

Abstract
Cancer cells rely on the enzyme telomerase (EC 2.7.7.49) to promote cellular immortality. Telomerase inhibitors (i.e., azidothymidine) can represent promising antitumor agents, although showing high toxicity when administered alone. Better outcomes were observed within a multipharmacological approach instead. In this context, we exploited the validated antitumor targets carbonic anhydrases (CAs; EC 4.2.1.1) IX and XII to attain the first proof of concept on CA–telomerase dual-hybrid inhibitors. Compounds 1b, 7b, 8b, and 11b showed good in vitro inhibition potency against the CAs IX and XII, with KI values in the low nanomolar range, and strong antitelomerase activity in PC-3 and HT-29 cells (IC50 values ranging from 5.2 to 9.1 μM). High-resolution X-ray crystallography on selected derivatives in the adduct with hCA II as a model study allowed to determine their binding modes and thus to set the structural determinants necessary for further development of compounds selectively targeting the tumoral cells.
Introduction
Eukaryotic cells do possess limited replicative potential as progressive shortage of the chromosome ends (i.e., the telomeres) takes place after every duplication cycle.1 Once the critical physical limits are reached, cellular senescence programs, that is, apoptosis, are triggered.2 Such an effect is properly referred as the “Hayflick limit”, who first reported experimentally the finite capacity of normal cells to replicate.1,3
The state-of-the-art knowledge on telomeres accounts for rather complicated and highly dynamic structures which are evolutionarily conserved among the eukaryotic cells.4 Human telomeres are composed of repetitive, noncoding hexameric nucleotide repeats in complex with the telomere-associated proteins (i.e., the shelterin proteins) and the telomerase.5−8 The former are mainly responsible for maintaining the telomere structure and its signaling functions, whereas the latter for synthetizing new telomeric DNA strands from its own RNA template.4,5 This enzyme is normally highly active in adult germ line and stem cells, whereas it is poorly or not expressed at all in the somatic ones.9,10 Besides the canonical function of telomere elongation, the telomerase enzymes (EC 2.7.7.49) were also found to act as transcriptional regulators of the Wnt/β-catenin signaling pathway, thus suggesting a role in determining cell growth, differentiation, and apoptosis via a nontelomer-dependent manner.11−13
The majority of malignant tumors in humans were demonstrated to depend on the telomerase activity, which resulted in increased telomerase activity when compared to the nontumorigenic counterpart cells.14 As a matter of fact, the catalytic subunit of the telomerase enzyme (i.e., hTERT) was found overexpressed in several tumors,15−18 and its regulatory role in metastatic events was also proved.19 In light of such data, the telomerase is properly considered a tumor marker,20 and still it is taken into consideration as a rational target for developing potent and effective anticancer drugs.15,20−22
By making use of the DNA polymerase activity of the telomerase, nucleoside and nucleotide analogues have been extensively investigated as potential inhibitors.23 In particular, chain-terminator reverse-transcriptase inhibitors have been explored as antitumor agents.23 The first study of this type was conducted by Blackburn in 1994 on the ciliated protozoan Tetrahymena thermophila which is quite rich in telomeres.24 Such studies revealed that azidothymidine (AZT) was able to decrease the de novo telomere addition, thus resulting in shortening of telomeres.24 Further studies showed that in spite of the low affinity of AZT for mammalian DNA polymerases, its triphosphate derivative (AZT-TP) was incorporated into the telomeric region of an eukaryotic genome through a process mediated by the telomerases.25,26 The efficiency of AZT in affecting tumor growth was properly assessed,27−29 and its association with cisplatin, paclitaxel, or 5-fluorouracil showed synergistic interactions.30,31 Although such promising results were obtained, AZT was dropped as an antitumor drug because of its potential tumorigenic properties and the tardiness of the drug to be fully functional, which may expose patients to dangerous side effects.32 Various drawbacks are associated with the use of telomerase inhibitors for cancer therapy.33 The tardiness to take action is the most critical issue, as cellular senescence is induced only when telomeres have reached their critical length and thus implying that such agents do require appropriate time to become effective.32,33 Induction of cellular senescence by telomeric dysfunction may also result in activation of oncogenes and/or silencing of tumor suppressor genes, thus promoting malignant transformations to occur instead.34 In addition, the use of inhibitors of the telomerases may interfere with highly proliferative cells such germ lines and stem cells.10,22 For all these reasons, the use of telomerase inhibitors (i.e., AZT, Imetelstat, BIBR1532, and antisense molecules) for the management of cancer is better envisaged within a polypharmacologically based approach, and the metalloenzyme carbonic anhydrase (CA; EC 4.2.1.1) IX is well suited.35−37 CA IX (and marginally CA XII) is selectively overexpressed in hypoxic solid tumors, and it actively participates in a complex pH regulation machinery tuned to warrant cancer cell survival within a metabolically driven pH-dysregulated environment.37−40 The paramount importance of CA IX in regulating proton dynamics by means of eq 1 was conclusively demonstrated, which allowed to validate such an enzyme as a druggable target for the management of hypoxic tumors.38,39
| 1 |
A recent contribution on the active involvement of CA IX in tumor physiology demonstrated such an enzyme to provide the H+ ions needed by the matrix metalloproteinase 14 to perform proteolytic cleavage of collagen, which in turn determines tumor invasiveness.41 In this context, during the last years, great interests have been turned to the CA IX “interactome”.42−45 A significant study conducted on HEK-293 cells showed that the ARM and/or HEAT-repeat domains are a feature of CA IX interacting partners.45 The majority of such proteins belong to the nuclear-cytoplasmic trafficking machinery, such as XPO1 exportin and TNPO 1 importin, and were found to interact with the CA IX C terminal region.45 These results strongly suggested that CA IX may play the role of a cell–surface signal transducer by undergoing nuclear translocation. This is in agreement with confocal immunofluorescence spectroscopy experiments, which showed nuclear distribution of CA IX in several cell lines, with a marked localization when experimental hypoxic conditions were established.45
In consideration of the robust antitumor effects observed when the telomerase and the CA IX were targeted, the research herein reported is aimed to obtain CA–telomerase dual small-molecule inhibitors (CAI–TI) that are able to (i) efficiently bind to the CA IX (XII) enzymes which is assumed as a discriminant feature between the tumor and normal cells and (ii) exert their antitumoral activity by inhibition of both the CA IX (or XII) and the telomerase. As a consequence, appropriate CAI–TI molecules will have the potential to achieve therapeutic performances far superior to the ones reached when coadministration of single therapeutic agents is considered. To the best of our knowledge, this is the first report on CAI–TI; dual-hybrid compounds designed to target two crucial players in cancer progression.
Results and Discussion
Design and Synthesis of Compounds
The hybridization strategy was performed by exploiting the versatile “click chemistry” approach, which allows to merge efficiently single chemical entities and thus grant easy access to wide molecular diversities.46,47 In this study, we performed a copper-catalyzed azide–alkyne cycloaddition (CuAAC) between the azide of the reverse-transcriptase inhibitor AZT with the terminal alkyne pendant installed on various CAI scaffolds (Figure 1). Our interest in establishing such a chemical connection was mainly based on (i) the rapid and regioselective formation of the 1,4-disubstituted-1,2,3-triazole ring under mild reaction conditions47,48 and (ii) the 1,2,3-triazole is among the most commonly used scaffolds in medicinal chemistry in the last decade because it is a bioisostere of the amide group and it shows good tolerance to metabolic processes as well as to pH fluctuations.49,50 In addition, the abundancy of electrons within the triazole ring allows it to establish H-bonds and π–π stacking interactions with biological targets and thus ensuring additional stabilization of the adducts formed.49,50
Figure 1.
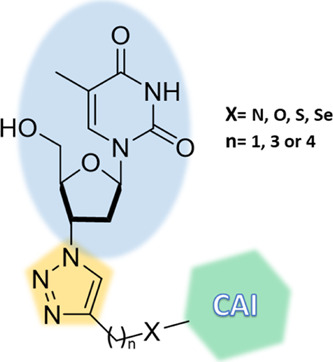
Schematic representation of the synthetized hybrids consisting of a CAI portion linked to AZT through the 1,2,3-triazole ring.
Figure 2.

Substrates for the synthesis of alkynes 1b–3b, 5b–10b, 14b–20b, 4d, and 13e.
The synthesis and characterization data of the appropriate alkyne precursors 1a–3a, 5a–10a, 14a–20a, and 4c, reported in Table 1, are descripted within the Experimental Protocols section. Both classical (i.e., sulfonamides) and nonclassical (i.e., coumarins and sulfocoumarins) CAIs have been included in our study. In particular, sulfonamide-based compounds 6a, 9a, and 10a and coumarin-based compounds 14a, 18a, and 19a are new.
Table 1. Reagents and Conditions for the Synthesis of Compounds 1a–3a, 5a–10a, 14a–20a, 4c, and 13d.

Compounds 13d, here reported for the first time, were obtained through a multistep synthetic approach, reported in Scheme 1.
Scheme 1. Synthesis of Compounds 1b–3b, 5b–10b, 14b–20b, 4d, and 13e.

Yields are reported in brackets.
The final compounds 1b–3b, 5b–12b, 14b–20b, 4d, and 13e, reported in Figure 3, were obtained by performing CuAAC by using Cu (0) nanosized, tetramethyl ammonium chloride (TMACl) as a phase-transfer agent in tBuOH/H2O 1:1 as a solvent at 40 °C (Scheme 2).
Figure 3.
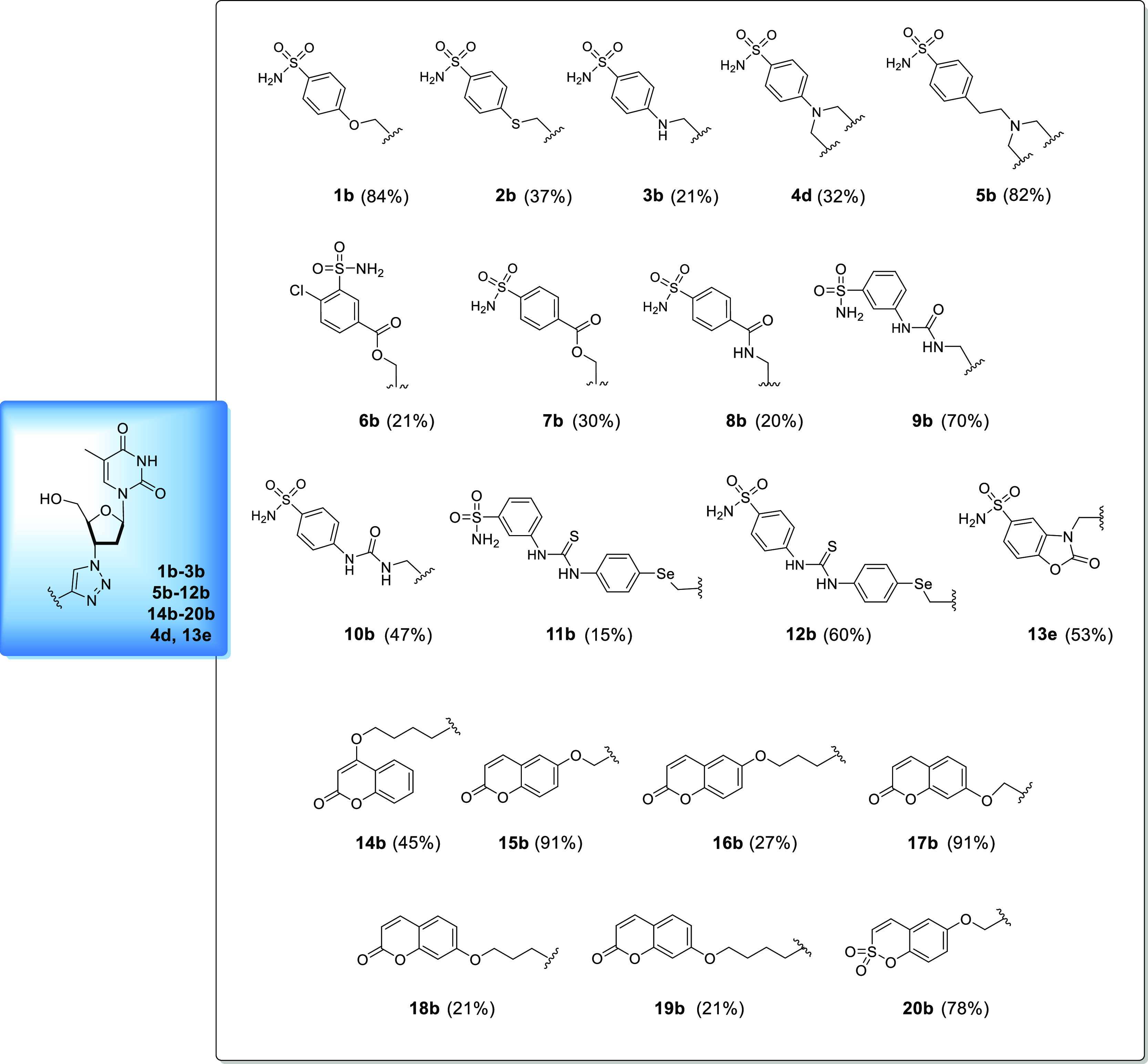
Chemical structures of compounds 1b–3b, 5b–12b, 14b–20b, 4d, and 13e. Yields are reported in brackets and are referred to the final coupling reaction.
Scheme 2. Synthesis of Compounds 1b–3b, 5b–10b, 14b–20b, 4d, and 13e.

Yields are reported in brackets.
The syntheses of compounds 11b and 12b are reported separately (Scheme 3), as for these compounds, CuAAC was not performed as the last reaction step. The synthesis started with the preparation of compound D, bearing the terminal alkyne pendant, obtained by reducing 4-selenocyanatoaniline C with NaBH4 and treating it in situ with propargyl bromide. The CuAAC reaction between the azide of AZT and the terminal alkyne of D was then performed to afford the common intermediate E, which was subsequently reacted with 3-isothiocyanatobenzenesulfonamide or 4-isothiocyanatobenzenesulfonamide to afford compounds 11b and 12b, respectively. All final compounds were obtained in good yields and with high-purity grade (i.e., ≥95%) as determined by high-performance liquid chromatography (HPLC). The structural characterization of both intermediates and final compounds was assessed by means of 1H NMR and 13C NMR as well as high-resolution mass spectroscopy (HRMS).
Scheme 3. Synthesis of Compounds 11b and 12b.
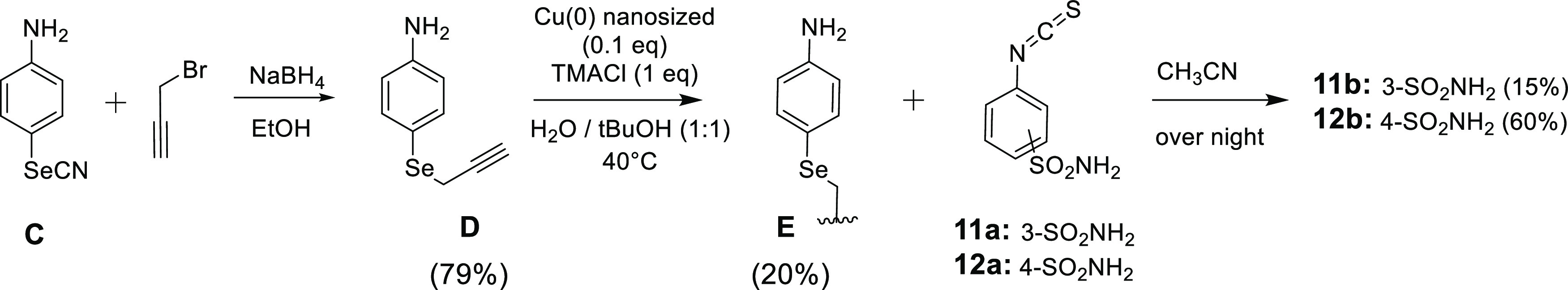
Yields are reported in brackets.
To the best of our knowledge, the AZT–coumarin derivative 17b was previously reported in the literature as part of a set of compounds intended to be used for their fluorescent properties. No biological applications were reported in such a study.51 In addition, ester-triazole-linked triterpenoid–AZT conjugates were also reported.52 Cytotoxic analysis of these hybrids and their triterpenoid precursors revealed moderate to good cytotoxic activities against two human tumor cell lines (KB and Hep-G2).52 However, no detailed studies on the specific targets responsible for the anticancer effects were conducted.
CA Inhibition Profiling
The library of compounds obtained, 1b–3b, 5b–12b, 14b–20b, 4d, and 13e, was evaluated for the inhibition properties against the human-expressed (h) CAs I, II, VA, VB, VII, IX, and XII isoforms by means of the stopped-flow technique applied to the CO2 hydrase assay.53 The inhibition data compared to those of the standard sulfonamide inhibitor acetazolamide (AAZ) are reported in Table 2.
