

Abstract
Obesity is a risk factor for developing several cancers. The dysfunctional metabolism and chronic activation of inflammatory pathways in obesity create a milieu that supports tumor initiation, progression, and metastasis. Obesity-associated metabolic, endocrine, and inflammatory mediators, besides interacting with cells leading to a malignant transformation, also modify the intrinsic metabolic and functional characteristics of immune myeloid cells. Here we discuss the evidence supporting the hypothesis that obesity metabolically primes and promotes the expansion of myeloid cells with immunosuppressive and pro-oncogenic properties. In consequence, the accumulation of these cells, such as myeloid-derived suppressor cells (MDSCs) and some subtypes of adipose-tissue macrophages (ATMs), creates a microenvironment conducive to tumor development. In this review we emphasize the role of lipids, insulin, and leptin, which are dysregulated in obesity, and dietary nutrients in metabolic reprogramming of these myeloid cells. Moreover, we also summarize emerging evidence indicating that obesity enhances immunotherapy response and hypothesized mechanisms. Priorities in deeper exploration involving the mechanisms of crosstalk between metabolic disorders and myeloid cells related to cancer risk in patients with obesity are highlighted.
Keywords: Obesity, cancer, myeloid immunosuppressive cells, MDSC, macrophages
INTRODUCTION
Obesity is associated with an elevated risk of developing or dying from at least 13 types of cancer. It is estimated that 11% of cancers in women and 5% of cancers in men are attributable to overweight and obesity. Data from animal models also demonstrate that obesity promotes tumorigenesis [1]; however, the specific biological mechanisms by which obesity stimulates carcinogenesis are incompletely understood. Tumorigenesis is influenced by several factors derived from dysfunctional and hypertrophic visceral adipose tissue. Excess adiposity induces systemic alterations in metabolism, immune, and endocrine systems that result in abnormal concentrations and signaling of insulin, insulin-like growth factors (IGF), sex hormones, lipids, cytokines, and adipokines including leptin. Additionally, impaired fat tissue promotes polarization shifts of myeloid cells and systemic and local chronic low-grade inflammation [2]. A feature of chronic inflammation is the induction of myeloid cells with the ability to restrain pro-inflammatory responses with carcinogenic properties. In obesity, the accumulation and polarization of myeloid cells towards an immune-regulatory phenotype are promoted to restore metabolic and inflammatory homeostasis; however, these myeloid cells are important drivers of immune suppression and inflammation that also facilitate tumor development [3, 4]. In this review, we examine the impact of obesity-primed myeloid immunosuppressive cells, specifically myeloid-derived suppressor cells (MDSCs) and the metabolically activated subtype of adipose-tissue macrophages (ATMs), in promoting cancer.
Chronic low-grade inflammation: a disorder that promotes cancer
The cellular and molecular mechanisms underlying chronic inflammation in obesity remain incompletely established. Adipocytes produce a diversity of inflammatory molecules that are dysregulated during adipose tissue expansion. Among these molecules include prostaglandin E2 (PGE2), cytokines (interleukin (IL)-1, IL-6, tumor necrosis factor-alpha (IFNα), IFNγ) chemokines (IL-8, Monocyte chemoattractant protein-1 (MCP-1/CCL2), and macrophage inflammatory protein 1 (MIP-1)), and hormones with pro-inflammatory properties (leptin and resistin) [5]. Several of these inflammatory molecules promote tumorigenesis by activating signaling cascades related to cellular proliferation, suppression of apoptosis and angiogenesis on nascent malignant cells [6]. Additionally, hypertrophic adipocytes exhibit increased rates of lipolysis, hypoxia, and frequency of adipocyte death [7], which enhances cytokine production, activates resident macrophages and facilitates the recruitment of circulating leukocytes. In inflamed tissue, the continuous infiltration of myeloid cells such as neutrophils and macrophages results in high local levels of reactive oxygen species (ROS) and reactive nitrogen intermediates (RNI), responsible for oxidative damage to DNA, proteins, and lipids [8]. Inflammation-associated oxidative stress causes genomic instability resulting in increased mutation rates that lead to cellular transformation into malignant cells [8, 9]. Together these events promote cellular damage, perpetuate and amplify the inflammatory process, support the proliferation of mutated cells, stimulate angiogenesis, and confer metastatic properties to tumor cells [10].
Myeloid cells in the obese milieu that foster cancer onset
It is generally accepted that with normal weight and metabolic homeostasis, ATMs with anti-inflammatory functions, traditionally termed as M2 macrophages, are the main macrophage subpopulation infiltrating adipose tissue. Once adiposity is increased (overweight), a switch from M2 towards M1 pro-inflammatory polarized macrophages occurs (Figure 1). This switch is considered critical for obesity-associated inflammation [11]. As obesity progresses and insulin resistance develops, ATMs proliferating within adipose tissue exhibit markers of an M2 alternatively activated state [12]. In the advanced stage of adiposity, the induction of other myeloid cells with immune-regulatory, angiogenic and tissue-remodeling activities, named MDSCs, also infiltrate the adipose tissue, perhaps in an attempt to resolve inflammation, repair tissue, and restore insulin sensitivity [3]. While these myeloid cells restore metabolic homeostasis and resolve inflammation, their sustained presence nurtures an immunosuppressive and pro-angiogenic microenvironment that facilitates immune escape of emerging malignant cells and tumor initiation [3]. The first key studies that established the role of these myeloid cells in promoting cancer development were originated in a mouse model by deletion of CCAAT/enhancer-binding protein β (C/EBPβ) [13], a transcription factor essential for myeloid M2-like polarization. In this model, when myeloid immunosuppressive cells were depleted, the host was protected from high-fat diet (HFD; 60% kcal fat, 6.8% kcal sucrose)-induced systemic inflammation, insulin resistance, and tumor development [14].
Figure 1. Role of obesity-induced inflammation in the evolution of tumor development.
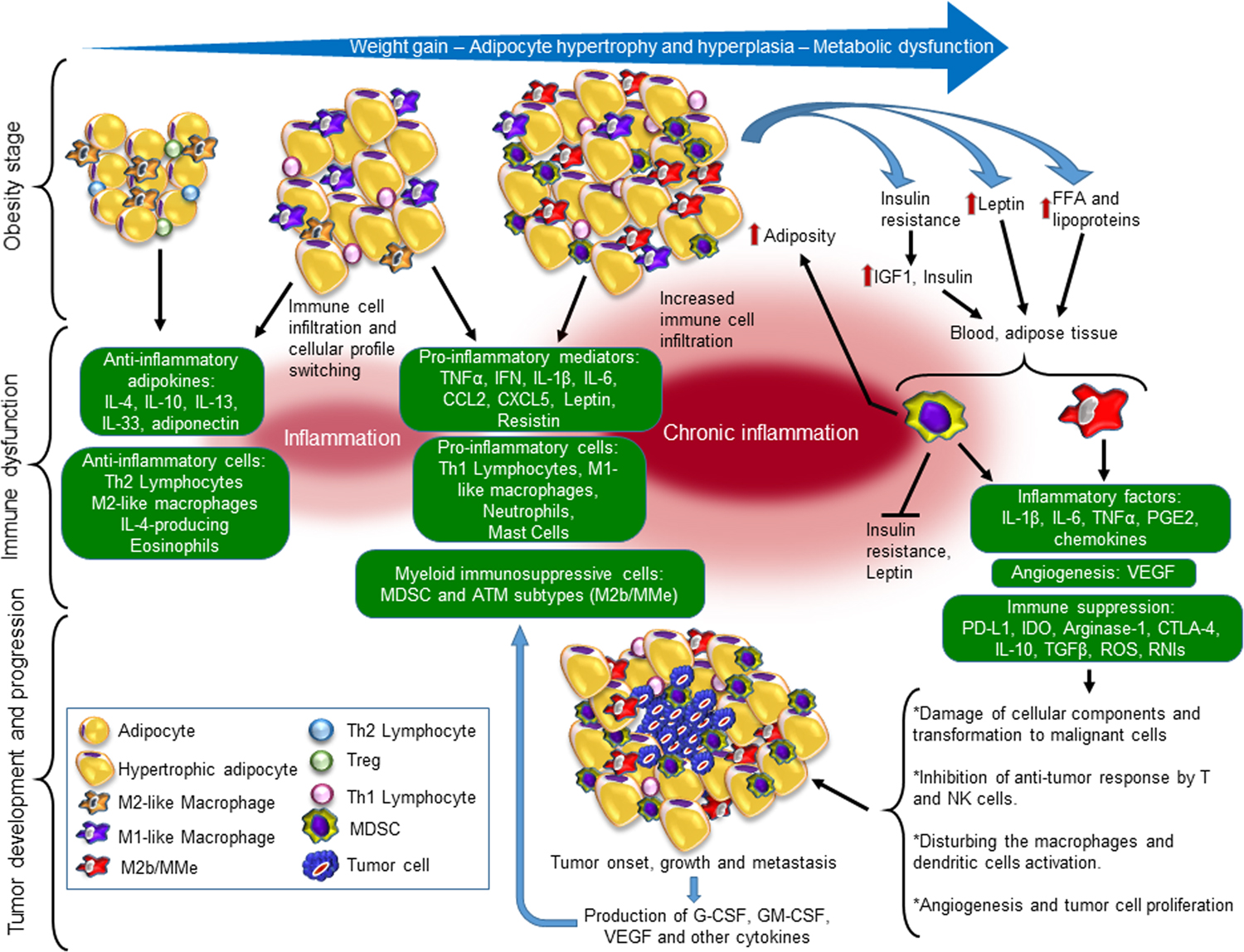
In the lean stage with metabolic homeostasis, scarce immune cells in the adipose tissue are characterized by adipose tissue-associated macrophages (ATMs) with an anti-inflammatory M2-like phenotype, Tregs and Th2 lymphocytes in an anti-inflammatory milieu. In early obesity, dysregulated inflammatory microenvironment polarizes M2-like macrophages towards M1-like phenotype producing pro-inflammatory cytokines. In turn, the cytokines impair adipocyte biology and perpetuate the unbalanced composition of leukocytes in the adipose tissue. As hypertrophy and hyperplasia of adipocytes progress with obesity, metabolic dysfunction and inflammation are exacerbated. Chronic inflammation comprises an accumulation of pro-inflammatory immune cells (i.e. Th1 and Th17 lymphocytes), subtypes of ATMs with pro- and anti-inflammatory functions and the metabolically activated phenotype (M2b/MMe), and MDSC. The molecules produced by both dysfunctional adipocytes and infiltrating immune cells perpetuate low-chronic inflammation and establish a proangiogenic condition. Chronic inflammation in fat tissue leads to insulin resistance, over-production of leptin, and increased levels of free fatty acids (FFA) that contribute to the expansion and enhance the immunosuppressive function of MDSCs and subtypes of ATMs, including MMe. MDSCs limit obesity-associated metabolic dysfunction and down-regulate leptin production while increase adiposity. When tumor onset occurs, infiltrating MDSC, different subtypes of ATMs, and tumor-associated macrophages (TAMs), may facilitate tumor progression and metastasis. Within the tumor microenvironment (TME), the enrichment of cytokines (i.e. G-CSF, IL-6 and GM-CSF) and FFA enhance the immunosuppressive potency of myeloid cells, which in consequence, the protective anti-tumor immune response becomes inhibited, and production of pro-inflammatory and proangiogenic mediators is enhanced, supporting tumor growth and metastasis. IL indicates interleukin; Th1/2, T helper type 1 or 2; TNFα, tumor necrosis factor-alpha; IFN, interferon; CCL2, C-C Motif Chemokine Ligand 2; CXCL5, C-X-C Motif Chemokine Ligand 5; IGF1, Insulin-Like Growth Factor 1; FFA, free fatty acid; GM-CSF, Granulocyte-macrophage colony-stimulating factor; G-CSF, colony-stimulating factor; PGE2, prostaglandin E2; VEGF, Vascular endothelial growth factor; PD-L1, Programmed death-ligand 1; IDO, indoleamine 2,3-dioxygenase; CTLA-4, Cytotoxic T-lymphocyte-associated Protein; TGFβ, transforming growth factor-beta; ROS, reactive oxygen species; RNIs, reactive nitrogen intermediates.
