

Abstract
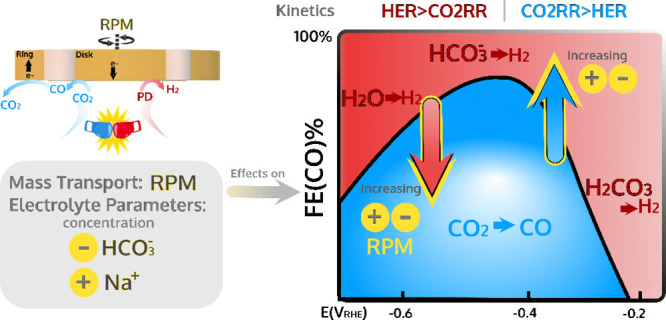
The electrochemical reduction of CO2 aims to be a central technology to store excess electricity generated by wind and solar energy. However, the reaction is hindered by the competition with the hydrogen evolution reaction. In this paper, we present a detailed quantitative study of the Faradaic efficiency (FE) to CO on a gold electrode under well-defined mass-transport conditions using rotating ring-disk electrode voltammetry. Varying the concentration of the bicarbonate and the electrolyte cation employing different rotation rates, we map out how these parameters affect the FE(CO). We identify two different potential regimes for the electrolyte effects, characterized by a different dependence on the cation and bicarbonate concentrations. For hydrogen evolution, we analyze the nature of the proton donor for an increasingly negative potential, showing how it changes from carbonic acid to bicarbonate and to water. Our study gives detailed insights into the role of electrolyte composition and mass transport, and helps defining optimized electrolyte conditions for a high FE(CO).
Keywords: electrochemical CO2 reduction, gold, electrolyte, proton donor, bicarbonate, cation, mass transport
Introduction
The ambition of reaching net-zero or even negative CO2 emissions drives the transition from a fossil fuel-based economy to a renewable-energy-based economy. A cornerstone in this energy transition is the storage of renewable energy into chemical bonds by means of electrochemical processes.1−4 An attractive option for the storage of excess renewable electricity is the production of simple carbon-containing molecules derived from the electrochemical reduction of CO2. Among heterogeneous electrocatalysts, gold is the metal exhibiting the highest activity for CO2 reduction to carbon monoxide (CO) (CO2RR) (eq 1) at low overpotential.5−9
| 1 |
However, in aqueous electrolytes, CO2RR occurs simultaneously with the hydrogen evolution reaction (HER), limiting the Faradaic efficiency (FE) for the CO2RR reaction [FE(CO)]. CO2RR is commonly carried out in bicarbonate electrolytes, given the desired near-neutral bulk pH (ca. 7) and its buffering ability.10 Considering the available proton donors (PDs) in bicarbonate electrolytes, we write the following possible electrochemical reactions leading to HER:
| 2 |
| 3 |
| 4 |
(the contribution of proton reduction at neutral pH is excluded here). Knowledge on the dominant pathway for HER between (2), (3), and (4) as a function of the applied potential is scarce. To design an optimal system, an understanding of the experimental conditions (e.g., concentration of the electrolyte species, pH, and mass transport) governing the competition between CO2RR and HER is essential to increase the FE(CO). Wuttig et al. investigated thoroughly the electrocatalytic activity of gold in bicarbonate electrolytes reporting that, while CO2RR is largely independent of the electrolyte environment, HER is strongly affected by the bicarbonate concentration, the pH, and the mass-transport conditions.11,12 In contrast to the results by Wuttig et al., who measured a 0th reaction order in bicarbonate for CO2RR,12 Dunwell et al. reported a reaction order of 1 and proposed that bicarbonate sustains the surface concentration of CO2 through homogeneous equilibria.13 Studies on the role of bicarbonate as PD for HER pointed to the importance of the pKa of the PD in relation to the electrolyte pH when evaluating the ability of a species to act as a PD.14,15 Previous estimates of the reaction order of HER in bicarbonates, which has been measured to increase from 0.05 to 0.5 with the overpotential, did not take into consideration the necessity of keeping the concentration of the cation constant over the examined bicarbonate concentration range.11 This may have led to a convolution of the bicarbonate effect with the cation effect. Cations are well-known to have an impact both on HER16,17 and on CO2RR18−23 rates. The cation effect on CO2RR has been hypothesized to highlight the relevance of the double-layer composition in screening the interfacial electric field.13,20,24 To the best of our knowledge, no study has focused on how the cation concentration influences the rates of both CO2RR and HER, in particular in relation to bicarbonate concentration and mass-transport conditions.
Complicating the understanding of the catalytic activity is the interplay of homogeneous reactions related to the bicarbonate equilibria25 together with the mass-transport characteristics of the electrochemical cell.26 The intricate relationship between the concentration of different species (CO2, HCO3–, and OH–) at the electrode surface couples with the current-induced build-up in alkalinity near the electrode surface.12,27 Elucidating mass-transport effects is essential when comparing the activity of different porous catalysts, where transport of species to the electroactive surface may be decisive.25,28−30 Multiple studies have examined the effect of mass transport, either qualitatively by magnetic stirring of the solution31 and more quantitatively by coupling rotating cone electrode measurements with online gas chromatography11,12,32 or by varying the flow rate of CO2.24,26 Generally, CO2RR is not mass transport limited at low cathodic currents, while it is at higher current densities.24−26,30−32 Due to the currently incomplete picture of possible PDs, the effect of mass transport on HER has remained unresolved.11,24,26,32
The rotating ring-disk electrode (RRDE) is a quantitative in situ technique to determine the product formed at the disk electrode while changing the mass transport conditions. Previous researchers adopted a RRDE setup with a Pt ring33,34 or a Au ring35 to probe qualitatively or semi-quantitatively, respectively, the CO2RR products evolved at the disk. Owing to the outstanding activity of gold as an electrocatalyst for CO electro-oxidation,36,37 we recently demonstrated that a RRDE with a gold ring can be used to quantitatively measure CO by carefully choosing the electrolyte conditions.38
Herein, we give a comprehensive study on the effect of bicarbonate (HCO3–) concentration, cation (Na+) concentration, and mass transport rate on CO2RR and HER, and thus on the FE(CO), by exploiting the previously described RRDE system.38 Notably, in a RRDE system, the diffusion layer thickness is in the range of 1 μm,39 which is at least 1-2 orders of magnitude smaller than that obtained by the stirring of the solution or by imposing flow rate.24 Hence, the RRDE greatly improved the mass-transport conditions allowing more confident measurements of the intrinsic catalytic activity of the electrochemical processes. Through the systematic study of gold activity by RRDE, we propose that the nature of the main PD for HER depends on the potential applied and on the current density, which in turn determines the surface pH. By elucidating the nature of the PD as a function of the potential, we can explain the universally observed bell-shaped curve for the FE(CO) as a function of potential in relation to the effect of PD, cation concentration, and mass transport on the rate of the competing HER.
Experimental Section
Chemicals and Materials
Electrolytes were prepared from H2SO4 (98 %, EMSURE, Merck), NaClO4(H2O)x (≥99.99 % trace metal basis, Sigma-Aldrich), and NaHCO3 anhydrous (≥99.7 % trace metal basis, Honeywell Fluka) using MilliQ water (resistivity > 18.2 MΩ cm). Prior to use, NaClO4(H2O)x was dried under vacuum to remove the water. Electrolytes were prepared using NaHCO3 and dehydrated NaClO4 by varying the bicarbonate concentration (see Table 1) and by varying the cation concentration (see Table 2).