Table 2. Inhibition Data of hCA I, hCA II, hCA VA, hCA VB, hCA VII, hCA IX, and hCA XII with Compounds 1b–3b, 5b–12b, 14b–20b, 4d, and 13e and the Standard Sulfonamide Inhibitor AAZ by a Stopped-Flow CO2 Hydrase Assay53.
| KI (nM)a | |||||||
|---|---|---|---|---|---|---|---|
| hCA I | hCA II | hCA VA | hCA VB | hCA VII | hCA IX | hCA XII | |
| 1b | 4666.7 | 9.3 | 59.1 | 141.3 | 51.6 | 6.2 | 78.9 |
| 2b | 4037.5 | 7.7 | 57.3 | 52.6 | 31.0 | 653.3 | 61.6 |
| 3b | >10,000 | 32.9 | 64.6 | 52.6 | 329.8 | 488.6 | 74.4 |
| 4d | >10,000 | 8.5 | 57.3 | 45.9 | 383.5 | 6557.1 | 74.0 |
| 5b | >10,000 | 70.7 | 59.4 | 42.9 | 281.1 | 8047.1 | 74.0 |
| 6b | 85.5 | 7.7 | 3217.2 | 22.6 | 688.5 | 240.1 | 40.4 |
| 7b | 28.0 | 1.3 | 4795.3 | 54.2 | 48.5 | 3.7 | 7.0 |
| 8b | 483.0 | 13.4 | 1469.3 | 29.0 | 9.5 | 85.5 | 7.8 |
| 9b | 289.7 | 6.3 | 3243.0 | 47.8 | 9.4 | >10,000 | 8.4 |
| 10b | 93.1 | 8.2 | 437.8 | 37.9 | 9.4 | 267.6 | 38.9 |
| 11b | 92.8 | 73.2 | 3972.2 | 45.0 | 66.0 | 373.2 | 9.0 |
| 12b | 62.3 | 5.6 | 6258.9 | 46.7 | 21.8 | >10,000 | 7.1 |
| 13e | 8771.0 | 21.3 | 3651.5 | 28.0 | 38.0 | >10,000 | 8.1 |
| 14b | >10,000 | >10,000 | 1725.8 | 44.0 | 0.7 | >10,000 | 8.7 |
| 15b | >10,000 | >10,000 | 57.8 | 161.0 | 9.3 | 6557.1 | 3.6 |
| 16b | >10,000 | >10,000 | 666.8 | 40.1 | 0.7 | 21.2 | 9.4 |
| 17b | >10,000 | >10,000 | 179.4 | 151.5 | 9.4 | 4885.7 | 3.5 |
| 18b | >10,000 | >10,000 | 301.8 | 42.7 | 0.6 | 2948.3 | 40.4 |
| 19b | >10,000 | >10,000 | 531.2 | 43.9 | 0.6 | >10,000 | 8.9 |
| 20b | >10,000 | >10,000 | 172.4 | 54.6 | 10.5 | 5852.3 | 2.8 |
| AAZ | 250.0 | 12.1 | 63.0 | 54.0 | 2.5 | 25.8 | 5.7 |
Mean from three different assays by a stopped-flow technique (errors were in the range of ±5–10% of the reported values).
As reported in Table 2, the compound series was investigated on the most relevant hCA isoforms such as the ubiquitous hCAs I and II, the mitochondria-expressed hCAs VA and VB, the abundantly central nervous system (CNS)-expressed hCA VII, and the tumor-associated hCAs IX and XII.
The structure–activity relationships (SARs) for the titled compounds are discussed below:
-
i)
Overall, the compound series screened in vitro against the ubiquitous hCAs I and II showed preferential inhibition in the low nanomolar range for the latter. In both cases, the coumarin- and sulfocoumarin-based derivatives (i.e., 14b–20b) resulted ineffective (i.e., KIs > 10,000 nM) in agreement with the previously reported data.54,55 The isosteric ethers 1b and 2b resulted in low micromolar inhibitors of hCA I with the latter being just a 1.2-fold more potent inhibitor (KIs 4666.7 and 4037.5 nM, respectively). Interestingly, the same kinetic profile for both compounds was retained for the hCA II isoform, although the kinetic data were in the low nanomolar range (KIs 9.3 and 7.7 nM, respectively). Further manipulations on the scaffold of the type reported in compounds 3b, 4d, and 5b resulted detrimental for the hCA I (KIs > 10,000 nM). As for the isoform II, the introduction of a N atom as in 3b and 5b determined enhancement of the KI values (32.9 and 70.7 nM, respectively), which were realigned to the previous ones when the N,N-bis-substituted aniline moiety was introduced instead (KI4d 8.5 nM). Compound 7b was the most potent inhibitor among the series against both the hCAs I and II (KIs 28.0 and 1.3 nM, respectively). Variations of the sulfonamide position (i.e., 6b) or of the linker connection (i.e., 8b) badly affected the potencies (see Table 2). Noteworthily, the switch of the sulfonamide moiety from 3- to 4-position as in 9b to 10b and 11b to 12b resulted in a decrease of the inhibition values for hCA I. As for hCA II, a similar profile was observed only for 11b and 12b, whereas the opposite was obtained for the regioisomers 9b and 10b (i.e., KIs 6.3 and 8.2 nM, respectively). Finally, compound 13e showed excellent discrimination between the isoforms tested, being 411.8-fold more potent against hCA II over hCA I.
-
ii)
Despite the high degree of similarity between the mitochondrially expressed hCAs VA and VB, the kinetic profile of the majority of the tested compounds accounted for the preferential inhibition of the latter. The ether derivative 1b was the only sulfonamide-bearing compound among the series which showed selective inhibition of hCA VA over VB up to 2.4-fold. The substitution of the ethereal oxygen in 1b with a sulfur or a nitrogen instead, as in compounds 2b and 3b, respectively, suppressed any isoform selectivity, which was maintained when N,N-disubstitution (i.e., 4d) or elongation (i.e., 5b) was applied (see Table 2). As for the remaining sulfonamide derivatives 6b–12b and 13e, their KI values against hCA VA were all in the micromolar range with compound 10b being the most potent among them (KI 437.8 nM). The same compounds were more effective in inhibiting the second mitochondrially expressed hCA as they showed medium nanomolar KI values. The derivatives 6b, 13e, and 8b were the most effective against the hCA VB and their KI values resulted up to 2.4-fold lower when compared to the reference AAZ (see Table 2). Interesting kinetic data were observed for the coumarin-containing CAIs. The 4-alkyl-substituted derivative 14b resulted quite effective in inhibiting hCA VB with a selectivity index (SI; KI hCA VA/hCA VB) of 39.2. Relocation of the chain to 7-position of the coumarin ring as in 19b did not change the kinetic profile but heavily reduced the SI for the preferential inhibition of the VB isoform (see Table 2). Regioisomeric effects on kinetics were also evident for compounds 15b and 17b. As reported in Table 2, the 6-methylenesubstituted coumarin derivative 15b resulted a 2.8-fold stronger inhibitor of hCA VA over hCA VB. The preferential inhibition for the former was lost when the chain in 15b was moved to the adjacent 7-position as in 17b (see Table 2). Interestingly, the same swapping position as in compounds 16b and 18b did not alter the SI, which was in favor of the hCA VB for both derivatives, and affected its intensity as it resulted halved. Finally, the sulfocoumarin prodrug 20b also reported preferential inhibition for the hCA VB isoform with KI values of 172.4 and 54.6 nM, respectively.
-
iii)
As for the CNS-expressed hCA VII, the majority of the compounds tested resulted low nanomolar inhibitors. On considering the SARs, it is worth noting that the ethers 1b and 2b showed KI values within the medium nanomolar range (51.6 and 31.0 nM, respectively). The introduction of a nitrogen atom instead (i.e., compounds 3b and 4d) or a tertiary amine with an alkyl spacer (i.e., compound 5b) spoiled the inhibition potency against the hCA VII and thus raising the inhibition values up to the high nanomolar range (see Table 2). Interestingly, the ester linkage seems to affect the inhibition potency for this isoform as demonstrated by the kinetic data for both compounds 6b and 7b. As a matter of fact, the insertion of the amide, as in compound 8b, or the ureido linker (i.e., 9b and 10b) resulted in a sensible enhancement of the hCA VII inhibition potency as reported in Table 2 for the corresponding KI values which are all comprised in the low nanomolar range (i.e., 9.5, 9.42, and 9.4 nM for 8b–10b, respectively). Interesting results were obtained for the seleno-containing compounds 11b and 12b as the regioisomer effect on kinetics was clearly observed. As reported in Table 2, the para-substituted benzenesulfonamide derivative 12b was a 3.0-fold more potent inhibitor of hCA VII when compared to the meta one 11b (KIs 21.8 and 66.0 nM, respectively). Finally, among the sulfonamide-containing CAIs is the 2-oxo-2,3-dihydrobenzo[d]oxazole derivative 13e which resulted in a medium hCA VII nanomolar inhibitor with a KI value of 38.0 nM. As for the coumarin-containing CAI moieties, the regioisomeric substitution seems to be ineffective on the kinetic profile of such compounds against the hCA VII isoform. As reported in Table 2, compounds 14b–19b resulted in low nanomolar inhibitors, and among them, the 6- and 7-methylene-substituted derivatives 15b and 17b were the less effective when compared to compounds bearing longer alkyne chain between the CAI portion and the AZT scaffold (14b, 16b, 18b, and 19b).
-
iv)
A very interesting inhibitory profile can be observed for all the synthetized compounds against the tumor-associated isoforms hCA IX and XII. In general, all of them acted as low nanomolar inhibitors of CA XII, with KI values ranging from 2.8 to 78.9 nM. As for CA IX, the different CAI moiety inserted within the scaffold (sulfonamide or coumarin) as well as the substitution patterns both turned out to deeply influence the inhibition potency against this isoform. Three main groups can be delineated on the basis of the observed KI values against CA IX. The first group has compounds that efficiently inhibit both tumor-associated isoforms, such as compounds 1b, 7b, 8b, and 16b (KI values < 100 nM against CA IX) and compounds 2b, 3b, 6b, 10b, and 11b (KI values < 1000 nM against CA IX). Except for compound 16b, which is a 6-substituted coumarin derivative, all the compounds belonging to this group are sulfonamide-based derivatives, in which only one AZT moiety is present within the scaffold. In the second group (KI values < 10,000 nM against CA IX), we can include disubstituted sulfonamide-based compounds 4d and 5b, in which two AZT moieties were “clicked” to the dipropargyl aminobenzensulfonamide and ethylaminobenzenesulfonamide, respectively. In particular, the ethylaminobenzenesulfonamide derivative 5b proved to be 1.23-fold less potent against CA IX then the shorter analogue 4d (Table 2). In the second group, we can also enumerate coumarin-based compounds 15b, 17b, and 18b and sulfocoumarin compound 20b, which inhibited CA IX in the micromolar range (KI values ranging from 2948.3 to 6557.1 nM). Interestingly, these compounds strongly inhibited CA XII in the low nanomolar range (Table 2). Finally, in the third group (KI values > 10,000 nM against CA IX), we can find compounds which selectively inhibited CA XII over CA IX. In particular, 4- and 7-substituted coumarins 14b and 19b, both bearing a four methylene alkyne chain between the coumarin scaffold and the AZT moiety, showed to be ineffective against CA IX in the concentration range considered, whereas a strong inhibition of CA XII can be observed (KI values of 8.7 and 8.9 nM for 14b and 19b, respectively). Meta-substituted ureido compound 9b, para-substituted thioureido compound 12b, and 2-oxo-2,3-dihydrobenzo[d]oxazole-5-sulfonamide compound 13e proved to be inactive in CA IX inhibition too (KI values > 10,000 nM). Again, a strong CA XII inhibition can be observed for all the compounds. Noteworthily, comparing homologous compounds such as meta- and para-substituted ureido compounds 9b and 10b and thioureido compounds 11b and 12b, the crucial impact of the regioisomer on the inhibition potency against CA IX can be appreciated, one isomer being about 30-fold more potent than the other. In particular, the meta-substituted ureido compound 10b proved to be more potent than the para-analogue 9b, whereas for the seleno-containing thioureido compounds 11b and 12b, the meta analogue 11b showed to be the most potent.
Cocrystallographic Studies
In light of the promising KI values observed against the tumor-associated isoforms CA IX and XII, the binding modes of compounds 1b and 3b within hCA II, used as a model study, were determined by means of X-ray experiments. The electron density maps of both 1b–hCA II and 3b–hCA II adducts accounted for both ligands placed well-ordered within the enzymatic cleft with their sulfonamide moieties deep buried up to the bottom of the cavity and coordinated to the zinc(II) ion in the canonical tetrahedral geometry. Again the additional interaction between the sulfonamidic oxygen with the T199 residue was conserved (Figures 4 and 5).56
Figure 4.

Inhibitor 1b bound within the active site of hCA II at 1.1 Å resolution and showing the σA-weighted |Fo – Fc| map contoured at 2.5σ. Ligand 1b is shown in cyan. Hydrogen bonds, van der Waals interactions, and water bridges are shown and labeled in red, blue, and green, respectively. Residues involved in the binding of inhibitors are also shown. PDB access code 6YPW.
Figure 5.

Inhibitor 3b bound within the active site of hCA II at 1.3 Å resolution and showing the σA-weighted |Fo – Fc| map contoured at 2.5σ. Ligand 3b is shown in magenta. Hydrogen bonds and van der Waals interactions are shown and labeled in red and blue. Residues involved in the binding of inhibitors are also shown. PDB access code 6WKA.
The ligand backbones of 1b and 3b are stabilized within the hCA II cavity site by means of a network of hydrogen bonds and van der Waals interactions with substantial differences of the tail orientations as clearly shown when superposition of two structures was performed as shown in Figure 6.
Figure 6.
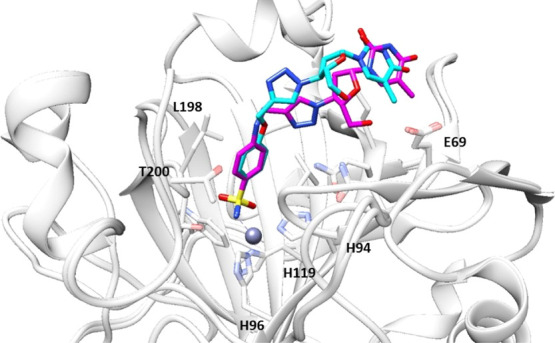
Superposition of inhibitors 1b and 3b bound in the active site of hCA II. Ligand 1b is shown in cyan and 3b in magenta.
The diverse spatial orientations of the tail sections must be ascribed to the replacement of the ethereal oxygen in 1b with the nitrogen atom instead as in 3b, which is the only structural difference among them. The tail in 1b is located toward the hydrophobic half of the catalytic cleft with the F131 residue acting as the major clipping point. The adduct is further stabilized by a network of hydrogen bonds, which connects the inner face of the inhibitor to the opposite hydrophilic half of the enzymatic cleft by means of bridged water molecules (Figures 4 and 6). As for the compound 3b tail, it resulted laid toward the hydrophilic section of the enzymatic cavity and directly stabilized by means of hydrogen bonds to the aminoacidic residues N67, E69, and Q92 (Figures 5 and 6). Such results were in agreement with the previously discussed CA kinetic data, which showed the strongly stabilized compound 1b being a 3.7-fold more potent inhibitor against hCA II when compared to 3b.
Telomerase Activity Assay
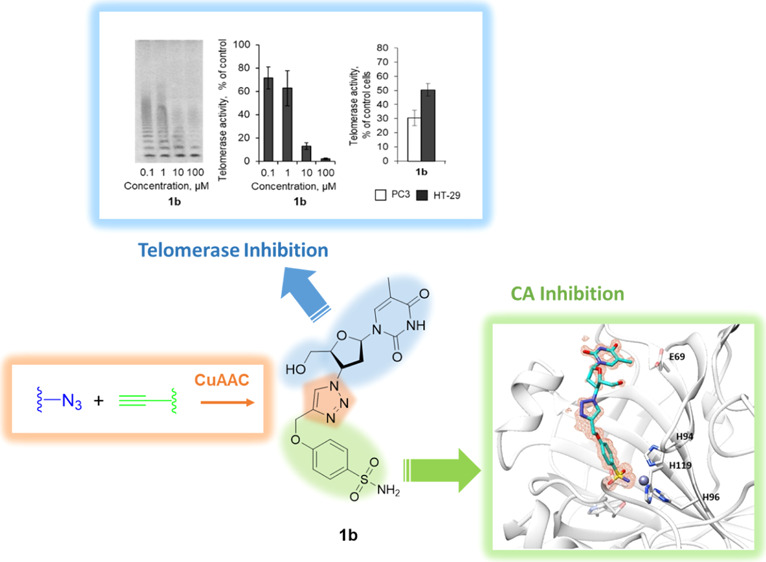
As mentioned above, AZT is known to be a potent telomerase inhibitor.57,58 To check whether our compounds can affect telomerase, we incubated PC3 and HT-29 cells with the most potent CA IX and XII CAI–TI compounds 1b, 7b, 8b, or 11b and measured telomerase activity. The results of the telomerase repeated amplification protocol (TRAP) assay showed that all the tested compounds suppressed telomerase in both PC3 and HT-29 cells (Figure 7A,B). The telomerase activity in PC3 cells was higher than in HT-29. Compounds 1b and 11b demonstrated the strongest antitelomerase activity, while 7b and 8b appeared to be less potent.
