

Abstract

Terminal unactivated alkynes are nowadays considered the golden standard for cysteine-reactive warheads in activity-based probes (ABPs) targeting cysteine deubiquitinating enzymes (DUBs). In this work, we study the versatility of the thiol–alkyne addition reaction in more depth. Contrary to previous findings with UCHL3, we now show that covalent adduct formation can progress with substituents on the terminal or internal alkyne position. Strikingly, acceptance of alkyne substituents is strictly DUB-specific as this is not conserved among members of the same subfamily. Covalent adduct formation with the catalytic cysteine residue was validated by gel analysis and mass spectrometry of intact ABP-treated USP16CDWT and catalytically inactive mutant USP16CDC205A. Bottom-up mass spectrometric analysis of the covalent adduct with a deuterated propargyl ABP provides mechanistic understanding of the in situ thiol–alkyne reaction, identifying the alkyne rather than an allenic intermediate as the reactive species. Furthermore, kinetic analysis revealed that introduction of (bulky/electron-donating) methyl substituents on the propargyl moiety decreases the rate of covalent adduct formation, thus providing a rational explanation for the commonly lower level of observed covalent adduct compared to unmodified alkynes. Altogether, our work extends the scope of possible propargyl derivatives in cysteine targeting ABPs from unmodified terminal alkynes to internal and substituted alkynes, which we anticipate will have great value in the development of ABPs with improved selectivity profiles.
Introduction
Ubiquitination is a post-translational modification (PTM) which regulates many cellular processes.1−3 Aberrant ubiquitination has been observed in numerous diseases, rendering the enzymes involved as attractive targets for drug design.4−8 Ubiquitination involves ligation of Ubiquitin (Ub), a small 76-amino-acid protein, onto the target protein by the E1–E2–E3 ligase machinery. Deubiquitinating enzymes (DUBs) reverse this process by cleavage of the native isopeptide bond between the Ub C-terminus and the target protein Lys (lysine) residue or between the distal and proximal Ub in poly-Ub chains.8,9 Cysteine DUBs are classified by their catalytic domain, which contains a catalytic cysteine residue essential for their proteolytic function. There are currently six known classes of human cysteine DUBs: USP, OTU, UCH, MJD, MINDY, and ZUFSP.1,10 Their proteolytic activity can be monitored with activity-based probes (ABPs), which covalently trap active enzymes by formation of a covalent bond between an electrophilic warhead on the ABP and the nucleophilic cysteine residue in the targeted enzyme.11−13 Cysteine DUB ABPs have been utilized to monitor DUB activity during infection, in disease and/or upon inhibitor treatment,14−17 to identify new DUB (classes) and catalytic cysteine residues in newly discovered DUBs,18−21 and to visualize Ub binding in crystal structures of covalent adducts.22,23
Terminal unactivated alkynes were believed to be unreactive toward (nontargeted) thiols under physiological conditions and are therefore widely applied as bioorthogonal handles.24−26 However, in 2013 two independent groups27,28 discovered that propargylamide on the C-terminus of ubiquitin (-like modifiers; Ubl) can act as a latent electrophile, forming an irreversible covalent adduct with the catalytic cysteine thiol of cysteine proteases that normally cleave the native Ub(l)–Lys isopeptide bond (Figure S1). The propargyl (Prg) moiety has since been utilized in various covalent Ub(l)-based ABPs and is considered the golden standard for DUB ABPs because of its high stability, ease of synthesis, and lack of intrinsic reactivity with nontargeted thiols.17,18,29 Formation of a Markovnikov-type thiovinyl bond between active site cysteine thiol and internal (quaternary) alkyne carbon has been confirmed with numerous crystal structures of Ub(l)–Prg ABPs bound to human and viral cysteine proteases (summarized in Table S1). Recently, we showed that the thiol–alkyne reaction can be extended to small molecule inhibitors; a small recognition element is sufficient to initiate covalent thiovinyl bond formation between the cathepsin K catalytic cysteine thiol and the inhibitor alkyne moiety.33
The covalent thiol–alkyne addition forming a Markovnikov-type thiovinyl adduct is a newly discovered reaction for which several reaction mechanisms have been proposed (Scheme 1). A radical-mediated thiol–yne mechanism was quickly excluded because covalent adduct formation was not prevented by absence of light and/or addition of radical scavengers and would have resulted in the anti-Markovnikov-type thiovinyl bond adduct with terminal C1 carbon (Scheme 1A).30,31 Ekkebus et al.27 and Sommer et al.28 both propose a proximity-driven in situ thiol(ate)–alkyne addition that involves direct nucleophilic attack of the catalytic cysteine thiol(ate) to the alkyne internal C2 carbon (Scheme 1B). However, it was not possible to exclude the possibility that nucleophilic addition actually occurs with a more reactive allenic isomer, present at the enzyme active site in equilibrium with the unreactive terminal alkyne (Scheme 1C).34,35 Alternatively, Arkona et al.32 propose an enzyme-templated stepwise reaction with stabilization of a secondary carbanion intermediate in the oxyanion hole to overcome the thermodynamically unfavored bond formation (Scheme 1D). This stepwise reaction mechanism would be similar to cysteine/serine protease-mediated proteolysis of native amide bonds that involves stabilization of the anion intermediate in the oxyanion hole via interactions with polar residues such as glutamine or by H-bonds with backbone amides.36,37
Scheme 1. Proposed Reaction Mechanisms for Nucleophilic Thiol–Alkyne Addition Forming Covalent Thiovinyl Bond between Cysteine Protease and Alkyne.

(A) Radical-mediated thiol–yne reaction. Excluded because this would form anti-Markovnikov-type product with alkyne C1 carbon atom.30,31 (B) Proximity-driven in situ thiol(ate)–alkyne addition.27,28 Direct nucleophilic attack on internal C2 alkyne by cysteine thiol is supported by mutagenesis experiments with SENP1; only catalytic Cys603 was essential to form covalent adduct with SUMO2-Prg.28 (C) Spontaneous or enzyme-initiated isomerization (tautomerization) of the terminal alkyne moiety to a thiol-reactive allenic intermediate prior to nucleophilic addition.27 Excluded in this work by mass spectrometric analysis. (D) Enzyme-templated thiol–alkyne addition via a secondary carbanion intermediate that is stabilized in the protease oxyanion hole, proposed by Arkona et al.32 Contradicted by mutagenesis with SENP1; Q597A mutation of important glutamine residue in oxyanion hole did not mitigate covalent adduct formation with SUMO2-Prg,28 but this does not exclude the role of stabilizing H-bonds with backbone amides.
To date, the scope of the thiol–alkyne addition with unactivated alkynes has been limited to unsubstituted terminal propargylamide; Ekkebus et al. report that substituting the hydrogens on either the terminal C1 carbon (CH) or the internal C3 carbon (CH2) of the propargyl moiety in Ub-Prg mitigates covalent bond formation with UCHL3 (Scheme 2).27 The lack of reactivity was contributed to mechanistic components, like an important role for the terminal alkyne proton, or formation of a reactive allene intermediate at the active site. Alternatively, we now hypothesize that the lack of reactivity with substituted propargyl derivatives is resultant from specific steric interactions at the UCHL3 active site and as such not representative for the prospective reactivity with other cysteine DUBs. Variation in the warhead has been reported to affect the adduct formation pattern in cell lysate while keeping the ubiquitin recognition element unchanged,19 although we would like to note that in those cases the nature of the warhead was changed rather than introduction of (bulky) substituents to the same electrophile.
Scheme 2. Covalent Adduct Formation between Catalytic Cysteine Thiol of Recombinant Cysteine Protease UCHL3 and the Alkyne Quaternary C2 Carbon of Ub-Prg Is Mitigated When Hydrogens Are Substituted on the Propargyl Terminal (C1) or Internal (C3) Carbon27.

In this work we show that restrictions on propargyl substitution are DUB-dependent rather than a general property of the in situ thiol–alkyne reaction. We selected a panel of substituted alkynes that are incorporated in DUB ABPs and explore their reactivity both in lysate and on an extensive set of recombinant cysteine DUBs. Formation of a covalent adduct with substituted alkynes is subsequently validated with USP16. Furthermore, we investigate the consequences of substituents on the rate of covalent adduct formation, since introduction of electron-donating substituents on internal and terminal alkyne carbons reduces alkyne electrophilicity. Together, these results illustrate the possibilities and flexibility of the in situ thiol–alkyne addition, thereby improving our understanding of its underlying reaction mechanism and expanding the scope of this reaction to substituted and internal alkynes.
Results and Discussion
Design and Synthesis of ABPs with Substituted Alkyne Warheads
Cysteine DUB activity can be probed by replacing the Ub C-terminal carboxylate (G76) with an electrophile positioned in alignment with the native isopeptide bond (Figure S1A), thus covalently trapping the catalytic cysteine residue.38−40 The binding affinity of the truncated C-terminal Ub peptide at the active site is low; therefore, full-length Ub is used as recognition element in cysteine DUB targeting ABPs.41,42 To elucidate the scope of alkyne substituents in the thiol–alkyne reaction, we prepared a panel of substituted alkynes that were coupled to the C-terminus of fully synthetic Rho-Ub1–75, thus generating new ABPs targeting cysteine DUBs (Figure 1 and Figure S1B). Substituents were introduced on the terminal C1 carbon (2 + 3) or internal C3 carbon (monosubstitution; 4 + 7, or double substitution; 5 + 8) as well as alterations on the Ub backbone (amide) (9 + 10).
