

Abstract
Artificial metalloenzymes (ArMs) are created by embedding a synthetic metal catalyst into a protein scaffold. ArMs have the potential to merge the catalytic advantages of natural enzymes with the reaction scope of synthetic catalysts. The choice of the protein scaffold is of utmost importance to tune the activity of the ArM. Herein, we show the repurposing of HaloTag, a self-labeling protein widely used in chemical biology, to create an ArM scaffold for metathesis. This monomeric protein scaffold allows for covalent attachment of metathesis cofactors, and the resulting ArMs are capable of catalyzing ring-closing metathesis. Both chemical and genetic engineering were explored to determine the evolvability of the resulting ArM. Additionally, exploration of the substrate scope revealed a reaction with promising turnover numbers (>48) and conversion rates (>96%).
Keywords: artificial metalloenzymes, aqueous catalysis, metathesis, enzyme engineering, HaloTag
New methods for catalysis are of increasing importance in synthetic chemistry. Enzymatic catalysis is becoming more central to organic synthesis in both academia and industry.1,2 The advantages of enzymes are multifold and include selectivity, activity at ambient temperature and pressure, aqueous catalysis, and access to catalytic cascades that can be challenging to realize with synthetic catalysts.
Artificial metalloenzymes (ArMs) are hybrid catalysts that are created by embedding a metal catalyst into a protein scaffold.3 The resulting hybrid catalysts have the potential to impart the advantages of enzymes and retain the reaction versatility of synthetic catalysts. Because of these advantages, ArMs represent potential tools for novel biocatalysis. Thus, identifying a suitable scaffold protein is essential for improving ArMs.4 Multiple scaffolds have been explored for ArMs, including heme proteins,5,6 (strept)avidin,7 human carbonic anhydrase,8 glycosylated albumin,9 lactococcal multidrug resistance regulator (LmrR),10 an oligopeptidase,11 FhuA,12 and so on.13 Increasing the number of viable scaffolds will eventually enable chemists to use ArMs as a plug-and-play strategy, in which several scaffolds can be screened to identify the best catalytic starting point. Accordingly, the exploration of more scaffold proteins is critical to advancing the versatility of the field.
For ArMs, an important factor for the design is the anchoring method of the synthetic catalyst to the scaffold protein. This anchoring can be achieved by three methods: (i) supramolecular interaction, (ii) dative coordination, and (iii) covalent binding. Each of these methods has distinct advantages. Although covalently linked ArMs have the potential to create the most stable bioconjugates, only a few examples of covalently linked ArMs have been reported.11,13−15 Thus, we sought to explore a new scaffold for covalent anchoring of catalysts. To this end, we examined a small monomeric protein, HaloTag (version 7, Promega, HT hereafter), as a novel scaffold for ArM engineering.
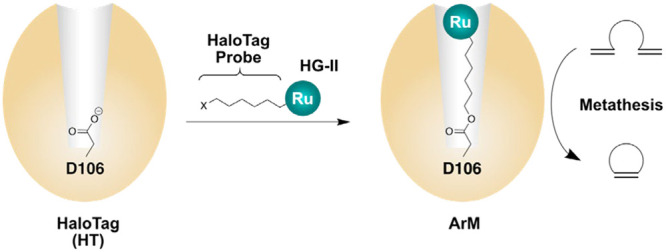
HT is a 34-kDa protein that contains a reactive aspartic acid residue deep within the protein (Figure 1).16 This aspartic acid residue can react with haloalkanes via nucleophilic substitution between the electrophilic haloalkane and the nucleophilic aspartate side chain. The result of this substitution reaction is a stable ester bond. Various haloalkanes can be used in this reaction, including bromoalkanes, chloroalkanes, and haloalkanes of varying chain lengths.16−18 Additionally, the haloalkane chain can be terminally functionalized to add cargo, most commonly a fluorophore16,19,20 but also other cargo such as a metal complex, used for magnetic resonance imaging.21 On the basis of this work, we posited that by functionalizing a haloalkane chain with a metal catalyst, we could create a stable ArM. An HT-based ArM could have several advantages: (i) the distance between the catalyst and protein surface could be easily optimized by changing the alkane linker length; (ii) the bioconjugation reaction is biocompatible, which would allow for whole-cell catalysis; and (iii) the monomeric nature offers engineering advantages. To assess the potential of HT as an ArM scaffold, we were interested in exploring a reaction that has no known equivalent in biology. As an archetypal bio-orthogonal reaction, metathesis has been often used as a test reaction for ArMs.9,12,22−29
Figure 1.