Figure 7.

Suppression of telomerase activity by CAI–TI compounds. (A) Representative TRAP gel electrophoresis for PC3 or HT-29 cells incubated with 20 μM CAI–TI for 48 h. (B) Quantification of TRAP for living cells. (C) hTERT expression in incubated PC3 or HT-29 cells. Levels of hTERT mRNA were normalized relative to the levels of reference 18S RNA. (D,F,H,J) Representative TRAP gel electrophoresis for cell lysates treated with different concentrations of CAI–TI. (E,G,I,K) Quantification of TRAP for treated cell lysates. One representative TRAP gel of total four for each of the experiment is shown. The results are presented as the mean ± standard error of the mean. Con., control intact cells.
Telomerase activity is strongly regulated by the expression of its catalytic subunit hTERT, and inhibition of its expression can be one of the ways of how CAI–TI suppresses telomerase in cells.59
We investigated hTERT expression in cells incubated with CAI–TI. In general, hTERT expression in PC3 cells was higher than in HT-29, which corresponds to increased telomerase activity in such cells (Figure 7C). We found that the compounds have no effect on hTERT gene expression in both types of cells. Another possible way of telomerase inhibition is the binding of substance to hTERT protein subunit.60 As it is shown in Figure 7A, PC3 cells have more active telomerase, that is why their lysates were used for telomerase testing in cell-free experiments. All the compounds demonstrated dose-dependent activity to inhibit telomerase within the rage of concentrations 0.1–100 μM (Figure 7D–K). The IC50 and IC90 values for each compound are shown in Table 3. Compounds 1b and 11b had the lowest IC50, that is in accordance to telomerase inhibition in living cells.
Table 3. Determined IC50 and IC90 Values for Telomerase Inhibitors (CAI–TI).
Mean from four different assays by RTQ-TRAP (errors were in the range of ±5% of the reported values).
Conclusions
To the best of our knowledge, this work is the proof-of-concept study about the concomitant use of CAIs and TIs merged within the same molecular scaffold and able to act on two validated targets for the management of cancer. Molecular hybridization is a powerful tool in medicinal chemistry with extensive and several successful applications reported so far.61 Herein, a series of 20 CAI–TI of the AZT-type compounds has been synthetized and fully characterized. Then, inhibition potencies against the two designed targets have been assessed. CA inhibition data against seven hCA isoforms revealed that all the titled compounds 1b–3b, 5b–12b, 14b–20b, 4d, and 13e strongly inhibit hCA XII, whereas some of them (1b–3b, 6b–8b, 10b, 11b, and 16b) showed medium–high inhibition potency against hCA IX.
The evaluation of telomerase activity in cell lysates or in cells incubated with the CA IX and XII most potent inhibitors 1b, 7b, 8b, and 11b showed their strong antitelomerase properties, which rely on the ability to suppress processivity of the enzyme rather than the suppression of hTERT expression.
High-resolution X-ray crystallography on compounds 1b and 3b in adduct with hCA II as a model study allowed to properly assess their binding mode. In particular, we (i) highlighted the crucial role played by a single heteroatom in determining CA isoform selectivity by means of diverse space orientation of the tail and (ii) first determined the molecular features of the CAI–TI molecules, which may be useful to address CA selectivity once proper chemical manipulation is operated.
Overall, the preliminary results obtained in this study fully sustained our strategy and gave us a strong background to further proceed in developing ad hoc designed CAI–TI molecules, which will be considered in appropriate tumor cell lines.
Experimental Protocols
Chemistry
Anhydrous solvents and all reagents were purchased from Sigma-Aldrich (Milan, Italy), Alfa Aesar (Milan, Italy), and TCI (Milan, Italy). All reactions involving air- or moisture-sensitive compounds were used under a nitrogen atmosphere using dried glassware and syringes to transfer solutions. Nuclear magnetic resonance spectra (1H NMR: 400 MHz; 13C NMR: 100 MHz) were recorded in DMSO-d6 using an Avance III 400 MHz spectrometer (Bruker, Milan, Italy). Chemical shifts are reported in parts per million (ppm) and the coupling constants (J) are expressed in hertz (Hz). Splitting patterns are designated as follows: s, singlet; d, doublet; t, triplet; q, quadruplet; m, multiplet; br s, broad singlet; and dd, double of doublets. The assignment of exchangeable protons (OH and NH) was confirmed by the addition of D2O.
The purity of the final compounds was determined in high-purity grade (i.e., ≥95%) by HPLC using an Agilent 1200 liquid chromatography system composed by an autosampler, binary pumps, a column oven, and a diode-array detector (LC–DAD) operating in UV range (210–400 nm). The operating conditions were reported within the Supporting Information file.
The solvents used in MS measures were acetone and acetonitrile (CHROMASOLV grade), purchased from Sigma-Aldrich, and mQ water 18 MΩ cm, obtained from Millipore’s Simplicity system (Milan, Italy). The HRMS analysis was performed with a Thermo Finnigan LTQ Orbitrap mass spectrometer equipped with an electrospray ionization (ESI) source. The accurate mass measure was carried out by introducing, via a syringe pump at 10 μL min–1, the sample solution (1.0 μg mL–1 in mQ water/acetonitrile 50:50), and the signal of the positive ions was acquired. The proposed experimental conditions allowed to monitor the protonated molecules of studied compounds ([M + H]+ species), such that they were measured with a proper dwell time to achieve 60,000 units of resolution at full width at half-maximum.
Synthesis of Final Compounds 1b–3b, 5b–12b, 14b–20b, 4d, and 13e and Their Intermediates
General Procedure A
Proper alkyl halide (1.2 equiv) was added to a suspension of either 1, 14, 15, or 17–20 (0.5 g, 1.0 equiv) and K2CO3 (2.0 equiv) in dry dimethylformamide (DMF) (4 mL) under a N2 atmosphere. The mixture was stirred at room temperature (r.t.) or 60 or 100 °C depending on the alkyl halide until consumption of the starting material [5 h, thin-layer chromatography (TLC) monitoring]. The reaction mixture was cooled at r.t. and quenched with slush. The mixture was extracted with EtOAc (×3), and the combined organic layers were washed with H2O and brine solution, dried over Na2SO4, filtered-off, and then concentrated under vacuum.
General Procedure B
The proper carbamate derivative 9 or 10 (0.5 g, 1 equiv) was dissolved in EtOH, and propargylamine (1.2 equiv) was added. The reaction was refluxed for 16 h, then cooled at r.t., and quenched with slush. The mixture was extracted with EtOAc (×3), dried over Na2SO4, filtered-off, and concentrated under vacuum to afford a solid that was purified by silica gel column chromatography eluting with 60% EtOAc/Hx.
General Procedure C
To a suspension of azidonucleoside AZT (1.1 equiv or 2.2 equiv) in H2O/t-BuOH 1:1 (4 mL), the appropriate alkyne (0.12 g, 1.0 equiv) was added at r.t., followed by copper (0) nanosized (0.1 equiv) and TMACl (1.0 equiv). The suspension was stirred at 40 °C until starting materials were consumed (TLC monitoring), then diluted with MeOH (20 mL), and filtered through Celite 521. The solvent was evaporated, affording to a residue that was triturated from EtOAc, to give a white powder.
4-(Prop-2-ynyloxy)benzenesulfonamide (1a)
Compound 1a was synthetized according to the general procedure A using 4-hydroxybenzenesulfonamide 1 and propargyl bromide 80% in toluene at 60 °C. It was purified by silica gel column chromatography eluting with 50% ethyl acetate in n-hexane to afford the titled compound 1a as a white powder. 47% yield; δH (400 MHz, DMSO-d6): 3.67 (1H, br s, CH), 4.94 (2H, br s, CH2), 7.17 (2H, d, J = 7.2, Ar-H), 7.28 (2H, s, exchange with D2O, SO2NH2), 8.01 (2H, d, J = 7.2, Ar-H). Experimental data in agreement with reported data.62
4-((1-(2-(Hydroxymethyl)-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-3-yl)-1H-1,2,3-triazol-4-yl)methoxy)benzenesulfonamide (1b)
Compound 1b was obtained according to the general procedure A using 1a as the starting material to afford the title compound 1b as a light yellow solid: 84% yield; δH (400 MHz, DMSO-d6): 1.85 (3H, s, CH3), 2.73 (2H, m, CH2), 3.70 (2H, m, CH2), 4.27 (1H, q, J = 3.5, CH), 5.29 (2H, s, CH2), 5.34 (1H, br t, exchange with D2O, OH), 5.45 (1H, m, CH), 6.46 (1H, t, J = 6.5, CH), 7.24 (4H, m, overlapped signals, 2× ArH, exchange with D2O, SO2NH2), 7.80 (2H, d, J = 8.8, ArH), 7.86 (1H, s, CH), 8.50 (1H, s, CH), 11.36 (1H, br s, exchange with D2O, NH); δC (100 MHz, DMSO-d6): 13.1, 38.0, 55.2, 60.3, 62.0, 84.5, 85.4, 110.5, 115.6, 125.4, 128.5, 137.1, 137.4, 143.2, 151.3, 161.2, 164.6; ESI-HRMS (m/z): calcd for [M + H]+ ion species C19H23N6O7S, 479.1343; found, 479.1336.
4-(Prop-2-ynylthio)benzenesulfonamide (2a)
NaBH4 (23 mg, 0.60 mmol, 3.0 equiv) was added portionwise to a freshly prepared solution of 4,4′-disulfanediyldibenzenesulfonamide 2 (75 mg, 0.20 mmol, 1.0 equiv) in EtOH (2 mL) at r.t. under a N2 atmosphere. After 2 h, propargyl chloride (0.42 mmol, 2.1 equiv) was slowly added, and the reaction mixture was stirred at r.t. for 3 h, until complete consumption of the starting material was observed by TLC. The reaction was quenched by addition of saturated NH4Cl aqueous solution (2 mL) and diluted with EtOAc (5 mL). The layers were separated, and the aqueous layer was extracted with EtOAc (2× 5 mL), dried over Na2SO4, filtered, and concentrated under vacuum. The crude material was purified by silica gel flash chromatography to afford the titled compound 2a as a white solid. 83% yield; δH (400 MHz, DMSO-d6): 3.22 (1H, t, J = 2.6, CH), 4.02 (2H, d, J = 2.6, CH2), 7.37 (2H, s, exchange with D2O, SO2NH2), 7.56 (2H, dd, J = 2.0, 6.7, Ar-H), 7.79 (2H, dd, J = 2.0, 6.7, Ar-H). Experimental data in agreement with reported data.63
4-(((1-(2-(Hydroxymethyl)-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-3-yl)-1H-1,2,3-triazol-4-yl)methyl)thio)benzenesulfonamide (2b)
Compound 2b was obtained according to the general procedure C using 2a as starting material to afford the title compound 2b as a white solid: 37% yield; δH (400 MHz, DMSO-d6): 1.84 (3H, s, CH3), 2.69 (2H, m, CH2), 3.64 (2H, m, CH2), 4.19 (1H, q, J = 3.5, CH), 4.45 (2H, s, CH2), 5.38 (2H, m, overlapped signals, 1× CH, exchange with D2O, 1× OH), 6.43 (1H, t, J = 6.5, CH), 7.36 (2H, s, exchange with D2O, SO2NH2), 7.57 (2H, d, J = 8.4, ArH), 7.76 (2H, d, J = 8.4, ArH), 7.85 (1H, s, CH), 8.29 (1H, s, CH), 11.35 (1H, br s, exchange with D2O, NH); δC (100 MHz, DMSO-d6): 13.1, 27.0, 37.9, 60.2, 61.6, 84.8, 85.5, 110.5, 124.2, 127.1, 127.7, 137.1, 141.9, 142.1, 144.1, 151.3, 164.6; ESI-HRMS (m/z): calcd for [M + H]+ ion species C19H23N6O6S2, 495.1115; found, 495.1118.
4-(Prop-2-ynylamino)benzenesulfonamide (3a)
Propargyl bromide 80% in toluene (1.2 equiv) was added to a suspension of sulfanilamide A (0.5 g, 1.0 equiv) and pyridine (1.2 equiv) in dry DMF (2 mL) under N2 atmosphere and the mixture was stirred at 70 °C (TLC monitoring). The reaction was quenched with H2O (10 mL) and extracted with EtOAc (3× 15 mL). The combined organic layers were washed with H2O (3× 15 mL) and brine (3× 15 mL), dried over anhydrous Na2SO4, filtered-off, and concentrated under vacuum to give a solid that was purified by silica gel column chromatography eluting with 50% ethyl acetate in n-hexane to afford the desired product 65 as a yellow solid. 33% yield; δH (400 MHz, DMSO-d6): 3.13 (1H, t, J = 2.4, CH), 3.97 (2H, dd, J = 2.4, 6.0, CH2), 6.73 (2H, d, J = 8.8, Ar-H), 6.76 (1H, br t, exchange with D2O, NH), 6.98 (2H, br s, exchange with D2O, SO2NH2), 7.59 (2H, d, J = 8.8, Ar-H). Experimental data in agreement with reported data.64
4-(((1-(2-(Hydroxymethyl)-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-3-yl)-1H-1,2,3-triazol-4-yl)methyl)amino)benzenesulfonamide (3b)
Compound 3b was obtained according to the general procedure C using 3a as the starting material to afford the title compound 3b as a white solid. 21% yield; δH (400 MHz, DMSO-d6): 1.84 (3H, s, CH3), 2.69 (2H, m, CH2), 3.68 (2H, m, CH2), 4.23 (1H, q, J = 3.5, CH), 4.40 (2H, d, J = 5.7, CH2), 5.32 (1H, br t, 1H, exchange with D2O, OH), 5.38 (1H, m, CH), 6.45 (1H, t, J = 6.5, CH), 6.74 (2H, d, J = 8.8, 2× Ar-H), 6.92 (1H, t, J = 5.7, exchange with D2O, NH), 6.98 (2H, s, exchange with D2O, SO2NH2), 7.55 (2H, d, J = 8.8, 2× Ar-H), 7.84 (1H, s, CH), 8.24 (1H, s, CH), 11.4 (1H, br s, exchange with D2O, NH); δC (100 MHz, DMSO-d6):12.2, 36.9, 37.9, 59.1, 60.7, 83.8, 84.5, 110.1, 111.2, 122.6, 127.2, 130.4, 136.3, 145.2, 150.4, 150.9, 163.7; ESI-HRMS (m/z): calcd for [M + H]+ ion species C19H24N7O6S, 478.1503; found, 478.1508.
4-(Diprop-2-ynylamino)benzenesulfonamide (4c)
Sulfanilamide A (0,5 g, 1.0 equiv) was dissolved in DMF and the solution was cooled to 0 °C. Then, dimethoxy-N,N-dimethylmethanamine (1.2 equiv) was added. The solution was stirred at r.t. until consumption of the starting material (2 h). The reaction was quenched with dichloromethane, and the precipitate formed was filtered-off and dried to afford N′-((4-aminophenyl)sulfonyl)-N,N-dimethylformimidamide 4a, which was used for the next step without further purification. N′-((4-Aminophenyl)sulfonyl)-N,N-dimethylformimidamide 4a (1.0 equiv) was solubilized in dry DMF and K2CO3 (3.0 equiv) was added. Then, propargyl bromide 80% in toluene (4.0 equiv) was added, and the mixture was stirred at 80 °C until consumption of the starting material. Then, the reaction was quenched with H2O (20 mL) and extracted with EtOAc (3× 15 mL). The combined organic layers were washed with H2O (3× 15 mL) and brine(3× 15 mL), dried over Na2SO4, filtered-off, and concentrated under vacuum to give a residue (4b) that was suspended in isopropylamine in a sealed tube and stirred at r.t. The solvent was removed in vacuo, obtaining a residue that was purified by silica gel column chromatography eluting with 50% ethyl acetate in n-hexane to afford a sticky residue which was triturated from Et2O to afford the titled compound 4c as a white powder: 10% yield; δH (400 MHz, DMSO-d6): 3.21 (2H, t, J = 2.4, 2× CH), 4.29 (4H, d, J = 2.4, 2× CH2), 7.03 (2H, d, J = 8.8, Ar-H), 7.08 (2H, s, exchange with D2O, SO2NH2), 7.70 (2H, d, J = 8.8, Ar-H). Experimental data in agreement with reported data.65
4-(Bis((1-(2-(hydroxymethyl)-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-3-yl)-1H-1,2,3-triazol-4-yl)methyl)amino)benzenesulfonamide (4d)
Compound 4d was obtained according to the general procedure C using 4c as the starting material to afford the title compound 4d as a light yellow solid. 32% yield; δH (400 MHz, DMSO-d6): 1.84 (6H, s, 2× CH3), 2.69 (4H, m, 2× CH2), 3.68 (4H, m, 2× CH2), 4.21 (2H, q, J = 3.8, 2× CH), 4.79 (4H, s, 2× CH2), 5.35 (2H, br t, exchange with D2O, 2× OH), 5.40 (2H, m, 2× CH), 6.45 (2H, t, J = 6.4, 2× CH), 7.03 (4H, m, overlapped signals, 2× ArH, exchange with D2O, SO2NH2), 7.60 (2H, d, J = 8.9, 2× ArH), 7.85 (2H, s, 2× CH), 8.28 (2H, s, 2× CH), 11.38 (2H, br s, exchange with D2O, 2× NH). δC (100 MHz, DMSO-d6): 12.2, 37.9, 45.4, 59.1, 60.7, 83.8, 84.5, 110.1, 111.2, 122.6, 127.2, 130.4, 136.3, 145.2, 150.4, 150.9, 163.7; ESI-HRMS (m/z): calcd for [M + H]+ ion species C32H39N12O10S, 783.2627; found, 783.2632.
