

Abstract
With a renewed and growing interest in therapeutic oligonucleotides across the pharmaceutical industry, pressure is increasing on drug developers to take more seriously the sustainability ramifications of this modality. With 12 oligonucleotide drugs reaching the market to date and hundreds more in clinical trials and preclinical development, the current state of the art in oligonucleotide production poses a waste and cost burden to manufacturers. Legacy technologies make use of large volumes of hazardous reagents and solvents, as well as energy-intensive processes in synthesis, purification, and isolation. In 2016, the American Chemical Society (ACS) Green Chemistry Institute Pharmaceutical Roundtable (GCIPR) identified the development of greener processes for oligonucleotide Active Pharmaceutical Ingredients (APIs) as a critical unmet need. As a result, the Roundtable formed a focus team with the remit of identifying green chemistry and engineering improvements that would make oligonucleotide production more sustainable. In this Perspective, we summarize the present challenges in oligonucleotide synthesis, purification, and isolation; highlight potential solutions; and encourage synergies between academia; contract research, development and manufacturing organizations; and the pharmaceutical industry. A critical part of our assessment includes Process Mass Intensity (PMI) data from multiple companies to provide preliminary baseline metrics for current oligonucleotide manufacturing processes.
1. Introduction
Oligonucleotides are a novel class of therapeutic molecule for the treatment of a wide variety of diseases. They are short pieces of modified DNA, typically around 20 nucleotides in length. The fact that they can bind to complementary sequences through Watson–Crick base pairing gives them a unique ability to treat diseases that have been considered untreatable by traditional small molecule therapeutics. For example, they can bind to target ribonucleic acid (RNA) molecules and induce degradation or splice-switching in order to treat genetic diseases or viral infections. Alternatively, they can fold up on themselves to form three-dimensional structures called aptamers which can bind to target molecules with high specificity. The in vivo properties of oligonucleotides can be improved by introducing various modifications to the structure, some of which are summarized in Figure 1. Incorporating phosphorothioate linkages into the backbone increases nuclease stability and cellular uptake. Introducing 2′-modified nucleotides [e.g., 2′-OMe, 2′-F, 2′-O-methoxyethyl (MOE)] and conformationally constrained nucleotides [e.g., locked nucleic acid (LNA), constrained ethyl (cEt)] improves binding affinity and nuclease resistance.1−3
Figure 1.

Some common oligonucleotide modifications (BASE = nucleobase; R = H, OH, OMe, F, O-methoxyethyl; X = H, Me; Y = O, S).
Another advance in the field has been the development of drug delivery systems to target delivery of oligonucleotides to specific organs and tissue types. For example, the covalent attachment (conjugation) of N-acetylgalactosamine (GalNAc) ligands to target liver hepatocytes2,3 and the Glucagon-like peptide-1 (GLP-1) ligand to target pancreatic beta cells.4 A lipid nanoparticle formulation has also been used to target delivery to the liver.5,6 As a result of these advances, there are now numerous oligonucleotides in clinical development, and at the time of writing, 12 compounds have been approved for commercial use.7
2. Current Oligonucleotide Manufacturing Process
2.1. Process Overview
The manufacture of single-stranded therapeutic oligonucleotides typically consists of four key components: (1) oligonucleotide synthesis on a solid support; (2) cleavage from the solid support and removal of protecting groups; (3) purification by preparative chromatography; and (4) isolation by lyophilization (Figure 2).
Figure 2.

Overview of the current oligonucleotide manufacturing process.
2.2. Synthesis
Oligonucleotides are synthesized using the solid-supported phosphoramidite method which has been the mainstay of oligonucleotide synthesis for almost 40 years.8 The basic starting materials are phosphoramidite monomers derived from protected nucleosides. Synthesis occurs in a column charged with a solid support, historically controlled-pore glass (CPG) with the first nucleotide preloaded, but more recently polystyrene with a UnyLinker molecule attached.9 The synthesis is computer-controlled and fully automated, and the reagents are flowed through the column sequentially to perform the reactions on the solid support. The synthesis typically proceeds in a 3′ to 5′ direction by a four-step synthesis cycle (Figure 3) with each step followed by a solvent wash:
-
(1)
Detritylation: The dimethoxytrityl (DMT) protecting group on the 5′-hydroxyl group is removed with a solution of an organic acid, typically dichloroacetic acid (DCA) in toluene.
-
(2)
Coupling: The next nucleoside phosphoramidite in the sequence is activated with an acidic activator and coupled with the 5′ hydroxyl group of the growing oligonucleotide chain.
-
(3)
Oxidation/Sulfurization: Oxygen or sulfur is added to the phosphorus atom using iodine or a sulfurizing reagent to give either a phosphodiester or a phosphorothioate group.
-
(4)
Capping: Unreacted 5′ hydroxyl groups are capped with acetic anhydride to prevent further chain extension of coupling failures and minimize N-1 impurities.
Figure 3.
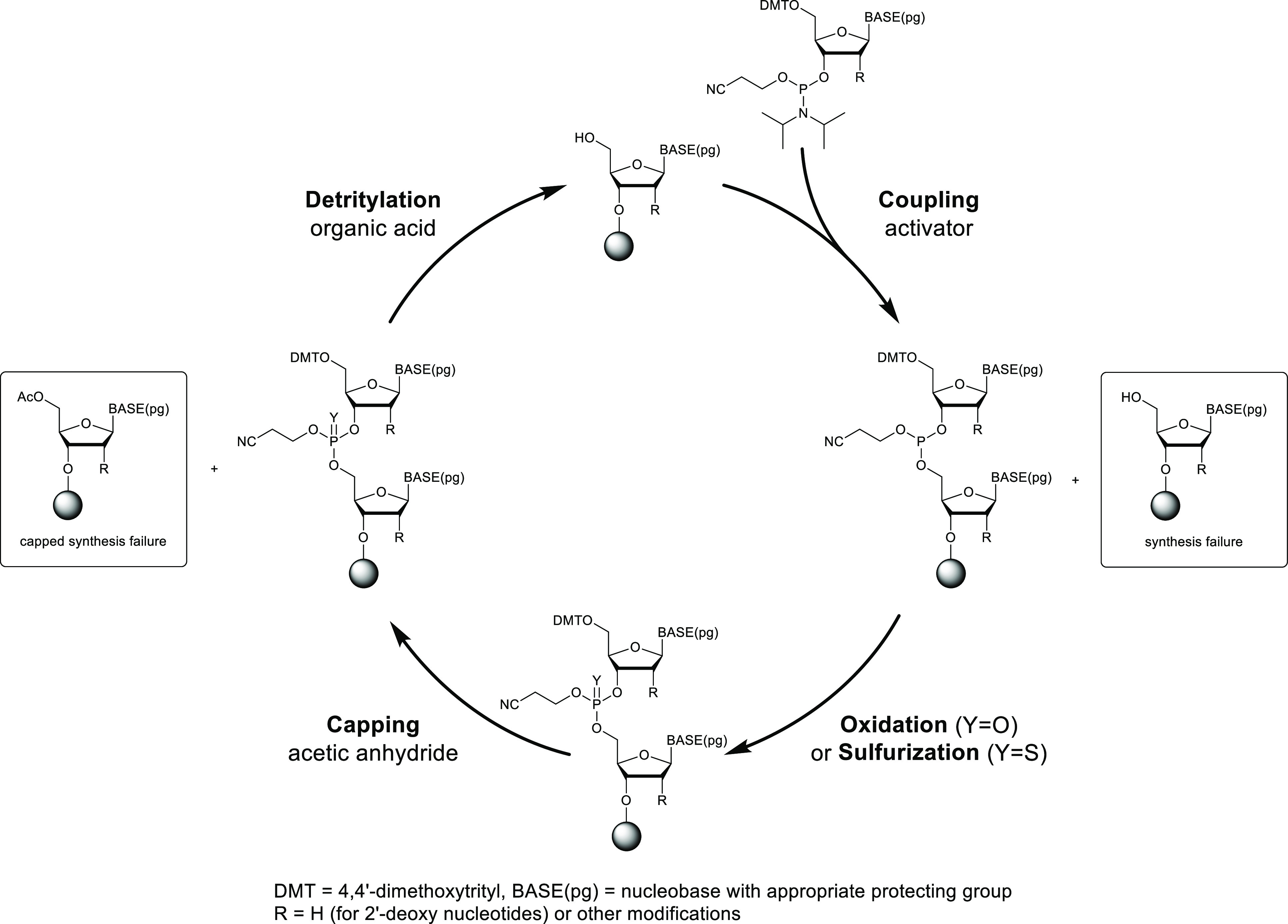
Oligonucleotide synthesis cycle.
This synthesis cycle is repeated for each nucleotide in the sequence until the fully protected oligonucleotide has been assembled on the solid support. All reactions in the cycle are rapid: a single cycle takes approximately 45 min and a full synthesis of a 20-mer is complete in less than 24 h. The final step of the synthesis is the removal of the cyanoethyl protecting groups from the phosphodiester/phosphorothioate backbone by washing with an amine solution.
2.3. Cleavage and Deprotection
Once the synthesis is complete, the full-length product is cleaved from the solid support and the base-labile protecting groups are removed, typically by heating with aqueous ammonium hydroxide. For RNA-containing oligonucleotides, the removal of the 2′-O-silyl protecting groups requires treatment with fluoride. Once cleaved from the solid support, the crude oligonucleotide can be analyzed for concentration, identity, and purity; crude purities of greater than 80% are not uncommon.
2.4. Purification
Several different modes of preparative chromatography can be used to purify oligonucleotides,10,11 the most common of which are reversed-phase and anion exchange chromatography. Often, the final 5′-DMT protecting group is left attached to the oligonucleotide as its hydrophobic properties can be exploited to assist with chromatographic resolution. Sample chromatograms for various oligonucleotide purification methods are shown in Figure 4.
Figure 4.