Use of HT as a scaffold for ArMs. A substitution reaction at an aspartic acid residue buried within HT generates a covalent linkage upon reaction with a haloalkane chain. The reactive haloalkane chain can be equipped with a synthetic metal cofactor to create an ArM.
Thus, we sought to create metathesis-catalyzing ArMs with HT as scaffold, two cofactors were synthesized for Ru-catalyzed metathesis (Figure 2A).30 Both cofactors were based on a Hoveyda–Grubbs II type catalyst (HG-II). The catalysts differed in the placement of the HT linker, which was appended at either the para-position of one mesityl group (Mes8) or the NHC core (N8). The difference in linker placement was designed to allow for different orientations upon binding to HT. The resulting cofactors both bind HT (Figures 2B and S2). Catalyst N8 resulted in higher yield of the ArM within the incubation time, suggesting that the position of catalyst N8 may fit better into the cleft of HT. Additionally, cofactors with shorter linker length were also tested but did not bind adequately to HT (Figure S2).
Figure 2.
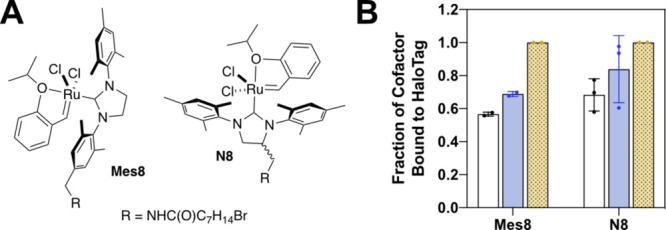
Cofactors and bioconjugation assay. Two metathesis cofactors—N8 and Mes8—based on HG-II derived catalysts were synthesized (A). Binding analysis at varying ratios of HT to cofactor: (white with black outline) 50 μM HT: 50 μM cofactor; (blue) 50 μM HT: 110 μM cofactor; and (yellow with dots) 40 μM HT: 160 μM cofactor. All of the reactions were conducted in 20 mM MOPS, pH 7.0 at room temperature for 2 h (B).
With the two ArMs at hand, the catalytic activities of the protein–cofactor conjugates (N8-HT and Mes8-HT) were examined. A pro-fluorescent substrate (Np7HC) system was used to characterize rapidly the metathesis activity of the ArM.31 Upon reaction with the synthetic catalysts or ArM, Np7HC can be converted into a fluorescent product, 7-hydroxycoumarin, and naphthalene (Figure 3A). Because the elimination step is essentially spontaneous, the production of 7-hydroxycoumarin can be monitored by fluorescence spectroscopy, as a readout for ring-closing metathesis (RCM) activity. The production of naphthalene can be further confirmed by GC-MS. The product concentrations and turnover numbers (TON) were determined by comparing the fluorescence intensity with a calibration curve (Figure S3).
Figure 3.