4-(2-(Di-prop-2-ynylamino)ethyl)benzenesulfonamide (5a)
Propargyl bromide (80% in toluene) (2 equiv) and N, N-diisopropylethylamine (1.7 equiv) were added to a stirred solution of 4-(2-aminoethyl)benzensulfonamide 5 (0.5 g, 1.0 equiv) in CH3CN (8 mL) under a N2 atmosphere. The mixture was stirred at r.t. until consumption of the starting material (TLC monitoring). The solvent was removed under reduced pressure and the obtained residue was portioned between H2O and EtOAc, followed by extraction with EtOAc (3× 15 mL). The combined organic layers were washed with H2O (3× 15 mL) and brine (3× 15 mL), then dried over Na2SO4, filtered-off, and concentrated under vacuum to afford compound 5a as a dark oil. 70% yield; δH (400 MHz, DMSO-d6): 2.75 (2H, t, J = 6.8, CH2), 2.84 (2H, t, J = 6.8, CH2), 3.21 (2H, br t, 2× CH), 3.44 (4H, d, J = 2.0, 2× CH2), 7.37 (2H, s, exchange with D2O, SO2NH2), 7.45 (2H, d, J = 8.4, Ar-H), 7.76 (2H, d, J = 8.4, Ar-H); δC (100 MHz, DMSO-d6): 32.6, 41.5, 53.3, 75.8, 79.1, 125.6, 129.1, 141.9, 144.4; ESI-HRMS (m/z): calcd for [M + H]+ ion species C14H17N2O2S, 277.1005; found, 277.1009. Experimental data in agreement with reported data.66
4-(2-(Bis((1-(2-(hydroxymethyl)-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-3-yl)-1H-1,2,3-triazol-4-yl)methyl)amino)ethyl)benzenesulfonamide (5b)
Compound 5b was obtained according to the general procedure C using 5a as the starting material to afford the title compound 5b as a white solid. 82% yield; δH (400 MHz, DMSO-d6): 1.85 (6H, s, 2× CH3), 2.72 (6H, m, overlapped signals, 3× CH2), 2.93 (2H, t, J = 7.2, CH2), 3.70 (4H, m, 2× CH2), 3.80 (4H, s, 2× CH2), 4.24 (2H, q, J = 3.5, 2× CH), 5.40 (4H; m, overlapped signals, 2× CH, exchange with D2O, 2× OH), 6.48 (2H, t, J = 6.4, 2× CH), 7.32 (2H, br s, exchange with D2O, SO2NH2), 7.41 (2H, d, J = 8.3, 2× Ar-H), 7.75 (2H, d, J = 8.3, 2× Ar-H), 7.87 (2H, s, 2× CH), 8.22 (2H, s, 2× CH), 11.40 (2H, br s, exchange with D2O, 2× NH). δC (100 MHz, DMSO-d6): 13.1, 33.4, 37.9, 48.2, 54.7, 60.1, 61.6, 84.8, 85.5, 110.5, 124.5, 126.5, 130.0, 137.2, 142.6, 144.6, 145.7, 151.4, 164.6; ESI-HRMS (m/z): calcd for [M + H]+ ion species C34H43N12O10S, 811.2940; found, 811.2951.
Prop-2-yn-1-yl 4-Chloro-3-sulfamoylbenzoate (6a)
To a stirring solution of 4-chloro-3-sulfamoylbenzoic acid 6 (1 equiv) in dry DMF, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) HCl (1.2 equiv) was added at 0 °C. After 30 min, propargyl alcohol (1.2 equiv) and 4-dimethylaminopyridine (1.2 equiv) were added. The mixture was stirred at r.t. under N2 for an additional 3 h until consumption of the starting material. The reaction was quenched with slush and extracted with EtOAc (×3). The organic extract was washed with saturated aqueous NaHCO3, water, and brine; dried over Na2SO4; filtered-off; and concentrated under vacuum. The crude was purified by flash silica chromatography (40% EtOAc/Hx) to afford the title compound 6a as a white solid. 40% yield; δH (400 MHz, DMSO-d6): 3.68 (1H, t, J = 2.4, CH), 5.05 (2H, d, J = 2.5, CH2), 7.86 (3H, m, 1× Ar-H, 2× SO2NH2), 8.16 (1H, dd, J = 2.2, 8.2, Ar-H), 8.56 (1H, d, J = 2.1, Ar-H); δC (100 MHz, DMSO-d6): 53.6, 79.0, 79.3, 129.0, 130.6, 133.4, 134.5, 136.7, 142.4, 165.5; ESI-HRMS (m/z): calcd for [M + H]+ ion species C15H16N3O2, 270.1237; found, 270.1237.
(1-((2-(Hydroxymethyl)-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-3-yl)methyl)-1H-1,2,3-triazol-4-yl)methyl 4-Chloro-3-sulfamoylbenzoate (6b)
Compound 6b was obtained according to the general procedure C using 6a as a starting material to afford the title compound 6b as a white solid. 21% yield; δH (400 MHz, DMSO-d6): 1.83 (3H, s, CH3), 2.69 (2H, m, CH2), 3.67 (2H, m, CH2), 4.22 (1H, q, J = 3.9, CH), 4.76 (2H, s, CH2), 5.29 (1H, br t, 1H, exchange with D2O, OH), 5.38 (1H, dt, J = 8.3, 5.29, CH), 6.43 (1H, t, J = 6.5, CH), 7.47 (2H, s, exchange with D2O, SO2NH2), 7.86 (2H, m, 2× Ar-H), 7.98 (1H, s, CH), 8.10 (1H, s, Ar-H), 8.38 (1H, s, CH), 11.3 (1H, br s, exchange with D2O, NH). δC (100 MHz, DMSO-d6):13.1, 27.5, 38.0, 60.4, 61.6, 84.8, 85.3, 110.5, 111.6, 116.9, 123.1, 124.6, 137.1, 142.0, 143.2, 151.3, 153.8, 163.2, 164.6, 167.1; ESI-HRMS (m/z): calcd for [M + H]+ ion species C20H22ClN6O8S, 541.0903; found, 541.0899.
Prop-2-ynyl 4-Sulfamoylbenzoate (7a)
To a stirring solution of 4-sulfamoylbenzoic acid 7 (2.0 g, 9.9 mmol) in dry DMF (40 mL) were successively added propargyl alcohol (1.17 mL, 19.8 mmol, 2.0 equiv), Et3N (2.8 mL, 19.9 mmol, 2.0 equiv), and EDC HCl (1.9 g, 9.9 mmol, 1.0 equiv). The solution was stirred at r.t. under N2 for an additional 4 h. The mixture was then concentrated under reduced pressure and ethyl acetate (40 mL) was added. The organic extract was washed with saturated aqueous NaHCO3 (40 mL) and back-extracted with ethyl acetate (40 mL). The organic layers were combined and washed with brine (40 mL), dried over Na2SO4, filtered, and evaporated. The crude oil was purified by flash silica chromatography (50% EtOAc/Hx) to afford the title compound 7a as a white crystalline solid. 38% yield; δH (400 MHz, DMSO-d6): 3.63 (1H, t, J = 2.4, CH), 4.97 (2H, d, J = 2.8, CH2), 7.55 (2H, br s, exchange with D2O, SO2NH2), 7.98 (4H, m, 4× ArH); δC (100 MHz, DMSO-d6): 53.0, 78.1, 78.3, 126.2, 130.0, 131.7, 148.3, 164.0. Experimental data in agreement with reported data.67
(1-(2-(Hydroxymethyl)-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-3-yl)-1H-1,2,3-triazol-4-yl)methyl 4-Sulfamoylbenzoate (7b)
Compound 7b was obtained according to the general procedure C using 7a as the starting material to afford the title compound 7b as a white solid. 30% yield; δH (400 MHz, DMSO-d6): 1.84 (3H, s, CH3), 2.74 (2H, m, CH2), 3.71 (2H, m, CH2), 4.28 (1H, m, CH), 5.32 (1H, t, J = 5.1, exchange with D2O, OH), 5.42 (1H, m, CH), 5.50 (2H, s, CH2), 6.46 (1H, t, J = 6.6, CH), 7.60 (2H, s, exchange with D2O, SO2NH2), 7.85 (1H, s, CH), 8.00 (2H, d, J = 8.4, 2× Ar-H), 8.18 (2H, d, J = 8.5, 2× Ar-H), 8.49 (1H, s, CH), 11.4 (1H, br s, exchange with D2O, NH); δC (100 MHz, DMSO-d6): 13.3, 38.0, 59.3, 60.5, 61.7, 84.8, 85.3, 110.5, 125.7, 127.3, 131.2, 133.0, 137.1, 142.8, 149.1, 151.3, 164.6, 165.4; ESI-HRMS (m/z): calcd for [M + H]+ ion species C20H23N6O8S, 507.1293; found, 507.1294.
N-(Prop-2-ynyl)-4-sulfamoylbenzamide (8a)
To a stirring solution of 4-sulfamoylbenzoic acid 7 (2.0 g, 9.9 mmol) and propargylamine (0.64 mL, 9.9 mmol, 1.0 equiv) in dry DMF (40 mL) were successively added N-hydroxybenzotriazole monohydrate (0.94 g, 6.6 mmol, 0.6 equiv), diisopropylethylamine (1.7 mL, 9.9 mmol, 1.0 equiv), and HBTU (3.8 g, 9.9 mmol, 1.0 equiv). The deep yellow solution was stirred at r.t. under N2 for 1 h when found complete by TLC. The mixture was concentrated under reduced pressure and ethyl acetate (40 mL) was added. The organic extract was washed with water (40 mL) and back-extracted with EtOAc (×3). The organic extracts were combined and washed with brine (50 mL). The organic layer was dried over Na2SO4, filtered, and evaporated to a crude white solid. Recrystallization from hot methanol/water (9:1) afforded the title compound 8a as a white crystalline solid. 82% yield; δH (400 MHz, DMSO-d6): 3.12 (1H, t, J = 2.4, CH), 4.05 (2H, d, J = 5.6, 2.8, CH2), 7.45 (2H, br s, exchange with D2O, SO2NH2), 7.92 (4H, m, 4× ArH), 9.09 (1H, t, J = 5.6, exchange with D2O, NH); δC (100 MHz, DMSO-d6): 29.0, 73.8, 81.7, 126.4, 128.6, 137.3, 147.1, 164.6. Experimental data in agreement with reported data.67
N-((1-(2-(Hydroxymethyl)-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-3-yl)-1H-1,2,3-triazol-4-yl)methyl)-4-sulfamoylbenzamide (8b)
Compound 8b was obtained according to the general procedure C using 8a as the starting material to afford the title compound 8b as a white solid. 20% yield; δH (400 MHz, DMSO-d6): 1.84 (3H, s, CH3), 2.71 (2H, m, CH2), 3.69 (2H, m, CH2), 4.25 (1H, t, J = 4.6, CH), 4.59 (2H, s, CH2), 5.30 (1H, m, exchange with D2O, OH), 5.38 (1H, m, CH), 6.45 (1H, t, J = 6.6, CH), 7.51 (2H, s, exchange with D2O, SO2NH2), 7.84 (1H, s, CH), 7.94 (2H, m, 2× Ar-H), 8.07 (2H, d, J = 8.5, 2× Ar-H), 8.24 (1H, s, CH), 9.25 (1H, br s, exchange with D2O, NH), 11.4 (1H, br s, exchange with D2O, NH); δC (100 MHz, DMSO-d6): 13.1, 35.9, 38.0, 60.1, 61.7, 84.8, 85.4, 110.5, 123.6, 126.5, 128.9, 137.1, 137.9, 145.9, 147.2, 151.3, 164.6, 166.0; ESI-HRMS (m/z): calcd for [M + H]+ ion species C20H24N7O7S, 506.1452; found, 506.1453.
3-(3-(Prop-2-yn-1-yl)ureido)benzenesulfonamide (9a)
Compound 9a was synthetized according to the general procedure B using phenyl (3-sulfamoylphenyl)carbamate 9 as the starting material. White solid, 65% yield; δH (400 MHz, DMSO-d6): 3.14 (1H, t, J = 2.4, CH), 3.93 (2H, dd, J = 2.2, 5.7, CH2), 6.59, (1H, t, J = 5.7, exchange with D2O, NH), 7.34 (2H, br s, exchange with D2O, SO2NH2), 7.40 (1H, dt, J = 8.2, 1.9, ArH), 7.45 (1H, d, J = 7.8, ArH), 7.57 (1H, dt, J = 8.1, 1.6, ArH), 8.02 (1H, t, J = 2.0, ArH), 8.98 (1H, br s, NH); δC (100 MHz, DMSO-d6): 29.7, 73.7, 82.8, 115.6, 119.3, 121.5, 130.2, 141.5, 145.5, 155.5; ESI-HRMS (m/z): calcd for [M + H]+ ion species C10H12N3O3S, 254.0594; found, 254.0592.
3-(3-((1-(2-(Hydroxymethyl)-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-3-yl)-1H-1,2,3-triazol-4-yl)methyl)ureido)benzenesulfonamide (9b)
Compound 9b was obtained according to the general procedure C using 9a as the starting material to afford the title compound 9b as a yellow solid. 70% yield; δH (400 MHz, DMSO-d6): 1.84 (3H, s, CH3), 2.72 (2H, m, CH2), 3.68 (2H, m, CH2), 4.24 (1H, q, J = 4.1, CH), 4.41 (2H, d, J = 5.6, CH2), 5.30 (1H, t, J = 5.2, exchange with D2O, OH), 5.39 (1H, m, CH), 6.45 (1H, t, J = 6.6, CH), 6.75 (1H, t, J = 5.7, exchange with D2O, NH), 7.33 (2H, s, exchange with D2O, SO2NH2), 7.42 (2H, m, 2× Ar-H), 7.56 (1H, d, J = 8.2, Ar-H), 7.84 (1H, s, CH), 8.03 (1H, d, J = 2.0, Ar-H), 8.19 (1H, s, CH), 8.96 (1H, br s, exchange with D2O, NH), 11.4 (1H, br s, exchange with D2O, NH); δC (100 MHz, DMSO-d6): 13.1, 35.7, 38.0, 60.0, 61.6, 84.7, 85.3, 110.5, 115.5, 119.1, 121.4, 123.3, 130.1, 137.1, 141.7, 145.4, 146.6, 151.3, 155.7, 164.6; ESI-HRMS (m/z): calcd for [M + H]+ ion species C20H25N8O7S, 521.1561; found, 521.1556.
4-(3-(Prop-2-yn-1-yl)ureido)benzenesulfonamide (10a)
Compound 10a was synthetized according to the general procedure B using phenyl (4-sulfamoylphenyl)carbamate 10 as the starting material. White solid, 60% yield; δH (400 MHz, DMSO-d6): 3.14 (1H, t, J = 2.4, CH), 3.37 (2H, d, J = 2.4, CH2), 6.67, (1H, br t, exchange with D2O, NH), 7.19 (2H, br s, exchange with D2O, SO2NH2), 7.58 (2H, d, J = 8.2, 2× ArH), 7.69 (2H, d, J = 8.2, 2× ArH), 9.04 (1H, br s, exchange with D2O, NH); δC (100 MHz, DMSO-d6): 29.7, 73.8, 82.8, 118.0, 127.7, 137.3, 144.2, 155.5; ESI-HRMS (m/z): calcd for [M + H]+ ion species C10H12N3O3S, 254.0594; found, 254.0593.
