

Abstract
In this study, we determined the crystal structure of an engineered human adenosine A2A receptor bound to a partial agonist and compared it to structures cocrystallized with either a full agonist or an antagonist/inverse agonist. The interaction between the partial agonist, belonging to a class of dicyanopyridines, and amino acids in the ligand binding pocket inspired us to develop a small library of derivatives and assess their affinity in radioligand binding studies and potency and intrinsic activity in a functional, label-free, intact cell assay. It appeared that some of the derivatives retained the partial agonist profile, whereas other ligands turned into inverse agonists. We rationalized this remarkable behavior with additional computational docking studies.
Introduction
Human adenosine receptors, of which there are four subtypes (hA1AR, hA2AAR, hA2BAR, and hA3AR), belong to the large superfamily of G protein-coupled receptors (GPCRs).1 The hA2AAR was one of the first GPCRs crystallized;2,3 multiple structures are available now, both with antagonists/inverse agonists or (full) agonists bound.4 Typically and unlike prototypic antagonists such as ZM241385, agonists such as adenosine itself bear a ribose moiety, which is deemed critical for agonist activity (Figure 1).5 The latter dogma was challenged by a series of dicyanopyridines without a ribose group, discovered, and further developed in the Bayer laboratories as partial agonists predominantly for the hA1AR.6,7 A recent phase 2b clinical trial with one of these compounds, neladenoson, failed to meet the primary endpoint in patients with heart failure, however.8 Depending on the substitution pattern of these pyridine derivatives, affinity for A2A and A2B receptors can be achieved as well. Colotta and co-workers synthesized a number of derivatives with high affinity for hA2BAR,9 while BAY 60-6583 was reported to be a partial agonist for A2BAR.10 We ourselves synthesized 8 (LUF5833) and LUF5834 (Figure 1) with appreciable affinity for the adenosine A2A receptor and partial agonistic activity on this receptor subtype.11,12
Figure 1.

Chemical structures of adenosine, the nonriboside partial agonists 8 (LUF5833) and LUF5834, inverse agonist/antagonist ZM241385, and the derivatives of 8 synthesized in this study (1–6).
In this study, we present the crystal structure of 8 bound to an engineered construct of the adenosine A2A receptor and compare this structure with those of antagonist- and agonist-bound receptors. We then synthesized a series of derivatives of 8 (Figure 1) inspired by the compound’s interaction with specific amino acids in the binding pocket of the receptor. These novel compounds were evaluated in radioligand binding studies to assess their affinity for the (four) adenosine receptor subtypes. We also tested these compounds in a label-free impedance-based assay to determine their intrinsic activity on the A2A receptor, revealing a huge variation in efficacy, in between agonism and inverse agonism. Further docking studies shed light on the atomic/structural features responsible for this behavior.
Results
Structure of the Compound 8–Receptor Complex
To obtain crystals of the compound 8–receptor complex, we used an existing thermostabilized receptor construct combined with a bRIL fusion protein, coined A2A-StaR2-bRIL,13 and a previously described approach in which crystals of A2A-StaR2-bRIL were formed with theophylline.14 After soaking the crystals with 8, they were exposed to synchrotron X-ray radiation and diffraction data collected to 3.1 Å resolution (Figure 2A and Figure S1A). Following initial rounds of refinement using the receptor only, both residual 2Fo – Fc and Fo – Fc electron density maps (Figure S1B) revealed clearly the entire pose of 8 and most surrounding side chains lining the binding site.
Figure 2.

(A) Crystal structure of stabilized hA2AAR-STaR2-bRIL in complex with partial agonist 8 (color-coded according to protein domains; TM: transmembrane domain; ECL: extracellular loop; Nt: N-terminus; Ct: C-terminus; bRIL structure omitted for clarity; compound 8 within TM cavity); (B) ligand binding site of 8 (LUF5833) surrounded by amino acids (three-letter code) within 4 Å distance; (C) two-dimensional representation of interacting amino acids and 8 docked into the hA2AAR crystal structure with all nine thermostabilizing mutations reverted to wild-type hA2AAR, showing the ligand binding cavity within the 4 Å distance of 8 (green circle: hydrophobic; red circle: negatively charged; blue circle: polar; gray circle: water; gradient gray circle: solvent exposed).
Compound 8 adopts an extended pose in the orthosteric binding site of hA2AAR, engaging many of the residues previously described as interacting with agonists and antagonists13 (Figure 2B). The imidazole substituent lies nearest the extracellular face in a pocket lined by the side chains of Leu2677.32, Met2707.35, Tyr2717.36, and Ile2747.39 (superscript numbering according to Ballesteros and Weinstein15). The pyridine core is positioned similarly to the adenine moiety in NECA, lying between Ile2747.39 and Phe1685.29, and the phenyl group lies deepest in the receptor, similar to the ribose of adenosine, contacting Trp246 and forming a π–π interaction with Phe1685.29. One cyano substituent forms a hydrogen bond with Asn2536.55 and the exocyclic amine interacts with Asn2536.55 and Glu169EL2.
Among the 19 amino acids close (<4 Å) to 8 (Figure 2C), Ser2777.42 (Ala in A2A-StaR2-bRIL; see Discussion and Conclusions) and His2787.43 have been found crucial for full agonist binding.16,17 For a further analysis of the latter, we compared this structure with two reference A2A receptor structures, one antagonist/inverse agonist (ZM241385)-bound (PDB ID: 5IU4)13 and the other agonist (NECA)-bound (PDB ID: 2YDV)18 (Figures S2 and S3). The overall compound 8 and ZM241385 receptor structures were very similar (protein–protein RMSD, 0.65 Å), not unexpectedly since the same receptor construct was used in both. However, the amino acids making up the binding pocket of ZM241385 do not include “agonistic” Ser2777.42 and His2787.43, validating the antagonistic nature of ZM241385 (Figures S2A and S3A). Contrarily, the binding pocket around NECA’s ribose moiety does include these two amino acids next to, e.g., Thr883.36, another amino acid known to be involved in full agonist activation16,19 (Figures 2B and 3B). Although far from being conclusive on the basis of 3D architecture alone, this comparison between the three receptor structures suggests that the binding pocket of 8 shares characteristics of both agonist- and antagonist-occupied receptors. Hence, we decided to synthesize six derivatives of 8 to shed further light on the compound’s agonistic/antagonistic behavior.
Synthesis of Compound 8 Derivatives
In these derivatives, the cyano groups and the number of nitrogen atoms in the core were varied (compounds 1–6, Figure 1). Due to the targeted change in the core scaffold, a different synthetic pathway was used for every analogue of 8. The synthesis routes toward the pyrimidine and triazine derivatives are shown in Scheme 1.
Scheme 1. Synthesis Route toward Pyrimidine and Triazine Derivatives 1–3.
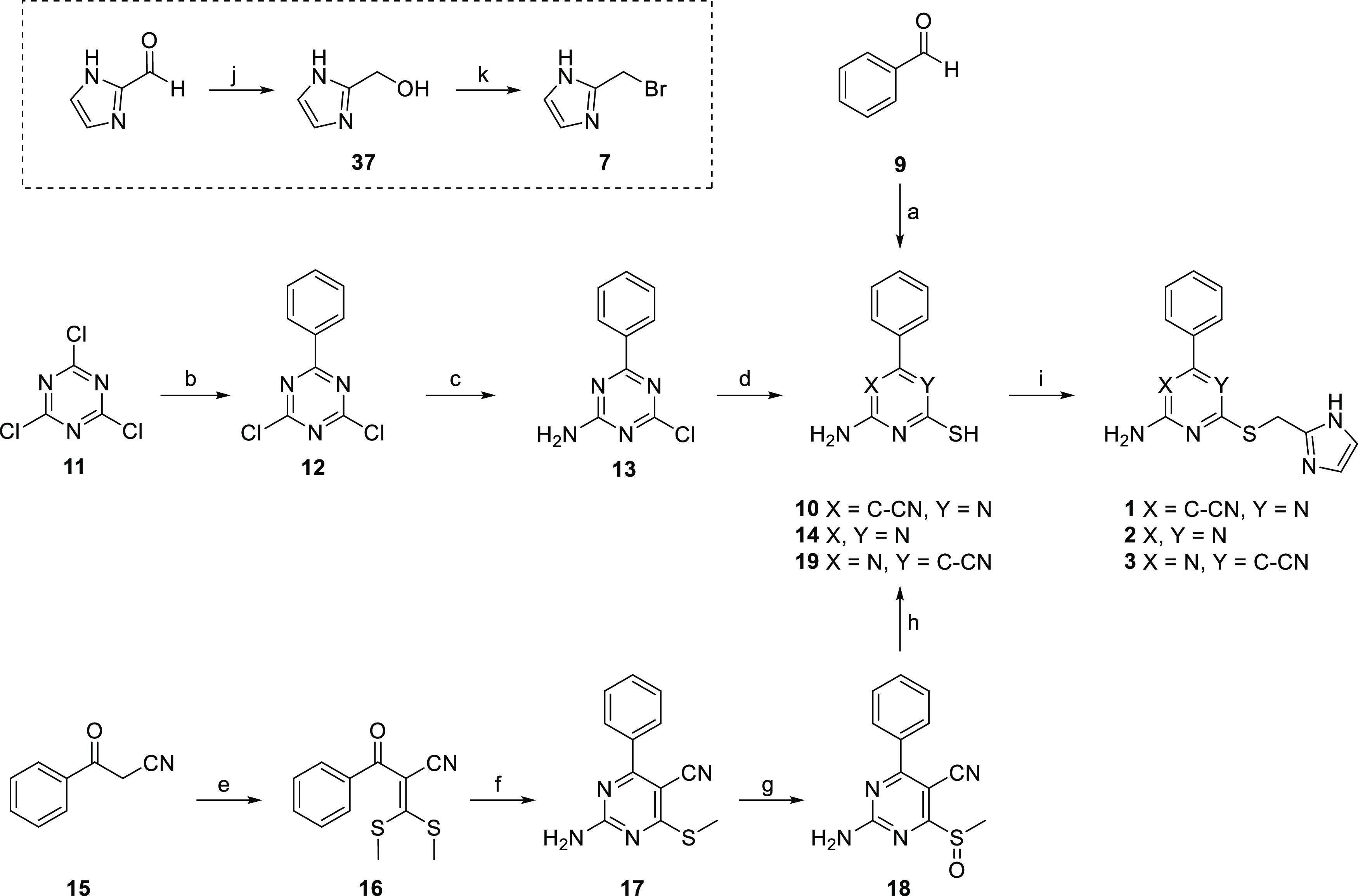
Reagents and conditions: (a) malononitrile, thiourea, K2CO3, EtOH, reflux, 21% (for 10); (b) R-phenylmagnesium bromide, dry THF, N2, rt, 52%; (c) NH3 (25%) in H2O, CH2Cl2, rt, 66%; (d) Na2S·9H2O, DMF, 80 °C, 28% (for 14); (e) NaH, carbon disulfide, iodomethane, dry DMSO, N2, rt, 82%; (f) guanidine hydrochloride, TEA, DMF, reflux, 42%; (g) mCPBA,CH2Cl2, rt, 66%; (h) potassium thioacetate, DMF, rt, 53% (for 19); (i) 7, Na2CO3, DMF, rt, 3–46%; (j) NaBH4, absolute EtOH, rt, 75%; (k) 33% HBr in CH3COOH, reflux, 65%.
Compound 1 was synthesized starting with a multicomponent reaction between benzaldehyde (9), malononitrile, and thiourea, forming intermediate 10.20 Then, 2-bromomethylimidazole (7) was synthesized by reducing the commercially available 2-imidazolecarboxaldehyde with sodium borohydride in absolute ethanol. Subsequent treatment with hydrobromic acid (33% in acetic acid) furnished the commonly used alkylating agent 7.11,21 Alkylation of intermediate 10 with agent 7 gave cyanopyrimidine 1 in a satisfactory 40% yield. The synthesis of 2 was started from the commercially available cyanuric chloride 11. Grignard alkylation with phenylmagnesium bromide provided 12 in a relatively good yield of 52%. Amination was then performed using 1.0 equiv of concentrated aqueous ammonia to give crude 13.22 This was treated with an excess of sodium sulfide nonahydrate to furnish the crystalline intermediate 14, which was alkylated using 7 to give the final compound 2. Although compounds 1 and 3 are both cyanopyrimidine derivatives, the synthesis of compound 3, in which the cyano group has an opposed topology, was slightly more laborious. Commercially available benzoyl acetonitrile (15) was treated with a strong base to form the enolate, which was reacted with carbon disulfide. The resulting dithiocarboxylic acid anions were reacted with iodomethane to form ketene dithioacetal 16. The following cyclization, using guanidine hydrochloride and an excess base, provided the cyanopyrimidine scaffold.23 Subsequent oxidation of the thioether gave sulfone 18, which was subjected to a substitution-hydrolysis sequence using potassium thioacetate followed by a strong base.24 The resulting free thiol (19) was then alkylated using 7 to provide the final compound 3 with a yield of 46%.
