

Abstract
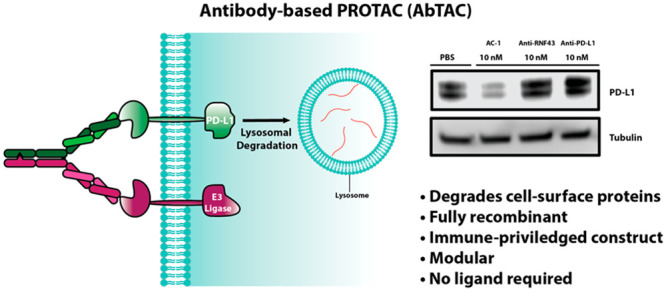
Targeted protein degradation has emerged as a new paradigm to manipulate cellular proteostasis. Proteolysis-targeting chimeras (PROTACs) are bifunctional small molecules that recruit an E3 ligase to a target protein of interest, promoting its ubiquitination and subsequent degradation. Here, we report the development of antibody-based PROTACs (AbTACs), fully recombinant bispecific antibodies that recruit membrane-bound E3 ligases for the degradation of cell-surface proteins. We show that an AbTAC can induce the lysosomal degradation of programmed death-ligand 1 by recruitment of the membrane-bound E3 ligase RNF43. AbTACs represent a new archetype within the PROTAC field to target cell-surface proteins with fully recombinant biological molecules.
Proteolysis targeting chimeras (PROTACs)1 and related degradation technologies, such as LYTACs,2,3 dTAGs,4 Trim-Away,5 and SNIPERs,6 have arisen as novel therapeutic modalities to target traditionally “undruggable” proteins. PROTACs are heterobifunctional molecules consisting of an E3 ligase binder and a substrate targeting ligand that exploit the cellular protein degradation machinery to selectively degrade target proteins.1 Unlike occupancy-based inhibition, PROTACs act catalytically.7 This enables them to target previously intractable proteins and to be effective even when resistance to inhibitors develops.8,9 Despite great promise for PROTACs, they require targets with cytosolic binding domains for small-molecule ligands, leaving many membrane proteins untargetable. Membrane proteins comprise ∼23% of encoded genes,10 and ∼70% of FDA-approved drugs target this important class.11 Therefore, a new strategy to degrade cell-surface proteins has the potential to be transformative to the field.
IgGs have long serum half-lives12 and can be rapidly generated as high-affinity binders to target proteins through phage display.13,14 Bispecific IgGs can bind to two proteins simultaneously, co-localizing them.15 We hypothesized that this biological construct could mimic PROTACs by recruiting membrane-bound E3 ligases to proteins of interest, inducing degradation. We have termed these antibody-based PROTACs (AbTACs). If successful, AbTACs would have several advantages over previous technologies. First, AbTACs are fully recombinant bispecific IgGs, allowing for their rapid and renewable generation. Next, we utilize standard phage display to generate multiple recombinant antibody binders, resulting in high affinity and high specificity. The AbTACs are recombinant by nature, allowing for simple genetic optimization of binding properties. Finally, AbTACs expand the PROTAC field’s attempts to target challenging membrane proteins.
The most commonly used E3 ligases by the PROTAC field are von Hippel–Lindau disease tumor suppressor (VHL)16 and Cereblon (CRBN).17 There have been numerous recent accounts of successfully recruiting different ligases for degradation;18−21 however, the use of a transmembrane E3 ligase (as required for our approach) has not been reported. We sought a single-pass E3 ligase with a structured ectodomain to facilitate phage display antibody generation. Ideally, it would be widely expressed across cell types to enable generalizability. RNF43 is a single-pass E3 ligase comprising a structured ectodomain and an intracellular RING domain that meets these requirements.22 RNF43 negatively regulates the Wnt signaling pathway by ubiquitinating Frizzled, a Wnt co-receptor, causing its endocytosis and degradation.23,24 We believe this could be quite general because only in rare cases does RNF43 act as a tumor suppressor due to mutation or transcriptional silencing.25 Here, we report the first AbTAC based on RNF43 targeting degradation of programmed death-ligand 1 (PD-L1) by recruitment of the membrane-bound E3 ligase, RNF43.