Macrophage subsets as key players in the inflamed fat tissue
Macrophages are plastic cells that easily adjust to different sources of stimuli in the local environment by changing their metabolic and functional properties, a process defined as polarization. Based on the function, genomic signature, and protein expression profiles, a wide spectrum of macrophage activation states between the conventional pro-inflammatory M1 (classical activated) and anti-inflammatory M2 (alternative activated) phenotypes have been identified (Table 1) [15]. Generally, these M2 macrophages are thought to participate in tissue homeostasis, remodeling, and wound healing; however, because of the heterogeneous subpopulations found in different microenvironment settings, those macrophages designated as M2 have rapidly expanded to include several functionally distinct subtypes. Based on the in vitro stimuli applied for their activation, M2 macrophages can be further divided into subsets called M2a, M2b, M2c and M2d [16]. M2a, often termed as wound-healing macrophages, are induced by IL-4 and IL-13 leading to the increased expression of IL-10, TGF-β, and CCL17/18/22. These macrophages activate an anti-parasite Th2 response by producing IL-10 and TGF-β. M2b cells, also known as regulatory macrophages, are activated by immune complexes and TLRs ligands (i.e. lipopolysaccharides or by IL-1β). M2b cells release both pro- and anti-inflammatory cytokines such as IL-1β, IL-6, TNFα, IL-10, CCL1 and TNF superfamily member 14 (TNFSF14), and regulate the immune responses and inflammatory reaction. IL-10 or the combination of glucocorticoids and TGFβ induces M2c macrophages. These cells produce IL-10, TGFβ, CCL16, and CCL18 and play a role in immune suppression, tissue repair, matrix remodeling and phagocytosis of apoptotic cells. M2d macrophages are induced by co-stimulation with TLR ligands and A2 adenosine receptor (A2R) agonists or by IL-6. This leads to the high release of IL-10, TGFβ and vascular endothelial growth factors (VEGF) and low production of IL-12, TNFα and IL1β which promotes angiogenesis and tumor progression. Within the tumor, infiltrating macrophages exhibit functions like those of M2 macrophages and can be classified as M2b and M2d subtypes [17, 18]. These cells display immunosuppressive and tumorigenic functions and express the enzyme arginase 1 (Arg-1), which depletes L-arginine thereby inhibiting anti-tumor T cell function [19] and promoting tumor initiation and progression. In addition to Arg-1 as the main signature of immunosuppressive macrophages, these cells also produce high levels of immune-regulatory cytokines such as IL-10 and TGFβ.
Table 1:
Phenotypic, functional, and metabolic characteristics of macrophages and MDSCs
| M1-like macrophages | M2-like macrophages | Metabolically activated macrophages (MMe) | Tumor-associated macrophages (TAMs) | Myeloid-derived suppressor cells (MDSCs) | ||
|---|---|---|---|---|---|---|
| Surface markers | CD11c, CD38, CD62L, CD274, CD319, CD80, CD86, HLA-DR, CCR2/5/7 | CD163, CD206, TFRC, CD36, CD204, CXCR1, CXCR2, CD206, CD209, CD163, FIZZ1, Ym1/2 | CD36+, CD11c-, CD163-, CD206- | CD36, CD204, CD206, Scavenger Receptor A, CD52, CD163, CD39, CD73 |
Mouse PMN-MDSC: CD11b+, Gr1+, Ly6Cdim, Ly6G+ Mouse M-MDSC: CD11b+, Gr1+, Ly6Chigh, Ly6G+ |
Human PMN-MDSC: CD11b+ (CD33+), CD14–, HLA-DR–, CD15+(CD66b+). Human M-MDSC: CD11b+(CD66b+), CD14+, HLA-DR-/low |
| Activators | TLR, TNFa, IFNg, GM-CSF, LPS | IL-4, IL-10, IL-13, TGFb, COX2-derived PGE2, VEGF, PPARg agonist | Metabolically dysfunctional adipose tissue: ↑Insulin, ↑glucose, ↑FFA | Tumor microenvironment: IL-1b, IL-4, IL-6, IL-8, IL-10, IL-13, IL-23, IL-27, COX2-derived PGE2, PDGF, MIF, CXCL12, CCL2/3/4/17/18/22; hypoxia, FFA | GM-CSF, G-CSF, IL-1b, IL-4, IL-6, IL-13, IL-17, IFNg, TNFa, PGE2, VEGF, TLR ligands, cellular stress, exogenous fatty acid uptake and lipid metabolism | |
| Functional and metabolic markers. Key transcription factors | ROS, iNOS, pro-inflammatory cytokines: TNFa, IFNg, IL-6, IL-12, IL-23. pSTAT1, HIF-1a, IRF5, SOCS3 | Arginase-1, Dectin-1, ABCA1, PLIN2, pSTAT6, IL-10, TGFb, IL-1β, IL-6, TNFα, IL-10, CCL1/16/18, TNFSF14, VEGF, IRF4, JMJD3, PPARg/PGC1b, PPARd. | ABCA1, CD36, PLIN2, p62, IL-1b, TNFa, arginase-1, PPARg | ↓iNOS, ↑Arginase-1, ↑ROS, ↑IDO, COX2, arginase-1, captesin B, MMP2, MMP7, MMP9, VEGF, EGF, PDGF, MIF, TNFa, TGFb, IL-8, IL-1b, TP, CCL2, CXCL8, PPARs, PI-3K/AKT/mTOR pathway | iNOS, IDO, Arginase-1, ROS, PD-L1, CTLA4, IL-1b, IL-6, MMPs, COX2-derived PGE2, pSTAT3, pSTAT6, C/EBPb, IRF8, Notch, HIF-1a, ER-stress response pathway | |
| Metabolic profile | Glycolysis, PPP, FA synthesis, truncated TCA cycle, triglycerides accumulated in lipid droplets | ↓Glycolysis, ↓PPP, OXPHOS, FAO, intact TCA, ↑Glutamine and lipid metabolism | ↑Glycolysis, ↑OXPHOS | Dynamic metabolic profile, although predominant glutamine and FA metabolism, ↓PPP, Glycolysis, ↑OXPHOS, FAO, broken TCA | Glycolysis, OXPHOS, ↑FAO | |
| Function | Pro-inflammatory, antimicrobial, and anti-tumor activity | Wound-healing and anti-parasite (M2a), regulatory cells and immunosuppression (M2b/c), tissue repair and matrix remodeling (M2c), angiogenesis and tumor progression (M2d) | Pro-and anti-inflammatory with potentially immunosuppressive abilities (M2b-like phenotype) | Immunosuppressive capabilities: Inhibit cytotoxic T lymphocytes (CTL) responses. Support angiogenesis, oncogenesis and metastasis, production of pro- and anti-inflammatory cytokines | ||
HLA-DR indicates Human Leukocyte Antigen – DR isotype; TLR, Toll-like receptor; TNFa, tumor necrosis factor alpha; IFNg, interferon gamma; GM-CSF, Granulocyte-macrophage colony-stimulating factor; LPS, lipopolysaccharide; ROS, reactive oxygen species; pSTAT, phosphorylated Signal transducer and activator of transcription; iNOS, inducible nitric oxide synthase; TFRC, transferrin receptor; CCR2, C-C Motif Chemokine Receptor 2; CXCR1/2, Chemokine (C-X-C motif) receptor 1/2; CXCL, C-X-C motif Chemokine Ligand; CCL, C-C Motif Chemokine Ligand; IL, interleukin; HIF1a, hypoxia-inducible factor 1-alpha; IRF, Interferon Regulatory Factor; SOCS3, Suppressor of cytokine signaling 3; PPP, pentose phosphate pathway; FA, fatty acid; TCA, tricarboxylic acid; TGFb, transforming growth factor beta; COX2, cyclooxygenase-2; PGE2, prostaglandin E2; VEGF, Vascular endothelial growth factor; ABCA1, ATP Binding Cassette Subfamily A Member 1; PLIN2, Perilipin 2; TNF superfamily member 14, TNFSF14; JMJD3, Jumonji domain-containing protein D3; PPARd/g, Peroxisome proliferator activated receptor delta/gamma; PGC1b, peroxisome proliferator-activated receptor-gamma coactivator 1-beta; OXPHOS, Oxidative phosphorylation pathway; FAO, fatty acid phosphorylation; FFA, free fatty acid; p62, 62 kDa protein; PDGF, platelet-derived growth factor; MIF, Macrophage Migration Inhibitory Factor; IDO, indoleamine 2,3-dioxygenase; MMP, metallopetidase; PI-3K, Phosphoinositide 3-kinase; AKT, Protein kinase B, PKB; mTOR, mechanistic Target Of Rapamycin Kinase; EGF, epidermal growth factor; PDGF, Platelet-derived growth factor; TP, Thymidine Phosphorylase; PMN-MDSC, Polymorphonuclear Myeloid-derived suppressor cell; M-MDSC, Monocytic Myeloid-derived suppressor cell; G-CSF, Granulocyte colony-stimulating factor; PD-L1, Programmed death-ligand 1; CTLA4, cytotoxic T-lymphocyte-associated protein 4; C/EBPb, CCAAT/enhancer-binding protein beta; ER, endoplasmic reticulum.