4-(3-((1-(2-(Hydroxymethyl)-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-3-yl)-1H-1,2,3-triazol-4-yl)methyl)ureido)benzenesulfonamide (10b)
Compound 10b was obtained according to the general procedure C using 10a as the starting material to afford the title compound 10b as a yellow solid. 47% yield; δH (400 MHz, DMSO-d6): 1.84 (3H, s, CH3), 2.71 (2H, m, CH2), 3.68 (2H, m, CH2), 4.24 (1H, q, J = 3.9, CH), 4.41 (2H, d, J = 5.6, CH2), 5.30 (1H, t, J = 5.2, exchange with D2O, OH), 5.39 (1H, dt, J = 8.3, 5.4, CH), 6.45 (1H, t, J = 6.6, CH), 6.84 (1H, t, J = 5.7, exchange with D2O, NH), 7.18 (2H, s, exchange with D2O, SO2NH2), 7.59 (2H, d, J = 8.9, 2× Ar-H), 7.71 (2H, d, J = 8.8, Ar-H), 7.84 (1H, s, CH), 8.20 (1H, s, CH), 9.04 (1H, br s, exchange with D2O, NH), 11.4 (1H, br s, exchange with D2O, NH); δC (100 MHz, DMSO-d6): 13.1, 35.7, 38.0, 60.0, 61.6, 84.7, 85.3, 110.5, 117.8, 123.2, 127.6, 137.8, 141.5, 144.3, 146.4, 151.3, 155.6, 164.6; ESI-HRMS (m/z): calcd for [M + H]+ ion species C20H25N8O7S, 521.1561; found, 521.1554.
4-(Prop-2-yn-1-ylselanyl)aniline (D)
4-Selenocyanatoaniline C (1 equiv) was dissolved in EtOH, and NaBH4 (4 equiv) was added. The reaction was stirred for 20 min. Then, propargyl bromide (1.2 equiv) was added, and the reaction was stirred until consumption of the starting material. The reaction was quenched with NH4Cl saturated solution, extracted with EtOAc (×3), dried over Na2SO4, filtered, and concentrated in reduced pressure to give the desired product D as a yellow solid. 79% yield; δH (400 MHz, DMSO-d6): 2.26 (1H, s, CH), 3.34 (2H, d, J = 2.4, CH2), 3.76 (2H, br s, exchange with D2O, NH2), 6.59 (2H, d, J = 8.3, 2× ArH), 7.44 (2H, d, J = 8.3, 2× ArH); δC (100 MHz, DMSO-d6): 13.7, 71.9, 81.7, 115.8, 116.2, 136.8, 147.2; ESI-HRMS (m/z): calcd for [M + H]+ ion species C9H10NSe, 211.9973; found, 211.9969.
1-(4-(4-(((4-Aminophenyl)selanyl)methyl)-1H-1,2,3-triazol-1-yl)-5-(hydroxymethyl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (E)
Synthetized according to the general procedure C using 4-(prop-2-yn-1-ylselanyl)aniline D as the starting material. Yellow solid. 20% yield; δH (400 MHz, DMSO-d6): 1.84 (3H, s, CH3), 2.69 (2H, m, CH2), 3.67 (2H, m, CH2), 4.04 (2H, s, CH2), 4.16 (2H, m, 2× CH), 5.34 (3H, br s, exchange with D2O, 1× OH, 2× NH2), 6.43 (1H, t, J = 6.6, CH), 6.49 (2H, d, J = 7.9, 2× ArH), 7.13 (2H, d, J = 7.9, 2× ArH), 7.84 (1H, s, CH), 7.96 (1H, s, CH), 11.4 (1H, br s, exchange with D2O, NH); δC (100 MHz, DMSO-d6): 13.2, 22.5, 38.0, 60.0, 61.5, 84.7, 85.3, 110.5, 113.5, 115.4, 123.6, 136.9, 137.1, 146.1, 149.2, 151.3, 164.6; ESI-HRMS (m/z): calcd for [M + H]+ ion species C19H23N6O4Se, 479.0942; found, 479.0932.
3-(3-(4-(((1-(2-(Hydroxymethyl)-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-3-yl)-1H-1,2,3-triazol-4-yl)methyl)selanyl)phenyl)thioureido)benzenesulfonamide (11b)
Compound 11b was obtained by reacting compound E (1 equiv) dissolved in CH3CN with 3-isothiocyanatobenzenesulfonamide 11a (1.1 equiv). The reaction was stirred and then quenched with H2O, assisting in the formation of a yellow precipitate that was filtered to afford the crude product. It was purified by silica gel column chromatography, eluting with 8% MeOH/DCM, to obtain the title compound 11b as a white solid. 15% yield; δH (400 MHz, DMSO-d6): 1.84 (3H, s, CH3), 2.72 (2H, m, CH2), 3.62 (2H, m, CH2), 4.14 (1H, s, CH), 4.23 (2H, s, CH2), 5.31 (1H, m, exchange with D2O, OH), 5.38 (1H, m, CH), 6.38 (1H, t, J = 6.3, CH), 7.49 (8H, m, exchange with D2O, 2× SO2NH2, 4× Ar-H, 2× Ar-H), 7.75 (1H, d, J = 7.9, Ar-H), 7.84 (1H, s, CH), 8.00 (1H, s, Ar-H), 8.11 (1H, s, CH), 10.16 (2H, br s, exchange with D2O, 2× NH), 11.37 (1H, br s, exchange with D2O, NH); δC (100 MHz, DMSO-d6): 13.1, 20.9, 37.9, 60.0, 61.6, 84.7, 85.3, 110.5, 121.4, 122.3, 123.6, 125.1, 126.2, 127.7, 129.8, 133.4, 137.1, 139.2, 140.9, 145.1, 145.5, 151.3, 164.6, 180.6; ESI-HRMS (m/z): calcd for [M + H]+ ion species C26H29N8O6S2Se, 693.0812; found, 693.0822.
4-(3-(4-(((1-(2-(Hydroxymethyl)-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-3-yl)-1H-1,2,3-triazol-4-yl)methyl)selanyl)phenyl)thioureido)benzenesulfonamide (12b)
Compound 12b was obtained by reacting compound E (1 equiv) dissolved in CH3CN with 4-isothiocyanatobenzenesulfonamide 12a (1.1 equiv). The reaction was stirred and then quenched with H2O, assisting in the formation of a yellow precipitate that was filtered to afford the crude product. It was purified by silica gel column chromatography, eluting with 8% MeOH/DCM, to obtain the title compound 12b as a white solid. 60% yield; δH (400 MHz, DMSO-d6): 1.84 (3H, s, CH3), 2.69 (2H, m, CH2), 3.67 (2H, m, CH2), 4.18 (1H, dd, J = 3.2, 5.7, CH), 4.28 (2H, s, CH2), 5.31 (1H, t, J = 5.3, exchange with D2O, OH), 5.34 (1H, m, CH), 6.43 (1H, t, J = 6.6, CH), 7.33 (2H, s, exchange with D2O, SO2NH2), 7.50 (4H, m, 4× Ar-H), 7.72 (2H, d, J = 8.7, 2× Ar-H), 7.80 (2H, d, J = 8.7, 2× Ar-H), 7.84 (1H, s, CH), 8.11 (1H, s, CH), 10.10 (1H, br s, exchange with D2O, NH), 10.15 (1H, br s, exchange with D2O, NH), 11.39 (1H, br s, exchange with D2O, NH); δC (100 MHz, DMSO-d6): 13.1, 20.9, 38.0, 60.0, 61.5, 84.7, 85.3, 110.5, 123.5, 125.1, 126.3, 127.1, 128.3, 133.3, 137.1, 139.2, 140.3, 143.5, 145.5, 151.3, 164.6, 180.3; ESI-HRMS (m/z): calcd for [M + H]+ ion species C26H29N8O6S2Se, 693.0812; found, 693.0819.
2-Oxo-2,3-dihydrobenzo[d]oxazole-5-sulfonamide (13a)
A solution of 3-amino-4-hydroxybenzenesulfonamide 13 (2.76 g, 1.0 equiv) in dry tetrahydrofuran (90 mL) was treated with dropwise phosgene solution (∼20% in toluene, 1.2 equiv) at 0 °C, and then, the reaction was warmed to r.t. and stirred overnight. After the consumption of the starting material (TLC monitoring), the reaction was quenched with slush and acidified with 1 M aqueous solution of HCl, extracted with EtOAc (3× 20 mL), and the combined organic layers were washed with H2O (3× 20 mL), dried over Na2SO4, filtered, and concentrated in reduced pressure to give the desired product as a brown solid. 83% yield; δH (400 MHz, DMSO-d6): 7.41 (2H, s, exchange with D2O, SO2NH2), 7.49 (1H, d, J = 8.4, Ar-H), 7.52 (1H, d, J = 1.9, Ar-H), 7.60 (1H, dd, J = 1.9, 8.4, Ar-H), 12.03 (1H, s, exchange with D2O, NH); δC (100 MHz, DMSO-d6): 108.2, 110.5, 121.0, 131.6, 140.8, 146.3, 155.1; ESI-HRMS (m/z): calcd for [M + H]+ ion species C7H7N2O4S, 215.0121; found, 215.0122.
N,N-Dimethyl-N′-((2-oxo-2,3-dihydrobenzo[d]oxazol-5-yl)sulfonyl)formimidamide (13b)
A solution of 13a (6.27 g, 1.0 equiv) in DMF (5 mL) was cooled to 0 °C and then treated with N,N-dimethylformamide dimethyl acetal (1.2 equiv). The reaction continued until the consumption of the starting material (TLC monitoring). The reaction was quenched with slush to obtain a precipitate that was filtered and washed with water (3× 5 mL) and dried under vacuum to afford 13b as a white solid. 41% yield; δH (400 MHz, DMSO-d6): 2.93 (3H, t, J = 0.6, CH3), 3.17 (3H, t, J = 0.6, CH3), 7.43 (2H, m, 2× Ar-H), 7.55 (1H, dd, J = 1.8, 8.4, Ar-H), 8.25 (1H, s, CH), 12.01 (1H, s, exchange with D2O, NH); δC (100 MHz, DMSO-d6): 35.9, 41.8, 108.3, 110.5, 121.3, 131.6, 139.6, 146.3, 155.1, 160.7; ESI-HRMS (m/z): calcd for [M + H]+ ion species C10H12N3O4S, 270.0543; found, 270.0538.
N-((Dimethylamino)methyl)-2-oxo-3-(prop-2-yn-1-yl)-2,3-dihydrobenzo[d]oxazole-5-sulfonamide (13c)
Compound 13b (2.0 g, 1.0 equiv) was treated with potassium carbonate (1.0 equiv) in dry DMF (5 mL) and the suspension was stirred at r.t. for 20 min. Then, propargyl bromide (1.2 equiv) was added and the reaction was stirred at r.t. until the starting material was consumed (TLC monitoring). The reaction was quenched with slush, and the precipitate formed was collected by filtration, washed with Et2O (3× 5 mL), and dried under vacuum to obtain the desired compound as a brown solid. 90% yield; δH (400 MHz, DMSO-d6): 2.94 (3H, t, J = 0.7, CH3), 3.19 (3H, t, J = 0.6, CH3), 3.53 (1H, t, J = 2.5, CH), 4.84 (2H, d, J = 2.5, CH2), 7.54 (1H, d, J = 8.4, Ar-H), 7.65 (1H, dd, J = 1.8, 8.4, Ar-H), 7.80 (1H, d, J = 1.8, Ar-H), 8.27 (1H, s, CH); δC (100 MHz, DMSO-d6): 32.6, 36.0, 41.8, 77.0, 77.7, 108.5, 111.0, 122.1, 131.3, 140.1, 144.8, 153.8, 160.7; ESI-HRMS (m/z): calcd for [M + H]+ ion species C13H14N3O4S, 308.0700; found, 308.0698.
2-Oxo-3-(prop-2-yn-1-yl)-2,3-dihydrobenzo[d]oxazole-5-sulfonamide (13d)
Compound 13c (3.4 g, 1.0 equiv) was dissolved in a 1.5 M HCl in MeOH solution (30 mL), and the reaction was stirred at 60 °C in a sealed tube for 4 h, concentrated under vacuum to give a precipitate that was washed water (3× 5 mL) and then with Et2O (3× 5 mL), and dried under vacuum to afford the desired product as a brown solid. 55% yield; δH (400 MHz, DMSO-d6): 3.53 (1H, t, J = 2.5, CH), 4.81 (2H, d, J = 2.5, CH2), 7.49 (2H, s, exchange with D2O, SO2NH2), 7.61 (1H, d, J = 8.4, Ar-H), 7.72 (1H, dd, J = 1.9, 8.4, Ar-H), 7.83 (1H, d, J = 1.9, Ar-H); δC (100 MHz, DMSO-d6): 32.7, 35.0, 77.3, 108.2, 111.1, 122.0, 131.1, 141.3, 144.8, 153.9; ESI-HRMS (m/z): calcd for [M + H]+ ion species C10H9N2O4S, 253.0278; found, 253.0280.
3-((1-(2-(Hydroxymethyl)-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-3-yl)-1H-1,2,3-triazol-4-yl)methyl)-2-oxo-2,3-dihydrobenzo[d]oxazole-5-sulfonamide (13e)
Compound 13e was obtained according to the general procedure C using 13d as the starting material to afford the title compound 13e as a white solid. 53% yield; δH (400 MHz, DMSO-d6): 1.84 (3H, s, CH3), 2.70 (2H, m, CH2), 3.68 (2H, m, CH2), 4.07 (1H, m, CH), 4.22 (1H, d, J = 5.4, CH), 5.22 (2H, s, CH2), 5.30 (1H, br t, exchange with D2O, OH), 6.44 (1H, t, J = 6.6, CH), 7.46 (2H, s, exchange with D2O, SO2NH2), 7.59 (1H, d, J = 8.4, Ar-H), 7.67 (1H, m, Ar-H), 7.78 (1H, s, Ar-H), 7.84 (1H, s, CH), 8.44 (1H, s, CH), 11.39 (1H, br s, exchange with D2O, NH); δC (100 MHz, DMSO-d6): 13.1, 37.9, 38.3, 60.3, 61.6, 84.8, 85.3, 108.1, 110.5, 110.8, 121.6, 124.3, 131.8, 137.0, 141.1, 142.1, 144.8, 151.3, 154.4, 164.6; ESI-HRMS (m/z): calcd for [M + H]+ ion species C20H22N7O8S, 520.1245; found, 520.1236.
4-(Hex-5-yn-1-yloxy)-2H-chromen-2-one (14a)
Compound 14a was synthetized according to the general procedure A using 4-hydroxy-2H-chromen-2-one 14 as the starting material and 6-chlorohex-1-yne as the alkyl halide at 100 °C. Compound 14a was obtained as a white powder. 80% yield; δH (400 MHz, DMSO-d6): 1.70 (2H, m, CH2), 1.95 (2H, m, CH2), 2.31 (2H, m, CH2), 2.84 (1H, t, J = 2.5, CH), 4.28 (2H, t, J = 6.2, CH2), 5.92 (1H, s, Ar-H), 7.39 (1H, d, J = 7.8, Ar-H), 7.43 (1H, d, J = 8.1, Ar-H), 7.69 (1H, t, J = 8.1, Ar-H), 7.85 (1H, d, J = 7.8, Ar-H); δC (100 MHz, DMSO-d6): 18.4, 25.6, 28.1, 70.0, 72.5, 85.2, 91.5, 116.3, 117.5, 123.8, 125.2, 133.7, 153.8, 162.7, 165.9; ESI-HRMS (m/z): calcd for [M + H]+ ion species C15H15O3, 243.1016; found, 243.1016.
1-(5-(Hydroxymethyl)-4-(4-(4-((2-oxo-2H-chromen-4-yl)oxy)butyl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (14b)
Compound 14b was obtained according to the general procedure C using 14a as the starting material to afford the title compound 14b as a white solid. 45% yield; δH (400 MHz, DMSO-d6): 1.84 (3H, s, CH3), 1.89 (2H, m, 2× CH2), 2.70 (2H, m, CH2), 2.78 (2H, m, CH2), 3.68 (2H, m, CH2), 4.22 (1H, m, CH), 4.29 (2H, m, CH2), 5.34 (2H, m, 1× CH, exchange with D2O, 1× OH), 5.93 (1H, br s, Ar-H), 6.45 (1H, br t, CH), 7.41 (2H, m, 1× Ar-H, 1× CH), 7.70 (1H, t, J = 7.9, Ar-H), 7.86 (2H, m, 1× Ar-H, 1× CH), 8.13 (1H, s, Ar-H), 11.4 (1H, br s, exchange with D2O, NH); δC (100 MHz, DMSO-d6):13.2, 25.5, 26.2, 28.4, 38.0, 59.9, 61.7, 70.1, 84.8, 85.4, 91.4, 110.5, 116.2, 117.4, 122.5, 123.7, 125.1, 133.6, 137.1, 147.8, 151.3, 153.7, 162.6, 164.6, 165.9; ESI-HRMS (m/z): calcd for [M + H]+ ion species C25H28N5O7, 510.1983; found, 510.1989.