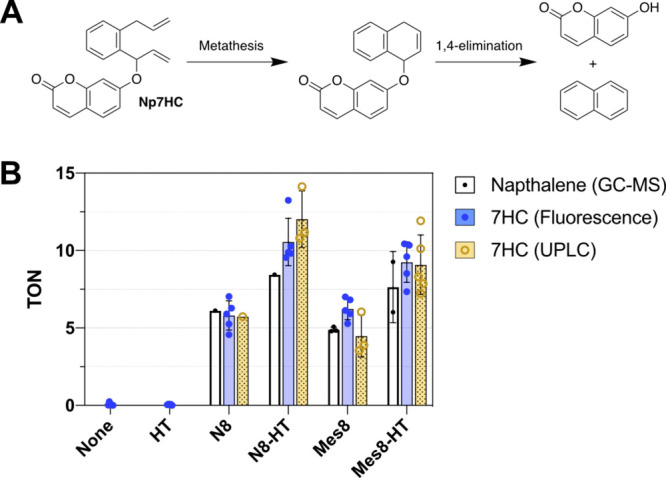
Activity of the cofactors and corresponding ArMs. A reaction that produces the fluorescent 7-hydroxycoumarin was selected for facile characterization (A). The TON for the cofactors N8 and Mes8 were determined by GC-MS, UPLC-MS, and fluorescence analysis (B). The bioconjugation reactions were conducted at 65 μM cofactor and 55 μM HT (Supporting Information, section 11). The metathesis reactions were conducted in 20 mM MOPS, 100 mM MgCl2, pH 7.0 at 25 °C. Each reaction contained 2 μM cofactor, 2 μM HT, and 100 μM substrate. Formation of 7-hydroxycomarin was determined by fluorescence (λex = 330 nm and λem = 450 nm) and UPLC-MS; formation of naphthalene was determined by GC-MS (Figure S3).
Using Np7HC in buffer, we found that the ArM produced a higher TON than the catalyst alone (Figures 3B and S4). The results were confirmed for both reaction products: 7-hydroxycoumarin and naphthalene. The benefit of the ArM is pH-dependent, with the ArM improving the TON more at pH 7.0 than pH 5.0 (Figure S5). This pH dependence could result from protonation changes in HT, low protein stability at pH 5.0, or improved free-cofactor activity at low pH, a frequently reported phenomenon.24,27 Comparison between the cofactors suggests that each cofactor alone has similar activity. Under the bioconjugation conditions used in Figure 3, the N8-HT yields slightly higher TONs than the Mes8-HT. However, when the purified ArMs were examined, both Mes8-HT and N8-HT exhibited similar activity (Figure S4), indicating that more complete bioconjugation increases the TON. Although the TONs were lower than conventional HG-II cofactors in organic solvent, they were comparable to those previously reported for other metathesis-catalyzing ArMs.9,12,15,22−27
Upon confirming that the ArM was competent for catalysis in buffer, we subsequently examined if mutagenesis and directed evolution could produce a more active catalyst, either by increasing the ArM activity or improving the bioconjugation. Both crystallographic data of HT (Figure 4A) and homology modeling (Figure S1) were used to identify suitable residues for mutagenesis.32−34 Residues lining the opening of the alkane binding cavity were the primary targets for mutagenesis. Of these residues, however, some were conserved in the family of dehalogenases from which HT is derived (Figure S1). These conserved residues were not selected for mutagenesis to reduce the chances that mutagenesis would alter the substitution reaction required to form the ArM.
Figure 4.
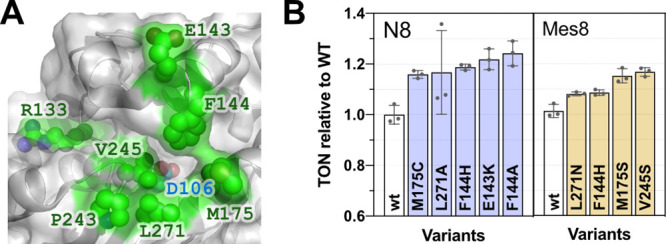
Genetic engineering of the ArM. (A) Analysis of amino acids for mutagenesis based on crystallography (PDB: 5vnp).34 Seven sites were targeted for saturation mutagenesis and activity profiling: E133, E143, F144, M175, P243, V245, and L271. Mutations at five of these sites increased the catalytic performance marginally. The best hits are displayed in panel B. The wt HT samples were completed as biological replicates, and the variants are replicates from the same protein purification batch. The bioconjugation and metathesis reactions were conducted as described for Figure 3.
