

Abstract
Gastric-type cervical adenocarcinoma (GCA) is an aggressive type of endocervical adenocarcinoma characterized by mucinous morphology, gastric-type mucin, lack of association with human papillomavirus (HPV) and resistance to chemo/radiotherapy. We characterized the landscape of genetic alterations in a large cohort of GCAs, and compared it with that of usual-type HPV-associated endocervical adenocarcinomas (UEAs), pancreatic adenocarcinomas (PAs) and intestinal-type gastric adenocarcinomas (IGAs).
GCAs (n=68) were subjected to massively parallel sequencing targeting 410–468 cancer-related genes. Somatic mutations and copy number alterations (CNAs) were determined using validated bioinformatics methods. Mutational data for UEAs (n=21), PAs (n=178) and IGAs (n=148) from The Cancer Genome Atlas (TCGA) were obtained from cBioPortal.
GCAs most frequently harbored somatic mutations in TP53 (41%), CDKN2A (18%), KRAS (18%) and STK11 (10%). Potentially targetable mutations were identified in ERBB3 (10%), ERBB2 (8%) and BRAF (4%). GCAs displayed low levels of CNAs with no recurrent amplifications or homozygous deletions. In contrast to UEAs, GCAs harbored more frequent mutations affecting cell cycle related genes including TP53 (41% vs 5%, p<0.01) and CDKN2A (18% vs 0%, p=0.01), and fewer PIK3CA mutations (7% vs 33%, p=0.01). TP53 mutations were less prevalent in GCAs compared to PAs (41% vs 56%, p<0.05) and IGAs (41% vs 57%, p<0.05). GCAs showed a higher frequency of STK11 mutations than PAs (10% vs 2%, p<0.05) and IGAs (10% vs 1%, p<0.05). GCAs harbored more frequent mutations in ERBB2 and ERBB3 (9% vs 1%, and 10% vs 0.5%, both p<0.01) compared to PAs, and in CDKN2A (18% vs 1%, p<0.05) and KRAS (18% vs 6%, p<0.05) compared to IGAs.
GCAs harbor recurrent somatic mutations in cell cycle-related genes and in potentially targetable genes, including ERBB2/3. Mutations in genes such as STK11 may be used as supportive evidence to help distinguish GCAs from other adenocarcinomas with similar morphology in metastatic sites.
Keywords: Cervix, Gastric-type adenocarcinoma, Genetics, Pathology
The 2020 World Health Organization classification of cervical adenocarcinomas includes HPV-associated adenocarcinoma, usual type and HPV-independent adenocarcinoma, gastric type.(1) Gastric-type cervical adenocarcinoma (GCA) characteristically exhibits gastric (pyloric)-type differentiation(2–6) and has been reported to account for 20–25% of all endocervical adenocarcinomas in Japan,(4, 7) but is less common in Western countries.(8)
In contrast to usual-type endocervical adenocarcinomas (UEAs), GCAs are not associated with human papillomavirus (HPV).(9) They are often situated in the upper endocervix and present with bleeding or profuse watery discharge with a clinically bulky cervix without a well-defined mass due to a highly infiltrative growth pattern.(10) GCAs exhibit aggressive behaviour, and are associated with poorer prognosis than UEAs.(4, 11–13)
Histologically, GCA shows a morphologic spectrum ranging from well-differentiated mucinous adenocarcinoma (previously referred to as “minimal deviation adenocarcinoma/adenoma malignum”) to poorly differentiated adenocarcinoma.(14, 15) The tumor cells typically contain moderate to large amounts of clear, foamy or pale eosinophilic cytoplasm with well-defined cell borders and variable nuclear atypia. Immunohistochemically, GCAs express markers of gastric differentiation, such as HIK1083, MUC6, trefoil factor 2 (TFF2)), pepsinogen II and lysozyme.(4, 6, 16, 17) The histopathologic features of GCA show some overlap with those of intestinal adenocarcinomas of the stomach (IGAs) and particularly pancreatic adenocarcinomas (PAs).(4, 18)
The genetic underpinnings of GCA are beginning to be elucidated, as illustrated by two recent small studies.(19, 20) One analyzed 14 GCAs using a targeted panel of 161 genes and found a heterogenous set of genetic findings, including alterations in TP53, DNA mismatch repair genes, CDKN2A/B, POLE, ARID1A, STK11 and KRAS,(19) while the other study evaluated 11 GCAs using a 447-gene panel and found similar results with alterations in TP53, KRAS, GNAS, STK11, ERBB2, PIK3CA and SMAD4.(20)
A subset of GCAs is associated with lobular endocervical glandular hyperplasia (LEGH), a rare benign, often incidentally encountered lesion that can be associated with Peutz-Jeghers syndrome (PJS).(21) LEGH is usually located in the upper endocervix, exhibits a lobular architecture and is positive for gastric markers including HIK1083 and MUC6.(15) The co-occurrence of some cases of GCA with LEGH(22, 23) and the identification of alterations in KRAS, STK11, TP53 and GNAS in LEGH(24) and GCA(19, 20) suggests that at least some cases of LEGH may represent precursors of GCA.(23, 24) A minority of LEGH, originally termed ‘atypical LEGH’, and more recently, postulated to be a variant of ‘gastric-type adenocarcinoma in situ’ (gAIS), exhibit variable cytoarchitectural atypia (loss of polarity, papillary or pseudopapillary arrangements, nuclear enlargement, conspicuous nucleoli, coarse chromatin, rare mitotic figures and apoptotic bodies) without evidence of stromal invasion.(25) gAIS with a normal (non-lobular) architecture has also recently been described (23). gAIS has been found in association with GCA(3) and is associated with genetic alterations (e.g. gains of chromosome 3p and loss of 1p) which may be found in GCA.(26) These findings collectively suggest that LEGH, gAIS and GCA represent a spectrum of HPV-independent cervical neoplasia associated with gastric differentiation.(26)
In this study, we sought to characterize the repertoire of somatic alterations affecting cancer-related genes in a large cohort of GCAs and to compare it with that found in UEAs and in cancers which share some histopathologic similarities with GCAs, including adenocarcinomas of the stomach and pancreas.
MATERIALS AND METHODS
Cases and histopathologic review
This study was approved by the Memorial Sloan Kettering Cancer Center (MSKCC) Institutional Review Board (protocol no. 16–237). We analyzed cervicovaginal neoplasms from 71 patients, whose tumors had been diagnosed as GCA by the original reporting pathologists, who are experienced specialist gynecologic pathologists. The samples were obtained from the Department of Pathology at MSKCC (n=35) and from the in-house cases and consultation files of eight co-authors (YH, TK, CM, WGM, YM, EO, KLT and EV; n=36).
Representative hematoxylin and eosin (H&E)-stained slides of each case were reviewed by two gynecologic pathologists (RM and KJP) prior to DNA extraction and downstream analyses. Each pathologist examined the slides independently, following which the cases were reviewed concurrently by both pathologists at a multi-head microscope and consensus with regard to the diagnoses and histologic parameters (detailed below) was reached.
The slides were first reviewed to confirm the diagnosis: GCA - invasive adenocarcinoma, ranging from well- to poorly-differentiated, with clear, foamy or pale eosinophilic cytoplasm and well-defined cytoplasmic borders; gAIS – LEGH-like glands exhibiting varying degrees of architectural atypia (papillary or pseudopapillary arrangements) and cytologic atypia (loss of polarity, nuclear enlargement, conspicuous nucleoli, coarse chromatin, mitotic figures and apoptotic bodies) but lacking stromal invasion; and LEGH – lobular arrangements of glands with abundant pale cytoplasm, well-defined cytoplasmic borders, lacking cytologic atypia. Only cases in which the reviewed slides showed GCA were included in the study cohort.
In cases with a diagnosis of GCA, several histopathologic parameters were evaluated: 1) Tumor-infiltrating lymphocytes (TILs) – lymphocytes infiltrating neoplastic glandular epithelium (scored as absent or present; rare if present but only focal or infrequent); 2) Peri-tumoral lymphocytes – lymphocytes present in tissue between neoplastic glands [scored as absent or present; if present and assessable, the intensity (mild, moderate or marked) and distribution (patchy or diffuse) of the lymphocytic infiltrate]; 3) neutrophils – presence or absence of neutrophils admixed with neoplastic glands or in tissue between neoplastic glands; 4) Mitoses in neoplastic epithelium (per 10 high-power fields, HPF); 5) Apoptotic bodies in neoplastic epithelium (per 10 HPF); and 6) Presence of associated gAIS or LEGH.