The synthetic routes toward the pyridine derivatives are shown in Scheme 2. The synthesis of compound 4 started with a Knoevenagel condensation between acetophenone and malononitrile.25 The thus formed intermediate 21 was reacted with dimethyl cyanocarbonimidodithioate to establish the cyanopyridine core.26 Similar to the synthesis route of compound 3, treatment with meta-chloroperbenzoic acid (mCPBA) provided the sulfone (23), which was converted to free thiol 24. Alkylation of the free thiol using 7 gave the final compound 4. Similar to the synthesis of compound 2, the synthesis of compound 5 started from a triple-halogen-substituted heteroaromatic system. Commercially available 2,6-dichloro-4-iodopyridine (25) was selectively alkylated on the 4-position using a Suzuki cross-coupling reaction to give intermediate 26 in quantitative yield.27 Subsequent sequential nucleophilic aromatic substitution of the two chloro-groups using equimolar tert-butylthiol and tert-butyl carbamate, respectively, gave intermediate 28.28,29 Treatment with trifluoroacetic acid (TFA), followed by treatment with conc. HCl, yielded the deprotected intermediate 30 in two steps. It was later found that this deprotection sequence could also be performed in one step by directly using hydrochloric acid. The final compound 5 was obtained in a yield of 33% by alkylation of 30 with 7 using the same procedure as mentioned before. Last, toward compound 6, commercially available ethylbenzoyl acetate (31) was reacted with 2-cyanoacetamide to give the cyanopyridine intermediate 32, which was subjected to double chlorination using phosphorus oxychloride to yield the asymmetric cyanopyridine dichloride 33.30,31 In an initial attempt to directly substitute one of the chlorides, tert-butylthiol was added to intermediate 33. However, even after varying the reaction temperature and substitution on the thiol, inseparable mixtures of 2-(tert-alkylthio)-6-chloro-substituted (desired compound), 6-(tert-alkylthio)-2-chloro-substituted, and disubstituted 2,6-bis(tert-alkylthio)-4-phenylnicotinonitrile were obtained. Instead, Buchwald–Hartwig amination using tert-butyl carbamate was attempted. Gratifyingly, using this procedure, intermediate 34 was obtained with the correct substitution and as a single product. Subsequent addition of tert-butylthiol followed by one-step deprotection of both protective groups using hydrochloric acid gave intermediate 36, which was alkylated using the procedure mentioned previously to produce the final compound 6 in 8% yield.
Scheme 2. Synthesis Route toward Pyridine Derivatives 4–6(25).
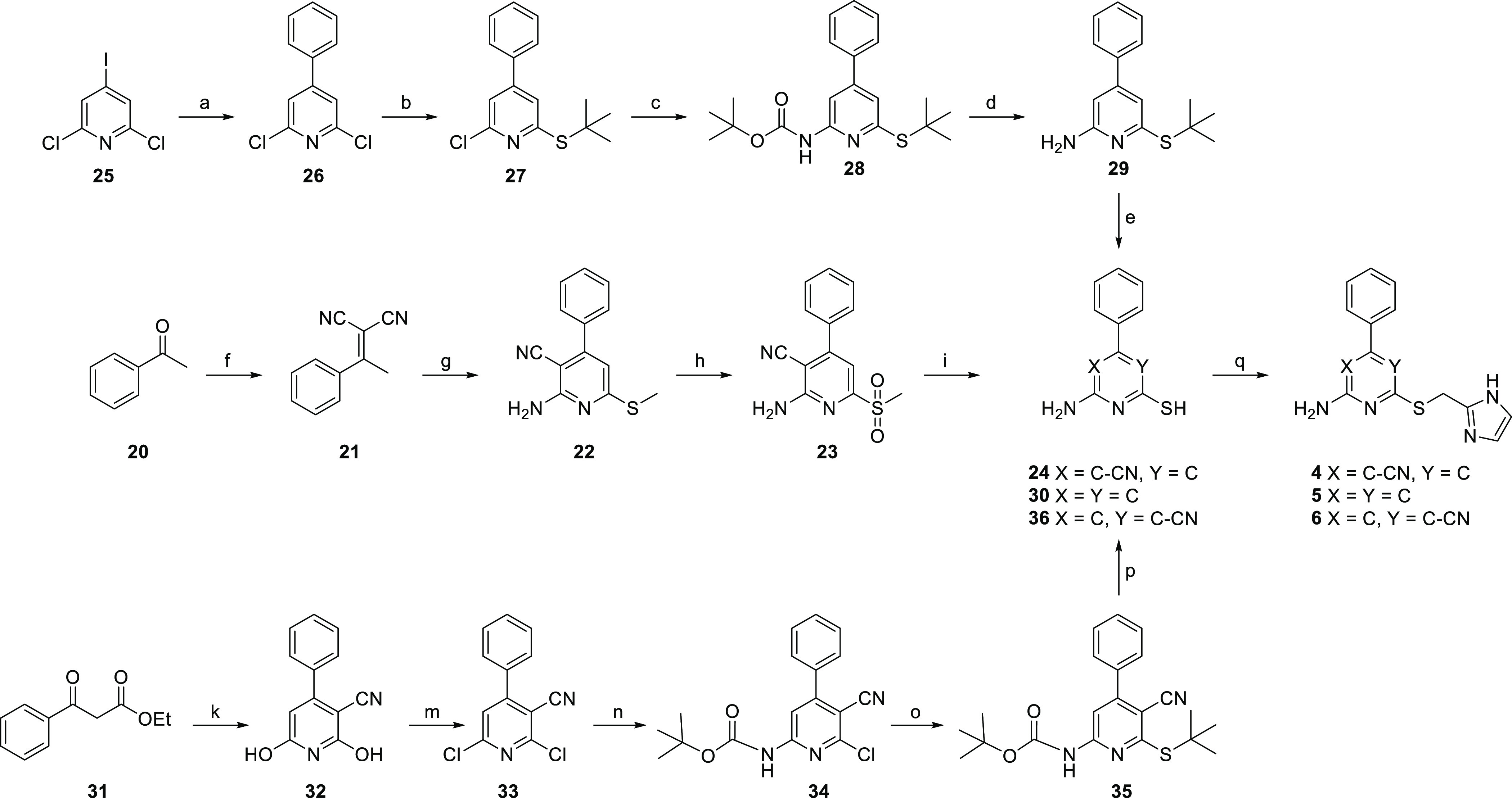
Reagents and conditions: (a) phenylboronic acid pinacol esther, Pd(PPh3)Cl2, Na2CO3, H2O/MeCN, N2, 70 °C, 92%; (b) 2-methyl-propanethiol, Cs2CO3, DMF, 80 °C, quantitative yield; (c) Pd(OAc)2, Xantphos, Cs2CO3, t-butyl carbamate, dry 1,4-dioxane, N2, 110 °C, 27%; (d) TFA, CH2Cl2, reflux, 75%; (e) 37% HCl, 100 °C; (f) malononitrile, ammonium acetate, toluene, reflux in Dean–Stark apparatus, 70%; (g) (i) dimethyl cyanocarbonimidodithioate, K2CO3, DMF, rt, (ii) piperidine, 80 °C, 62%; (h) mCPBA,CH2Cl2, rt, 56%; (i) potassium thioacetate, DMF, rt, 61% (for 24); (k) 2-cyanoacetamide, KOH, EtOH, reflux, 34%; (m) POCl3 in an autoclave, 180 °C, 70%; (n) Pd(OAc)2, Xantphos, Cs2CO3, t-butyl carbamate, dry 1,4-dioxane, N2, 40 °C, 33%; (o) 2-methyl-propanethiol, Cs2CO3, DMF, 90 °C, 89%; (p) 37% HCl, 100 °C, 37% (for 36); (q) 7, NaHCO3, DMF, rt, 8–57%.
Pharmacological Assessment of the Library of Compound 8 Derivatives
We first assessed the compounds’ affinities for all adenosine receptor subtypes in radiolabeled antagonist10 binding studies on cell membranes expressing the individual receptors (A1: [3H]DPCPX; A2A: [3H]ZM241385; A2B: [3H]PSB-603; A3: [3H]PSB-11). Single-point displacement assays at 1 μM on all four human adenosine receptor subtypes were performed as a preliminary assay for all compounds. Only compounds that showed greater than 50% radioligand displacement were evaluated in full-range concentration-dependent displacement assays to determine their affinities (Table 1 and Figure S4).
Table 1. Affinities of 8 and Derivatives (1–6) in Radioligand Binding Assays on the Human Adenosine Receptors.
| pKi or % displacementa |
||||
|---|---|---|---|---|
| compound | A1ARb | A2AARc | A2BARd | A3ARe |
| 8 | 8.52 ± 0.04 | 8.13 ± 0.05 | 7.40 ± 0.05 | 7.38 ± 0.01 |
| 1 | 7.07 ± 0.01 | 7.17 ± 0.08 | 51%f | 6.46 ± 0.06 |
| 2 | 6.54 ± 0.05 | 6.25 ± 0.08 | 2%f | 5.42 ± 0.06 |
| 3 | 7.16 ± 0.02 | 8.00 ± 0.05 | 50%f | 5.94 ± 0.05 |
| 4 | 6.38 ± 0.06 | 6.42 ± 0.01 | 6.43 ± 0.03 | 5.57 ± 0.05 |
| 5 | 12%g | –17%g | –12%f | 46%g |
| 6 | 6.95 ± 0.08 | 5.99 ± 0.12 | 37%f | 5.87 ± 0.10 |
Data are expressed as mean ± SEM of three separate experiments each performed in duplicate, unless indicated otherwise.
Displacement of [3H]DPCPX binding in CHO cells expressing hA1AR.
Displacement of [3H]ZM241385 binding in HEK293 cells expressing hA2AAR.
Displacement of [3H]PSB-603 binding in CHO cells expressing hA2BAR.
Displacement of [3H]PSB-11 binding in CHO cells expressing hA3AR.
Percent displacement (n = 2) of specific [3H]PSB-603 binding in CHO cells expressing hA2BAR at 1 μM ligand concentrations.
Percent displacement (n = 2) of specific radioligand binding at 10 μM ligand concentrations.
Of all compounds tested, the parent compound 8 displayed the highest affinity toward all human adenosine receptor subtypes, being slightly selective for the hA1AR with a Ki value of 3 nM. Modification of 8 by changing one cyano group at the thiol side for a nitrogen atom in the pyridine core yielded compound 1 with a significant, 10–30 fold, decreased affinity toward all adenosine receptor subtypes. Changing the other cyano group in 8 in the same way at the other, “left-hand” side provided compound 3 with similar affinity to 8 on hA2AAR (Ki = 10 nM), while it showed approximately 25-fold lower affinity at hA1AR and hA3AR, rendering this compound the more selective derivative at hA2AR. The non-nitrile triazine compound 2 displayed lower affinity at all adenosine receptor subtypes with minimal affinity toward hA2BAR (only 2% displacement of radioligand binding). The mono-cyano pyridine derivative 4 with structural resemblance to 1 displayed lower affinity than 1 on all adenosine receptor subtypes except hA2BAR, where it had a Ki value of 370 nM, rendering it the more nonselective derivative over all four adenosine receptor subtypes. The mono-cyano pyridine derivative 6 with the same cyano position as in pyrimidine 3 showed a significant, 100-fold lower affinity to hA2AAR compared to 3, although it had similar affinity to the other subtypes. Pyridine 5, lacking both cyano groups, was almost devoid of affinity at all adenosine receptor subtypes, as it showed no or negligible displacement of radioligand binding in the preliminary single-concentration assays (1 μM). Apparently, the number of nitrogen atoms in or around the scaffold is an important determinant of receptor binding, especially for hA2AAR.
Next, we examined the compounds’ efficacy/intrinsic activity in a label-free impedance-based assay on cells expressing hA2AAR. As the parent compound 8 had been identified as a partial agonist,11,12 we sought to characterize the new derivatives for their functional behavior along the agonist/antagonist spectrum. Intrinsic activities of 8 and its derivatives at hA2AAR were assessed using a label-free whole-cell assay with a real-time cell analysis (RTCA) system by measuring the flow of electrons transmitted between two gold electrodes at the bottom of a well in a 96-well plate. Upon receptor activation, the HEK293hA2AR cell morphology changes and covers more of the electrodes, which impedes the electron flow. The impedance produced is displayed as a unit-less number, the so-called cell index (CI).32 A reference full agonist of hA2AAR, ribose-containing CGS21680, was used to define the highest CI observed, and the peak values obtained were used to determine EC50 values. Concentration–response curves of CGS21680 and the compounds in the library are displayed in Figure 3. In this study, the parent compound 8 showed a lower potency than CGS21680 and acted as a partial agonist of the receptor, with an intrinsic activity (Emax value) of 66 ± 5% (Table 2). A similar observation had been reported in a previous study by Beukers et al.,11 focusing on cAMP production as a signaling readout.
Figure 3.

Concentration–response curves for CGS21680 and nonriboside compounds at HEK293hA2AR derived from the peak analysis of cell index (CI) changes obtained in the xCELLigence RTCA system. The cellular responses were normalized and shown as % of the maximal CI by 1 μM CGS21680. Representative graphs from one experiment performed in duplicate.
Table 2. Compound Potency and Intrinsic Activity Derived from the Label-Free Whole-Cell Assay Performed on HEK293-hA2AR Cellsa.
| compound | pEC50 | Emax, % |
|---|---|---|
| CGS21680 | 8.85 ± 0.08 | 100 ± 2 |
| 8 | 8.01 ± 0.22 | 66 ± 5 |
| 1 | 6.57 ± 0.37 | 41 ± 5 |
| 2 | 6.09 ± 0.23 | –48 ± 5 |
| 3 | 7.35 ± 0.28 | –50 ± 5 |
| 4 | 5.78 ± 0.38 | 37 ± 7 |
| 6 | N.D. | N.D. |
Data are shown as mean ± S.E.M. of three independent experiments performed in duplicate. Log potency (pEC50) and efficacy (Emax) were calculated from concentration–response curves derived from the peak analysis of CI changes. N.D., not determined.
Various responses were shown by the nonriboside derivatives. The pyrimidine compound 1 behaved as a partial agonist as well (Emax = 41 ± 5%), although it had lower potency than 8 (pEC50 of 6.57 ± 0.37 vs 8.01 ± 0.22). The pyridine derivative 4 was a partial agonist too (Emax = 37 ± 7%) but with the lowest measurable potency in the series (pEC50 = 5.78 ± 0.38). Remarkably, compound 3, the derivative with the highest selectivity for hA2AAR, had an opposite activity compared to 8. Figure 3 shows that this cyanopyrimidine compound acted as an inverse agonist at the receptor (Emax = −50 ± 5%) with a potency quite comparable to 8 (pEC50 = 7.35 ± 0.28). The absence of both cyano groups in the triazine compound 2 also led to inverse agonism at hA2AAR with similar efficacy (−48 ± 5%) to 3, albeit with significantly lower potency (pEC50 = 6.09 ± 0.23 vs 7.35 ± 0.28). Compound 6 did not show any measurable degree of intrinsic activity, as it neither activated nor inactivated the receptor (Figure 3). These results show that modification/deletion of the cyano groups alters the functional characteristics of the compounds. Keeping the cyano group at the amino, “left-hand” side maintained agonism, while the absence of this group abolished this feature, resulting in inverse agonism for 2 and 3 and, possibly, neutral antagonism for 6.