Signals found in the complex milieu of fat tissue from mice and humans with obesity also elicit multiple distinct subpopulations of ATM with distinctive function, transcriptome signature, and epigenetic landscapes [20]. For instance, macrophages in the peritoneal cavity and cecal tissue from mice with diet-induced obesity (DIO) exhibit an M2b polarization (TNFαhigh, IL-10high, CD206+, Dectin1+) and produce TNFα, IL-1β and IL-10 [21]. Also, ATM that are metabolically activated by dysregulated mediators found in adiposity, named metabolically activated macrophages (MMe) [22, 23], exhibit both M1 pro- and M2 anti-inflammatory characteristics (as described for M2b cells) with functional properties of M2-like macrophages [23]. Potential immunosuppressive properties in these MMe may be modulated by PPARγ, a transcription factor that regulates the gene expression of M2-like markers including Arg-1 [24], which confers the ability to limit the pro-inflammatory response of MMe [23]. Interestingly, adipose-derived mesenchymal stem cells (ADMSCs), which are suspected to promote tumor development and progression, have an important role in immunosuppression in the local tissue. Culture of peritoneal macrophages with conditioned media derived from ADMSCs induces macrophage polarization towards M2b/c phenotype via IL-6. Although M2b/c macrophages can produce several pro-inflammatory cytokines, the most prominent feature of these cells is the large production of IL-10 [25]. However, whether DIO-induced macrophage subsets can block the protective anti-tumor T cell immune response has not been convincingly demonstrated. These observations suggest that M2b/c and MMe macrophages may be critical cell populations within tumor-associated macrophages (TAMs) in obesity. Despite the critical roles of macrophage subsets in adiposity progression and comorbidities associated with obesity such as cancer, the specific molecular and metabolic markers of M2b and MMe macrophages, as well as their ability to block protective T cell immune response have not yet been definitively established.
MDSCs as major contributors to chronic inflammation
MDSCs are a heterogeneous population of immature myeloid cells with pro-inflammatory and immune-suppressor capacity. Two major subtypes have been identified, monocytic- (M-MDSCs) and granulocytic polymorphonuclear-MDSCs (PMN-MDSCs). A third recently described subset of human MDSCs, named early (e)-MDSCs, is considered precursors of M- and PMN-MDSCs [26]. The knowledge about MDSCs biology is derived mostly from studies in cancer, but they are also found in other chronic inflammatory conditions, such as autoimmunity, transplantation, infections, and pregnancy [27, 28]. MDSCs are powerful inhibitors of the protective anti-tumor immune response through diverse mechanisms. Those include i) the expression of the enzymes Arg-1, inducible nitric oxide synthase (iNOS), and indoleamine 2,3-dioxygenase (IDO) that deplete essential amino acids for T cell activation; ii) the production of ROS, and peroxynitrite (PNT) that cause oxidative stress; iii) by expressing molecules such as cytotoxic T-lymphocyte-associated protein 4 (CTLR4) and programmed death-ligand 1 (PD-L1) that block effector T cells responses; and iv) by secreting IL-10 and TGFβ that restrain the pro-inflammatory functions of immune cells. However, MDSCs also produce several pro-inflammatory molecules such as IL-1β and IL-6 that may contribute to chronic inflammation [29–31] (Table 1).
Obesity promotes an increase in circulating inflammatory cytokines leading to alteration of immune cell production, activation, and infiltration in tissues. Mechanisms of MDSCs expansion in obesity-associated inflamed tissue rely on VEGF, IL-1β, IL-6, TNFα, and PGE2 as well as growth factors that regulate myelopoiesis such as GM-CSF and G-CSF [32]. The role of MDSCs in the pathophysiology of obesity is still under extensive investigation. Recent studies showed an association between the elevated number of MDSCs with reduced insulin resistance [33], down-regulation of leptin [34], increased adiposity [34] and increased liver damage [35]. Furthermore, DIO tumor-bearing mice have increased accumulation and immunosuppressive activity of MDSCs and accelerated tumor growth compared to lean-tumor bearing counterparts in models of breast, renal and prostate cancer [34, 36, 37]. Meticulous dissection of the role of different dysfunctional factors in obesity on the emergence of MDSCs in cancer may provide important insight into the connection between obesity, tumor growth, and anti-tumor immunity.
Accumulation of MDSCs is observed in different tissues such as the liver, spleen, peripheral blood, and visceral fat tissue of mice fed with HFD (60% kcal fat, 6.8% kcal sucrose) [33, 35]. Furthermore, an increased number of MDSCs occurs in the peripheral blood of patients with overweight/obesity (BMI > 25) [38, 39]. Whether tissue residing MDSCs are involved in local or distant cancer development have not been explored. An increased number of MDSCs in different tissues could generate a systemic immunosuppressive environment attenuating the protective anti-tumor immune response in the site of tumor onset. In fact, increased levels of circulating Arg-1 have been shown in individuals with overweight [40] and higher Arg-1 activity in serum from women with obesity and metabolic syndrome [41]. Although it is unknown if MDSCs are the source of the increased levels of Arg-1 in circulation, it is plausible that Arg-1 produced by tissue infiltrating and circulating MDSCs creates a systemic immunosuppressive microenvironment. During cancer onset, these obesity-induced MDSCs could migrate to primary tumors in response to tumor-derived chemokines. Indeed, primary tumors and spleens of tumor-bearing mice are characterized by elevated CCL2 concentrations and hence increased infiltration of functionally immunosuppressive MDSCs [36].
Metabolic reprogramming of myeloid cells in obesity
The phenotype and function of macrophages rely on their flexibility to switch their metabolism in response to the local metabolic stimuli [42]. Macrophages with M1 pro-inflammatory function (M1-like) rely on glycolytic metabolism and pentose phosphate pathway (PPP) to achieve ATP requirements, with downregulation of oxidative phosphorylation (OXPHOS) and fatty acid beta-oxidation (FAO), and tricarboxylic acid (TCA) cycle interrupted. Conversely, macrophages with anti-inflammatory functions (M2-like) display increased uptake and catabolism of glutamine and lipids, enhanced FAO, and OXPHOS with an intact TCA cycle, while glycolysis is abrogated [42, 43]. The increased lipid uptake and metabolism result in signaling cascades that promote the expression of Arg-1 [24, 43]. In contrast, MMe in metabolically dysfunctional adipose tissue display a unique hypermetabolic profile with both enhanced glycolysis and OXPHOS compared to ATMs from lean adipose tissue [42, 44]. This metabolic phenotype is the result of combinatorial signaling induced by elevated concentrations of saturated fatty acids (SFAs), insulin, and glucose [23]. It is unclear whether MDSCs exhibit a different activation status than macrophages that could be metabolically modulated; however, it is known that glycolysis, OXPHOS, and FAO are increased in MDSCs [45], and the immunosuppressive capacity of tumor-infiltrating MDSCs is enhanced by FA uptake and FAO [45, 46]. Together, this suggests that microenvironments rich in lipids, such as obesity, could potentiate the immunosuppressive properties of MDSCs. Inflammatory and metabolic alterations that occur in obesity, besides directly affecting malignant cells, are critical for shaping the metabolic and functional phenotype of myeloid cells.