Massively parallel sequencing analysis
DNA extracted from formalin-fixed, paraffin-embedded GCA samples and matched normal tissues were subjected to MSK-IMPACT sequencing at MSK’s Integrated Genomics Operation (IGO), a massively parallel sequencing assay targeting 410–468 cancer-related genes,(27) as previously described.(28) DNA from matched normal tissue was obtained from macrodissected areas of non-neoplastic tissue spatially separated from the tumor. For cases in which matched normal tissue was not available, a pool of unmatched normal DNAs subjected to MSK-IMPACT sequencing served as normal control for mutation calling. Sequencing data were analyzed as previously described.(28) Briefly, somatic single nucleotide variants (SNVs) were called using MuTect (v1.17)(29) and small insertions and deletions (indels) were identified using Strelka (v1.0.15)(30) VarScan2 (v2.3.7)(31) Lancet (v1.0.0)(32) and Scalpel (v0.5.3)(33) and further curated by manual inspection. Somatic copy number alterations and loss of heterozygosity were obtained using FACETS.(34) The potential functional impact of each SNV was defined using a combination of in silico function prediction algorithms, as previously described.(35) Mutation hotspots were assigned according to Chang et al.(36) Lollipop plots were created using MutationMapper on cBioPortal, (37, 38) and manually curated.
Mutational signatures were ascribed to samples that harbored ≥5 SNVs using SigMa (39), as previously described.(40)
Mutation and copy number calls for UEAs (n=21), PAs (n=178), and IGAs (n=148) from The Cancer Genome Atlas (TCGA) were retrieved from cBioPortal.(37, 38) We restricted the comparison to the 410 genes targeted by the smallest MSK-IMPACT panel employed in this study.
Statistical analyses
Comparisons of mutation frequencies in GCAs with those in UEAs, IGAs and PAs were performed using Fisher’s exact tests. Resulting p-values were corrected for multiple testing using the Benjamini-Hochberg false discovery rate,(41) and two-tailed adjusted p-values <0.05 were deemed statistically significant.
RESULTS
Histopathologic findings
Representative hematoxylin-eosin-stained slides for all 71 tumors were available for review. Upon review of the available histologic slides (which corresponded to the tumor material from which DNA was extracted for sequencing), the consensus diagnoses were: GCA (n=68, of which 5 were associated with extensive gAIS or LEGH); gAIS only (n=2); and LEGH only (n=1) (Table 1). The slides from cases showing only gAIS or only LEGH were recuts of the original block, and this might be the reason they did not contain the invasive GCA component present in the original slides. The 68 women with GCA were aged 23–81 years (median 52 years; age at diagnosis was available in 65 patients). None of the patients were known to have PJS.
Table 1.
Histopathologic features of 68 GCAs
| Sample ID | TILs | Peri-tumoral lymphocytes | Neutrophils | Mitoses per 10HPF | Apoptoses per 10HPF | Additional findings and Comments |
|---|---|---|---|---|---|---|
| GCA-001 | Small amount of tumor | |||||
| GCA-002 | (+) | - | - | 1 | 5 | |
| GCA-004 | Small amount of tumor | |||||
| GCA-006 | Small amount of tumor | |||||
| GCA-007 | Small amount of tumor | |||||
| GCA-008 | - | - | - | 2 | 6 | GCA involving bowel |
| GCA-009 | - | - | - | 1 | 3 | Bizarre nuclear atypia |
| GCA-010 | Small amount of tumor | |||||
| GCA-011 | - | +, 1 | - | 1 | 2 | |
| GCA-012 | Small amount of tumor | |||||
| GCA-014 | - | +, 2 | - | 7 | 4 | |
| GCA-015 | - | +, 2 | + | 2 | 1 | |
| GCA-016 | + | +, (1) | + | 1 | 8 | |
| GCA-017 | - | +, (1) | - | 1 | 2 | |
| GCA-018 | + | +, (1) | (+) | 49 | 58 | |
| GCA-019 | - | +, (1) | - | 3 | 27 | |
| GCA-020 | + | - | + | 0 | 5 | |
| GCA-024 | (+) | +, 2 | + | 4 | 6 | |
| GCA-025 | + | +, 2 | + | 11 | 5 | |
| GCA-026 | + | +, 1 | + | 4 | 4 | |
| GCA-027 | (+) | - | + | 0 | 0 | Small amount of tumor |
| GCA-028 | - | +, (1) | (+) | 1 | 4 | |
| GCA-029 | - | - | (+) | 5 | 7 | |
| GCA-030 | + | +, 1 | + | 13 | 13 | |
| GCA-031 | + | +, (1) | - | 10 | 2 | |
| GCA-032 | + | +, 1 | - | 0 | 14 | Extensive gAIS |
| GCA-033 | (+) | - | 26 | 16 | Metastatic GCA involving lymph node | |
| GCA-034 | + | +, (1) | - | 0 | 0 | |
| GCA-035 | - | - | - | 7 | 13 | |
| GCA-036 | - | +, (1) | - | 7 | 7 | |
| GCA-037 | + | +, 1 | + | 2 | 3 | |
| GCA-039 | - | - | + | 3 | 6 | Metastatic GCA involving lymph node |
| GCA-041 | - | - | - | 4 | 6 | |
| GCA-042 | - | + | - | 0 | 0 | |
| GCA-043 | - | + | + | 3 | 6 | |
| GCA-044 | - | + | - | 3 | 0 | |
| GCA-045 | - | + | + | 3 | 4 | Extensive gAIS and LEGH |
| GCA-046 | Small amount of tumor | |||||
| GCA-047 | Small amount of tumor | |||||
| GCA-048 | - | + | - | 2 | 0 | |
| GCA-049 | - | - | + | 2 | 12 | |
| GCA-050 | - | + | - | 4 | 2 | |
| GCA-102 | - | +, (1) | + | 4 | 8 | |
| GCA-104 | - | - | (+) | 9 | 6 | |
| GCA-105 | (+) | +, (1) | +, dense | 12 | 4 | Exuberant desmoplasia |
| GCA-106 | (+) | +, (1) | - | 8 | 4 | |
| GCA-107 | + | +, (1) | - | 11 | 6 | |
| GCA-108 | (+) | +, (2) | - | 4 | 5 | |
| GCA-109 | (+) | - | - | 5 | 2 | |
| GCA-110 | - | - | - | 1 | 3 | Extensive LEGH and focal gAIS |
| GCA-112 | - | - | - | 6 | 46 | |
| GCA-113 | (+) | +, (1) | - | 7 | 4 | |
| GCA-115 | - | +, (1) | + | 6 | 5 | Associated extensive lymphovascular invasion and LEGH |
| GCA-116 | + | +, 2 | + | 26 | 8 | |
| GCA-117 | + | +, 2 | - | 8 | 0 | |
| GCA-118 | Extensive gAIS with focal invasion + associated LEGH | |||||
| GCA-119 | + | + | + | 4 | 9 | Extensive lymphovascular invasion |
| GCA-120 | - | +, (2) | (+) | 1 | 1 | |
| GCA-121 | - | +, (1) | +, dense | 3 | 4 | |
| GCA-122 | - | - | + | 14 | 2 | |
| GCA-123 | - | +, (1) | + | 1 | 7 | |
| GCA-124 | - | - | - | 1 | 5 | |
| GCA-125 | + | +, (1) | + | 5 | 7 | |
| GCA-127 | (+) | +, 2 | +, dense | 3 | 12 | |
| GCA-128 | - | - | + | 6 | 0 | |
| GCA-129 | - | +, (1) | - | 1 | 2 | |
| GCA-130 | - | +, 2 | +, dense | 3 | 2 | |
| GCA-131 | - | - | - | 24 | 4 | |
| GCA-132 | + | +, 3 | + | 10 | 9 |
GCA – gastric-type cervical adenocarcinoma
TILs - Tumor-infiltrating lymphocytes; + present, (+) rare, - absent
Peri-tumoral lymphocytes: + present; +, 1 mild, diffuse; +, (1) mild, patchy; +, 2 moderate, diffuse; +, (2) moderate, diffuse; +, 3 marked, diffuse; - absent
Neutrophils - + present, (+) rare, - absent
HPF - high-power field; LEGH - Lobular endocervical glandular hyperplasia; gAIS - gastric-type adenocarcinoma in situ
Histologic parameters were assessable in 60 GCAs; in the remaining cases, the available slides contained insufficient amounts of tumor to allow reliable evaluation of the parameters. GCAs displayed clear, foamy or pale eosinophilic cytoplasm and well-defined cytoplasmic borders. TILs were absent or only rarely found in 44 (73%) tumors, while peri-tumoral lymphocytes and neutrophils were present in 41 (82%) and 32 (53%) of tumors, respectively. Of 8 GCAs with STK11 mutations (see below), 6 had sufficient tumor for detailed pathologic evaluation. TILs were present in 3 of these tumors and peritumoral lymphocytes were noted in all cases; intraepithelial or interstitial neutrophils were identified in 5 of the 6 tumors. Mitotic figures (median 4 per 10 HPF, range 0–49 per 10 HPF) and apoptotic bodies (median 5 per 10 HPF, range 0–58 per 10 HPF) were variable, but generally infrequent in the majority of cases [≤5 mitoses per 10 HPF in 39 (65%) and ≤5 apoptoses per 10 HPF in 35 (58%)]. Mitoses and apoptoses numbered ≤10 per 10 HPF in 83% and 85%, respectively) (Table 2). The histopathologic appearances of selected tumors are illustrated in Figure 1.