Sample chromatograms for oligonucleotide purification methods.
RP-HPLC employs a hydrophobic stationary phase such as a polystyrene-divinylbenzene copolymer or a C18-modified silica to retain the oligonucleotide with the aid of its hydrophobic 5′-DMT group. Impurities without the DMT-group are washed out with an eluent of intermediate strength, and the product fraction is then recovered at higher solvent strength (Figure 4A). The DMT group can then be removed in solution by treatment with acid.
AEX chromatography is typically performed using a quaternary ammonium-functionalized stationary phase which binds the negatively charged oligonucleotide at low salt concentrations. Often, the interaction of the DMT group with the partially hydrophobic resin is exploited to remove some of the DMT-off impurities with a wash at intermediate salt concentration. Detritylation is then carried out on-column with an acid wash and the product is eluted by increasing the salt concentration (Figure 4B). Alternatively, the whole purification can be performed in DMT-off mode, relying on the salt gradient alone to separate impurities from the API.
2.5. Isolation
After purification, a dilute aqueous oligonucleotide solution is obtained which is desalted to remove elution buffer components and concentrated prior to either further processing steps or the final API isolation step. One of two processes can be employed to desalt: precipitation or tangential flow filtration (TFF).
Precipitation of oligonucleotides from aqueous solution can be achieved by adding ethanol or 2-propanol, either at reduced temperature or using salt promoters.12,13 Inorganic salts remain in the supernatant that is decanted off, and the residue is redissolved in water to the required concentration.
Alternatively, the desalting of oligonucleotides can be achieved by TFF across a membrane.14,15 In this process, small molecules such as water, inorganic salts, and organic molecules pass through channels in the membrane to the permeate, whereas larger species such as oligonucleotides do not and are concentrated in the retentate (Figure 5). Water is added to the retentate to help wash the small molecules through the membrane and achieve desalting.16 The combined process is often referred to as ultrafiltration/diafiltration (UF/DF) and leads to a reduction in inorganic concentration by several orders of magnitude while increasing the oligonucleotide concentration.
Figure 5.

Desalting via TFF.
Once desalting is complete, further processing steps may be required depending on the final structure of the API. An annealing step is required for double-stranded oligonucleotides, e.g., small interfering RNAs (siRNAs), to join the two complementary strands together. This usually involves mixing solutions of the two strands, heating the solution, and cooling to facilitate hybridization. A conjugation step may also be required for oligonucleotides covalently attached to targeting ligands such as GalNAc. Further purification, desalting, and concentration steps are usually employed after conjugation.
The API is isolated by lyophilization to give a low-density, hygroscopic, electrostatic, amorphous solid, typically as the all-sodium salt. This solid is usually stored frozen and shipped to the drug product facility for formulation, which will generally be an aqueous solution for parenteral delivery.
2.6. Call to Action
The general manufacturing process described above is a remarkable achievement, able to synthesize, purify, and isolate oligonucleotides in excellent yield (∼50%) and purity (∼90%); especially considering the size and complexity of the molecules and the number of chemical transformations involved (∼80). It is a flexible process, able to produce a large number of nucleic acid derivatives, such as those incorporating modifications, single- and double-stranded oligonucleotides, aptamers, and conjugates. Finally, the process can be readily scaled up from gram to multikilogram scale to produce oligonucleotides for clinical and commercial applications.
However, this achievement comes at an environmental cost in terms of waste, chemical hazards, and energy efficiency, and this burden will only grow as volumes of therapeutic oligonucleotides increase. In this Perspective, we17 aim to summarize the current challenges in oligonucleotide manufacture in terms of sustainability. We wish to review efforts to date to improve the “greenness” of oligonucleotide manufacture, highlight possible solutions and encourage collaboration between academia, the pharmaceutical industry, and contract manufacturing organizations in this field.
3. Assessing the Sustainability of the Current Process
There are several ways to evaluate the sustainability of the oligonucleotide manufacturing process. In this article we assess:
The greenness of the process against the 12 Principles of Green Chemistry
The material efficiency of the process using the PMI metric
The hazards of reagents used in the process by consulting REACH lists and solvent selection guides
3.1. Assessment against the 12 Principles of Green Chemistry
The 12 Principles of Green Chemistry were developed by Paul Anastas and John Warner as a way to “help reduce or eliminate the use or generation of hazardous substances in the design, manufacture and application of chemical products.”18 A traffic light assessment of the oligonucleotide process against these principles is presented in Table 1, highlighting six areas for improvement.
Table 1. Assessment of the Oligonucleotide Manufacturing Process against the 12 Principles of Green Chemistry.