Figure 1.

Panel of substituted alkynes incorporated in activity-based probes (ABPs) targeting cysteine DUBs. (A) Synthetic ubiquitin lacking the C-terminal glycine residue (Ub1–75 or UbΔG) was modified with fluorescent rhodamine (Rho) moiety on the N-terminus as reporter tag and with propargylamide (Prg) or propargylamide derivatives 2–10 as cysteine thiol-reactive electrophiles on the C-terminus (synthetic scheme shown in Figure S1B). (B) Substituents were introduced on the terminal C1 carbon, internal C3 carbon, and Ub backbone (amide). Propylamide (Prp) is a noncovalent control.
In detail, terminal methylated alkyne 2 and phenylated alkyne 3 were designed to investigate the importance of the terminal proton on C1. The monomethylated alkyne 4 and monophenylated alkyne 7 (increased bulkiness) with a single substituent on the C3 carbon were included to gain further insight into restrictions at the Ub C-terminus (P1 site in substrate nomenclature). The double-substituted quaternary C3 derivatives geminal-3,3-dimethylated alkyne 5 and cyclohexylated alkyne 8 were included to examine the option of a reactive allene intermediate rather than a reactive alkyne (as presented in Scheme 1C). Adduct formation with these quaternary C3 alkynes would exclude the formation of a reactive allene isomer prior to nucleophilic thiol addition, as it is not possible to deprotonate a quaternary C3 carbon. Furthermore, we included butargyl 9 and N-methylated alkyne 10 to examine the role of the Ub backbone (amide). The longer linker between the amide and the reactive carbon in butargyl 9 excludes conjugating effects by the Ub amide (but is also not optimally aligned with the native isopeptide bond, Figure S1A), whereas the role of the amide proton itself can be examined by replacing it with a methyl group in the N-methylated propargyl derivative 10. Finally, propylamide (Prp) was included as a control, as this compound lacks a reactive warhead and should be incapable of forming covalent adducts.
Rho-Ub-ABPs with Substituted Alkynes Form Covalent Adduct with DUBs in Lysate and Recombinant DUBs
To explore the reactivity of our panel of substituted alkyne ABPs, we explored DUB adduct formation both in lysates and against recombinant DUBs. Whole HEK293 lysate was incubated with the panel of Rho-Ub-ABPs to identify DUBs that form covalent adducts with substituted alkynes 2–10 (Figure 2A).43 In-gel fluorescence shows the typical labeling pattern for Rho-Ub-Prg and reveals that substituents on alkynes 2–10 do not fully mitigate covalent adduct formation (Figure 2B) as labeling, although to a lesser extent, can still be observed. A similar pattern was observed upon incubation of EL4 lysate (Figure S2).
Figure 2.
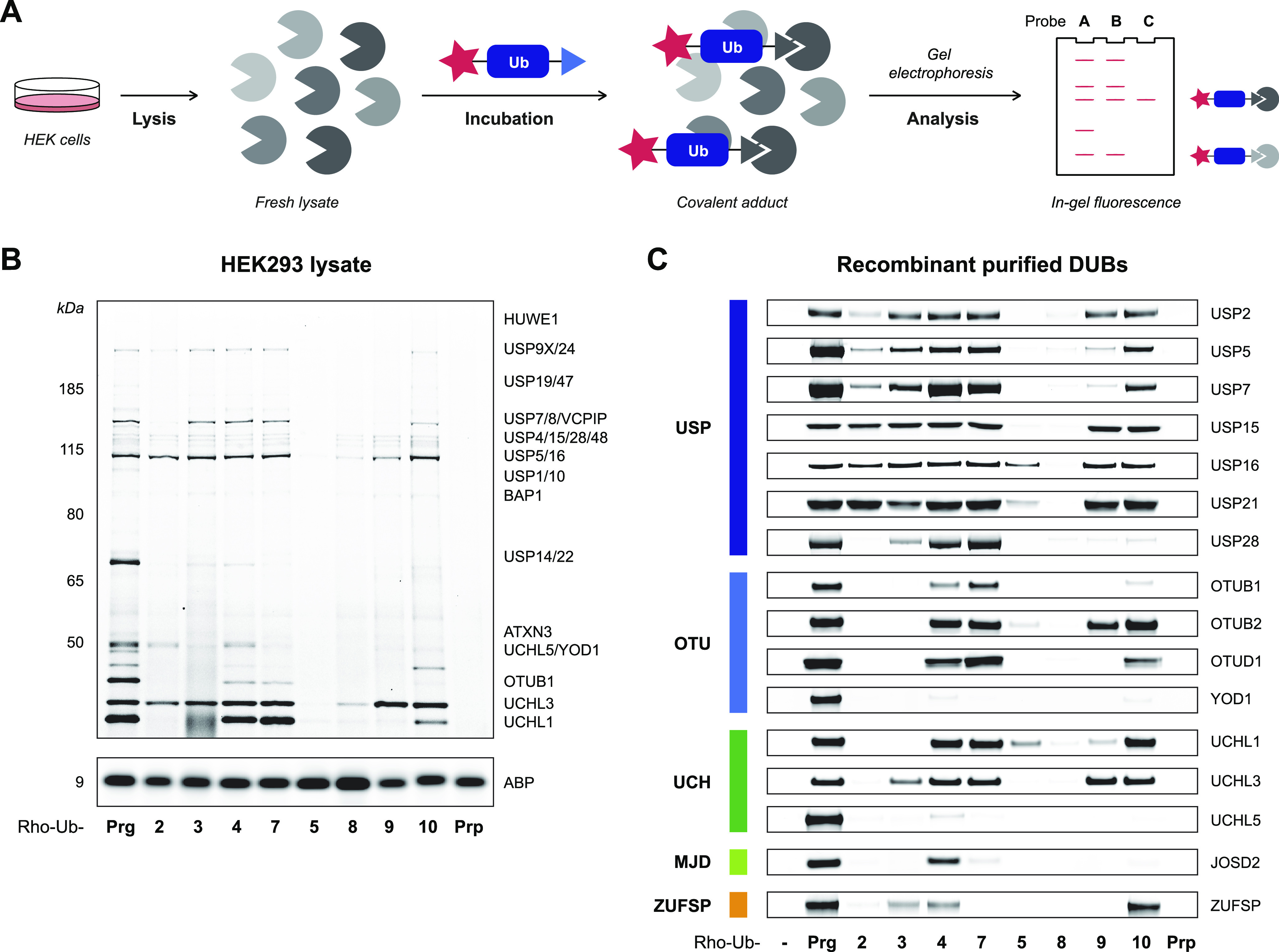
Incubation of whole lysate and purified recombinant DUBs with Rho-Ub-alkyne ABPs. (A) Incubation of whole lysate with Rho-Ub-ABPs to identify covalent ABP-DUB adducts. (B) Fluorescence scan of HEK293 lysate incubated with Rho-Ub-ABP (10 μM) reveals that acceptance of alkyne substituents is DUB-specific. Assignment of labeled DUBs based on proteomic analysis by Altun et al.14 Darker bands correlate with more covalent ABP–enzyme adduct, but the maximum intensity depends on total protein expression. Fluorescence scans of HEK293 or EL4 lysate incubated with 1 or 10 μM Rho-Ub-ABP are shown in Figure S2. Full gel scans and loading controls are provided in Figure S6. (C) Fluorescence scan of recombinant purified cysteine DUBs incubated with Rho-Ub-ABP (10 μM). Fluorescence intensity was normalized to Rho-Ub-Prg adduct. DUB conversion to covalent adduct (visualized with Coomassie protein stain) is shown in Figure S3. Constructs and source of recombinant purified cysteine DUBs are specified in Table S2. Full gel scans and loading controls are provided in Figures S9 and S10.