Recent studies have shown that myeloid cells can adopt a long-term memory, named trained immunity, induced by sustained exposure to a stimulus that leads to long-term changes in intracellular metabolism and histone remodeling [47]. In consequence, this trained immunity phenotype yields an enhanced response upon a second triggering signal. Obesity-induced trained immunity in monocytes has been associated with increased cardiovascular risk [48]. However, whether obesity confers a trained immunity phenotype in MDSCs and ATMs subtypes is unknown. It is plausible that such training by obesity leads to an increased immunosuppressive response following a secondary stimulation such as tumor initiation contributing to cancer progression.
Although progress has been achieved in characterizing the intrinsic metabolic-associated profiles of macrophages and MDSCs, little is known about the impact of systemic metabolic disturbances in adiposity that play a role in obesity-related reprogramming of myeloid cells towards immunosuppressive pro-tumor cells. Next, we provide some evidence of the putative role of lipids, insulin, and leptin in mediating metabolic reprogramming of MDSCs and macrophages (Figure 1).
Role of lipids in the immunosuppressive function of myeloid cells
Alteration of lipid homeostasis is common in metabolic diseases that are characterized by chronic inflammation such as obesity, hepatic steatosis, and cardiovascular disease. Chronic intake of dietary fats and excessive release of lipids, including FFAs, triglycerides, and cholesterol, as a result of increased lipolysis and adipocyte cell death, is observed in obesity [49]. Hyperplastic and hypertrophic adipocytes provide a continuous source of lipid species including SFAs, which propagate the pro-inflammatory condition of obesity [50]. The elevated levels of FFAs and lipoproteins may cause a profound metabolic and functional reprogramming of myeloid cells priming their phenotype as immune-regulatory cells. In macrophages, SFAs, particularly lauric acid and palmitic acid, stimulate a pro-inflammatory response through the TLR4 signaling pathway priming macrophages towards an M1-like phenotype [51]. On contrary, polyunsaturated acids (ω−3 fatty acid docosahexaenoic acid (DHA), eicosapentaenoic acid (EPA), linoleic and) and the monosaturated oleic acid inhibit the expression of inflammatory genes, bind and activate PPAR signaling pathway, that results in metabolic reprogramming of macrophages towards an anti-inflammatory M2-like profile [52].
Dietary linoleic acid, particularly consumed from refined omega-6 vegetable oils, is incorporated into lipoproteins increasing the susceptibility to be oxidized and may increase cardiovascular risk [53]. It has been shown that the exposition of M2-like macrophages to oxidized LDL enhances a pro-inflammatory response [54]. However, the exposition to oxidized phospholipids, that is accumulated in atherosclerotic lesions, instead activate macrophages towards a functionally distinct phenotype from the conventional M1 or M2 polarization [55]. In the same context of atherosclerosis, macrophages treated with desmosterol, the most prevalent sterol in atherosclerotic plaques, or macrophages that contain high cholesterol content which also leads to accumulation of desmosterol, have down-regulated gene expression involved in pro-inflammatory responses [56].
Furthermore, intracellular accumulation of triglycerides, in form of lipid droplets, drives macrophages towards an M1-like phenotype [57], while enhanced lipid catabolism by FAO promotes the anti-inflammatory M2-like activation in mouse [58], but partially, in human macrophages [59]. Besides FAO, ATMs from DIO mice also activate the lysosomal lipid metabolism following a program towards M2-like polarization [60]. In MDSCs, lipid uptake and FAO is closely associated with functional responses [46, 61]. The high lipid content in the tumor microenvironment fuels the oxidative metabolism of MDSCs and increases the expression of immunosuppressive molecules in tumor-resident MDSCs [46]. Whether MDSCs in obesity-related dyslipidemia and lipolysis are functionally and phenotypically distinct to tumor-resident MDSCs remains uncharacterized. These findings suggest that in addition to promoting systemic insulin resistance [62], lipids differentially activate diverse polarization programs in myeloid cells depending on lipid species, modifications of their chemical structure by oxidation, and lipid catabolism.
Impact of insulin in myeloid cells polarization
Obesity is commonly associated with insulin resistance, which leads to increased concentrations of insulin, and amplified bioactivity of insulin growth factor-1 (IGF-1). Insulin mediates its signal through the insulin receptor (IR), but also by its highly homologous Insulin-like growth factor 1 receptor (IGF-1R). It is well known the important role of insulin signaling in adipocytes and cancer cells. In adipocytes, insulin receptor stimulation promotes glucose and FFA uptake, stimulates de novo lipogenesis, and inhibits lipolysis; however, these effects are dependent on the insulin resistance status [63]. Many cancer cells overexpress isoform A of the insulin receptor (IR-A) which has a higher affinity for insulin/IGF-1 compared to isoform B. Signaling through IR-A promotes a mitogenic response of cancer cells and inhibits apoptosis leading to enhanced cellular proliferation and support of tumor cell proliferation [64].
The insulin signaling in myeloid lineage cells may differentially influence cell activation and its effect may depend on the level and ability to trigger signaling pathways. Some studies showed the contribution of insulin on macrophage metabolism which promotes glycolysis causing polarization towards an M1-like profile [23, 43]. The pro-inflammatory polarization of Insulin/IGF-1 in macrophages has been demonstrated in vitro [65] and in vivo in the context of atherosclerosis [66], obesity-induced inflammation and systemic insulin resistance [67]. Expression of IRS2, an adaptor protein that mediates both insulin and IGF-1 signaling, suppresses the M2-like polarization of macrophages in vivo in a model of allergic lung inflammation [68]. Macrophages also express other molecules involved in the insulin signaling cascade, including receptors (IR and IGF-1R), and Akt kinase isoforms which differentially influence the functional phenotypes; for example, the ablation of Akt2 renders macrophages prone to M2-like, whereas the absence of Akt1 renders them towards M1-like polarization [69].
Myeloid lineage-restricted insulin resistance has also been suggested. Peripheral macrophages from mice with systemic insulin resistance or in vitro exposure to elevated insulin become insulin resistant and exhibit a subtype of M2-like macrophages [69], termed as insulin resistance macrophages (M-InsR). These cells have a reduced expression of IR and IGF-1R, defective IR signaling, and increased glycolysis in response to insulin [69]. Genetic ablation of IR in macrophages protected against inflammation and systemic insulin resistance in HFD (55.2% kcal fat) fed mice [67], and also decreased pro-inflammatory gene expression in macrophages [70], suggesting a polarization towards M2-like profile. Certainly, M-InsR macrophages presented a reduced response to lipopolysaccharide (LPS; inducer of M1-like polarization), but express Arg-1 and found in inflammatory zone 1 (Fizz1) (M2 markers) [69]. Together, these findings suggest that defective IR signaling could promote MMe phenotype.
The biological functions of insulin signaling on MDSCs metabolism and its contribution to their immune-regulatory capacity are poorly studied. Although it is unknown whether insulin directly modulates transcriptional control on MDSCs, systemic insulin resistance appears to induce their expansion as a physiological response to promote insulin sensitivity. This was observed in a mouse model of DIO, where depletion of MDSCs exacerbated insulin resistance, while adoptive transfer of MDSCs significantly improved glucose tolerance and reduced inflammation [33]. This observation suggests that MDSCs may contribute to the maintenance of systemic metabolic functions. However, following the theory of trained immunity by obesity-related metabolic dysfunction, the permanent accumulation of MDSCs to restore insulin sensitivity also may impair immune surveillance and antitumor immunity contributing to tumor development.
Leptin as an immunomodulatory molecule with a potential role in polarizing myeloid cells
Leptin is another hormone found in high concentrations in obesity and has additional biological properties besides the regulation of energy balance. Leptin is a pro-inflammatory and pro-angiogenic adipokine that promotes survival, proliferation, and modulates the function of several immune cells [71]. Although the mechanism is not yet identified, the crosstalk between leptin and MDSCs was recently described in vivo. The over-production of leptin was associated with an increased frequency of MDSCs in DIO mice and counteracted by the inoculation of soluble leptin receptor [34]. In this study, the leptin-dependent accumulation of MDSCs was associated with suppression of anti-tumor CD8+ T cell response and further tumor progression and metastasis in DIO tumor-bearing mice. These findings suggest that higher levels of leptin drive MDSCs accumulation and immunosuppressive function fostering tumor growth and cancer progression. The effect of leptin on reprogramming myeloid cells has not been rigorously explored; however, leptin receptor (ObR) is ubiquitously expressed on the surface of immune cells, regulates both innate and adaptive immunity [72], and stimulates differentiation and leukocyte migration [73]. Activation of leptin/ObR signaling on macrophages elicits the secretion of several pro-inflammatory and pro-angiogenic cytokines such as IL-1, TNFα, IL-6, IL-11 and facilitates the pro-inflammatory polarization [74]. These cytokines, in combination with other molecules such as prostaglandin E2 (PGE2), which is also elevated in obesity, could indirectly favor the induction of MDSCs [32]. Besides the impact of leptin through induction of cytokine production, leptin itself may have a direct effect on myeloid cells by activating the leptin/ObR/STAT3 signaling cascade. STAT3 is a transcription factor critical for MDSCs expansion and function and confers immunosuppressive abilities of TAMs [75], suggesting a plausible role of leptin in inducing a myeloid cell activation towards immunosuppressive cells.