Molecular Modeling
Intrigued by the findings in the pharmacological experiments, we decided to perform induced-fit docking studies of compounds 1 and 3, the two compounds with the most obvious differences in receptor activation patterns while having appreciable affinity for the adenosine A2A receptor. To prepare the receptor structure for this analysis, the bRIL fusion partner was removed from the crystal structure and the third intracellular loop (ICL3) was implemented again.33 Mutated amino acids used to thermostabilize the structure in the crystallization process, such as M1P, A54L, T88A, R107A, K122A, N154A, L235A, and V239A, were changed back to the wild-type amino acids. Missing water molecules were retrieved from a previous high-resolution crystal structure (PDB ID: 4EIY)2 to provide stability of the receptor–ligand interaction in the docking simulation. First, as a control experiment, compound 8 was docked into this wild-type hA2AAR model. Induced flexible docking allowed the imidazole moiety to adopt slightly different positions, not surprisingly in view of its orientation toward the more flexible extracellular loops. The dicyanopyridine scaffold, however, did hardly move (Figure S5). Next, compounds 1 and 3 were flexibly docked34 into the binding pocket with the binding pose of 8 as a reference (Figures 4–6, respectively). The partial agonist 1 showed a similar binding pose as 8 after convergence of the docking procedure, with two hydrogen bonds with Asn2536.55, one hydrogen bond with Glu169ECL2, a π–π stacking interaction with Phe1685.29, and its phenyl moiety surrounded by Ser2777.42 and His2787.43 (Figures 4 and 6). The inverse agonist 3 took a slightly different pose due to the absence of the cyano substituent on the “left-hand” side of the molecule. The ligand moved closer to Asn2536.55 as to maintain hydrogen bonding, now to the nitrogen atom in the ligand’s core. As a result, it also forms two hydrogen bonds with Asn2536.55, one hydrogen bond with Glu169ECL2, and a π–π stacking interaction between its pyrimidine core and Phe1685.29. However, this movement also caused a shift of the phenyl moiety away from Ser2777.42 and His2787.43, bringing this moiety closer to His2506.52 (Figures 5 and 6). A similar alignment is also present in the binding pocket of antagonist/inverse agonist ZM241385. Its furan ring forms a π–π stacking interaction with His2506.52 (Figure S2A).
Figure 4.
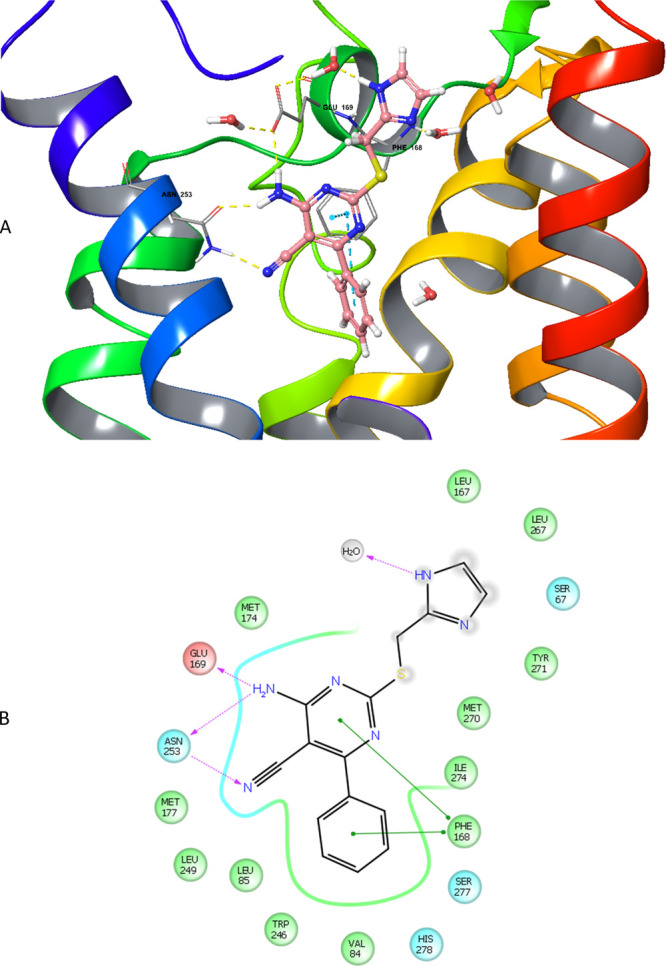
(A) Proposed binding mode of compound 1 in the hA2AAR crystal structure with all nine thermostabilizing mutations reverted to wild-type hA2AAR, obtained by induced-fit docking. It shows the interactions formed between 1 and the receptor, in particular three hydrogen bond interactions with Asn2536.55 and Glu1695.30, and two π–π stacking interactions with Phe1685.29. A part of helix 7 was omitted for better visualization. (B) Two-dimensional representation of the interaction between 1 and hA2AAR in the ligand binding cavity within a 4 Å distance (green circle: hydrophobic; red circle: negatively charged; blue circle: polar; gray circle: water; gradient gray circle: solvent exposed). The phenyl moiety is surrounded by Ser2777.42 and His2787.43, key residues in receptor activation.
Figure 6.

Superimposition of compounds 1 (orange), 3 (pink), and 8 (green) in the hA2AAR crystal structure with all nine thermostabilizing mutations reverted to wild-type hA2AAR, obtained by induced-fit docking. For reasons of clarity, only five helices and the amino acid Asn2536.55 are shown.
Figure 5.
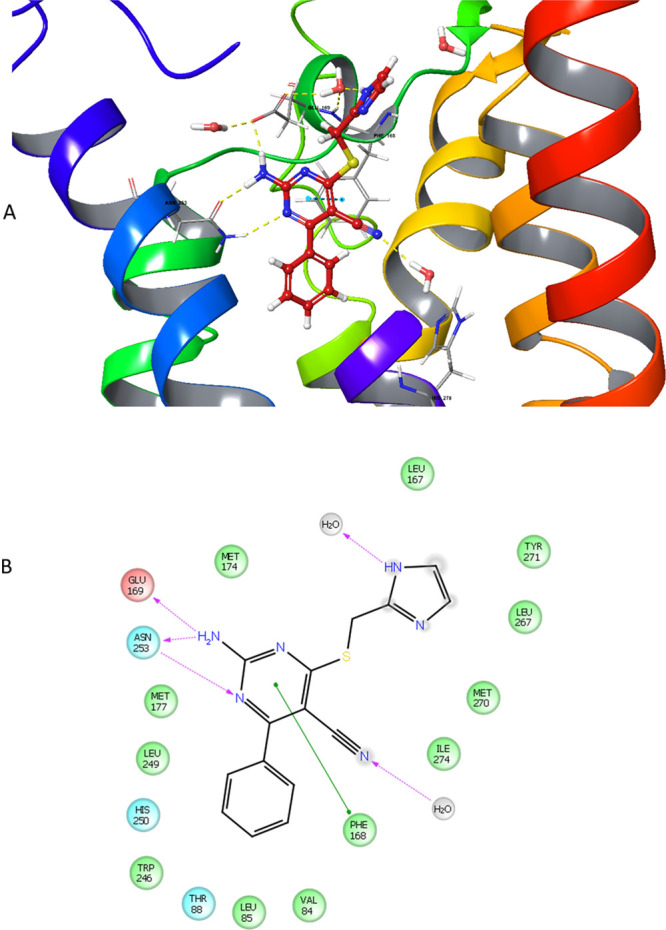
(A) Proposed binding mode of compound 3 in the hA2AAR crystal structure with all nine thermostabilizing mutations reverted to wild-type hA2AAR, obtained by induced-fit docking. Compared to Figure 4 (compound 1), the hydrogen bond interaction between the nitrogen atom in the pyrimidine ring and Asn2536.55 causes a shift of 3’s position in the binding cavity. A part of helix 7 was omitted for better visualization. (B) Two-dimensional representation of the interaction between 3 and hA2AAR in the ligand binding cavity within a 4 Å distance (green circle: hydrophobic; red circle: negatively charged; blue circle: polar; gray circle: water; gradient gray circle: solvent exposed). Most key interactions for binding to the receptor are maintained, but the phenyl moiety is moved away from Ser2777.42 and His2787.43.
Discussion and Conclusions
The ligand binding pocket of 8 encompasses amino acids that have been subjected to a mutagenesis study by Lane et al. in which the reference agonist CGS21680 and LUF5834 (Figure 1), a close analogue of 8, were compared.16 In fact, within the scope of the present study, we also attempted to obtain a receptor crystal structure with LUF5834 but were unsuccessful. The authors performed cAMP measurements to demonstrate that the potencies of both CGS21680 and LUF5834 were dramatically (>100-fold) reduced in the Phe1685.29 alanine mutant, in accordance with the pivotal role this amino acid plays in coordinating the scaffold and phenyl substituent of 8 in the crystal structure. The Asn2536.55 alanine mutation negatively affected CGS2160 potency and LUF5834 intrinsic activity, with the latter losing its partial agonist activity and turning into an antagonist. The Ala mutant of Glu169EL2 on the contrary slightly increased the potency and intrinsic activity of LUF5834 with no effect on CGS21680. This amino acid has been shown to interact with a histidine (His2646.66) residue to form a lid above the binding pocket of the receptor, affecting the residence time of AR ligands.13 Alanine mutation of Ser2777.42, as present in the crystal structure, reduced the potency of CGS21680 by two orders of magnitude, while LUF5834’s potency and affinity increased somewhat and the compound turned into a full agonist.
Rather than reiterating this mutagenesis study to include 8, we decided to synthesize and test six derivatives of the compound for a further analysis of its agonistic characteristics. The decoration of the scaffold was altered in a one-by-one step fashion to compare the effect of relatively minor changes on both potency and intrinsic activity. These structural alterations caused changes in affinity, as assessed on all four adenosine receptor subtypes through radioligand binding studies. All compounds were less active than 8, in particular compound 5, bearing the pyridine scaffold without cyano substituents. Interestingly, compound 3 was the most selective for hA2AAR; it kept its affinity on this receptor subtype, while its affinity for the other adenosine receptor subtypes dropped substantially (Table 1).
We then examined the functional characteristics of the compounds in label-free functional experiments on intact HEK293 cells overexpressing hA2AAR. We had reported previously that the endogenous expression of hA2BAR on these cells does not compromise the functional evaluation of hA2AAR occupancy.16 Hence, we were able to record the pEC50 values of all compounds in this cell system (Table 2 and Figure 4) and compared these with the radioligand binding data. The pKi and pEC50 values were strongly correlated (r2 = 0.91, P = 0.01, Figure S6) yet another indication of the unequivocal involvement of hA2AAR. It also suggests there is no or negligible receptor reserve, in line with the partial nature of the compounds’ efficacy.
We were quite surprised to record huge changes in intrinsic activity. Here, some of the compounds (2 and 3) were turned into inverse agonists instead of (partial) agonists. Only compounds 1 and 4, both retaining the cyano substituent on the “left-hand” side of the molecule, kept the partial agonist profile as in 8. Induced-fit docking studies helped interpret our pharmacological findings. Subtle ligand movements in the binding pocket carried some ligands away from “agonistic” amino acids to negate their activation potential, while other ligands kept a partial agonistic profile. These ligand movements may also be dependent on the presence of explicit water molecules. In the hA2AAR-ZM241385 crystal structure, three water molecules occupy a region next to the furan ring of ZM241385,2,3 which is taken by the riboside moiety in an hA2AAR-agonist structure.35 It is entirely plausible that 8 and derivatives take intermediate positions with varying amounts of water molecules, reflecting their antagonistic vs agonistic nature. In a previous study focusing on the hA1AR, we had synthesized a series of quite different monocyanopyrimidines but also with the cyano substituent on either the “left hand” or “right hand” side of the scaffold.36 These compounds were all inverse agonists, however, suggesting that depending on the hAR subtype, subtle changes at various parts of these molecules can alter the activation profile dramatically and differently.
In conclusion, in this study, we elucidated the structure of the adenosine A2A receptor bound to a chemically distinct partial agonist, compound 8. The interaction profile between the ligand and the receptor included amino acids that had been shown to be involved in receptor activation in earlier, e.g., mutagenesis studies. The synthesis and molecular docking of a series of derivatives of the partial agonist allowed us to further investigate the requirements for receptor activation. With this newly solved structure, we significantly expand the repertoire of A2A receptor structures, allowing a more elaborate analysis of the ligand binding pocket than in any other GPCR.
Experimental Section
Crystal Structure Determination
A2A-Star2 bRIL Construct
The A2A-StaR2 construct consists of nine thermostabilizing mutations: A54L2.52, T88A3.36, R107A3.55, K122A4.43, N154A, L202A5.63, L235A6.37, V239A6.41, and S277A7.42. The first three amino acids at the N-terminus were deleted and the protein had a C-terminal truncation after L315 where 3 additional alanines and 10 histidines were added. The apocytochrome b 562 RIL (bRIL) was inserted into the third intracellular loop (ICL3) between residues L208 and G218.
Expression
The A2A-StaR2-bRIL was expressed using the baculovirus system. Tni PRO cells were grown in suspension in flasks up to a maximum volume of 500 mL in 2 L roller bottles at 27 °C with shaking. Cells were grown in ESF921 (Expression Systems) medium supplemented with 5% (v/v) FBS and 1% (v/v) penicillin/streptomycin. Cells were infected at a density of 2.6 × 106 cells/mL with recombinant virus at an approximate multiplicity of infection of 1. Cells were harvested by centrifugation 48 h post infection.
Membranes were prepared from a pellet of 2 L cells resuspended in 40 mM Tris–HCl (pH 7.6), 1 mM EDTA, and Complete EDTA-free protease inhibitor cocktail tablets (Roche). Cells were disrupted through a microfluidizer (processor M-110L Pneumatic, Microfluidics) cooled down with ice (lysis pressure, ∼15,000 psi) and membranes were pelleted by centrifugation at 200,000g for 50 min. Membranes were washed with 40 mM Tris–HCl (pH 7.6), 1 M NaCl, and Complete EDTA-free protease inhibitor cocktail tablets and centrifuged at 200,000g for 50 min. After removal of the supernatant, membranes were resuspended in 50 mL with 40 mM Tris–HCl (pH 7.6) and Complete EDTA-free protease inhibitor cocktail tablets and frozen at −80 °C.