Table 2.
Summary of pathologic features of GCAs (n=60, unless stated otherwise)
| Parameter | Level | N | % |
|---|---|---|---|
| TILs | Absent | 34 | 57 |
| Rare | 10 | 17 | |
| Present | 16 | 27 | |
| Peri-tumoral lymphocytes (n=57)* | Absent | 16 | 28 |
| Present | 41 | 82 | |
| Mild, patchy | 18 | ||
| Mild, diffuse | 5 | ||
| Moderate, patchy | 2 | ||
| Moderate, diffuse | 8 | ||
| Marked, diffuse | 1 | ||
| Neutrophils | Absent | 28 | 47 |
| Present | 32 | 53 | |
| Mitoses per 10 HPF | Median = 4 | ||
| Range = 0–49 | |||
| ≤5 per 10 HPF | 39 | 65 | |
| ≤10 per 10 HPF | 50 | 83 | |
| Apoptoses per 10 HPF | Median = 5 | ||
| Range = 0–58 | |||
| ≤5 per 10 HPF | 35 | 58 | |
| ≤10 per 10 HPF | 51 | 85 |
Distribution of peritumoral lymphocytes could not be reliably assessed in some tumors in which it was present, due to insufficient volume of peritumoral tissue in the H&E slide for reliable evaluation
Figure 1. Histopathologic appearances of selected cases of GCA with associated genetic alterations.
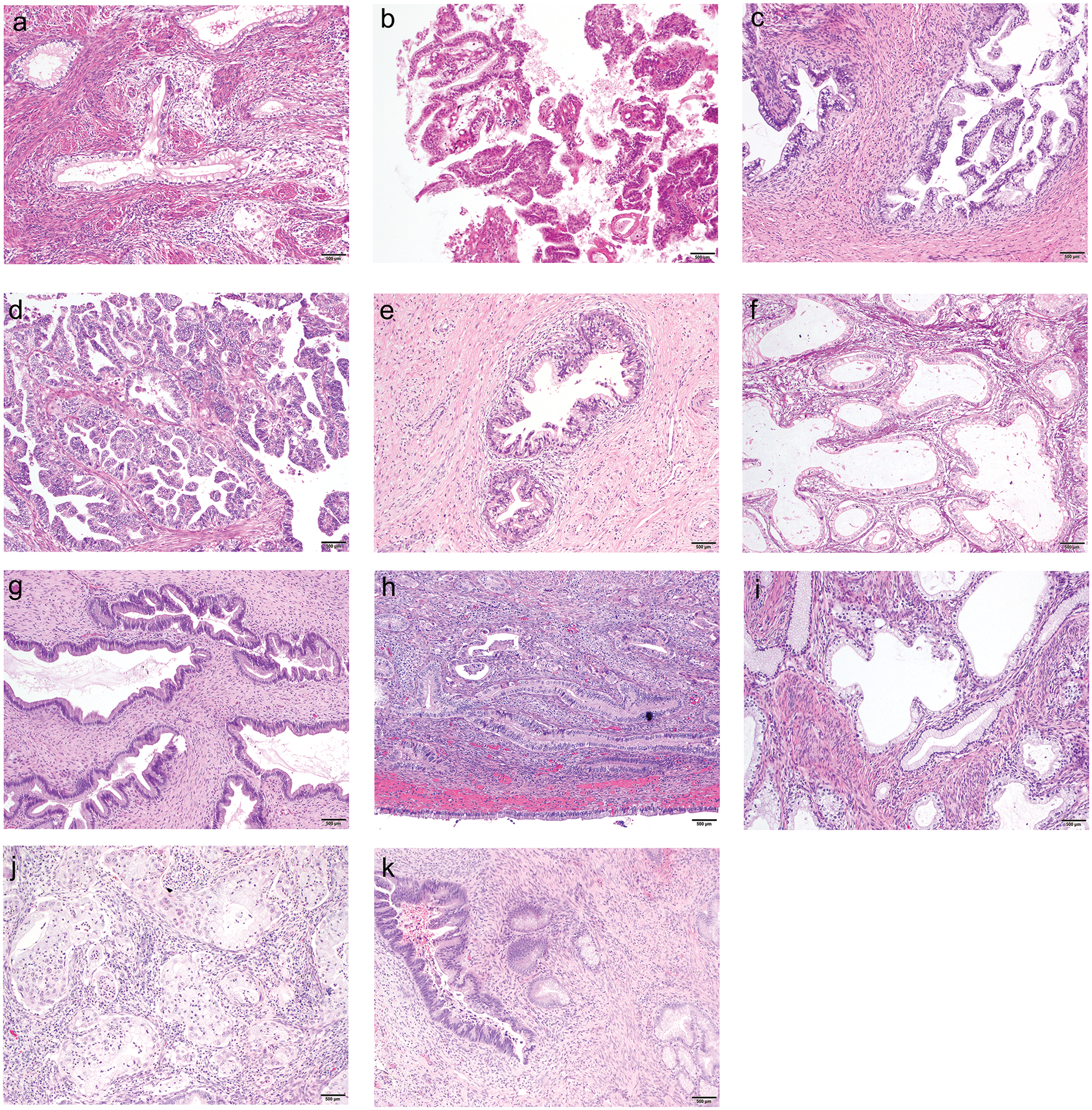
a) GCA-011. Adenocarcinoma with clear and vacuolated cytoplasm, associated with peritumoral lymphocytes; no mutations identified (hematoxylin-eosin, x100).
b) GCA-018. Adenocarcinoma, with clear and vacuolated cytoplasm, associated with scant tumor-infiltrating lymphocytes; TP53 hotspot mutation with loss of heterozygosity (hematoxylin-eosin, x100).
c) GCA-111. Gastric-type adenocarcinoma in situ, with clear and vacuolated cytoplasm; CDKN2A hotspot mutation (hematoxylin-eosin, x100).
d) GCA-022. Adenocarcinoma with focally clear cytoplasm, without peritumoral lymphocytes; KRAS hotspot mutation with loss of heterozygosity (hematoxylin-eosin, x100).
e) GCA-012. Gastric-type adenocarcinoma in situ, with clear and vacuolated cytoplasm; STK11 truncating SNV (hematoxylin-eosin, x100).
f) GCA-019. Adenocarcinoma with ERBB3 missense SNV with loss of heterozygosity (hematoxylin-eosin, x100).
g) GCA-002. Adenocarcinoma with GNAS hotspot mutation. (hematoxylin-eosin, x100).
h) GCA-102.. Adenocarcinoma associated with patchy peritumoral lymphocytes; TP53 hotspot mutation with CDKN2A SNV (hematoxylin-eosin, x100).
i) GCA-037. Adenocarcinoma with foamy cytoplasm, associated with tumor-infiltrating and peritumoral lymphocytes; CDKN2A hotspot mutation with STK11 SNV (hematoxylin-eosin, x100).
j) GCA-015. Adenocarcinoma with foamy cytoplasm, associated with peritumoral lymphocytes and neutrophils; ERBB3 and ERBB2 hotspot mutations (hematoxylin-eosin, x100).
k) GCA-110. Adenocarcinoma with KRAS and GNAS hotspot mutations (hematoxylin-eosin, x100).
Repertoire of somatic mutations and copy number alterations
GCA samples (n=68) and matched normal tissues (n=60) or pooled unmatched normal DNA (n=8) were subjected to MSK-IMPACT sequencing, at a median depth of 509x (210x-1114x) and 359x (69x-1128x) for tumor and normal tissue, respectively.