On the basis of these considerations, seven positions were identified for mutagenesis, and a library of 84 single mutants was designed and screened for TON (Figure 4B). Some of the single mutants displayed up to 120% of wild-type (wt) ArM activity. However, neither recombination of the best mutants nor random mutagenesis further improved activity (Figure S6).
The modest improvements suggest that these ArMs may not be ideally suited for directed evolution. We identified two possible reasons for this observation: (i) the linker length projects the cofactor too far from the protein surface or (ii) the protein provides minimal interaction with the transition state, limiting the effect of the second coordination sphere provided by HT on the catalytic event. Cofactors with shorter linkers were evaluated, but these did not bind efficiently to HT (Figure S2), lending more support to the lack of transition state stabilization.
To examine further the catalytic activity of the wt ArM, the N8-HT was evaluated with several RCM substrates (Figure 5). Characterization of the TON for these substrates was completed by UPLC-MS or 1H NMR (Figures S7–S10). A comparison of these reactions suggests that the N8-HT is capable of catalyzing RCM with multiple substrates. Catalysis with the N8-HT yield both five-membered (pyrrole or cyclopentene) and six-membered rings (naphthalene from Np7HC). However, when provided with a substrate that can undergo RCM to form either a five- or six-membered ring, the ArM yields exclusively the five-membered ring (TenDA) at 34% conversion. The five- and six-membered rings are the kinetically and thermodynamically favored products, respectively.35 The N8-HT was most effective with the substrates BzDA and EnDA. Notably, conversion with the alkyne-based EnDA substrate was near complete (Figure S10). This trend in reactivity is similar to the previously reported ArM using albumin as the scaffold.9
Figure 5.

Substrate scope for metathesis reactions. The reaction TON and percent conversion were determined for wt HT with the N8 cofactor.
In summary, we have identified a new scaffold system for creation of artificial metalloproteins. We have shown that these artificial metalloproteins can act as ArMs for metathesis in aqueous systems at pH 7.0. Additionally, we have shown that chemical optimization and enzyme engineering lead to improvements in the ArM activity. Finally, we have shown that the ArM is capable of catalyzing RCM with diverse substrates. On the basis of these findings, we are currently exploring the HT scaffold as a scaffold for additional reaction types and loop designs to provide stabilization of the transition state.
Acknowledgments
T.R.W. acknowledges the Swiss National Science Foundation (Grant 200020_182046), the NCCR Molecular Systems Engineering and the ERC (DrEAM-Advanced Grant 694424) for their generous financial support. A.D.L. thanks the EU for Marie Skłodowska-Curie fellowship (H2020-MSCA-IF-2017) for funding.
Glossary
ABBREVIATIONS
- ArM
artificial metalloenzyme
- HT
HaloTag
- HG-II
Hoveyda-Grubbs II style catalyst
- RCM
ring-closing metathesis
- TON
turnover number
- λex
excitation wavelength
- λem
emission wavelength
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.1c01470.
Synthetic methods, NMR data, detailed experimental, and additional data (PDF)
Author Contributions
The manuscript was written through contributions of all authors.
T.R.W. acknowledges the Swiss National Science Foundation (Grant 200020_182046), the NCCR Molecular Systems Engineering and the ERC (DrEAM-Advanced Grant 694424) for their generous financial support. A.D.L. thanks the EU for Marie Skłodowska-Curie fellowship (H2020-MSCA-IF-2017) for funding.
The authors declare no competing financial interest.