Purification and Lipidic Cubic Phase Crystallization
Cells from 2 L cultures were resuspended and disrupted using a microfluidizer (Processor M-110L Pneumatic, Microfluidics). Membranes pelleted by ultracentrifugation were subjected to a high-salt wash in a buffer containing 1 M NaCl before being resuspended in 50 mL of 40 mM Tris (pH 7.6) supplemented with Complete EDTA-free protease inhibitor cocktail tablets and stored at −80 °C until further use. Membranes were thawed, resuspended in a total volume of 150 mL, and incubated with 3 mM theophylline (Sigma-Aldrich) for 2 h at room temperature. Membranes were then solubilized by addition of 1.5% n-decyl-β-d-maltopyranoside (DM, Anatrace) and incubation for 2 h at 4 °C, followed by centrifugation at 145000g for 60 min to harvest a solubilized material. The solubilized material was applied to a 5 mL Ni-NTA (nickel-nitrilotriacetic acid) Superflow cartridge (Qiagen) and the protein was eluted with a buffer containing 1 mM theophylline. Collected fractions were pooled, concentrated, and applied to a Superdex200 size exclusion column (GE Healthcare). Fractions containing the protein were then concentrated to ∼35 mg/mL and subjected to an ultracentrifugation at 436,000g prior to crystallization. Protein concentrations were measured using the detergent compatible (DC) protein assay (Bio-Rad).
The A2A-StaR2-bRIL was crystallized first in complex with theophylline in the lipidic cubic phase at 20 °C. The final protein/lipid ratio was 40:60 (w/w), and 40 nL of boli was dispensed using a Mosquito LCP crystallization robot (TTP Labtech) and overlaid with 800 nL of precipitant solution. Crystals grew within 2 weeks in 0.1 M trisodium citrate (pH 5.3–5.4), 0.05 M sodium thiocyanate, 29–32% PEG400, 2% (v/v) 2,5-hexanediol, and 0.5 mM theophylline. Then, incisions were made into the Laminex cover over base wells containing crystals, and wells were flooded with 10 μL of mother liquor supplemented by compound 8 (1 mM). Flooded wells were then resealed and plates were incubated for 24 h at 20 °C. Single crystals were mounted in LithoLoops (Molecular Dimensions Ltd.) and flash-frozen in liquid nitrogen without the addition of further cryoprotectant.
Data Collection and Structure Solution
X-ray diffraction data (0.25°/frame; ∼500 frames) were collected on an Eiger 16M detector at beamline X06SA (Swiss Light Source) in a single swipe from one crystal. Integration was carried out using XDS37 and scaled using aP scale38 (Global Phasing, Cambridge, U.K.). The structure of the A2A-StaR2-bRIL complex with compound 8 was solved by molecular replacement (MR) with Phaser39 within the CCP4 interface using the previously reported A2A-StaR2-bRIL-theophylline complex structure as the search model (PDB code: 5MZJ). Model refinement was performed initially using phenix.refine40 and then using BUSTER41 (Global Phasing), including TLS refinement for two groups corresponding to the receptor and bRIL562, respectively. The crystallographic data reduction and refinement statistics are presented in Table 3.
Table 3. Data Collection and Refinement Statistics.
| A2A-LUF5833 (compound 8) | |
|---|---|
| data collection | |
| number of crystals | 1 |
| space group | C2221 |
| cell dimensions | |
| a, b, c (Å) | 39.54, 181.03, 140.85 |
| α, β, γ (°) | 90, 90, 90 |
| resolution (Å) | 43.09–3.12 (3.31–3.12)a |
| no. of reflections | 7990 (399)a |
| Rpim | 0.134 (0.934)a |
| I/σ(I) | 5.2 (1.1)a |
| CC1/2 | 0.992 (0.309)a |
| completeness (%) | |
| spherical | 84.5 (26.3)a |
| ellipsoidal | 87.2 (32.5)a |
| redundancy | 3.9 (4.0)a |
| refinement | |
| resolution (Å) | 43.09–3.12 |
| Rwork/Rfree | 0.223/0.254 |
| no. of atoms | |
| protein | 2937 |
| ligand | 24 |
| other | 69 |
| B factors | |
| protein | 77.21 |
| ligand | 62.56 |
| other | 67.69 |
| r.m.s. deviations | |
| bond lengths (Å) | 0.007 |
| bond angles (°) | 0.81 |
Values in parentheses are for the highest-resolution shell.
Chemistry
All solvents and reagents were of analytical grade and purchased from commercial sources. Demineralized water was simply referred to as H2O and used in all cases, unless stated otherwise (i.e., brine). Thin-layer chromatography (TLC), aluminum-coated Merck silica gel F254 plates were used to monitor the progression of the reactions. The 1H NMR spectra were recorded on a Bruker AV400 liquid spectrometer (1H NMR, 400 MHz; 13C NMR, 100 MHz) at ambient temperature. Chemical shifts are reported in parts per million (ppm), designated by δ and downfield to the internal standard tetramethylsilane (TMS) in CDCl3. Coupling constants are reported in Hz and designated as J. The analytical purity of the final compounds was determined by high-performance liquid chromatography (HPLC) with a Phenomenex Gemini 3 μm C18 110 Å column (50 × 4.6 mm, 3 μm), measuring UV absorbance at 254 nm. Sample preparation and HPLC method were as follows: 0.3–1.0 mg of compound was dissolved in 1 mL of a 1:1:1 mixture of MeCN/H2O/tBuOH and eluted from the column within 15 min at a flow rate of 1.3 mL/min with a three-component system of H2O/MeCN/1% TFA in H2O. The elution method was set up as follows: in a 1–4 min isocratic system of H2O/MeCN/1% TFA in H2O, 80:10:10, from the 4th min, a gradient was applied from 80:10:10 to 0:90:10 within 9 min, followed by 1 min of equilibration at 0:90:10 and 1 min at 80:10:10. All final compounds showed a single peak at the designated retention time and were thus considered at least 95% pure. Liquid chromatography–mass spectrometry (LC–MS) analyses were performed on a Thermo Finnigan Surveyor–LCQ Advantage Max LC–MS system, Shimadzu LCMS-2020 system, and Phenomenex Gemini C18 110A columns (50 × 4.6 mm, 3 μm; 50 × 3 mm, 3 μm). The sample preparation was the same as for HPLC analysis. The compounds were eluted from the column within 15 min after injection, with a three-component system of H2O/MeCN + 0.1% FA, while decreasing the polarity of the solvent mixture in time from 90:10 to 10:90. Purification by column chromatography was achieved by use of Grace Davison Davisil silica column material (LC60A 30–200 micron). Solutions were concentrated using a Heidolph Laborota W8 2000 efficient rotary evaporation apparatus. All reactions in the synthetic routes were performed without a nitrogen atmosphere, unless stated otherwise.
(1H-Imidazol-2-yl)methanol (37)11
To a suspension of NaBH4 (0.787 g, 20.8 mmol, 2.0 equiv) in absolute EtOH (15 mL) was added commercially available 1H-imidazole-2-carbaldehyde (1 g, 10.4 mmol, 1.0 equiv) portion-wise. The mixture was stirred at room temperature for 3 h. After the completion of the reaction, the mixture was quenched with water and concentrated in vacuo. The crude compound was purified by flash column chromatography (10% MeOH in CHCl3) to obtain 37 as a pale yellow solid (769 mg, 7.85 mmol, 75%). 1H NMR (400 MHz, MeOD): δ 6.97 (s, 2H, imidazoyl), 4.61 (s, 2H, CH2).
2-(Bromomethyl)-1H-imidazole hydrobromide (7)11
A suspension of 37 (0.55 g, 5.6 mmol) in HBr 33% (w/v) acetic acid solution (10 mL) was heated at 100 °C for 5 h. The mixture was concentrated in vacuo and treated with 25% PET in diethyl ether. The suspended solid was collected by filtration and dried to give 7 as a pale brown solid (885 mg, 3.66 mmol, 65%). 1H NMR (400 MHz, MeOD): δ 7.60 (s, 2H, imidazolyl), 4.82 (s, 2H, CH2). HNMR is consistent with the reported literature data.
4-Amino-2-sulfanyl-6-phenylpyrimidine-5-carbonitrile (10)20
To malononitrile (0.99 g, 15 mmol, 1.0 equiv) dissolved in ethanol (7.5 mL) were added benzaldehyde (1.528 mL, 15 mmol, 1.0 equiv), thiourea (1.14 g, 15 mmol, 1.0 equiv), and K2CO3 (2.07 g, 15 mmol, 1.0 equiv). The mixture was stirred at reflux for 5 h. Upon the completion of the reaction, the precipitate formed in the mixture was filtered and then stirred with warm water (50 °C). The filtrate was acidified with acetic acid to pH below 7. The precipitate formed was filtrated and dried in vacuo to obtain 10 as a white solid (716 mg, 3.14 mmol, 21%). 1H NMR (400 MHz, DMSO): δ 13.07 (br s, 1H, SH), 8.45 (br s, 1H, 0.5 NH2), 7.93 (br s, 1H, 0.5 NH2), 7.68 (d, J = 6.8 Hz, 2H, Ph), 7.63–7.53 (m, 3H, Ph).
2-(1H-Imidazol-2-yl)methylthio-4-amino-6-phenylpyrimidine-5-carbonitrile (1)
Alkylation was performed according to previously reported procedures.11 To a solution of free thiol 10 (0.684 g, 3 mmol, 1.0 equiv) in DMF (12 mL) were added 2-bromomethylimidazole 7 (0.483 g, 3 mmol, 1.0 equiv) and Na2CO3 (0.318 g, 3 mmol, 1.0 equiv). The reaction was stirred at 50 °C for 4 h. Water (60 mL) was added to force the crude product to precipitate, which was collected by filtration. The precipitate was washed with DMF (6 mL) and EtOAc (6 mL), and water (30 mL) was added to cause precipitation, which was filtered and dried in vacuo to obtain the desired product as a white powder (370 mg; 1.20 mmol; yield, 40%). Mp 237 °C. 1H NMR (850 MHz, DMSO): δ 11.85 (br s, 1H, NH), 8.30 (br s, 1H, imidazolyl), 7.86–7.84 (m, 2H, Ph), 7.80 (br s, 1H, imidazolyl), 7.58 (tt, J = 7.6, 1.7 Hz, 1H, Ph), 7.54 (tt, J = 6.8, 1.7 Hz, 2H, Ph), 7.10–6.80 (m, 2H, NH2), 4.43 (s, 2H, CH2). MS (ESI): calcd for C15H12N6S [M + H]+, 309.08; found: 309.00. HPLC: 3.107 min; purity: 99%.
2,4-Dichloro-6-phenyl-1,3,5-triazine (12)42
To a stirred suspension of commercially available cyanuric chloride 11 (0.5 g, 2.71 mmol, 1.0 equiv) in dry THF (20 mL) at 0 °C under a nitrogen atmosphere was added a 3 M solution of phenylmagnesium bromide (0.993 mL, 2.98 mmol, 1.1 equiv) in THF (10 mL) dropwise (over 30 min). The reaction mixture was stirred at room temperature for 6 h. Upon the completion of the reaction (monitored by TLC), the mixture was treated with 10% aqueous HCl (50 mL) and extracted with EtOAc (40 mL × 3). The combined organic layer was washed with water (30 mL), dried on MgSO4, and evaporated under reduced pressure to give the intermediate as a brown solid (318 mg, 1.42 mmol, 52%). The crude product was used for the next reaction without further purification. 1H NMR (400 MHz, CDCl3) δ 8.50 (dd, J = 8.0, 1.6 Hz, 2H, Ph), 7.66 (tt, J = 7.6, 1.2 Hz, 1H, Ph), 7.54 (t, J = 8.0 Hz, 2H, Ph).
4-Chloro-6-phenyl-1,3,5-triazine-2-amine (13)43
Aqueous ammonia (25%) was added dropwise (0.1 mL, 1.327 mmol, 1.0 equiv) to a stirred solution of 12 (0.3 g, 1.327 mmol, 1.0 equiv) in CH2Cl2 (5 mL). The resulting mixture was stirred at room temperature for 8 h. The reaction progress was monitored by TLC. The precipitate formed was filtered, rinsed with CH2Cl2 (30 mL), and dried under reduced pressure, affording a pale orange solid of 13 (180 mg, 0.87 mmol, 66%). The compound was used in the next reaction without further purification. 1H NMR (400 MHz, CDCl3) δ 8.40 (d, J = 8.0, 1.6 Hz, 2H, Ph), 7.57 (tt, J = 7.6, 1.6 Hz, 1H, Ph), 7.51 (t, J = 8.0 Hz, 2H, Ph), 5.71 (br s, 2H, NH2). MS (ESI) calcd for C9H7ClN4 [M + H]+: 207.04; found, 207.10.
4-Amino-6-phenyl-1,3,5-triazine-2-thiol (14)44
A suspension of 13 (0.68 g, 3.29 mmol, 1.0 equiv) and sodium sulfide nonahydrate (1.185 g, 4.93 mmol, 1.5 equiv) in DMF (3 mL) was stirred at 80 °C for 3 h. The reaction progress was monitored by TLC. DMF was evaporated under reduced pressure at 80 °C. The resulting residue was treated carefully with HCl in EtOAc (10 mL). The suspended solid was collected by filtration and extracted with boiling EtOH (20 mL × 5). The collected organic layers were concentrated in vacuo and washed with warm DCM (15 mL × 3) to afford a pale yellow solid (190 mg, 0.92 mmol, 28%). 1H NMR (400 MHz, DMSO) δ 12.84 (br s, 1H, SH), 8.22 (d, J = 7.6 Hz, 2H, Ph), 8.20 (br s, 1H, 0.5 NH2), 7.77 (br s, 1H, 0.5 NH2), 7.59 (t, J = 7.6 Hz, 1H, Ph), 7.51 (t, J = 8.0 Hz, 2H, Ph). MS (ESI) calcd for C9H8N4S [M + H]+, 205.05; found, 205.10.