GCAs were found to harbor a median of 4 non-synonymous somatic mutations (range 0–23) most frequently involving TP53 (41%), CDKN2A (18%), KRAS (18%), STK11 (10%) and ERBB3 (10%) (Figure 2A). Hotspot and potentially targetable mutations were identified in a subset of the cases analysed, including ERBB2 and ERBB3 hotspot mutations (7/68, 11%). In particular, we identified ERBB2 p.S310F, p.R678Q, p.G776V and p.T862A hotspot mutations, as well as ERBB3 p.V104L, p.F219I, p.G284R and p.E332K hotspot mutations (Figure 2B). Additionally, we found hotspot mutations in other oncogenes such as GNAS (9%), PIK3CA (6%), BRAF (4%) and SMAD4 (4%); the BRAF mutations (p.G469V, p.D594G and p.G596R) were classified as potentially targetable by OncoKB (https://www.oncokb.org). TP53 and ERBB3 mutations were mutually exclusive in the 35 GCAs with mutations in either gene (p<0.05; Supplementary Table). In addition, only rare GCAs that harbored mutations in either of the following gene pairs harbored a mutation in both genes: TP53/ERBB2 (1/33, 3%), TP53/STK11 (1/34, 3%), TP53/GNAS (1/33, 3%) and CDKN2A/KRAS (1/23, 4%) (Supplementary Table). There were no statistically significant associations between individual mutations and histologic features of GCA.
Figure 2. Somatic mutations in 68 GCAs.
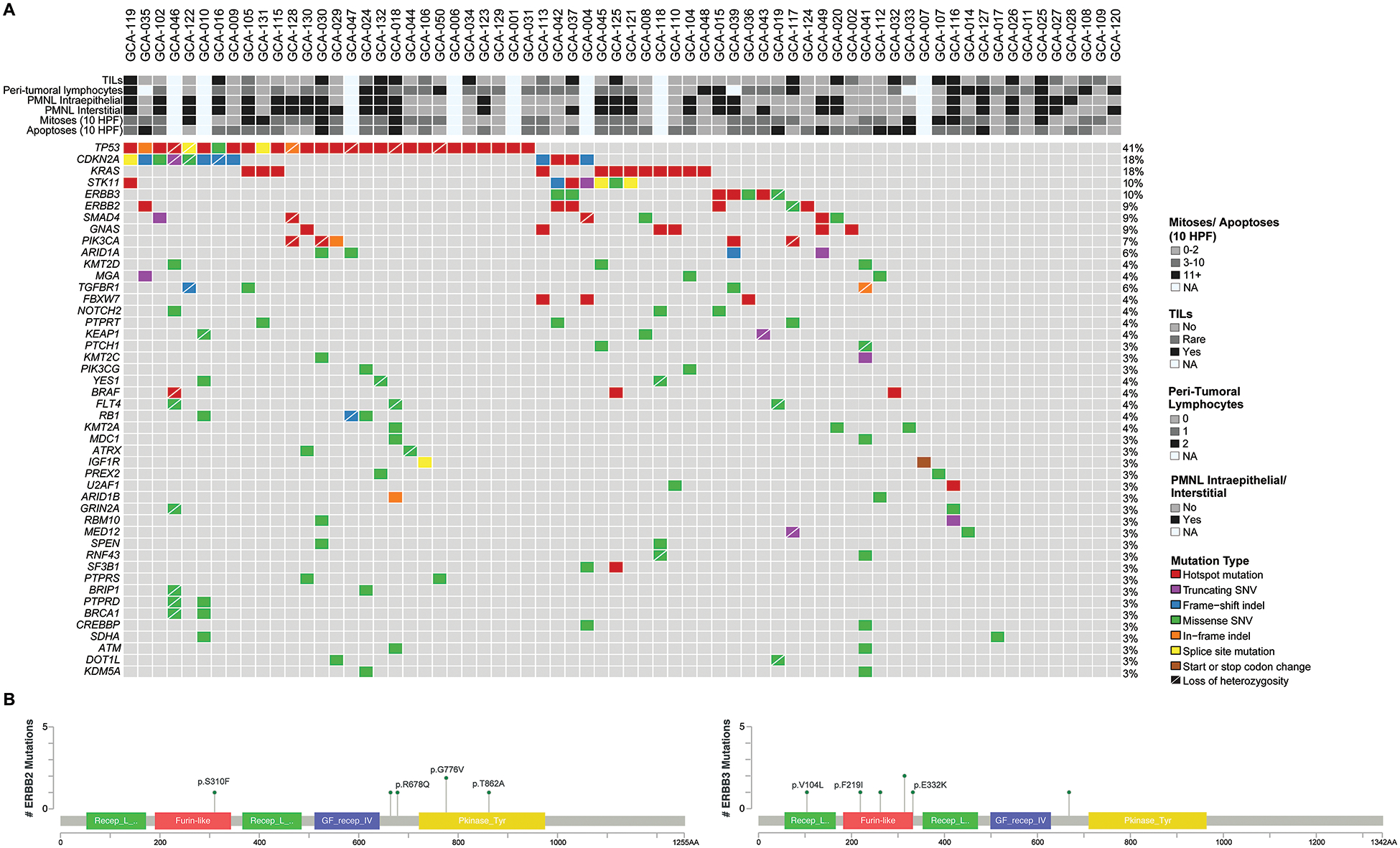
A) Recurrent non-synonymous somatic mutations affecting 410–468 cancer-related genes in 68 GCAs are shown, color-coded in the legend. The most common mutations involved TP53 (41%) and CDKN2A (18%). Clinicopathologic features are shown in the phenobar. TILs, tumor-infiltrating lymphocytes; PMNL, polymorphonuclear leukocytes; Indel, small insertion and deletion; SNV, single nucleotide variant. B) Schematic representation of the protein domains of ERBB2 (left) and ERBB3 (right) and the somatic mutations identified in GCAs. Mutational frequency is represented by the height of each lollipop (y-axis). Hotspot mutations are annotated showing the amino acid changes.
Note: In addition, three other cases that were not diagnosed as GCA were also sequenced. Two were gastric-type adenocarcinomas in situ (gAIS), one of which harbored a nonsense mutation in STK11 and missense mutations in ERBB3 and TP53, while a CDKN2A nonsense mutation was identified in the other case. One was a case of lobular endocervical glandular hyperplasia, in which a frameshift mutation in CDKN2A and a hotspot mutation in GNAS were identified.
Most GCAs displayed low levels of copy number alterations; the median fraction of genome altered was 6.1% (range, 0–67%). While no recurrent high-level amplifications or homozygous deletions were detected, individual cases displayed amplifications in genes such as CCND1, MYC or ERBB2 (Figure 3). A subset of tumors showed no copy number alterations or non-synonymous mutations (n=8); in some of the cases, this may be due to their low tumor-cell content (<20%), whereas those with higher tumor-cell content may be driven by somatic mutations in genes not tested in our sequencing panel, by epigenetic alterations or structural variants/gene fusions.
Figure 3. DNA copy number alterations in 68 GCAs.