We first tested if recruitment of RNF43 to a model protein, GFP, would lead to its internalization and lysosomal degradation (Figure 1a). We fused GFP via a transmembrane domain to a NanoLuc domain26 to serve as an orthogonal expression and localization reporter. As an initial test for recruitment, we next fused an anti-GFP single chain Fab (scFab) to the N-terminus of RNF43;27 for an isotype control, we used an anti-GCN4 scFab. Upon expression of these constructs in HeLa cells, confocal microscopy showed GFP localized to the cell surface in the isotype control and to reporter cells treated with an anti-GFP Fab (Figures 1b,c and S1a,b). Only the anti-GFP-RNF43 fusion caused the internalization of the GFP with co-localization in the lysosome (Figures 1d and S1c). We performed a Nanoluciferase assay to quantify the amount of reporter which showed a modest ∼20% reduction (Figure S2). This value is comparable to degradation levels seen for other overexpression systems.28 These data suggested that RNF43 can be used to induce protein degradation of endogenous proteins.
Figure 1.

AbTACs recruit RNF43 to internalize cell-surface proteins. (a) Graphical representation of the AbTAC mode of action. (b–d) Engineered RNF43 constructs and the GFP-Nanoluciferase reporter with their corresponding confocal microscopy images showing GFP localization for each experimental condition. (b) Anti-GCN4-RNF43 fusion as an isotype control. (c) Soluble anti-GFP Fab to control for Fab binding effects. (d) Anti-GFP-RNF43 (green, GFP reporter protein; red, Lysosome Tracker; blue, DAPI).
We next generated a recombinant antibody for the ectodomain of RNF43 using phage display. Figure S3 outlines our selection strategy. After four rounds, we used a Fab-phage ELISA to triage clones with predicted affinities greater than 20 nM (Figure 2a). Following the sequencing of passing clones, we identified four unique Fab-phage sequences. One of these clones, R3, bound well to RNF43 in vitro and on cells. Biolayer interferometry confirmed that the Fab derived from R3 had an in vitroKD of 12.5 nM (Figure 2b). To assess on-cell binding, we overexpressed full-length RNF43 in HEK293T cells and used flow cytometry to verify Fab binding (Figure 2c).
Figure 2.

Generaration of an AbTAC bispecific IgG that can simultaneously bind to RNF43 and PD-L1. (a) Fab-phage ELISA data showing binders from a phage display campaign with RNF43 Fc fusion as the target antigen. (b) BLI trace showing Fab R3 has a Kd of 12.5 nM. (c) Flow cytometry showing Fab R3 binds to RNF43 on cells. (d) Conditions for in vitro assembly of separately expressed half IgGs to form a bispecific IgG using the described point mutations. (e) Two-step BLI experiment showing that AC-1 can simultaneously bind to both purified ecto-domains of RNF43 and PD-L1.
We chose PD-L1 for an initial degradation target. PD-L1 is a protein overexpressed in numerous cancers, causing the suppression of the T-cell response via binding to the inhibitory receptor PD-1 on T cells.29 PD-L1 has a small, 31 amino acid long cytoplasmic domain with no known small-molecule ligands, making it challenging to target with conventional small-molecule PROTAC approaches.30 PD-L1 has previously been degraded using the LYTAC approach,2 and there are glyco-form specific antibodies that induce its degradation.31 The AbTAC method offers a fully genetically encoded alternative for degrading cell-surface proteins based on E3 ligases.