4-(1H-Imidazol-2-yl)methylthio-6-phenyl-1,3,5-triazin-2-amine (2)
Similar procedure as synthesis of 1 using 14 (0.148 g, 0.725 mmol, 1.0 equiv) as a starting material. The final compound 2 was obtained as a white solid (7 mg; 0.02 mmol; yield, 3%). Mp 209 °C. 1H NMR (400 MHz, MeOD): δ 8.34 (dd, J = 8.4, 1.2 Hz, 2H, Ph), 7.53 (tt, J = 7.6, 1.2 Hz, 1H, Ph), 7.45 (t, J = 7.6 Hz, 2H, Ph), 6.97 (br s, 2H, imidazolyl), 4.49 (s, 2H, CH2). MS (ESI): calcd for C13H12N6S [M + H]+, 285.08; found, 285.00. HPLC: 6.906 min; purity, 98%.
2-Benzoyl-3,3-bis(methylthio)acrylonitrile (16)45
A solution of benzoylacetonitrile 15 (1.189 g, 8.2 mmol, 1.0 equiv) in 16 mL of dry DMSO was added dropwise to a stirred suspension of 60% NaH dispersion in mineral oil (0.656 g, 16.4 mmol, 2.0 equiv) in 16 mL of dry DMSO under the N2 condition at room temperature. Carbon disulfide (0.49 mL, 8.2 mmol, 1.0 equiv) was added dropwise under external water bath cooling, and the mixture was stirred for 2 h. Iodomethane (1.0 mL, 16.4 mmol, 2.0 equiv) was then added to the mixture dropwise under external water bath cooling, and the reaction was stirred overnight. The reaction progress was monitored by TLC. After the completion of the reaction, the mixture was poured into 600 mL of ice-cold water, and the precipitate formed was filtered and dried in vacuo to provide 16 in good yield (1.677 g, 6.72 mmol, 82%). 1H NMR (400 MHz, DMSO) δ 7.85 (d, J = 7.2 Hz, 2H, Ph), 7.67 (t, J = 7.6 Hz, 1H, Ph), 7.55 (t, J = 7.6 Hz, 2H, Ph), 2.80 (s, 3H, CH3), 2.53 (s, 3H, CH3).
2-Amino-4-(methylthio)-6-phenylpyrimidine-5-carbonitrile (17)46
A solution of 16 (0.168 g, 6.73 mmol, 1.0 equiv), guanidine hydrochloride (0.643 g, 6.73 mmol, 1.0 equiv), and triethylamine (2.34 mL, 16.8 mmol, 2.5 equiv) in 21 mL of DMF was heated at reflux for 6 h. The reaction progress was monitored by TLC. After the completion of the reaction, the mixture was cooled down to room temperature and 32 mL of water was added to cause precipitation. The precipitate was collected by filtration and washed with water and methanol to give compound 17 as a pale yellow solid (680 mg, 2.83 mmol, 42%). 1H NMR (400 MHz, DMSO) δ 7.88 (br s, 1H, 0.5 NH2), 7.81 (br s, 1H, 0.5 NH2) 7.80–7.78 (m, 2H, Ph), 7.57–7.51 (m, 3H, Ph), 2.58 (s, 3H, CH3).
2-Amino-4-(methylsulfinyl)-6-phenylpyrimidine-5-carbonitrile (18)46
Intermediate 17 (0.706 g, 2.92 mmol, 1.0 equiv) was dissolved in 10 mL of CH2Cl2 at room temperature, and mCPBA (0.808 g, 4.68 mmol, 1.6 equiv) was added to the mixture. The mixture was stirred at room temperature for 3.5 h and monitored by TLC. The precipitate formed in the reaction was filtered, washed by CH2Cl2, and dried under reduced pressure to obtain 18 (495 mg, 1.93 mmol, 66%). 1H NMR (400 MHz, DMSO) δ 8.39 (br s, 1H, 0.5 NH2), 8.32 (br s, 1H, 0.5 NH2), 7.84 (d, J = 7.2 Hz, 2H, Ph), 7.64–7.54 (m, 3H, Ph), 2.93 (s, 3H, CH3).
2-Amino-4-sulfanyl-6-phenylpirimidine-5-carbonitrile (19)47
To a stirred suspension of 18 (0.495 g, 1.92 mmol, 1.0 equiv) in 17 mL of DMF was added potassium thioacetate (0.376 g, 3.84 mmol, 2.0 equiv), and the mixture was stirred at room temperature for 19 h. The reaction progress was monitored by TLC. After the completion of the reaction, the mixture was cooled down in an external ice water bath, and 17 mL of 2 M aqueous NaOH was added to the cold solution. The mixture was stirred for another 1.5 h and diluted with 17 mL of H2O, followed by pH adjustment to 5–6 with the addition of 1 M HCl (±50 mL). The mixture was diluted with 70 mL of H2O and stirred for 1 h at room temperature. The precipitate formed in the mixture was collected by filtration, washed with H2O (25 mL × 3), and dried under reduced pressure to give 19 in good yield (232 mg, 1.02 mmol, 53%). 1H NMR (400 MHz, DMSO) δ 12.91 (br s, 1H, SH), 8.67 (br s, 1H, 0.5 NH2), 7.78 (d, J = 7.6 Hz, 2H, Ph), 7.61–7.48 (m, 3H, Ph), 6.95 (br s, 1H, 0.5 NH2).
4-((1H-Imidazol-2-yl)methylthio)-2-amino-6-phenylpyrimidine-5-carbonitrile (3)
Similar procedure as synthesis of 1 using 19 (0.187 g, 0.82 mmol, 1.0 equiv) as a starting material to afford a light yellow solid (116 mg; 0.38 mmol; yield, 46%). Mp 224 °C. 1H NMR (400 MHz, DMSO) δ 11.86 (br s, 1H, NH), 7.97 (m, 2H, NH2), 7.79 (d, J = 6.8 Hz, 2H, Ph), 7.60–7.50 (m, 3H, Ph), 7.08 (br s, 1H, imidazolyl), 6.85 (br s, 1H, imidazolyl), 4.53 (s, 2H, CH2). MS (ESI): calcd for C15H12N6S [M + H]+, 309.08; found, 309.00. HPLC: 4.377 min; purity: 99%.
2-(1-Phenylethylidene)malononitrile (21)25
The synthesis of this compound was adapted from the conditions previously described by Longstreet et al.25 A mixture of acetophenone 20 (2.80 mL, 24 mmol, 1.0 equiv), malononitrile (1.586 g, 24.00 mmol, 1 equiv), and ammonium acetate (0.370 g, 4.80 mmol, 0.2 equiv) in toluene (20 mL) was heated at reflux under Dean–Stark conditions for 5 h. The reaction progress was monitored by TLC. After the completion of the reaction, the mixture was cooled down to room temperature. The reaction was diluted with EtOAc (50 mL), washed with saturated NaHCO3 solution (50 mL × 3) followed by water (50 mL), dried over MgSO4, and concentrated in vacuo to obtain a brown oil, which solidified at room temperature after 30 min. Purification was performed by column chromatography (CH2Cl2) to obtain 21 as a white solid (2.82 g, 16.8 mmol, 70%). 1H NMR (400 MHz, CDCl3) δ 7.58–7.26 (m, 5H, Ph), 2.64 (s, 3H, CH3). HNMR is consistent with the reported literature data.
2-Amino-6-(methylthio)-4-phenylnicotinonitrile (22)48
A mixture of 21 (2.5 g, 14.86 mmol, 1.0 equiv), dimethyl cyanocarbonimidodithioate (3.69 g, 25.3 mmol, 1.7 equiv), and potassium carbonate (2.465 g, 17.84 mmol, 1.2 equiv) in DMF (30 mL) was stirred at room temperature for 6 h. After the completion of the reaction, piperidine (2.4 mL, 23.78 mmol, 1.6 equiv) was added and the mixture was stirred at 80 °C overnight. The reaction progress was monitored by TLC. The mixture was then concentrated in vacuo, diluted with water, and extracted three times with CH2Cl2. The organic layer was dried over MgSO4 and concentrated in vacuo. The brown oil obtained was purified by column chromatography (CH2Cl2), yielding 22 as a yellow solid (2.21 g, 9.21 mmol, 62%). 1H NMR (400 MHz, CDCl3) δ 7.56–7.52 (m, 2H, Ph), 7.49–7.47 (m, 3H, Ph), 6.64 (s, 1H, pyridinyl), 5.27 (br s, 2H, NH2), 2.54 (s, 3H, CH3).
2-Amino-6-(methylsulfonyl)-4-phenylnicotinonitrile (23)
A mixture of 22 (1 g, 4.14 mmol, 1 equiv) and mCPBA (1.114 g, 4.97 mmol, 1 equiv) in 120 mL of CH2Cl2 was stirred at room temperature for 5 min. The mixture was then washed twice with NaHCO3 solution followed by water, dried over MgSO4, and concentrated in vacuo to yield a mixture of sulfoxide and sulfone product. Without further separation, it was then reacted with 77 wt % mCPBA (0.674 g, 3.90 mmol, 0.94 equiv) in CH2Cl2 (70 mL) and stirred at room temperature for another 5 min. The mixture was washed twice with NaHCO3 solution followed by water, dried over MgSO4, and concentrated in vacuo. Purification was performed by column chromatography (2% MeOH in CH2Cl2), yielding 23 as a yellow solid (0.631 g, 2.31 mmol, 56%). 1H NMR (400 MHz, DMSO) δ 7.68–7.65 (m, 4H, NH2 + Ph), 7.59–7.57 (m, 3H, Ph), 7.17 (s, 1H, pyridinyl), 3.27 (s, 3H, CH3).
2-Amino-6-sulfanyl-4-phenylnicotinonitrile (24)
A mixture of 23 (0.631 g, 2.31 mmol, 1 equiv) and potassium ethanethioate (0.527 g, 4.62 mmol, 2 equiv) in DMF (8 mL) was stirred at 80 °C for 4 h. After the completion of the reaction as shown by TLC, 2 M NaOH solution was added and the mixture was stirred at room temperature for another 2 h. The mixture was then diluted with water (80 mL) and 1 M HCl solution was added to adjust pH to 1. The precipitate formed was filtered and dried in vacuo to obtain a brown crude solid. Purification was performed by column chromatography (3% MeOH in CH2Cl2) to obtain the product as a yellow solid (0.321 g, 1.41 mmol, 61%). 1H NMR (400 MHz, DMSO) δ 12.58 (s, 1H, SH), 7.58–7.48 (m, 6H), 7.31 (br s, 2H, NH2), 6.47 (s, 1H). MS (ESI): calcd for C12H9N3S [M + H]+, 228.05; found, 228.13.
6-(((1H-Imidazol-2-yl)methyl)thio)-2-amino-4-phenylnicotinonitrile (4)
A mixture of 24 (0.12 g, 0.66 mmol, 1 equiv), 7 (0.16 g, 0.66 mmol, 1 equiv), and NaHCO3 (0.166 g, 1.98 mmol, 2 equiv) in DMF (3 mL) was stirred at 50 °C for 4 h. After the completion of the reaction as shown by TLC, the mixture was concentrated in vacuo and dissolved in EtOAc followed by washing three times with water and brine. The organic phase was dried over MgSO4, concentrated in vacuo to give a crude yellow oil, and purified by dry-loaded column chromatography (5% MeOH in CH2Cl2) as a column chromatography eluent. Compound 4 was obtained as a white solid (0.115 g; 0.376 mmol; yield, 57%). Mp 220 °C. 1H NMR (400 MHz, DMSO) δ 11.86 (br s, 1H, NH), 7.587.53 (m, 2H, Ph), 7.52–7.49 (m, 3H, Ph), 7.17 (br s, 2H, NH2), 7.05 (br s, 1H, imidazolyl), 6.82 (br s, 1H, imidazolyl), 6.68 (s, 1H, pyridinyl), 4.39 (s, 2H, CH2). MS (ESI): calcd for C16H13N5S [M + H]+, 308.09; found, 308.05. HPLC: 6.33 min; purity: 99%.
2,6-Dichloro-4-phenylpyridine (26)49
A stirred suspension of 2,6-dichloro-4-iodopyridine 25 (1 g, 3.65 mmol, 1.0 equiv), Na2CO3 (1.16 g, 10.95 mmol, 3.0 equiv), phenylboronic acid pinacol ester (0.745 g, 3.65 mmol, 1.0 equiv), and Pd(PPh3)2Cl2 (0.128 g, 0.183 mmol, 0.05 equiv) in a mixture of acetonitrile (12 mL) and H2O (8 mL) under a N2 atmosphere was heated at 70 °C for 16 h. The reaction progress was monitored by TLC (2% EtOAc in PET) and HPLC. The mixture was diluted with EtOAc (50 mL) and washed with brine (30 mL × 3). The organic layer was dried over MgSO4 and evaporated under reduced pressure. The resulting brown oil was purified by column chromatography (2% EtOAc in PET) to afford a white solid (750 mg, 3.36 mmol, 92%). 1H NMR (400 MHz, CDCl3) δ 7.61–7.57 (m, 2H, Ph), 7.53–7.48 (m, 3H, Ph), 7.47 (s, 2H, pyridinyl). HNMR in accordance with the literature.