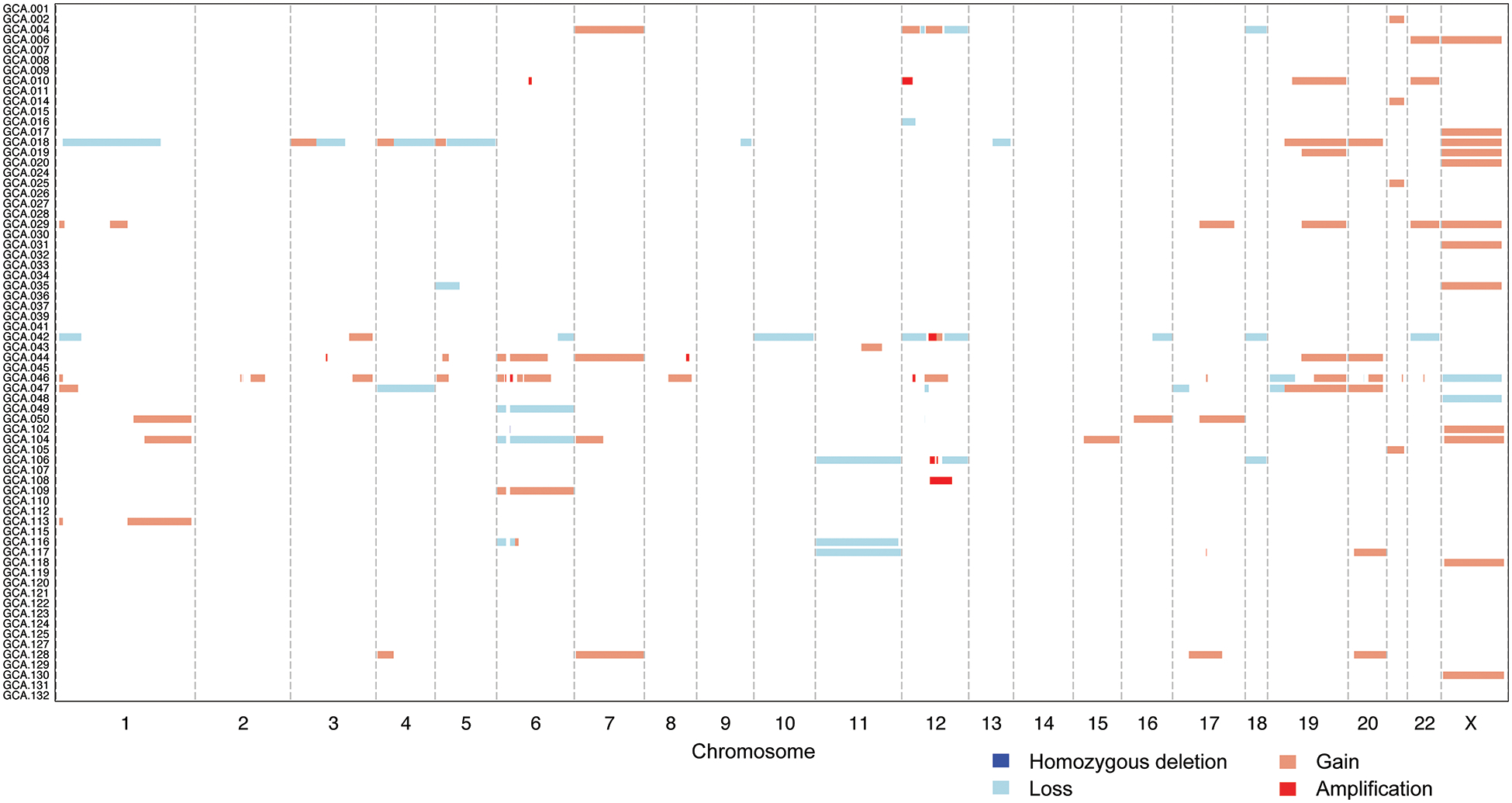
DNA copy number alterations in 68 GCAs subjected to targeted massively parallel sequencing showed that most tumors displayed low levels of copy number alterations; the median fraction of genome altered was 6.1% (range, 0–67%). While no recurrent high-level amplifications or homozygous deletions were detected, individual cases displayed amplifications in genes such as CCND1, MYC and ERBB2. A subset of cases showed no copy number alterations or non-synonymous mutations. Chromosomes are shown along the x-axis, and cases along the y-axis. Homozygous deletions, dark blue; losses, light blue; gains, orange; amplifications, red.
Assessment of the mutated genes revealed that the pathways most frequently altered in GCA included cell cycle (TP53, CDKN2A); PI3K-AKT (PIK3CA, STK11) and Notch (FBXW7, NOTCH2, CREBBP, SPEN) (Figure 4).
Figure 4. Signaling pathways affected by somatic mutations in 68 GCAs.
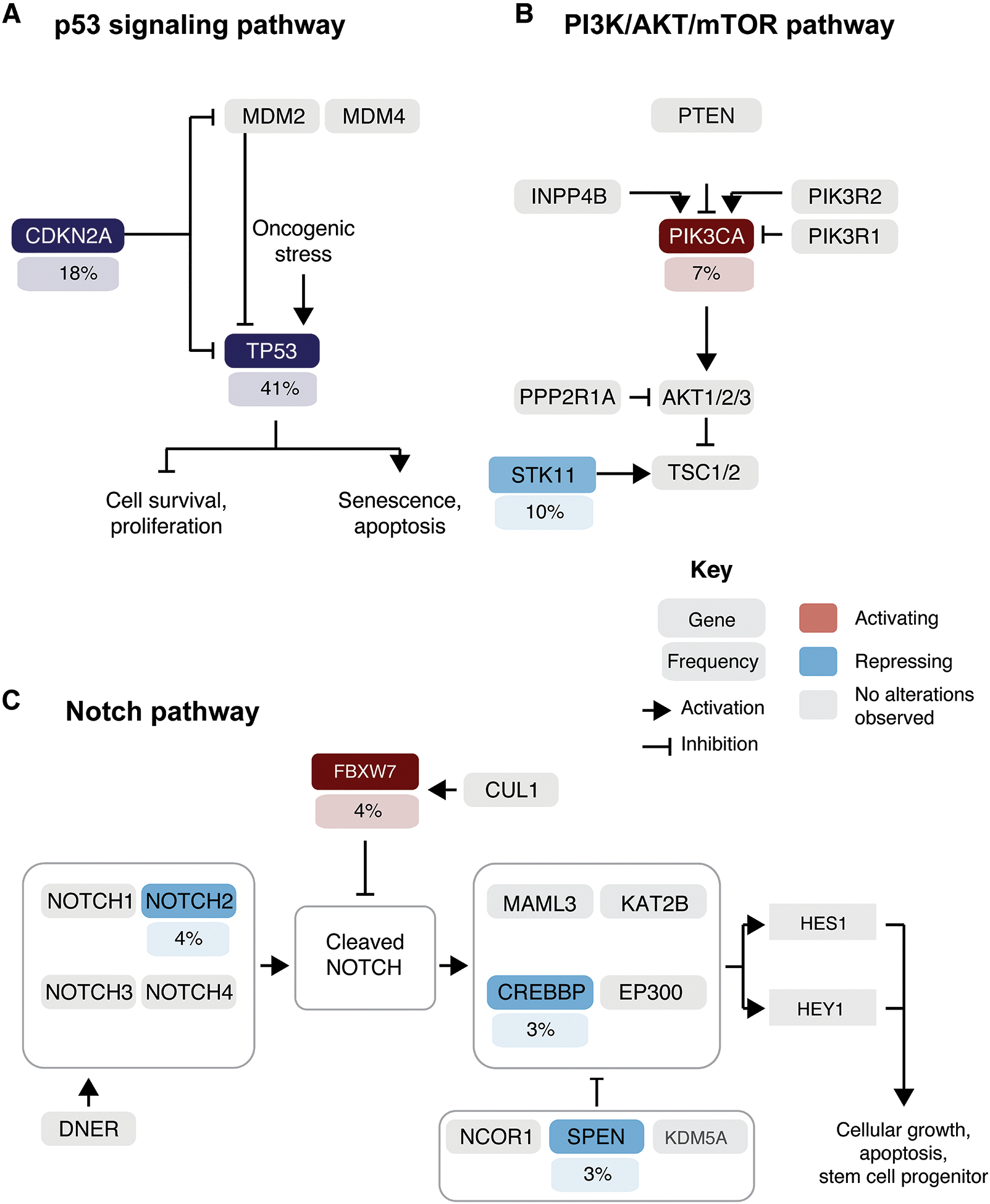
Frequency of activating (red) or loss-of-function (blue) somatic mutations affecting genes in the canonical A) p53, B) PI3K/AKT/mTOR and C) Notch signaling pathways. The prevalence of GCAs harboring a given somatic mutations is depicted under the gene name.
We next explored the mutational processes driving GCAs. Mutational signature analysis using SigMA (39) of 30 GCAs that harbored at least 5 SNVs revealed that the majority (n=24; 80%) displayed the aging-related signature 1. In addition, 3 (10%) GCAs had the APOBEC signatures 2 or 13, 2 (7%) the homologous recombination deficiency (HRD)-related signature 8, and 1 (3%) had an MSI-related signature but was not found to harbor a pathogenic mutation in a mismatch repair (MMR) related gene and did not display any genomic features of MMR-deficiency such as a high mutational burden or enrichment for short insertions and deletions.
Comparison of the repertoire of somatic mutations of GCAs with HPV-associated usual type endocervical adenocarcinomas (UEAs), intestinal-type gastric adenocarcinomas (IGAs) and pancreatic adenocarcinomas (PAs)
We next sought to define whether GCAs differed from UEAs at the molecular level, and whether there were similarities in the repertoire of genomic alterations of GCAs and IGAs and PAs, which share some morphologic similarities. This comparison was restricted to the smallest sequencing panel (i.e. 410 cancer-related genes from MSK-IMPACT).
We compared the mutational profiles of GCAs with UEAs (n=21), PAs (n=178) and IGAs (n=148) from The Cancer Genome Atlas (Figure 5). Compared to HPV-associated UEAs, GCAs were found to harbor a statistically significantly higher frequency of mutations affecting the cell cycle genes TP53 (41% vs 5%, p<0.01) and CDKN2A (18% vs 0%, p=0.01) and fewer mutations in PIK3CA (7% vs 25%, p=0.01).