With an RNF43 binder in hand, we chose to use Tecentriq (atezolizumab) as our PD-L1 binder.32 To generate the bispecific IgG (BsIgG), we utilized the highly validated knobs-into-holes Fc construct to ensure correct heavy chain pairing (Figure 2d).33 To prevent light chain mismatch pairing, we expressed the antibodies as half IgGs, followed by their in vitro assembly to construct AC-1 bispecific (Figure 2d).34 We introduced a His-tag on the knob half IgG to purify away unwanted hole–hole homodimer. BsIgGs bind to both antigens simultaneously. To verify that both arms of the BsIgG were functional, we immobilized PD-L1 on a BLI tip, followed by subsequent addition of AbTAC and the ecto domain of RNF43. Indeed, we observed two sequential increases in signal, confirming that AC-1 can bind to both RNF43 and PD-L1 concurrently (Figure 2e). To determine if the bispecific format altered affinity, we compared the in vitro binding affinities for AC-1 to its Fab components; indeed, AC-1 bound to RNF43 with the same 12.5 nM Kd as the Fab (Figure S4). These BLI experiments validated thatAC-1 is a BsIgG that can bind to both antigens simultaneously.
We next sought to test if AC-1 can degrade PD-L1 on cells. We first chose a triple-negative breast cancer cell line, MDA-MB-231, due to the high expression of PD-L1. Flow cytometry confirmed the presence of RNF43 on these cells (Figure S5). PD-L1 stains as a doublet in Western blot analysis due to glycosylation heterogeneity.35 Treatment of MDA-MB-231 cells with 10 nM AC-1 induced substantial degradation of both forms of PD-L1 within 12 h, with maximal degradation by 24 h (Figure 3a). We found that AC-1 effectively induced the degradation of PD-L1 with a DC50 = 3.4 nM and maximal percent degradation Dmax = 63% at 24 h (Figure 3b). Treatment with either arm of the BsIgG individually or simultaneously at 10 nM did not affect PD-L1 levels, indicating both targets must be brought together to cause degradation (Figures 3c and S6).
Figure 3.

AC-1 causes the degradation of PD-L1 in MDA-MB-241 in an RNF43- and lysosomal-dependent manner. (a) Time course experiment showing degradation of PD-L1 after 12 h, with maximum degradation after 24 h. (b) Dose escalation experiment showing maximum degradation of PD-L1 at 20 nM bispecific IgG treatment. Densitometry was used to quantify degradation levels. Error bars represents SD of three biological replicates. (c) Western blot analysis showing AC-1 can degrade PD-L1 at 10 nM after 24 h, whereas each component of the bispecific has no effect on protein levels. (d) CRISPRi knockdown of RNF43 rescues degradation, demonstrating degradation is RNF43 dependent. (e) Pretreatment with either bafilomycin or MG132 indicates that AC-1 degrades PD-L1 in a lysosomal-dependent manner. The presented data are representative of three independent replicates.
To measure proteomic changes that might result from loss of PD-L1 or engagement of RNF43, we ran whole-cell proteomics, which showed no large cellular perturbations (Figure S7). Due to the relatively low abundance of cell-surface proteins in a cell, PD-L1 peptides were not observed in either sample. We also found that PD-L1 levels recovered within 24 h after wash-out (Figure S8), consistent with our findings that AC-1 does not produce irreversible cellular perturbations.
To test if PD-L1 degradation by AC-1 is dependent on RNF43, we knocked RNF43 down in MDA-MB-231 cells using CRISPR interference (CRISPRi) and confirmed knock down via RT-qPCR (Figure S9). AC-1 did not induce degradation of PD-L1 in these RNF43 knock down cells after 24 h of 10 nM treatment (Figure 3d). To evaluate the degradation pathway, we pretreated with either bafilomycin (a lysosome acidification inhibitor) or MG-132 (a proteasome inhibitor). Bafilomycin mitigated the degradation of PD-L1, whereas MG-132 did not (Figure 3e). These data suggest that AC-1 causes degradation of PD-L1 in an RNF43- and lysosomal-dependent manner.
