

Abstract
Ice binding proteins modulate ice nucleation/growth and have huge (bio)technological potential. There are few synthetic materials that reproduce their function, and rational design is challenging due to the outstanding questions about the mechanisms of ice binding, including whether ice binding is essential to reproduce all their macroscopic properties. Here we report that nanoparticles obtained by polymerization-induced self-assembly (PISA) inhibit ice recrystallization (IRI) despite their constituent polymers having no apparent activity. Poly(ethylene glycol), poly(dimethylacrylamide), and poly(vinylpyrrolidone) coronas were all IRI-active when assembled into nanoparticles. Different core-forming blocks were also screened, revealing the core chemistry had no effect. These observations show ice binding domains are not essential for macroscopic IRI activity and suggest that the size, and crowding, of polymers may increase the IRI activity of “non-active” polymers. It was also discovered that poly(vinylpyrrolidone) particles had ice crystal shaping activity, indicating this polymer can engage ice crystal surfaces, even though on its own it does not show any appreciable ice recrystallization inhibition. Larger (vesicle) nanoparticles are shown to have higher ice recrystallization inhibition activity compared to smaller (sphere) particles, whereas ice nucleation activity was not found for any material. This shows that assembly into larger structures can increase IRI activity and that increasing the “size” of an IRI does not always lead to ice nucleation. This nanoparticle approach offers a platform toward ice-controlling soft materials and insight into how IRI activity scales with molecular size of additives.
Introduction
Ice binding proteins (IBPs)1,2 (and also polysaccharides3,4) are a diverse class of biological macromolecules that achieve the molecular recognition of ice crystals in the presence of a large excess of water. INPs include those classed as antifreeze proteins (AFPs), which can depress the freezing point of water (and can also raise the melting point)5 and are ice recrystallization (growth) inhibitors (IRI). Ice nucleating proteins (INPs) that promote ice formation are another distinct class.6−8 There are a huge range of technological challenges where IBPs (or synthetic mimics) could have impact, spanning the cryopreservation of donor cells and tissues,9−13 protecting infrastructure against freeze/thaw damage,14,15 and improving the texture of frozen foods.16,17 Inspired by IBPs, there has been significant interest in the development of synthetic materials that can modulate ice growth and/or materials that can enhance cellular cryopreservation.18−22 Synthetic materials have advantages in terms of scalability, cost, and tunability compared to biological ones, but there is real need to understand their function and to address questions around how the size and nanoscale dimensions may impact activity.23−26
Poly(vinyl alcohol) (PVA) is established as a uniquely potent polymeric IRI, even though its exact mechanism of action is still under study.27,28 Graphene oxide has been reported to interact with ice,29−31 and the self-assembly of saffranine leads to ice binding aggregates.32,33 Ben and co-workers have shown that short glycopeptides,34 or even some surfactants,35 can inhibit ice growth but do not lead to altered ice crystal morphologies. It has been discovered that thermal hysteresis activity (a macroscopic property which requires ice face binding) does not scale with IRI activity in several ice binding proteins,36,37 and hence there is increasing evidence that ice binding is not a prerequisite for the macroscopic effect of IRI. Removing the need for ice binding (which needs precision placement of the interacting groups) would simplify the design process and enable a new range of IRI-active materials to emerge with potentially easier design rules.
In the absence of ice binding, there is emerging evidence that materials with hydrophobic patches can display moderate IRI (but less than AFPs)38 such as self-assembled metallohelices,39 ROMP-derived polymers,40 or nanocellulose.41 Balcerzak et al. synthesized a library of sequentially modified lysine derivatives showing increased hydrophobicity lead to more IRI.42 In many cases, IRI activity increases with molecular weight, observed for proteins43 and polymers,27 and in self-assembly.32,44 However, it is more complicated than “more polymers on a surface, or larger structures, are always better”; the incorporation of PVA (IRI active and ice binder) as the corona of micelles45,46 or grafted to gold nanoparticles47 led to no increase in activity, potentially due to the low surface density of the polymers. Similarly, dendrimer or gold nanoparticle conjugated AFPs showed no enhancement on a per-protein subunit basis but on a molar basis led to large enhancements in both IRI and thermal hysteresis activity.48,49 Ice nucleating proteins (a distinct class of IBPs) have to be sufficiently large to match the minimum ice cluster size,50,51 and it is proposed that a key difference in activity between larger ice nucleating proteins and smaller antifreeze proteins is their size.23,52,53 Considering this evidence, it is clear that the impact of macromolecular size, but also crowding, plays a role in understanding IRI (but also other properties such as thermal hysteresis) but that there are size limits on what can be obtained by using linear polymers; increasing the size of proteins (e.g., linear assemblies of repeat units) is not practical and will not lead to identical folding or display of surfaces. Hence, there is a need to discover new platforms where nanoscale dimensions and composition can be tuned.
Polymerization-induced self-assembly (PISA) is a powerful approach for in situ development of block copolymer nanoparticles to generate multivalent nanomaterials at high concentrations of controlled morphology and size.54−56 During aqueous PISA, a water-soluble corona-forming block is chain-extended by using specific water-miscible (dispersion PISA) or water-immiscible (emulsion PISA) monomers that gradually form a second, insoluble core block (as the length of the core-forming block increases), resulting in nanoparticle formation. Higher-order morphologies such as worm-like micelles and polymersomes are typically being observed, resulting in higher packing density for the copolymer chains compared to spherical (micellar) particles.57,58 The versatility of this method has allowed the synthesis of nanoparticles for drug delivery,59−62 cell storage,63−65 and permeable nanoreactors,66−69 and has been extensively reviewed.70−73 PVA-graft-macroinitiators have been reported to lead to polymer nanoparticles with enhanced IRI (compared to the already very potent IRI activity of PVA), potentially due to the high local PVA density arising from PISA.74 However, the use of nanoparticle engineering to discover emergent AFP mimetic materials from components with no intrinsic IRI activity has not been explored.
Considering the above, we herein report the unexpected enhancement of IRI activity of polymerization-induced self-assembly derived nanoparticles from constituent components with no IRI activity of their own. A panel of nanoparticles with variation in size, corona-forming, and core-forming block chemistry/functionality were synthesized. In each case IRI activity was unexpectedly increased relative to the homopolymer, but only in the largest (vesicular) nanoparticles, providing a previously unreported mechanism to generate IRI-active materials. By use of complementary ice nucleation assays, it was found that the size increase did not substantially increase ice nucleation activity, showing that making IRI-active materials larger does not lead to ice nucleation in all cases. This demonstrates that self-assembled nanomaterials and multivalent presentation is a crucial tool to understand, and translate to application, ice growth modifiers.
Results and Discussion
To investigate the hypothesis that aggregation and crowding of non-ice binding polymers in a nanoparticle format may induce IRI activity, a PISA strategy was followed to obtain polymer nanoparticles with tunable sizes and morphologies. Deionized water gives false positives in the ice growth assays used (below) as a eutectic phase where ice/water coexistence is essential, and hence saline solutes (or other solutes) are required.38 Therefore, to perform PISA, we first chose to use diacetone acrylamide (DAAm) as the core-forming monomer, which we have recently discovered to promote saline stability in PISA formulations (Figure 1A), unlike many other monomers.74 A series of aqueous dispersion polymerizations in the presence of [NaCl] = 0.05 M were performed for DAAm via thermally initiated RAFT polymerization using a PEG-based macromolecular chain transfer agent (macro-CTA, PEG45 mCTA, Mn,SEC = 4.1 kg mol–1, ĐM = 1.1) as the corona-forming polymer. For these experiments the concentration of monomer was held constant at 10% w/w, the [macro-CTA]:[initiator] ratio was maintained at 1:0.1, and the [monomer]/[macro-CTA] ratio was varied to target different core-block degrees of polymerization. The polymerizations were carried out at 60 °C with 2,2′-azobis(2-methylpropionamidine) dihydrochloride (V-50) as the water-soluble initiator. A gradual turbidity increase was noticed for polymerization solutions with increasing DPPDAAm, indicating the onset of particle formation typically observed in PISA. Quantitative monomer conversions (>95%) were achieved in all cases, as determined by 1H NMR spectroscopic analysis in methanol-d4 of the crude samples (Figure S5).
Figure 1.

Synthesis of PEG-corona PISA-derived nanoparticles. (A) Synthesis of PEG45-b-PDAAmn diblock copolymer nanoparticles via aqueous RAFT-mediated PISA using a commercially available PEG45 macro-CTA ([solids] = 10% w/w, [NaCl] = 0.05 M). (B) Normalized SEC RI molecular weight distributions. Mn,SEC and ĐM values were calculated from PMMA standards with DMF + 5 mM NH4BF4 as the eluent. (C) Evolution of Mn (black circles) and ĐM (red circles) values with increasing targeted DPPDAAm. (D) Zeta potential values for PEG45-b-PDAAmn diblock copolymer nanoparticles. (E) Representative dry-state images of PEG45-b-PDAAmn diblock copolymer nanoparticles, stained with 1 wt % uranyl acetate (UA) solution. (F) Intensity-weighted size distributions along with average Dh and PD values (the error shows the standard deviation from five repeat measurements).