Dietary immunomodulation
A recent study has revealed that different nutritional components, besides influencing metabolism, also affect critical pathways to inflammation [76]. This could be explained by the ability of dietary patterns and nutrients to modulate immune cells response via several broad mechanisms related to circulating hormones, excess of nutrients, and alteration in the gastrointestinal (GI) tract. The GI tract is the initial intersection between diet and immunity. Gut-associated lymphoid tissue, a component of the mucosa-associated lymphoid tissue, is the largest immune organ in the body [77, 78]. Complex interactions among diet and microbiome alter activation and function of resident immune cells. Also, diet can alter gut microbiota diversity and function. Microbial dysbiosis is related to local and systemic inflammation [79] and alteration of the host immune system composition [80]. Maintaining gut microbiota diversity is important as gut microbiota produce short-chain fatty acids (SCFA) as a byproduct of fiber fermentation. These SCFA have an important role in regulating and maintaining normal function of the innate and adaptive immune system. Indeed, SCFA activate anti-inflammatory effects including reduction of pro-inflammatory cytokines, modulating the proliferation, differentiation, and activation of T cells, and downregulate the pro-inflammatory response of myeloid cells [81, 82]. In consequence, the anti-inflammatory high fiber diet improves host immunity by maintaining microbial diversity, increasing SCFA production and lowering local and systemic inflammation all while slowing glucose and decreasing cholesterol absorption [82].
Mouse models have increased understanding of the role different diets have in the alteration of metabolism, inflammation, and obesity. Pro-inflammatory diets including the commonly used HFD, in which the proportion of calories deriving from different nutrients is 60% fat, 20% protein, 20% carbohydrate (6.8% sucrose) or high-fat high sucrose diet (HFHSD; 45% fat, 24% protein, 35% carbohydrate (17% sucrose)) are highly obesogenic. Conversely, ketogenic diets (KD; 89–94% fat, 5–10% protein, 1% carbohydrate (0% sucrose)) have an anti-inflammatory effect by activating metabolically protective γδT cells and lowering pro-inflammatory cytokines (e.g. TNFα, IFN, IL-1, and IL-6) [83, 84]. In some preclinical rodent models, KD has shown to slow tumor growth, reduce angiogenesis, inflammation, migration, invasion by boosting tumor-reactive immune response in mice (i.e. increased CD4+ T cell infiltration and increased cytokine production by tumor-reactive CD8+ T cells) and increase survival [85, 86]. The decrease of inflammatory cytokines and increase in CD4+ T cells infiltration into the tumor suggests that manipulation of metabolism by this specific diet may abrogate the function of immunosuppressive myeloid cells immunotherapy allowing the anti-tumor T cell response. In fact, KD depletes MDSCs and Tregs, thereby improving the immunological profile of pancreatic tumor-bearing animals [87]. Interestingly, the impact of nutrition and systemic metabolism on myeloid cells function also influences the responses to cancer therapy. Several animal studies with fasting, hypocaloric, ketogenic and western diets have shown a different effect on the efficacy of chemotherapy and toxicity of cancer therapies [88]. However, more research is warranted to understand the molecular, metabolic, or epigenetic mechanisms by which macro- and micronutrients influence immune cell functions and response to anticancer therapies.
In humans, the impact of diets on inflammation has been calculated by the dietary inflammatory index (DII), which has been used to evaluate the contribution of diet to cancer risk. Women who consumed diets with high inflammatory potential indicated by the DII score, had an increased risk of breast cancer, compared to women who consumed more anti-inflammatory diets [89]. The promising effect of dietary intervention on cancer risk was also observed in African Americans when increasing dietary fibers changed the microbiome and increased SCFA production, mainly butyrate, resulting in a reduction of biomarkers of cancer risk [90]. The positive impact of dietary intervention on inducting protracted anticancer immune response after chemotherapy has also been observed [91]. The mechanisms involved in the response are still under investigation; however, patients with a low-calorie KD have shown improvements in levels of insulin and glucose suggesting an important impact on cellular metabolism. The consumption of a SFA-rich diet results in a pro-inflammatory gene expression profile in subcutaneous adipose tissue (i.e., IL-1β, IL-6, and TNF-α), while a monounsaturated fatty acid-rich diet causes different profiles depending on the category of subjects [92]. Whether such dietary effects might involve a major impact on immunity and immunotherapy response has been poorly explored. Nevertheless, different nutritional interventions to boost the efficacy of different cancer therapy in patients with a variety of advanced solid malignancies have begun [91].
The intriguing finding of obesity improve response to immunotherapy
Surprisingly, while augmented weight is associated with increased cancer risk and patients with obesity and certain types of cancer have a poorer prognosis, obesity seems also to confer survival advantages to some cancer treatments [93]. The improved outcome was recently shown in patients with obesity and cancer receiving programmed death receptor-1/programmed death-ligand (PD-1/PD-L1) blockade therapy (checkpoint blockade immunotherapy. PD-L1 is expressed by tumor and immune cells that interact with the corresponding receptor PD-1 which is expressed on T and NK cells. Sustained PD-1 stimulation by PD-L1/PD-1 interaction interferes with T cell receptor signal transduction causing T cell exhaustion and apoptosis [94], supporting tumor cell ability to evade the immune system [95]. The mechanisms for the efficiency of the immunotherapy in obesity are incompletely understood; however, the expression of PD-L1 on MDSCs [96], macrophages [97], and the surface of tumor cells [95] could be higher in obesity which may facilitate an enhanced immunotherapy response. In fact, infiltrated MDSCs in HFD (60% kcal fat, 6.8% kcal sucrose) fed mice have elevated immunosuppression capacity by expressing higher levels of PD-L1 compared to tumor-infiltrating MDSCs in mice fed with low-fat diet [34]. Although the mechanisms of obesity to amplify the expression of PD-L1 on cells are under investigation, pro-inflammatory molecules such as IFNγ and leptin are potentially involved [93, 98]. These findings indicate that IFNγ and leptin upregulate the expression of PD-L1 and PD-1 providing a broader target for checkpoint blockade immunotherapy in patients with obesity. Although the evidence supporting improved outcomes is markedly strong for PD-1/PD-L1 blockade therapies, anti-Cytotoxic T-lymphocyte-associated Protein (CTLA)-4 therapy in patients with obesity has also been shown to be beneficial. CTLA-4 is expressed by activated T-cells and a subset of regulatory T-cells and acts as a competitive inhibitor by counteracting the signaling through costimulatory receptors during antigen-presenting cell (APC) and T cell interaction. Recently, a review of studies demonstrated improved survival in men with obesity and metastatic melanoma who were treated with anti-CTL4 with or without combination with chemotherapy [93]. Interestingly, the response to anti-CTL4 therapy in DIO mice was only achieved when leptin levels were reduced [99], inferring a potential inverse relationship between leptin concentration and CTLA-4 expression.
Concluding remarks and future directions.
There is compelling evidence linking obesity to cancer; however, the underlying molecular mechanisms driving this association remain incompletely established. One of the major factors associated with the metabolic inflammation of obesity is the expansion of myeloid cells with immunosuppressive and pro-oncogenic abilities. Obesity is a complex disease with a malfunction of multiple factors that independently, or in combination, lead to the accumulation of myeloid cells. Although it is partly defined, several alterations that promote low-grade inflammation and expansion of myeloid immunosuppressive cells may include i) nutrient over-supply, ii) tissue hypoxia, iii) excess of growth factors, cytokines and lipolysis, and iv) metabolic and endocrine abnormalities, such as dyslipidemia, deregulated signaling by insulin and leptin, altered levels of resistin and adiponectin, elevated bioavailable estrogen and hypovitaminosis due to sequestration of fat-soluble vitamins such as the immuno-modulator vitamin D in body fat depots. All together may create the milieu for obesity-induced trained immunity.
The sustained presence of inflammatory myeloid cells keeps the vicious cycle skewed towards adipogenesis, perpetuating chronic inflammation, and creates an ideal microenvironment that supports cancer initiation, growth, and metastasis. Myeloid immunosuppressive cells inhibit anti-tumor immune response and present an obstacle to cancer immunotherapy. In this review, we focused on the view towards MDSCs and subtypes of macrophages as critical contributors to cancer development induced by obesity-associated inflammation and metabolic disorder. We discussed the role of insulin, IGF-1, lipids and leptin in metabolically priming, or training, macrophages and MDSCs in obesity. These cells may adopt an enhanced immunosuppressive capacity followed by a subsequent stimulation derived from the tumor onset.