2-(tert-Butylthio)-6-chloro-4-phenylpyridine (27)
A suspension of 2,6-dichloro-4-phenylpyridine 26 (0.2 g, 0.89 mmol, 1.0 equiv), Cs2CO3 (0.58 g, 1.78 mmol, 2.0 equiv), and 2-methylpropane-2-thiol (0.084 g, 0.93 mmol, 1.04 equiv) in DMF was heated at 80 °C overnight. The reaction progress was monitored by TLC (10% EtOAc in PET) and HPLC. The mixture was diluted with EtOAc (50 mL) and washed with brine (30 mL × 5). The organic phase was dried on MgSO4 and evaporated to afford a pale yellow oil (249 mg, 0.89 mmol, quantitative yield). 1H NMR (400 MHz, CDCl3) δ 7.69–7.54 (m, 2H, Ph), 7.50–7.43 (m, 3H, Ph), 7.37 (d, J = 1.2 Hz, 1H, pyridinyl), 7.26 (d, J = 1.2 Hz, 1H, pyridinyl) 1.59 (s, 9H, t-butyl). MS (ESI): calcd for C15H16ClNS [M + H]+, 278.07; found, 277.90.
tert-Butyl-(6-(tert-butylthio)-4-phenylpyridin-2-yl)-carbamate (28)
A suspension of 2-(tert-butylthio)-6-chloro-4-phenylpyridine 27 (0.23 g, 0.83 mmol, 1.0 equiv), Cs2CO3 (0.54 g, 1.66 mmol, 2.0 equiv), Xantphos (0,144 g, 0.25 mmol, 0.3 equiv), Pd(OAc)2 (0.028 g, 0.124 mmol, 0.15 equiv), and t-butyl carbamate (0.097 g, 0.83 mmol, 1.0 equiv) in dry 1,4-dioxane (2.8 mL) under a N2 atmosphere was heated at 110 °C overnight. The reaction progress was monitored by TLC (2% EtOAc in PET) and HPLC. The mixture was treated with warm acetone (25 mL) and filtered. The organic layer was evaporated and purified by column chromatography (20% EtOAc in PET) to afford 28 as a yellow solid (80 mg, 0.22 mmol, 27%). 1H NMR (400 MHz, DMSO) δ 9.96 (s, 1H, NH), 7.84 (d, J = 1.6 Hz, 1H), 7.67 (d, J = 6.8 Hz, 2H, Ph), 7.54–7.44 (m, 3H, Ph), 7.15 (d, J = 1.6 Hz, 1H), 1.53 (s, 9H), 1.50 (s, 9H). MS (ESI): calcd for C20H26N2O2S [M + H]+, 359.17; found, 358.9.
6-(tert-Butylthio)-4-phenylpyridin-2-amine (29)
To a solution of tert-butyl (6-(tert-butylthio)-4-phenylpyridin-2-yl)-carbamate 28 (0.25 g, 0.697 mmol, 1.0 equiv) in CH2Cl2 (5 mL) was added TFA (0.266 mL, 3.48 mmol, 5.0 equiv). The mixture was refluxed overnight. The reaction progress was monitored by TLC (6% MeOH in CH2Cl2) and HPLC. The mixture was diluted with EtOAc (50 mL) and washed with H2O (25 mL × 4). The organic phase was dried on MgSO4 and evaporated to afford a brown oil. The crude compound was purified by liquid chromatography (50% EtOAc in PET) to give 29 as a pale yellow oil (135 mg, 0.523 mmol, 75%). 1H NMR (400 MHz, DMSO) δ 7.66 (dd, J = 6.8, 2.0 Hz, 2H, Ph), 7.54–7.47 (m, 3H, Ph), 6.96 (s br, 1H, 0.5 NH2), 6.76 (s br, 1H, 0.5 NH2), 1.46 (s, 9H, t-butyl). MS (ESI): calcd for C15H18N2S [M + H]+, 259.12; found, 259.0 [M + H]+.
6-Amino-4-phenylpyridine-2-thiol (30)
A stirred solution of 6-(tert-butylthio)-4-phenylpyridin-2-amine 29 (0.445 g, 1.722 mmol) in 37% HCl (15 mL) was heated at 100 °C for 10 h. The reaction progress was monitored by HPLC. The mixture was carefully treated with NaHCO3 saturated solution to pH = 7 and extracted with EtOAc (40 mL × 5). The organic phase was dried over MgSO4 and evaporated to afford an orange solid (135 mg, mixture of the desired compound and the corresponding dimer). The compound was used for the next reaction without further purification. 1H NMR (400 MHz, DMSO) δ 12.25 (s br, 1H, SH), 7.59–7.55 (m, 2H, Ph), 7.49–7.43 (m, 3H, Ph), 6.85 (s br, 1H, 0.5 NH2), 6.40 (s br, 1H, 0.5 NH2). MS (ESI): calcd for C11H10N2S [M + H]+, 203.06; found, 203.1.
6-(((1H-Imidazol-2-yl)methyl)thio)-4-phenylpyridin-2-amine (5)
To a suspension of 6-amino-4-phenylpyridine-2-thiol 30 (0.145 g, 0.716 mmol, 1.0 equiv) and NaHCO3 (0.061 g, 0.57 mmol, 0.8 equiv) in dry DMF (2 mL) was added 7 (0.225 g, 0.931 mmol, 1.3 equiv). The mixture was stirred at room temperature for 23 h. The reaction progress was monitored by HPLC. DMF was evaporated under reduced pressure (water bath at 70 °C). The resulting residue was treated with H2O (20 mL) and extracted with EtOAc (30 mL × 4). The organic phase was dried over MgSO4 and evaporated to afford an orange oil. The compound was purified by liquid chromatography (10% MeOH in CH2Cl2) to obtain pure 5 as a pale red solid (67 mg; 0.24 mmol; yield, 33%). Mp 109 °C. 1H NMR (400 MHz, MeOD) δ 7.55–7.50 (m, 2H, Ph), 7.46–7.32 (m, 3H, Ph), 6.94 (s, 2H, pyridinyl), 6.70 (d, J = 1.2 Hz, 1H, imidazolyl), 6.50 (d, J = 1.2 Hz, 1H, imidazolyl), 4.39 (s, 2H, CH2). MS (ESI): calcd for C15H14N4S [M + H]+, 283.09; found, 283.00. HPLC: 3.722 min; purity: 97%.
2,6-Dihydroxy-4-phenylnicotinonitrile (32)30
A suspension of commercially available ethylbenzoyl acetate 31 (3 g, 15.6 mmol, 1.0 equiv), 2-cyanoacetamide (1.31 g, 15.6 mmol, 1.0 equiv), and KOH (0.96 g, 15.6 mmol, 1.0 equiv) in EtOH (20 mL) was refluxed for 24 h. The reaction progress was monitored by HPLC and TLC (20% MeOH in CH2Cl2). After the completion of the reaction, the mixture was cooled down to 0 °C. The suspended solid was collected by filtration and subsequently dissolved in warm water (60 mL). The alkaline solution was carefully treated with 37% HCl solution to adjust the pH to 1. The precipitate was collected by filtration and dried to give 32 in good yield (1.3 g, 5.3 mmol, 34%). 1H NMR (400 MHz, DMSO) δ 7.52 (s, 5H, Ph), 5.80 (s, 1H, pyridinyl). MS (ESI): calcd for C12H8N2O2 [M + H]+, 213.06; found, 213.1. HNMR is consistent with the reported literature data.
2,6-Dichloro-4-phenylnicotinonitrile (33)
A suspension of 2,6-dihydroxy-4-phenylnicotinonitrile 32 (1.2 g, 5.657 mmol, 1.0 equiv) in POCl3 (5.3 mL, 56.6 mmol, 10.0 equiv) was heated at 180 °C in an autoclave for 16 h. The reaction progress was monitored by HPLC. The mixture was cooled down to 0 °C and treated with crushed ice. The suspended solid was collected by filtration, rinsed with petroleum ether (30 mL), and dried under reduced pressure to afford 33 as a gray solid (0.990 g, 3.96 mmol, 70%). 1H NMR (400 MHz, CDCl3) δ 7.63–7.60 (m, 5H, Ph), 7.45 (s, 1H, pyridinyl). HNMR is consistent with the reported literature data.
tert-Butyl-(6-chloro-5-cyano-4-phenylpyridin-2-yl)-carbamate (34)
To a suspension of 2,6-dichloro-4-phenylnicotinonitrile 33 (0.2 g, 0.806 mmol, 1.0 equiv), t-butyl carbamate (0.095 g, 0.806 mmol, 1.0 equiv), Cs2CO3 (0.54 g, 1.66 mmol, 2.06 equiv), and Xantphos (0.139 g, 0.242 mmol, 0.3 equiv) in dry 1,4-dioxane (2.7 mL) under a N2 atmosphere was added Pd(OAc)2 (0.027 g, 0.121 mmol, 0.15 equiv). The mixture was heated at 40 °C for 23 h. The reaction progress was monitored by TLC (2% EtOAc in PET) and HPLC. The mixture was then treated with warm acetone (25 mL) and filtered. The organic layer was evaporated to afford a pale brown solid (90 mg, 0.26 mmol, 33%). The product was used for the next reaction without further purification. 1H NMR (300 MHz, CDCl3) δ 8.07 (s, 1H, pyridinyl), 7.64–7.58 (m, 2H, Ph), 7.55–7.47 (m, 4H, Ph + NH), 1.53 (s, 9H, t-butyl). MS (ESI): calcd for C17H16ClN3O2 [M + H]+, 330.09; found, 329.92.
tert-Butyl (6-(tert-butylthio)-5-cyano-4-phenylpyridin-2-yl)-carbamate (35)
A suspension of tert-butyl (6-chloro-5-cyano-4-phenylpyridin-2-yl)-carbamate 34 (0.09 g, 0.273 mmol, 1.0 equiv), 2-methyl-propanethiol (0.03 mL, 0.273 mmol, 1.0 equiv), and Cs2CO3 (0.178 g, 0.56 mmol, 2 equiv) in DMF (2 mL) was heated at 90 °C for 20 h. The reaction progress was monitored by HPLC. The mixture was cooled down to room temperature, diluted with EtOAc (50 mL), and washed with brine (25 mL × 5). The organic layer was dried on MgSO4 and evaporated to afford an orange oil (93 mg, 0.211 mmol, 89%). The compound was used for the next reaction without further purification. 1H NMR (400 MHz, CDCl3) δ 7.81 (s, 1H, pyridinyl), 7.59–7.55 (m, 2H, Ph), 7.48–7.44 (m, 3H, Ph), 7.32 (s br, 1H, NH), 1.65 (s, 9H, t-butyl), 1.54 (s, 9H, t-butyl). MS (ESI): calcd for C21H25N3O2S [M + H]+, 384.17; found, 284.00.
6-Amino-2-sulfanyl-4-phenylnicotinonitrile (36)
A suspension of 35 (0.7 g, 1.825 mmol) in 37% HCl (7 mL) was heated at 100 °C for 2 h. The reaction progress was monitored by HPLC. The mixture was cooled down to room temperature and diluted with NaHCO3 saturated solution (7 mL). The resulting suspension was treated carefully with solid NaHCO3 to pH 7 and extracted with EtOAc (30 mL × 5). The organic layer was dried over MgSO4 and evaporated to afford an orange solid, which was purified by column chromatography (40% EtOAc in PET and 10% MeOH in CH2Cl2). Intermediate 36 was obtained as a pale yellow solid (156 mg, 0.675 mmol, 37%). 1H NMR (400 MHz, MeOD) δ 7.56–7.51 (m, 2H, Ph), 7.51–7.47 (m, 3H, Ph), 6.05 (s, 1H, pyridinyl). MS (ESI): calcd for C12H9N3S [M + H]+, 228.05; found, 228.08.
2-(((1H-Imidazol-2-yl)methyl)thio)-6-amino-4-phenylnicotinonitrile (6)
A suspension of 36 (0.096 g, 0.422 mmol, 1.0 equiv), NaHCO3 (0.020 g, 0.422 mmol, 1.0 equiv), and 7 in dry DMF (2.5 mL) was stirred at room temperature for 4 h. The reaction progress was monitored by TLC (10% MeOH in CH2Cl2). DMF was evaporated under reduced pressure (water bath at 70 °C). The resulting residue was treated with water (10 mL) and extracted with EtOAc (30 mL × 5). The organic layer was dried over MgSO4 and evaporated to afford a pale brown oil. The final compound 6 was obtained after purification by column chromatography (PET/EtOAc/MeOH, 1:7.8:1.2) and recrystallized using diethyl ether and MeOH to yield 6 as a pale yellow solid (10 mg; 0.032 mmol; yield, 8%). Mp 222 °C. 1H NMR (400 MHz, MeOD) δ 7.53–7.45 (m, 5H, Ph), 6.97 (s, 2H, imidazolyl), 6.28 (s, 1H, pyridinyl), 4.53 (s, 2H, CH2). MS (ESI): calcd for C16H13N5S [M + H]+, 308.09; found, 308.00. HPLC: 7.490 min; purity: 99%.
Pharmacology
Cell Lines and Chemicals
Chinese hamster ovary (CHO) cells stably expressing the human adenosine A1 receptor (CHOhA1R) were kindly provided by Prof. S. J. Hill (University of Nottingham, U.K.), human embryonic kidney 293 cells stably expressing the human adenosine A2A receptor (HEK293hA2AR) were kindly provided by Dr. J. Wang (Biogen/IDEC, Cambridge, MA), CHO-spap cells stably expressing the wild-type (WT) hA2B receptor (CHO-spap-hA2BR) were kindly provided by S. J. Dowell (GlaxoSmithKline, U.K.), and CHO cells stably expressing the human adenosine A3 receptor (CHOhA3R) were a gift from Dr. K.-N. Klotz (University of Würzburg, Germany). [3H]1,3-Dipropyl-8-cyclopentyl-xanthine ([3H]DPCPX; specific activity, 120 Ci/mmol) was purchased from ARC, Inc. (St. Louis, USA), [3H]4-(2-[7-amino-2-(furan-2-yl)-[1,2,4]triazolo[1,5-a][1,3,5]triazin-5-ylamino)ethyl) phenol ([3H]-ZM241385; specific activity, 50 Ci/mmol) was purchased from ARC, Inc. (St. Louis, USA), [3H]8-(4-(4-(4-chlorophenyl)piperazide-1-sulfonyl)phenyl)-1-propylxanthine ([3H]PSB-603; specific activity, 79 Ci/mmol) was purchased from Quotient Bioresearch (Waltham, MA), and [3H]8-ethyl-4-methyl-2-phenyl-(8R)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]-purin-5-one ([3H]PSB-11; specific activity, 56 Ci/mmol) was obtained with the kind help of Prof. C. E. Müller (University of Bonn, Germany). 5′-N-Ethylcarboxamidoadenosine (NECA), N6-cyclopentyladenosine (CPA), and adenosine deaminase (ADA) were purchased from Sigma-Aldrich (Steinheim, Germany). Unlabeled 4-(2-[7-amino-2-(furan-2-yl)-[1,2,4]triazolo[1,5-a][1,3,5]triazin-5-ylamino)ethyl) phenol (ZM241385) was a kind gift from Dr. S. M. Poucher (Astra Zeneca, Manchester, U.K.). 3-[4-[2-[[6-Amino-9-[(2R,3R,4S,5S)-5-(ethylcarbamoyl)-3,4-dihydroxy-oxolan-2-yl]purin-2-yl]amino]ethyl]phenyl] propanoic acid (CGS21680) was purchased from Ascent Scientific (Bristol, U.K.). Bicinchoninic acid (BCA) protein assay reagents were obtained from Pierce Chemical Company (Rockford, IL, USA). All other chemicals were of analytical grade and were obtained from standard commercial sources.