Table 1. Electrolytes Used for Studying the Bicarbonate Dependence.
| [NaHCO3]/M | [NaClO4]/M | [Na+]/M | pH (CO2 sat.) |
|---|---|---|---|
| 0.01 | 0.49 | 0.5 | 5.7 |
| 0.05 | 0.45 | 0.5 | 6.2 |
| 0.1 | 0.4 | 0.5 | 6.7 |
| 0.25 | 0.25 | 0.5 | 7.0 |
| 0.5 | 0.5 | 7.4 |
Table 2. Electrolytes Used for Studying the Cation Dependence.
| [NaHCO3]/M | [NaClO4]/M | [Na+]/M | pH (CO2 sat.) |
|---|---|---|---|
| 0.1 | 0.1 | 6.7 | |
| 0.1 | 0.1 | 0.2 | 6.7 |
| 0.1 | 0.2 | 0.3 | 6.7 |
| 0.1 | 0.4 | 0.5 | 6.7 |
The electrolytes were purged for 20 min before electrochemical experiments with Ar (6.0 purity, Linde) or CO2 (4.5 purity, Linde).
For the coating of the RRDE tip (E6/E5 ChangeDisk, Pine Research Instrumentation), dopamine hydrochloride (Sigma-Aldrich) was used to suppress bubble attachment at the spacer between the disk and ring.40
Three electrochemical cells in a three-electrode configuration were used to perform the experiments at room temperature (see Figure S3 in the Supporting Information). The first cell (cell 1) was used to remove the dopamine coating from the gold electrode and for electrode characterization, the second cell (cell 2) to study the electrocatalytic activity of gold in bicarbonate electrolytes, and the third cell (cell 3) served to determine the RRDE collection efficiency factor. All three cells featured a gold counter electrode (99.99% purity). In cell 2, the counter electrode was separated by a glass frit from the main compartment. For the reference electrode, in cell 1, a home-made reversible hydrogen electrode (RHE) was adopted, while in cells 2 and 3, a Ag/AgCl reference electrode (KCl-saturated, Pine Research Instrumentation) was employed. In all cases, the reference electrode was located in a Luggin capillary. All measurements were performed using an IviumStat bipotentiostat (Ivium Technologies) and a Modulated Speed Rotator (Pine Research Instrumentation).
Experimental Procedure
The glassware was stored overnight in a 1 g L–1 KMnO4 solution to assure the removal of organic contaminants. Before experiments, the KMnO4 and the as-formed MnO2 were removed by adding acidified H2O2. The glassware was then thoroughly rinsed with MilliQ water and boiled a total of five times. The RRDE tip was assembled by inserting an interchangeable gold disk (⌀ = 5.0 mm, Pine Research Instrumentation) in a tip with a fixed gold ring embedded in a PEEK shroud. The as-assembled tip was polished with diamond suspensions of decreasing particle sizes (3, 1, 0.25, and 0.05 μm).41 In order to remove the residual diamond particles, the tip was placed in an ultrasonication bath in a mixture of MilliQ water and acetone for 15 min. Afterward, the tip was mounted on the shaft of the rotator and immersed in a solution of 0.002 g/mL dopamine hydrochloride in 0.1 M NaHCO3 heated at ca. 50 °C (to speed up the polymerization process). To obtain a homogeneous dopamine coating, the RRDE tip was rotated at 800 rpm for ca. 1 h. The coating by this hydrophilic polymer (dopamine) makes the PEEK parts of the RRDE less hydrophobic, preventing the attachment of bubbles.40 Bubbles may lead to an irreproducible collection efficiency (see Figures S4 and S5 in the Supporting Information).
The dopamine stripping from the electroactive surfaces was performed by electrooxidation. In brief, we short-circuited the disk and the ring, and we performed cyclic voltammetry (CV) in Ar-saturated 0.1 M H2SO4 at 1 Vs–1 between 0.08 and 1.75 V vs RHE (cell 1) until complete removal of the dopamine. Then, in the same potential range, we characterized the disk and the ring, separately, by CV at 20 mVs–1 (see Figure S1 in the Supporting Information).
The electrocatalytic activity of gold in CO2-saturated bicarbonate solutions was tested in cell 2. The electrolyte pH was checked after 20 min of CO2-purging with a pH-meter (SI Analytics Lab 855 Benchtop Meter). First, the Ohmic resistance was measured by electrochemical impedance spectroscopy, and CVs were then corrected for 85% of the measured Ohmic drop. Second, the disk and ring were short-circuited and scanned at 1 Vs–1 between 0.08 and 1.75 V vs RHE to assure a pristine electrode surface after the transfer of the RRDE tip from cell 1 to cell 2. Next, the activity of the gold disk was measured by CV at 20 mVs–1 between 0.0 and −0.8 V vs RHE starting at 0.08 V, while the ring potential was set to 1.0 V vs RHE. Since a low CV scan rate leads to more bubble accumulation and a decrease in the ring collection efficiency, we choose a scan rate of 20 mV s–1. For our experimental setup rotating at 2500 rpm, we did not detect major differences in currents upon changing the scan rate (see Figures S6 and S7 in the Supporting Information), hence we can assume that the measured currents approach the steady state. The CVs were taken at 400 and 2500 rpm, and the ring and the disk were re-activated in between each rotation by fast scanning in the double-layer and oxide region (0.08–1.75 V vs RHE).
Finally, the RRDE tip was transferred to cell 3 to determine the experimental collection efficiency factor in Ar-saturated 0.1 MKNO3/0.01 M K3Fe(CN)6. Starting from 0.6 V vs Ag/AgCl, the gold disk was scanned to −0.2 V vs Ag/AgCl keeping the ring at an oxidation potential of 0.6 V vs Ag/AgCl. The measurement was performed for both rotation rates, that is, 400 and 2500 rpm.
The first scan of the voltammetry was used, and the reproducibility of the system was confirmed after re-electroactivation of the disk. Each measurement to determine the reaction order in HCO3– and in Na+ was repeated three times. Data points give the average of these measurements.
Processing of the RRDE Data
The disk currents were normalized by the electrochemically active surface area (ECSAdisk) to obtain jdisk. The ECSAdisk was determined by integrating the reduction peak from the characterization CV of the gold disk between 0.93 and 1.35 V vs RHE divided by the charge corresponding to a gold monolayer (390 μC cm–2).42 A detailed study on the selectivity of the gold ring to CO electro-oxidation over hydrogen oxidation, and Koutecky–Levich analysis of CO electro-oxidation in a bicarbonate electrolyte can be found in our previous work.38 Having determined the experimental collection efficiency N (see eq S1 in the Supporting Information), we calculated the current density for CO2RR to CO as
| 5 |
where iring (mA) is the oxidation current measured at the ring. We corrected jCO for the experimental time delay, which is the time that a species takes to transfer from the ring to the disk.39 The time delay is rotation dependent: it is negligible at 2500 rpm, while at 400 rpm it is ca. 0.45 s (i.e., 9 mV at 20 mVs–1).39 Additionally, the initial background-capacitive current of the ring was set to zero. In this way, only the Faradaic ring current corresponding to the CO oxidation process was taken into account. Hence, we extracted the current density for HER as
| 6 |
The FE(CO) can be simply calculated from
| 7 |
considering that at the potentials studied in our work, the majority of the investigations of CO2RR on gold surfaces reported CO as the only product.11,20,25 Throughout the paper, the data are reported on the RHE scale. For comparison to the literature, in the Supporting Information, the data are also reported on the SHE scale. The experimental potentials measured on the Ag/AgCl scale have been converted according to
| 8 |
| 9 |
where the pH corresponds to the bulk pH as reported in Table 1 and in Table 2.