Waste is highlighted as a significant issue for oligonucleotide manufacturing. Excess starting materials and reagents are used to drive the reactions to completion, and wash solvents are used to remove them. Purification by preparative chromatography uses large volumes of mobile phase which are discarded as waste. Atom economy for the starting materials is poor (for example an average of 36% for the four deoxyribonucleoside phosphoramidites) due to the extensive use of protecting groups; consequently the majority of atoms in these materials are not incorporated into API but are discarded as waste. The nucleoside cores of the phosphoramidite starting materials can be derived from renewable feedstocks, but the protecting groups that they possess and the majority of the other reagents and solvents are derived from nonrenewable petroleum-based sources. All of the chemical reactions in the process are stoichiometric rather than catalytic, with many reagents used in vast excess. Finally, although some real-time data is generated during processing (for example UV, conductivity, temperature and pressure) it is not used for pollution prevention purposes.
3.2. PMI Assessment
The ACS GCIPR has developed a series of green chemistry tools to help chemists and engineers assess and improve the sustainability of their chemical processes.19 These include reagent and solvent guides, a Principal Component Analysis (PCA) solvent selection tool, and a PMI calculator, among other items. Application of these tools to the current oligonucleotide manufacturing process provides a baseline assessment of sustainability from which to improve and enables comparison with other classes of therapeutic molecules such as small molecules, peptides, and biopharmaceuticals.
PMI is a measure of material efficiency and is a key metric for assessing the sustainability of a process.20,21 Put simply, it is a measure of the quantity of raw materials required to produce one kilogram of isolated API (Figure 6).
Figure 6.

PMI calculation.
PMI is a good metric for measuring the sustainability of a process. It takes into consideration all of the materials used in a process, including water, and incorporates other metrics like yield and atom economy. It can be calculated with readily available data and has the advantage of being simple and easy to understand. However, it does not take into account other factors such as energy usage, environmental impact or starting material complexity.
For this article, five ACS GCIPR member companies compiled PMI metrics on eight oligonucleotide processes in development. These compounds included representatives from several oligonucleotide classes (antisense, siRNA) possessing different modifications (MOE, LNA, cEt) and in different phases of development. Some processes included a capping step; others did not.
The oligonucleotide manufacturing process was divided into stages (synthesis, purification, and isolation) and these were further subdivided into steps as required. The materials were classified as reagent, reaction solvent, wash solvent, or water according to their use in each step/stage. The quantities of materials for the whole process were summed to give the total “quantity of raw materials in (kg)” figure. This was divided by the “quantity of API out (kg)” to give a PMI figure for each oligonucleotide.
Only materials used in the oligonucleotide manufacturing processes were included in this analysis; materials used in the synthesis of the phosphoramidite nucleoside starting materials were not included. It is acknowledged that the complexity of the phosphoramidites will affect the mass efficiency of the overall process. However, the phosphoramidites are small molecules synthesized by conventional batch chemistry and their PMIs can be improved using standard small molecule process development approaches. Materials used in the synthesis of targeting ligands (such as the GalNAc intermediate) and the processing steps to incorporate them were also not included for similar reasons. A summary of the output of this assessment is shown in Table 2.
Table 2. PMI Data for Eight Oligonucleotides in Development.
| compd | phase of development | no. of nucleotides | PMI | PMI per nucleotide |
|---|---|---|---|---|
| A | 1 | 20 | 4841 | 242 |
| B | 1 | 20 | 3369 | 168 |
| C | 2 | 21 | 4134 | 197 |
| D | 2 | 40 | 7023 | 176 |
| E | 2 | 19 | 3385 | 178 |
| F | 2 | 16 | 3581 | 224 |
| G | 3 | 20 | 3035 | 152 |
| H | 3 | 20 | 5028 | 251 |
PMIs were obtained in the range of 3035–7023 with an average of 4299. These values sit between the PMI figures calculated for small molecule (median 168–308)22 and biopharmaceutical (∼8300)23 APIs and are probably comparable to similarly sized peptides, although little information is published for peptides at present.24
There are different ways to express PMI for oligonucleotides. For comparison with other oligonucleotides, PMI per nucleotide would be a fair comparison as it takes chain length into consideration. These values are shown in Table 2 and range from 152 to 251 with an average of 199. For comparison with small molecule APIs, quoting PMI per synthesis step or molecular weight might be more appropriate as it would factor in molecular complexity and size. Perhaps the best way to compare sustainability across all classes of therapeutic molecule would be to express PMI as a function of a complete dosing regimen as it would reflect the total amount of waste generated per patient?
The data can be visualized in various ways. Plotting PMI versus development phase shows no clear downward trend with time as is typically seen with small molecule APIs22 (Figure 7). This may be because oligonucleotide manufacturing is a platform approach where the current best practice conditions from previous projects are applied to new compounds, resulting in fewer opportunities to reduce PMIs during the development process.
Figure 7.

PMI per nucleotide across phase of development.
The data can be analyzed in more detail by plotting the contribution to PMI by material type, process stage, and synthesis step (Figure 8, graphs A–C). The colors of the graphs give an approximate indication of the material type: shades of blue represent aqueous materials and shades of yellow represent organic materials. Graph A (by material type) shows that approximately equal amounts of organic materials and water are used in the process, and that wash solvent makes up half of the organic materials. There is a higher percentage of water used in oligonucleotide processing (51%) compared with small molecule processing (28%),22 probably a consequence of the aqueous-based purification techniques employed. Graph B (by process stage) shows that the synthesis and purification stages make the most significant contributions to the PMI. Graph C (by synthesis step) shows that the detritylation step makes the biggest contribution in the synthesis stage, accounting for almost half of the material used.
Figure 8.

Percentage contribution to PMI by material type, stage and synthesis step.
3.3. Assessment of Compounds Used in Oligonucleotide Manufacture
A list of compounds used in oligonucleotide manufacture by the member companies was compiled and assessed against EU REACH lists and published solvent guides to highlight any materials of concern from a green chemistry perspective.
At the time of writing, none of the compounds were on the list of substances included in Annex XIV of REACH (the “Authorization List”).25 However, disodium tetraborate has been highlighted as toxic for reproduction and is on the REACH Candidate List of substances of very high concern for Authorization.26 This compound is used as a buffer for oligonucleotide conjugation reactions, and it may be worth investigating replacements in anticipation of it being added to the authorization list. Toluene and dichloromethane (DCM) appear in the REACH Annex XVII table (Substances restricted under REACH),27 limiting their use to <0.1% by weight in products available to the general public.
Regarding solvent guides, the ACS GCIPR recommends using the CHEM21 solvent selection guide28 which is based on a recent survey of publicly available information. The guide scores compounds against safety, health, and environmental criteria and classifies them as recommended, problematic, or hazardous after consideration of other factors such as occupational exposure limits. Table 3 shows the compounds in oligonucleotide manufacture that are present in the guide and their classification.
Table 3. Assessment of Compounds Used in Oligonucleotide Manufacture against the CHEM21 Solvent Guide.