Cell Culture and Membrane Preparation
CHOhA1R and CHOhA3R cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) and Ham’s F12 medium (1:1) supplemented with 10% (v/v) newborn calf serum, 50 μg·mL–1 streptomycin, 50 IU·mL–1 penicillin, and 200 μg·mL–1 G418 at 37 °C and 5% CO2. CHOhA1R cells were subcultured twice a week at a ratio of 1:20 on 10 cm Ø plates and 15 cm Ø plates. CHOhA3 cells were subcultured twice a week at a ratio of 1:8 on 10 cm Ø plates and 15 cm Ø plates. HEK293hA2AR cells were grown in a culture medium consisting of Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% newborn calf serum, 50 μg·mL–1 streptomycin, 50 IU·mL–1 penicillin, and 500 μg·mL–1 G418 at 37 °C and 7% CO2. Cells were subcultured twice a week at a ratio of 1:8 on 10 cm Ø plates and 15 cm Ø plates. CHO-spap-hA2BR cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) and Ham’s F12 medium (1:1) supplemented with 10% (v/v) newborn calf serum, 100 μg·mL–1 streptomycin, 100 IU·mL–1 penicillin, 1 mg·mL–1 G418, and 0.4 mg·mL–1 hygromycin at 37 °C and 5% CO2. Cells were subcultured at a ratio of 1:20 twice weekly.
All cells were grown to 80–90% confluency and detached from plates by scraping them into 5 mL of PBS. Detached cells were collected and centrifuged at 200g for 5 min. Pellets derived from 100 15 cm Ø plates were pooled and resuspended in 70 mL of ice-cold 50 mM Tris–HCl buffer (pH 7.4). A Heidolph Diax 900 homogenizer was used to homogenize the cell suspension. Membranes and the cytosolic fraction were separated by centrifugation at 100,000g in a Beckman Optima LE-80K ultracentrifuge (Beckman Coulter, Fullerton, CA) at 4 °C for 20 min. The pellet was resuspended in 35 mL of Tris–HCl buffer, and the homogenization and centrifugation steps were repeated. Tris–HCl buffer (25 mL) was used to resuspend the pellet, and ADA was added (0.8 U/mL) to break down endogenous adenosine. Membranes were stored in 250 and 500 μL aliquots at −80 °C. Total protein concentrations were measured using the BCA method.50
Radioligand Binding Studies
Membrane aliquots containing 5 μg (CHOhA1R) or 30 μg (HEK293hA2AR) of total protein were incubated in a total volume of 100 μL assay buffer (50 mM Tris–HCl, pH 7.4) at 25 °C for 1 h. Membrane aliquots containing 30 μg (CHO-spap-hA2BR) of total protein were incubated in a total volume of 100 μL assay buffer (0.1% CHAPS in 50 mM Tris–HCl, pH 7.4) at 25 °C for 2 h, or those containing 15 μg (CHOhA3R) of total protein were incubated in a total volume of 100 μL assay buffer (50 mM Tris–HCl, pH 8.0, supplemented with 10 mM MgCl2, 1 mM EDTA, and 0.01% (w/v) CHAPS) at 25 °C for 2 h. Radioligand displacement experiments were performed using six concentrations of the competing ligand in the presence of 1.6 nM [3H]DPCPX for CHOhA1R, 5.5 nM [3H]ZM241385 for HEK293hA2AR, 1.5 nM [3H]PSB-603 for CHO-spap-hA2BR, and 10 nM [3H]PSB-11 for CHOhA3R. At these concentrations, the total radioligand binding did not exceed 10% of that added to prevent ligand depletion. Nonspecific binding was determined in the presence of 100 μM CPA for CHOhA1R, 100 μM NECA for HEK293hA2AR and CHOhA3R, and 10 μM ZM241385 for CHO-spap-hA2BR. Incubations were terminated by rapid vacuum filtration to separate the bound and free radioligands through prewetted 96-well GF/B filter plates using a PerkinElmer Filtermate-harvester (PerkinElmer, Groningen, The Netherlands). Filters were subsequently washed 12 times with ice-cold wash buffer: 50 mM Tris–HCl (pH 7.4) for CHOhA1R and HEK293hA2AR, 0.1% BSA in 50 mM Tris–HCl (pH 7.4) for CHO-spap-hA2BR, and 50 mM Tris–HCl supplemented with 10 mM MgCl2 and 1 mM EDTA (pH 8.0) for CHOhA3R. The plates were dried at 55 °C after which MicroScint-20 cocktail was added (PerkinElmer, Groningen, The Netherlands). After 3 h, the filter-bound radioactivity was determined by scintillation spectrometry using a 2450 MicroBeta Microplate Counter (PerkinElmer, Groningen, The Netherlands).
Label-Free Impedance-Based Assay
Label-free whole-cell assays were performed on the xCELLigence real-time cell analyzer (RTCA) system.51,52 In this system, the cells are adhered to arrayed gold electrodes embedded at the bottom of microelectronic E-plate 96 (Bioké, Leiden, NL), which is compatible with the xCELLigence RTCA system (ACEA Bioscience, San Diego, CA, USA). Upon activation of GPCR-mediated signaling, the cell morphology changes and therefore affects the local ionic environment at the cell–electrode interface. This leads to an increased electronic readout of cell-sensor impedance (Z), which is displayed in real time as the cell index (CI). Specifically, the CI value at each time point is defined as (Zi – Z0)Ω/15Ω, where Zi is the impedance at each individual time point, and Z0 is the impedance derived from the electrode/solution interface in the absence of cells prior to the start of the experiment. HEK293hA2AR cells were cultured as a monolayer on 10 cm Ø culture plates to 80–90% confluency and subsequently harvested and centrifuged two times at 1000 rpm for 5 min. Initially, 45 μL of culture media was added to wells in E-plates 96 to obtain background readings (Z0) followed by the addition of 50 μL of cell suspension containing 20,000 cells/well. The E-plate containing the cells was left at room temperature for 30 min before being placed on the recording device station in the incubator at 37 °C in 7% CO2. Afterward, cell attachment, spreading, and proliferation were continuously monitored every 15 min. The cells were cultured until the end of log phase (∼18–20 h) to obtain an optimal assay window. Prior to agonist application (either the reference compound CGS21680, 8, or newly synthesized compounds), the interval between two measurements was adjusted to 15 s. Subsequently, 5 μL of compound solution (final concentration of 0.1% DMSO) or vehicle control (0.1% DMSO) was added to each well, after which the CI was recorded for 60 min.
Molecular Modeling
All preparations and simulations were performed using Schrödinger Suite.53 The crystal structure of hA2AAR in complex with compound 8 was obtained in-house (vide supra) and used for the in silico experiment. All thermostabilizing mutations were reverted to wild-type hA2AAR, the bRIL fusion protein was removed from the crystal structure, and the third intracellular loop (ICL3) was filled in using Prime.3,33,54 Missing water molecules in the binding pocket of the crystal structure were added from 4EIY.3 The protein was prepared by the “Protein Preparation Wizard” at default settings. The compounds (1 and 3) were built and prepared using LigPrep.55 Induced-fit docking34 was performed flexibly with the crystal ligand 8 as the center of workspace in the standard protocol and OPLS3 as the force field. Figures were rendered using PyMol.56
Data Analysis
All experimental data were analyzed using the nonlinear regression curve fitting program GraphPad Prism 7.0 (GraphPad Software Inc., San Diego, CA). IC50 values obtained from competition displacement binding data were converted into Ki values using the Cheng–Prusoff equation.57 The KD value of [3H]DPCPX (1.6 nM) at CHOhA1R membranes was taken from Kourounakis et al.58 The KD value (1.0 nM) of [3H]ZM241385 at hA2AR membranes, the KD value (1.71 nM) of [3H]PSB-603 at CHO-spap-hA2BR membranes, and the KD value (17.3 nM) of [3H]PSB-11 at CHOhA3R membranes were taken from in-house determinations. Efficacy data from xCELLigence were obtained using RTCA software 1.2 (Roche, Germany) by normalizing CI traces to define peak responses within 60 min after compound addition. Peak values were used further for the determination of concentration–effect curves, yielding EC50 (concentration causing a half-maximum effect) and Emax (maximum effect relative to CGS21680) values.
Acknowledgments
T.A. acknowledges financial support by the Indonesia Endowment Fund for Education (LPDP). Eelke B. Lenselink is acknowledged for his help with the computational aspects of the study.
Glossary
Abbreviations Used
- bRIL
thermostabilized apocytochrome b562RIL
- GPCR
G protein-coupled receptor
- hAR
human adenosine receptor
- mCPBA
meta-chloroperoxybenzoic acid
- RTCA
real-time cell analysis
- StaR
(thermo)stabilized receptor
- TFA
trifluoroacetic acid
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01856.
Author Present Address
# Present address: LeadXpro AG, Park InnovAARE, Villigen 5234, Switzerland (R.K.Y.C.).
Author Present Address
⊥ Present address: Department of Biotechnology, University of Verona, Strada Le Grazie 15, 37134 Verona, Italy (E.S.).
Author Present Address
∥ Present address: Department of Neuroscience, Pharmaceutical and Nutraceutical Section, University of Florence, 50019 Firenze, Italy (M.F.).
Author Contributions
T.A., L.H.H., G.J.P.v.W., D.v.d.E., R.M.C., and A.P.IJ. were involved in the study design. E.S., G.V., and R.K.Y.C. carried out the crystal structure determination. T.A., J.P.D.v.V., M.F., and D.v.d.E. conducted synthetic chemistry experiments. R.L., L.H.H., and A.P.IJ. performed pharmacology experiments. T.A. and G.J.P.v.W. carried out the computational study. T.A., J.P.D.v.V., L.H.H., G.J.P.v.W., E.S., G.V., R.M.C., D.v.d.E., and A.P.IJ. wrote the manuscript.
The authors declare no competing financial interest.
Notes
The 8–hA2AAR complex structure coordinates and structure factors are available via the Protein Data Bank, accession code (PDB ID) 7ARO. Receptor models with compounds 1 and 3 are available as well. The authors will release the atomic coordinates and experimental data upon article publication.
Supplementary Material
References
- Fredholm B. B.; IJzerman A. P.; Jacobson K. A.; Linden J.; Müller C. E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors-an update. Pharmacol. Rev. 2011, 63, 1–34. 10.1124/pr.110.003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaakola V.-P.; Griffith M. T.; Hanson M. A.; Cherezov V.; Chien E. Y. T.; Lane J. R.; IJzerman A. P.; Stevens R. C. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 2008, 322, 1211–1217. 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Chun E.; Thompson A. A.; Chubukov P.; Xu F.; Katritch V.; Han G. W.; Roth C. B.; Heitman L. H.; IJzerman A. P.; Cherezov V.; Stevens R. C. Structural basis for allosteric regulation of GPCRs by sodium ions. Science 2012, 337, 232–236. 10.1126/science.1219218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazayeri A.; Andrews S. P.; Marshall F. H. Structurally enabled discovery of adenosine A2A receptor antagonists. Chem. Rev. 2017, 117, 21–37. 10.1021/acs.chemrev.6b00119. [DOI] [PubMed] [Google Scholar]
- Fredholm B. B.; IJzerman A. P.; Jacobson K. A.; Klotz K.-N.; Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 2001, 53, 527–552. [PMC free article] [PubMed] [Google Scholar]
- Rosentreter U.; Henning R.; Bauser M.; Kraemer T.; Vaupel A.; Hübsch W.; Dembowsky K.; Salcher-Schräufstaetter O.; Stasch J.-P.; Krahn T.; Perzborn E.. Substituted 2-thio-3,5-dicyano-4-aryl-6-aminopyridines and the use thereof as adenosine receptor ligands. World patent WO2001025210, April 12, 2001.
- Rosentreter U.; Kraemer T.; Shimada M.; Huebsch W.; Diedrichs N.; Krahn T.; K H.; Stasch J.. Preparation of 2-heteroarylmethylthio-3,5-dicyano-4-phenyl-6-aminopyridines as adenosine receptor selective ligands. World patent WO03008384, January 30, 2003.