SEC analysis of PEG45-b-PDAAmn diblock copolymers in DMF + 5 mM NH4BF4 revealed the controlled character of the aqueous RAFT-PISA process (Figure 1B), with symmetric, monomodal molecular weight distributions shifting linearly toward higher molecular weight (Mn) values upon increasing the DP of PDAAm. A small low Mw shoulder of unconsumed macro-CTA was apparent, due to a small amount of unfunctionalized PEG. Calculated Mn values agreed well with theoretically expected values, while dispersity values remained relatively low for the polymerizations targeting DPs of 12, 25, 50, and 100 (ĐM ≤ 1.6) but was higher for DP 150 (ĐM = 2.1) throughout (Figure 1C and Table S1). The absence of charges on the outer surface of the obtained nanoparticles was confirmed by electrophoretic analysis at neutral pH (measured zeta potential <±5 mV) (Figure 1D).
DLS analysis of PEG45-b-PDAAmn formulations revealed the formation of particles with multiple populations and high polydispersity (PD) values for the lower DPs of PDAAm, consistent with worm-like micelles or nanoparticles with mixed morphologies.54,75 Single particle populations with low PD values were observed for DPPDAAm ≥ 50, indicating the formation of uniform assemblies (Figure 1F). A single-exponential decay, smooth autocorrelation function with optimal signal-to-noise ratio was also recorded (Y-intercept >0.9) for all formulations (Figure S6). Dry-state TEM supported the DLS findings and showed an evolution in morphology as the DP of the core-forming PDAAm block increased from spheres (DPPDAAm = 12) to mixed morphologies of worms and spheres (DPPDAAm = 25) to mixed worms, bilayer lamella and vesicles (DPPDAAm = 50) to perforated vesicles (DPPDAAm = 100), and finally to single-phase vesicles of uniform size (DPPDAAm = 150) (Figure 1E and Figure S7). Cryogenic TEM analysis (cryo-TEM) was also performed on PEG45-b-PDAAM100 vesicles and confirmed a perforation with uniform pores on the surface of the vesicles.76,77 It has previously been reported that PISA diblock formulations with PDAAm as core-forming block have a composition-dependent thermoresponsive behavior.78−80 Therefore, PEG45-b-PDAAm100 was also synthesized by using 2,2′-azobis[2-(2-imidazolin-2-yl)propane] dihydrochloride (VA-044) at lower temperature, and its morphology was studied via TEM imaging (see Figure S8). This revealed uniform spherical vesicles being formed with perforated bilayer surface with fewer and less uniform pores but indicates that it is not a temperature-induced effect, but its investigation is beyond the scope of this work.
Next, IRI activity was assessed by the “splat” assay. This assay involves seeding a large number of small ice crystals, which are then allowed to grow at −8 °C.27,81 IRI-active materials will slow ice growth, resulting in smaller crystals relative to the negative control (saline), and the results are plotted as the mean grain size (MGS). To conduct these assays, it is essential to have some salt to prevent false negatives (any additive in pure water in this assay leads to ice growth inhibition).38,81 To ensure colloidal stability, 0.05 M NaCl was used here (as opposed to the more common 0.137 M), which was validated to not produce false positive/negatives in the IRI assays, but this lower concentration must be considered when comparing to other reported activities as it is non-identical for each saline concentration (see Figure S9). Figure 2A shows dose–response curves for the PEG-based particles, compared to the PEG macroinitiator, with concentrations normalized to PEG concentration (as the coronal component) to enable comparison of multivalent effects. Remarkably, all the formulations showed some IRI activity. Considering PEG is widely used as a negative control (as it does not bind ice), the induction of observable IRI was unexpected. It is important to note that any material of sufficiently high concentration can inhibit ice growth, and this is not an on/off property.38 Larger particles (Dh = 350 and 730 nm corresponding to DPPDAAm of 100 and 150, respectively) showed higher activity than the smaller (Dh = 42 nm for DPPDAAm = 12), demonstrating that size and potential packing of the corona (due to less curvature for large particles compared to small) are crucial to IRI activity, although this cannot be proved at this stage. The largest particles inhibited all growth at a PEG concentration of 5 mg mL–1, placing them in the medium activity range of antifreeze protein mimetics (less active than PVA or antifreeze proteins).38,43 Cryomicrographs of annealed ice wafers (Figure 2B) visualize this unexpected activity and clearly demonstrate that a self-assembly strategy to induce IRI activity into soft materials is possible and that particle size is a tool to control the magnitude of activity.38 In the case of PEG45-b-PDAAm100 vesicles with perforated topologies, their IRI activity was compared with a formulation synthesized at lower temperature and showed no significant difference in IRI activities (Figure S10). To further explore the dynamics of this system, PEG45-b-PDAAM100 block copolymer vesicles were annealed for different time points (t = 30 and 60 min) showing complete inhibition even after this time (Figure S13). Colloidal stability for PEG45-b-PDAAm150 vesicles upon multiple freeze–thaw cycles was assessed by DLS analysis and showed the particles were recovered with no significant changes in their hydrodynamic diameter (Dh) (Figure S11). The IRI activity as a function of total solid (polymer) concentration is also plotted in Figure S12. This shows that even when the core-polymer mass is included (which is not in contact with ice/water), the nanoparticles are still more active than a PEG (corona) alone on a mass basis. The larger particles were also still more active, and hence the activity is not an artifact of concentration calculations and the nanoparticle size is a key criteria. To further demonstrate the IRI activity, an alternative assay was conducted in 45 wt % sucrose (“sucrose sandwich”) which has a lower ice fraction.43Figure S14 shows nucleated ice crystals, which after 30 min have grown significantly (recrystallized) in the presence of PEG45 macro-CTA, while Figure S15 shows that a solution containing 10 mg mL–1 of PEG45-b-PDAAm150 nanoparticles displays inhibition of ice recrystallization, demonstrating activity in a range of different formulations.
Figure 2.
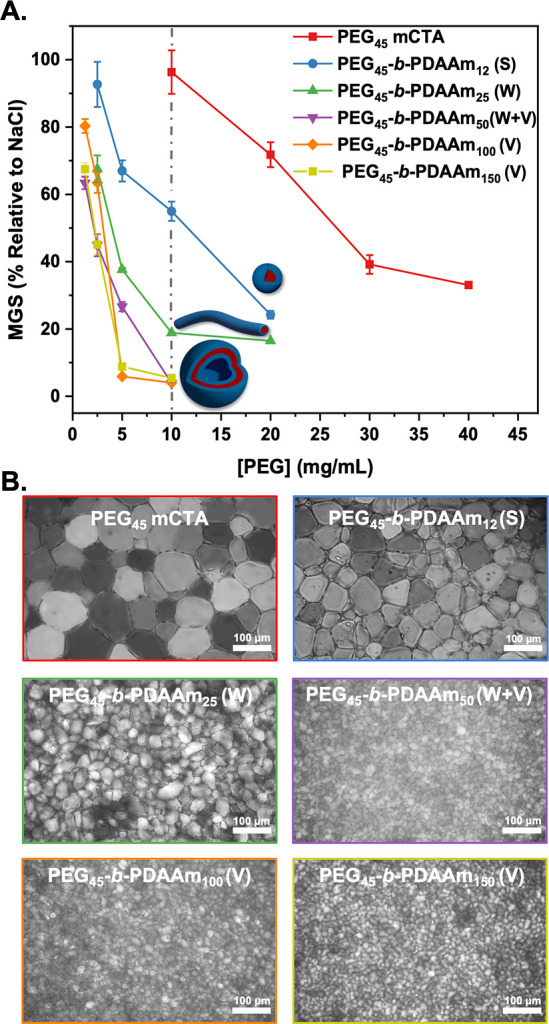
Assessment of ice recrystallization inhibition activity for PEG-based nanoparticles. (A) IRI activity summary of PEG45-b-PDAAmn nanoparticles corrected to [PEG]. Error bars are ± SD from a minimum of three repeats (key: S, spherical micelles; W, worm-like micelles; V, vesicles). The percent mean grain size (MGS) was reported relative to saline control ([NaCl] = 0.05 M). (B) Example cryomicrographs of ice wafers from the “splat” assay of PEG45-b-PDAAmn nanoparticles, [PEG] = 10 mg mL–1.