Of the compounds highlighted as hazardous, DCM has been used as a solvent for the detritylation step but has largely been replaced with toluene in large-scale manufacture. Pyridine and triethylamine act as bases in the oxidation, sulfurization, capping, and backbone deprotection steps. Both compounds have low occupational exposure limit values resulting in their classification as hazardous. It may be worth investigating replacements for these compounds to remove all solvents used in oligonucleotide manufacture from the hazardous category.
Other potentially problematic or hazardous compounds are used or generated during oligonucleotide manufacture that do not appear on these lists. For example, DCA (suspected of causing cancer, very toxic to aquatic life); ammonium hydroxide (very toxic to aquatic life); and acrylonitrile (generated during the backbone deprotection step, may cause cancer, suspected of damaging fertility or the unborn child) which may be a concern from a safety, health, or environment perspective.
4. Improving Sustainability in the Short Term
Several approaches could be proposed to improve the sustainability of the current oligonucleotide manufacturing process following on from the assessment in section 3.
4.1. Synthesis
Significant progress to improve the sustainability of oligonucleotide synthesis has been made in recent decades. Work began in the late 1990s as the first compounds approached late phase clinical studies and commercialization. Early efforts focused on minimizing waste through the recovery and reuse of reagents (amidites,29 activator,29 DMT group30) and solvents (DCM,30 toluene31) and the increase of reagent concentration (detritylation step).32−34 Progress was also made in replacing hazardous solvents and reagents such as DCM35,36 and tetrazole.37 These innovations have been summarized in an early review article.38
Subsequent developments focused on the reduction, recovery or replacement of solvents used, perhaps due in part to the global acetonitrile shortage in 2008.39 Acetone has been shown to be an alternative greener wash solvent for oligonucleotide synthesis,40 and acetonitrile itself can be recovered and reused in oligonucleotide synthesis.41 Acetonitrile containing up to 200 ppm water can be used in sulfurization, capping and wash steps, facilitating its reuse.42 Varying the column height throughout a synthesis using dynamic axial compression columns eliminates the headspace, reducing the amount of wash solvent required for the detritylation step.43,44 Recently, it has been shown that the byproducts of the sulfurization reaction can act as capping agents for coupling failures, allowing for the removal of the capping step and reducing the PMI across the synthesis stage by 14%.45,46
Despite this previous work, significant opportunities remain to improve the sustainability of the synthesis. The PMI data in Figure 8 shows that the synthesis stage is still responsible for approximately half of the materials used in the process and that they are predominantly organic materials. Efforts could focus on reducing the volumes of synthesis wash solvents as they represent half of the organic materials used and contribute 25% to the total PMI. Some wash steps are less important than others, for example, those following the coupling and sulfurization steps, and these could be minimized. Other possibilities to reduce wash volumes could include using nitrogen pressure to displace reagents from the column, or collecting the cleaner solvent from the end of a wash and reusing it at the beginning of the next equivalent wash. The detritylation step accounts for approximately half of the materials in the synthesis stage and could therefore be a focus of attention. Alternative acids could be investigated that remove the DMT group more efficiently and require less wash solvent to remove. Other ways to improve the PMI could be to decrease reagent equivalents; increase reagent concentrations; use catalytic rather than stoichiometric reagents; and maximize yield by preventing impurity formation. Solvent recycling also has a very important part to play in minimizing waste.
To address the poor atom economy of the phosphoramidite starting materials, alternative protecting groups with smaller molecular weights could be investigated. The DMT group in particular corresponds to approximately one-third of the mass of the deoxy phosphoramidites, and its replacement may also help to minimize the amounts of reagents and wash solvents required for removal. However, it would still be advantageous to use a DMT-protected phosphoramidite in the final cycle in order to use the group as a purification handle. An inventory of reagents used in oligonucleotide synthesis could be compiled and the origin of the compounds determined in an effort to obtain reagents from renewable sources. Finally, although on line analysis has been used to monitor oligonucleotide synthesis,47,48 efforts could be made to apply it to pollution prevention and waste minimization.
4.2. Purification
The PMI data in Figure 8 shows that the purification stage is responsible for approximately half of the material used in the process; therefore, improvements to this stage would make a significant contribution to creating a greener process.
From a green chemistry perspective, the use of reversed-phase purification comes with the cost of hazardous waste disposal since all waste from the process contains organic solvent. At high production volumes, solvent recovery could be economically and environmentally beneficial; however, roughly 50% of the waste volume would still need to be discarded as aqueous-rich hazardous waste. In contrast, AEX chromatography has a lower potential environmental impact because the waste streams produced are aqueous and thus more easily disposed of than hazardous waste.
Recently, HIC has been developed as a green alternative to RP-HPLC.49 The technique is performed on DMT-on material and affords very similar selectivity and performance to reversed-phase chromatography (Figure 4C). It employs high salt concentrations (typically ammonium sulfate) in an aqueous buffer to bind the oligonucleotide to a weakly hydrophobic resin, followed by elution at a lower salt gradient. The flow-through fraction is typically treated as hazardous waste since it contains organic fragments from the cleavage and deprotection step, but subsequent waste can be disposed of as aqueous streams (subject to local ordinances). Another advantage is that elution occurs at very low salt concentrations. The product can then be detritylated and, if needed, directly loaded onto an AEX column without significant buffer adjustment for a second polishing chromatography step.
Notwithstanding the reduced environmental impact of employing aqueous-based purifications, there is room to improve the chromatographic techniques employed. AEX chromatography in displacement mode was suggested several years ago.50−52 However, the development was performed in an era when full understanding of the various process impurities was not available, and no further efforts have been made since. Displacement chromatography can be very efficient and generate concentrated, purified product, potentially with significant reduction in solvent usage, so a revisit may be warranted. While there has been a study showing the benefits of oligonucleotide purification using a simulated moving-bed multicolumn chromatography system,53 more sophisticated continuous multicolumn gradient technologies54 have not been adequately explored. These too hold the promise of increased yield and thus lower solvent/buffer consumption.
4.3. Isolation
Inspection of the PMI data in Figure 8 shows that the isolation stage accounts for approximately 10% of the materials used in the process, the majority of which is water. This is typically highly purified water rather than conventional process water and so carries increased economic and environmental cost.
With respect to TFF, water consumption is related to the mode of operation of diafiltration, the most efficient of which is continuous diafiltration at constant volume.55 In this process, the volume of the retentate is kept constant throughout by diluting with water at the same rate as permeate is produced. Continuous diafiltration uses approximately 70% of the water of alternative methods and therefore has a positive impact on PMI.
Further improvements could come from running the diafiltration at a higher oligonucleotide concentration. This would require a lower volume of water to achieve an equal number of exchanges, and therefore equivalent salt removal, compared to a lower concentration solution (Table 4). However, factors such as solution viscosity and membrane permeability may limit the maximum operable concentration. Another approach to minimizing diafiltration volumes would be to use on line monitoring of permeate conductivity to determine the exact end point of the operation.
Table 4. Impact of Solution Concentration on PMI during TFF.
| PMI
contribution from TFF |
||||
|---|---|---|---|---|
| solution concentration during continuous DF (mg/mL) | solution volume per kg oligo (L) | 7 diavolumes | 8 diavolumes | 9 diavolumes |
| 5 | 200 | 1400 | 1600 | 1800 |
| 20 | 50 | 350 | 400 | 450 |
| 40 | 25 | 175 | 200 | 225 |
Lyophilization is an energy-intensive operation (>2 kWh/kg56) due to the refrigeration system and vacuum pumps required to freeze the solution and induce sublimation and deposition. It represents one of the main process bottlenecks in oligonucleotide manufacturing, requiring several days to complete. One improvement would be to increase the amount of API lyophilized in a single batch. This would involve combining several batches after TFF and concentrating, for example using thin film evaporation (TFE), and would have the added benefit of improving the throughput of the process. However, there may be a limit to this approach since increased oligonucleotide concentration depresses the freezing point of the solution while also increasing viscosity. This increases time to freeze the solution and also increases resistance of the solid to permeation of water vapor.
Spray-drying is a lower energy alternative to lyophilization for isolation of oligonucleotide APIs. It can be operated continuously, increasing throughput, and delivers a free-flowing, amorphous powder with higher bulk density. However, the yield is significantly lower compared with lyophilization, which by extension increases the PMI. These losses can be minimized by processing larger batch sizes as the losses are related to equipment hold-up rather than batch size.
The oligonucleotide drug product is often an aqueous solution, and the process begins by dissolving the solid API in water. Therefore, an interesting option would be to remove the drying step altogether and isolate the API as a solution.57−59 This would remove the energy-intensive drying step and reduce the amount of water used in the drug product process. A careful assessment of other factors such as shipping of larger quantities of solution, relative clean room energy usage costs, and processing times is required to determine the full sustainability impact of such a change.
5. Improving Sustainability in the Long Term
The approaches discussed so far are short- to medium-term improvements to the current manufacturing process. Longer term goals could involve improving compound design or investigating alternative methods of synthesis and purification.
5.1. Compound Design
The advances in molecular design and targeted delivery (for example, the use of cEt nucleosides and GalNAc ligands) described in the Introduction have significantly increased potency and stability of oligonucleotide therapeutics, resulting in lower efficacious doses and less frequent administration. From a green chemistry perspective, these reductions in dose and frequency may result in reductions in oligonucleotide production volumes and therefore waste generated, providing the gains are not offset by the extra efforts required to incorporate the modifications.
A recent area of interest is the diastereoselective synthesis of phosphorothioate oligonucleotides.60−63 Traditional synthesis using the phosphoramidite method is not stereoselective, resulting in a mixture of diastereoisomers at each phosphorus atom in the backbone. There is evidence that phosphorothioate stereochemistry affects the metabolic stability and activity of oligonucleotides. If stereospecific compounds can be designed that require lower doses relative to stereorandom compounds, and if they can be manufactured with comparable sustainability metrics, then this may result in less waste generated for the reasons stated above.
5.2. Liquid-Phase Oligonucleotide Synthesis (LPOS)
One approach that has seen renewed interest recently is the concept of oligonucleotide synthesis in organic solution, termed LPOS.64 In this technology, the growing oligonucleotide is attached to a soluble anchor molecule and the reactions are performed in homogeneous solution. Compounds that would interfere with subsequent transformations are removed either by membrane filtration or precipitation after each step. While primarily developed to address scalability issues with the current solid-supported method, there may be sustainability benefits to these approaches. For example, because the intermediates are soluble, it is possible to monitor and optimize each individual step in a similar manner to standard small molecule chemistry, minimizing the use of excess reagents. Telescoping several steps of the synthetic cycle could facilitate lower wash volumes thereby decreasing solvent consumption. If crude purity can be increased, then purification steps can be streamlined or omitted entirely, having a positive impact on PMI.
5.3. Enzyme-Catalyzed Synthetic Cycle
A longer-term goal could be to develop an alternative synthesis cycle based on the enzymes involved in the synthesis of DNA in naturally occurring systems. Progress has been made toward this goal by a number of groups using the template-independent polymerase terminal deoxynucleotidyl transferase (TdT).65−67 The enzyme can add deoxynucleotide triphosphates (dNTPs) to the 3′-end of an oligonucleotide primer in an indiscriminate fashion. With appropriate protection of the 3′-hydroxyl group of the dNTPs, a single controlled addition can be performed. The enzyme and excess dNTPs are then removed, followed by a deprotection step, establishing a synthetic cycle (Figure 9).
Figure 9.