Supplementary Material
References
- Devine P. N.; Howard R. M.; Kumar R.; Thompson M. P.; Truppo M. D.; Turner N. J. Extending the Application of Biocatalysis to Meet the Challenges of Drug Development. Nat. Rev. Chem. 2018, 2, 409–421. 10.1038/s41570-018-0055-1. [DOI] [Google Scholar]
- de Souza R. O. M. A.; Miranda L. S. M.; Bornscheuer U. T. A Retrosynthesis Approach for Biocatalysis in Organic Synthesis. Chem. - Eur. J. 2017, 23, 12040–12063. 10.1002/chem.201702235. [DOI] [PubMed] [Google Scholar]
- Liang A. D.; Serrano-Plana J.; Peterson R. L.; Ward T. R. Artificial Metalloenzymes Based on the Biotin-Streptavidin Technology: Enzymatic Cascades and Directed Evolution. Acc. Chem. Res. 2019, 52 (3), 585–595. 10.1021/acs.accounts.8b00618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong W. J.; Yu J.; Song W. J. Proteins as Diverse, Efficient, and Evolvable Scaffolds for Artificial Metalloenzymes. Chem. Commun. 2020, 56 (67), 9586–9599. 10.1039/D0CC03137B. [DOI] [PubMed] [Google Scholar]
- Sommer D. J.; Vaughn M. D.; Ghirlanda G. Protein Secondary-Shell Interactions Enhance the Photoinduced Hydrogen Production of Cobalt Protoporphyrin IX. Chem. Commun. 2014, 50 (100), 15852–15855. 10.1039/C4CC06700B. [DOI] [PubMed] [Google Scholar]
- Oohora K.; Onoda A.; Hayashi T. Hemoproteins Reconstituted with Artificial Metal Complexes as Biohybrid Catalysts. Acc. Chem. Res. 2019, 52 (4), 945–954. 10.1021/acs.accounts.8b00676. [DOI] [PubMed] [Google Scholar]
- Heinisch T.; Ward T. R. Artificial Metalloenzymes Based on the Biotin-Streptavidin Technology: Challenges and Opportunities. Acc. Chem. Res. 2016, 49 (9), 1711–1721. 10.1021/acs.accounts.6b00235. [DOI] [PubMed] [Google Scholar]
- Monnard F. W.; Heinisch T.; Nogueira E. S.; Schirmer T.; Ward T. R. Human Carbonic Anhydrase II as a Host for Piano-Stool Complexes Bearing a Sulfonamide Anchor. Chem. Commun. 2011, 47 (29), 8238–8240. 10.1039/c1cc10345h. [DOI] [PubMed] [Google Scholar]
- Eda S.; Nasibullin I.; Vong K.; Kudo N.; Yoshida M.; Kurbangalieva A.; Tanaka K. Biocompatibility and Therapeutic Potential of Glycosylated Albumin Artificial Metalloenzymes. Nat. Catal. 2019, 2 (9), 780–792. 10.1038/s41929-019-0317-4. [DOI] [Google Scholar]
- Chordia S.; Narasimhan S.; Lucini Paioni A.; Baldus M.; Roelfes G. In Vivo Assembly of Artificial Metalloenzymes and Application in Whole-Cell Biocatalysis. Angew. Chem., Int. Ed. 2021, 60 (11), 5913–5920. 10.1002/anie.202014771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava P.; Yang H.; Ellis-Guardiola K.; Lewis J. C. Engineering a Dirhodium Artificial Metalloenzyme for Selective Olefin Cyclopropanation. Nat. Commun. 2015, 6 (7789), 1–8. 10.1038/ncomms8789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippart F.; Arlt M.; Gotzen S.; Tenne S.-J.; Bocola M.; Chen H.-H.; Zhu L.; Schwaneberg U.; Okuda J. A Hybrid Ring-Opening Metathesis Polymerization Catalyst Based on an Engineered Variant of the β-Barrel Protein FhuA. Chem. - Eur. J. 2013, 19 (41), 13865–13871. 10.1002/chem.201301515. [DOI] [PubMed] [Google Scholar]
