihydrofolate reductase by cloned Escherichia coli and its application to asymmetric synthesis of l-leucovorin. Biosci., Biotechnol., Biochem. 1992, 56, 437–440. 10.1271/bbb.56.437. [DOI] [PubMed] [Google Scholar]
- Meilleur F.; Munshi P.; Robertson L.; Stoica A. D.; Crow L.; Kovalevsky A.; Koritsanszky T.; Chakoumakos B. C.; Blessing R.; Myles D. A. The IMAGINE instrument: first neutron protein structure and new capabilities for neutron macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2013, 69, 2157–2160. 10.1107/S0907444913019604. [DOI] [PubMed] [Google Scholar]
- Meilleur F.; Coates L.; Cuneo M. J.; Kovalevsky A.; Myles D. The Neutron Macromolecular Crystallography Instruments at Oak Ridge National Laboratory: Advances, Challenges, and Opportunities. Crystals 2018, 8, 388–398. 10.3390/cryst8100388. [DOI] [Google Scholar]
- Meilleur F.; Kovalevsky A.; Myles D. IMAGINE: The neutron protein crystallography beamline at the high flux isotope reactor. Methods Enzymol. 2020, 634, 69–85. 10.1016/bs.mie.2019.11.016. [DOI] [PubMed] [Google Scholar]
- Niimura N.; Karasawa Y.; Tanaka I.; Miyahara J.; Takahashi K.; Saito H.; Koizumi S.; Hidaka M. An elastically bent silicon monochromator for a neutron diffractometer. Nucl. Instrum. Methods Phys. Res., Sect. A 1994, 349, 521–525. 10.1016/0168-9002(94)91220-3. [DOI] [Google Scholar]
- Minor W.; Cymborowski M.; Otwinowski Z.; Chruszcz M. HKL-3000: the integration of data reduction and structure solution – from diffraction images to an initial model in minutes. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2006, D62, 859–866. 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joosten R. P.; Long F.; Murshudov G. N.; Perrakis A. The PDB_REDO server for macromolecular structure model optimization. IUCrJ 2014, 1, 213–220. 10.1107/S2052252514009324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnley B. T.; Afonine P. V.; Adams P. D.; Gros P. Modelling dynamics in protein crystal structures by ensemble refinement. eLife 2012, 1, e00311 10.7554/eLife.00311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards F. M.; Kundrot C. E. Identification of structural motifs from protein coordinate data: secondary structure and first-level supersecondary structure. Proteins: Struct., Funct., Genet. 1988, 3, 71–84. 10.1002/prot.340030202. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, D60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Brooks B. R.; Brooks C. L. 3rd; Mackerell A. D. Jr; Nilsson L.; Petrella R. J.; Roux B.; Won Y.; Archontis G.; Bartels C.; Boresch S.; Caflisch A.; Caves L.; Cui Q.; Dinner A. R.; Feig M.; Fischer S.; Gao J.; Hodoscek M.; Im W.; Kuczera K.; Lazaridis T.; Ma J.; Ovchinnikov V.; Paci E.; Pastor R. W.; Post C. B.; Pu J. Z.; Schaefer M.; Tidor B.; Venable R. M.; Woodcock H. L.; Wu X.; Yang W.; York D. M.; Karplus M. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaus M.; Cui Q.; Elstner M. DFTB3: Extension of the Self-Consistent-Charge Density-Functional Tight-Binding Method (SCC-DFTB). J. Chem. Theory Comput. 2011, 7, 931–948. 10.1021/ct100684s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimme S. Accurate calculation of the heats of formation for large main group compounds with spin-component scaled MP2 methods. J. Phys. Chem. A 2005, 109, 3067–3077. 10.1021/jp050036j. [DOI] [PubMed] [Google Scholar]
- Chai J.-D.; Head-Gordon M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. 10.1039/b810189b. [DOI] [PubMed] [Google Scholar]
- Rappoport D.; Furche F. Property-optimized Gaussian basis sets for molecular response calculations. J. Chem. Phys. 2010, 133, 134105. 10.1063/1.3484283. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Haser M.; Patzelt H.; Ahlrichs R. RI-MP2: optimized auxiliary basis sets and demonstration of efficiency. Chem. Phys. Lett. 1998, 294, 143. 10.1016/S0009-2614(98)00862-8. [DOI] [Google Scholar]