Enzyme-catalyzed single base extension using TdT.
A related approach involves the use of DNA polymerases and reverse transcriptases to synthesize single-stranded oligonucleotides controlled by transient hybridization to neighboring strands.68
Further development of these methods would be required to enable kilogram-scale synthesis of the kinds of therapeutic oligonucleotides illustrated in Figure 1, such as the toleration of modified nucleosides and incorporation of phosphorothioate linkages. However, this remains a very promising area of research, and one which has the potential to bring sustainability benefits such as the use of aqueous systems and the removal of several protecting groups, thus minimizing the use of hazardous organic solvents and reagents and improving atom economy.
5.4. Enzyme-Catalyzed Templated Ligation
Another enzyme-based technology under development is oligonucleotide synthesis by templated ligation.69,70 This approach involves the assembly of short, unpurified 5′-(thio)phosphorylated oligonucleotide fragments on a complementary template in aqueous solution. Several of these template strands are attached to a soluble chemical “hub” molecule. Once the fragments are assembled, an engineered DNA ligase joins them together to form the full-length product. Uncoupled fragments (impurities) are selectively detached from the hub by heating and removed by a nanofiltration/diafiltration operation while the product remains attached to the hub and template. At higher temperature, the product detaches from the template and is recovered in a similar fashion, and the hub is recycled for further use (Figure 10).
Figure 10.

Enzyme-catalyzed templated ligation.
Reported purities are high enough to enable the removal of further purification steps which would have a significant impact on the PMI. Such a technique could be coupled with the single base extension work above for the synthesis of the fragments to give an entirely aqueous-based method of oligonucleotide synthesis.
6. Summary
Oligonucleotides are an exciting class of potential therapeutics, offering the possibility of treating disease through direct targeting of genes and thereby influencing proteins at the level of initial expression. Although this concept has been under investigation for several decades, new technologies (sugar, base, and phosphate modifications; conjugation of oligonucleotides with functional or targeting ligands; as well as techniques useful in assembling, purifying, and isolating the desired species) have advanced the field to the point of commercial approval. With this scientific maturation come opportunities to drive further efficiency into the process of oligonucleotide discovery, development and manufacturing. These include improving the sustainability of all operations for a greener and more economical process. This article has presented practices and tools that can be used to measure and improve the sustainability footprint and allow further advances in delivering high-quality oligonucleotide APIs into the market for the benefit of patients.
Acknowledgments
This manuscript was developed with the support of the American Chemical Society (ACS) Green Chemistry Institute Pharmaceutical Roundtable (GCIPR) (www.acsgcipr.org). The ACS GCI is a not-for-profit organization whose mission is to catalyze and enable the implementation of green and sustainable chemistry throughout the global chemistry enterprise. The ACS GCI Pharmaceutical Roundtable, composed of pharmaceutical and related industries, was established in 2005 to encourage innovation while catalyzing the integration of green chemistry and green engineering in the pharmaceutical industry. The activities of the Roundtable reflect its members’ shared belief that the pursuit of green chemistry and engineering is imperative for business and environmental sustainability.
Biographies

Ben Andrews is a process chemist in Chemical Development at GlaxoSmithKline (Stevenage, UK). He has a background in organic chemistry with a Ph.D. and postdoc in the field of asymmetric synthesis. In his 17 years at GSK he has contributed to the development of several mid- to late-phase compounds, from small molecules to oligonucleotides. He has 10 years’ experience in oligonucleotide manufacturing with expertise in synthesis, purification, and isolation. He is a member of the European Pharma Oligonucleotide Consortium (EPOC) purge factor team and currently leads the ACS GCIPR oligonucleotide team.

Firoz Antia is Director, Antisense Oligonucleotide Process Development at Biogen. In this and prior roles at Palatin Technologies, Merck & Co., J&J, and Sandoz, he has worked on and managed the synthesis, purification, and analysis of oligonucleotides, peptides, small molecules and natural products. He holds a Ph.D. in Chemical Engineering from Yale University.

Shawn Brueggemeier is a Director in Chemical Process Development, Drug Substance Operations and Supply at Bristol Myers Squibb and has experience with oligonucleotide process development and scale-up across clinical phases of pharmaceutical development. He holds a Ph.D. in Chemical Engineering from the University of Wisconsin—Madison.

Louis Diorazio received his B.Sc. and a Ph.D. in aromatic fluorination from Imperial College, London before undertaking postdoctoral studies with Prof. Phil Magnus at the University of Texas at Austin on natural product synthesis. He has 27 years of industry experience in areas ranging from early discovery investigations through to commercial application and is a Principal Scientist in Chemical Development at AstraZeneca. He is also a cochair of the American Chemical Society Green Chemistry Institute Pharmaceutical Roundtable, and his interests include oligonucleotides, synthetic Route Design, and delivering sustainability in pharma processing.