To evaluate whether the observations made above were only due to the PEG corona (as the component with most solvent interactions), a higher Mw PEG macro-CTA (PEG113 mCTA) was employed, and the composition of the core-forming block was separately examined. Few PISA core-forming monomers are reported to be saline-tolerant.82 We employed an acrylamide-based monomer that can undergo PISA, N-(isobutoxymethyl)acrylamide (iBuOMAm), which was selected based on in silico computation/experimental study by O’Reilly and co-workers.83iBuoMAm was polymerized via aqueous RAFT in the presence of [NaCl] = 0.05 M by using PEG45 mCTA at 10% w/w solids content targeting a DPPiBuoMAm of 100. The prepared PEG45-b-PiBuOMAm100 diblock copolymer possessed bimodal molecular weight distribution due to unconsumed PEG mCTA with relatively low dispersity value, as determined by SEC analysis (Mn,SEC = 25.1 kg mol–1, ĐM = 1.6) (Figure S16). Dynamic light scattering (DLS) analysis revealed the formation of nanoparticles with a monomodal size distribution and mean hydrodynamic diameter (Dh) of 357.9 ± 1.3 nm and low polydispersity (PD = 0.24 ± 0.01) (Figure 3A). Dry-state transmission electron microscopy (TEM) imaging confirmed the development of uniform PEG45-b-PiBuOMAm100 porous spherical nanoparticles and uniform size (Figure S12D). Cryo-TEM imaging of PEG45-b-PiBuOMAm100 nanoparticles in solution confirmed the formation of perforated spherical nanoparticles (Figure S16E). A higher Mw PEG macroinitiator (PEG113 mCTA—to probe effect of coronal chain length) was subsequently used for the polymerization of DAAm to yield PEG113-b-PDAAm100 and PEG113-b-PDAAm400 (to enable comparison of the same morphology with PEG45-b-PDAAM100 vesicles) diblock copolymer nanoparticles. TEM and DLS analyses showed the formation of uniform spherical nanoparticles and unilamellar vesicles for DPPDAAm = 100 and 400, respectively (Figure 3B, Figures S17 and S18), with monomodal size distributions, mean hydrodynamic diameters (Dh) of 96.6 ± 1.8 nm (DPPDAAm = 100) and 280.8 ± 8.2 nm (DPPDAAm = 400), and low polydispersities (PD = 0.20 ± 0.04 and 0.08 ± 0.04). These particles were tested for IRI and were found to show nearly identical activity to those that had PDAAm cores (above). Additionally, PEG113-b-PDAAm400 vesicles showed higher IRI activities compared to PEG45-b-PDAAm100 vesicles. This experiment proves that the nanoparticle size and morphologies were crucial for IRI activity, but not the nature of the core-forming block (other than its role in ensuring saline stability), primarily because of its inaccessibility by the solvent molecules (Figure 3C).
Figure 3.

Synthesis, characterization, and ice recrystallization inhibition activity of PEG-coronal nanoparticles using PiBuOMAm- and PDAAm-based cores. (A) PEG45-b-PiBuOMAm100, (B) PEG113-b-PDAAm100 (i), and PEG113-b-PDAAm400 vesicles (ii) synthesized at [solids] = 10% w/w via aqueous RAFT-mediated PISA in the presence of [NaCl] = 0.05 M with commercially available PEG45 and PEG113 macro-RAFT agents. Insets show intensity-weighted size distributions along with average Dh and PD values (the error shows the standard deviation from five repeat measurements) and representative dry-state TEM images stained with 1 wt % uranyl acetate (UA) solution. (C) IRI activity summary corrected to [PEG].
With the above observation of PEG activity and confirmation that the nature of core was not crucial, we envisioned that introducing a different corona composition, as it is the component that is in direct contact with the aqueous/ice phases, would result in different IRI activities. PEG is not known to present IRI, so as it was important to compare it to other PISA-derived particles with a linear non-ionic hydrophilic corona-forming polymer, with similar repeat unit dimensions (to avoid comb-type PEG-(meth)acrylates which might affect packing density). Here, poly(vinylpyrrolidone) (PVP) was employed for the first time as a steric stabilizing block for performing PISA by using DAAm as the core-forming monomer (Figure 4). Initially, N-vinylpyrrolidone (NVP) was polymerized by using xanthate chain transfer of 2-(ethoxycarbonothioyl)sulfanyl propanoate (EXEP) (see Supporting Information for full synthetic procedure, Figures S3 and S4) via photoinitiated RAFT/MADIX to ensure maximum end-group fidelity (PVP40, Mn,SEC RI = 4600 g mol–1, ĐM = 1.3) (Figure S19).84,85 Then, a series of aqueous dispersion polymerizations in the presence of [NaCl] = 0.05 M were performed for DAAm via RAFT by using the synthesized PVP40 macromolecular chain transfer agent. The concentration of monomer was held constant at 20% w/w, and the [monomer]/[macro-CTA] ratio was varied to target DPPDAAm = 25, 50, 100, and 200. A gradual turbidity increase was noticed for polymerization solutions with increasing DPPDAAm, indicating the onset of particle formation. Quantitative monomer conversions (>97%) were achieved in all cases after 6 h, as determined by 1H NMR spectroscopic analysis in methanol-d4 of the crude samples (Figure S20 and Table S2).
Figure 4.

Summary of PISA formulation using non-PEG-based coronas. (A) Synthesis of PVP40-b-PDAAmn diblock copolymer nanoparticles via aqueous RAFT/MADIX-mediated PISA. PVP40 macro-CTA was synthesized by photoinitiated RAFT polymerization ([solids] = 20% w/w, [NaCl] = 0.05 M). (B) Representative dry-state TEM images of PVP40-b-PDAAmn diblock copolymer nanoparticles, stained with 1 wt % uranyl acetate (UA) solution. (C) Normalized intensity-weighted size distributions obtained by DLS. (D) Zeta potential values for PVP40-b-PDAAmn diblock copolymer nanoparticles. (E) Synthesis of PDMAC40-b-PDAAm200 diblock copolymer vesicles via aqueous RAFT-mediated PISA using a PDMAC40 macro-CTA ([solids] = 20% w/w, [NaCl] = 0.05 M). (F) (i) Representative dry state and (ii) cryo-TEM images; (iii) intensity-weighted size distribution along with average Dh and PD values (the error shows the standard deviation from five repeat measurements). Inset: autocorrelation function.
SEC analysis of PVP40-b-PDAAmn diblock copolymers revealed a rather uncontrolled character of chain extensions of PVP40 macro-CTA with high dispersity values (ĐM > 2.0) as would be expected for this macroinitiator (Figure S21).86 PVP is known to undergo undesirable side reactions in aqueous media, leading to poor RAFT control and incomplete monomer conversions,87,88 and it can only be readily copolymerized with a limited set of other lesser activated monomers (LAM), such as vinyl acetate or acrylics,89,90 but no LAM dispersion PISA monomer is known, necessitating this approach. It is crucial to note that a relatively high proportion of PVP stabilizer chains remained unreacted at the end of the polymerization, leading to bimodal molecular weight distributions and is discussed in terms of the IRI activity below. Peak deconvolution was also performed on SEC curves for resulting block copolymer nanoparticles (Figures S23–S26), and the ratios of the areas between chain extended and unconsumed PVP macro-CTA are summarized in Table S3.
Dry-state TEM analysis showed an evolution in morphology upon increasing the DP of the core-forming block from spheres (DPPDAAm = 25 and 50) to mixed morphologies of spheres and vesicles (DPPDAAm = 100) to pure single-phase vesicles of uniform size (Figure 4B). For DPPDAAm = 100, the appearance of mixed spheres and vesicles can be attributed to the high dispersity values with a large proportion of PVP stabilizer chains being chain-extended to different degrees. Cryo-TEM analysis further confirmed the resulting morphologies (Figure S27). DLS analysis supported TEM findings and revealed the stable formation of particles with single populations and low polydispersity (PD) values (Figure 4C and Figure S28). The absence of charges on the outer surface of the obtained nanoparticles was also confirmed by electrophoretic analysis at neutral pH (measured zeta potential <±5 mV) (Figure 4D).
A stabilizing block of poly(N-dimethylacrylamide) (PDMAC) was also employed to perform PISA by using DAAm as the core-forming monomer (Figure 4E). DMAC was initially polymerized in dioxane at 70 °C by using 2-(((butylthio)carbonothiolyl)thio)propanoic acid as the chain transfer agent (see Supporting Information for full synthetic procedure, Figures S5 and S6). The resulting homopolymer was analyzed by 1H NMR and SEC (PDMAC40, Mn,SEC RI = 4800 g mol–1, ĐM = 1.1) (Figure S30). This water-soluble polymer precursor was chain-extended with DAAm via RAFT aqueous dispersion polymerization at 60 °C and 20% w/w solids in the presence of [NaCl] = 0.05 M, targeting a final DPPDAAm of 200. This system has been extensively studied in the past, and previous synthetic procedures were followed to achieve vesicular morphologies.79,80,91−93 Quantitative monomer conversion (>98%) was achieved after 6 h, as determined by 1H NMR spectroscopic analysis in methanol-d4 of the crude sample (Figure S31). The prepared PDMAC40-b-PDAAm200 diblock copolymer possessed monomodal molecular weight with relatively low dispersity value, as determined by SEC analysis in DMF + 5 mM NH4BF4 (Mn,SEC = 31600 g mol–1, ĐM = 1.5) (Figure S32). Dynamic light scattering (DLS) analysis revealed the formation of nanoparticles with a monomodal size distribution and mean hydrodynamic diameter (Dh) of 308.8 ± 5.5 nm and low polydispersity (PD = 0.12 ± 0.02) (Figure 4Fiii). Dry-state and cryo-TEM imaging finally confirmed the development of single-phase unilamellar vesicles (Figure 4Fi,ii).