Figure 5. Comparison of the mutational profiles of 68 GCAs in this study with those reported in usual endocervical adenocarcinomas, pancreatic adenocarcinomas and intestinal/tubular gastric adenocarcinomas.
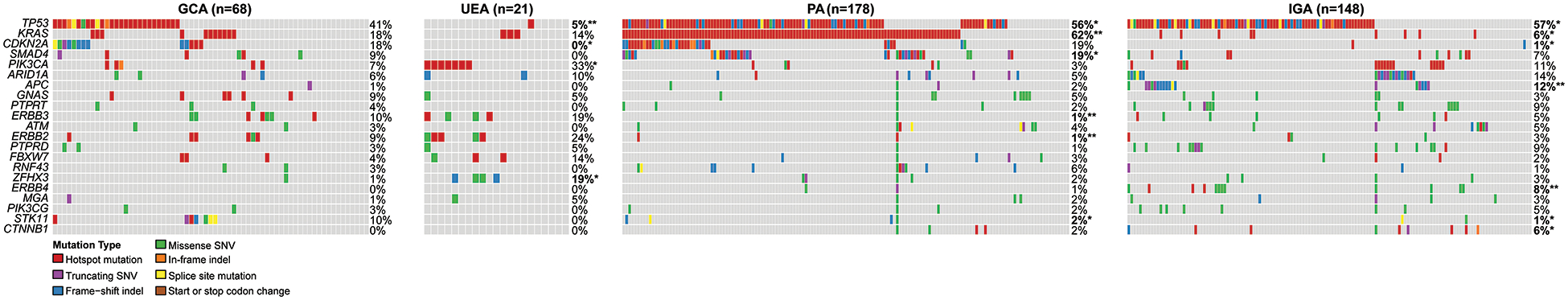
Comparison of somatic non-synonymous mutations identified in GCAs in the present study (n=68) with those of HPV-associated usual endocervical adenocarcinomas (UEAs), pancreatic adenocarcinomas (PAs) and intestinal gastric adenocarcinomas (IGAs), as reported in The Cancer Genome Atlas (TCGA), retrieved from cBioPortal.(37, 38). Fisher’s exact test, *p<0.05; ** p<0.01. Indel, small insertion and deletion; SNV, single nucleotide variant.
In contrast, TP53 mutations were found to be less prevalent in GCAs (41%) compared to both PAs (56%, p<0.05) and IGAs (57%, p<0.05). GCAs were also found to display a higher frequency of mutations in STK11 than both PAs (10% vs 2%, p<0.01) and IGAs (10% vs 1%, p<0.01). Additionally, mutations in ERBB3 (10% vs 1%, p<0.01) and ERBB2 (9% vs 1%, p<0.01) were enriched in GCAs compared to PAs, whereas CDKN2A mutations (18% vs 1%, p<0.01) were enriched in GCAs compared to IGAs. Compared to GCAs, KRAS mutations were considerably more frequent in PAs (62% vs 18%, p<0.01), but were less frequent in IGAs (6% vs 18%, p<0.05). SMAD4 mutations were significantly more frequent in PAs (19%) than GCAs (9%, p<0.05) (Figure 5, Table 3).
Table 3.
Mutation frequencies in 68 GCAs (this study) compared to UEAs, PAs and IGAs.#
| Gene | GCA (n=68) | UEA (n=21) | PA (n=178) | IGA (n=148) |
|---|---|---|---|---|
| TP53 | 41% | 5%** | 56%* | 57%* |
| CDKN2A | 18% | 0%* | 19% | 1%** |
| KRAS | 18% | 11% | 62%** | 6%* |
| STK11 | 10% | 0% | 2%** | 1%** |
| ERBB3 | 10% | 14% | 1%** | 5% |
| GNAS | 9% | 5% | 5% | 3% |
| ERBB2 | 9% | 18% | 1%** | 3% |
| SMAD4 | 9% | 0% | 19%* | 7% |
| PIK3CA | 7% | 25%* | 3% | 11% |
| ARID1A | 6% | 10% | 5% | 14% |
p<0.05,
p<0.01,
Fisher’s exact test (two-tailed, comparing mutation frequencies in GCA with those in each of the other cancer types).
GCA = gastric-type cervical adenocarcinoma; UEA = usual-type (HPV-associated) endocervical adenocarcinoma; PA = pancreatic adenocarcinoma; IGA = intestinal/tubular gastric adenocarcinoma
DISCUSSION
The clinico-pathologic features of GCA have been well documented in the literature.(12, 14–16, 25, 26) The primary aim of this study was to describe the repertoire of genomic alterations in a large cohort of GCAs. Two recent studies of 11 and 14 cases(19, 20) have investigated the genomic alterations in these neoplasms, but ours is by far the largest study to date. All of the tumors included in this study were classified as GCA by the original reporting pathologists, who are experienced specialist gynecologic pathologists. Based on pathologic review of the available slides, 68 cases were classified as GCA (associated in some cases with conspicuous gAIS and/or LEGH).
The spectrum of histopathologic appearances of the GCAs in our cohort is similar to that in prior reports.(12, 14–16, 25, 26) GCAs ranged from well- to poorly-differentiated, and displayed clear, foamy or pale eosinophilic cytoplasm and well-defined cytoplasmic borders. Although we did not have staging or follow-up data in our study, it has previously been shown that grading is of no prognostic value in GCA.(12)
From a molecular viewpoint, GCAs displayed very low levels of copy number alterations with no recurrent amplifications or homozygous deletions. These neoplasms were found to be heterogenous at the mutational level. The most common somatic mutations were seen in TP53, CDKN2A, KRAS and STK11. Mutations in ERBB2 and ERBB3 were seen in 8% and 11% of tumors, respectively. Alterations in ERBB2/3, which are mutated at roughly similar frequencies in UEAs, provide potential avenues for targeted therapy, particularly in patients with locally advanced or metastatic tumors since GCAs are refractory to conventional systemic agents.(42–45) Other somatic mutations we identified in GCA that are potentially targetable, based on data from a range of tumors harboring mutations in these genes, include those involving KRAS, STK11 and BRAF (discussed below), and PIK3CA,(46) and TGFBR1.(47) Further studies of patients with genomically-defined subsets of GCA, perhaps in the context of basket trials,(48, 49) will be required to determine which of these mutations confer susceptibility to targeted therapies with clinical benefit.
Although KRAS has long been considered a challenging therapeutic target, recent studies of KRASG12C inhibitors have shown promising results in the treatment of KRAS-mutant malignancies.(50, 51) Since KRAS hotspot mutations (8 KRASG12D, 2 KRASG12V, and 1 each of KRASG12C, KRASG12V and KRASG13D) were identified in 17% of GCAs in our cohort, a significant subset of these neoplasms may prove to be amenable to KRAS inhibitor therapy.
STK11/LKB1 mutations are associated with PJS.(52) STK11 encodes a serine threonine kinase which plays a role in the regulation of cellular metabolism/energy homeostasis, growth, and polarity through phosphorylation of adenosine monophosphate-activated protein (AMP) kinase and related kinases.(53) Preclinical studies have shown promising effects on tumor growth after abrogation of AMP kinase activity and downstream mTOR inhibition.(54) Mutational inactivation of STK11 has been shown to be associated with neutrophil recruitment and reduced density of infiltrating cytotoxic CD8+ T lymphocytes resulting in an inert or “cold” tumor immune microenvironment.(55) In our cohort, TILs were present in 3 of 6 STK11-mutant tumors and peritumoral lymphocytes were noted in all cases; intraepithelial or interstitial neutrophils were identified in 5 of the 6 tumors. Three of the STK11-mutant tumors also harbored a coexisting KRAS hotspot mutation, and TILs were absent in two of these tumors. It remains to be seen whether STK11-mutant GCAs are associated with an inert immune microenvironment and predict lack of clinical benefit from PD-1/PD-L1 blockade, as shown recently in KRAS-mutant lung adenocarcinomas.(56)
BRAF inhibitors, alone or in combination with MEK inhibitors, have shown great efficacy in the treatment of advanced stage BRAF-mutant malignancies, prototypically melanoma.(57) Following the discovery of resistance mechanisms to BRAF inhibition, this field is rapidly evolving, with the emergence of novel BRAF inhibitors and combination therapies (such as the addition of MEK inhibitors) targeting the mitogen activated protein kinase (MAPK) pathway.(57–59) The BRAF hotspot mutations found in 4% of the GCAs in our cohort were classified as potentially targetable, and MAPK pathway inhibition may prove to be helpful in some patients with locally advanced or metastatic GCA.