Stefan Koenig is a process chemist at Genentech (South San Francisco, CA), where he has contributed to several R&D projects, including small molecules, antibody–drug conjugates, and new modalities. He started his industrial career at Sepracor/Sunovion (Marlboro, MA) after earning his Ph.D. from Yale University (New Haven, CT) and a postdoc at the ETH Zurich (Switzerland). He has served as cochair of the American Chemical Society (ACS) Green Chemistry Institute Pharmaceutical Roundtable (GCIPR) and is an elected member of the executive committee of the ACS Division of Organic Chemistry (DOC).

Michael Kopach earned a Ph.D. in Organic Chemistry from the University of Virginia in 1995 under the guidance of Prof. W. D. Harman. He then completed postdoctoral studies in natural product synthesis at Colorado State University with Prof. A. I. Meyers. Mike began his industrial career in 1997 at Roche, where his primary responsibilities included development and implementation of commercial processes for nelfinavir, Xenical, and enfuvirtide—the first synthetic peptides produced on tonnage scale. In 2001, Mike joined Eli Lilly and Company and has led several small-molecule phase 1 to phase 3 R&D projects. Throughout his industrial career, Mike has led research teams applying green chemistry principles to product research and development and has published several collaborative articles in this field. For the past decade, Mike has represented Eli Lilly and Company at the ACS Green Chemistry Institute Pharmaceutical Roundtable including serving a two-year term as cochair from 2011 to 2013 and another from 2017 to 2019. After a 14-year career within Lilly Small Molecule R&D, Mike now has responsibility for synthetic peptide projects. In this area, Mike has led an internal infrastructure build to bring peptide synthesis capability in house.

Heewon Lee is a director in the Chemical Development department at Boehringer Ingelheim Pharmaceuticals, Inc., in Ridgefield, CT. She earned a B.S. degree in chemistry and an M.S. degree in physical chemistry at Seoul National University in Seoul, Korea. She earned her Ph.D. in analytical chemistry at the University of Michigan, Ann Arbor, MI. After working for a couple of years at ArQule, she joined the Medicinal Chemistry department at Boehringer Ingelheim and then moved to the Chemical Development department. She is a member of the Pharmaceutical IQ (Innovation and Quality) Working Groups, Enabling Technologies Consortium (ETC), ACS Green Chemistry Institute Pharmaceutical Roundtable (GCIPR), and USP Expert Committee on Small Molecules.

Martin Olbrich obtained his Ph.D. in Organic Chemistry (2014) from the Ludwig Maximilian University of Munich under the supervision of Professor Dirk Trauner. In 2015, he joined the Process Chemistry & Catalysis group at F. Hoffmann-La Roche, Switzerland, where he is now responsible for the late-stage development and manufacturing of oligonucleotide processes. He is currently serving as the Roche Green Chemistry team lead.