Despite the progressive evidence about metabolic and functional characteristics of macrophages and MDSCs in different diseases, including cancer and obesity, important gaps remain. As research continues, there are outstanding questions to be considered:
Is the elevated cancer risk in obesity due to the reprogramming of immune cells by the systemic metabolic abnormalities? If so, do patients with obesity without metabolic syndrome have less accumulation of myeloid immunosuppressive cells and therefore a reduced risk of cancer? Conversely, are normal-weight individuals with metabolic syndrome predisposed to the oncogenic microenvironment because of the increased myeloid immunosuppressive cells? Efforts are needed to compare metabolically healthy versus unhealthy individuals. Also, the detailed molecular mechanisms and crosstalk of cytokines, hormones, lipids, among others, to expand and activate myeloid immunosuppressive cells in the settings of obesity-associated disturbances, are critically needed.
Several studies have shown that patients who have had bariatric surgery have a reduction in cancer incidence and mortality [100]. Does bariatric surgery lower the risk of cancer by reversing the accumulation or function of immunosuppressive myeloid cells in patients with obesity? Is the normal-weight inflammatory (untrained) phenotype restored upon weight loss and resolution of metabolic disorders induced by bariatric surgery? Do patients resistant to anti-obesity interventions or those having difficulty achieving sustained weight loss have increased accumulation of metabolically trained myeloid cells? Biomarkers to properly identify these cells in peripheral blood or fat tissue residents are required.
Following our hypothesis that the risk of cancer is driven by the expansion and function of myeloid immunosuppressive cells in obesity, could the use of drugs known to target MDSCs and TAMs prevent cancer initiation in patients with morbid obesity? However, strategies for partially blocking the MDSCs functions should be counted to assure the maintenance of their ability to restore insulin sensitivity while simultaneously preventing their capacity to restrain the anti-tumor immune response.
What is the immunological profile of primary, advanced, and metastatic tumors in patients with obesity? Is there any degree and specific activation subtypes of infiltrating myeloid cells in the tumor milieu that predict a better response to checkpoint-inhibitors therapy in patients with obesity? Enrollment of patients across the body mass index (BMI), metabolic, and inflammatory spectrum is crucial to study the response of immunotherapy. These studies can facilitate the identification of potential biomarkers to distinguish patients that will respond to immunotherapy. It will also allow the detection of patients that may need an initial therapeutic approach that enables immunologic and metabolic tumor profile modification before the immunotherapy initiation to improve outcomes.
Many more research questions remain: What is the causal relationship between metabolic alterations, epigenetics, and functional phenotype in obesity? Can any of these mechanisms be targeted by therapies to reprogram the immune-regulatory myeloid cells? How is this relationship at the molecular level relevant for the development of effective anti-cancer therapeutics in obesity? What is the interaction between metabolic factors and the intrinsic molecular characteristics of myeloid immunosuppressive cells that facilitate their anti-tumor hallmarks?
We acknowledge that the relationships between obesity, cancer, and the immune system are very complex. We propose a hypothesis that the multi-factorial metabolic and inflammatory abnormalities in obesity, independently or in combination, lead to the expansion and activation of metabolically primed myeloid immunosuppressive cells. Although their induction seems to be promoted to restore the metabolic homeostasis and curtail overt immune responses, obesity may also prime these myeloid cells that enhance their pro-oncogenic properties promoting tumor growth. Furthermore, although the maintenance of a healthy weight is an essential principle to prevent cancer, obesity, within the context of a certain metabolic and inflammatory phenotype that remains to be defined, may be advantageous for the response of immune checkpoint therapy.
Study importance questions:
What major reviews have already been published on this subject?
The majority of reviews have been focused on obesity inducing cellular transformation into malignant cells and broadly the role of obesity-related inflammatory mediators on cancer.
What are the new findings in your manuscript?
We discuss in the manuscript a perspective of the pro-tumor impact of obesity where myeloid cells with immunosuppressive capacity are the key players.
The concepts of reprogramming and trained immunity are used to elucidate the molecular mechanisms of metabolic disturbances in obesity for enhancing the pro-oncogenic capabilities of MDSCs and subtypes of ATMs with immune-regulatory properties.
Finally, the recent controversial finding of obesity improving immunotherapy response is reviewed, and our hypothesis is offered.
How might your results change the direction of research or the focus of clinical practice?
The manuscript highlights several questions where is emphasized the importance of finding biomarkers related to the identification of myeloid immunosuppressive cell activation status, as well as the systemic metabolic and inflammatory grades in patients with obesity to predict outcomes in terms of anti-obesity interventions such as bariatric surgery, cancer progression, and immunotherapy.
Funding:
This work was supported by NIH funds 5P30GM114732-02, P20CA233374 and the Obesity / Cancer Pilot and Feasibility Grant (Grant # DK072476 Nutrition Obesity Research Center (NORC) from Pennington Biomedical Research center and Louisiana Cancer Research Center (LCRC-Louisiana State University) to MDSP; by the National Center for Advancing Translational Sciences of the National Institutes of Health under award number KL2TR003097 to LAG, by the National Institute of General Medicine Sciences of the National Institutes of Health (U54-GM104940), the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health (P30-DK072476), by the National Cancer Institute of the National Institutes of Health (R00-CA218603 and R25-CA203650) and Susan G. Komen Foundation to JB; and by NIH funds (5P30GM114732-02, P20CA233374) to AO. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The funding agency had no role in the design and conduct of the study; collection, management, analysis and interpretation of the data; preparation, review or approval of the manuscript; and decision to submit the manuscript for publication.
Footnotes
Disclosures: The authors declared no conflict of interest
References:
- 1.Xu YXZ and Mishra S, Obesity-Linked Cancers: Current Knowledge, Challenges and Limitations in Mechanistic Studies and Rodent Models. Cancers (Basel). 2018;10:523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Renehan AG, Zwahlen M, and Egger M, Adiposity and cancer risk: new mechanistic insights from epidemiology. Nat Rev Cancer. 2015;15:484–98. [DOI] [PubMed] [Google Scholar]
- 3.Okwan-Duodu D, Umpierrez GE, Brawley OW, and Diaz R, Obesity-driven inflammation and cancer risk: role of myeloid derived suppressor cells and alternately activated macrophages. Am J Cancer Res. 2013;3:21–33. [PMC free article] [PubMed] [Google Scholar]
- 4.Ugel S, De Sanctis F, Mandruzzato S, and Bronte V, Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest. 2015;125:3365–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muñoz JL, Ortega J, Gutiérrez O, Villalobos PT, Contreras JF, and Ventura J, Adipose Tissue and Inflammation, Adipose Tissue, Leszek Szablewski, IntechOpen. 2018. https://www.intechopen.com/books/adipose-tissue/adipose-tissue-and-inflammation.
- 6.Grivennikov SI, Greten FR, and Karin M, Immunity, inflammation, and cancer. Cell. 2010;140:883–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matthews SB and Thompson HJ, The Obesity-Breast Cancer Conundrum: An Analysis of the Issues. Int J Mol Sci. 2016;17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Azad N, Rojanasakul Y, and Vallyathan V, Inflammation and lung cancer: roles of reactive oxygen/nitrogen species. J Toxicol Environ Health B Crit Rev. 2008;11:1–15. [DOI] [PubMed] [Google Scholar]
- 9.Lin R, Zhang C, Zheng J, Tian D, Lei Z, Chen D, et al. , Chronic inflammation-associated genomic instability paves the way for human esophageal carcinogenesis. Oncotarget. 2016;7:24564–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deng T, Lyon CJ, Bergin S, Caligiuri MA, and Hsueh WA, Obesity, Inflammation, and Cancer. Annu Rev Pathol. 2016;11:421–49. [DOI] [PubMed] [Google Scholar]
- 11.Ray I, Mahata SK, and De RK, Obesity: An Immunometabolic Perspective. Front Endocrinol (Lausanne). 2016;7:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Braune J, Weyer U, Hobusch C, Mauer J, Bruning JC, Bechmann I, et al. , IL-6 Regulates M2 Polarization and Local Proliferation of Adipose Tissue Macrophages in Obesity. J Immunol. 2017;198:2927–2934. [DOI] [PubMed] [Google Scholar]
- 13.Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, et al. , Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity. 2010;32:790–802. [DOI] [PubMed] [Google Scholar]