6-(Prop-2-ynyloxy)-2H-chromen-2-one (15a)
Compound 15a was synthetized according to the general procedure A using 6-hydroxy-2H-chromen-2-one 15 as the starting material and propargyl bromide 80% in toluene as the alkyl halide. The reaction was performed at r.t. Compound 15a was obtained as a white powder: 65% yield; δH (400 MHz, DMSO-d6): 3.64 (1H, br t, CH), 4.90 (2H, d, J = 2.1, CH2), 6.54 (1H, d, J = 9.6, Ar-H), 7.30 (1H, dd, J = 2.9, 9.0, Ar-H), 7.38 (1H, d, J = 2.9, Ar-H), 7.41 (1H, d, J = 9.0, Ar-H), 8.06 (1H, d, J = 9.6, Ar-H); δC (100 MHz, DMSO-d6): 56.0, 78.6, 78.9, 112.3, 116.8, 117.4, 119.2, 120.0, 144.0, 148.3, 153.4, 160.1; ESI-HRMS (m/z): calcd for [M + H]+ ion species C12H9O3, 201.0546; found, 201.0543. Experimental data in agreement with reported data.68
1-(5-(Hydroxymethyl)-4-(4-(((2-oxo-2H-chromen-6-yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (15b)
Compound 15b was obtained according to the general procedure C using 15a as the starting material to afford the title compound 15b as a white solid. 91% yield; δH (400 MHz, DMSO-d6): 1.85 (3H, s, CH3), 2.74 (2H, m, CH2), 3.70 (2H, m, CH2), 4.26 (1H, q, J = 3.5, CH), 5.25 (2H, s, CH2), 5.45 (2H, m, overlapped signals, 1× CH, exchange with D2O, 1× OH), 6.47 (1 H, t, J = 6.5, CH), 6.53 (1H, d, J = 9.6, ArH), 7.33 (1H, dd, J = 2.8, 9.0, ArH), 7.39 (1H, d, J = 9.0, ArH), 7.48 (1H, d, J = 2.8, ArH), 7.88 (1H, s, CH), 8.07 (1H, d, J = 9.6, ArH), 8.54 (1H, s, CH), 11.37 (1H, br s, exchange with D2O, NH). δC (100 MHz, DMSO-d6):13.1, 38.0, 60.3, 61.6, 62.6, 84.8, 85.4, 110.5, 112.9, 117.5, 118.3, 120.1, 120.9, 125.4, 137.1, 143.4, 144.9, 148.9, 151.3, 155.2, 161.0, 164.6; ESI-HRMS (m/z): calcd for [M + H]+ ion species C22H22N5O7, 468.1514; found, 468.1508.69
6-(Pent-4-yn-1-yloxy)-2H-chromen-2-one (16a)
Compound 16a was synthetized according to the general procedure A using 6-hydroxy-2H-chromen-2-one 15 as the starting material and 5-chloropent-1-yne as the alkyl halide at 100 °C. Compound 16a was obtained as a white powder. 79% yield; δH (400 MHz, DMSO-d6): 1.94 (2H, m, CH2), 2.38 (2H, dd, J = 4.9, 6.8, CH2), 2.87 (1H, s, CH), 4.11 (2H, t, J = 6.1, CH2), 6.53 (1H, d, J = 9.6, Ar-H), 7.24 (1H, dd, J = 2.6, 9.0, Ar-H), 7.34 (1H, d, J = 2.4, Ar-H), 7.37 (1H, d, J = 9.0, Ar-H), 8.04 (1H, d, J = 9.6, Ar-H); δC (100 MHz, DMSO-d6):15.6, 28.7, 67.7, 72.7, 84.6, 112.5, 117.6, 118.4, 120.3, 120.9, 145.1, 148.9, 155.9, 161.2; ESI-HRMS (m/z): calcd for [M + H]+ ion species C14H13O3, 229.0859; found, 229.0861. Experimental data in agreement with reported data.70
1-(5-(Hydroxymethyl)-4-(4-(3-((2-oxo-2H-chromen-6-yl)oxy)propyl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (16b)
Compound 16b was obtained according to the general procedure C using 16a as the starting material to afford the title compound 16b as a white solid. 27% yield; δH (400 MHz, DMSO-d6): 1.84 (3H, s, CH3), 2.14 (2H, q, J = 6.8, CH2), 2.71 (2H, m, CH2), 2.85 (2H, t, J = 7.5, CH2), 3.69 (2H, m, CH2), 4.12 (2H, t, J = 6.3, CH2), 4.23 (1H, q, J = 4.1, CH), 5.34 (2H, m, 1× CH, exchange with D2O, 1× OH), 6.45 (1H, t, J = 6.6, CH), 6.52 (1H, d, J = 9.5, 1× Ar-H), 7.25 (1H, dd, J = 2.9, 9.0, 1× Ar-H), 7.32 (1H, d, J = 2.9, 1× Ar-H), 7.38 (1H, d, J = 8.9, 1× Ar-H), 7.85 (1H, s, CH), 8.04 (1H, d, J = 9.5, 1× Ar-H), 8.15 (1H, s, CH), 11.4 (1H, br s, exchange with D2O, NH); δC (100 MHz, DMSO-d6):13.1, 22.5, 29.2, 38.0, 59.9, 61.6, 68.3, 84.8, 85.4, 110.5, 112.3, 117.4, 118.2, 120.1, 120.8, 122.5, 137.1, 144.9, 147.3, 148.7, 151.3, 155.8, 161.0, 164.1; ESI-HRMS (m/z): calcd for [M + H]+ ion species C24H26N5O7, 496.1827; found, 496.1830.
7-(Prop-2-ynyloxy)-2H-chromen-2-one (17a)
Compound 17a was synthetized according to the general procedure A using 7-hydroxy-2H-chromen-2-one 17 as the starting material and propargyl bromide 80% in toluene as the alkyl halide. The reaction was performed at r.t. Compound 17a was obtained as a white powder. 73% yield; δH (400 MHz, DMSO-d6): 3.69 (1H, t, J = 2.4, CH), 4.97 (2H, d, J = 2.4, CH2), 6.36 (1H, d, J = 9.6, Ar-H), 7.03 (1H, dd, J = 2.4, 8.6, Ar-H), 7.09 (1H, d, J = 2.4, Ar-H), 7.70 (1H, d, J = 8.6, Ar-H), 8.04 (1H, d, J = 9.6, Ar-H). Experimental data in agreement with reported data.70
1-(5-(Hydroxymethyl)-4-(4-(((2-oxo-2H-chromen-7-yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (17b)
Compound 17b was obtained according to the general procedure C using 17a as the starting material to afford the title compound 17b as a white solid. 91% yield; δH (400 MHz, DMSO-d6): 1.85 (3H, s, CH3), 2.73 (2H, m, CH2), 3.69 (2H, m, CH2), 4.27 (1H, q, J = 3.5, CH), 5.32 (2H, s, CH2), 5.36 (1H, t, J = 5.0, exchange with D2O, OH), 5.46 (1 H, m, CH), 6.34 (1H, d, J = 9.5, ArH), 6.47 (1 H, t, J = 6.5, CH), 7.07 (1H, dd, J = 2.4, 8.6, ArH), 7.21 (1H, d, J = 2.4, ArH), 7.69 (1H, d, J = 8.6, ArH), 7.86 (1H, s, CH), 8.04 (1H, d, J = 9.5, ArH), 8.53 (1H, s, CH), 11.38 (1H, br s, exchange with D2O, NH). δC (100 MHz, DMSO-d6): 13.1, 38.1, 60.4, 61.7, 62.6, 84.9, 85.5, 102.5, 110.6, 113.5, 113.6, 113.8, 125.7, 130.5, 137.2, 143.1, 145.2, 151.4, 156.2, 161.2, 162.0, 164.0; ESI-HRMS (m/z): calcd for [M + H]+ ion species C22H22N5O7, 468.1514; found, 468.1520.51
7-(Pent-4-yn-1-yloxy)-2H-chromen-2-one (18a)
Compound 18a was synthetized according to the general procedure A using 7-hydroxy-2H-chromen-2-one 17 as the starting material and 5-chloropent-1-yne as the alkyl halide at 100 °C. Compound 18a was obtained as a white powder. 71% yield; δH (400 MHz, DMSO-d6): 1.95 (2H, m, CH2), 2.38 (2H, m, CH2), 2.88 (1H, d, J = 1.8, CH), 4.18 (2H, t, J = 6.0, CH2), 6.33 (1H, d, J = 9.4, Ar-H), 6.99 (1H, d, J = 8.6, Ar-H), 7.04 (1H, s, Ar-H), 7.67 (1H, d, J = 8.5, Ar-H), 8.03 (1H, d, J = 9.5, Ar-H); δC (100 MHz, DMSO-d6): 15.5, 28.5, 67.8, 72.7, 84.5, 102.2, 113.4, 113.5, 113.7, 130.6, 145.3, 156.4, 161.3, 162.7; ESI-HRMS (m/z): calcd for [M + H]+ ion species C14H13O3, 229.0859; found, 229.0857.
1-(5-(Hydroxymethyl)-4-(4-(3-((2-oxo-2H-chromen-7-yl)oxy)propyl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (18b)
Compound 18b was obtained according to the general procedure C using 18a as the starting material to afford the title compound 18b as a white solid. 21% yield; δH (400 MHz, DMSO-d6): 1.84 (3H, s, CH3), 2.13 (2H, m, CH2), 2.70 (2H, m, CH2), 2.86 (2H, m, CH2), 3.69 (2H, m, CH2), 4.21 (3H, m, 1× CH2, 1× CH), 5.34 (2H, m, 1× CH, exchange with D2O, 1× OH), 6.32 (1H, d, J = 9.47, 1× Ar-H), 6.45 (1H, m, CH), 7.01 (2H, m, 1× CH, 1× Ar-H), 7.66 (1H, m, Ar-H), 7.85 (1H, s, CH), 8.03 (1H, m, Ar-H), 8.15 (1H, s, CH), 11.4 (1H, br s, exchange with D2O, NH); δC (100 MHz, DMSO-d6):13.2, 22.4, 29.1, 38.0, 59.9, 61.6, 68.4, 84.8, 85.4, 102.1, 110.5, 113.2, 113.3, 113.6, 122.6, 130.4, 137.1, 145.2, 147.3, 151.3, 156.3, 161.2, 162.7, 164.1; ESI-HRMS (m/z): calcd for [M + H]+ ion species C24H26N5O7, 496.1827; found, 496.2000.
7-(Hex-5-yn-1-yloxy)-2H-chromen-2-one (19a)
Compound 19a was synthetized according to the general procedure A using 7-hydroxy-2H-chromen-2-one 17 as the starting material and 6-chlorohex-1-yne as the alkyl halide at 100 °C. Compound 19a was obtained as a white powder. 80% yield; δH (400 MHz, DMSO-d6): 1.64 (2H, m, CH2), 1.86 (2H, m, CH2), 2.28 (2H, m, CH2), 2.82 (1H, t, J = 2.5, CH), 4.13 (2H, t, J = 6.4, CH2), 6.31 (1H, d, J = 9.5, Ar-H), 6.97 (1H, dd, J = 2.1, 8.6, Ar-H), 7.01 (1H, d, J = 2.1, Ar-H), 7.65 (1H, d, J = 8.6, Ar-H), 8.02 (1H, d, J = 9.5, Ar-H); δC (100 MHz, DMSO-d6):18.7, 25.9, 28.8, 68.8, 72.4, 85.3, 102.3, 113.3, 113.4, 113.7, 130.5, 145.3, 156.5, 161.3, 162.9; ESI-HRMS (m/z): calcd for [M + H]+ ion species C15H15O3, 243.1016; found, 243.1012.
1-(5-(Hydroxymethyl)-4-(4-(4-((2-oxo-2H-chromen-7-yl)oxy)butyl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (19b)
Compound 19b was obtained according to the general procedure C using 19a as the starting material to afford the title compound 19b as a white solid. 21% yield; δH (400 MHz, DMSO-d6): 1.83 (7H, m, 2× CH2, 1× CH3), 2.70 (2H, m, CH2), 2.76 (2H, m, CH2), 3.71 (2H, m, CH2), 4.15 (2H, m, CH2), 4.23 (1H, m, CH), 5.34 (2H, m, 1× CH, exchange with D2O, 1× OH), 6.31 (1H, d, J = 9.4, Ar-H), 6.45 (1H, t, J = 6.5, CH), 6.96 (1H, m, Ar-H), 7.0 (1H, m, Ar-H), 7.64 (1H, d, J = 8.6, 1× Ar-H), 7.85 (1H, s, CH), 8.01 (1H, d, J = 9.4, 1× Ar-H), 8.11 (1H, s, CH), 11.4 (1H, br s, exchange with D2O, NH). δC (100 MHz, DMSO-d6):13.2, 25.6, 26.3, 28.9, 38.0, 59.9, 61.7, 68.9, 84.8, 85.4, 102.0, 110.5, 113.2, 113.4, 113.6, 122.4, 130.4, 137.1, 145.2, 147.8, 151.3, 156.3, 161.2, 162.7, 164.6; ESI-HRMS (m/z): calcd for [M + H]+ ion species C25H28N5O7, 510.1983; found, 510.1989.
6-Prop-2-ynyloxy-benzo-[e][1,2]-oxathiine 2,2-dioxide (20a)
Compound 20a was synthetized according to the general procedure A using 6-hydroxybenzo[e][1,2]oxathiine 2,2-dioxide 20 as the starting material and propargyl bromide 80% in toluene as the alkyl halide. Reaction performed at r.t. Compound 20a was obtained as a white powder, pure: 85% yield; δH (400 MHz, DMSO-d6): 3.66 (1H, t, J = 2.4, CH), 4.90 (2H, d, J = 2.4, CH2), 7.24 (1H, dd, J = 3.0, 9.0, Ar-H), 7.38 (1H, d, J = 3.0, Ar-H), 7.45 (1H, d, J = 9.0, Ar-H), 7.55 (1H, d, J = 10.3, Ar-H), 7.68 (1H, d, J = 10.3, Ar-H); δC (100 MHz, DMSO-d6): 56.1, 78.8, 78.9, 115.2, 119.1, 119.6, 119.7, 123.3, 136.4, 145.0, 154.6; ESI-HRMS (m/z): calcd for [M + H]+ ion species C11H9O4S, 237.0216; found, 237.0212. Experimental data in agreement with reported data.68
1-(4-(4-(((2,2-Dioxidobenzo[e][1,2]oxathiin-6-yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)-5-(hydroxymethyl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (20b)
Compound 20b was obtained according to the general procedure C using 20a as the starting material to afford the title compound 20b as a white solid. 78% yield; δH (400 MHz, DMSO-d6): 1.85 (3H, s, CH3), 2.74 (2H, m, CH2), 3.70 (2H, m, CH2), 4.26 (1H, q, J = 3.5, CH), 5.26 (2H, s, CH2), 5.37 (1H, br t, exchange with D2O, OH), 5.45 (1H, m, CH), 6.47 (1H, t, J = 6.5, CH), 7.29 (1H, dd, J = 3.0, 9.0, ArH), 7.44 (1H, d, J = 9.0, ArH), 7.47 (1H, d, J = 3.0, ArH), 7.54 (1H, d, J = 10.3, ArH), 7.68 (1H, d, J = 10.3, ArH), 7.87 (1H, s, CH), 8.51 (1H, s, CH), 11.38 (1H, br s, exchange with D2O, NH). δC (100 MHz, DMSO-d6):13.1, 38.0, 60.3, 61.6, 62.7, 84.8, 85.4, 110.5, 115.7, 119.9, 120.4, 120.5, 124.0, 125.5, 137.1, 137.3, 143.3, 145.6, 151.4, 156.4, 164.9; ESI-HRMS (m/z): calcd for [M + H]+ ion species C21H22N5O8S, 504.1184; found, 504.1183.