Figure 5 shows the IRI activity of the non-PEG-based corona formulations. For PVP, the same particle size dependence on activity as seen for PEG was observed with larger particles (vesicles) being more active than smaller ones (micelles), and both were more active than the macro-CTA alone. It is crucial to note that due to inefficient macroinitiation (see above), the PVP particles as synthesized included a population of free polymer chains. The particle IRI activity (shown in Figure 5) was determined after removal of unreacted PVP40 macro-CTA (by centrifugation, Figures S21 and S22). The presented PVP concentration was determined from deconvolution of the SEC traces (Figures S23–S26 and Table S3). IRI activity before removing excess PVP40 is also shown in Figure S29 for comparison. PDMAC-based particles also showed enhancement relative to the homopolymer alone, with the magnitude similar to the PVP-based particles (Figure 5C). Comparing vesicular particles whereby the PDAAm core-block DP was similar (Figure 5D) shows that all presented similar activity, and hence the formation of self-assembled structures rather than the chemical nature of the corona-forming block is the key requirement. All the data are summarized (independent of chemical composition) as MGS versus diameter in Figure S33, confirming that particle size correlated to activity. We should highlight this does prove a mechanism but does show that nanoparticle engineering is a tool to control the macroscopic IRI activity.
Figure 5.

Ice recrystallization inhibition (IRI) activity of PVP and PDMAC-based nanoparticles. (A) PVP40-b-PDAAmn nanoparticles corrected to total PVP concentration in particle format. (B) PVP40-b-PDAAmn nanoparticles at [PVP] = 10 mg.mL–1. (C) PDMAC40-b-PDAAm200 diblock copolymer vesicles and PDMAC40 corrected to [PDMAC]. (D) Comparison of IRI activity for diblock copolymer vesicles of PVP40-b-PDAAm200, PDMAC40-b-PDAAm200, and PEG45-b-PDAAm150. Error bars are ± SD from a minimum of three repeats. The percent mean grain size (MGS) was reported relative to saline control. [PVP] was recalibrated based on chain extended PVP40 present particle format after estimating amount of unconsumed PVP40 macro-CTA based on Table S3 data.
The key property investigated here, IRI, is a macroscopic observation which could be caused by a range of molecular-level mechanisms from single, or multiple, ice face binding or disruption of water transfer at the ice/water interface without actual binding.35,41,94 IRI is also known to increase with molecular weight for many materials, and hence the effective very high Mw of a nanoparticle could provide a simple reason for the observations here. It is important to again highlight that IRI is not a binary on/off property but that sufficient concentrations of any material can induce it.38 To investigate whether the particles were affecting ice crystal morphology, which may indicate ice binding (as opposed to a non-specific “size” related effect), crystals were grown in sucrose solution (which leads to segregated ice crystals whose morphology can be viewed). Figure 6A shows ice crystals grown with a PEG-coronal nanoparticle formulation (PEG45-b-PDAAm100) did not induce significant faceting (Figure S34). This fits a hypothesis that the PEG-coronal nanoparticle activity enhancement is not due to ice binding. In contrast, PVP alone (Figure 6B and Figure S35) showed some weak faceting of the crystals and formation of dendritic morphologies. However, the PVP-coronal nanoparticles (PVP40-b-PDAAm200) resulted in extensive dendritic ice crystals (Figure 6C and Figure S36). This would imply that PVP, while itself only having weak (or no) IRI activity,27 when assembled in the correct format can engage ice or affect the morphology of ice produced. The increased faceting could be due to the nanoparticles spanning between multiple ice faces due to their large size. This is an exciting observation as if ice crystal morphology can be probed or modulated by these easy-to-make synthetic soft materials, it opens doors to fundamental studies on the ice interface, without needing protein engineering as well as potential applications. It also suggests that the enhanced activity seen for PEG/PDMAC coronas (without ice engagement) still apply when using coronas with some affinity for ice.
Figure 6.

Ice binding and ice nucleation analysis of nanoparticles. (A) Modified “sucrose sandwich” ice shaping assay images for 1 mg mL–1 of PEG45-b-PDAAm100 nanoparticles in 45 wt % sucrose solution; (B) 1 mg mL–1 of PVP40 and (C) 1 mg mL–1 of PVP40-b-PDAAm200. (D) Assessment of ice nucleation activity in microliter droplets showing immersion mode fraction frozen curves as a function of temperature for droplets containing 1 mg mL–1 PEG45-b-PDAAm150, PDMAC40-b-PDAAm200, and PVP40-b-PDAAm200 along with their corresponding macro-CTAs (E) and PVP40-b-PDAAmn (n = 25, 50, 100, 200) nanoparticle series. Example temperature errors bars are included on selected data points on panels D and E.
With the above observations that the PISA particle format led to increased IRI activity and ice shaping, the ability of the particles to nucleate ice was next studied. On cooling, pure water can persist as liquid to temperatures as low as −40 °C. This phenomenon is known as supercooling, and some substances such as INPs are capable of inducing a stable ice nucleus in supercooled water: known as heterogeneous ice nucleation. A feature of ice nucleating proteins, INPs (as discussed in the Introduction), is that assembly of small domains into larger ones is essential for activity, as macromolecules must be of a similar scale to the classical nucleation theory derived critical ice cluster size to nucleate ice at a given temperature.50,51 The ice critical cluster size increases rapidly as temperature approaches the melting point. For example, the critical ice cluster has a diameter of about 20 nm at −5 °C.51 This means large aggregates of (for example) bacterial ice nucleating proteins are required to induce ice nucleation close to 0 °C. There is theoretical evidence that molecular size is a key criteria for ice nucleation,23 and it has been observed experimentally that some AFPs can also nucleate ice, with the size of the AFP being an essential descriptor.52,53 Given that some of the polymer particles synthesized bind ice, all show IRI and are large compared to ice critical nuclei; it might be thought that they would be capable of nucleating ice at warm temperatures as seen for higher molecular weight antifreeze proteins. To test this, microliter ice nucleation assays were undertaken by using a bespoke apparatus described in the Supporting Information. This instrument freezes around 50 droplets per experiment to establish the spread of likely nucleation temperatures for a given heterogeneous nucleator. The ice nucleation temperatures of 1 mg mL–1 nanoparticle and polymer solution are reported in Figure 6D,E. It was found that the PEG- and PDMAC-based formulations nucleated ice at average temperatures of −25.8 and −26.8 °C, respectively, only slightly warmer than the average freezing temperature of pure water on the instrument, −28.1 °C. PVP and the various PVP-based nanoparticles tested nucleated at average temperatures between −23.6 and −24.5 °C. Notably, the fraction frozen curves for PVP and PVP40-b-PDAAm200 were very similar. While the PEG- and PDMAC-derived nanoparticles do nucleate ice at slightly warmer temperatures than their precursor polymers, the difference is not large. Figure 6E shows a slight increase in nucleation temperature with increasing particle size for PVP-derived nanoparticles; however, the enhancement is not large. The nucleation data show that aggregation of polymers into particles does not substantially enhance ice nucleation effectiveness. This supports the theory that correct long- and short-range structure, as well as sufficient size, is required for a nucleant to be highly effective. These combined observations support emerging evidence that the observation macroscopic properties of ice binding proteins are not always caused by the same underlying molecular mechanisms36−38 and that a particular property can be dialled into a material. There is an opportunity to achieve desirable levels of ice growth inhibition (for a specific application) by deploying nanoparticle formulations such as those presented here, even if their overall activity is substantially weaker than IBPs, and indeed their mechanism is non-specific.