Results and Discussion
Bicarbonate Concentration Dependence
Using the ring of the RRDE to probe the amount of CO evolved at the disk electrode, we measured the bicarbonate dependence of CO2RR current (jCO) and of the HER current (jH2). Figure 1A,B displays the CO2RR and HER currents, respectively, for different bicarbonate concentrations as measured by RRDE at 2500 rpm with the Na+ concentration fixed at 0.5 M. Comparison of Figure 1A,B shows that both CO2RR and HER are favored, to a different extent, by an increase in bicarbonate concentration. From jCO, we calculated the FE(CO) at 2500 rpm between 0.0 and −0.8 V vs RHE, as depicted in Figure 1C. Figure 1D displays the effect of rotation rate (400 and 2500 rpm) on the FE(CO) for a given electrolyte.
Figure 1.

(A) CO2RR and (B) HER currents and (C) FE(CO) in CO2-saturated bicarbonate electrolytes with different concentrations of NaHCO3 and a constant concentration of Na+ (0.5 M), as measured by RRDE voltammetry at 20 mVs–1 and 2500 rpm. (D) FE(CO) for different bicarbonate concentrations at 400 and 2500 rpm.
The FE(CO)-potential curve exhibits a distinct bell shape independent of the bicarbonate concentration. In the potential window examined, we can distinguish two opposite regimes for the bicarbonate dependence of the FE(CO), separated by a potential region (−0.35/–0.45 V vs RHE) where the FE(CO) is maximum. In the first regime (E > −0.4 V vs RHE), the FE(CO) is promoted by an increase in the bicarbonate concentration. In the second regime (E < −0.5 V vs RHE), the FE(CO) is suppressed by an increase in the bicarbonate concentration. The trend gained in the second regime agrees with the reported dependence of FE(CO) on the bicarbonate concentration as determined by steady-state measurements at a few selected potentials, even though in these previous studies the bicarbonate effect was convoluted with the cation effect.11,43,44
To understand the trend observed for the FE(CO), we analyzed the dependence of CO2RR and HER on bicarbonate concentration. From the experimental data, we calculated the reaction order in bicarbonate (mHCO3–) for CO2RR and HER, which is defined as
| 10 |
where x = H2 and CO, and E(V) is the electrode potential. The reaction order is calculated on a constant potential on the RHE scale for both CO2RR and HER.
Figure 2A shows the experimentally measured reaction order in bicarbonate for CO2RR at 2500 rpm, which is positive ca. 0.7 at −0.4 V vs RHE and it approaches zero for increasingly negative potential. The high positive reaction order measured for CO2RR close to the onset potential may be rationalized by a reaction mechanism consisting of a coupled proton–electron transfer.45 For more negative applied potential, the decrease in the bicarbonate reaction order for CO2RR may be attributed to the build-up of a pH and concentration gradient for increasing total current density, which scales with the bicarbonate concentration (see Figure S17 in the Supporting Information). Previous studies measured a reaction order of 0 in bicarbonate for CO2RR on the SHE scale under stagnant conditions.11,12 Extracting the reaction order in bicarbonate for CO2RR on the SHE scale, we also obtained a similar value (see Figure S11 in the Supporting Information).
Figure 2.
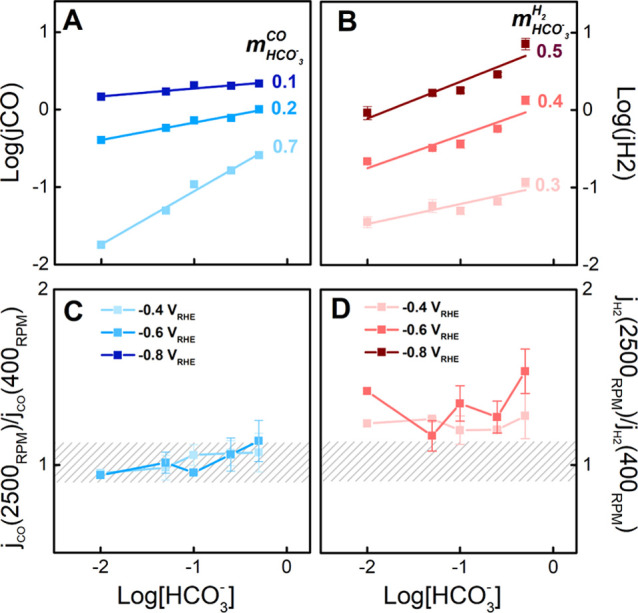
Reaction order in bicarbonate for (A) CO2RR and (B) HER, as extracted from RRDE measurements in CO2-saturated electrolytes at 20 mV s–1 and 2500 rpm, as reported in Figure 1. Rotation dependence expressed as the ratio of the current at 2500 rpm to the current at 400 rpm for (C) CO2RR and (D) HER as a function of bicarbonate concentration.
As shown in Figure 2B, HER has a positive reaction order in bicarbonate, which increases from 0.3 to 0.5 with more negative applied potential. The positive effect of the bicarbonate concentration on the HER currents is still observed when plotting the results on a pH-independent scale (see Figure S10B in the Supporting Information). Besides the difference in the experimental conditions, a similar reaction order of ca. 0.5 at −1.0 V vs SHE for HER in bicarbonate was previously reported.11
To summarize, the two different potential regimes
observed for
the FE(CO) dependence on bicarbonate are resolved in terms of the
overall beneficial role of bicarbonate for CO2RR in the first regime
( = 0.7 and
= 0.7 and  = 0.3) and for HER in the second regime
(
= 0.3) and for HER in the second regime
( = 0.1 and
= 0.1 and  = 0.5).
= 0.5).
Considering the effect of mass transport at constant bicarbonate concentration, the FE(CO) is independent of the rotation speed in the first regime and decreases with the rotation speed in the second regime (Figure 1D). Figure 2C,D shows the mass-transport dependence for CO2RR and HER, respectively, expressed as the current at 2500 rpm divided by the current at 400 rpm for the respective electrochemical processes. CO2RR shows a negligible rotation dependence, as the value for jCO (2500 rpm)/jCO (400 rpm) is ca. 1 independently of the applied potential and bicarbonate concentration.11,28 Previous reports observed an increase in CO2RR currents with mass transport. This effect may be ascribed to the larger CO2RR currents measured (jCO > 3 mA cm–2) combined with a larger diffusion layer thickness (>40 μm) compared to our experimental conditions.24,26,30 Analogously, in our study for jCO > 2 mA cm–2, we observe an improvement of CO2RR currents with mass transport (see Figures S4 and S5 in the Supporting Information). On the other hand, HER exhibits a positive rotation dependence which intensifies at larger overpotentials.11,28 Essentially, at these current densities and diffusion layer thicknesses, in the second regime the improved mass transport favors HER over CO2RR and, hence, the FE(CO) drops. We note that, in comparison to the literature, our current densities are normalized by the real surface area, whereas practical current densities relate to high surface area catalysts normalized by geometrical area. For higher current density, we suggest that improved mass transport is needed to sustain CO2RR (see Figures S4 and S5 in the Supporting Information). To operate CO2RR at a high current density, the gas diffusion electrode (GDE) is widely used, which circumvents the issue of mass-transport limitation in CO2RR.46 The GDE allows to selectively improve the mass transport of the feeding gas (CO2) but not that of the electrolyte species (e.g., HCO3–). Still, finding the right concentration of HCO3 is crucial to suppress the increase in pH at the electrode surface through its fast buffering ability, without leading to an increase in HER. In this way, bicarbonate would help to minimize the issue of mass balance in GDE cells,47 which may lead to CO2 consumption by its homogeneous reaction.
Cation Concentration Dependence
In this section, we will discuss the effect of the Na+ cation concentration on the CO2RR, HER, and FE(CO) at constant bicarbonate concentration. Figure 3A shows jCO and Figure 3B shows jH2 for increasing Na+ concentration at 2500 rpm. For increasing cation concentration, both jCO and jH2 increase to a different extent. The resulting FE(CO) at 2500 rpm is shown in Figure 3C, and the effect of the rotation speed (400 and 2500 rpm) on the FE(CO) in a given electrolyte is shown in Figure 3D.