- Schwizer F.; Okamoto Y.; Heinisch T.; Gu Y.; Pellizzoni M. M.; Lebrun V.; Reuter R.; Köhler V.; Lewis J. C.; Ward T. R. Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem. Rev. 2018, 118 (1), 142–231. 10.1021/acs.chemrev.7b00014. [DOI] [PubMed] [Google Scholar]
- Davies R. R.; Distefano M. D. A Semisynthetic Metalloenzyme Based on a Protein Cavity That Catalyzes the Enantioselective Hydrolysis of Ester and Amide Substrates. J. Am. Chem. Soc. 1997, 119 (48), 11643–11652. 10.1021/ja970820k. [DOI] [Google Scholar]
- Matsuo T.; Imai C.; Yoshida T.; Saito T.; Hayashi T.; Hirota S. Creation of an Artificial Metalloprotein with a Hoveyda-Grubbs Catalyst Moiety through the Intrinsic Inhibition Mechanism of α-Chymotrypsin. Chem. Commun. 2012, 48 (11), 1662–1664. 10.1039/c2cc16898g. [DOI] [PubMed] [Google Scholar]
- Los G. V.; Encell L. P.; Mcdougall M. G.; Hartzell D. D.; Karassina N.; Zimprich C.; Wood M. G.; Learish R.; Ohana R. F.; Urh M.; Simpson D.; Mendez J.; Zimmerman K.; Otto P.; Vidugiris G.; Zhu J.; Darzins A.; Klaubert D. H.; Bulleit R. F.; Wood K. V. HaloTag: A Novel Protein Labeling Technology for Cell Imaging and Protein Analysis. ACS Chem. Biol. 2008, 3 (6), 373–382. 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- Liu D. S.; Phipps W. S.; Loh K. H.; Howarth M.; Ting A. Y. Quantum Dot Targeting with Lipoic Acid Ligase and HaloTag for Single-Molecule Imaging on Living Cells. ACS Nano 2012, 6 (12), 11080–11087. 10.1021/nn304793z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark S. A.; Singh V.; Mendoza D. V.; Margolin W.; Kool E. T. Light-Up “ Channel Dyes ” for Haloalkane-Based Protein Labeling in Vitro and in Bacterial Cells. Bioconjugate Chem. 2016, 27, 2839–2843. 10.1021/acs.bioconjchem.6b00613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm J. B.; English B. P.; Chen J.; Slaughter J. P.; Zhang Z.; Revyakin A.; Patel R.; Macklin J. J.; Normanno D.; Singer R. H.; Lionnet T.; Lavis L. D. SI: A General Method to Improve Fluorophores for Live-Cell and Single-Molecule Microscopy. Nat. Methods 2015, 12 (3), 244–250. 10.1038/nmeth.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Tran M.; D’Este E.; Roberti J.; Koch B.; Xue L.; Johnsson K. A General Strategy to Develop Cell Permeable and Fluorogenic Probes for Multicolour Nanoscopy. Nat. Chem. 2020, 12 (2), 165–172. 10.1038/s41557-019-0371-1. [DOI] [PubMed] [Google Scholar]
- Strauch R. C.; Mastarone D. J.; Sukerkar P. A.; Song Y.; Ipsaro J. J.; Meade T. J. Reporter Protein-Targeted Probes for Magnetic Resonance Imaging. J. Am. Chem. Soc. 2011, 133 (41), 16346–16349. 10.1021/ja206134b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeschek M.; Reuter R.; Heinisch T.; Trindler C.; Klehr J.; Panke S.; Ward T. R. Directed Evolution of Artificial Metalloenzymes for in Vivo Metathesis. Nature 2016, 537 (7622), 661–665. 10.1038/nature19114. [DOI] [PubMed] [Google Scholar]
- Zhao J.; Kajetanowicz A.; Ward T. R. Carbonic Anhydrase II as Host Protein for the Creation of a Biocompatible Artificial Metathesase. Org. Biomol. Chem. 2015, 13 (20), 5652–5655. 10.1039/C5OB00428D. [DOI] [PubMed] [Google Scholar]