Next, we validated the labeling observed in whole lysates by incubation of purified recombinant cysteine DUBs with Rho-Ub-ABPs (Figure 2C). Strikingly, substituted alkynes 2 and 5, which were previously reported unreactive toward UCHL3,27 showed reactivity toward other cysteine DUBs. A closer look into our data reveals that Rho-Ub-ABPs with terminally modified alkyne 2 or 3 generally do not form covalent adduct with our set of recombinant OTU, UCH, MJD, and ZUFSP DUBs, but labeling is observed for several USP DUBs. Moreover, labeling patterns in lysate and recombinant DUBs show that monosubstituted alkynes 4 (Ala mimic) and 7 are generally accepted, highlighting that variants at the Ub-ABP P1 position are acceptable. There are some controversies in the field on this matter as DUBs are believed to be sensitive to modifications at P1; available crystal structures show Gly76 occupies a restricted tunnel.44 The most-described example here is mutant UbG76A, which renders poly-Ub chains resistant to DUB cleavage while still posing as a substrate for E1 ligases.45,46 However, Wilkinson et al.47 report that poly-Ub chains with UbG76A at the distal position, although processed slower than UbWT chains, are not resistant to USP5-mediated proteolysis. This is in agreement with our findings that Ala mimic 4 forms a covalent adduct with recombinant USP5 (Figure 2C). Furthermore, even double-substituted alkynes 5 and 8 are accepted by some DUBs. Adduct formation of Rho-Ub-5 with recombinant purified USP16 was evident (Figure 2C), but labeling of endogenous USP16 in HEK lysate was hard to observe (Figure 2B). However, adduct formation of Rho-Ub-5 with USP16 in lysate could be detected when the fluorescence exposure was increased (Figure S2C) as well as by incubation of HeLa lysate overexpressing FLAG-HA-USP16 (Figure S2D). In addition, lysate treatment reveals that UCHL3 is one of the few DUBs that has enough flexibility at its active site to accommodate the longer linker of butargyl 9. Even close family members, UCHL1 and UCHL5, do not accommodate the longer linker length, confirming the deciding role of the cysteine protease in adduct formation. Methylation of amide nitrogen in alkyne 10 is accepted by the majority of DUBs included in our panel. As expected, covalent adducts with Rho-Ub-Prp were not observed, since this compound lacks an electrophilic warhead.
On the basis of these results, we can conclude that substituents on the alkyne warhead do not generally block covalent adduct formation. Mitigation of covalent adduct formation with the cysteine thiol by introduction of substituents is DUB-specific and could be the result of electronic or steric effects or a combination thereof. Two alkyne ABPs were selected for validation of the covalent bond formation: terminal modified alkyne 2 to gain insight into the role of the terminal proton (or steric hindrance) and gem-dimethylated alkyne 5 as isomerization to a more reactive allene intermediate prior to nucleophilic thiol addition (Scheme 1C) is not possible for this substituted alkyne.
ABPs Form Covalent Adducts with Catalytic Cysteine Residue in Recombinant USP16
USP16 (Ubp-M) was selected for validation of covalent adduct formation as it forms a covalent adduct with both Rho-Ub-2 and Rho-Ub-5 (Figure 2C and Figure S2). We selected the catalytic USP domain rather than full length USP16 for validation because of its higher stability and compatibility with top-down mass spectrometry (MS). First, covalent adduct formation with Rho-Ub-Prg, Rho-Ub-2, and Rho-Ub-5 was validated by incubation of recombinant USP16CDWT and resolved by SDS-PAGE under denaturing conditions (Figure 3A, left). As expected, a higher running band corresponding to the fluorescent covalent ABP–enzyme adduct (+8.9 kDa) was revealed by in-gel fluorescence scanning and protein staining. Preincubation of USP16CDWT with the thiol-alkylating reagent N-ethylmaleimide (NEM) prior to incubation with ABPs abolishes adduct formation (Figure 3A, middle), indicative of adduct formation with a cysteine thiol. Catalytically inactive mutant USP16CDC205A was generated to validate modification of catalytic Cys205 rather than one of the 13 noncatalytic cysteine residues present in USP16CD,48,49 as covalent adduct formation of ABP Ub-VS (vinyl sulfone) with less nucleophilic noncatalytic cysteine residues has been reported for UCHL1 and OTUB1.18 Covalent adduct formation was not observed upon incubation of USP16CDC205A mutant (Figure 3A, right).
Figure 3.

Validation of covalent adduct between Rho-Ub-alkyne ABPs and catalytic Cys205 in USP16CD. (A) In-gel fluorescence (top) and Coomassie stain (bottom) of purified recombinant USP16CDWT or mutant USP16CDC205A incubated with ABP (Rho-Ub-Prg, Rho-Ub-2, or Rho-Ub-5). Adduct is formed with USP16CDWT, but preincubation with the thiol-alkylating reagent N-ethylmaleimide (NEM) prior to incubation with ABPs blocks adduct formation, indicating cysteine thiol is required for adduct formation. Adduct is not observed with USP16CDC205A, identifying catalytic Cys205 as the modified cysteine residue. (B) Deconvoluted mass from intact protein MS confirms the covalent adduct (+8.9 kDa) of USP16CDWT with Rho-Ub-Prg, Rho-Ub-2, and Rho-Ub-5. (C) Covalent adduct is not observed in deconvoluted mass from intact protein MS for catalytically inactive mutant USP16CDC205A with Rho-Ub-Prg, Rho-Ub-2, or Rho-Ub-5.
Mass spectrometry (MS) of intact protein (adducts)50,51 confirms covalent ABP–enzyme adduct formation with USP16CDWT (Figure 3B), but covalent adducts are not observed with inactive mutant USP16CDC205A (Figure 3C). Together, these findings confirm that USP16 is covalently modified by the Rho-Ub-alkyne ABPs on catalytic Cys205.
Bottom-Up Mass Spectrometric Analysis Identifies Alkyne and Not Allene as the Reactive Group
Covalent adduct formation of gem-dimethylated alkyne 5 with USP16 does not only illustrate the important role of the cysteine DUB in the in situ thiol–alkyne addition; the retained ability to form a covalent adduct also has mechanistic implications. Adduct formation with Rho-Ub-5 cannot occur through isomerization to an allene intermediate prior to nucleophilic addition (Scheme 1C); deprotonation of the quaternary C3 carbon atom to form the allene is not possible. To confirm our hypothesis, we synthesized Rho-Ub-[D2]-Prg with deuterated propargylamine [D2]-Prg as warhead (Figure 4A). The covalent adduct of a recombinant DUB with Rho-Ub-[D2]-Prg will contain two deuterium atoms if the alkyne is indeed the reactive species (Scheme 1B,D) while isomerization to an allene intermediate (Scheme 1C) would result in replacement of one deuterium atom by a hydrogen atom. Covalent adducts of Rho-Ub-[D2]-Prg and Rho-Ub-Prg with UCHL3 (unreactive toward 5) and USP16 (reactive toward 5) were submitted to alkylation and trypsin digestion to generate peptides for bottom-up mass spectrometric analysis. Peptides of different lengths containing the QTISNACGTIGLIHAIANNK stretch were detected for UCHL3 adducts modified with Prg or [D2]-Prg, with a mass difference of 2 Da between deuterated and protonated adducts (Figure 4B). Peptide GLSNLGNTCFFNAVM(ox)QNLSQTPVLR (with oxidized methionine) was detected for both USP16 adducts, with a mass difference of 2 Da between deuterated and protonated adducts (Figure 4B). Peptides corresponding with isomerization were not detected for the [D2]-Prg adducts. Furthermore, tandem mass spectrometric analysis of both modified UCHL3 peptides confirms that the 2 Da mass difference can be attributed to a modification on the catalytic cysteine residue (Figure 4C,D and Table S6).
Figure 4.