Anna Watson is a Process Engineer in Pharmaceutical Technology & Development at AstraZeneca and joined the company in 2015. She primarily works on drug substance process development for late-stage oligonucleotide and peptide projects. This includes leading oligonucleotide downstream development and supporting clinical drug substance manufactures.
The authors declare no competing financial interest.
References
- Dirin M.; Winkler J. Influence of diverse chemical modifications on the ADME characteristics and toxicology of antisense oligonucleotides. Expert Opin. Biol. Ther. 2013, 13 (6), 875–88. 10.1517/14712598.2013.774366. [DOI] [PubMed] [Google Scholar]
- Wan W. B.; Seth P. P. The Medicinal Chemistry of Therapeutic Oligonucleotides. J. Med. Chem. 2016, 59 (21), 9645–9667. 10.1021/acs.jmedchem.6b00551. [DOI] [PubMed] [Google Scholar]
- Khvorova A.; Watts J. K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017, 35 (3), 238–248. 10.1038/nbt.3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammala C.; Drury W. J. 3rd; Knerr L.; Ahlstedt I.; Stillemark-Billton P.; Wennberg-Huldt C.; Andersson E. M.; Valeur E.; Jansson-Lofmark R.; Janzen D.; Sundstrom L.; Meuller J.; Claesson J.; Andersson P.; Johansson C.; Lee R. G.; Prakash T. P.; Seth P. P.; Monia B. P.; Andersson S. Targeted delivery of antisense oligonucleotides to pancreatic beta-cells. Sci. Adv. 2018, 4 (10), eaat3386. 10.1126/sciadv.aat3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowdy S. F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 2017, 35 (3), 222–229. 10.1038/nbt.3802. [DOI] [PubMed] [Google Scholar]
- Hoy S. M. Patisiran: First Global Approval. Drugs 2018, 78 (15), 1625–1631. 10.1007/s40265-018-0983-6. [DOI] [PubMed] [Google Scholar]
- Vitravene (fomivirsen), Macugen (pegaptanib), Kynamro (mipomersen), Defitelio (defibrotide), Exondys 51 (eteplirsen), Spinraza (nusinersen), Heplisav-B, (hepatitis B vaccine, adjuvanted), Onpattro (patisiran), Tegsedi (inotersen), Waylivra (volanesorsen), Givlaari (givosiran), and Vyondys 53 (golodirsen).
- Brown T.Nucleic Acids Book. https://www.atdbio.com/nucleic-acids-book (accessed 2020-01-09).
- Ravikumar V. T.; Kumar R. K.; Olsen P.; Moore M. N.; Carty R. L.; Andrade M.; Gorman D.; Zhu X.; Cedillo I.; Wang Z.; Mendez L.; Scozzari A. N.; Aguirre G.; Somanathan R.; Berneès S. UnyLinker: An Efficient and Scaleable Synthesis of Oligonucleotides Utilizing a Universal Linker Molecule: A Novel Approach To Enhance the Purity of Drugs. Org. Process Res. Dev. 2008, 12 (3), 399–410. 10.1021/op8000178. [DOI] [Google Scholar]
- Capaldi D. C.; Scozzari A. N., Manufacturing and Analytical Processes for 2′-O-(2-Methoxyethyl)-Modified Oligonucleotides. In Antisense Drug Technology: Principles, Strategies and Applications, 2nd ed.; Crooke S. T., Ed.; CRC Press: Boca Raton, 2008; pp 401–434. [Google Scholar]
- Paredes E.; Aduda V.; Ackley K. L.; Cramer H., Manufacturing of Oligonucleotides. In Comprehensive Medicinal Chemistry III; Elsevier, 2017; Vol. 6, pp 233–279. [Google Scholar]
- Cleaver J. E.; Boyer H. W. Solubility and dialysis limits of DNA oligonucleotides. Biochim. Biophys. Acta, Nucleic Acids Protein Synth. 1972, 262 (2), 116–24. 10.1016/0005-2787(72)90224-9. [DOI] [PubMed] [Google Scholar]
- Moore M. N.; Arthur J. C.; Vansooy K.; Scozzari A. N.. Use of salts and alcohols in precipitation and purification of oligonucleotides. WO2002100873A1, 2002.
- Schwartz L.; Seeley K.. Introduction to Tangential Flow Filtration for Laboratory and Process Development Applications. https://laboratory.pall.com/content/dam/pall/laboratory/literature-library/non-gated/id-34212.pdf (accessed 2020-01-09).
- novasep Industrial cross-flow filtration technology. https://www.novasep.com/technologies/industrial-cross-flow-filtration-technology.html (accessed 2020-01-09).
- Schwartz L.Diafiltration: A Fast, Efficient Method for Desalting, or Buffer Exchange of Biological Samples. https://laboratory.pall.com/content/dam/pall/laboratory/literature-library/non-gated/02.0629_Buffer_Exchange_STR.pdf (accessed 2020-01-09).
- The members of the American Chemical Society (ACS) Green Chemistry Institute Pharmaceutical Roundtable (GCIPR) oligonucleotide subteam.
- Anastas P. T.; Warner J. C.. Green Chemistry: Theory and Practice; Oxford University Press: New York, 1998; p 30. [Google Scholar]
- ACS Green Chemistry Institute Pharmaceutical Roundtable Tools for Innovation in Chemistry. https://www.acsgcipr.org/tools-for-innovation-in-chemistry/ (accessed 2020-01-09).
- PMI is closely related to Complete E-Factor where Complete E-factor = PMI – 1.
- Jimenez-Gonzalez C.; Ponder C. S.; Broxterman Q. B.; Manley J. B. Using the Right Green Yardstick: Why Process Mass Intensity Is Used in the Pharmaceutical Industry To Drive More Sustainable Processes. Org. Process Res. Dev. 2011, 15 (4), 912–917. 10.1021/op200097d. [DOI] [Google Scholar]
- Roschangar F.; Sheldon R. A.; Senanayake C. H. Overcoming barriers to green chemistry in the pharmaceutical industry - the Green Aspiration Level concept. Green Chem. 2015, 17 (2), 752–768. 10.1039/C4GC01563K. [DOI] [Google Scholar]
- Budzinski K.; Blewis M.; Dahlin P.; D’Aquila D.; Esparza J.; Gavin J.; Ho S. V.; Hutchens C.; Kahn D.; Koenig S. G.; Kottmeier R.; Millard J.; Snyder M.; Stanard B.; Sun L. Introduction of a process mass intensity metric for biologics. New Biotechnol. 2019, 49, 37–42. 10.1016/j.nbt.2018.07.005. [DOI] [PubMed] [Google Scholar]
- Isidro-Llobet A.; Kenworthy M. N.; Mukherjee S.; Kopach M. E.; Wegner K.; Gallou F.; Smith A. G.; Roschangar F. Sustainability Challenges in Peptide Synthesis and Purification: From R&D to Production. J. Org. Chem. 2019, 84 (8), 4615–4628. 10.1021/acs.joc.8b03001. [DOI] [PubMed] [Google Scholar]
- European Chemicals Agency List of substances included in Annex XIV of REACH (“Authorisation List”). https://echa.europa.eu/authorisation-list (accessed 2020-01-10).
- European Chemicals Agency Candidate List of substances of very high concern for Authorisation. https://echa.europa.eu/candidate-list-table (accessed 2020-01-10).
- European Chemicals Agency Substances restricted under REACH. https://echa.europa.eu/substances-restricted-under-reach (accessed 2020-01-10).
- Prat D.; Wells A.; Hayler J.; Sneddon H.; McElroy C. R.; Abou-Shehada S.; Dunn P. J. CHEM21 selection guide of classical- and less classical-solvents. Green Chem. 2016, 18 (1), 288–296. 10.1039/C5GC01008J. [DOI] [Google Scholar]
- Scremin C. L.; Zhou L.; Srinivasachar K.; Beaucage S. L. Stepwise Regeneration and Recovery of Deoxyribonucleoside Phosphoramidite Monomers During Solid-Phase Oligonucleotide Synthesis. J. Org. Chem. 1994, 59 (8), 1963–6. 10.1021/jo00087a005. [DOI] [Google Scholar]
- Guo Z.; Pfundheller H. M.; Sanghvi Y. S. Process for the capture and reuse of the 4,4′-dimethoxytriphenylmethyl group during manufacturing of oligonucleotides. Org. Process Res. Dev. 1998, 2 (6), 415–417. 10.1021/op980048l. [DOI] [Google Scholar]
- Krotz A. H.; Carty R. L.; Scozzari A. N.; Cole D. L.; Ravikumar V. T. Large-Scale Synthesis of Antisense Oligonucleotides without Chlorinated Solvents. Org. Process Res. Dev. 2000, 4 (3), 190–193. 10.1021/op990183d. [DOI] [Google Scholar]
- Septak M. Kinetic studies on depurination and detritylation of CPG-bound intermediates during oligonucleotide synthesis. Nucleic Acids Res. 1996, 24 (15), 3053–8. 10.1093/nar/24.15.3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul C. H.; Royappa A. T. Acid binding and detritylation during oligonucleotide synthesis. Nucleic Acids Res. 1996, 24 (15), 3048–52. 10.1093/nar/24.15.3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheruvallath Z. S.; Carty R. L.; Andrade M.; Moore M. N.; Song Q.; Rentel C.; Cole D. L.; Ravikumar V. T. Efficient Synthesis of Antisense Phosphorothioate Oligonucleotides: Evaluation of Dichloroacetic Acid at Higher Concentration to Reduce Cycle Time. Org. Process Res. Dev. 2003, 7 (6), 917–920. 10.1021/op030006j. [DOI] [Google Scholar]