Next, we wanted to test if AC-1 could degrade PD-L1 on other cell lines. Tecentriq is approved for combination therapy in three indications: triple-negative breast cancer, non-small-cell lung cancer, and advanced bladder cancer. We chose three cell lines representing each of these indications that are known to have high PD-L1 levels. In each cell line (MDA-MB-231, HCC827, and T24), treatment with AC-1 at 10 nM induced degradation of PD-L1 after 24 h, suggesting broad cellular applicability (Figure 4).
Figure 4.

To demonstrate the applicability of AC-1 to degrade PD-L1, we chose three cancer cell lines that correspond to indications for which anti-PD-L1 checkpoint inhibitors have been approved. AC-1 is able to degrade PD-L1 in each of these cell lines at 10 nM after 24 h. The presented data are representative of three independent replicates.
PROTACs have provided an important alternative to traditional small-molecule inhibitors by their ability to deplete a protein of interest. Despite the great potential of PROTACs for targeted degradation, the examples so far have only been applied to cytosolic E3 ligases. Here, using the AbTAC technology, we expand targeted degradation to transmembrane E3 ligases, allowing depletion of a cell-surface protein without a known small-molecule ligand.
As this work was underway, the Bertozzi laboratory reported a clever method, termed LYTACs,2 to degrade cell-surface proteins by hijacking lysosomal targeting receptors. This elegant platform technology focused on degrading surface proteins via chemical conjugation of glycans to an antibody directed to the victim protein. In the first LYTAC iteration the glycan targets the mannose-6-phosphate receptor, which upon binding drags the cargo to the lysosome via a receptor internalization mechanism. We envision that the AbTACs can act as a complementary approach by providing fully recombinant and renewable parts to induce membrane protein degradation using membrane bound E3 ligases.
In summary, AC-1 recruits RNF43, a cell-surface E3 ligase, to degrade PD-L1, an important cell-surface check point protein with a small intracellular domain and no reported small-molecule ligands required for PROTACs. The lysosomal degradation is caused by recruitment of the two proteins, and there are no large cellular proteomic perturbations that occur, as was similarly reported for LYTACs. AC-1 is a fully recombinant BsIgG, making it renewable. The AbTAC is built of human parts, making it less likely to illicit an immune response. It is possible that engaging RNF43 for degrading PD-L1 could compromise its function in the Wnt signaling pathways. However, it has been shown that inhibiting RNF43 or ZNRF3 (a close homologue that is involved in the Wnt pathway) alone has very little effect on Wnt activity.36
AC-1 achieves a DMax of 63%, which represents the steady state between synthesis and degradation rates. We envision that multiple factors can affect this steady-state process, including binding properties, cell-surface levels, E3-target stoichiometry, endocytosis kinetics upon antibody binding, turnover rate of the protein of interest, and the specific E3 ligase. With a deeper understanding of these factors we hope to expand the AbTAC toolbox to degrade a range of cell-surface proteins and to rationally optimize hits to test their phenotypic effects in vivo. The modular nature and genetic tractability of AbTACs offer promise for academic and translational applications.
Acknowledgments
We thank the members of the Wells laboratory and Antibiome (past and present) for helpful discussions, M. Trieu and E. Grumish for expression of IgG constructs, M. Hornsby and J. Liu for performing phage display selections on the antibot, J. Byrnes for assistance with mass spectrometry experiments, and P. Solomon for input on RT-qPCR assays. Microscopy images in this study were acquired at the Nikon Imaging Center at UCSF. J.A.W. is grateful for funding from the Harry and Dianna Hind Endowed Professorship in Pharmaceutical Sciences, NCI P41CA196276, and R35GM122451.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c10008.
Detailed experimental procedures and additional experimental data (PDF)
The authors declare the following competing financial interest(s): A.D.C., D.P.N., and J.A.W. and the Regents of the University of California have filed a patent application (International Application No. PCT/US20/58328) related to this project.