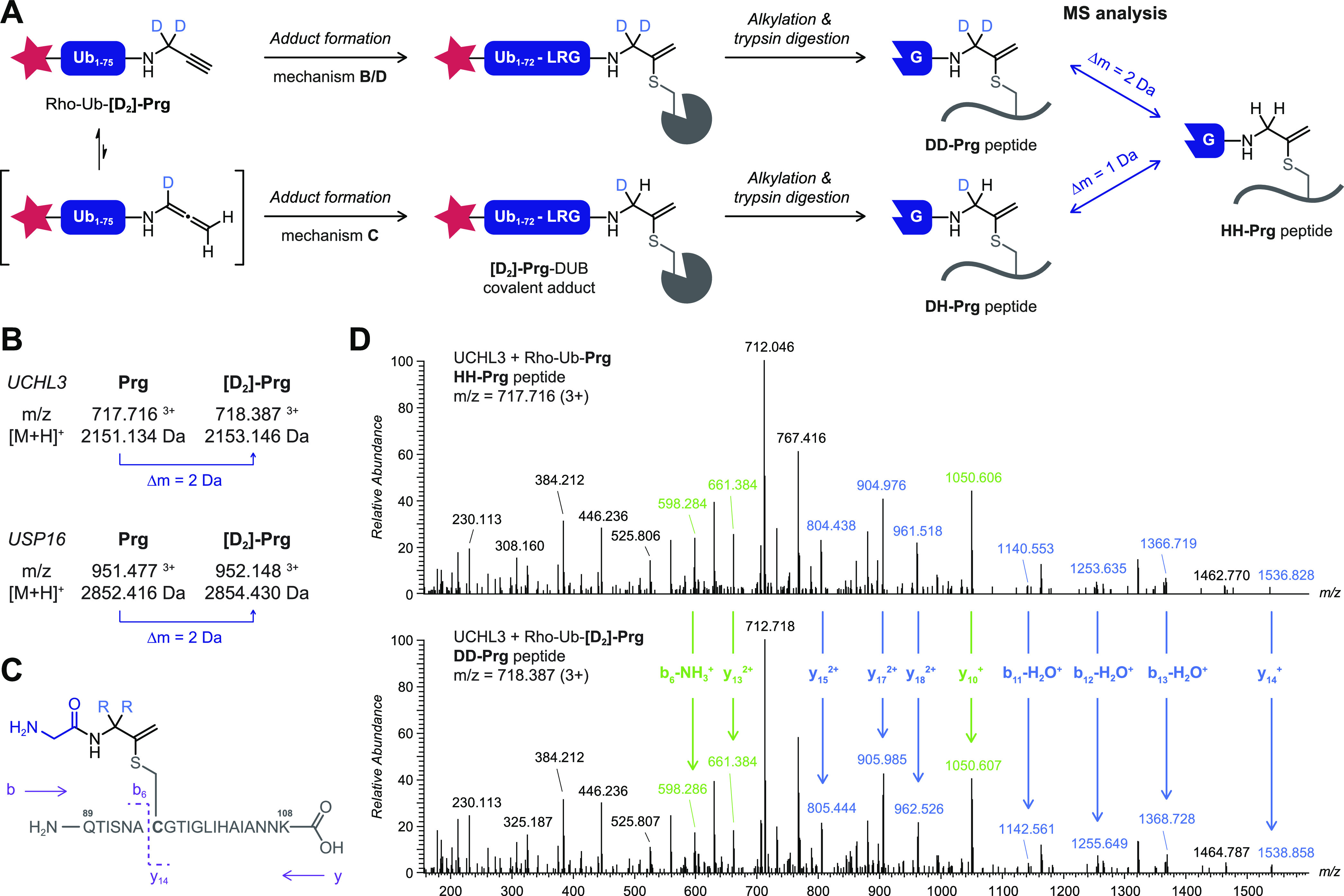
Bottom-up mass spectrometric analysis of covalent adduct with Rho-Ub-[D2]-Prg excludes allene intermediate in mechanism of in situ thiol–alkyne addition. Details on chemical synthesis of deuterated propargylamine [D2]-Prg are provided in Scheme S1. (A) Schematic overview of methodology. Incubation of recombinant DUB with Rho-Ub-[D2]-Prg and Rho-Ub-Prg is followed by alkylation and trypsin digestion to generate modified peptides for mass spectrometric analysis. Isomerization to an allene intermediate (Scheme 1C) will result in replacement of one deuterium atom in the covalent adduct, while both deuterium atoms remain for the other mechanisms (Scheme 1B,D). (B) Modified peptides detected for UCHL3WT (QTISNACGTIGLIHAIANNK) and USP16CDWT (GLSNLGNTCFFNAVM(ox)QNLSQTPVLR) adducts with Rho-Ub-[D2]-Prg and Rho-Ub-Prg have a mass difference of 2 Da, corresponding with mechanisms B and D. (C) Tandem MS fragmentation of trypsin-digested UCHL3 peptide QTISNAC*GTIGLIHAIANNK (residues 89–108). (D) Full MS2 spectrum for UCHL3 peptide QTISNAC*GTIGLIHAIANNK modified with Gly-HH-Prg (top) or Gly-DD-Prg (bottom). Relevant fragment ions are assigned in blue (contains cysteine residue) or green (does not contain cysteine residue), confirming modification on the cysteine residue with 2 Da mass difference. The m/z values of expected and detected fragment ions are provided in Table S6.
Together, this clearly shows that the in situ thiol–alkyne addition to unsubstituted alkynes does not involve isomerization to an allene intermediate, thereby excluding mechanism C (Scheme 1C). It is more challenging to conclude whether nucleophilic addition to the alkyne moiety is exclusively proximity-driven (Scheme 1B) or goes through enzyme-templated stabilization of a carbanion intermediate in the oxyanion hole (Scheme 1D). To our knowledge, all cysteine residues targeted by unactivated alkynes are located at the active site of cysteine proteases (or ligases), which could stabilize a carbanion intermediate in an oxyanion hole (Scheme 3). We cannot exclude nor confirm this mechanism based on our current data, but we would like to note that the inductive effect of the electron-donating methyl group in alkyne 2 contributes negatively to the internal stabilization of the negative charge, thus reducing the stability of the tertiary carbanion compared to the already unfavored secondary carbanion intermediate that is formed with terminal alkynes (Scheme 3B). It is possible that enzyme oxyanion hole sufficiently stabilizes the tertiary carbanion to progress with covalent bond formation, but the proximity-driven reaction seems more likely for internal alkyne 2.
Scheme 3. Stabilization of Anionic Intermediates for (Enzymatic) Reactions with Cysteine DUBs.

(A) Cysteine DUB-mediated isopeptide bond proteolysis. Stabilization of anionic tetrahedral intermediate in the oxyanion hole. Release of ubiquitin, (ubiquitinated) substrate, and cysteine protease. (B) Proposed enzyme-templated thiol–alkyne addition with stabilization of unfavored carbanion intermediate in the oxyanion hole. Terminal alkynes such as Prg would form a secondary carbanion, but internal alkynes such as terminally methylated alkyne 2 would form a tertiary carbanion intermediate that is internally destabilized if R is an electron-donating group (EDG). (C) Nonenzymatic internal stabilization of a carbanion intermediate by inductive effect of electron-withdrawing group. Details on chemical synthesis of trifluoromethylated alkyne 18 are provided in Scheme S2.60
Kinetic Analysis of Covalent Adduct Formation with USP16
Next, we examined whether introduction of bulky/electron-donating substituents on the alkyne terminal C1 or internal C3 carbon atom reduces the rate of covalent adduct formation. Incubation of USP16CDWT with 10 μM Rho-Ub-ABP does indeed show time-dependent increase of the higher running covalent adduct and a decrease of the lower running noncovalent/unbound USP16 for Rho-Ub-2 and Rho-Ub-5 (Figure 5A). Adduct formation does not progress beyond the first time point for Rho-Ub-Prg, indicating that reaction completion was reached before the first sample was quenched (within 15 min). This finding is in agreement with exceptionally fast adduct formation reported for Ub(l)-Prg ABPs (reaction completion within minutes).27,28
Figure 5.
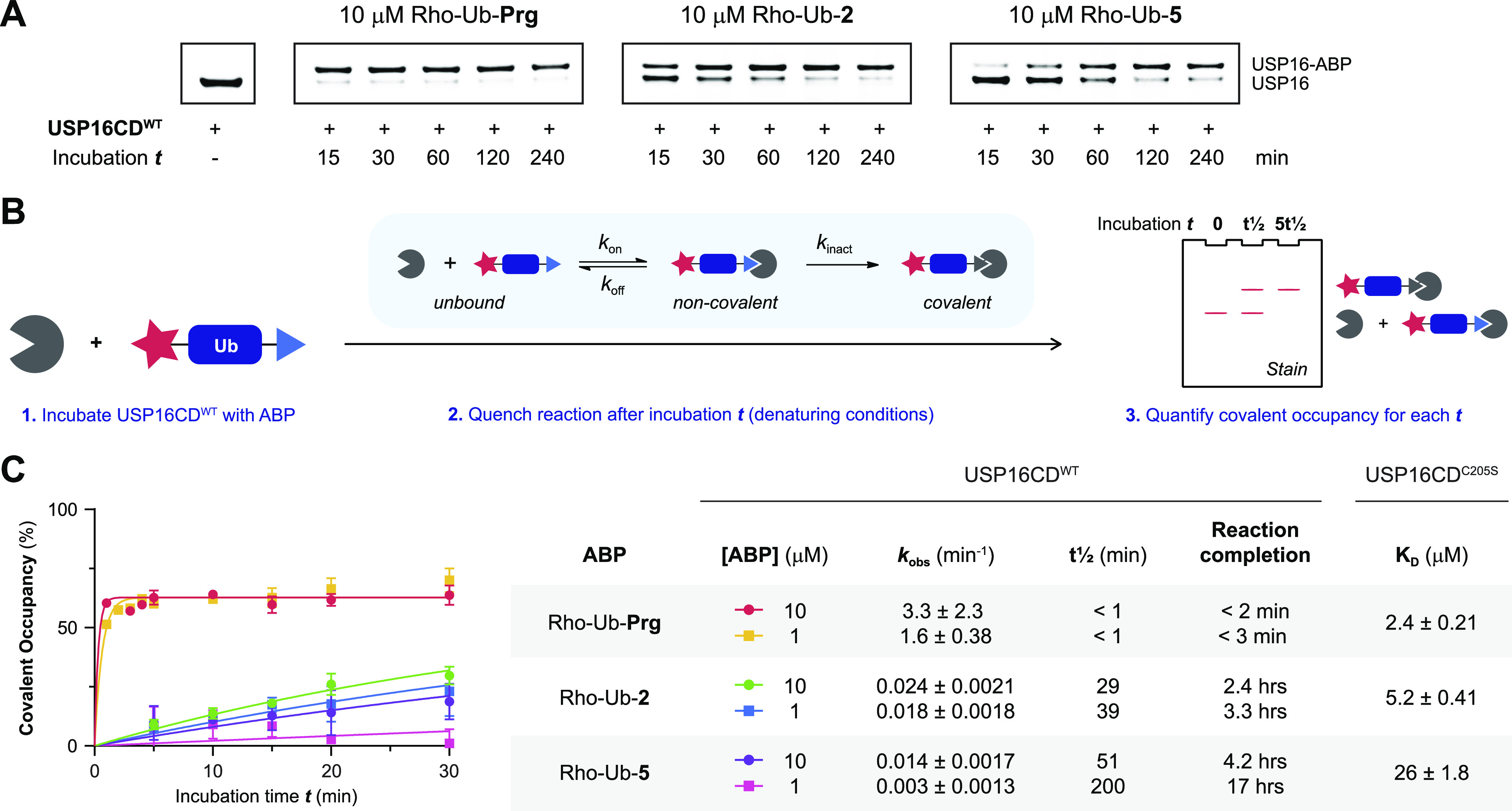
Kinetic analysis of covalent adduct formation with Rho-Ub-alkyne ABPs. (A) Incubation time-dependent covalent adduct formation of Rho-Ub-alkyne ABPs with USP16CDWT visualized by Coomassie stain after gel electrophoresis (denaturing conditions). Intensity of covalent USP16-ABP adduct band increases upon longer incubation time for substituted alkyne ABPs Rho-Ub-2 and Rho-Ub-5, but reaction completion is reached before the first time point for Rho-Ub-Prg. (B) General method to obtain kinetic parameters for covalent ligands from incubation time-dependent covalent occupancy. (C) Kinetic analysis of covalent adduct formation between USP16CD and Rho-Ub-ABP. USP16CDWT was incubated with excess ABP, and samples were quenched after various incubation times. Covalent occupancy was quantified from gel analysis (triplicate measurement) to obtain the rate of covalent adduct formation kobs, reaction half-life t1/2, and reaction completion (details in the Supporting Information). The maximum occupancy is less than 100%, which can be attributed to commonly observed inactive subpopulations in (recombinant) enzyme.61,62 Adduct formation with Rho-Ub-Prg was completed within 5 min; therefore, the measurement was repeated with shorter intervals. Reliable estimates for the kinetic parameters could not be obtained because reaction completion was still reached too quickly. KD values for noncovalent binding were obtained in a binding assay based on fluorescence polarization (FP) of Rho-Ub-ABPs with excess USP16CDC205S mutant.