- Krotz A. H.; Cole D. L.; Ravikumar V. T. Synthesis of an antisense oligonucleotide targeted against C-raf kinase: efficient oligonucleotide synthesis without chlorinated solvents. Bioorg. Med. Chem. 1999, 7 (3), 435–439. 10.1016/S0968-0896(98)00253-3. [DOI] [PubMed] [Google Scholar]
- Krotz A. H.; Carty R. L.; Moore M. N.; Scozzari A. N.; Cole D. L.; Ravikumar V. T. Synthesis of antisense oligonucleotides. Green Chem. 1999, 1 (6), 277–282. 10.1039/a907685i. [DOI] [Google Scholar]
- Research G.Glen Report 19.29: Technical Brief - About Activators: Now and Tomorrow. https://www.glenresearch.com/reports/gr19-29 (accessed 2020-05-31).
- Sanghvi Y. S.; Ravikumar V. T.; Scozzari A. N.; Cole D. L. Applications of green chemistry in the manufacture of oligonucleotide drugs. Pure Appl. Chem. 2001, 73 (1), 175–180. 10.1351/pac200173010175. [DOI] [Google Scholar]
- Lowe D.The Great Acetonitrile Shortage. https://blogs.sciencemag.org/pipeline/archives/2009/01/22/the_great_acetonitrile_shortage (accessed 2020-03-11).
- Gaytan P. Chemical synthesis of oligonucleotides using acetone as a washing solvent. BioTechniques 2009, 47 (2), 701–702. 10.2144/000113206. [DOI] [PubMed] [Google Scholar]
- Gajda G. J.; Riley M. G.; Mohan V.; Lorenz S. M.; Cohen A. P.. Methods and systems for purifying an acetonitrile waste stream and methods for synthesizing oligonucleotides using purified acetonitrile waste streams. WO2015126713A1, 2015.
- Capaldi D. C.; Scozzari A. N.; Cole D. L.; Ravikumar V. T. Is It Essential to Use Anhydrous Acetonitrile in the Manufacture of Phosphorothioate Oligonucleotides?. Org. Process Res. Dev. 1999, 3 (6), 485–487. 10.1021/op9900333. [DOI] [Google Scholar]
- Cedillo I. E.; Moore M. N.; Ring F. J.. Apparatus for synthesizing oligonucleotides and methods of use. WO2010141361A1, 2010.
- Cedillo I. E.Considerations for Ton-scale Oligonucleotide Manufacturing via Solid-phase synthesis, Preparing for the Future. TIDES Europe, Amsterdam, 2019.
- Yang J.; Stolee J. A.; Jiang H.; Xiao L.; Kiesman W. F.; Antia F. D.; Fillon Y. A.; Ng A.; Shi X. Solid-Phase Synthesis of Phosphorothioate Oligonucleotides Using Sulfurization Byproducts for in Situ Capping. J. Org. Chem. 2018, 83 (19), 11577–11585. 10.1021/acs.joc.8b01553. [DOI] [PubMed] [Google Scholar]
- Antia F. D., Bringing Oligonucleotides into a Biologics Manufacturing Company: Designing Greener Processes. 23rd Green Chemistry & Engineering Conference and 9th International Conference on Green and Sustainable Chemistry, Reston, VA, 2019.
- Rydzak J. W.; White D. E.; Airiau C. Y.; Sterbenz J. T.; York B. D.; Clancy D. J.; Dai Q. Real-Time Process Analytical Technology Assurance for Flow Synthesis of Oligonucleotides. Org. Process Res. Dev. 2015, 19 (1), 203–214. 10.1021/op500035j. [DOI] [Google Scholar]
- McElderry J.-D.Real-time Sequencing by FTIR in GMP Oligonucleotide Synthesis. https://www.brighttalk.com/webcast/10519/375145/real-time-sequencing-by-ftir-in-gmp-oligonucleotide-synthesis (accessed 2020-05-31).
- Gronke R. S.; Joshi R.; Ruanjaikaen K.; Fillon Y.; Tran C.; Antia F. D., Re-designing Purification Processes for Oligonucleotides; What’s Achievable? Recovery of Biological Products Conference XVII; Bermuda, 2016.
- Shukla A. A.; Deshmukh R. R.; Moore J. A.; Cramer S. M. Purification of oligonucleotides by high affinity, low molecular weight displacers. Biotechnol. Prog. 2000, 16 (6), 1064–70. 10.1021/bp0000860. [DOI] [PubMed] [Google Scholar]
- Tugcu N.; Deshmukh R. R.; Sanghvi Y. S.; Cramer S. M. Displacement chromatography of anti-sense oligonucleotide and proteins using saccharin as a non-toxic displacer. React. Funct. Polym. 2003, 54 (1–3), 37–47. 10.1016/S1381-5148(02)00181-5. [DOI] [Google Scholar]
- Tugcu N.; Deshmukh R. R.; Sanghvi Y. S.; Moore J. A.; Cramer S. M. Purification of an oligonucleotide at high column loading by high affinity, low-molecular-mass displacers. J. Chromatogr A 2001, 923 (1–2), 65–73. 10.1016/S0021-9673(01)00954-2. [DOI] [PubMed] [Google Scholar]
- Schulte M.; Lühring N.; Keil A.; Sanghvi Y. S. Purification of DMT-On Oligonucleotide by Simulated Moving-Bed (SMB) Chromatography. Org. Process Res. Dev. 2005, 9 (2), 212–215. 10.1021/op050006e. [DOI] [Google Scholar]
- Aumann L.; Morbidelli M. A continuous multicolumn countercurrent solvent gradient purification (MCSGP) process. Biotechnol. Bioeng. 2007, 98 (5), 1043–55. 10.1002/bit.21527. [DOI] [PubMed] [Google Scholar]
- As opposed to discontinuous diafiltration involving sequential concentrations and dilutions.
- Cuddon FD300 Freeze Dryer Specifications. https://www.cuddonfreezedry.com/products/fd300-freeze-dryer/ (accessed 2020-01-10).
- Mueller C., Technical Considerations for use of Oligonucleotide Liquid API. In AAPS PharmSci 360, San Antonio, TX 2019.
- Muslehiddinoglu J.; Simler R.; Hill M. L.; Mueller C.; Amery J. H. A.; Dixon L.; Watson A.; Storch K.; Gazziola C.; Gielen F.; Lange S. A.; Prail J. D.; Nesta D. P. Technical Considerations for Use of Oligonucleotide Solution API. Nucleic Acid Ther. 2020, 30, 189. 10.1089/nat.2020.0846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simler R.Is Solution API Right for You? Strategic and Practical Considerations. TIDES Europe, Amsterdam, The Netherlands, 2018. [Google Scholar]
- Iwamoto N.; Butler D. C. D.; Svrzikapa N.; Mohapatra S.; Zlatev I.; Sah D. W. Y.; Meena; Standley S. M.; Lu G.; Apponi L. H.; Frank-Kamenetsky M.; Zhang J. J.; Vargeese C.; Verdine G. L. Control of phosphorothioate stereochemistry substantially increases the efficacy of antisense oligonucleotides. Nat. Biotechnol. 2017, 35 (9), 845–851. 10.1038/nbt.3948. [DOI] [PubMed] [Google Scholar]
- Knouse K. W.; deGruyter J. N.; Schmidt M. A.; Zheng B.; Vantourout J. C.; Kingston C.; Mercer S. E.; McDonald I. M.; Olson R. E.; Zhu Y.; Hang C.; Zhu J.; Yuan C.; Wang Q.; Park P.; Eastgate M. D.; Baran P. S. Unlocking P(V): Reagents for chiral phosphorothioate synthesis. Science 2018, 361 (6408), 1234–1238. 10.1126/science.aau3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.; Lightfoot H. L.; Halloy F.; Malinowska A. L.; Berk C.; Behera A.; Schumperli D.; Hall J. Synthesis and cellular activity of stereochemically-pure 2′-O-(2-methoxyethyl)-phosphorothioate oligonucleotides. Chem. Commun. (Cambridge, U. K.) 2017, 53 (3), 541–544. 10.1039/C6CC08473G. [DOI] [PubMed] [Google Scholar]
- Oka N.; Yamamoto M.; Sato T.; Wada T. Solid-phase synthesis of stereoregular oligodeoxyribonucleoside phosphorothioates using bicyclic oxazaphospholidine derivatives as monomer units. J. Am. Chem. Soc. 2008, 130 (47), 16031–7. 10.1021/ja805780u. [DOI] [PubMed] [Google Scholar]
- Molina A. G.; Sanghvi Y. S. Liquid-Phase Oligonucleotide Synthesis: Past, Present, and Future Predictions. Curr. Protoc Nucleic Acid Chem. 2019, 77 (1), e82. 10.1002/cpnc.82. [DOI] [PubMed] [Google Scholar]
- Palluk S.; Arlow D. H.; de Rond T.; Barthel S.; Kang J. S.; Bector R.; Baghdassarian H. M.; Truong A. N.; Kim P. W.; Singh A. K.; Hillson N. J.; Keasling J. D. De novo DNA synthesis using polymerase-nucleotide conjugates. Nat. Biotechnol. 2018, 36 (7), 645–650. 10.1038/nbt.4173. [DOI] [PubMed] [Google Scholar]
- Sarac I.; Hollenstein M. Terminal Deoxynucleotidyl Transferase in the Synthesis and Modification of Nucleic Acids. ChemBioChem 2019, 20 (7), 860–871. 10.1002/cbic.201800658. [DOI] [PubMed] [Google Scholar]
- Laqua M. Pioneers in the synbio revolution. European Biotechnology 2018, 48–54. [Google Scholar]
- Hoff K.; Halpain M.; Garbagnati G.; Edwards J. S.; Zhou W. Enzymatic Synthesis of Designer DNA Using Cyclic Reversible Termination and a Universal Template. ACS Synth. Biol. 2020, 9 (2), 283–293. 10.1021/acssynbio.9b00315. [DOI] [PubMed] [Google Scholar]
- Crameri A.; Tew D. G.. Novel processes for the production of therapeutic oligonucleotides. WO2019121500A1, 2019.
- Tew D. G.Complete Enzyme Catalysed Oligonucleotide Synthesis: From Single Nucleotides to Final Product. TIDES Europe, Amsterdam, 2019.