Conclusions
We report that biomimetic ice recrystallization inhibition activity can be introduced into polymer nanoparticles from components which themselves have no ice binding or associated activity. Saline-tolerant polymerization induced self-assembly (PISA) was exploited to obtain nanoparticles of controlled size and morphology by using a library of different corona-forming polymers [poly(ethylene glycol), poly(vinylpyrrolidone) (for the first time via dispersion PISA), and poly(dimethylacrylamide)] and core-forming blocks [poly(diacetone acrylamide) and poly((isobutoxymethyl)acrylamide)]. In all cases, ice recrystallization inhibition was observed despite the constituent homopolymers (i.e., corona-forming blocks) having no activity themselves, suggesting that size and confinement of the coronal blocks can enhance this macroscopic property. Larger particles (vesicles) were more active than smaller ones (micelles/spheres and intermediate morphologies), confirming that the size and nanoarchitecture of the particles were crucial for inducing activity. The activity enhancement relative to linear polymers was seen both in terms of the total coronal mass (as the solvent-contacting component) or as the total mass (including the core), showing this was not simply due to the increased mass applied in a nanoparticle formulation. The core-forming blocks were also varied, and this had minimal or no impact on the observable ice recrystallization inhibition activity, confirming this was a general effect. In the case of poly(vinylpyrrolidone) coronal particles, there was observation of ice shaping, suggesting these particles may engage specific ice faces, which was not seen with the polymers alone. This additional binding did not lead to significantly higher IRI activity though, suggesting IRI was dominated by their size and packing density for each morphology. Ice nucleation was also studied, but no activity was seen. This was a crucial observation as it has been reported that larger assemblies of antifreeze proteins can lead to ice nucleation activity but that the assembly of these polymers does not lead to ice nucleation. This may suggest a unique mechanism, but the huge structural differences between the particles and AFPs make a real comparison difficult. The data here also supports emerging evidence that the magnitude of each macroscopic property associated with ice binding proteins mimics does not scale equally between all materials. The exact molecular mechanism for this increased activity is not clear and may be simply related to the overall size and molecular weight of the particles, which exceeds what is accessible with linear polymers. However, the PVP data supports that increased ice binding can occur in these assemblies. These observations show that coronal confinement of polymers can induce the macroscopic effect of ice recrystallization inhibition but that the same is not true for ice-nucleation activity, providing evidence that simply enlarging IRI-active components does not guarantee ice nucleation. While the overall magnitude of activity here is less than ice binding proteins, there is huge technological potential in the development of freeze-stable colloids, exploiting this size-driven enhancement to translate to applications, as well as a tool to study the fundamentals of ice-binding/modifying materials.
Acknowledgments
M.I.G. thanks the ERC for a Consolidator Grant (866056) and the Royal Society for an Industry Fellowship (191037) joint with Cytivia. This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie Grant Agreement No. 814236. BBSRC-funded MIBTP program (BB/M01116X/1) and Cytivia are thanked for supporting H.M. T.F.W. thanks the Leverhulme Trust and the University of Warwick for supporting an Early Career Fellowship (ECF-2018-127). T.R.C. thanks the BBSRC Emerging Innovations Grant ref BB/S506783/1. The Warwick Polymer Research Technology Platform is acknowledged for SEC analysis, and the Warwick Electron Microscopy Research Technology Platform is acknowledged for TEM. We also acknowledge the University of Warwick Advanced Bioimaging Research Technology Platform supported by BBSRC ALERT14 Award BB/M01228X/1 and Dr. S. Bakker for cryo-TEM. The BBSRC-funded MIBTP program (BB/M01116X/1) and Iceni Diagnostics Ltd. are thanked for a studentship for A.N.B.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c01963.
Full synthetic details and characterization of polymers and nanomaterials (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Bar Dolev M.; Braslavsky I.; Davies P. L. Ice-Binding Proteins and Their Function. Annu. Rev. Biochem. 2016, 85 (1), 515–542. 10.1146/annurev-biochem-060815-014546. [DOI] [PubMed] [Google Scholar]
- Guo S.; Stevens C. A.; Vance T. D. R.; Olijve L. L. C.; Graham L. A.; Campbell R. L.; Yazdi S. R.; Escobedo C.; Bar-Dolev M.; Yashunsky V.; et al. Structure of a 1.5-MDa Adhesin That Binds Its Antarctic Bacterium to Diatoms and Ice. Sci. Adv. 2017, 3 (8), e1701440. 10.1126/sciadv.1701440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters K. R.; Serianni A. S.; Sformo T.; Barnes B. M.; Duman J. G. A Nonprotein Thermal Hysteresis-Producing Xylomannan Antifreeze in the Freeze-Tolerant Alaskan Beetle Upis Ceramboides. Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (48), 20210–20215. 10.1073/pnas.0909872106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreischmeier K.; Budke C.; Wiehemeier L.; Kottke T.; Koop T. Boreal Pollen Contain Ice-Nucleating as Well as Ice-Binding ‘Antifreeze’ Polysaccharides. Sci. Rep. 2017, 7, 41890. 10.1038/srep41890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celik Y.; Graham L. A.; Mok Y.-F.; Bar M.; Davies P. L.; Braslavsky I. Superheating of Ice Crystals in Antifreeze Protein Solutions. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (12), 5423–5428. 10.1073/pnas.0909456107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzner M.; Sosso G. C.; Cox S. J.; Michaelides A. The Many Faces of Heterogeneous Ice Nucleation: Interplay between Surface Morphology and Hydrophobicity. J. Am. Chem. Soc. 2015, 137 (42), 13658–13669. 10.1021/jacs.5b08748. [DOI] [PubMed] [Google Scholar]
- Duman J. G. Antifreeze and Ice Nucleator Proteins in Terrestrial Arthropods. Annu. Rev. Physiol. 2001, 63 (1), 327–357. 10.1146/annurev.physiol.63.1.327. [DOI] [PubMed] [Google Scholar]
- Cochet N.; Widehem P. Ice Crystallization by Pseudomonas Syringae. Appl. Microbiol. Biotechnol. 2000, 54 (2), 153–161. 10.1007/s002530000377. [DOI] [PubMed] [Google Scholar]
- Fowler A.; Toner M. Cryo-Injury and Biopreservation. Ann. N. Y. Acad. Sci. 2005, 1066, 119–135. 10.1196/annals.1363.010. [DOI] [PubMed] [Google Scholar]
- Deller R. C. R. C.; Vatish M.; Mitchell D. A. D. A.; Gibson M. I. M. I. Synthetic Polymers Enable Non-Vitreous Cellular Cryopreservation by Reducing Ice Crystal Growth during Thawing. Nat. Commun. 2014, 5, 3244. 10.1038/ncomms4244. [DOI] [PubMed] [Google Scholar]
- Matsumura K.; Hyon S. H. Polyampholytes as Low Toxic Efficient Cryoprotective Agents with Antifreeze Protein Properties. Biomaterials 2009, 30 (27), 4842–4849. 10.1016/j.biomaterials.2009.05.025. [DOI] [PubMed] [Google Scholar]
- Chao H.; Davies P. L.; Carpenter J. F. Effects of Antifreeze Proteins on Red Blood Cell Survival during Cryopreservation. J. Exp. Biol. 1996, 199, 2071–2076. 10.1242/jeb.199.9.2071. [DOI] [PubMed] [Google Scholar]