Covalent adduct formation of USP16 with Rho-Ub-ABPs is slower with substituted alkynes 2 and 5 than with Prg, requiring a longer incubation time to reach maximum covalent occupancy. We performed a kinetic evaluation of covalent adduct formation to calculate the minimum incubation time to reach reaction completion at a specific ABP concentration (Figure 5B). Covalent adduct formation between ABP and cysteine protease is a two-step process: the noncovalent ABP–enzyme complex is formed rapidly, followed by covalent adduct formation as the rate-determining step.52−54 Time-dependent covalent occupancy of irreversible covalent ligands can be directly detected (in the absence of competing substrate/ligand) by separation of covalent adduct from noncovalent complex and unbound enzyme on LCMS or gel and subsequent quantification of signals.55−59 Here we incubated USP16CDWT with excess Rho-Ub-ABPs and quantified incubation time-dependent covalent occupancy by gel analysis (Figure 5C). Estimates for the rate of covalent adduct formation kobs, reaction half-life t1/2 and incubation time to reach reaction completion were obtained by assuming maximum covalent occupancy is shared among all ABPs. Adduct formation with all ABPs is concentration-dependent; reaction completion is reached faster at the high ABP concentration. However, covalent adduct formation with Rho-Ub-Prg is unusually fast; maximum covalent occupancy is reached within a few minutes at both concentrations, and the reaction rates might be even faster than what we reported here. The half-life and extrapolated incubation time to reach maximum covalent adduct formation provide valuable insights into the reduced reactivity of Rho-Ub-5 in incubation experiments (Figure 2 and Figure S2); reaction completion is reached after more than 4 h, which well exceeds the common incubation time for ABPs with lysate or recombinant protein. Incomplete adduct formation is observed as a band with (significantly) lower intensity than the band with Rho-Ub-Prg which does reach maximum intensity.
Overall, introduction of substituents on propargylamide decreases the rate of covalent adduct formation with USP16CDWT by >30-fold for methylation of the terminal C1 carbon (Rho-Ub-2) and >100-fold for gem-dimethylation of the internal C3 carbon (Rho-Ub-5). This dramatic reduction in reaction rate explains the low reactivity of substituted alkynes upon incubation of lysate or recombinant protein as adduct formation is not completed within the standard incubation time of 30–60 min. Rho-Ub-ABPs with slower covalent adduct formation than Rho-Ub-Prg could be desirable as they are more suited to study (ir)reversible inhibitor potency in kinetic competition assays.12,63,64
Next, a binding assay based on fluorescence polarization (FP) of the Rho-Ub-ABPs with excess catalytic inactive USP16C205S mutant was performed to determine KD values independent of electronic factors as covalent adduct formation with USP16C205S does not occur (Figure 5C). Introduction of methyl substituents clearly reduced the noncovalent affinity (reflected in higher KD), indicative of disfavored steric interactions. However, electronic effects cannot be disregarded as the rate of adduct formation (kobs) with Rho-Ub-2 is more than 30-fold slower than with Rho-Ub-Prg where the noncovalent affinity (KD) is less than 3-fold lower. This shows that disfavored steric interactions as well as electronic effects contribute to the reduced rate of covalent adduct formation with methylated alkynes.
Contribution of Steric and Electronic Effects on DUB Reactivity toward Substituted Alkynes
Substituents introduced on the alkyne C1 or C3 positions (Figure 1) were designed to have a minimal electronic effect, but kinetic evaluation of covalent adduct formation (kobs) and noncovalent affinity (reflected in the KD) with USP16 revealed that the role of steric and electronic effects cannot be separated completely (Figure 5C). To further study the individual contribution of steric and electronic components, we included electron-deficient alkyne 18, with an electron-withdrawing −CF3 group on the terminal alkyne carbon (Scheme 3C and Figure S4A). Introduction of an electron-withdrawing group (EWG) on the terminal position of an alkyne significantly increases the thiol reactivity as the inductive effect contributes positively to the stabilization of a negative charge, thereby enabling nonenzymatic internal stabilization of a carbanion intermediate (Scheme 3C). The increased electrophilicity was indeed reflected in the observation of significant adduct formation with nontargeted thiol glutathione (GSH) (Figure S4B,C). Incubation of HEK293T lysate (Figure S4D) showed that most DUBs form a covalent adduct with Rho-Ub-18, indicating an electronic rather than steric component driving the lack of reactivity with alkyne 2. Faint covalent adduct formation with Rho-Ub-18 was observed upon incubation of USP16CDC205A mutant, indicating a preference for the catalytic cysteine residue over other (nontargeted) cysteine residues (Figure S4E,F). Altogether, we can conclude that (disfavored) steric as well as electronic properties of the substituent affect DUB reactivity with substituted alkynes.
Implications on the Scope of the In Situ Thiol–Alkyne Addition
Introduction of bulky and/or electron-donating substituents can reduce the rate of covalent bond formation, but it is DUB-dependent whether modifications are allowed. We foresee this might be used for the development of ABPs with improved selectivity for a specific DUB. Here, introduction of electron-donating and electron-withdrawing substituents on the C1 or C3 positions would tune alkyne reactivity (electronic effect) while simultaneously modulating selectivity (steric effect). Another possibility would be to introduce primed site recognition peptide fragments on the terminal alkyne position to improve selectivity and/or affinity.10,65,66
The reaction mechanism (Scheme 1B or 1D) has extensive consequences for the scope of the in situ alkyne–thiol addition. Enzyme-templated stabilization of a carbanion intermediate (Scheme 1D) would restrict the applicability in drug design to targeting catalytic cysteine residues with unactivated alkynes, but it also mitigates the risk of covalent adduct formation with nontargeted thiols. A covalent adduct is not formed with noncatalytic cysteine residues because the carbanion intermediate cannot be stabilized as there is no oxyanion hole present in their vicinity, resulting in a mechanism-based selectivity for the targeted thiol. To date, only electron-deficient (activated) alkynes such as propiolamides, propiolonitriles, and alkynylated heteroarenes have been reported to form covalent adducts with noncatalytic cysteine residues (in kinase targets).25,35,59,67−69 The inductive effect of (conjugated) electron-withdrawing groups sufficiently stabilizes the carbanion intermediate to progress with covalent bond formation, with targeted as well as (undesired) nontargeted thiols (Scheme 3C). Unfortunately, electron-deficient alkyne 18 is not suited to study whether the in situ thiol addition to unactivated alkynes involves enzymatic stabilization of a carbanion in the oxyanion hole (Schemes 1D and 3B) because thiol addition can progress through an alternative, nonenzymatic mechanism (Scheme 3C). We believe further research to elucidate the mechanism of thiol addition to unactivated (internal) alkynes should be directed toward computational studies with enzymes for which structural data are available or by successfully targeting noncatalytic cysteines with unactivated alkynes.