Of the 6 sequenced GCAs which harbored GNAS hotspot mutations, two were associated with LEGH and gAIS. These findings and published data (22–24) are consistent with LEGH, gAIS and GCA being a continuum of HPV-independent cervical neoplasia with LEGH and gAIS representing the precursor lesions of GCA.(23, 24, 26) In our study, generally only a single tumor section was available for histologic review and often only a small biopsy specimen was available. It is highly likely that the precursor lesions were present in association with GCA in other cases.
The differential mutational profiles of HPV-associated UEAs and HPV-independent GCAs, the latter displaying a higher prevalence of TP53 and CDKN2A mutations but a lower prevalence of PIK3CA mutations,(60) (Table 3) supports distinct pathogenetic mechanisms in these two groups of cervical adenocarcinomas. Abrogation of p53 function is mediated by the HPV E6 protein in UEAs,(61–64) and by TP53 mutations in GCAs. GCAs show morphologic features that overlap with, and can closely mimic, IGAs and PAs (especially the latter), and this can present diagnostic problems in metastatic settings, especially since GCAs commonly metastasize to abdominal sites such as the omentum.(4, 18) Immunohistochemically, PAX8 positivity can help distinguish GCAs from IGAs and PAs since GCAs are usually positive while IGAs and PAs are typically negative; however, some GCAs (32% in one study (6)) are negative for this marker. There is also marked immunophenotypic overlap with regard to other markers such as cytokeratin 7 and 20, CEA, CA19.9 and CDX2 (5). At the genetic level, there is also considerable overlap among these tumor types, with many of the same genes exhibiting mutations in all three tumors, albeit at varying frequencies (Figure 4). Compared to IGAs, GCAs displayed a higher prevalence of mutations in CDKN2A, KRAS and STK11 but a lower prevalence of TP53 mutations. GCAs displayed a higher frequency of mutations in ERBB2/3 and STK11 but fewer TP53, KRAS, and SMAD4 mutations compared to PAs. Although the mutational frequencies differ, the considerable overlap precludes the use of mutational profiles to classify tumor type in an individual case; a possible exception is STK11 mutation, which we identified in 10% of GCAs, but which is seen in 2% or less of UEAs, PAs and IGAs, suggesting that GCA should at least be a diagnostic consideration in an adenocarcinoma harboring this mutation.
Our study has some limitations. Given the limited tissue availability in some of our cases, we restricted our sequencing analysis to a targeted panel of 410–468 cancer-related genes. Therefore, we cannot rule out the possibility that other genes (not represented in these panels) harbor somatic mutations or copy number alterations in GCAs; to explore this possibility, further studies using whole-exome or whole-genome sequencing approaches are warranted. As discussed, generally only a single tumor section was available for histologic review. In addition, given the multi-institutional nature of our study, treatment and outcome data were not consistently available and survival analyses could not be performed.
In summary, in this largest genomic analysis of GCAs to date, we found that these tumors harbor a spectrum of somatic mutations, which differ from those seen in HPV-associated UEAs, and from adenocarcinomas of the stomach and pancreas which can show morphologic overlap with GCAs. Some of these alterations (e.g. STK11 mutations) may provide supportive evidence of tumor origin, especially in the setting of metastatic adenocarcinoma of unknown primary. Potentially targetable mutations in several genes, including ERBB2/3, may be helpful in the treatment of these clinically aggressive tumors which are often unresponsive to conventional treatment.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to the Integrated Genomics Operation at Memorial Sloan Kettering Cancer Center for their assistance with sequencing.
DISCLOSURE/CONFLICTS OF INTEREST
This work was funded in part by a Cycle for Survival grant (to RM, KJP and BW), and in part by the NIH/NCI Cancer Center Support Grant P30 CA008748. BW is supported in part by Breast Cancer Research Foundation and Stand Up to Cancer grants.
Footnotes
The authors have no other relevant disclosures or conflicts of interest.
REFERENCES
- 1.WHO Classification of Tumours Editorial Board. Female Genital Tumours. Vol 4. 5th edition ed. IARC Press: Lyon, 2020. [Google Scholar]
- 2.Ishii K, Hosaka N, Toki T, Momose M, Hidaka E, Tsuchiya S, et al. A new view of the so-called adenoma malignum of the uterine cervix. Virchows Arch 1998;432:315–22. [DOI] [PubMed] [Google Scholar]
- 3.Mikami Y, Kiyokawa T, Hata S, Fujiwara K, Moriya T, Sasano H, et al. Gastrointestinal immunophenotype in adenocarcinomas of the uterine cervix and related glandular lesions: a possible link between lobular endocervical glandular hyperplasia/pyloric gland metaplasia and ‘adenoma malignum’. Mod Pathol 2004;17:962–72. [DOI] [PubMed] [Google Scholar]
- 4.Kojima A, Mikami Y, Sudo T, Yamaguchi S, Kusanagi Y, Ito M, et al. Gastric morphology and immunophenotype predict poor outcome in mucinous adenocarcinoma of the uterine cervix. Am J Surg Pathol 2007;31:664–72. [DOI] [PubMed] [Google Scholar]
- 5.Kurman RJ, Carcangiu M-L, Herrington CS, Young RH. World Health Organization classification of tumours of female reproductive organs. IARC Press: Lyon, 2014. [Google Scholar]
- 6.Carleton C, Hoang L, Sah S, Kiyokawa T, Karamurzin YS, Talia KL, et al. A Detailed Immunohistochemical Analysis of a Large Series of Cervical and Vaginal Gastric-type Adenocarcinomas. Am J Surg Pathol 2016;40:636–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kusanagi Y, Kojima A, Mikami Y, Kiyokawa T, Sudo T, Yamaguchi S, et al. Absence of high-risk human papillomavirus (HPV) detection in endocervical adenocarcinoma with gastric morphology and phenotype. Am J Pathol 2010;177:2169–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holl K, Nowakowski AM, Powell N, McCluggage WG, Pirog EC, Collas De Souza S, et al. Human papillomavirus prevalence and type-distribution in cervical glandular neoplasias: Results from a European multinational epidemiological study. Int J Cancer 2015;137:2858–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.An HJ, Kim KR, Kim IS, Kim DW, Park MH, Park IA, et al. Prevalence of human papillomavirus DNA in various histological subtypes of cervical adenocarcinoma: a population-based study. Mod Pathol 2005;18:528–34. [DOI] [PubMed] [Google Scholar]
- 10.Kido A, Mikami Y, Koyama T, Kataoka M, Shitano F, Konishi I, et al. Magnetic resonance appearance of gastric-type adenocarcinoma of the uterine cervix in comparison with that of usual-type endocervical adenocarcinoma: a pitfall of newly described unusual subtype of endocervical adenocarcinoma. Int J Gynecol Cancer 2014;24:1474–9. [DOI] [PubMed] [Google Scholar]
- 11.Karamurzin Y, Parkash V, Kiyokawa R, Soslow RA, Park KJ. Gastric type endocervical adenocarcinoma – an aggressive histologic subtype. Mod Pathol 2012;25:1171A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karamurzin YS, Kiyokawa T, Parkash V, Jotwani AR, Patel P, Pike MC, et al. Gastric-type Endocervical Adenocarcinoma: An Aggressive Tumor With Unusual Metastatic Patterns and Poor Prognosis. Am J Surg Pathol 2015;39:1449–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nishio S, Mikami Y, Tokunaga H, Yaegashi N, Satoh T, Saito M, et al. Analysis of gastric-type mucinous carcinoma of the uterine cervix - An aggressive tumor with a poor prognosis: A multi-institutional study. Gynecol Oncol 2019;153:13–9. [DOI] [PubMed] [Google Scholar]
- 14.McCluggage WG. New developments in endocervical glandular lesions. Histopathology 2013;62:138–60. [DOI] [PubMed] [Google Scholar]
- 15.Mikami Y, McCluggage WG. Endocervical glandular lesions exhibiting gastric differentiation: an emerging spectrum of benign, premalignant, and malignant lesions. Advances in anatomic pathology 2013;20:227–37. [DOI] [PubMed] [Google Scholar]
- 16.Turashvili G, Morency EG, Kracun M, DeLair DF, Chiang S, Soslow RA, et al. Morphologic Features of Gastric-type Cervical Adenocarcinoma in Small Surgical and Cytology Specimens. Int J Gynecol Pathol 2019;38:263–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takako K, Hoang L, Terinte C, Pesci A, Aviel-Ronen S, Alvarado-Cabrero I, et al. Trefoil Factor 2 (TFF2) as a Surrogate Marker for Endocervical Gastric-type Carcinoma. Int J Gynecol Pathol 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pirog EC, Park KJ, Kiyokawa T, Zhang X, Chen W, Jenkins D, et al. Gastric-type Adenocarcinoma of the Cervix: Tumor With Wide Range of Histologic Appearances. Advances in anatomic pathology 2019;26:1–12. [DOI] [PubMed] [Google Scholar]