- Neese F. Software update: the ORCA program system, version 4. 0. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2018, 8, e1327. 10.1002/wcms.1327. [DOI] [Google Scholar]
- Field M. The pDynamo Program for Molecular Simulations using Hybrid Quantum Chemical and Molecular Mechanical Potentials. J. Chem. Theory Comput. 2008, 4, 1151–1161. 10.1021/ct800092p. [DOI] [PubMed] [Google Scholar]
- Rod T.; Radkiewicz J.; Brooks C. Correlated motion and the effect of distal mutations on dihydrofolate reductase. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 6980–6985. 10.1073/pnas.1230801100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez A.; Tuckerman M.; Hjalmarson H.; von Lilienfeld O. A. Enol Tautomers of Watson–Crick Base Pair Models Are Metastable Because of Nuclear Quantum Effects. J. Am. Chem. Soc. 2010, 132, 11510–11515. 10.1021/ja102004b. [DOI] [PubMed] [Google Scholar]
- Oksanen E.; Chen J.-C.; Fisher S. Z. Neutron Crystallography for the Study of Hydrogen Bonds in Macromolecules. Molecules 2017, 22, 596. 10.3390/molecules22040596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon H.; Basran J.; Devos J.; Suardiaz R.; van der Kamp M.; Mulholland A.; Schrader T. E.; Ostermann A.; Blakeley M. P.; Moody P.; Raven E. Visualizing the protons in a metalloenzyme electron proton transfer pathway. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 6484–6490. 10.1073/pnas.1918936117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTigue M. A.; Davies J. F. 2nd; Kaufman B. T.; Kraut J. Crystal structure of chicken liver dihydrofolate reductase complexed with NADP+ and biopterin. Biochemistry 1992, 31, 7264–7273. 10.1021/bi00147a009. [DOI] [PubMed] [Google Scholar]
- Kossiakoff A. A. Protein dynamics investigated by the neutron diffraction-hydrogen exchange technique. Nature 1982, 296, 713–721. 10.1038/296713a0. [DOI] [PubMed] [Google Scholar]
- Bennett B. C.; Gardberg A. S.; Blair M. D.; Dealwis C. G. On the determinants of amide backbone exchange in proteins: a neutron crystallographic comparative study. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2008, D64, 764–783. 10.1107/S0907444908012845. [DOI] [PubMed] [Google Scholar]
- Banco M. T.; Mishra V.; Ostermann A.; Schrader T. E.; Evans G. B.; Kovalevsky A.; Ronning D. R. Neutron structures of the Helicobacter pylori 5′-methylthioadenosine nucleosidase highlight proton sharing and protonation states. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 13756–13761. 10.1073/pnas.1609718113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P.; Agarwal P. K.; Cuneo M. J. On the Case of the Misplaced Hydrogens. ChemBioChem 2021, 22, 288. 10.1002/cbic.202000376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhabha G.; Ekiert D. C.; Jennewein M.; Zmasek C. M.; Tuttle L. M.; Kroon G.; Dyson H. J.; Godzik A.; Wilson I. A.; Wright P. E. Divergent evolution of protein conformational dynamics in dihydrofolate reductase. Nat. Struct. Mol. Biol. 2013, 20, 1243–1249. 10.1038/nsmb.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cody V.; Galitsky N.; Rak D.; Luft J. R.; Pangborn W.; Queener S. F. Ligand-induced conformational changes in the crystal structures of Pneumocystis carinii dihydrofolate reductase complexes with folate and NADP+. Biochemistry 1999, 38, 4303–4312. 10.1021/bi982728m. [DOI] [PubMed] [Google Scholar]
- Oefner C.; Parisi S.; Schulz H.; Lociuro S.; Dale G. E. Inhibitory properties and X-ray crystallographic study of the binding of AR-101, AR-102 and iclaprim in ternary complexes with NADPH and dihydrofolate reductase from Staphylococcus aureus. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2009, 65, 751–757. 10.1107/S0907444909013936. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.