Conclusion
To conclude, this work shows that the in situ thiol–alkyne reaction is more flexible and versatile than previously assumed. A panel of substituted propargylamide derivatives was incorporated into Rho-Ub-ABPs as the electrophilic warhead, and treatment of lysate or recombinant cysteine DUBs showed that covalent adducts can also be formed with internal alkynes and terminal alkynes with (double) substituents on the internal C3 carbon. Covalent adduct formation of terminally methylated alkyne 2 and gem-dimethylated alkyne 5 with catalytic Cys205 of USP16 was validated by gel analysis and mass spectrometry of intact covalent adducts. Adduct formation was mitigated by preincubation with thiol-alkylating reagent NEM or by C205A mutation, thus confirming catalytic Cys205 as the targeted amino acid residue. Mechanistically, acceptance of gem-dimethylated alkyne 5 together with mass spectrometric analysis of covalent adducts with deuterated ABP Rho-Ub-[D2]-Prg validates the alkyne moiety rather than an allenic isomer as the reactive species in the in situ thiol–alkyne addition. Kinetic analysis revealed reaction completion was reached within in a few minutes for Rho-Ub-Prg, while electron-donating/bulky methyl substituents on alkynes 2 and 5 significantly reduced the rate of covalent adduct formation resultant from a combination of (disfavored) steric interactions and electronic effects, reaching maximum covalent occupancy after (several) hours. Whether nucleophilic addition of the catalytic cysteine thiol to the alkyne moiety is solely proximity driven or involves enzymatic stabilization of a carbanion intermediate could not be concluded definitively.
Altogether, we extended the scope of the in situ thiol–alkyne reaction from unmodified terminal alkynes to substituted (internal) alkynes, provided mechanistic insight, and discovered that acceptance of alkyne substituents is DUB-dependent. We anticipate substituted unactivated alkynes not to be restricted to bioorthogonal handles but also to be of great value as electrophiles in future development of cysteine-targeting covalent inhibitors and activity-based probes with improved selectivity profiles.
Acknowledgments
In memory of Prof. Dr. Huib Ovaa. His passion for science will always be an inspiration to us. We thank Henk Hilkmann, Dris el Atmioui, and Cami Talavera Ormeño for the chemical synthesis of ubiquitin and Dris el Atmioui for his support with ABP synthesis. Angeliki Moutsiopoulou, Patrick Celie, Ruud Wijdeven, Aysegul Sapmaz, Gabrielle van Tilburg, and Remco Merkx are thanked for expression and purification of recombinant DUBs. Recombinant YOD1 was kindly provided by Tycho Mevissen and David Komander, and recombinant ZUFSP was a gift from Kay Hofmann. Sabina van der Zanden, Yves Leestemaker, Merve Öyken, and Lorina Gjonaj are thanked for culturing HEK293, EL4, HEK293T, and HeLa cells, respectively. We thank Annemarie Otte for mass spectrometric measurements. This work was supported by a VICI grant (no. 724.013.002) from The Netherlands Organization for Scientific Research (NWO) to H.O.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c10513.
Detailed experimental procedures and figures (PDF)
The authors declare the following competing financial interest(s): H.O. was founder and shareholder of the company UbiQ that markets reagents in the Ub field.
Author Status
⊥ H.O.: Deceased 19 May 2020.
Supplementary Material
References
- Clague M. J.; Urbé S.; Komander D. Breaking the chains: deubiquitylating enzyme specificity begets function. Nat. Rev. Mol. Cell Biol. 2019, 20 (6), 338–352. 10.1038/s41580-019-0099-1. [DOI] [PubMed] [Google Scholar]
- Ebner P.; Versteeg G. A.; Ikeda F. Ubiquitin enzymes in the regulation of immune responses. Crit. Rev. Biochem. Mol. Biol. 2017, 52 (4), 425–460. 10.1080/10409238.2017.1325829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L.; Meng T.; Chen L.; Wei W.; Wang P. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduction Targeted Ther. 2020, 5 (1), 11. 10.1038/s41392-020-0107-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovic D.; Vucic D.; Dikic I. Ubiquitination in disease pathogenesis and treatment. Nat. Med. 2014, 20 (11), 1242–1253. 10.1038/nm.3739. [DOI] [PubMed] [Google Scholar]
- Huang X.; Dixit V. M. Drugging the undruggables: exploring the ubiquitin system for drug development. Cell Res. 2016, 26 (4), 484–498. 10.1038/cr.2016.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veggiani G.; Gerpe M. C. R.; Sidhu S. S.; Zhang W. Emerging drug development technologies targeting ubiquitination for cancer therapeutics. Pharmacol. Ther. 2019, 199, 139–154. 10.1016/j.pharmthera.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertz I. E.; Wang X. From Discovery to Bedside: Targeting the Ubiquitin System. Cell Chem. Biol. 2019, 26 (2), 156–177. 10.1016/j.chembiol.2018.10.022. [DOI] [PubMed] [Google Scholar]
- Kemp M. Chapter Three - Recent Advances in the Discovery of Deubiquitinating Enzyme Inhibitors. Prog. Med. Chem. 2016, 55, 149–192. 10.1016/bs.pmch.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B.; Tsai Y. C.; Jin B.; Wang B.; Wang Y.; Zhou H.; Carpenter T.; Weissman A. M.; Yin J. Protein Engineering in the Ubiquitin System: Tools for Discovery and Beyond. Pharmacol. Rev. 2020, 72 (2), 380–413. 10.1124/pr.118.015651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor N. C.; McGouran J. F. Strategies to Target Specific Components of the Ubiquitin Conjugation/Deconjugation Machinery. Front. Chem. 2020, 7, 914. 10.3389/fchem.2019.00914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewings D. S.; Flygare J. A.; Bogyo M.; Wertz I. E. Activity-based probes for the ubiquitin conjugation–deconjugation machinery: new chemistries, new tools, and new insights. FEBS J. 2017, 284 (10), 1555–1576. 10.1111/febs.14039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H.; Lei Q.; Wu Y.; He Y.; Li W. Activity-based protein profiling: Recent advances in medicinal chemistry. Eur. J. Med. Chem. 2020, 191, 112151. 10.1016/j.ejmech.2020.112151. [DOI] [PubMed] [Google Scholar]
- Chakrabarty S.; Kahler J. P.; van de Plassche M. A. T.; Vanhoutte R.; Verhelst S. H. L. Recent Advances in Activity-Based Protein Profiling of Proteases. Curr. Top. Microbiol. Immunol. 2019, 420, 253–281. 10.1007/82_2018_138. [DOI] [PubMed] [Google Scholar]
- Altun M.; Kramer H. B.; Willems L. I.; McDermott J. L.; Leach C. A.; Goldenberg S. J.; Kumar K. G. S.; Konietzny R.; Fischer R.; Kogan E.; Mackeen M. M.; McGouran J.; Khoronenkova S. V.; Parsons J. L.; Dianov G. L.; Nicholson B.; Kessler B. M. Activity-Based Chemical Proteomics Accelerates Inhibitor Development for Deubiquitylating Enzymes. Chem. Biol. 2011, 18 (11), 1401–1412. 10.1016/j.chembiol.2011.08.018. [DOI] [PubMed] [Google Scholar]
- Love K. R.; Pandya R. K.; Spooner E.; Ploegh H. L. Ubiquitin C-Terminal Electrophiles Are Activity-Based Probes for Identification and Mechanistic Study of Ubiquitin Conjugating Machinery. ACS Chem. Biol. 2009, 4 (4), 275–287. 10.1021/cb9000348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnick E.; Bradley A.; Gan J.; Douangamath A.; Krojer T.; Sethi R.; Geurink P. P.; Aimon A.; Amitai G.; Bellini D.; Bennett J.; Fairhead M.; Fedorov O.; Gabizon R.; Gan J.; Guo J.; Plotnikov A.; Reznik N.; Ruda G. F.; Díaz-Sáez L.; Straub V. M.; Szommer T.; Velupillai S.; Zaidman D.; Zhang Y.; Coker A. R.; Dowson C. G.; Barr H. M.; Wang C.; Huber K. V. M.; Brennan P. E.; Ovaa H.; von Delft F.; London N. Rapid Covalent-Probe Discovery by Electrophile-Fragment Screening. J. Am. Chem. Soc. 2019, 141 (22), 8951–8968. 10.1021/jacs.9b02822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto-Fernández A.; Davis S.; Schofield A. B.; Scott H. C.; Zhang P.; Salah E.; Mathea S.; Charles P. D.; Damianou A.; Bond G.; Fischer R.; Kessler B. M. Comprehensive Landscape of Active Deubiquitinating Enzymes Profiled by Advanced Chemoproteomics. Front. Chem. 2019, 7, 592. 10.3389/fchem.2019.00592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewings D. S.; Heideker J.; Ma T. P.; AhYoung A. P.; El Oualid F.; Amore A.; Costakes G. T.; Kirchhofer D.; Brasher B.; Pillow T.; Popovych N.; Maurer T.; Schwerdtfeger C.; Forrest W. F.; Yu K.; Flygare J.; Bogyo M.; Wertz I. E. Reactive-site-centric chemoproteomics identifies a distinct class of deubiquitinase enzymes. Nat. Commun. 2018, 9 (1), 1162. 10.1038/s41467-018-03511-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borodovsky A.; Ovaa H.; Kolli N.; Gan-Erdene T.; Wilkinson K. D.; Ploegh H. L.; Kessler B. M. Chemistry-Based Functional Proteomics Reveals Novel Members of the Deubiquitinating Enzyme Family. Chem. Biol. 2002, 9 (10), 1149–1159. 10.1016/S1074-5521(02)00248-X. [DOI] [PubMed] [Google Scholar]