- 14.Rahman SM, Janssen RC, Choudhury M, Baquero KC, Aikens RM, de la Houssaye BA, et al. , CCAAT/enhancer-binding protein beta (C/EBPbeta) expression regulates dietary-induced inflammation in macrophages and adipose tissue in mice. J Biol Chem. 2012;287:34349–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mosser DM and Edwards JP, Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roszer T, Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediators Inflamm. 2015;2015:816460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang LX, Zhang SX, Wu HJ, Rong XL, and Guo J, M2b macrophage polarization and its roles in diseases. J Leukoc Biol. 2019;106:345–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Correa LH, Correa R, Farinasso CM, de Sant’Ana Dourado LP, and Magalhaes KG, Adipocytes and Macrophages Interplay in the Orchestration of Tumor Microenvironment: New Implications in Cancer Progression. Front Immunol. 2017;8:1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arlauckas SP, Garren SB, Garris CS, Kohler RH, Oh J, Pittet MJ, et al. , Arg1 expression defines immunosuppressive subsets of tumor-associated macrophages. Theranostics. 2018;8:5842–5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hill DA, Lim HW, Kim YH, Ho WY, Foong YH, Nelson VL, et al. , Distinct macrophage populations direct inflammatory versus physiological changes in adipose tissue. Proc Natl Acad Sci U S A. 2018;115:E5096–E5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lefevre L, Gales A, Olagnier D, Bernad J, Perez L, Burcelin R, et al. , PPARgamma ligands switched high fat diet-induced macrophage M2b polarization toward M2a thereby improving intestinal Candida elimination. PLoS One. 2010;5:e12828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coats BR, Schoenfelt KQ, Barbosa-Lorenzi VC, Peris E, Cui C, Hoffman A, et al. , Metabolically Activated Adipose Tissue Macrophages Perform Detrimental and Beneficial Functions during Diet-Induced Obesity. Cell Rep. 2017;20:3149–3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kratz M, Coats BR, Hisert KB, Hagman D, Mutskov V, Peris E, et al. , Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. 2014;20:614–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gallardo-Soler A, Gomez-Nieto C, Campo ML, Marathe C, Tontonoz P, Castrillo A, et al. , Arginase I induction by modified lipoproteins in macrophages: a peroxisome proliferator-activated receptor-gamma/delta-mediated effect that links lipid metabolism and immunity. Mol Endocrinol. 2008;22:1394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun M, Sun L, Huang C, Chen BC, and Zhou Z, Induction of Macrophage M2b/c Polarization by Adipose Tissue-Derived Mesenchymal Stem Cells. J Immunol Res. 2019;2019:7059680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. , Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7:12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ostrand-Rosenberg S, Sinha P, Figley C, Long R, Park D, Carter D, et al. , Frontline Science: Myeloid-derived suppressor cells (MDSCs) facilitate maternal-fetal tolerance in mice. J Leukoc Biol. 2017;101:1091–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pawelec G, Verschoor CP, and Ostrand-Rosenberg S, Myeloid-Derived Suppressor Cells: Not Only in Tumor Immunity. Front Immunol. 2019;10:1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Veglia F, Perego M, and Gabrilovich D, Myeloid-derived suppressor cells coming of age. Nat Immunol. 2018;19:108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagaraj S and Gabrilovich DI, Myeloid-derived suppressor cells in human cancer. Cancer J. 2010;16:348–53. [DOI] [PubMed] [Google Scholar]
- 31.Ostrand-Rosenberg S and Fenselau C, Myeloid-Derived Suppressor Cells: Immune-Suppressive Cells That Impair Antitumor Immunity and Are Sculpted by Their Environment. J Immunol. 2018;200:422–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ostrand-Rosenberg S, Myeloid derived-suppressor cells: their role in cancer and obesity. Curr Opin Immunol. 2018;51:68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xia S, Sha H, Yang L, Ji Y, Ostrand-Rosenberg S, and Qi L, Gr-1+ CD11b+ myeloid-derived suppressor cells suppress inflammation and promote insulin sensitivity in obesity. J Biol Chem. 2011;286:23591–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clements VK, Long T, Long R, Figley C, Smith DMC, and Ostrand-Rosenberg S, Frontline Science: High fat diet and leptin promote tumor progression by inducing myeloid-derived suppressor cells. J Leukoc Biol. 2018;103:395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deng ZB, Liu Y, Liu C, Xiang X, Wang J, Cheng Z, et al. , Immature myeloid cells induced by a high-fat diet contribute to liver inflammation. Hepatology. 2009;50:1412–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hale M, Itani F, Buchta CM, Wald G, Bing M, and Norian LA, Obesity triggers enhanced MDSC accumulation in murine renal tumors via elevated local production of CCL2. PLoS One. 2015;10:e0118784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Turbitt WJ, Collins SD, Meng H, and Rogers CJ, Increased Adiposity Enhances the Accumulation of MDSCs in the Tumor Microenvironment and Adipose Tissue of Pancreatic Tumor-Bearing Mice and in Immune Organs of Tumor-Free Hosts. Nutrients. 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bao Y, Mo J, Ruan L, and Li G, Increased monocytic CD14(+)HLADRlow/- myeloid-derived suppressor cells in obesity. Mol Med Rep. 2015;11:2322–8. [DOI] [PubMed] [Google Scholar]
- 39.Fernandez-Ruiz JC, Galindo-De Avila JC, Martinez-Fierro ML, Garza-Veloz I, Cervantes-Villagrana AR, Valtierra-Alvarado MA, et al. , Myeloid-Derived Suppressor Cells Show Different Frequencies in Diabetics and Subjects with Arterial Hypertension. J Diabetes Res. 2019;2019:1568457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jung C, Figulla HR, Lichtenauer M, Franz M, and Pernow J, Increased levels of circulating arginase I in overweight compared to normal weight adolescents. Pediatr Diabetes. 2014;15:51–6. [DOI] [PubMed] [Google Scholar]
- 41.Uslu S, Ozcelik E, Kebapci N, Temel HE, Demirci F, Ergun B, et al. , Effects of serum uric acid levels on the arginase pathway in women with metabolic syndrome. Ir J Med Sci. 2016;185:259–63. [DOI] [PubMed] [Google Scholar]
- 42.Boutens L, Hooiveld GJ, Dhingra S, Cramer RA, Netea MG, and Stienstra R, Unique metabolic activation of adipose tissue macrophages in obesity promotes inflammatory responses. Diabetologia. 2018;61:942–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Viola A, Munari F, Sanchez-Rodriguez R, Scolaro T, and Castegna A, The Metabolic Signature of Macrophage Responses. Front Immunol. 2019;10:1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu X, Tu Y, Chen H, Jackson AO, Patel V, and Yin K, Micro-environment and intracellular metabolism modulation of adipose tissue macrophage polarization in relation to chronic inflammatory diseases. Diabetes Metab Res Rev. 2018;34:e2993. [DOI] [PubMed] [Google Scholar]
- 45.Yan D, Adeshakin AO, Xu M, Afolabi LO, Zhang G, Chen YH, et al. , Lipid Metabolic Pathways Confer the Immunosuppressive Function of Myeloid-Derived Suppressor Cells in Tumor. Front Immunol. 2019;10:1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Al-Khami AA, Zheng L, Del Valle L, Hossain F, Wyczechowska D, Zabaleta J, et al. , Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology. 2017;6:e1344804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kar UK and Joosten LAB, Training the trainable cells of the immune system and beyond. Nat Immunol. 2020;21:115–119. [DOI] [PubMed] [Google Scholar]
- 48.Bekkering S, Saner C, Riksen NP, Netea MG, Sabin MA, Saffery R, et al. , Trained Immunity: Linking Obesity and Cardiovascular Disease across the Life-Course? Trends Endocrinol Metab. 2020;31:378–389. [DOI] [PubMed] [Google Scholar]
- 49.Nicklas BJ, Rogus EM, Colman EG, and Goldberg AP, Visceral adiposity, increased adipocyte lipolysis, and metabolic dysfunction in obese postmenopausal women. Am J Physiol. 1996;270:E72–8. [DOI] [PubMed] [Google Scholar]
- 50.Nguyen MT, Favelyukis S, Nguyen AK, Reichart D, Scott PA, Jenn A, et al. , A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J Biol Chem. 2007;282:35279–92. [DOI] [PubMed] [Google Scholar]
- 51.Lancaster GI, Langley KG, Berglund NA, Kammoun HL, Reibe S, Estevez E, et al. , Evidence that TLR4 Is Not a Receptor for Saturated Fatty Acids but Mediates Lipid-Induced Inflammation by Reprogramming Macrophage Metabolism. Cell Metab. 2018;27:1096–1110 e5. [DOI] [PubMed] [Google Scholar]
- 52.Bouhlel MA, Derudas B, Rigamonti E, Dievart R, Brozek J, Haulon S, et al. , PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007;6:137–43. [DOI] [PubMed] [Google Scholar]
- 53.DiNicolantonio JJ and O’Keefe JH, Omega-6 vegetable oils as a driver of coronary heart disease: the oxidized linoleic acid hypothesis. Open Heart. 2018;5:e000898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Tits LJ, Stienstra R, van Lent PL, Netea MG, Joosten LA, and Stalenhoef AF, Oxidized LDL enhances pro-inflammatory responses of alternatively activated M2 macrophages: a crucial role for Kruppel-like factor 2. Atherosclerosis. 2011;214:345–9. [DOI] [PubMed] [Google Scholar]