- John Morris G.; Acton E. Controlled Ice Nucleation in Cryopreservation - A Review. Cryobiology 2013, 66 (2), 85–92. 10.1016/j.cryobiol.2012.11.007. [DOI] [PubMed] [Google Scholar]
- Frazier S. D.; Matar M. G.; Osio-Norgaard J.; Aday A. N.; Delesky E. A.; Srubar W. V. Inhibiting Freeze-Thaw Damage in Cement Paste and Concrete by Mimicking Nature’s Antifreeze. Cell Reports Phys. Sci. 2020, 1 (6), 100060. 10.1016/j.xcrp.2020.100060. [DOI] [Google Scholar]
- Valarezo W. O.; Lynch F. T.; McGhee R. J. Aerodynamic Performance Effects Due to Small Leading-Edge Ice (Roughness) on Wings and Tails. J. Aircr. 1993, 30 (6), 807–812. 10.2514/3.46420. [DOI] [Google Scholar]
- Sidebottom C.; Buckley S.; Pudney P.; Twigg S.; Jarman C.; Holt C.; Telford J.; McArthur A.; Worrall D.; Hubbard R.; et al. Heat-Stable Antifreeze Protein from Grass. Nature 2000, 406 (6793), 256. 10.1038/35018639. [DOI] [PubMed] [Google Scholar]
- Regand A.; Goff H. D. Ice Recrystallization Inhibition in Ice Cream as Affected by Ice Structuring Proteins from Winter Wheat Grass. J. Dairy Sci. 2006, 89 (1), 49–57. 10.3168/jds.S0022-0302(06)72068-9. [DOI] [PubMed] [Google Scholar]
- Stubbs C.; Bailey T. L.; Murray K.; Gibson M. I. Polyampholytes as Emerging Macromolecular Cryoprotectants. Biomacromolecules 2020, 21 (7), 7–17. 10.1021/acs.biomac.9b01053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham B.; Bailey T. L.; Healey J. R. J.; Marcellini M.; Deville S.; Gibson M. I. Polyproline as a Minimal Antifreeze Protein Mimic That Enhances the Cryopreservation of Cell Monolayers. Angew. Chem. 2017, 129 (50), 16157–16160. 10.1002/ange.201706703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voets I. K. From Ice-Binding Proteins to Bio-Inspired Antifreeze Materials. Soft Matter 2017, 13 (28), 4808–4823. 10.1039/C6SM02867E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z.; Liu K.; Wang J. Bioinspired Materials for Controlling Ice Nucleation, Growth, and Recrystallization. Acc. Chem. Res. 2018, 51 (5), 1082–1091. 10.1021/acs.accounts.7b00528. [DOI] [PubMed] [Google Scholar]
- Gibson M. I. Slowing the Growth of Ice with Synthetic Macromolecules: Beyond Antifreeze(Glyco) Proteins. Polym. Chem. 2010, 1 (8), 1141–1152. 10.1039/c0py00089b. [DOI] [Google Scholar]
- Qiu Y.; Hudait A.; Molinero V. How Size and Aggregation of Ice-Binding Proteins Control Their Ice Nucleation Efficiency. J. Am. Chem. Soc. 2019, 141 (18), 7439–7452. 10.1021/jacs.9b01854. [DOI] [PubMed] [Google Scholar]
- Biggs C. I.; Bailey T. L.; Ben Graham; Stubbs C.; Fayter A.; Gibson M. I. Polymer Mimics of Biomacromolecular Antifreezes. Nat. Commun. 2017, 8 (1), 1546. 10.1038/s41467-017-01421-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun T.; Lin F.-H.; Campbell R. L.; Allingham J. S.; Davies P. L. An Antifreeze Protein Folds with an Interior Network of More than 400 Semi-Clathrate Waters. Science 2014, 343 (6172), 795–798. 10.1126/science.1247407. [DOI] [PubMed] [Google Scholar]
- Marshall C. B.; Daley M. E.; Sykes B. D.; Davies P. L. Enhancing the Activity of a β-Helical Antifreeze Protein by the Engineered Addition of Coils. Biochemistry 2004, 43 (37), 11637–11646. 10.1021/bi0488909. [DOI] [PubMed] [Google Scholar]
- Congdon T.; Notman R.; Gibson M. I. Antifreeze (Glyco)Protein Mimetic Behavior of Poly(Vinyl Alcohol): Detailed Structure Ice Recrystallization Inhibition Activity Study. Biomacromolecules 2013, 14 (5), 1578–1586. 10.1021/bm400217j. [DOI] [PubMed] [Google Scholar]
- Naullage P. M.; Lupi L.; Molinero V. Molecular Recognition of Ice by Fully Flexible Molecules. J. Phys. Chem. C 2017, 121 (48), 26949–26957. 10.1021/acs.jpcc.7b10265. [DOI] [Google Scholar]
- Whale T. F.; Rosillo-Lopez M.; Murray B. J.; Salzmann C. G. Ice Nucleation Properties of Oxidized Carbon Nanomaterials. J. Phys. Chem. Lett. 2015, 6 (15), 3012–3016. 10.1021/acs.jpclett.5b01096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng H.; Liu X.; Shi G.; Bai G.; Ma J.; Chen J.; Wu Z.; Song Y.; Fang H.; Wang J. Graphene Oxide Restricts Growth and Recrystallization of Ice Crystals. Angew. Chem., Int. Ed. 2017, 56 (4), 997–1001. 10.1002/anie.201609230. [DOI] [PubMed] [Google Scholar]
- Biggs C. I.; Packer C.; Hindmarsh S.; Walker M.; Wilson N. R.; Rourke J. P.; Gibson M. I. Impact of Sequential Surface-Modification of Graphene Oxide on Ice Nucleation. Phys. Chem. Chem. Phys. 2017, 19 (33), 21929–21932. 10.1039/C7CP03219F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drori R.; Li C.; Hu C.; Raiteri P.; Rohl A. L.; Ward M. D.; Kahr B. A Supramolecular Ice Growth Inhibitor. J. Am. Chem. Soc. 2016, 138 (40), 13396–13401. 10.1021/jacs.6b08267. [DOI] [PubMed] [Google Scholar]
- Fayter A.; Huband S.; Gibson M. I. X-Ray Diffraction to Probe the Kinetics of Ice Recrystallization Inhibition. Analyst 2020, 145, 3666–3677. 10.1039/C9AN02141H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balcerzak A. K.; Capicciotti C. J.; Briard J. G.; Ben R. N. Designing Ice Recrystallization Inhibitors: From Antifreeze (Glyco)Proteins to Small Molecules. RSC Adv. 2014, 4 (80), 42682–42696. 10.1039/C4RA06893A. [DOI] [Google Scholar]
- Capicciotti C. J.; Leclere M.; Perras F. A.; Bryce D. L.; Paulin H.; Harden J.; Liu Y.; Ben R. N. Potent Inhibition of Ice Recrystallization by Low Molecular Weight Carbohydrate-Based Surfactants and Hydrogelators. Chem. Sci. 2012, 3 (5), 1408–1416. 10.1039/c2sc00885h. [DOI] [Google Scholar]
- Gruneberg A. K.; Graham L. A.; Eves R.; Agrawal P.; Oleschuk R. D.; Davies P. L. Ice Recrystallization Inhibition Activity Varies with Ice-Binding Protein Type and Does Not Correlate with Thermal Hysteresis. Cryobiology 2021, 99, 28–39. 10.1016/j.cryobiol.2021.01.017. [DOI] [PubMed] [Google Scholar]
- Olijve L. L. C.; Meister K.; DeVries A. L.; Duman J. G.; Guo S.; Bakker H. J.; Voets I. K. Blocking Rapid Ice Crystal Growth through Nonbasal Plane Adsorption of Antifreeze Proteins. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (14), 3740–3745. 10.1073/pnas.1524109113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggs C. I.; Stubbs C.; Graham B.; Fayter A. E. R.; Hasan M.; Gibson M. I. Mimicking the Ice Recrystallization Activity of Biological Antifreezes. When Is a New Polymer “Active”?. Macromol. Biosci. 2019, 19 (7), 1900082. 10.1002/mabi.201900082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell D. E.; Clarkson G.; Fox D. J.; Vipond R. A.; Scott P.; Gibson M. I. Antifreeze Protein Mimetic Metallohelices with Potent Ice Recrystallization Inhibition Activity. J. Am. Chem. Soc. 2017, 139 (29), 9835–9838. 10.1021/jacs.7b05822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham B.; Fayter A. E. R.; Houston J. E.; Evans R. C.; Gibson M. I. Facially Amphipathic Glycopolymers Inhibit Ice Recrystallization. J. Am. Chem. Soc. 2018, 140 (17), 5682–5685. 10.1021/jacs.8b02066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T.; Zhao Y.; Zhong Q.; Wu T. Inhibiting Ice Recrystallization by Nanocelluloses. Biomacromolecules 2019, 20 (4), 1667–1674. 10.1021/acs.biomac.9b00027. [DOI] [PubMed] [Google Scholar]
- Balcerzak A. K.; Febbraro M.; Ben R. N. The Importance of Hydrophobic Moieties in Ice Recrystallization Inhibitors. RSC Adv. 2013, 3 (10), 3232–3236. 10.1039/c3ra23220d. [DOI] [Google Scholar]