Figure 3.

(A) CO2RR and (B) HER currents and (C) FE(CO) in CO2-saturated bicarbonate electrolytes with different concentrations of Na+ and a constant concentration of NaHCO3 (0.1 M), as measured by RRDE voltammetry at 20 mV s–1 and 2500 rpm. (D) FE(CO) for different cation concentrations at 400 and 2500 rpm.
Two different regimes for the effect of Na+ concentration on the FE(CO) are distinguishable. In the first regime (E > −0.4 V vs RHE), the FE(CO) slightly increases with the increasing cation concentration. Inversely, in the second regime (E < −0.4 V vs RHE), the FE(CO) decreases with the increasing cation concentration. The maximum FE(CO) is ca. 85% in 0.1 M NaHCO3 at ca. −0.5 V vs RHE. Similarly to the bicarbonate dependence, we evaluated the reaction order in sodium cation (mNa+x) for CO2RR and HER, which is defined as
| 11 |
Figure 4A,B
displays
the reaction order in the cation for CO2RR and HER, respectively.
For CO2RR, the reaction order in cation decreases from a positive
value of 0.3 to 0.1 as the potential becomes more negative. By contrast,
the cation dependence of HER becomes more pronounced for more negative
potential, being 0.2 at −0.4 V and 0.5 at −0.8 V vs
RHE. Close to the CO2RR onset potential (−0.2/–0.4 V
vs RHE), the cation promotes CO2RR ( = 0.3) in agreement with the positive effect
of the cation on the CO2RR kinetics reported by Liu et al.19 At more cathodic potential, however, the beneficial
effect of the cation on CO2RR may be counterbalanced by the larger
promotion of HER. This promotion of HER by the cation suggests a compelling
dependence of the PD activity on the composition of the electrode
double layer.15 We exclude that the large
differences observed for reaction rates upon changing in the cation
concentration can mainly be ascribed to migration effects. Differences
in electrolyte conductivity are corrected by Ohmic drop compensation.
Furthermore, as most of the current arises due to concentration gradients
in the thin diffusion layer of the RRDE, we expect a negligible contribution
of migration compared to the diffusion component to the current.39 Metals impurities have been reported to adsorb
on the electrode surface at cathodic potentials, enhancing the HER
activity at the expense of CO2RR rates.24,48 Hence, to
exclude that the observed cation concentration dependence is an effect
of a higher concentration of metal impurities in the electrolyte,
we performed the RRDE measurements in electrolytes prepurified by
means of a chelating agent (see Figure S20 in the Supporting Information). The results obtained in the prepurified
electrolyte agree with the cation concentration dependence observed
in Figure 3.
= 0.3) in agreement with the positive effect
of the cation on the CO2RR kinetics reported by Liu et al.19 At more cathodic potential, however, the beneficial
effect of the cation on CO2RR may be counterbalanced by the larger
promotion of HER. This promotion of HER by the cation suggests a compelling
dependence of the PD activity on the composition of the electrode
double layer.15 We exclude that the large
differences observed for reaction rates upon changing in the cation
concentration can mainly be ascribed to migration effects. Differences
in electrolyte conductivity are corrected by Ohmic drop compensation.
Furthermore, as most of the current arises due to concentration gradients
in the thin diffusion layer of the RRDE, we expect a negligible contribution
of migration compared to the diffusion component to the current.39 Metals impurities have been reported to adsorb
on the electrode surface at cathodic potentials, enhancing the HER
activity at the expense of CO2RR rates.24,48 Hence, to
exclude that the observed cation concentration dependence is an effect
of a higher concentration of metal impurities in the electrolyte,
we performed the RRDE measurements in electrolytes prepurified by
means of a chelating agent (see Figure S20 in the Supporting Information). The results obtained in the prepurified
electrolyte agree with the cation concentration dependence observed
in Figure 3.
Figure 4.

Reaction order in the Na+ cation for (A) CO2RR and (B) HER, as extracted from RRDE measurements in CO2-saturated electrolytes at 20 mV s–1 and 2500 rpm. Rotation dependence expressed as the ratio of the current at 2500 rpm to the current at 400 rpm for (C) CO2RR and (D) HER.
Considering the mass-transport effect, we observe a switch in the FE(CO)-rotation dependence trend for increasing cation concentration. At low Na+ concentration (0.1 M), the FE(CO) is larger for a higher rotation rate, but for concentrations of 0.5 M Na+, the opposite is true. Figure 4C,D shows the effect of the rotation rate on CO2RR and HER separately. At these applied potentials, CO2RR is independent of mass transport. On the other hand, HER exhibits a rotation dependence which depends on the cation concentration. The ratio jH2 (2500 rpm)/jH2 (400 rpm) is lower than 1 in 0.1 M Na+, while it increases to a value of ca. 1.2 in 0.5 M Na+. This intricate rotation dependence points to the sensitivity of the HER to the electrode–electrolyte environment, whose compositional changes may lead to a variation in the branching of the HER. In conclusion, the observed FE(CO)-rotation rate dependence as a function of Na+ concentration is predominantly a reflection of the effect of rotation on the jH2.
Nature of the PD
As recently reported by Zhang et al.,25 we can distinguish three regimes in the FE(CO)-potential plot. The first regime is observed at low overpotential E ≥ −0.4 V vs RHE, in which the FE(CO) increases from 0 to maximum. In the second regime, between −0.4 and −0.5 V vs RHE, the FE(CO) is maximum. In the third regime, by further polarizing the cathodic potential to E ≤ −0.5 V vs RHE, the FE(CO) drops proportionally with the bicarbonate concentration. In this section, we will discuss the nature of the PD for HER in these three regimes of the FE(CO), considering that the total HER current is given by
| 12 |
The hydronium ion is not considered as a viable PD under these experimental conditions, as its concentration is too low, that is ≤10–6M.
Notably, in the first regime, the onset potential for HER (i.e., increase in jH2) is less negative in CO2-saturated electrolytes than in Ar-saturated. In Figure 5A the HER current at low overpotential in Ar and in CO2-saturated electrolytes are compared. This low HER onset potential cannot be explained in terms of H2O reduction. Various previous papers have indicated that H2O reduction on gold electrodes is favored by increased alkalinity.24,38,51−53 Hence, in bicarbonate electrolytes, H2O reduction rates should be higher at pH ca. 9 (Ar-saturated) than at pH 5.7–7.4 (CO2-saturated).
Figure 5.

(A) HER currents in Ar and in CO2-saturated electrolytes with varying bicarbonate concentrations and a constant sodium concentration of 0.5 M at 20 mV s–1 and 2500 rpm. (B) Tafel slope for HER in CO2-saturated (empty) and Ar-saturated (solid) 0.5 M NaHCO3 derived from CV.