- 55.Kadl A, Meher AK, Sharma PR, Lee MY, Doran AC, Johnstone SR, et al. , Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ Res. 2010;107:737–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Spann NJ, Garmire LX, McDonald JG, Myers DS, Milne SB, Shibata N, et al. , Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell. 2012;151:138–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rosas-Ballina M, Guan XL, Schmidt A, and Bumann D, Classical Activation of Macrophages Leads to Lipid Droplet Formation Without de novo Fatty Acid Synthesis. Front Immunol. 2020;11:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Malandrino MI, Fucho R, Weber M, Calderon-Dominguez M, Mir JF, Valcarcel L, et al. , Enhanced fatty acid oxidation in adipocytes and macrophages reduces lipid-induced triglyceride accumulation and inflammation. Am J Physiol Endocrinol Metab. 2015;308:E756–69. [DOI] [PubMed] [Google Scholar]
- 59.Namgaladze D and Brune B, Fatty acid oxidation is dispensable for human macrophage IL-4-induced polarization. Biochim Biophys Acta. 2014;1841:1329–35. [DOI] [PubMed] [Google Scholar]
- 60.Xu X, Grijalva A, Skowronski A, van Eijk M, Serlie MJ, and Ferrante AW Jr., Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab. 2013;18:816–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yan D, Yang Q, Shi M, Zhong L, Wu C, Meng T, et al. , Polyunsaturated fatty acids promote the expansion of myeloid-derived suppressor cells by activating the JAK/STAT3 pathway. Eur J Immunol. 2013;43:2943–55. [DOI] [PubMed] [Google Scholar]
- 62.Boden G, Interaction between free fatty acids and glucose metabolism. Curr Opin Clin Nutr Metab Care. 2002;5:545–9. [DOI] [PubMed] [Google Scholar]
- 63.Cignarelli A, Genchi VA, Perrini S, Natalicchio A, Laviola L, and Giorgino F, Insulin and Insulin Receptors in Adipose Tissue Development. Int J Mol Sci. 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vella V, Milluzzo A, Scalisi NM, Vigneri P, and Sciacca L, Insulin Receptor Isoforms in Cancer. Int J Mol Sci. 2018;19. [DOI] [PMC free article] [PubMed]
- 65.Renier G, Clement I, Desfaits AC, and Lambert A, Direct stimulatory effect of insulin-like growth factor-I on monocyte and macrophage tumor necrosis factor-alpha production. Endocrinology. 1996;137:4611–8. [DOI] [PubMed] [Google Scholar]
- 66.Baumgartl J, Baudler S, Scherner M, Babaev V, Makowski L, Suttles J, et al. , Myeloid lineage cell-restricted insulin resistance protects apolipoproteinE-deficient mice against atherosclerosis. Cell Metab. 2006;3:247–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mauer J, Chaurasia B, Plum L, Quast T, Hampel B, Bluher M, et al. , Myeloid cell-restricted insulin receptor deficiency protects against obesity-induced inflammation and systemic insulin resistance. PLoS Genet. 2010;6:e1000938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dasgupta P, Dorsey NJ, Li J, Qi X, Smith EP, Yamaji-Kegan K, et al. , The adaptor protein insulin receptor substrate 2 inhibits alternative macrophage activation and allergic lung inflammation. Sci Signal. 2016;9:ra63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ieronymaki E, Theodorakis EM, Lyroni K, Vergadi E, Lagoudaki E, Al-Qahtani A, et al. , Insulin Resistance in Macrophages Alters Their Metabolism and Promotes an M2-Like Phenotype. J Immunol. 2019;202:1786–1797. [DOI] [PubMed] [Google Scholar]
- 70.Senokuchi T, Liang CP, Seimon TA, Han S, Matsumoto M, Banks AS, et al. , Forkhead transcription factors (FoxOs) promote apoptosis of insulin-resistant macrophages during cholesterol-induced endoplasmic reticulum stress. Diabetes. 2008;57:2967–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Naylor C and Petri WA Jr., Leptin Regulation of Immune Responses. Trends Mol Med. 2016;22:88–98. [DOI] [PubMed] [Google Scholar]
- 72.Fernandez-Riejos P, Najib S, Santos-Alvarez J, Martin-Romero C, Perez-Perez A, Gonzalez-Yanes C, et al. , Role of leptin in the activation of immune cells. Mediators Inflamm. 2010;2010:568343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lang K and Ratke J, Leptin and Adiponectin: new players in the field of tumor cell and leukocyte migration. Cell Commun Signal. 2009;7:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Saxena NK and Sharma D, Multifaceted leptin network: the molecular connection between obesity and breast cancer. J Mammary Gland Biol Neoplasia. 2013;18:309–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rebe C, Vegran F, Berger H, and Ghiringhelli F, STAT3 activation: A key factor in tumor immunoescape. JAKSTAT. 2013;2:e23010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tabung FK, Steck SE, Liese AD, Zhang J, Ma Y, Johnson KC, et al. , Patterns of change over time and history of the inflammatory potential of diet and risk of breast cancer among postmenopausal women. Breast Cancer Res Treat. 2016;159:139–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mason KL, Huffnagle GB, Noverr MC, and Kao JY, Overview of gut immunology. Adv Exp Med Biol. 2008;635:1–14. [DOI] [PubMed] [Google Scholar]
- 78.Wershil BK and Furuta GT, 4. Gastrointestinal mucosal immunity. J Allergy Clin Immunol. 2008;121:S380–3; quiz S415. [DOI] [PubMed] [Google Scholar]
- 79.Brandsma E, Kloosterhuis NJ, Koster M, Dekker DC, Gijbels MJJ, van der Velden S, et al. , A Proinflammatory Gut Microbiota Increases Systemic Inflammation and Accelerates Atherosclerosis. Circ Res. 2019;124:94–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zheng D, Liwinski T, and Elinav E, Interaction between microbiota and immunity in health and disease. Cell Res. 2020;30:492–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Correa-Oliveira R, Fachi JL, Vieira A, Sato FT, and Vinolo MA, Regulation of immune cell function by short-chain fatty acids. Clin Transl Immunology. 2016;5:e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Makki K, Deehan EC, Walter J, and Backhed F, The Impact of Dietary Fiber on Gut Microbiota in Host Health and Disease. Cell Host Microbe. 2018;23:705–715. [DOI] [PubMed] [Google Scholar]
- 83.Goldberg EL, Shchukina I, Asher JL, Sidorov S, Artyomov MN, and Dixit VD, Ketogenesis activates metabolically protective gammadelta T cells in visceral adipose tissue. Nat Metab. 2020;2:50–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dupuis N, Curatolo N, Benoist JF, and Auvin S, Ketogenic diet exhibits anti-inflammatory properties. Epilepsia. 2015;56:e95–8. [DOI] [PubMed] [Google Scholar]
- 85.Lussier DM, Woolf EC, Johnson JL, Brooks KS, Blattman JN, and Scheck AC, Enhanced immunity in a mouse model of malignant glioma is mediated by a therapeutic ketogenic diet. BMC Cancer. 2016;16:310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Poff AM, Ari C, Seyfried TN, and D’Agostino DP, The ketogenic diet and hyperbaric oxygen therapy prolong survival in mice with systemic metastatic cancer. PLoS One. 2013;8:e65522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Husain Z, Seth P, and Sukhatme VP, Tumor-derived lactate and myeloid-derived suppressor cells: Linking metabolism to cancer immunology. Oncoimmunology. 2013;2:e26383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Porta C, Marino A, Consonni FM, Bleve A, Mola S, Storto M, et al. , Metabolic influence on the differentiation of suppressive myeloid cells in cancer. Carcinogenesis. 2018;39:1095–1104. [DOI] [PubMed] [Google Scholar]
- 89.Seiler A, Chen MA, Brown RL, and Fagundes CP, Obesity, Dietary Factors, Nutrition, and Breast Cancer Risk. Curr Breast Cancer Rep. 2018;10:14–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.O’Keefe SJ, Li JV, Lahti L, Ou J, Carbonero F, Mohammed K, et al. , Fat, fibre and cancer risk in African Americans and rural Africans. Nat Commun. 2015;6:6342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Levesque S, Pol JG, Ferrere G, Galluzzi L, Zitvogel L, and Kroemer G, Trial watch: dietary interventions for cancer therapy. Oncoimmunology. 2019;8:1591878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Conti L, Del Corno M, Scazzocchio B, Vari R, D’Archivio M, Verano B, et al. , Dietary fatty acids and adipose tissue inflammation at the crossroad between obesity and colorectal cancer. J Cancer Metastasis Treat. 2019;5:64. [Google Scholar]
- 93.Woodall MJ, Neumann S, Campbell K, Pattison ST, and Young SL, The Effects of Obesity on Anti-Cancer Immunity and Cancer Immunotherapy. Cancers (Basel). 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Arasanz H, Gato-Canas M, Zuazo M, Ibanez-Vea M, Breckpot K, Kochan G, et al. , PD1 signal transduction pathways in T cells. Oncotarget. 2017;8:51936–51945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jiang X, Wang J, Deng X, Xiong F, Ge J, Xiang B, et al. , Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol Cancer. 2019;18:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ballbach M, Dannert A, Singh A, Siegmund DM, Handgretinger R, Piali L, et al. , Expression of checkpoint molecules on myeloid-derived suppressor cells. Immunol Lett. 2017;192:1–6. [DOI] [PubMed] [Google Scholar]
- 97.Oldenhove G, Boucquey E, Taquin A, Acolty V, Bonetti L, Ryffel B, et al. , PD-1 Is Involved in the Dysregulation of Type 2 Innate Lymphoid Cells in a Murine Model of Obesity. Cell Rep. 2018;25:2053–2060 e4. [DOI] [PubMed] [Google Scholar]
- 98.Mimura K, Teh JL, Okayama H, Shiraishi K, Kua LF, Koh V, et al. , PD-L1 expression is mainly regulated by interferon gamma associated with JAK-STAT pathway in gastric cancer. Cancer Sci. 2018;109:43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Murphy KA, James BR, Sjaastad FV, Kucaba TA, Kim H, Brincks EL, et al. , Cutting Edge: Elevated Leptin during Diet-Induced Obesity Reduces the Efficacy of Tumor Immunotherapy. J Immunol. 2018;201:1837–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tee MC, Cao Y, Warnock GL, Hu FB, and Chavarro JE, Effect of bariatric surgery on oncologic outcomes: a systematic review and meta-analysis. Surg Endosc. 2013;27:4449–56. [DOI] [PMC free article] [PubMed] [Google Scholar]