- Abdul Rehman S. A.; Kristariyanto Y. A.; Choi S.-Y.; Nkosi P. J.; Weidlich S.; Labib K.; Hofmann K.; Kulathu Y. MINDY-1 Is a Member of an Evolutionarily Conserved and Structurally Distinct New Family of Deubiquitinating Enzymes. Mol. Cell 2016, 63 (1), 146–155. 10.1016/j.molcel.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermanns T.; Woiwode I.; Guerreiro R. F. M.; Vogt R.; Lammers M.; Hofmann K. An evolutionary approach to systematic discovery of novel deubiquitinases, applied to Legionella. Life Sci. Alliance 2020, 3 (9), e202000838. 10.26508/lsa.202000838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert A. F.; Nguyen J. V.; Franklin T. G.; Geurink P. P.; Roberts C. G.; Sanderson D. J.; Miller L. N.; Ovaa H.; Hofmann K.; Pruneda J. N.; Komander D. Identification and characterization of diverse OTU deubiquitinases in bacteria. EMBO J. 2020, 39 (15), e105127. 10.15252/embj.2020105127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausman J. M.; Kenny S.; Iyer S.; Babar A.; Qiu J.; Fu J.; Luo Z.-Q.; Das C. The Two Deubiquitinating Enzymes from Chlamydia trachomatis Have Distinct Ubiquitin Recognition Properties. Biochemistry 2020, 59 (16), 1604–1617. 10.1021/acs.biochem.9b01107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker C. G.; Pratt M. R. Click Chemistry in Proteomic Investigations. Cell 2020, 180 (4), 605–632. 10.1016/j.cell.2020.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talele T. T. Acetylene Group, Friend or Foe in Medicinal Chemistry. J. Med. Chem. 2020, 63 (11), 5625–5663. 10.1021/acs.jmedchem.9b01617. [DOI] [PubMed] [Google Scholar]
- Wright M. H.; Sieber S. A. Chemical proteomics approaches for identifying the cellular targets of natural products. Nat. Prod. Rep. 2016, 33 (5), 681–708. 10.1039/C6NP00001K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekkebus R.; van Kasteren S. I.; Kulathu Y.; Scholten A.; Berlin I.; Geurink P. P.; de Jong A.; Goerdayal S.; Neefjes J.; Heck A. J. R.; Komander D.; Ovaa H. On Terminal Alkynes That Can React with Active-Site Cysteine Nucleophiles in Proteases. J. Am. Chem. Soc. 2013, 135 (8), 2867–2870. 10.1021/ja309802n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer S.; Weikart N. D.; Linne U.; Mootz H. D. Covalent inhibition of SUMO and ubiquitin-specific cysteine proteases by an in situ thiol–alkyne addition. Bioorg. Med. Chem. 2013, 21 (9), 2511–2517. 10.1016/j.bmc.2013.02.039. [DOI] [PubMed] [Google Scholar]
- Jia Y.; Claessens L. A.; Vertegaal A. C. O.; Ovaa H. Chemical Tools and Biochemical Assays for SUMO Specific Proteases (SENPs). ACS Chem. Biol. 2019, 14 (11), 2389–2395. 10.1021/acschembio.9b00402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairbanks B. D.; Sims E. A.; Anseth K. S.; Bowman C. N. Reaction Rates and Mechanisms for Radical, Photoinitated Addition of Thiols to Alkynes, and Implications for Thiol–Yne Photopolymerizations and Click Reactions. Macromolecules 2010, 43 (9), 4113–4119. 10.1021/ma1002968. [DOI] [Google Scholar]
- Jayasree E. G.; Reshma S. A computational study on the reaction mechanism and energetics of Markovnikov and anti-Markovnikov addition in alkyne hydrothiolation reactions. Comput. Theor. Chem. 2016, 1098, 13–21. 10.1016/j.comptc.2016.10.012. [DOI] [Google Scholar]
- Arkona C.; Rademann J. Propargyl Amides as Irreversible Inhibitors of Cysteine Proteases—A Lesson on the Biological Reactivity of Alkynes. Angew. Chem., Int. Ed. 2013, 52 (32), 8210–8212. 10.1002/anie.201303544. [DOI] [PubMed] [Google Scholar]
- Mons E.; Jansen I. D. C.; Loboda J.; van Doodewaerd B. R.; Hermans J.; Verdoes M.; van Boeckel C. A. A.; van Veelen P. A.; Turk B.; Turk D.; Ovaa H. The Alkyne Moiety as a Latent Electrophile in Irreversible Covalent Small Molecule Inhibitors of Cathepsin K. J. Am. Chem. Soc. 2019, 141 (8), 3507–3514. 10.1021/jacs.8b11027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashmi A. S. K. Synthesis of Allenes by Isomerization Reactions. Mod. Allene Chem. 2004, 2–50. 10.1002/9783527619573.ch1. [DOI] [Google Scholar]
- Gehringer M.; Laufer S. A. Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2019, 62 (12), 5673–5724. 10.1021/acs.jmedchem.8b01153. [DOI] [PubMed] [Google Scholar]
- Menard R.; Storer A. C. Oxyanion Hole Interactions in Serine and Cysteine Proteases. Biol. Chem. Hoppe-Seyler 1992, 373 (2), 393–400. 10.1515/bchm3.1992.373.2.393. [DOI] [PubMed] [Google Scholar]
- Shokhen M.; Traube T.; Vijayakumar S.; Hirsch M.; Uritsky N.; Albeck A. Differentiating Serine and Cysteine Protease Mechanisms by New Covalent QSAR Descriptors. ChemBioChem 2011, 12 (7), 1023–1026. 10.1002/cbic.201000459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heal W. P.; Dang T. H. T.; Tate E. W. Activity-based probes: discovering new biology and new drug targets. Chem. Soc. Rev. 2011, 40 (1), 246–257. 10.1039/C0CS00004C. [DOI] [PubMed] [Google Scholar]
- de Jong A.; Merkx R.; Berlin I.; Rodenko B.; Wijdeven R. H. M.; El Atmioui D.; Yalçin Z.; Robson C. N.; Neefjes J. J.; Ovaa H. Ubiquitin-Based Probes Prepared by Total Synthesis To Profile the Activity of Deubiquitinating Enzymes. ChemBioChem 2012, 13 (15), 2251–2258. 10.1002/cbic.201200497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui X.; Wang Y.; Du Y.-X.; Liang L.-J.; Zheng Q.; Li Y.-M.; Liu L. Development and application of ubiquitin-based chemical probes. Chem. Sci. 2020, 11, 12633–12646. 10.1039/D0SC03295F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hameed D. S.; Sapmaz A.; Ovaa H. How Chemical Synthesis of Ubiquitin Conjugates Helps To Understand Ubiquitin Signal Transduction. Bioconjugate Chem. 2017, 28 (3), 805–815. 10.1021/acs.bioconjchem.6b00140. [DOI] [PubMed] [Google Scholar]
- Rut W.; Zmudzinski M.; Snipas S. J.; Bekes M.; Huang T. T.; Drag M. Engineered unnatural ubiquitin for optimal detection of deubiquitinating enzymes. Chem. Sci. 2020, 11 (23), 6058–6069. 10.1039/D0SC01347A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leestemaker Y.; de Jong A.; Ovaa H. Profiling the Activity of Deubiquitinating Enzymes Using Chemically Synthesized Ubiquitin-Based Probes. Methods Mol. Biol. 2017, 1491, 113–130. 10.1007/978-1-4939-6439-0_9. [DOI] [PubMed] [Google Scholar]
- Drag M.; Mikolajczyk J.; Bekes M.; Reyes-Turcu F. E.; Ellman J. A.; Wilkinson K. D.; Salvesen G. S. Positional-scanning fluorigenic substrate libraries reveal unexpected specificity determinants of DUBs (deubiquitinating enzymes). Biochem. J. 2008, 415 (3), 367–375. 10.1042/BJ20080779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgins R. R.; Ellison K. S.; Ellison M. J. Expression of a ubiquitin derivative that conjugates to protein irreversibly produces phenotypes consistent with a ubiquitin deficiency. J. Biol. Chem. 1992, 267 (13), 8807–8812. 10.1016/S0021-9258(19)50351-9. [DOI] [PubMed] [Google Scholar]