Consequently, at a low overpotential, the PD must be H2CO3 or HCO3–. To compare the ability of these two species to act as a PD, we consider the pKa, the bulk concentration, and the mass transport-limiting currents (Table 3). The pKa of a PD has been proposed to be related to the thermodynamic driving force for HER.14,15,25,54 Given that the pKa of H2CO3 is 6.3 and that of HCO3 is 10.3, we would expect that in CO2-saturated electrolytes, H2CO3 reduction to H2 will be thermodynamically more favorable than HCO3– reduction. In CO2-saturated bicarbonate electrolytes, the concentration of H2CO3 is several orders of magnitude lower than the concentration of HCO3, while in Ar-saturated electrolytes, due to the higher pH, the concentration of H2CO3 is insignificant and H2CO3 is not a viable PD. In CO2-purged electrolytes, the low concentration of H2CO3 leads to low limiting currents, in the order of 0.11–0.17 mA cm–2, while the HCO3– limiting currents are much higher than the experimentally measured currents. Figure 5B shows the Tafel slope (TS) for HER in CO2 and Ar-saturated 0.5 M NaHCO3 (a similar potential trend for the TS is obtained from chronoamperometry measurements as seen in Figure S21). Clearly, the HER onset potential is lower in CO2-saturated bicarbonate electrolyte (ca. −0.3 V vs RHE), and the TS is ca. 120 mV dec–1 in accordance with a first electron-transfer step being rate-determining. We suggest that the PD for HER in this low cathodic potential range is H2CO3 (eq 2), explaining the shift in the onset potential between CO2 and Ar-saturated solutions. As the overpotential increases, the TS increases to a value larger than 120 mV dec–1 probably because the H2CO3 reduction becomes limited by mass transport. Interestingly, in CO2-saturated 0.01 M HCO3, the HER current results in a plateau at ca. −0.35 V. The HER current plateau increases with rotation rate and scan rate (see Figure S22 in the Supporting Information), pointing to a mass transport-limiting process. Comparison of the experimentally observed plateau current for HER and the theoretically limiting currents for different PD (Table 3) supports the hypothesis of H2CO3 being the PD at these potentials. The plateau current for HER disappears when increasing the bicarbonate concentration. Because of the slow kinetics of CO2 hydration,12 we suggest that the surface concentration of H2CO3 is sustained by the faster equilibrium with bicarbonate, explaining the larger HER current for increasing bicarbonate concentration. Furthermore, the low reduction potential of H2CO3 to H2 would explain the universally observed bell-shaped FE(CO) in bicarbonate electrolytes, and particularly, why at low current density, initially the FE(CO) is close to zero [and FE(H2) is close to 100%]. We propose that the competition between CO2RR and HER from H2CO3 reduction results in the FE(CO) increasing from 0 to the maximum in the first regime, as shown in Figures 1C and 3C.
Table 3. PDs in CO2-Saturated Bicarbonate Electrolytes.
The concentrations were calculated by employing the method described in Ref (49) for bicarbonate concentrations in the range 0.01–0.5 M (with [CO2]aq = 33 mM)50 using the measured experimental pH.
Limiting currents39 at 2500 rpm for the calculated bulk species concentrations. Details of the calculations can be found in the Supporting Information.
Interestingly, the onset of the second regime (E ≤ −0.43 V vs RHE) coincides with the onset potential of HER in Ar-saturated electrolytes. The TS of ca. 120 mV dec–1 in an Ar-saturated electrolyte again suggests a first electron-transfer step being rate-determining. We propose that at potentials more negative than ca. −0.43 V, HCO3– becomes the dominant PD for HER (eq 3). In the range −0.4/–0.5 V vs RHE, the TS remains ca. 120 mV dec–1 in an Ar-purged bicarbonate solution where HCO3 is the only PD, while in a CO2-containing solution, a larger TS is observed because of the mixed reduction of H2CO3 and HCO3–. The hypothesis of HCO3 being a PD agrees with the observed reaction order of ca. 0.4–0.5 in bicarbonate for HER. As a larger HCO3– concentration corresponds to higher buffer capacity, thus lower surface pH, the increase in HER current cannot be explained in terms of alkalinity-driven H2O reduction. In a survey of the effect of different electrolyte anions on HER, Resasco et al. highlighted that HCO3 may outcompete H2O as PD for HER because in spite of the higher H2O concentration, the pKa of HCO3– is almost 4 orders of magnitude lower than that of H2O (Table 3).14 Additionally, the positive reaction order in Na+ concentration (ca. 0.5) points to a HER mechanism which benefits from a large cation surface concentration. As suggested by Jackson et al., the formation of a PD complex in the double layer may profit from the presence of surface cations, especially in the case of a negatively charged PD (e.g., HCO3).15
Ultimately, in the third potential window, the TS reaches a value of ca. 210 mV dec–1 in both electrolytes (Ar and CO2-saturated) in accordance with a previous report.11 At potentials more cathodic than −0.5 V vs RHE, the TS increases in Ar and stops decreasing in CO2-saturated electrolytes, which may be explained by the onset of H2O reduction leading to a larger surface pH gradient according to eq 4. This onset potential for H2O reduction agrees with the suppression of HER currents upon increased mass transport, attributed to the apparently pH-dependent H2O reduction branch of HER in 0.1 M NaHCO3.38
In conclusion, at a more negative potential, the dominant branch of HER between HCO3– and H2O reduction appears to be related to the Na+ cation concentration, as shown by the switch in the rotation dependence for different concentrations of Na+ and on the concentration of HCO3. In an electrolyte concentration of 0.1 M Na+, H2O reduction may be preponderant, while for a concentration of 0.5 M Na+, HCO3– reduction is the dominant branch of HER. In the latter case, an increasing rotation rate favors HER, indicating that the PD surface concentration is affected by the enhanced mass transport. This rotation dependence further corroborates the hypothesis that for concentrations of 0.5 M Na+, the main branch of HER is attributed to HCO3 reduction. However, the main difference is observed once the concentration of Na+ goes from 0.1 to 0.2 M, suggesting that the dominant branch of HER switches from H2O to HCO3– reduction for [Na+] > [HCO3]. As the cathodic polarization induces an increase in the surface pH, HCO3– conversion into CO3 (eq 13) becomes relevant.
| 13 |
Larger rotation rates (smaller diffusion layer thickness) suppress the surface pH.27 Hence, the surface concentration of HCO3– will be higher than that at a lower rotation rate, resulting in larger HCO3 reduction current, that is, larger HER currents for larger rotation rates. As explained in our previous paper, for H2O reduction, the rotation dependence is the opposite.38 H2O reduction on gold prefers high (local) pH, which is suppressed by enhanced mass transport. The change in the nature of the PD for HER as a function of the Na+ concentration indicates a distinct role of the cation in the mechanism of H2O and HCO3– reduction. In the first case, the cation may be involved in the stabilization of the reaction product (OH–), while in the second case, the cation may assist the formation of an anion–cation complex prior to the electrochemical reduction.15 Indeed, a different dependence on the nature of the alkali cation was reported for HER from H2O and from the buffering anion. For the increasing cation size on the gold electrode, H2O reduction is promoted,17 while the reaction order of HER in phosphate decreases going from Na+ to K+.15 Hence, increasing the concentration of Na+ results in a larger promotion of HCO3 over H2O reduction.
Alternatively, we could also consider that bicarbonate may not be a direct PD for HER but a promoter of water reduction. In other words, as water reduction is extremely sensitive to the electrolyte composition,55 we may consider that not only the cation concentration but also the anion concentration may define water reactivity.
Influence of CO2RR and HER Kinetics on the FE(CO)
As previously discussed, we identified two different potential regimes for the effect of the electrolyte on the FE(CO). In the first regime, we observe an increase in FE(CO) by increasing both bicarbonate and cation concentrations, while in the second regime, the dependence is reserved. This opposite dependence of the FE(CO) may be illustrated in terms of the kinetics for CO2RR and HER in the different electrolytes. The following kinetics analyses are not meant to give mechanistic insights but rather to highlight the presence of different kinetics regimes. Hence, we discuss the potential dependence of the TS and not the TS absolute value. Figure 6 shows the TS for CO2RR and HER superimposed on the FE(CO) in electrolytes of different bicarbonate concentrations (0.5, 0.1, and 0.1 M HCO3–).
Figure 6.