- Shah S. J.; Voors A. A.; McMurray J. J. V.; Kitzman D. W.; Viethen T.; Bomfim Wirtz A.; Huang E.; Pap A. F.; Solomon S. D. Effect of neladenoson bialanate on exercise capacity among patients with heart failure with preserved ejection fraction: a randomized clinical trial. JAMA 2019, 321, 2101–2112. 10.1001/jama.2019.6717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betti M.; Catarzi D.; Varano F.; Falsini M.; Varani K.; Vincenzi F.; Dal Ben D.; Lambertucci C.; Colotta V. The aminopyridine-3,5-dicarbonitrile core for the design of new non-nucleoside-like agonists of the human adenosine A2B receptor. Eur. J. Med. Chem. 2018, 150, 127–139. 10.1016/j.ejmech.2018.02.081. [DOI] [PubMed] [Google Scholar]
- Eckle T.; Krahn T.; Grenz A.; Köhler D.; Mittelbronn M.; Ledent C.; Jacobson M. A.; Osswald H.; Thompson L. F.; Unertl K.; Eltzschig H. K. Cardioprotection by ecto-5′-nucleotidase (CD73) and A2B adenosine receptors. Circulation 2007, 115, 1581–1590. 10.1161/CIRCULATIONAHA.106.669697. [DOI] [PubMed] [Google Scholar]
- Beukers M. W.; Chang L. C. W.; von Frijtag Drabbe Künzel J. K.; Mulder-Krieger T.; Spanjersberg R. F.; Brussee J.; IJzerman A. P. New, non-adenosine, high-potency agonists for the human adenosine A2B receptor with an improved selectivity profile compared to the reference agonist N-ethylcarboxamidoadenosine. J. Med. Chem. 2004, 47, 3707–3709. 10.1021/jm049947s. [DOI] [PubMed] [Google Scholar]
- Guo D.; Mulder-Krieger T.; IJzerman A. P.; Heitman L. H. Functional efficacy of adenosine A2A receptor agonists is positively correlated to their receptor residence time. Br. J. Pharmacol. 2012, 166, 1846–1859. 10.1111/j.1476-5381.2012.01897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segala E.; Guo D.; Cheng R. K. Y.; Bortolato A.; Deflorian F.; Doré A. S.; Errey J. C.; Heitman L. H.; IJzerman A. P.; Marshall F. H.; Cooke R. M. Controlling the dissociation of ligands from the adenosine A2A receptor through modulation of salt bridge strength. J. Med. Chem. 2016, 59, 6470–6479. 10.1021/acs.jmedchem.6b00653. [DOI] [PubMed] [Google Scholar]
- Rucktooa P.; Cheng R. K. Y.; Segala E.; Geng T.; Errey J. C.; Brown G. A.; Cooke R. M.; Marshall F. H.; Doré A. S. Towards high throughput GPCR crystallography: In meso soaking of adenosine A2A receptor crystals. Sci. Rep. 2018, 8, 41. 10.1038/s41598-017-18570-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros J. A.; Weinstein H. [19] Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci 1995, 25, 366–428. 10.1016/S1043-9471(05)80049-7. [DOI] [Google Scholar]
- Lane J. R.; Klein Herenbrink C.; van Westen G. J. P.; Spoorendonk J. A.; Hoffmann C.; IJzerman A. P. A novel nonribose agonist, LUF5834, engages residues that are distinct from those of adenosine-like ligands to activate the adenosine A2A receptor. Mol. Pharmacol. 2012, 81, 475–487. 10.1124/mol.111.075937. [DOI] [PubMed] [Google Scholar]
- Gao Z.-G.; Jiang Q.; Jacobson K. A.; IJzerman A. P. Site-directed mutagenesis studies of human A2A adenosine receptors: involvement of Glu13 and His278 in ligand binding and sodium modulation. Biochem. Pharmacol. 2000, 60, 661–668. 10.1016/S0006-2952(00)00357-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebon G.; Warne T.; Edwards P. C.; Bennett K.; Langmead C. J.; Leslie A. G. W.; Tate C. G. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 2011, 474, 521–525. 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertheleme N.; Singh S.; Dowell S. J.; Hubbard J.; Byrne B. Loss of constitutive activity is correlated with increased thermostability of the human adenosine A2A receptor. Br. J. Pharmacol. 2013, 169, 988–998. 10.1111/bph.12165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louvel J.; Guo D.; Agliardi M.; Mocking T. A. M.; Kars R.; Pham T. P.; Xia L.; de Vries H.; Brussee J.; Heitman L. H.; IJzerman A. P. Agonists for the adenosine A1 receptor with tunable residence time. A case for nonribose 4-amino-6-aryl-5-cyano-2-thiopyrimidines. J. Med. Chem. 2014, 57, 3213–3222. 10.1021/jm401643m. [DOI] [PubMed] [Google Scholar]
- Chaplen P.; Slack R.; Wooldridge K. R. H. 843. Isothiazoles. Part VII. Quaternary isothiazoles. J. Chem. Soc. 1965, 4577–4578. 10.1039/jr9650004577. [DOI] [Google Scholar]
- Viira B.; Selyutina A.; García-Sosa A. T.; Karonen M.; Sinkkonen J.; Merits A.; Maran U. Design, discovery, modelling, synthesis, and biological evaluation of novel and small, low toxicity s-triazine derivatives as HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg. Med. Chem. 2016, 24, 2519–2529. 10.1016/j.bmc.2016.04.018. [DOI] [PubMed] [Google Scholar]
- Rudorf W.-D.; Augustin M. Acylketen-S,S- und Acylketen-S,N-acetale als Bausteine für Heterocyclen: 5-Cyanopyrimidine. J. Prakt. Chem. 1978, 320, 576–584. 10.1002/prac.19783200411. [DOI] [Google Scholar]
- Suda A.; Kawasaki K.-I.; Komiyama S.; Isshiki Y.; Yoon D.-O.; Kim S.-J.; Na Y.-J.; Hasegawa K.; Fukami T. A.; Sato S.; Miura T.; Ono N.; Yamazaki T.; Saitoh R.; Shimma N.; Shiratori Y.; Tsukuda T. Design and synthesis of 2-amino-6-(1H,3H-benzo[de]isochromen-6-yl)-1,3,5-triazines as novel Hsp90 inhibitors. Bioorg. Med. Chem. 2014, 22, 892–905. 10.1016/j.bmc.2013.11.036. [DOI] [PubMed] [Google Scholar]
- Longstreet A. R.; Campbell B. S.; Gupton B. F.; McQuade D. T. Improved synthesis of mono- and disubstituted 2-halonicotinonitriles from alkylidene malononitriles. Org. Lett. 2013, 15, 5298–5301. 10.1021/ol4025265. [DOI] [PubMed] [Google Scholar]
- Teague S. J. Synthesis of heavily substituted 2-aminopyridines by displacement of a 6-methylsulfinyl group. J. Org. Chem. 2008, 73, 9765–9766. 10.1021/jo801303v. [DOI] [PubMed] [Google Scholar]
- Doebelin C.; Wagner P.; Bertin I.; Simonin F.; Schmitt M.; Bihel F.; Bourguignon J.-J. Trisubstitution of pyridine through sequential and regioselective palladium cross-coupling reactions affording analogs of known GPR54 antagonists. RSC Adv. 2013, 3, 10296–10300. 10.1039/c3ra41985a. [DOI] [Google Scholar]
- Capaldi C.; Armani E.; Hurley C.; Avitabile-Woo B. G.; Lanaro R.; Jennings N. S.. (4-(([1,2,4]Triazolo[4,3-A]pyridine-6-yl)oxy)-1,2,3,4-tetrahydronaphthalen-1-yl) ureido derivatives as anti-inflammatory P38 MAPK inhibitors for treating diseases of the respiratory tract. World Patent WO2018224423, December 13, 2018.
- Kim Y. K.; Park S. Y.; Joo H. W.; Choi E. S.. Biaryl derivatives as GPR120 agonists. World Patent WO2014209034, December 31, 2014.
- Pevet I.; Brulé C.; Tizot A.; Gohier A.; Cruzalegui F.; Boutin J. A.; Goldstein S. Synthesis and pharmacological evaluation of thieno[2,3-b]pyridine derivatives as novel c-Src inhibitors. Bioorg. Med. Chem. 2011, 19, 2517–2528. 10.1016/j.bmc.2011.03.021. [DOI] [PubMed] [Google Scholar]
- Roch J.; Muller E.; Narr B.; Nickl J.; Haarmann W.. 3-Amino-4-phenyl-6-piperidino-1H-pyrazolo[3,4-B]-pyridines and salts thereof. U.S. Patent US4,260,621, April 7, 1981.
- Hamidi H.; Lilja J.; Ivaska J. Using xCELLigence RTCA instrument to measure cell adhesion. Bio-Protoc. 2017, 7, e2646 10.21769/BioProtoc.2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun E.; Thompson A. A.; Liu W.; Roth C. B.; Griffith M. T.; Katritch V.; Kunken J.; Xu F.; Cherezov V.; Hanson M. A.; Stevens R. C. Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure 2012, 20, 967–976. 10.1016/j.str.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman W.; Day T.; Jacobson M. P.; Friesner R. A.; Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- Xu F.; Wu H.; Katritch V.; Han G. W.; Jacobson K. A.; Gao Z.-G.; Cherezov V.; Stevens R. C. Structure of an agonist-bound human A2A adenosine receptor. Science 2011, 332, 322–327. 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Veldhoven J. P. D.; Chang L. C. W.; von Frijtag Drabbe Künzel J. K.; Mulder-Krieger T.; Struensee-Link R.; Beukers M. W.; Brussee J.; IJzerman A. P. I. A new generation of adenosine receptor antagonists: from di- to trisubstituted aminopyrimidines. Bioorg. Med. Chem. 2008, 16, 2741–2752. 10.1016/j.bmc.2008.01.013. [DOI] [PubMed] [Google Scholar]
- Kabsch W. Integration, scaling, space-group assignment and post-refinement. Acta Cryst. 2010, 66, 133–144. 10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonrhein C.; Tickle I. J.; Flensburg C.; Keller P.; Paciorek W.; Sharff A.; Bricogne G. Advances in automated data analysis and processing within autoPROC, combined with improved characterisation, mitigation and visualisation of the anisotropy of diffraction limits using STARANISO. Acta Cryst. 2018, A74, a360. [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser crystallographic software. J. Appl. Cryst. 2007, 40, 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine P. V.; Grosse-Kunstleve R. W.; Echols N.; Headd J. J.; Moriarty N. W.; Mustyakimov M.; Terwilliger T. C.; Urzhumtsev A.; Zwart P. H.; Adams P. D. Towards automated crystallographic structure refinement with phenix.refine. Acta Cryst. 2012, 68, 352–367. 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricogne G.; Blanc E.; Brandl M.; Flensburg C.; Keller P.; Paciorek W.; Roversi P.; Sharff A.; Smart O. S.; Vonrhein C.; Womack T. O.. BUSTER version 2.11.7; Global Phasing Ltd.: Cambridge, United Kingdom: 2017.
- Park J. H.; Kim Y. K.; Lui J. H.; Yu E. S.; Lee H. I.; Jung H. K.. Organic optoelectronic device and display apparatus. U.S. Patent US2,017,331,067, November 16, 2017.
- Reimschuessel H. K.; McDevitt N. T. Infrared spectra of some 1,3,5-triazine derivatives. J. Am. Chem. Soc. 1960, 82, 3756–3762. 10.1021/ja01499a071. [DOI] [Google Scholar]
- Joshua C. P.; Rajan V. P. Interaction of dicyandiamides with thioamides: Formation of 6-Alkyl(or aryl)-4-amino(or arylamino)-1,3,5-triazine-2-thiols and amidino(or aryamidino)thioureas. Aust. J. Chem. 1972, 27, 2627–2634. 10.1071/CH9742627. [DOI] [Google Scholar]
- Fryszman O. M.; Lang H.; Lan J.; Chang E.; Fang Y.. 5-Membered heterocycle-based P38 kinase inhibitors. World Patent WO2005009973, February 3, 2005.
- Borroni E.; Hubert-Trottmann G.; Kilpatrick G.; Norcross R.. Adenosine receptor ligands and their use in the treatment of disease. U.S. Patent US20,010,027,196, October 4, 2001.
- Perez M. A.; Soto J. L.; Carrillo J. R. A simple, unambiguous synthesis of 2-amino-4-oxo- and −4-thioxo-3,4-dihydropyrimidine-5-carbonitriles. Synthesis 1983, 5, 402–404. [Google Scholar]
- Peseke K.; Suarez J.; Quincoces J. Push-pull-butadiene; Synthese von 2-Amino-4-aryl(fur-2-yl)-6-methylthio-pyridin-3-carbonitrilen. Zeitschrift fur Chemie 1983, 23, 404–405. [Google Scholar]
- Mello J. V.; Finney N. S. Convenient synthesis and transformation of 2,6-dichloro-4-iodopyridine. Org. Lett. 2001, 3, 4263–4265. 10.1021/ol0169269. [DOI] [PubMed] [Google Scholar]
- Smith P. K.; Krohn R. I.; Hermanson G. T.; Mallia A. K.; Gartner F. H.; Provenzano M. D.; Fujimoto E. K.; Goeke N. M.; Olson B. J.; Klenk D. C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- Xi B.; Yu N.; Wang X.; Xu X.; Abassi Y. The application of cell-based label-free technology in drug discovery. Biotechnol. J. 2008, 3, 484–495. 10.1002/biot.200800020. [DOI] [PubMed] [Google Scholar]
- Yu N.; Atienza J. M.; Bernard J.; Blanc S.; Zhu J.; Wang X.; Xu X.; Abassi Y. A. Real-time monitoring of morphological changes in living cells by electronic cell sensor arrays: an approach to study G protein-coupled receptors. Anal. Chem. 2006, 78, 35–43. 10.1021/ac051695v. [DOI] [PubMed] [Google Scholar]
- Schrödinger, LLC . Small-Molecule Drug Discovery Suite 2018-4; Schrödinger, LLC: New York, 2018. [Google Scholar]
- Jacobson M. P.; Pincus D. L.; Rapp C. S.; Day T. J. F.; Honig B.; Shaw D. E.; Friesner R. A. A hierarchical approach to all-atom protein loop prediction. Proteins 2004, 55, 351–367. 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- Schrödinger, LLC . LigPrep Schrödinger Release 2019–1; Schrödinger, LLC: New York, 2019. [Google Scholar]
- Schrödinger, LLC . The PyMOL Molecular Graphics System, 2.2.2; Schrödinger, LLC: New York, 2018. [Google Scholar]
- Cheng Y.-C.; Prusoff W. H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Kourounakis A.; Visser C.; de Groote M.; IJzerman A. P. Differential effects of the allosteric enhancer (2-amino-4,5-dimethyl-trienyl)[3-trifluoromethyl phenyl]methanone (PD81,723) on agonist and antagonist binding and function at the human wild-type and a mutant (T277A) adenosine A1 receptor. Biochem. Pharmacol. 2001, 61, 137–144. 10.1016/S0006-2952(00)00536-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.