- Budke C.; Dreyer A.; Jaeger J.; Gimpel K.; Berkemeier T.; Bonin A. S.; Nagel L.; Plattner C.; Devries A. L.; Sewald N.; et al. Quantitative Efficacy Classification of Ice Recrystallization Inhibition Agents. Cryst. Growth Des. 2014, 14 (9), 4285–4294. 10.1021/cg5003308. [DOI] [Google Scholar]
- Adam M. K.; Jarrett-Wilkins C.; Beards M.; Staykov E.; MacFarlane L. R.; Bell T. D. M.; Matthews J. M.; Manners I.; Faul C. F. J.; Moens P. D. J.; et al. 1D Self-Assembly and Ice Recrystallization Inhibition Activity of Antifreeze Glycopeptide-Functionalized Perylene Bisimides. Chem. - Eur. J. 2018, 24 (31), 7834–7839. 10.1002/chem.201800857. [DOI] [PubMed] [Google Scholar]
- Olijve L. L. C.; Hendrix M. M. R. M.; Voets I. K. Influence of Polymer Chain Architecture of Poly(Vinyl Alcohol) on the Inhibition of Ice Recrystallization. Macromol. Chem. Phys. 2016, 217 (8), 951–958. 10.1002/macp.201500497. [DOI] [Google Scholar]
- Sproncken C. C. M.; Surís-Valls R.; Cingil H. E.; Detrembleur C.; Voets I. K. Complex Coacervate Core Micelles Containing Poly(Vinyl Alcohol) Inhibit Ice Recrystallization. Macromol. Rapid Commun. 2018, 39, 1700814. 10.1002/marc.201700814. [DOI] [PubMed] [Google Scholar]
- Stubbs C.; Wilkins L. E.; Fayter A. E. R.; Walker M.; Gibson M. I. Multivalent Presentation of Ice Recrystallization Inhibiting Polymers on Nanoparticles Retains Activity. Langmuir 2019, 35 (23), 7347–7353. 10.1021/acs.langmuir.8b01952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens C. A.; Drori R.; Zalis S.; Braslavsky I.; Davies P. L. Dendrimer-Linked Antifreeze Proteins Have Superior Activity and Thermal Recovery. Bioconjugate Chem. 2015, 26 (9), 1908–1915. 10.1021/acs.bioconjchem.5b00290. [DOI] [PubMed] [Google Scholar]
- Wilkins L. E.; Hasan M.; Fayter A. E. R.; Biggs C.; Walker M.; Gibson M. I. Site-Specific Conjugation of Antifreeze Proteins onto Polymer-Stabilized Nanoparticles. Polym. Chem. 2019, 10, 2986–2990. 10.1039/C8PY01719K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa S.; Koga M.; Osanai S. Anomalous Ice Nucleation Behavior in Aqueous Polyvinyl Alcohol Solutions. Chem. Phys. Lett. 2009, 480 (1), 86–89. 10.1016/j.cplett.2009.08.046. [DOI] [Google Scholar]
- Pummer B. G.; Budke C.; Augustin-Bauditz S.; Niedermeier D.; Felgitsch L.; Kampf C. J.; Huber R. G.; Liedl K. R.; Loerting T.; Moschen T.; et al. Ice Nucleation by Water-Soluble Macromolecules. Atmos. Chem. Phys. 2015, 15 (8), 4077–4091. 10.5194/acp-15-4077-2015. [DOI] [Google Scholar]
- Eickhoff L.; Dreischmeier K.; Zipori A.; Sirotinskaya V.; Adar C.; Reicher N.; Braslavsky I.; Rudich Y.; Koop T. Contrasting Behavior of Antifreeze Proteins: Ice Growth Inhibitors and Ice Nucleation Promoters. J. Phys. Chem. Lett. 2019, 10 (5), 966–972. 10.1021/acs.jpclett.8b03719. [DOI] [PubMed] [Google Scholar]
- Bissoyi A.; Reicher N.; Chasnitsky M.; Arad S.; Koop T.; Rudich Y.; Braslavsky I. Ice Nucleation Properties of Ice-Binding Proteins from Snow Fleas. Biomolecules 2019, 9 (10), 532. 10.3390/biom9100532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanazs A.; Madsen J.; Battaglia G.; Ryan A. J.; Armes S. P. Mechanistic Insights for Block Copolymer Morphologies: How Do Worms Form Vesicles?. J. Am. Chem. Soc. 2011, 133 (41), 16581–16587. 10.1021/ja206301a. [DOI] [PubMed] [Google Scholar]
- Warren N. J.; Armes S. P. Polymerization-Induced Self-Assembly of Block Copolymer Nano-Objects via RAFT Aqueous Dispersion Polymerization. J. Am. Chem. Soc. 2014, 136 (29), 10174–10185. 10.1021/ja502843f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren N. J.; Mykhaylyk O. O.; Ryan A. J.; Williams M.; Doussineau T.; Dugourd P.; Antoine R.; Portale G.; Armes S. P. Testing the Vesicular Morphology to Destruction: Birth and Death of Diblock Copolymer Vesicles Prepared via Polymerization-Induced Self-Assembly. J. Am. Chem. Soc. 2015, 137 (5), 1929–1937. 10.1021/ja511423m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doncom K. E. B.; Blackman L. D.; Wright D. B.; Gibson M. I.; O’Reilly R. K. Dispersity Effects in Polymer Self-Assemblies: A Matter of Hierarchical Control. Chem. Soc. Rev. 2017, 46 (14), 4119–4134. 10.1039/C6CS00818F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czajka A.; Armes S. P. In Situ SAXS Studies of a Prototypical RAFT Aqueous Dispersion Polymerization Formulation: Monitoring the Evolution in Copolymer Morphology during Polymerization-Induced Self-Assembly. Chem. Sci. 2020, 11 (42), 11443–11454. 10.1039/D0SC03411H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu L.; Xu C.-R.; Zhong F.; Hong C.-Y.; Pan C.-Y. Fabrication of Functional Nano-Objects through RAFT Dispersion Polymerization and Influences of Morphology on Drug Delivery. ACS Appl. Mater. Interfaces 2016, 8 (28), 18347–18359. 10.1021/acsami.6b04693. [DOI] [PubMed] [Google Scholar]
- Hinde E.; Thammasiraphop K.; Duong H. T. T.; Yeow J.; Karagoz B.; Boyer C.; Gooding J. J.; Gaus K. Pair Correlation Microscopy Reveals the Role of Nanoparticle Shape in Intracellular Transport and Site of Drug Release. Nat. Nanotechnol. 2017, 12 (1), 81–89. 10.1038/nnano.2016.160. [DOI] [PubMed] [Google Scholar]
- Noy J.-M.; Chen F.; Akhter D. T.; Houston Z. H.; Fletcher N. L.; Thurecht K. J.; Stenzel M. H. Direct Comparison of Poly(Ethylene Glycol) and Phosphorylcholine Drug-Loaded Nanoparticles In Vitro and In Vivo. Biomacromolecules 2020, 21 (6), 2320–2333. 10.1021/acs.biomac.0c00257. [DOI] [PubMed] [Google Scholar]
- Zhang W.-J.; Hong C.-Y.; Pan C.-Y. Polymerization-Induced Self-Assembly of Functionalized Block Copolymer Nanoparticles and Their Application in Drug Delivery. Macromol. Rapid Commun. 2019, 40 (2), 1800279. 10.1002/marc.201800279. [DOI] [PubMed] [Google Scholar]
- Canton I.; Warren N. J.; Chahal A.; Amps K.; Wood A.; Weightman R.; Wang E.; Moore H.; Armes S. P. Mucin-Inspired Thermoresponsive Synthetic Hydrogels Induce Stasis in Human Pluripotent Stem Cells and Human Embryos. ACS Cent. Sci. 2016, 2 (2), 65–74. 10.1021/acscentsci.5b00370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binch A. L. A.; Ratcliffe L. P. D.; Milani A. H.; Saunders B. R.; Armes S. P.; Hoyland J. A. Site-Directed Differentiation of Human Adipose-Derived Mesenchymal Stem Cells to Nucleus Pulposus Cells Using an Injectable Hydroxyl-Functional Diblock Copolymer Worm Gel. Biomacromolecules 2021, 22 (2), 837–845. 10.1021/acs.biomac.0c01556. [DOI] [PubMed] [Google Scholar]
- Mitchell D. E.; Lovett J. R.; Armes S. P.; Gibson M. I. Combining Biomimetic Block Copolymer Worms with an Ice-Inhibiting Polymer for the Solvent-Free Cryopreservation of Red Blood Cells. Angew. Chem., Int. Ed. 2016, 55 (8), 2801–2804. 10.1002/anie.201511454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackman L. D.; Varlas S.; Arno M. C.; Fayter A.; Gibson M. I.; O’Reilly R. K. Permeable Protein-Loaded Polymersome Cascade Nanoreactors by Polymerization-Induced Self-Assembly. ACS Macro Lett. 2017, 6 (11), 1263–1267. 10.1021/acsmacrolett.7b00725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackman L. D.; Varlas S.; Arno M. C.; Houston Z. H.; Fletcher N. L.; Thurecht K. J.; Hasan M.; Gibson M. I.; O’Reilly R. K. Confinement of Therapeutic Enzymes in Selectively Permeable Polymer Vesicles by Polymerization-Induced Self-Assembly (PISA) Reduces Antibody Binding and Proteolytic Susceptibility. ACS Cent. Sci. 2018, 4 (6), 718–723. 10.1021/acscentsci.8b00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varlas S.; Foster J. C.; Georgiou P. G.; Keogh R.; Husband J. T.; Williams D. S.; O’Reilly R. K. Tuning the Membrane Permeability of Polymersome Nanoreactors Developed by Aqueous Emulsion Polymerization-Induced Self-Assembly. Nanoscale 2019, 11 (26), 12643–12654. 10.1039/C9NR02507C. [DOI] [PubMed] [Google Scholar]