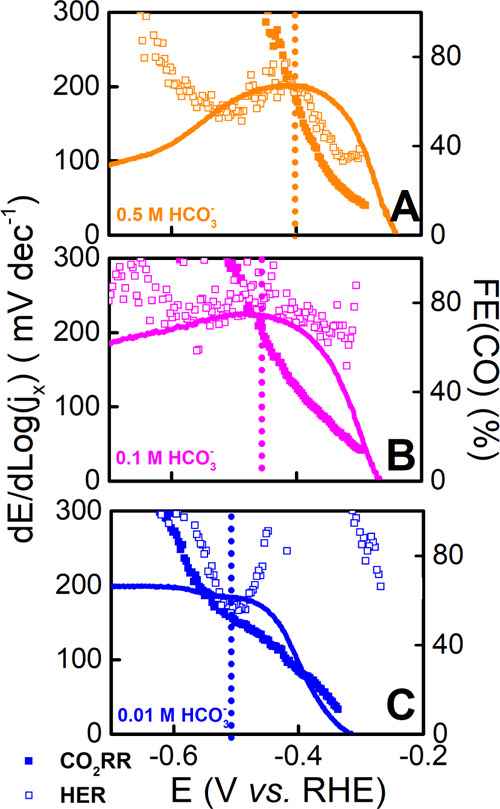
TS for CO2RR (solid symbols) and HER (open symbols) and FE(CO) in CO2-saturated NaHCO3 at (A) 0.5, (B) 0.1, and (C) 0.01 M concentrations and at a constant 0.5 M Na+ concentration derived from CVs.
In the first regime, the TS for CO2RR is lower than that for HER. Hence, after the onset of CO2RR, the FE(CO) increases as the kinetics of CO2RR is more strongly potential-dependent than the one of HER. At these potential values, we proposed that the PD is H2CO3. Due to the low concentration of H2CO3, the HER current becomes mass transport-limited leading to an increase in the TS, which scales with the buffer capacity. As the potential becomes more negative, the TS of CO2RR keeps increasing and reaches a similar value to the one of HER. The FE(CO) is maximum at this potential value, which is more negative for lower bicarbonate concentration. In the second regime, the FE(CO) drops as the kinetics of HER is more strongly potential-dependent than the one of CO2RR. Moreover, a lower TS for HER is measured for increasing bicarbonate concentration resulting in a lower FE(CO). In 0.01 M HCO3– in the second regime, the kinetics for CO2RR and HER are comparable; as a consequence, the FE(CO) remains more or less constant. We would like to remark that for CO2RR, the practice of extracting TS value to gain mechanistic insights, even from chronoamperometry measurements, is complicated. Besides the TS being strongly dependent on the chosen potential interval, CO2RR kinetics is convoluted by the role of CO2 in the suppression of the pH gradient at the electrode surface.
Conclusions
In this paper, we have used RRDE voltammetry to quantify the effect of bicarbonate and cation concentrations on the CO2RR and HER currents, and thus on the FE(CO), on a gold disk. This systematic study allowed us to extend and complete the discussion on the nature of the PD for HER in bicarbonate electrolytes stated in our previous paper,38 as illustrated schematically in Figure 7. The sensitivity of the RRDE measurements revealed for the first time an early onset potential for HER in CO2-saturated vs Ar-saturated bicarbonate electrolytes propounding for H2CO3 being the PD for HER at a low overpotential and explaining the initially low FE(CO). At more negative potential, HCO3– is the dominant PD for HER. The reaction order in HCO3 for HER was measured to be ca. 0.4; hence, increasing the concentration of HCO3– results in a larger current for HER and a decrease in the FE(CO). Only at a potential more negative than ca. −0.5 V RHE, H2O reduction initiates and the FE(CO) declines. At this large negative potential, HCO3 and H2O are both available PDs for HER and the dominant branch for HER is related to both local bicarbonate concentration and (local) cation concentration, and both concentrations can be varied independently. Notably, the rotation dependence of HER depends on the concentration of the electrolyte cation. At a low cation concentration (Na+ ≤ 0.1 M), the HER rate decreases for an increasing rotation rate. Oppositely, for a larger cation concentration (Na+ ≥0.2 M), HER current increases with the rotation rate. Thus, our study suggests that increasing the cation concentration favors the HCO3– reduction branch of HER over the H2O reduction branch.
Figure 7.

Schematic of the bell-shaped FE(CO) as a function of the applied potential illustrating the different PDs for HER in bicarbonate electrolytes and how FE(CO) shifts with electrolyte parameters.
Concerning the nature of the PD for CO2RR, little is known, as the rate-limiting step for CO2RR is believed to be pH-independent.11,12,14,45 Still, valuable considerations about the nature of the PD for CO2RR as a function of the applied potential can be derived from the discussion for HER in bicarbonate electrolytes.
In a broader sense, our findings may help to explain the differences in the HER rates for porous electrodes28,29 in relation to variations in the local properties, for example, local concentration of the cation, as well as differences in the mass-transport effect in different electrode geometries and different electrolytes.11,24,26 Our results will also provide an ideal platform for kinetic modeling because mass transport and electrolyte parameters in our study are very well-defined.
Finally, our investigation of factors governing the kinetics of HER and CO2RR gives guidelines for the choice of optimized reaction conditions to enhance the FE(CO). We identified two different regimes, characterized by an opposite interplay of CO2RR and HER kinetics, for the electrolyte effect on the FE(CO). For low negative potential (first regime), larger HCO3– and Na+ concentration is beneficial for FE(CO). By contrast, at more negative potential (second regime), a low concentration of HCO3, as well as a low concentration of Na+, is preferred to hinder HER. An ideal electrocatalyst for CO2RR should work at a potential less negative than −0.5 V vs RHE, where the kinetics of CO2RR surpasses HER and before the onset of H2O reduction. In this way, building up high surface alkalinity would be greatly suppressed, leading to higher FE(CO).
Acknowledgments
The authors thank Vladislav Antonovich Mints for his contribution to the development of the experimental setup. This project is part of the Solar-to-Products program and of the Advanced Research Center for Chemical Building Blocks (ARC-CBBC) consortium, co-financed by The Netherlands Organization for Scientific Research (NWO) and by “Shell Global Solutions International B.V”.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.1c00272.