CA Inhibition
An Applied Photophysics stopped-flow instrument has been used for assaying the CA-catalyzed CO2 hydration activity.53 Phenol red (at a concentration of 0.2 mM) has been used as an indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 7.5) as a buffer, and 20 mM Na2SO4 (for maintaining the ionic strength constant), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of the inhibitor (0.1 mM) were prepared in distilled deionized water, and dilutions up to 0.01 nM were done thereafter with the assay buffer. The inhibitor and enzyme solutions were preincubated together for 15 min for sulfonamide derivatives and 6 h for coumarin and sulfocoumarin derivatives at r.t. prior to the assay, in order to allow for the formation of the E–I complex. The inhibition constants were obtained by nonlinear least-squares methods using PRISM 3 and the Cheng–Prusoff equation, as reported earlier, and represent the mean from at least three different determinations. All CA isoforms were recombinant ones obtained in-house as reported earlier.53
Cocrystallization and X-ray Data Collection
The crystals of hCA II were obtained using the hanging drop vapor diffusion method using a 24-well Linbro plate. Two microliters of 10 mg/mL solution of hCA II in Tris-HCl 20 mM pH 8.0 was mixed with 2 μL of a solution of 1.5 M sodium citrate and 0.1 M Tris pH 8.0 and was equilibrated against the same solution at 296 K. The crystals of the protein grew in 1 week. Afterward, hCA II crystals were soaked in 5 mM inhibitor solution for 3 days. The crystals were flash-frozen at 100 K using a solution obtained by adding 15% (v/v) glycerol to the mother liquor solution as a cryoprotectant. Data on the crystal of the complex with 1b was collected using synchrotron radiation at the ID-11.2C beamline at Elettra (Trieste, Italy) with a wavelength of 1.000 Å and a Pilatus3_6M Dectris CCD detector. Data on the crystal of the complex with 3b was collected using synchrotron radiation at the MX1 beamline of the Australian Synchrotron. Data were integrated and scaled using the program XDS.71
Structure Determination
The crystal structure of hCA II (PDB accession code: 3P58) without solvent molecules and other heteroatoms was used to obtain the initial phases of the structures using Refmac5.72 Unique reflections (5%) were selected randomly and excluded from the refinement data set for the purpose of Rfree calculations. The initial |Fo – Fc| difference electron density maps unambiguously showed the inhibitor molecules. Atomic models for inhibitors were calculated and energy-minimized using the program JLigand 1.0.40.73 Refinements were proceeded using normal protocols of positional, isotropic atomic displacement parameters alternating with manual building of the models using COOT.74 Solvent molecules were introduced automatically using the program ARP.75 The quality of the final models was assessed with COOT and RAMPAGE.76 Atomic coordinates were deposited in the Protein Data Bank (PDB accession code: 6YPW, 6WKA). Graphical representations were generated with Chimera.77
In Vitro Telomerase Activity Assay
Human prostate cancer PC3 and human colorectal adenocarcinoma HT-29 cell lines (both from ATCC, Manassas, VA) were cultivated in RPMI-1640 cell media supplemented with 10% fetal bovine serum (Hyclone Laboratories, Logan, UK) at 37 °C in the presence of 5% CO2 and 95% humidity. Cell lines have been tested for mycoplasma contamination before the experiment using the Mycoplasma Detection Kit PlasmoTest (InvivoGen, San Diego, CA). The most potent CA IX and XII inhibitors 1b, 7b, 8b, or 11b were diluted to a final concentration of 20 μM and incubated with cells for 48 h. Telomerase activity was determined using the TRAP assay15 with modifications previously described by us.78,79 Briefly, cells were lysed in 10 mM Tris-HCl, pH 7.5, 1 mM MgCl2, 1 mM ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetracetic acid (EGTA), 0.1 mM phenylmethylsulfonylfluoride, 5 mM 2-mercaptoethanol, 0.5% 3-[(3-cholamidopropyl)dimethylammonium]-1-propanesulfonate hydrate, and 10% glycerol (all from Sigma-Aldrich, St. Louis, MO) and centrifuged for 30 min at 12,000g. Supernatants were stored at −80 °C. The protein concentration of cell extracts was determined using the BCA-1 protein assay kit (Sigma-Aldrich, St. Louis, MO). For elongation reaction, 5 μg of total protein and CAI–TI within the range of concentrations 0–100 μM were added to 30 μL of the reaction mixture containing 67 mM Tris-HCl, pH 8.8, 16.6 mM (NH4)2SO4, 0.01% Tween-20, 1.5 mM MgCl2, 1 mM EGTA (all from Sigma-Aldrich, St. Louis, MO), 0.25 mM each dNTPs (Evrogen, Moscow, Russia), and the telomerase substrate primer (TS-primer—AATCCGTCGAGCAGAGTT). Elongation was performed for 30 min at 37 °C and 10 min at 96 °C to inactivate the telomerase. Copy-extended primer 0.1 μL (CX-primer—CCCTTACCCTTACCCTTACCCTAA) and 2.5 units of Taq-polymerase were added to the elongation mixture, followed by the following PCR reaction: 94 °C—5 min; 30 cycles of 94 °C—30 s, 50 °C—30 s, and 72 °C—40 s; and 72 °C—5 min. PCR product visualization was performed using 12% nondenaturing PAAG electrophoresis and TBE buffer. Each sample (10 μL) was added to each well of the gel comb. Gels were stained with SYBR Green I (Invitrogen, Grand Island, NY), photographed under UV light in a ChemiDoc XRS imaging system, and analyzed using a GelAnalyzer 2010a. Statistical analysis involving the Student’s t-test was implemented with the Statistica 6.0 software (StatSoft, Tulsa, OK). To determine the IC50 and IC90 values (inhibitor concentration where the response is reduced by 50 and 90%, respectively), 1 μL of the reaction mixture was subjected to the real-time quantitative TRAP assay (RTQ-TRAP) as described by Hou and co-authors.80
RNA Isolation and Real-Time RT-PCR
A previously described protocol was followed.81 Briefly, total RNA from cells was extracted using a PureLink RNA Mini kit (Life Technologies, Carlsbad, CA). Five micrograms of total RNA were reverse-transcribed using the RevertAid RT Kit (Invitrogen, Grand Island, NY) in a 25 μL reaction mixture, followed by real-time RT-PCR using DTprime5 (DNA Technology, Protvino, Russia). The reaction mix was prepared using Platinum SYBR Green qPCR Supermix-UDG (Invitrogen, Grand Island, NY) according to the manufacturer’s recommendations using the following primers (5′–3′). hTERT sense: GTCCGAGGTGTCCCTGAGTA; hTERT antisense: CAGGGCCTCGTCTTCTACAG; 18S sense: GGATCCATTGGAGGGCAAGT; 18S antisense: ACGAGCTTTTTAACTGCAGCAA (all primers were from Evrogen, Moscow, Russia). Two temperature cycles for annealing/extension were used. The fluorescence was measured at the end of each annealing step, and the melting curve analysis was performed at the end of the reaction (after the 35th cycle), between 60 and 95 °C, to assess the quality of the final PCR products. The standard curves indicating reaction effectiveness were generated using four serial dilutions (1:40, 1:80, 1:160, and 1:320) of total cDNAs. The relative level of hTERT mRNA was calculated using DTprime5 software. The levels of mRNA were normalized relative to the expression of the reference gene 18S. The data are presented as normalized mRNA levels of the studied genes using the averaged expression values of the reference gene.
Statistical Analysis
Telomerase activity assay and measurement of hTERT gene expression were performed in quadruplicate. Statistical analysis using Student’s t-test was completed using Statistica 9.0 software (StatSoft, Tulsa, OK). Differences described as p ≤ 0.05 were considered significant. The values of IC50 and IC90 were calculated using Prism 6 software (GraphPad, San Diego, CA) according to the recommendations by Sebaugh.82
Glossary
Abbreviations
- CA
carbonic anhydrase
- CAI(s)
carbonic anhydrase inhibitor(s)
- AAZ
acetazolamide
- AZT
azidothymidine
- TERT
telomerase reverse transcriptase
- TERC
telomerase RNA component
- RTQ-TRAP
real-time quantitative telomeric repeat amplification protocol
- RT
reverse transcriptase
- EGTA
ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetracetic acid
- PMSF
phenylmethylsulfonylfluoride
- CHAPS
3-[(3-cholamidopropyl)dimethylammonium]-1-propanesulfonate hydrate
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c00636.
SMILES representation for compounds (CSV), data collection and atomic model refinement statistics, HPLC–DAD method for purity analysis, and HPLC traces (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. Telomerase assays and hTERT gene expression were performed by D.D.Z. and A.P.K.
F.C. is grateful to “Bando di Ateneo per il Finanziamento di Progetti Competitivi per Ricercatori a Tempo Determinato (RTD) dell’Università di Firenze—2020–2021”, which partially funded this work. D.D.Z. and A.P.K. are grateful to “Program for Basic Research of the State Academies of Sciences” for 2013–2020 and Ministry of Science and Higher Education of the Russian Federation. Program no. 0518-2018-0003.
The authors declare no competing financial interest.
Supplementary Material
References
- Harley C. B.; Futcher A. B.; Greider C. W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- Bodnar A. G.; Ouelette M.; Frolkis M.; Holt S. E.; Chiu C.; Morin G. B.; Harley C. B.; Shay J. W.; Lichtsteiner S.; Wright W. E. Extension of Life-Span by Introduction of Telomerase into Normal Human Cells. Science 1998, 279, 349–352. 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965, 37, 614–636. 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- Blackburn E. H. Telomeres and telomerase: the means to the end (Nobel lecture). Angew. Chem., Int. Ed. 2010, 49, 7405–7421. 10.1002/anie.201002387. [DOI] [PubMed] [Google Scholar]
- Blackburn E. H. Structure and function of telomeres. Nature 1991, 350, 569–573. 10.1038/350569a0. [DOI] [PubMed] [Google Scholar]
- Moyzis R. K.; Buckingham J. M.; Cram L. S.; Dani M.; Deaven L. L.; Jones M. D.; Meyne J.; Ratliff R. L.; Wu J. R. A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc. Natl. Acad. Sci. U.S.A. 1988, 85, 6622–6626. 10.1073/pnas.85.18.6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan R. J.; Karlseder J. Telomeres: protecting chromosomes against genome instability. Nat. Rev. Mol. Cell Biol. 2010, 11, 171–181. 10.1038/nrm2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason M.; Schuller A.; Skordalakes E. Telomerase structure function. Curr. Opin. Struct. Biol. 2011, 21, 92–100. 10.1016/j.sbi.2010.11.005. [DOI] [PubMed] [Google Scholar]
- Broccoli D.; Young J. W.; de Lange T. Telomerase activity in normal and malignant hematopoietic cells. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 9082–9086. 10.1073/pnas.92.20.9082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masutomi K.; Yu E. Y.; Khurts S.; Ben-Porath I.; Currier J. L.; Metz G. B.; Brooks M. W.; Kaneko S.; Murakami S.; DeCaprio J. A.; Weinberg R. A.; Stewart S. A.; Hahn W. C. Telomerase maintains telomere structure in normal human cells. Cell 2003, 114, 241–253. 10.1016/s0092-8674(03)00550-6. [DOI] [PubMed] [Google Scholar]
- Koh C. M.; Khattar E.; Leow S. C.; Liu C. Y.; Muller J.; Ang W. X.; Li Y.; Franzoso G.; Li S.; Guccione E.; Tergaonkar V. Telomerase regulates MYC-driven oncogenesis independent of its reverse transcriptase activity. J. Clin. Invest. 2015, 125, 2109–2122. 10.1172/jci79134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.-I.; Venteicher A. S.; Hong J. Y.; Choi J.; Jun S.; Shkreli M.; Chang W.; Meng Z.; Cheung P.; Ji H.; McLaughlin M.; Veenstra T. D.; Nusse R.; McCrea P. D.; Artandi S. E. Telomerase modulates Wnt signalling by association with target gene chromatin. Nature 2009, 460, 66–72. 10.1038/nature08137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez P.; Blasco M. A. Telomeric and extra-telomeric roles for telomerase and the telomere-binding proteins. Nat. Rev. Cancer 2011, 11, 161–176. 10.1038/nrc3025. [DOI] [PubMed] [Google Scholar]
- Harley C. B. Telomerase and cancer therapeutics. Nat. Rev. Cancer 2008, 8, 167–179. 10.1038/nrc2275. [DOI] [PubMed] [Google Scholar]
- Kim N.; Piatyszek M.; Prowse K.; Harley C.; West M.; Ho P.; Coviello G.; Wright W.; Weinrich S.; Shay J. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- Zhang A.; Zheng C.; Lindvall C.; Hou M.; Ekedahl J.; Lewensohn R.; Yan Z.; Yang X.; Henriksson M.; Blennow E.; Nordenskjöld M.; Zetterberg A.; Björkholm M.; Gruber A.; Xu D. Frequent amplification of the telomerase reverse transcriptase gene in human tumors. Cancer Res. 2000, 60, 6230–6235. [PubMed] [Google Scholar]
- Zhang A.; Zheng C.; Hou M.; Lindvall C.; Wallin K.-L.; Ångström T.; Yang X.; Hellström A.-C.; Blennow E.; Björkholm M.; Zetterberg A.; Gruber A.; Xu D. Amplification of thetelomerase reverse transcriptase(hTERT) gene in cervical carcinomas. Genes Chromosom. Cancer 2002, 34, 269–275. 10.1002/gcc.10071. [DOI] [PubMed] [Google Scholar]
- Takuma Y.; Nouso K.; Kobayashi Y.; Nakamura S.; Tanaka H.; Matsumoto E.; Fujikawa T.; Suzuki M.; Hanafusa T.; Shiratori Y. Telomerase reverse transcriptase gene amplification in hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2004, 19, 1300–1304. 10.1111/j.1440-1746.2004.03447.x. [DOI] [PubMed] [Google Scholar]
- Teralı K.; Yilmazer A. New surprises from an old favourite: the emergence of telomerase as a key player in the regulation of cancer stemness. Biochimie 2016, 121, 170–178. 10.1016/j.biochi.2015.12.001. [DOI] [PubMed] [Google Scholar]
- Low K. C.; Tergaonkar V. Telomerase: central regulator of all of the hallmarks of cancer. Trends Biochem. Sci. 2013, 38, 426–434. 10.1016/j.tibs.2013.07.001. [DOI] [PubMed] [Google Scholar]
- Rousseau P.; Autexier C. Telomere biology: rationale for diagnostics and therapeutics in cancer. RNA Biol. 2015, 12, 1078–1082. 10.1080/15476286.2015.1081329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shay J. W.; Wright W. E. Telomeres and telomerase in normal and cancer stem cells. FEBS Lett. 2010, 584, 3819–3825. 10.1016/j.febslet.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hukezalie K. R.; Thumati N. R.; Côté H. C. F.; Wong J. M. Y. In vitro and ex vivo inhibition of human telomerase by anti-HIV nucleoside reverse transcriptase inhibitors (NRTIs) but not by non-NRTIs. PLoS One 2012, 7, e47505 10.1371/journal.pone.0047505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl C.; Blackburn E. H. The effects of nucleoside analogs on telomerase and telomeres inTetrahymena. Nucleic Acids Res. 1994, 22, 893–900. 10.1093/nar/22.6.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraj A.; El Alaoui A. M.; Gosselin G.; Imbach J.-L.; Morrow C.; Sommadossi J.-P. Effects of β-l-3′-azido-3′-deoxythymidine 5′-triphosphate on host and viral DNA polymerases. Antiviral Res. 2000, 47, 97–102. 10.1016/s0166-3542(00)00095-4. [DOI] [PubMed] [Google Scholar]
- Gomez D.; Kassim A.; Olivero O. Preferential incorporation of 3′-azido-2′,3′-dideoxythymidine (AZT) in telomeric sequences of CHO cells. Int. J. Oncol. 1995, 7, 1057–1060. 10.3892/ijo.7.5.1057. [DOI] [PubMed] [Google Scholar]