Supplementary Material
References
- Sakamoto K. M.; Kim K. B.; Kumagai A.; Mercurio F.; Crews C. M.; Deshaies R. J. Protacs: Chimeric Molecules That Target Proteins to the Skp1-Cullin-F Box Complex for Ubiquitination and Degradation. Proc. Natl. Acad. Sci. U. S. A. 2001, 98 (15), 8554–8559. 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banik S. M.; Pedram K.; Wisnovsky S.; Ahn G.; Riley N. M.; Bertozzi C. R. Lysosome-Targeting Chimaeras for Degradation of Extracellular Proteins. Nature 2020, 584 (7820), 291–297. 10.1038/s41586-020-2545-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn G.; Banik S.; Miller C. L.; Riley N.; Cochran J. R.; Bertozzi C. Lysosome Targeting Chimeras (LYTACs) That Engage a Liver-Specific Asialoglycoprotein Receptor for Targeted Protein Degradation. Chemrxiv 2020, 10.26434/chemrxiv.12736778.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabet B.; Roberts J. M.; Buckley D. L.; Paulk J.; Dastjerdi S.; Yang A.; Leggett A. L.; Erb M. A.; Lawlor M. A.; Souza A.; Scott T. G.; Vittori S.; Perry J. A.; Qi J.; Winter G. E.; Wong K.-K.; Gray N. S.; Bradner J. E. The DTAG System for Immediate and Target-Specific Protein Degradation. Nat. Chem. Biol. 2018, 14 (5), 431–441. 10.1038/s41589-018-0021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clift D.; McEwan W. A.; Labzin L. I.; Konieczny V.; Mogessie B.; James L. C.; Schuh M. A Method for the Acute and Rapid Degradation of Endogenous Proteins. Cell 2017, 171 (7), 1692–1706. 10.1016/j.cell.2017.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito M.; Ohoka N.; Shibata N. SNIPERs—Hijacking IAP Activity to Induce Protein Degradation. Drug Discovery Today: Technol. 2019, 31, 35–42. 10.1016/j.ddtec.2018.12.002. [DOI] [PubMed] [Google Scholar]
- Bondeson D. P.; Mares A.; Smith I. E. D.; Ko E.; Campos S.; Miah A. H.; Mulholland K. E.; Routly N.; Buckley D. L.; Gustafson J. L.; Zinn N.; Grandi P.; Shimamura S.; Bergamini G.; Faelth-Savitski M.; Bantscheff M.; Cox C.; Gordon D. A.; Willard R. R.; Flanagan J. J.; Casillas L. N.; Votta B. J.; den Besten W.; Famm K.; Kruidenier L.; Carter P. S.; Harling J. D.; Churcher I.; Crews C. M. Catalytic in Vivo Protein Knockdown by Small-Molecule PROTACs. Nat. Chem. Biol. 2015, 11 (8), 611–617. 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salami J.; Alabi S.; Willard R. R.; Vitale N. J.; Wang J.; Dong H.; Jin M.; McDonnell D. P.; Crew A. P.; Neklesa T. K.; Crews C. M. Androgen Receptor Degradation by the Proteolysis-Targeting Chimera ARCC-4 Outperforms Enzalutamide in Cellular Models of Prostate Cancer Drug Resistance. Communications Biology 2018, 1, 1. 10.1038/s42003-018-0105-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burslem G. M.; Smith B. E.; Lai A. C.; Jaime-Figueroa S.; McQuaid D. C.; Bondeson D. P.; Toure M.; Dong H.; Qian Y.; Wang J.; Crew A. P.; Hines J.; Crews C. M. The Advantages of Targeted Protein Degradation Over Inhibition: An RTK Case Study. Cell Chemical Biology 2018, 25 (1), 67–77.e3. 10.1016/j.chembiol.