- Pickart C. M.; Kasperek E. M.; Beal R.; Kim A. Substrate properties of site-specific mutant ubiquitin protein (G76A) reveal unexpected mechanistic features of ubiquitin-activating enzyme (E1). J. Biol. Chem. 1994, 269 (10), 7115–7123. 10.1016/S0021-9258(17)37255-1. [DOI] [PubMed] [Google Scholar]
- Wilkinson K. D.; Tashayev V. L.; O’Connor L. B.; Larsen C. N.; Kasperek E.; Pickart C. M. Metabolism of the polyubiquitin degradation signal: structure, mechanism, and role of isopeptidase T. Biochemistry 1995, 34 (44), 14535–14546. 10.1021/bi00044a032. [DOI] [PubMed] [Google Scholar]
- Kathman S. G.; Span I.; Smith A. T.; Xu Z.; Zhan J.; Rosenzweig A. C.; Statsyuk A. V. A Small Molecule That Switches a Ubiquitin Ligase From a Processive to a Distributive Enzymatic Mechanism. J. Am. Chem. Soc. 2015, 137 (39), 12442–12445. 10.1021/jacs.5b06839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.-J.; Du L.; Wang J.; Vega R.; Lee T. D.; Miao Y.; Aldana-Masangkay G.; Samuels E. R.; Li B.; Ouyang S. X.; Colayco S. A.; Bobkova E. V.; Divlianska D. B.; Sergienko E.; Chung T. D. Y.; Fakih M.; Chen Y. Allosteric Inhibition of Ubiquitin-like Modifications by a Class of Inhibitor of SUMO-Activating Enzyme. Cell Chem. Biol. 2019, 26 (2), 278–288. 10.1016/j.chembiol.2018.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tailor A.; Waddington J. C.; Meng X.; Park B. K. Mass Spectrometric and Functional Aspects of Drug–Protein Conjugation. Chem. Res. Toxicol. 2016, 29 (12), 1912–1935. 10.1021/acs.chemrestox.6b00147. [DOI] [PubMed] [Google Scholar]
- Backus K. M. Applications of Reactive Cysteine Profiling. Curr. Top. Microbiol. Immunol. 2019, 420, 375–417. 10.1007/82_2018_120. [DOI] [PubMed] [Google Scholar]
- Copeland R. A.Irreversible Enzyme Inactivators. In Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, 2nd ed.; John Wiley & Sons: Hoboken, NJ, 2013; pp 345–382. [PubMed] [Google Scholar]
- Tuley A.; Fast W. The Taxonomy of Covalent Inhibitors. Biochemistry 2018, 57 (24), 3326–3337. 10.1021/acs.biochem.8b00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J.; Petter R. C.; Baillie T. A.; Whitty A. The resurgence of covalent drugs. Nat. Rev. Drug Discovery 2011, 10 (4), 307–317. 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- Johansson H.; Isabella Tsai Y.-C.; Fantom K.; Chung C.-W.; Kümper S.; Martino L.; Thomas D. A.; Eberl H. C.; Muelbaier M.; House D.; Rittinger K. Fragment-Based Covalent Ligand Screening Enables Rapid Discovery of Inhibitors for the RBR E3 Ubiquitin Ligase HOIP. J. Am. Chem. Soc. 2019, 141 (6), 2703–2712. 10.1021/jacs.8b13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan X.; Yang T.; Cuesta A.; Pang X.; Balius T. E.; Irwin J. J.; Shoichet B. K.; Taunton J. Discovery of Lysine-Targeted eIF4E Inhibitors through Covalent Docking. J. Am. Chem. Soc. 2020, 142 (11), 4960–4964. 10.1021/jacs.9b10377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klüter S.; Simard J. R.; Rode H. B.; Grütter C.; Pawar V.; Raaijmakers H. C. A.; Barf T. A.; Rabiller M.; van Otterlo W. A. L.; Rauh D. Characterization of Irreversible Kinase Inhibitors by Directly Detecting Covalent Bond Formation: A Tool for Dissecting Kinase Drug Resistance. ChemBioChem 2010, 11 (18), 2557–2566. 10.1002/cbic.201000352. [DOI] [PubMed] [Google Scholar]
- Hansen R.; Firdaus S. J.; Li S.; Janes M. R.; Zhang J.; Liu Y.; Zarrinkar P. P. An Internally Controlled Quantitative Target Occupancy Assay for Covalent Inhibitors. Sci. Rep. 2018, 8 (1), 14312. 10.1038/s41598-018-32683-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson S. H.; Liu Q.; Beaudoin Bertrand M.; Batt D. G.; Li L.; Pattoli M. A.; Skala S.; Cheng L.; Obermeier M. T.; Moore R.; Yang Z.; Vickery R.; Elzinga P. A.; Discenza L.; D’Arienzo C.; Gillooly K. M.; Taylor T. L.; Pulicicchio C.; Zhang Y.; Heimrich E.; McIntyre K. W.; Ruan Q.; Westhouse R. A.; Catlett I. M.; Zheng N.; Chaudhry C.; Dai J.; Galella M. A.; Tebben A. J.; Pokross M.; Li J.; Zhao R.; Smith D.; Rampulla R.; Allentoff A.; Wallace M. A.; Mathur A.; Salter-Cid L.; Macor J. E.; Carter P. H.; Fura A.; Burke J. R.; Tino J. A. Discovery of Branebrutinib (BMS-986195): A Strategy for Identifying a Highly Potent and Selective Covalent Inhibitor Providing Rapid in Vivo Inactivation of Bruton’s Tyrosine Kinase (BTK). J. Med. Chem. 2019, 62 (7), 3228–3250. 10.1021/acs.jmedchem.9b00167. [DOI] [PubMed] [Google Scholar]
- Tresse C.; Guissart C.; Schweizer S.; Bouhoute Y.; Chany A.-C.; Goddard M.-L.; Blanchard N.; Evano G. Practical Methods for the Synthesis of Trifluoromethylated Alkynes: Oxidative Trifluoromethylation of Copper Acetylides and Alkynes. Adv. Synth. Catal. 2014, 356 (9), 2051–2060. 10.1002/adsc.201400057. [DOI] [Google Scholar]
- Bovet C.; Zenobi R. Determination of active enzyme concentration using activity-based probes and direct mass spectrometric readout. Anal. Biochem. 2008, 373 (2), 380–382. 10.1016/j.ab.2007.11.003. [DOI] [PubMed] [Google Scholar]
- Brocklehurst K.; Resmini M.; Topham C. M. Kinetic and Titration Methods for Determination of Active Site Contents of Enzyme and Catalytic Antibody Preparations. Methods 2001, 24 (2), 153–167. 10.1006/meth.2001.1176. [DOI] [PubMed] [Google Scholar]
- Miyahisa I.; Sameshima T.; Hixon M. S. Rapid Determination of the Specificity Constant of Irreversible Inhibitors (kinact/KI) by Means of an Endpoint Competition Assay. Angew. Chem., Int. Ed. 2015, 54 (47), 14099–14102. 10.1002/anie.201505800. [DOI] [PubMed] [Google Scholar]
- Sameshima T.; Tanaka Y.; Miyahisa I. Universal and Quantitative Method To Evaluate Inhibitor Potency for Cysteinome Proteins Using a Nonspecific Activity-Based Protein Profiling Probe. Biochemistry 2017, 56 (23), 2921–2927. 10.1021/acs.biochem.7b00190. [DOI] [PubMed] [Google Scholar]
- Kim R. Q.; Geurink P. P.; Mulder M. P. C.; Fish A.; Ekkebus R.; El Oualid F.; van Dijk W. J.; van Dalen D.; Ovaa H.; van Ingen H.; Sixma T. K. Kinetic analysis of multistep USP7 mechanism shows critical role for target protein in activity. Nat. Commun. 2019, 10 (1), 231. 10.1038/s41467-018-08231-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocek M.; Fojta M. Cross-coupling reactions of nucleoside triphosphates followed by polymerase incorporation. Construction and applications of base-functionalized nucleic acids. Org. Biomol. Chem. 2008, 6 (13), 2233–2241. 10.1039/b803664k. [DOI] [PubMed] [Google Scholar]
- Wood E. R.; Shewchuk L. M.; Ellis B.; Brignola P.; Brashear R. L.; Caferro T. R.; Dickerson S. H.; Dickson H. D.; Donaldson K. H.; Gaul M.; Griffin R. J.; Hassell A. M.; Keith B.; Mullin R.; Petrov K. G.; Reno M. J.; Rusnak D. W.; Tadepalli S. M.; Ulrich J. C.; Wagner C. D.; Vanderwall D. E.; Waterson A. G.; Williams J. D.; White W. L.; Uehling D. E. 6-Ethynylthieno[3,2-d]- and 6-ethynylthieno[2,3-d]pyrimidin-4-anilines as tunable covalent modifiers of ErbB kinases. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (8), 2773–2778. 10.1073/pnas.0708281105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAulay K.; Hoyt E. A.; Thomas M.; Schimpl M.; Bodnarchuk M. S.; Lewis H. J.; Barratt D.; Bhavsar D.; Robinson D. M.; Deery M. J.; Ogg D. J.; Bernardes G. J. L.; Ward R. A.; Waring M. J.; Kettle J. G. Alkynyl Benzoxazines and Dihydroquinazolines as Cysteine Targeting Covalent Warheads and Their Application in Identification of Selective Irreversible Kinase Inhibitors. J. Am. Chem. Soc. 2020, 142 (23), 10358–10372. 10.1021/jacs.9b13391. [DOI] [PubMed] [Google Scholar]
- Barf T.; Covey T.; Izumi R.; van de Kar B.; Gulrajani M.; van Lith B.; van Hoek M.; de Zwart E.; Mittag D.; Demont D.; Verkaik S.; Krantz F.; Pearson P. G.; Ulrich R.; Kaptein A. Acalabrutinib (ACP-196): A Covalent Bruton Tyrosine Kinase Inhibitor with a Differentiated Selectivity and In Vivo Potency Profile. J. Pharmacol. Exp. Ther. 2017, 363 (2), 240–252. 10.1124/jpet.117.242909. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.