Experimental details, electrochemical characterization, scan rate dependence, bicarbonate and cation concentration dependence at 400 rpm, and additional voltammograms (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Whipple D. T.; Kenis P. J. A. Prospects of CO2 Utilization via Direct Heterogeneous Electrochemical Reduction. J. Phys. Chem. Lett. 2010, 1, 3451–3458. 10.1021/jz1012627. [DOI] [Google Scholar]
- Kortlever R.; Shen J.; Schouten K. J. P.; Calle-Vallejo F.; Koper M. T. M. Catalysts and Reaction Pathways for the Electrochemical Reduction of Carbon Dioxide. J. Phys. Chem. Lett. 2015, 6, 4073–4082. 10.1021/acs.jpclett.5b01559. [DOI] [PubMed] [Google Scholar]
- Seh Z. W.; Kibsgaard J.; Dickens C. F.; Chorkendorff I.; Nørskov J. K.; Jaramillo T. F. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 2017, 355, eaad4998 10.1126/science.aad4998. [DOI] [PubMed] [Google Scholar]
- Birdja Y. Y.; Pérez-Gallent E.; Figueiredo M. C.; Göttle A. J.; Calle-Vallejo F.; Koper M. T. M. Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels. Nat. Energy 2019, 4, 732–745. 10.1038/s41560-019-0450-y. [DOI] [Google Scholar]
- Hori Y.; Kikuchi K.; Suzuki S. Production Of CO and CH4 in Electrochemical Reduction of CO2 at metal elctrodes in aqueous hydrogencarbonate solution. Chem. Lett. 1985, 14, 1695–1698. 10.1246/cl.1985.1695. [DOI] [Google Scholar]
- Hori Y.; Murata A.; Kikuchi K.; Suzuki S. Electrochemical reduction of carbon dioxides to carbon monoxide at a gold electrode in aqueous potassium hydrogen carbonate. J. Chem. Soc., Chem. Commun. 1987, 728–729. 10.1039/c39870000728. [DOI] [Google Scholar]
- Noda H.; Ikeda S.; Oda Y.; Imai K.; Maeda M.; Ito K. Electrochemical Reduction of Carbon Dioxide at Various Metal Electrodes in Aqueous Potassium Hydrogen Carbonate Solution. Bull. Chem. Soc. Jpn. 1990, 63, 2459–2462. 10.1246/bcsj.63.2459. [DOI] [Google Scholar]
- Hori Y.; Wakebe H.; Tsukamoto T.; Koga O. Electrocatalytic process of CO selectivity in electrochemical reduction of CO2 at metal electrodes in aqueous media. Electrochim. Acta 1994, 39, 1833–1839. 10.1016/0013-4686(94)85172-7. [DOI] [Google Scholar]
- Hansen H. A.; Varley J. B.; Peterson A. A.; Nørskov J. K. Understanding Trends in the Electrocatalytic Activity of Metals and Enzymes for CO2 Reduction to CO. J. Phys. Chem. Lett. 2013, 4, 388–392. 10.1021/jz3021155. [DOI] [PubMed] [Google Scholar]
- Hori Y. i.Electrochemical CO2 Reduction on Metal Electrodes. Modern aspects of electrochemistry; Springer, 2008; Vol. 42, pp 89–189. [Google Scholar]
- Wuttig A.; Yaguchi M.; Motobayashi K.; Osawa M.; Surendranath Y. Inhibited proton transfer enhances Au-catalyzed CO2-to-fuels selectivity. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, E4585–E4593. 10.1073/pnas.1602984113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuttig A.; Yoon Y.; Ryu J.; Surendranath Y. Bicarbonate Is Not a General Acid in Au-Catalyzed CO2 Electroreduction. J. Am. Chem. Soc. 2017, 139, 17109–17113. 10.1021/jacs.7b08345. [DOI] [PubMed] [Google Scholar]
- Dunwell M.; Lu Q.; Heyes J. M.; Rosen J.; Chen J. G.; Yan Y.; Jiao F.; Xu B. The Central Role of Bicarbonate in the Electrochemical Reduction of Carbon Dioxide on Gold. J. Am. Chem. Soc. 2017, 139, 3774–3783. 10.1021/jacs.6b13287. [DOI] [PubMed] [Google Scholar]
- Resasco J.; Lum Y.; Clark E.; Zeledon J. Z.; Bell A. T. Effects of Anion Identity and Concentration on Electrochemical Reduction of CO2. ChemElectroChem 2018, 5, 1064–1072. 10.1002/celc.201701316. [DOI] [Google Scholar]
- Jackson M. N.; Jung O.; Lamotte H. C.; Surendranath Y. Donor-Dependent Promotion of Interfacial Proton-Coupled Electron Transfer in Aqueous Electrocatalysis. ACS Catal. 2019, 9, 3737–3743. 10.1021/acscatal.9b00056. [DOI] [Google Scholar]
- Hamelin A.; Weaver M. J. Dependence of the kinetics of proton reduction at gold electrodes on the surface cyrstallographic orientation. J. Electroanal. Chem. Interfacial Electrochem. 1987, 223, 171–184. 10.1016/0022-0728(87)85258-0. [DOI] [Google Scholar]
- Xue S.; Garlyyev B.; Watzele S.; Liang Y.; Fichtner J.; Pohl M. D.; Bandarenka A. S. Influence of Alkali Metal Cations on the Hydrogen Evolution Reaction Activity of Pt, Ir, Au, and Ag Electrodes in Alkaline Electrolytes. ChemElectroChem 2018, 5, 2326–2329. 10.1002/celc.201800690. [DOI] [Google Scholar]
- Murata A.; Hori Y. Product Selectivity Affected by Cationic Species in Electrochemical Reduction of CO2 and CO at a Cu Electrode. Bull. Chem. Soc. Jpn. 1991, 64, 123–127. 10.1246/bcsj.64.123. [DOI] [Google Scholar]
- Liu M.; Pang Y.; Zhang B.; De Luna P.; Voznyy O.; Xu J.; Zheng X.; Dinh C. T.; Fan F.; Cao C.; de Arquer F. P. G.; Safaei T. S.; Mepham A.; Klinkova A.; Kumacheva E.; Filleter T.; Sinton D.; Kelley S. O.; Sargent E. H. Enhanced electrocatalytic CO2 reduction via field-induced reagent concentration. Nature 2016, 537, 382–386. 10.1038/nature19060. [DOI] [PubMed] [Google Scholar]
- Ringe S.; Clark E. L.; Resasco J.; Walton A.; Seger B.; Bell A. T.; Chan K. Understanding cation effects in electrochemical CO2 reduction. Energy Environ. Sci. 2019, 12, 3001–3014. 10.1039/c9ee01341e. [DOI] [Google Scholar]
- Resasco J.; Chen L. D.; Clark E.; Tsai C.; Hahn C.; Jaramillo T. F.; Chan K.; Bell A. T. Promoter Effects of Alkali Metal Cations on the Electrochemical Reduction of Carbon Dioxide. J. Am. Chem. Soc. 2017, 139, 11277–11287. 10.1021/jacs.7b06765. [DOI] [PubMed] [Google Scholar]
- Pérez-Gallent E.; Marcandalli G.; Figueiredo M. C.; Calle-Vallejo F.; Koper M. T. M. Structure- and Potential-Dependent Cation Effects on CO Reduction at Copper Single-Crystal Electrodes. J. Am. Chem. Soc. 2017, 139, 16412–16419. 10.1021/jacs.7b10142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayemoba O.; Cuesta A. Spectroscopic Evidence of Size-Dependent Buffering of Interfacial pH by Cation Hydrolysis during CO2 Electroreduction. ACS Appl. Mater. Interfaces 2017, 9, 27377–27382. 10.1021/acsami.7b07351. [DOI] [PubMed] [Google Scholar]
- Clark E. L.; Bell A. T. Direct Observation of the Local Reaction Environment during the Electrochemical Reduction of CO2. J. Am. Chem. Soc. 2018, 140, 7012–7020. 10.1021/jacs.8b04058. [DOI] [PubMed] [Google Scholar]
- Zhang B. A.; Ozel T.; Elias J. S.; Costentin C.; Nocera D. G. Interplay of Homogeneous Reactions, Mass Transport, and Kinetics in Determining Selectivity of the Reduction of CO2 on Gold Electrodes. ACS Cent. Sci. 2019, 5, 1097–1105. 10.1021/acscentsci.9b00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark E. L.; Resasco J.; Landers A.; Lin J.; Chung L.-T.; Walton A.; Hahn C.; Jaramillo T. F.; Bell A. T. Standards and Protocols for Data Acquisition and Reporting for Studies of the Electrochemical Reduction of Carbon Dioxide. ACS Catal. 2018, 8, 6560–6570. 10.1021/acscatal.8b01340. [DOI] [Google Scholar]