- 19.Garg S, Nagaria TS, Clarke B, Freedman O, Khan Z, Schwock J, et al. Molecular characterization of gastric-type endocervical adenocarcinoma using next-generation sequencing. Mod Pathol 2019;32:1823–33. [DOI] [PubMed] [Google Scholar]
- 20.Hodgson A, Howitt BE, Park KJ, Lindeman N, Nucci MR, Parra-Herran C. Genomic Characterization of HPV-related and Gastric-type Endocervical Adenocarcinoma: Correlation With Subtype and Clinical Behavior. Int J Gynecol Pathol 2020;39:578–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mikami Y, Hata S, Melamed J, Fujiwara K, Manabe T. Lobular endocervical glandular hyperplasia is a metaplastic process with a pyloric gland phenotype. Histopathology 2001;39:364–72. [DOI] [PubMed] [Google Scholar]
- 22.Nucci MR, Clement PB, Young RH. Lobular endocervical glandular hyperplasia, not otherwise specified: a clinicopathologic analysis of thirteen cases of a distinctive pseudoneoplastic lesion and comparison with fourteen cases of adenoma malignum. Am J Surg Pathol 1999;23:886–91. [DOI] [PubMed] [Google Scholar]
- 23.Nishio S, Tsuda H, Fujiyoshi N, Ota S, Ushijima K, Sasajima Y, et al. Clinicopathological significance of cervical adenocarcinoma associated with lobular endocervical glandular hyperplasia. Pathol Res Pract 2009;205:331–7. [DOI] [PubMed] [Google Scholar]
- 24.Matsubara A, Sekine S, Ogawa R, Yoshida M, Kasamatsu T, Tsuda H, et al. Lobular endocervical glandular hyperplasia is a neoplastic entity with frequent activating GNAS mutations. Am J Surg Pathol 2014;38:370–6. [DOI] [PubMed] [Google Scholar]
- 25.Talia KL, Stewart CJR, Howitt BE, Nucci MR, McCluggage WG. HPV-negative Gastric Type Adenocarcinoma In Situ of the Cervix: A Spectrum of Rare Lesions Exhibiting Gastric and Intestinal Differentiation. Am J Surg Pathol 2017;41:1023–33. [DOI] [PubMed] [Google Scholar]
- 26.Kawauchi S, Kusuda T, Liu XP, Suehiro Y, Kaku T, Mikami Y, et al. Is lobular endocervical glandular hyperplasia a cancerous precursor of minimal deviation adenocarcinoma?: a comparative molecular-genetic and immunohistochemical study. Am J Surg Pathol 2008;32:1807–15. [DOI] [PubMed] [Google Scholar]
- 27.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn 2015;17:251–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Da Cruz Paula A, da Silva EM, Segura SE, Pareja F, Bi R, Selenica P, et al. Genomic profiling of primary and recurrent adult granulosa cell tumors of the ovary. Mod Pathol 2020;33:1606–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013;31:213–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 2012;28:1811–7. [DOI] [PubMed] [Google Scholar]
- 31.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 2012;22:568–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Narzisi G, Corvelo A, Arora K, Bergmann EA, Shah M, Musunuri R, et al. Genome-wide somatic variant calling using localized colored de Bruijn graphs. Commun Biol 2018;1:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Narzisi G, O’Rawe JA, Iossifov I, Fang H, Lee YH, Wang ZH, et al. Accurate de novo and transmitted indel detection in exome-capture data using microassembly. Nature Methods 2014;11:1033–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic acids research 2016;44:e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martelotto LG, Baslan T, Kendall J, Geyer FC, Burke KA, Spraggon L, et al. Whole-genome single-cell copy number profiling from formalin-fixed paraffin-embedded samples. Nat Med 2017;23:376–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang MT, Bhattarai TS, Schram AM, Bielski CM, Donoghue MTA, Jonsson P, et al. Accelerating Discovery of Functional Mutant Alleles in Cancer. Cancer Discov 2018;8:174–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013;6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018;174:1034–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gulhan DC, Lee JJ, Melloni GEM, Cortes-Ciriano I, Park PJ. Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nat Genet 2019;51:912–9. [DOI] [PubMed] [Google Scholar]
- 40.Smith ES, Da Cruz Paula A, Cadoo KA, Abu-Rustum NR, Pei X, Brown DN, et al. Endometrial Cancers in BRCA1 or BRCA2 Germline Mutation Carriers: Assessment of Homologous Recombination DNA Repair Defects. JCO Precision Oncology 2019:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res 2001;125:279–84. [DOI] [PubMed] [Google Scholar]
- 42.Gala K, Chandarlapaty S. Molecular pathways: HER3 targeted therapy. Clin Cancer Res 2014;20:1410–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hyman DM, Piha-Paul SA, Won H, Rodon J, Saura C, Shapiro GI, et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature 2018;554:189–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ruiz-Saenz A, Moasser MM. Targeting HER2 by Combination Therapies. J Clin Oncol 2018;36:808–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oh DY, Bang YJ. HER2-targeted therapies - a role beyond breast cancer. Nat Rev Clin Oncol 2020;17:33–48. [DOI] [PubMed] [Google Scholar]
- 46.Patnaik A, Appleman LJ, Tolcher AW, Papadopoulos KP, Beeram M, Rasco DW, et al. First-in-human phase I study of copanlisib (BAY 80–6946), an intravenous pan-class I phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors and non-Hodgkin’s lymphomas. Ann Oncol 2016;27:1928–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Herbertz S, Sawyer JS, Stauber AJ, Gueorguieva I, Driscoll KE, Estrem ST, et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des Devel Ther 2015;9:4479–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cunanan KM, Gonen M, Shen R, Hyman DM, Riely GJ, Begg CB, et al. Basket Trials in Oncology: A Trade-Off Between Complexity and Efficiency. J Clin Oncol 2017;35:271–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tao JJ, Schram AM, Hyman DM. Basket Studies: Redefining Clinical Trials in the Era of Genome-Driven Oncology. Annu Rev Med 2018;69:319–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019;575:217–23. [DOI] [PubMed] [Google Scholar]
- 51.Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov 2020;10:54–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beggs AD, Latchford AR, Vasen HF, Moslein G, Alonso A, Aretz S, et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut 2010;59:975–86. [DOI] [PubMed] [Google Scholar]
- 53.Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer 2009;9:563–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Momcilovic M, Shackelford DB. Targeting LKB1 in cancer - exposing and exploiting vulnerabilities. Br J Cancer 2015;113:574–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koyama S, Akbay EA, Li YY, Aref AR, Skoulidis F, Herter-Sprie GS, et al. STK11/LKB1 Deficiency Promotes Neutrophil Recruitment and Proinflammatory Cytokine Production to Suppress T-cell Activity in the Lung Tumor Microenvironment. Cancer Res 2016;76:999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov 2018;8:822–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ribas A, Flaherty KT. BRAF targeted therapy changes the treatment paradigm in melanoma. Nat Rev Clin Oncol 2011;8:426–33. [DOI] [PubMed] [Google Scholar]
- 58.Holderfield M, Deuker MM, McCormick F, McMahon M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer 2014;14:455–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Proietti I, Skroza N, Michelini S, Mambrin A, Balduzzi V, Bernardini N, et al. BRAF Inhibitors: Molecular Targeting and Immunomodulatory Actions. Cancers (Basel) 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ojesina AI, Lichtenstein L, Freeman SS, Pedamallu CS, Imaz-Rosshandler I, Pugh TJ, et al. Landscape of genomic alterations in cervical carcinomas. Nature 2014;506:371–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li X, Coffino P. High-risk human papillomavirus E6 protein has two distinct binding sites within p53, of which only one determines degradation. J Virol 1996;70:4509–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zimmermann H, Degenkolbe R, Bernard HU, O’Connor MJ. The human papillomavirus type 16 E6 oncoprotein can down-regulate p53 activity by targeting the transcriptional coactivator CBP/p300. J Virol 1999;73:6209–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Crook T, Fisher C, Masterson PJ, Vousden KH. Modulation of transcriptional regulatory properties of p53 by HPV E6. Oncogene 1994;9:1225–30. [PubMed] [Google Scholar]
- 64.Lechner MS, Laimins LA. Inhibition of p53 DNA binding by human papillomavirus E6 proteins. J Virol 1994;68:4262–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.