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M.; Fagerberg L.; Hallstrom B. M.; Lindskog C.; Oksvold P.; Mardinoglu A.; Sivertsson A.; Kampf C.; Sjostedt E.; Asplund A.; Olsson I.; Edlund K.; Lundberg E.; Navani S.; Szigyarto C. A.-K.; Odeberg J.; Djureinovic D.; Takanen J. O.; Hober S.; Alm T.; Edqvist P.-H.; Berling H.; Tegel H.; Mulder J.; Rockberg J.; Nilsson P.; Schwenk J. M.; Hamsten M.; von Feilitzen K.; Forsberg M.; Persson L.; Johansson F.; Zwahlen M.; von Heijne G.; Nielsen J.; Ponten F. Tissue-Based Map of the Human Proteome. Science 2015, 347 (6220), 1260419–1260419. 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- Lundstrom K. An Overview on GPCRs and Drug Discovery: Structure-Based Drug Design and Structural Biology on GPCRs. Methods Mol. Biol. 2009, 552, 51–66. 10.1007/978-1-60327-317-6_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo E. D.; Hansen R. J.; Balthasar J. P. Antibody Pharmacokinetics and Pharmacodynamics. J. Pharm. Sci. 2004, 93 (11), 2645–2668. 10.1002/jps.20178. [DOI] [PubMed] [Google Scholar]
- Lowman H. B.; Bass S. H.; Simpson N.; Wells J. A. Selecting High-Affinity Binding Proteins by Monovalent Phage Display. Biochemistry 1991, 30 (45), 10832–10838. 10.1021/bi00109a004. [DOI] [PubMed] [Google Scholar]
- Hornsby M.; Paduch M.; Miersch S.; Sääf A.; Matsuguchi T.; Lee B.; Wypisniak K.; Doak A.; King D.; Usatyuk S.; Perry K.; Lu V.; Thomas W.; Luke J.; Goodman J.; Hoey R. J.; Lai D.; Griffin C.; Li Z.; Vizeacoumar F. J.; Dong D.; Campbell E.; Anderson S.; Zhong N.; Gräslund S.; Koide S.; Moffat J.; Sidhu S.; Kossiakoff A.; Wells J. A High Through-Put Platform for Recombinant Antibodies to Folded Proteins. Mol. Cell. Proteomics 2015, 14 (10), 2833–2847. 10.1074/mcp.O115.052209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontermann R. Dual Targeting Strategies with Bispecific Antibodies. mAbs 2012, 4 (2), 182–197. 10.4161/mabs.4.2.19000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Gonzalez A.; Cyrus K.; Salcius M.; Kim K.; Crews C. M.; Deshaies R. J.; Sakamoto K. M. Targeting Steroid Hormone Receptors for Ubiquitination and Degradation in Breast and Prostate Cancer. Oncogene 2008, 27 (57), 7201–7211. 10.1038/onc.2008.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; Qian Y.; Altieri M.; Dong H.; Wang J.; Raina K.; Hines J.; Winkler J. D.; Crew A. P.; Coleman K.; Crews C. M. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22 (6), 755–763. 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward C. C.; Kleinman J. I.; Brittain S. M.; Lee P. S.; Chung C. Y. S.; Kim K.; Petri Y.; Thomas J. R.; Tallarico J. A.; McKenna J. M.; Schirle M.; Nomura D. K. Covalent Ligand Screening Uncovers a RNF4 E3 Ligase Recruiter for Targeted Protein Degradation Applications. ACS Chem. Biol. 2019, 14 (11), 2430–2440. 