- Cheng G.; Pérez-Mercader J. Polymerization-Induced Self-Assembly for Artificial Biology: Opportunities and Challenges. Macromol. Rapid Commun. 2019, 40 (2), 1970006. 10.1002/marc.201970006. [DOI] [PubMed] [Google Scholar]
- Canning S. L.; Smith G. N.; Armes S. P. A Critical Appraisal of RAFT-Mediated Polymerization-Induced Self-Assembly. Macromolecules 2016, 49, 1985–2001. 10.1021/acs.macromol.5b02602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khor S. Y.; Quinn J. F.; Whittaker M. R.; Truong N. P.; Davis T. P. Controlling Nanomaterial Size and Shape for Biomedical Applications via Polymerization-Induced Self-Assembly. Macromol. Rapid Commun. 2019, 40 (2), 1800438. 10.1002/marc.201800438. [DOI] [PubMed] [Google Scholar]
- D’Agosto F.; Rieger J.; Lansalot M. RAFT-Mediated Polymerization-Induced Self-Assembly. Angew. Chem., Int. Ed. 2020, 59 (22), 8368–8392. 10.1002/anie.201911758. [DOI] [PubMed] [Google Scholar]
- Penfold N. J. W.; Yeow J.; Boyer C.; Armes S. P. Emerging Trends in Polymerization-Induced Self-Assembly. ACS Macro Lett. 2019, 8 (8), 1029–1054. 10.1021/acsmacrolett.9b00464. [DOI] [PubMed] [Google Scholar]
- Georgiou P. G.; Kontopoulou I.; Congdon T. R.; Gibson M. I. Ice Recrystallisation Inhibiting Polymer Nano-Objects: Via Saline-Tolerant Polymerisation-Induced Self-Assembly. Mater. Horiz. 2020, 7 (7), 1883–1887. 10.1039/D0MH00354A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren N. J.; Mykhaylyk O. O.; Mahmood D.; Ryan A. J.; Armes S. P. RAFT Aqueous Dispersion Polymerization Yields Poly(Ethylene Glycol)-Based Diblock Copolymer Nano-Objects with Predictable Single Phase Morphologies. J. Am. Chem. Soc. 2014, 136 (3), 1023–1033. 10.1021/ja410593n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida E. Perforated Vesicles Composed of Amphiphilic Diblock Copolymer: New Artificial Biomembrane Model of Nuclear Envelope. Soft Matter 2019, 15 (48), 9849–9857. 10.1039/C9SM01832H. [DOI] [PubMed] [Google Scholar]
- Yoshida E. Fission of Giant Vesicles Accompanied by Hydrophobic Chain Growth through Polymerization-Induced Self-Assembly. Colloid Polym. Sci. 2014, 292 (6), 1463–1468. 10.1007/s00396-014-3216-x. [DOI] [Google Scholar]
- Scheutz G. M.; Touve M. A.; Carlini A. S.; Garrison J. B.; Gnanasekaran K.; Sumerlin B. S.; Gianneschi N. C. Probing Thermoresponsive Polymerization-Induced Self-Assembly with Variable-Temperature Liquid-Cell Transmission Electron Microscopy. Matter 2021, 4 (2), 722–736. 10.1016/j.matt.2020.11.017. [DOI] [Google Scholar]
- Byard S. J.; Williams M.; McKenzie B. E.; Blanazs A.; Armes S. P. Preparation and Cross-Linking of All-Acrylamide Diblock Copolymer Nano-Objects via Polymerization-Induced Self-Assembly in Aqueous Solution. Macromolecules 2017, 50 (4), 1482–1493. 10.1021/acs.macromol.6b02643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Zhou J.; Lv X.; Zhang B.; An Z. Temperature-Induced Morphological Transitions of Poly(Dimethylacrylamide)–Poly(Diacetone Acrylamide) Block Copolymer Lamellae Synthesized via Aqueous Polymerization-Induced Self-Assembly. Macromolecules 2017, 50 (18), 7222–7232. 10.1021/acs.macromol.7b01644. [DOI] [Google Scholar]
- Knight C. A.; Hallett J.; DeVries A. L. Solute Effects on Ice Recrystallization: An Assessment Technique. Cryobiology 1988, 25 (1), 55–60. 10.1016/0011-2240(88)90020-X. [DOI] [PubMed] [Google Scholar]
- Byard S. J.; Blanazs A.; Miller J. F.; Armes S. P. Cationic Sterically Stabilized Diblock Copolymer Nanoparticles Exhibit Exceptional Tolerance toward Added Salt. Langmuir 2019, 35 (44), 14348–14357. 10.1021/acs.langmuir.9b02789. [DOI] [PubMed] [Google Scholar]
- Foster J. C.; Varlas S.; Couturaud B.; Jones J. R.; Keogh R.; Mathers R. T.; O’Reilly R. K. Predicting Monomers for Use in Polymerization-Induced Self-Assembly. Angew. Chem., Int. Ed. 2018, 57 (48), 15733–15737. 10.1002/anie.201809614. [DOI] [PubMed] [Google Scholar]
- Yamago S.; Nakamura Y. Recent Progress in the Use of Photoirradiation in Living Radical Polymerization. Polymer 2013, 54 (3), 981–994. 10.1016/j.polymer.2012.11.046. [DOI] [Google Scholar]
- McKenzie T. G.; Fu Q.; Uchiyama M.; Satoh K.; Xu J.; Boyer C.; Kamigaito M.; Qiao G. G. Beyond Traditional RAFT: Alternative Activation of Thiocarbonylthio Compounds for Controlled Polymerization. Adv. Sci. 2016, 3 (9), 1500394. 10.1002/advs.201670047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binauld S.; Delafresnaye L.; Charleux B.; D’Agosto F.; Lansalot M. Emulsion Polymerization of Vinyl Acetate in the Presence of Different Hydrophilic Polymers Obtained by RAFT/MADIX. Macromolecules 2014, 47 (10), 3461–3472. 10.1021/ma402549x. [DOI] [Google Scholar]
- Pound G.; Eksteen Z.; Pfukwa R.; McKenzie J. M.; Lange R. F. M.; Klumperman B. Unexpected Reactions Associated with the Xanthate-Mediated Polymerization of N-Vinylpyrrolidone. J. Polym. Sci., Part A: Polym. Chem. 2008, 46 (19), 6575–6593. 10.1002/pola.22968. [DOI] [Google Scholar]
- Guinaudeau A.; Coutelier O.; Sandeau A.; Mazières S.; Nguyen Thi H. D.; Le Drogo V.; Wilson D. J.; Destarac M. Facile Access to Poly(N-Vinylpyrrolidone)-Based Double Hydrophilic Block Copolymers by Aqueous Ambient RAFT/MADIX Polymerization. Macromolecules 2014, 47 (1), 41–50. 10.1021/ma4017899. [DOI] [Google Scholar]
- Wan D.; Satoh K.; Kamigaito M.; Okamoto Y. Xanthate-Mediated Radical Polymerization of N -Vinylpyrrolidone in Fluoroalcohols for Simultaneous Control of Molecular Weight and Tacticity. Macromolecules 2005, 38 (25), 10397–10405. 10.1021/ma0515230. [DOI] [Google Scholar]
- Kang H. U.; Yu Y. C.; Shin S. J.; Kim J.; Youk J. H. One-Pot Synthesis of Poly(N-Vinylpyrrolidone)-b-Poly(ε-Caprolactone) Block Copolymers Using a Dual Initiator for RAFT Polymerization and ROP. Macromolecules 2013, 46 (4), 1291–1295. 10.1021/ma302372h. [DOI] [Google Scholar]
- Zhou W.; Qu Q.; Xu Y.; An Z. Aqueous Polymerization-Induced Self-Assembly for the Synthesis of Ketone-Functionalized Nano-Objects with Low Polydispersity. ACS Macro Lett. 2015, 4 (5), 495–499. 10.1021/acsmacrolett.5b00225. [DOI] [PubMed] [Google Scholar]
- Parkinson S.; Hondow N. S.; Conteh J. S.; Bourne R. A.; Warren N. J. All-Aqueous Continuous-Flow RAFT Dispersion Polymerisation for Efficient Preparation of Diblock Copolymer Spheres Worms and Vesicles. React. Chem. Eng. 2019, 4 (5), 852–861. 10.1039/C8RE00211H. [DOI] [Google Scholar]
- Figg C. A.; Carmean R. N.; Bentz K. C.; Mukherjee S.; Savin D. A.; Sumerlin B. S. Tuning Hydrophobicity To Program Block Copolymer Assemblies from the Inside Out. Macromolecules 2017, 50 (3), 935–943. 10.1021/acs.macromol.6b02754. [DOI] [Google Scholar]
- Tam R. Y.; Ferreira S. S.; Czechura P.; Ben R. N.; Chaytor J. L. Hydration Index-a Better Parameter for Explaining Small Molecule Hydration in Inhibition of Ice Recrystallization. J. Am. Chem. Soc. 2008, 130 (7), 17494–17501. 10.1021/ja806284x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.