- Gupta N.; Gattrell M.; MacDougall B. Calculation for the cathode surface concentrations in the electrochemical reduction of CO2 in KHCO3 solutions. J. Appl. Electrochem. 2006, 36, 161–172. 10.1007/s10800-005-9058-y. [DOI] [Google Scholar]
- Hall A. S.; Yoon Y.; Wuttig A.; Surendranath Y. Mesostructure-Induced Selectivity in CO2 Reduction Catalysis. J. Am. Chem. Soc. 2015, 137, 14834–14837. 10.1021/jacs.5b08259. [DOI] [PubMed] [Google Scholar]
- Chen C.; Zhang B.; Zhong J.; Cheng Z. Selective electrochemical CO2 reduction over highly porous gold films. J. Mater. Chem. A 2017, 5, 21955–21964. 10.1039/c7ta04983h. [DOI] [Google Scholar]
- Lobaccaro P.; Singh M. R.; Clark E. L.; Kwon Y.; Bell A. T.; Ager J. W. Effects of temperature and gas–liquid mass transfer on the operation of small electrochemical cells for the quantitative evaluation of CO2 reduction electrocatalysts. Phys. Chem. Chem. Phys. 2016, 18, 26777–26785. 10.1039/c6cp05287h. [DOI] [PubMed] [Google Scholar]
- Dunwell M.; Yang X.; Setzler B. P.; Anibal J.; Yan Y.; Xu B. Examination of Near-Electrode Concentration Gradients and Kinetic Impacts on the Electrochemical Reduction of CO2 using Surface-Enhanced Infrared Spectroscopy. ACS Catal. 2018, 8, 3999–4008. 10.1021/acscatal.8b01032. [DOI] [Google Scholar]
- Lim C. F. C.; Harrington D. A.; Marshall A. T. Effects of mass transfer on the electrocatalytic CO2 reduction on Cu. Electrochim. Acta 2017, 238, 56–63. 10.1016/j.electacta.2017.04.017. [DOI] [Google Scholar]
- Zhang J.; Pietro W. J.; Lever A. B. P. Rotating ring-disk electrode analysis of CO2 reduction electrocatalyzed by a cobalt tetramethylpyridoporphyrazine on the disk and detected as CO on a platinum ring. J. Electroanal. Chem. 1996, 403, 93–100. 10.1016/0022-0728(95)04270-9. [DOI] [Google Scholar]
- Zhu X.; Gupta K.; Bersani M.; Darr J. A.; Shearing P. R.; Brett D. J. L. Electrochemical reduction of carbon dioxide on copper-based nanocatalysts using the rotating ring-disc electrode. Electrochim. Acta 2018, 283, 1037–1044. 10.1016/j.electacta.2018.07.025. [DOI] [Google Scholar]
- Lates V.; Falch A.; Jordaan A.; Peach R.; Kriek R. J. An electrochemical study of carbon dioxide electroreduction on gold-based nanoparticle catalysts. Electrochim. Acta 2014, 128, 75–84. 10.1016/j.electacta.2013.10.162. [DOI] [Google Scholar]
- Lin A. S.; Lin J.; Huang J. C. Electrochemical oxidation of dissolved carbon monoxide on gold electrode in alkaline medium. Gold Bull. 2007, 40, 82–85. 10.1007/bf03215297. [DOI] [Google Scholar]
- Rodriguez P.; Garcia-Araez N.; Koper M. T. M. Self-promotion mechanism for CO electrooxidation on gold. Phys. Chem. Chem. Phys. 2010, 12, 9373–9380. 10.1039/b926365a. [DOI] [PubMed] [Google Scholar]
- Goyal A.; Marcandalli G.; Mints V. A.; Koper M. T. M. Competition between CO2 Reduction and Hydrogen Evolution on a Gold Electrode under Well-Defined Mass Transport Conditions. J. Am. Chem. Soc. 2020, 142, 4154–4161. 10.1021/jacs.9b10061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard A. J.; Faulkner L. R.. Electrochemical Methods: Fundamentals and Applications, 2nd ed; John Wiley & Sons, 2000; p 331. [Google Scholar]
- Vos J. G.; Koper M. T. M. Examination and prevention of ring collection failure during gas-evolving reactions on a rotating ring-disk electrode. J. Electroanal. Chem. 2019, 850, 113363. 10.1016/j.jelechem.2019.113363. [DOI] [Google Scholar]
- Monteiro M. C. O.; Koper M. T. M. Alumina contamination through polishing and its effect on hydrogen evolution on gold electrodes. Electrochim. Acta 2019, 325, 134915. 10.1016/j.electacta.2019.134915. [DOI] [Google Scholar]
- Lukaszewski M.; Soszko M.; Czerwiński A. Electrochemical Methods of Real Surface Area Determination of Noble Metal Electrodes – an Overview. Int. J. Electrochem. Sci. 2016, 11, 4442–4469. 10.20964/2016.06.71. [DOI] [Google Scholar]
- Ahangari H. T.; Portail T.; Marshall A. T. Comparing the electrocatalytic reduction of CO2 to CO on gold cathodes in batch and continuous flow electrochemical cells. Electrochem. Commun. 2019, 101, 78–81. 10.1016/j.elecom.2019.03.005. [DOI] [Google Scholar]
- Hori Y.; Murata A.; Takahashi R. Formation of hydrocarbons in the electrochemical reduction of carbon dioxide at a copper electrode in aqueous solution. J. Chem. Soc., Faraday Trans. 1 1989, 85, 2309–2326. 10.1039/f19898502309. [DOI] [Google Scholar]
- Ringe S.; Morales-Guio C. G.; Chen L. D.; Fields M.; Jaramillo T. F.; Hahn C.; Chan K. Double layer charging driven carbon dioxide adsorption limits the rate of electrochemical carbon dioxide reduction on Gold. Nat. Commun. 2020, 11, 33. 10.1038/s41467-019-13777-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdyny T.; Smith W. A. CO2 reduction on gas-diffusion electrodes and why catalytic performance must be assessed at commercially-relevant conditions. Energy Environ. Sci. 2019, 12, 1442–1453. 10.1039/c8ee03134g. [DOI] [Google Scholar]
- Rabinowitz J. A.; Kanan M. W. The future of low-temperature carbon dioxide electrolysis depends on solving one basic problem. Nat. Commun. 2020, 11, 5231. 10.1038/s41467-020-19135-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuttig A.; Surendranath Y. Impurity Ion Complexation Enhances Carbon Dioxide Reduction Catalysis. ACS Catal. 2015, 5, 4479–4484. 10.1021/acscatal.5b00808. [DOI] [Google Scholar]
- Zhong H.; Fujii K.; Nakano Y.; Jin F. Effect of CO2 Bubbling into Aqueous Solutions Used for Electrochemical Reduction of CO2 for Energy Conversion and Storage. J. Phys. Chem. C 2015, 119, 55–61. 10.1021/jp509043h. [DOI] [Google Scholar]
- Sander R. Compilation of Henry’s law constants (version 4.0) for water as solvent. Atmos. Chem. Phys. 2015, 15, 4399–4981. 10.5194/acp-15-4399-2015. [DOI] [Google Scholar]
- Takeshi S.; Akiya M. Mechanism of Hydrogen Evolution Reaction On Gold in Aqueous Sulfuric Acid and Sodium Hydroxide. J. Res. Inst. Catal. Hokkaido Univ. 1982, 29, 113–132. [Google Scholar]
- Ohmori T.; Enyo M. Hydrogen evolution reaction on gold electrode in alkaline solutions. Electrochim. Acta 1992, 37, 2021–2028. 10.1016/0013-4686(92)87118-j. [DOI] [Google Scholar]
- Pentland N.; Bockris J. O. M.; Sheldon E. Hydrogen Evolution Reaction on Copper, Gold, Molybdenum, Palladium, Rhodium, and Iron: Mechanism and Measurement Technique under High Purity Conditions. J. Electrochem. Soc. 1957, 104, 182–194. 10.1149/1.2428530. [DOI] [Google Scholar]
- Mayer J. M. Proton-Coupled Electron Transfer: A Reaction Chemist’s View. Annu. Rev. Phys. Chem. 2004, 55, 363–390. 10.1146/annurev.physchem.55.091602.094446. [DOI] [PubMed] [Google Scholar]
- Dubouis N.; Serva A.; Berthin R.; Jeanmairet G.; Porcheron B.; Salager E.; Salanne M.; Grimaud A. Tuning water reduction through controlled nanoconfinement within an organic liquid matrix. Nat. Catal. 2020, 3, 656–663. 10.1038/s41929-020-0482-5. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.