10.1021/acschembio.8b01083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradlin J. N.; Hu X.; Ward C. C.; Brittain S. M.; Jones M. D.; Ou L.; To M.; Proudfoot A.; Ornelas E.; Woldegiorgis M.; Olzmann J. A.; Bussiere D. E.; Thomas J. R.; Tallarico J. A.; McKenna J. M.; Schirle M.; Maimone T. J.; Nomura D. K. Harnessing the Anti-Cancer Natural Product Nimbolide for Targeted Protein Degradation. Nat. Chem. Biol. 2019, 15 (7), 747–755. 10.1038/s41589-019-0304-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Crowley V. M.; Wucherpfennig T. G.; Dix M. M.; Cravatt B. F. Electrophilic PROTACs That Degrade Nuclear Proteins by Engaging DCAF16. Nat. Chem. Biol. 2019, 15 (7), 737–746. 10.1038/s41589-019-0279-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M.; Liu T.; Jiao Q.; Ji J.; Tao M.; Liu Y.; You Q.; Jiang Z. Discovery of a Keap1-Dependent Peptide PROTAC to Knockdown Tau by Ubiquitination-Proteasome Degradation Pathway. Eur. J. Med. Chem. 2018, 146, 251–259. 10.1016/j.ejmech.2018.01.063. [DOI] [PubMed] [Google Scholar]
- Zebisch M.; Xu Y.; Krastev C.; MacDonald B. T.; Chen M.; Gilbert R. J. C.; He X.; Jones E. Y. Structural and Molecular Basis of ZNRF3/RNF43 Transmembrane Ubiquitin Ligase Inhibition by the Wnt Agonist R-Spondin. Nat. Commun. 2013, 4 (1), 2787. 10.1038/ncomms3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo B.-K.; Spit M.; Jordens I.; Low T. Y.; Stange D. E.; van de Wetering M.; van Es J. H.; Mohammed S.; Heck A. J. R.; Maurice M. M.; Clevers H. Tumour Suppressor RNF43 Is a Stem-Cell E3 Ligase That Induces Endocytosis of Wnt Receptors. Nature 2012, 488 (7413), 665–669. 10.1038/nature11308. [DOI] [PubMed] [Google Scholar]
- Tsukiyama T.; Fukui A.; Terai S.; Fujioka Y.; Shinada K.; Takahashi H.; Yamaguchi T. P.; Ohba Y.; Hatakeyama S. Molecular Role of RNF43 in Canonical and Noncanonical Wnt Signaling. Mol. Cell. Biol. 2015, 35 (11), 2007–2023. 10.1128/MCB.00159-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakis M.; Hodis E.; Jasmine Mu X.; Yamauchi M.; Rosenbluh J.; Cibulskis K.; Saksena G.; Lawrence M. S.; Qian Z. R.; Nishihara R.; Van Allen E. M.; Hahn W. C.; Gabriel S. B.; Lander E. S.; Getz G.; Ogino S.; Fuchs C. S.; Garraway L. A. RNF43 Is Frequently Mutated in Colorectal and Endometrial Cancers. Nat. Genet. 2014, 46 (12), 1264–1266. 10.1038/ng.3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall M. P.; Unch J.; Binkowski B. F.; Valley M. P.; Butler B. L.; Wood M. G.; Otto P.; Zimmerman K.; Vidugiris G.; Machleidt T.; Robers M. B.; Benink H. A.; Eggers C. T.; Slater M. R.; Meisenheimer P. L.; Klaubert D. H.; Fan F.; Encell L. P.; Wood K. V. Engineered Luciferase Reporter from a Deep Sea Shrimp Utilizing a Novel Imidazopyrazinone Substrate. ACS Chem. Biol. 2012, 7 (11), 1848–1857. 10.1021/cb3002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koerber J. T.; Hornsby M. J.; Wells J. A. An Improved Single-Chain Fab Platform for Efficient Display and Recombinant Expression. J. Mol. Biol. 2015, 427 (2), 576–586. 10.1016/j.jmb.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riching K. M.; Mahan S.; Corona C. R.; McDougall M.; Vasta J. D.; Robers M. B.; Urh M.; Daniels D. L. Quantitative Live-Cell Kinetic Degradation and Mechanistic Profiling of PROTAC Mode of Action. ACS Chem. Biol. 2018, 13 (9), 2758–2770. 10.1021/acschembio.8b00692. [DOI] [PubMed] [Google Scholar]