- Datta A.; Bellon M.; Sinha-Datta U.; Bazarbachi A.; Lepelletier Y.; Canioni D.; Waldmann T. A.; Hermine O.; Nicot C. Persistent inhibition of telomerase reprograms adult T-cell leukemia to p53-dependent senescence. Blood 2006, 108, 1021–1029. 10.1182/blood-2006-01-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falchetti A.; Franchi A.; Bordi C.; Mavilia C.; Masi L.; Cioppi F.; Recenti R.; Picariello L.; Marini F.; Del Monte F.; Ghinoi V.; Martineti V.; Tanini A.; Brandi M. L. Azidothymidine induces apoptosis and inhibits cell growth and telomerase activity of human parathyroid cancer cells in culture. J. Bone Miner. Res. 2004, 20, 410–418. 10.1359/jbmr.041123. [DOI] [PubMed] [Google Scholar]
- Fang J.-L.; Beland F. A. Long-term exposure to zidovudine delays cell cycle progression, induces apoptosis, and decreases telomerase activity in human hepatocytes. Toxicol. Sci. 2009, 111, 120–130. 10.1093/toxsci/kfp136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami J.; Nagai N.; Shigemasa K.; Ohama K. Inhibition of telomerase activity and cell proliferation by a reverse transcriptase inhibitor in gynaecological cancer cell lines. Eur. J. Cancer 1999, 35, 1027–1034. 10.1016/s0959-8049(99)00037-4. [DOI] [PubMed] [Google Scholar]
- Johnston J. S.; Johnson A.; Gan Y.; Wientjes M. G.; Au J. L. S. Synergy between 3-azido-3-deoxythymidine and paclitaxel in human pharynx FaDu cells. Pharm. Res. 2003, 20, 957–961. 10.1023/a:1024431218327. [DOI] [PubMed] [Google Scholar]
- Olivero O. A.; Yuspa S. H.; Poirier M. C.; Anderson L. M.; Jones A. B.; Wang C.; Diwan B. A.; Haines D. C.; Logsdon D.; Harbaugh S. W.; Moskal T. J.; Rice J. M.; Riggs C. W. Transplacental Effects of 3′-Azido-2′,3′-Dideoxythymidine (AZT): Tumorigenicity in Mice and Genotoxicity in Mice and Monkeys. J. Natl. Cancer Inst. 1997, 89, 1602–1608. 10.1093/jnci/89.21.1602. [DOI] [PubMed] [Google Scholar]
- Shay J. W.; Wright W. E. Telomerase: a target for cancer therapeutics. Cancer Cell 2002, 2, 257–265. 10.1016/s1535-6108(02)00159-9. [DOI] [PubMed] [Google Scholar]
- Artandi S. E.; Chang S.; Lee S.-L.; Alson S.; Gottlieb G. J.; Chin L.; DePinho R. A. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature 2000, 406, 641–645. 10.1038/35020592. [DOI] [PubMed] [Google Scholar]
- Lee S.-H.; McIntyre D.; Honess D.; Hulikova A.; Pacheco-Torres J.; Cerdán S.; Swietach P.; Harris A. L.; Griffiths J. R. Carbonic anhydrase IX is a pH-stat that sets an acidic tumour extracellular pH in vivo. Br. J. Cancer 2018, 119, 622–630. 10.1038/s41416-018-0216-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Türeci O.; Sahin U.; Vollmar E.; Siemer S.; Göttert E.; Seitz G.; Parkkila A.-K.; Shah G. N.; Grubb J. H.; Pfreundschuh M.; Sly W. S. Human carbonic anhydrase XII: cDNA cloning, expression, and chromosomal localization of a carbonic anhydrase gene that is overexpressed in some renal cell cancers. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 7608–7613. 10.1073/pnas.95.13.7608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Švastová E.; Hulíková A.; Rafajová M.; Zat’ovicová M.; Gibadulinová A.; Casini A.; Cecchi A.; Scozzafava A.; Supuran C. T.; Pastorek J.; Pastoreková S. Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH. FEBS Lett. 2004, 577, 439–445. 10.1016/j.febslet.2004.10.043. [DOI] [PubMed] [Google Scholar]
- Berrino E.; Supuran C. T. Novel approaches for designing drugs that interfere with pH regulation. Expert Opin. Drug Discovery 2019, 14, 231–248. 10.1080/17460441.2019.1567488. [DOI] [PubMed] [Google Scholar]
- Neri D.; Supuran C. T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discovery 2011, 10, 767–777. 10.1038/nrd3554. [DOI] [PubMed] [Google Scholar]
- Schwartz L.; Supuran C.; Alfarouk K. The Warburg effect and the hallmarks of cancer. Anti-Cancer Agents Med. Chem. 2017, 17, 164–170. 10.2174/1871520616666161031143301. [DOI] [PubMed] [Google Scholar]
- Swayampakula M.; McDonald P. C.; Vallejo M.; Coyaud E.; Chafe S. C.; Westerback A.; Venkateswaran G.; Shankar J.; Gao G.; Laurent E. M. N.; Lou Y.; Bennewith K. L.; Supuran C. T.; Nabi I. R.; Raught B.; Dedhar S. The interactome of metabolic enzyme carbonic anhydrase IX reveals novel roles in tumor cell migration and invadopodia/MMP14-mediated invasion. Oncogene 2017, 36, 6244–6261. 10.1038/onc.2017.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Švastová E.; Zilka N.; Zat’ovicová M.; Gibadulinová A.; Ciampor F.; Pastorek J.; Pastoreková S. Carbonic anhydrase IX reduces E-cadherin mediated adhesion of MDCK cells via interaction with beta-catenin. Exp. Cell Res. 2003, 290, 332–345. 10.1016/s0014-4827(03)00351-3. [DOI] [PubMed] [Google Scholar]
- Svastova E.; Witarski W.; Csaderova L.; Kosik I.; Skvarkova L.; Hulikova A.; Zatovicova M.; Barathova M.; Kopacek J.; Pastorek J.; Pastorekova S. Carbonic anhydrase IX interacts with bicarbonate transporters in lamellipodia and increases cell migration via its catalytic domain. J. Biol. Chem. 2012, 287, 3392–3402. 10.1074/jbc.m111.286062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buonanno M.; Langella E.; Zambrano N.; Succoio M.; Sasso E.; Alterio V.; Di Fiore A.; Sandomenico A.; Supuran C. T.; Scaloni A.; Monti S. M.; De Simone G. Disclosing the Interaction of Carbonic Anhydrase IX with Cullin-Associated NEDD8-Dissociated Protein 1 by Molecular Modeling and Integrated Binding Measurements. ACS Chem. Biol. 2017, 12, 1460–1465. 10.1021/acschembio.7b00055. [DOI] [PubMed] [Google Scholar]
- Buanne P.; Renzone G.; Monteleone F.; Vitale M.; Monti S. M.; Sandomenico A.; Garbi C.; Montanaro D.; Accardo M.; Troncone G.; Zatovicova M.; Csaderova L.; Supuran C. T.; Pastorekova S.; Scaloni A.; De Simone G.; Zambrano N. Characterization of carbonic anhydrase IX interactome reveals proteins assisting its nuclear localization in hypoxic cells. J. Proteome Res. 2013, 12, 282–292. 10.1021/pr300565w. [DOI] [PubMed] [Google Scholar]
- Thirumurugan P.; Matosiuk D.; Jozwiak K. Click chemistry for drug development and diverse chemical-biology applications. Chem. Rev. 2013, 113, 4905–4979. 10.1021/cr200409f. [DOI] [PubMed] [Google Scholar]
- Meldal M.; Tornøe C. W. Cu-Catalyzed Azide–Alkyne Cycloaddition. Chem. Rev. 2008, 108, 2952–3015. 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]
- Musumeci F.; Schenone S.; Desogus A.; Nieddu E.; Deodato D.; Botta L. Click chemistry, a potent tool in medicinal sciences. Curr. Med. Chem. 2015, 22, 2022–2050. 10.2174/0929867322666150421110819. [DOI] [PubMed] [Google Scholar]
- Bozorov K.; Zhao J.; Aisa H. A. 1,2,3-Triazole-containing hybrids as leads in medicinal chemistry: A recent overview. Bioorg. Med. Chem. 2019, 27, 3511–3531. 10.1016/j.bmc.2019.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonandi E.; Christodoulou M. S.; Fumagalli G.; Perdicchia D.; Rastelli G.; Passarella D. The 1,2,3-triazole ring as a bioisostere in medicinal chemistry. Drug Discovery Today 2017, 22, 1572–1581. 10.1016/j.drudis.2017.05.014. [DOI] [PubMed] [Google Scholar]
- Kosiova I.; Kovackova S.; Kois P. Synthesis of coumarin-nucleoside conjugates via Huisgen 1,3-dipolar cycloaddition. Tetrahedron 2007, 63, 312–320. 10.1016/j.tet.2006.10.075. [DOI] [Google Scholar]
- Dang Thi T. A.; Kim Tuyet N. T.; Pham The C.; Thanh Nguyen H.; Ba Thi C.; Doan Duy T.; D’hooghe M.; Van Nguyen T. Synthesis and cytotoxic evaluation of novel ester-triazole-linked triterpenoid-AZT conjugates. Bioorg. Med. Chem. Lett. 2014, 24, 5190–5194. 10.1016/j.bmcl.2014.09.079. [DOI] [PubMed] [Google Scholar]
- Khalifah R. G. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J. Biol. Chem. 1971, 246, 2561–2573. [PubMed] [Google Scholar]
- Maresca A.; Temperini C.; Vu H.; Pham N. B.; Poulsen S.-A.; Scozzafava A.; Quinn R. J.; Supuran C. T. Non-Zinc Mediated Inhibition of Carbonic Anhydrases: Coumarins Are a New Class of Suicide Inhibitors#. J. Am. Chem. Soc. 2009, 131, 3057–3062. 10.1021/ja809683v. [DOI] [PubMed] [Google Scholar]
- Tars K.; Vullo D.; Kazaks A.; Leitans J.; Lends A.; Grandane A.; Zalubovskis R.; Scozzafava A.; Supuran C. T. Sulfocoumarins (1,2-benzoxathiine-2,2-dioxides): a class of potent and isoform-selective inhibitors of tumor-associated carbonic anhydrases. J. Med. Chem. 2013, 56, 293–300. 10.1021/jm301625s. [DOI] [PubMed] [Google Scholar]
- Alterio V.; Di Fiore A.; D’Ambrosio K.; Supuran C. T.; De Simone G. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms?. Chem. Rev. 2012, 112, 4421–4468. 10.1021/cr200176r. [DOI] [PubMed] [Google Scholar]
- Brown T.; Sigurdson E.; Rogatko A.; Broccoli D. Telomerase inhibition using azidothymidine in the HT-29 colon cancer cell line. Ann. Surg Oncol. 2003, 10, 910–915. 10.1245/aso.2003.03.032. [DOI] [PubMed] [Google Scholar]
- Gomez D. E.; Armando R. G.; Alonso D. F. AZT as a telomerase inhibitor. Front. Oncol. 2012, 2, 113. 10.3389/fonc.2012.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerson M.; Counter C. M.; Eaton E. N.; Ellisen L. W.; Steiner P.; Caddle S. D.; Ziaugra L.; Beijersbergen R. L.; Davidoff M. J.; Liu Q.; Bacchetti S.; Haber D. A.; Weinberg R. A. hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell 1997, 90, 785–795. 10.1016/s0092-8674(00)80538-3. [DOI] [PubMed] [Google Scholar]
- Mengual Gomez D. L.; Armando R. G.; Cerrudo C. S.; Ghiringhelli P. D.; Gomez D. E. Telomerase as a cancer target. Development of new molecules. Curr. Top. Med. Chem. 2016, 16, 2432–2440. 10.2174/1568026616666160212122425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viegas-Junior C.; Danuello A.; da Silva Bolzani V.; Barreiro E. J.; Fraga C. A. Molecular hybridization: a useful tool in the design of new drug prototypes. Curr. Med. Chem. 2007, 14, 1829–1852. 10.2174/092986707781058805. [DOI] [PubMed] [Google Scholar]
- Nocentini A.; Ferraroni M.; Carta F.; Ceruso M.; Gratteri P.; Lanzi C.; Masini E.; Supuran C. T. Benzenesulfonamides incorporating flexible triazole moieties are highly effective carbonic anhydrase inhibitors: synthesis and kinetic, crystallographic, computational, and intraocular pressure lowering investigations. J. Med. Chem. 2016, 59, 10692–10704. 10.1021/acs.jmedchem.6b01389. [DOI] [PubMed] [Google Scholar]
- Angeli A.; Tanini D.; Capperucci A.; Malevolti G.; Turco F.; Ferraroni M.; Supuran C. T. Synthesis of different thio-scaffolds bearing sulfonamide with subnanomolar carbonic anhydrase II and IX inhibitory properties and X-ray investigations for their inhibitory mechanism. Bioorg. Chem. 2018, 81, 642–648. 10.1016/j.bioorg.2018.09.028. [DOI] [PubMed] [Google Scholar]
- Nocentini A.; Vullo D.; Bartolucci G.; Supuran C. T. N-Nitrosulfonamides: a new chemotype for carbonic anhydrase inhibition. Bioorg. Med. Chem. 2016, 24, 3612–3617. 10.1016/j.bmc.2016.05.072. [DOI] [PubMed] [Google Scholar]
- Carta F.; Di Cesare Mannelli L.; Pinard M.; Ghelardini C.; Scozzafava A.; McKenna R.; Supuran C. T. A class of sulfonamide carbonic anhydrase inhibitors with neuropathic pain modulating effects. Bioorg. Med. Chem. 2015, 23, 1828–1840. 10.1016/j.bmc.2015.02.027. [DOI] [PubMed] [Google Scholar]
- Babich J. W.; Zimmerman C.; Joyal J.; Lu S.; Hilliger S.; Maresca K. P.; Sheem J. M.. Metal Complexes of Poly(carboxyl)amine-Containing Ligands Having an Affinity for Carbonic Anhydrase IX. EP 2800471 A4, 2015.
- Wilkinson B. L.; Bornaghi L. F.; Houston T. A.; Innocenti A.; Supuran C. T.; Poulsen S.-A. A Novel Class of Carbonic Anhydrase Inhibitors: Glycoconjugate Benzene Sulfonamides Prepared by “Click-Tailing”. J. Med. Chem. 2006, 49, 6539–6548. 10.1021/jm060967z. [DOI] [PubMed] [Google Scholar]
- Berrino E.; Milazzo L.; Micheli L.; Vullo D.; Angeli A.; Bozdag M.; Nocentini A.; Menicatti M.; Bartolucci G.; di Cesare Mannelli L.; Ghelardini C.; Supuran C. T.; Carta F. Synthesis and evaluation of carbonic anhydrase inhibitors with carbon monoxide releasing properties for the management of rheumatoid arthritis. J. Med. Chem. 2019, 62, 7233–7249. 10.1021/acs.jmedchem.9b00845. [DOI] [PubMed] [Google Scholar]
- Supuran C. T.; Dedhar S.; Carta F.. Carbonic Anhydrase Inhibitors With Antimetastatic Activity. WO 2012070024 A1, 2012.
- Nocentini A.; Carta F.; Ceruso M.; Bartolucci G.; Supuran C. T. Click-tailed coumarins with potent and selective inhibitory action against the tumor-associated carbonic anhydrases IX and XII. Bioorg. Med. Chem. 2015, 23, 6955–6966. 10.1016/j.bmc.2015.09.041. [DOI] [PubMed] [Google Scholar]
- Leslie A. G. W.; Powell H. R.. Processing Diffraction Data with Mosflm. In Evolving Methods for Macromolecular Crystallography; Read R. J., Sussman J. L., Eds.; NATO Science Series; Springer: Dordrecht, 2007; pp 41–51. [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1997, 53, 240–255. 10.1107/s0907444996012255. [DOI] [PubMed] [Google Scholar]
- Lebedev A. A.; Young P.; Isupov M. N.; Moroz O. V.; Vagin A. A.; Murshudov G. N. JLigand: a graphical tool for the CCP4 template-restraint library. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2012, 68, 431–440. 10.1107/s090744491200251x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. 10.1107/s0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamzin V. S.; Perrakis A.; Wilson K. S.. The ARP/wARP Suite for Automated Construction and Refinement of Protein Models. In International Tables for Crystallography, 1st ed.; Rossmann M. G., Arnold E., Eds.; Kluwer Academic: Dordrecht, 2001; p 311. [Google Scholar]
- Lovell S. C.; Davis I. W.; Arendall W. B. III; de Bakker P. I. W.; Word J. M.; Prisant M. G.; Richardson J. S.; Richardson D. C. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins 2003, 50, 437–450. 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Zhdanov D. D.; Vasina D. A.; Grachev V. A.; Orlova E. V.; Orlova V. S.; Pokrovskaya M. V.; Alexandrova S. S.; Sokolov N. N. Alternative splicing of telomerase catalytic subunit hTERT generated by apoptotic endonuclease EndoG induces human CD4 + T cell death. Eur. J. Cell Biol. 2017, 96, 653–664. 10.1016/j.ejcb.2017.08.004. [DOI] [PubMed] [Google Scholar]
- Kovalenko N. A.; Zhdanov D. D.; Bibikova M. V.; Gotovtseva V. Y. The influence of compound aITEL1296 on telomerase activity and growth of cancer cells. Biomed. Khim. 2011, 57, 501–510. 10.18097/pbmc20115705501. [DOI] [PubMed] [Google Scholar]
- Hou M.; Xu D.; Björkholm M.; Gruber A. Real-Time Quantitative Telomeric Repeat Amplification Protocol Assay for the Detection of Telomerase Activity2. Clin. Chem. 2001, 47, 519–524. 10.1093/clinchem/47.3.519. [DOI] [PubMed] [Google Scholar]
- Vasina D. A.; Zhdanov D. D.; Orlova E. V.; Orlova V. S.; Pokrovskaya M. V.; Aleksandrova S. S.; Sokolov N. N. Apoptotic endonuclease EndoG inhibits telomerase activity and induces malignant transformation of human CD4+ T cells. Biochemistry 2017, 82, 24–37. 10.1134/s0006297917010035. [DOI] [PubMed] [Google Scholar]
- Sebaugh J. L. Guidelines for accurate EC50/IC50 estimation. Pharm. Stat. 2011, 10, 128–134. 10.1002/pst.426. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.