- Mayer C.; Gillingham D. G.; Ward R.; Hilvert D. An Artificial Metalloenzyme for Olefin Metathesis. Chem. Commun. 2011, 47, 12068–12070. 10.1039/c1cc15005g. [DOI] [PubMed] [Google Scholar]
- Lo C.; Ringenberg M. R.; Gnandt D.; Wilson Y.; Ward T. R. Artificial Metalloenzymes for Olefin Metathesis Based on the Biotin-(Strept)Avidin Technology. Chem. Commun. 2011, 47, 12065–12067. 10.1039/c1cc15004a. [DOI] [PubMed] [Google Scholar]
- Basauri-Molina M.; Verhoeven D. G. A.; Van Schaik A. J.; Kleijn H.; Klein Gebbink R. J. M. Ring-Closing and Cross-Metathesis with Artificial Metalloenzymes Created by Covalent Active Site-Directed Hybridization of a Lipase. Chem. - Eur. J. 2015, 21 (44), 15676–15685. 10.1002/chem.201502381. [DOI] [PubMed] [Google Scholar]
- Sauer D. F.; Himiyama T.; Tachikawa K.; Fukumoto K.; Onoda A.; Mizohata E.; Inoue T.; Bocola M.; Schwaneberg U.; Hayashi T.; Okuda J. A Highly Active Biohybrid Catalyst for Olefin Metathesis in Water: Impact of a Hydrophobic Cavity in a β-Barrel Protein. ACS Catal. 2015, 5 (12), 7519–7522. 10.1021/acscatal.5b01792. [DOI] [Google Scholar]
- Sauer D. F.; Schiffels J.; Hayashi T.; Schwaneberg U.; Okuda J. Olefin Metathesis Catalysts Embedded in β-Barrel Proteins: Creating Artificial Metalloproteins for Olefin Metathesis. Beilstein J. Org. Chem. 2018, 14, 2861–2871. 10.3762/bjoc.14.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo T.; Miyake T.; Hirota S. Recent Developments on Creation of Artificial Metalloenzymes. Tetrahedron Lett. 2019, 60 (45), 151226. 10.1016/j.tetlet.2019.151226. [DOI] [Google Scholar]
- Garber S. B.; Kingsbury J. S.; Gray B. L.; Hoveyda A. H. Efficient and Recyclable Monomeric and Dendritic Ru-Based Metathesis Catalysts. J. Am. Chem. Soc. 2000, 122 (34), 8168–8179. 10.1021/ja001179g. [DOI] [Google Scholar]
- Sabatino V.; Rebelein J. G.; Ward T. R. Close-to-Release”: Spontaneous Bioorthogonal Uncaging Resulting from Ring-Closing Metathesis. J. Am. Chem. Soc. 2019, 141 (43), 17048–17052. 10.1021/jacs.9b07193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang M.; Lee H.; Kim H.; Dunbayev Y.; Seo J. K.; Lee C.; Rhee H.-W. Structure-Guided Synthesis of a Protein-Based Fluorescent Sensor for Alkyl Halides. Chem. Commun. 2017, 53, 9226–9229. 10.1039/C7CC03714G. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Miao K.; Dunham N. P.; Liu H.; Fares M.; Boal A. K.; Li X.; Zhang X. SI: The Cation-π Interaction Enables a Halo-Tag Fluorogenic Probe for Fast No-Wash Live Cell Imaging and Gel-Free Protein Quantification. Biochemistry 2017, 56, 1585–1595. 10.1021/acs.biochem.7b00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Fares M.; Dunham N. P.; Gao Z.; Miao K.; Jiang X.; Bollinger S. S.; Boal A. K.; Zhang X. AgHalo: A Facile Fluorogenic Sensor to Detect Drug-Induced Proteome Stress. Angew. Chem., Int. Ed. 2017, 56, 8672–8676. 10.1002/anie.201702417. [DOI] [PubMed] [Google Scholar]
- Yoshida K.; Kano Y.; Takahashi H.; Yanagisawa A. Ring Size-Selective Ring-Closing Olefin Metathesis: Taking Advantage of the Deleterious Effect of Ethylene Gas. Adv. Synth. Catal. 2011, 353 (8), 1229–1233. 10.1002/adsc.201000944. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.