- Freeman G. J.; Long A. J.; Iwai Y.; Bourque K.; Chernova T.; Nishimura H.; Fitz L. J.; Malenkovich N.; Okazaki T.; Byrne M. C.; Horton H. F.; Fouser L.; Carter L.; Ling V.; Bowman M. R.; Carreno B. M.; Collins M.; Wood C. R.; Honjo T. Engagement of the PD-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. J. Exp. Med. 2000, 192 (7), 1027–1034. 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escors D.; Gato-Cañas M.; Zuazo M.; Arasanz H.; García-Granda M. J.; Vera R.; Kochan G. The Intracellular Signalosome of PD-L1 in Cancer Cells. Sig Transduct Target Ther 2018, 3 (1), 26. 10.1038/s41392-018-0022-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.-W.; Lim S.-O.; Chung E. M.; Kim Y.-S.; Park A. H.; Yao J.; Cha J.-H.; Xia W.; Chan L.-C.; Kim T.; Chang S.-S.; Lee H.-H.; Chou C.-K.; Liu Y.-L.; Yeh H.-C.; Perillo E. P.; Dunn A. K.; Kuo C.-W.; Khoo K.-H.; Hsu J. L.; Wu Y.; Hsu J.-M.; Yamaguchi H.; Huang T.-H.; Sahin A. A.; Hortobagyi G. N.; Yoo S. S.; Hung M.-C. Eradication of Triple-Negative Breast Cancer Cells by Targeting Glycosylated PD-L1. Cancer Cell 2018, 33 (2), 187–201.e10. 10.1016/j.ccell.2018.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F.; Qi X.; Wang X.; Wei D.; Wu J.; Feng L.; Cai H.; Wang Y.; Zeng N.; Xu T.; Zhou A.; Zheng Y. Structural Basis of the Therapeutic Anti-PD-L1 Antibody Atezolizumab. Oncotarget 2017, 8 (52), 90215–90224. 10.18632/oncotarget.21652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridgway J. B. B.; Presta L. G.; Carter P. ‘Knobs-into-Holes’ Engineering of Antibody CH3 Domains for Heavy Chain Heterodimerization. Protein Eng., Des. Sel. 1996, 9 (7), 617–621. 10.1093/protein/9.7.617. [DOI] [PubMed] [Google Scholar]
- Ovacik A. M.; Li J.; Lemper M.; Danilenko D.; Stagg N.; Mathieu M.; Ellerman D.; Gupta V.; Kalia N.; Nguy T.; Plaks V.; Johnson C. D.; Wang W.; Brumm J.; Fine B.; Junttila T.; Lin K.; Carter P. J.; Prabhu S.; Spiess C.; Kamath A. V. Single Cell-Produced and in Vitro-Assembled Anti-FcRH5/CD3 T-Cell Dependent Bispecific Antibodies Have Similar in Vitro and in Vivo Properties. mAbs 2019, 11 (2), 422–433. 10.1080/19420862.2018.1551676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.-W.; Lim S.-O.; Xia W.; Lee H.-H.; Chan L.-C.; Kuo C.-W.; Khoo K.-H.; Chang S.-S.; Cha J.-H.; Kim T.; Hsu J. L.; Wu Y.; Hsu J.-M.; Yamaguchi H.; Ding Q.; Wang Y.; Yao J.; Lee C.-C.; Wu H.-J.; Sahin A. A.; Allison J. P.; Yu D.; Hortobagyi G. N.; Hung M.-C. Glycosylation and Stabilization of Programmed Death Ligand-1 Suppresses T-Cell Activity. Nat. Commun. 2016, 7 (1), 12632. 10.1038/ncomms12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luca V. C.; Miao Y.; Li X.; Hollander M. J.; Kuo C. J.; Garcia K. C. Surrogate R-spondins for tissue-specific potentiation of Wnt Signaling. PLoS One 2020, 15, e0226928. 10.1371/journal.pone.0226928. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.