

Abstract
Immunity against different Mycobacteria species targeting the lung requires distinctly different pulmonary immune responses for bacterial clearance. Many parameters of acquired and regulatory immune responses differ quantitatively and qualitatively from immunity during infection with Mycobacteria species. Nontuberculosis Mycobacterial species (NTM) includes Mycobacterium avium- (M. avium), and Mycobacterium abscessus (M. abscessus). However, the more virulent Mycobacterial species by far are Mycobacterium tuberculosis-(Mtb). Herein, we discuss the potential implications of acquired and regulatory immune responses in the context of animal and human studies, as well as future directions for efforts to treat Mycobacteria diseases.
Keywords: Immunity, Mycobacterium avium, Mycobacterium abscessus, Mycobacterium tuberculosis
Introduction:
A deeper understanding of the acquired and suppressive immunity against M. tuberculosis, M. avium, and M. abscessus is required to shed light on protective immunity enabling effective vaccine and therapeutic compound treatment. Tuberculosis (Mtb) is caused by M. tuberculosis and causes more deaths worldwide than any other infectious disease before the covid-19 pandemic. However, COVID-19 alone may have caused more deaths worldwide than TB annually.1 This fact highlights the heightened virulence of Mtb compared to moderate virulence of M. avium and low virulence of M. abscessus, as demonstrated in animal models. 2–4
It has been approximated that a third of the world’s population is infected through aerosol droplets from an infected person with Mtb. However, only around 10% of exposed individuals develop the Tuberculosis (TB) disease.5–6 Moreover, Mtb remains one of the world’s most devastating diseases. Although most TB forms are curable and advances in the combination regimen of bedaquiline, pretomanid, and linezolid have helped reduce MDR-TB.7 However, the development of Mtb drug resistance still occurs, and an effective vaccine is urgently needed.
In the 1920s, the attenuated strain of Mycobacterium Bovis Bacillus Calmette-Guerin (BCG) was to be used as a vaccine against TB. Nowadays, it remains the only existing vaccine against TB.8 The BCG vaccine has been used in many geographical areas of the world – but not the U.S., Canada, or several European countries – for well over 60 years.9 BCG is delivered as an intradermal injection and has been shown to reduce the acquisition of meningitis and miliary forms of childhood Mtb disease.10 The goal of vaccination is to establish a stable population of long-lived memory T cells. However, BCG provides limited and geographically variable protective immunity against pulmonary Mtb disease in adults.11
An extensive range of ideas exists regarding why BCG efficacy in individuals diminishes in adulthood (reviewed in )12 Several proposed theories explain the range of efficacy seen with BCG. Such hypothesis include the (i) the theoretical concern of genetic variations of BCG strains that have been developed as a consequence of hundreds of passages and the differences in BCG growth protocols employed by different manufacturers, (ii) high background prevalence of non-protective TH2 response (that may antagonize the desired immune response to BCG) in areas of the world endemic for helminths- which may be related to reduced efficacy in countries closer to the equator), (iii) waning BCG-induced immunity with time, (iv) less of a response to BCG in older individuals, (v) socioeconomic / psychosocial factors, nutritional status, and (vi) possible interference of BCG-induced immunity by NTM illustrated that prior sensitization with NTMs can stop BCG multiplication and thereby prevent the induction of an effective BCG immune response and ultimately protect against Mtb infections as shown by the finding that prior sensitization with NTMs can stop BCG multiplication and thereby prevent the induction of an effective BCG immune response and ultimately protect infections.13–14
Immunity against Mycobacteria species requires a delicate balance between acquired immune responses to stop bacterial replication and the limitation of potentially tissue-damaging inflammation due to immune activation. Regulatory T cells (Tregs) express the transcription factor Foxp3+ and constitute an essential counterbalance of inflammatory Th1 responses and maintain immune homeostasis. The first reports describing Foxp3-expressing CD4+ Treg cells in tuberculosis (Mtb) emerged in 200615 , and additional investigations also suggested human anti-inflammatory macrophage indicated Foxp3+GITR+CD25+ regulatory T cells, which suppress by membrane bound TGFbeta.16 Different Treg cell subsets, most likely specialized for different microenvironments and tissues, have been shown to expand in human Mtb and animal models of Mtb. New functions for Treg cells have been demonstrated during different stages and spectrums of Mtb disease. For example, Foxp3+ regulatory cells can expand quickly during early infection, and slow down cellular immunity onset and continue to persist during chronic Mtb infection. Increased frequencies of Tregs have been associated with a detrimental outcome of active Mtb. They may depend on the M. tuberculosis strain, local environment, animal model, and infection stage. However, it’s more plausible that some investigations also suggest that Tregs are required together with effector T cell responses to obtain reduced pathology and sterilizing immunity.17
This manuscript will first provide an overview of the regulatory cells and mechanisms that control the defense against different Mycobacteria infections, followed by a review of what we know about Tregs’ phenotype and function from studies in TB caused by Mtb. Secondly, this manuscript will provide an overview of the global emergence of Mycobacterium avium complex disease (MAC) and Mycobacterium abscessus complex (MABC) disease. Finally, we will discuss Treg cells’ potential role in the progression of Mycobacteria disease in human and experimental animal models and the relevance of this knowledge for future efforts to prevent, modulate, and treat Mycobacteria pathogens.
Supporting Data for a role in Acquired T cell Immunity in the Defense against M. tuberculosis in the lung.
Effector T-cell functions contributing to the protection against M. tuberculosis
Immunity against Mtb requires a balance between adaptive TH1 immune responses to reduce bacterial replication and the prevention of potentially damaging immune activation and inflammation.17–18 The CD4+ T cell plays a critical role in this process by attempting to control disseminated disease while helping the development of the lung’s granuloma that limits M. tuberculosis (i) infection of healthy lung tissue, (ii) dissemination from the lung to other organs, and (iii) ultimately aerosol transmission. In light of Mycobacteria pathogen’s ability to infect and evade the human immune response, it is not surprising that our ability to vaccinate against TB has not been successful.19–20
The CD4+ T cells mediate protection determined by host factors that regulate these cells’ effector function. In this review, we describe T cells’ initiation and expression during M. tuberculosis infection and the fulminant inflammatory response that can compromise effective T-cell function and allow for bacterial survival. Many cell types, such as epithelial cells, dendritic cells, macrophages, neutrophils, eosinophils, and B and T lymphocytes, contribute to lung immunity. We focus on the recent advances in understanding how T lymphocytes mediate pulmonary host defenses against Mycobacteria pathogens.21
T cells play a critical role in pulmonary defense against Mtb.22 CD4+ T cells help B cells to mount antibody responses, provide feedback to dendritic cells (DCs) via costimulatory molecules and the elaboration of cytokines, and enhance and maintain responses of CD8+ (cytotoxic) T cells. Moreover, CD4+ T cells have direct effector activity, including performing cytotoxic functions, mediating macrophage activation, and inducing genes in mucosal tissues that contribute to host defense. CD8+ T cells contribute to cytokine production and, more importantly, kill infected Mycobacteria cells, releasing the bacilli to hopefully be engulfed by a bystander macrophage better able to kill it directly. Also, γδ T cells and invariant natural killer T (iNKT) cells and the newly discovered innate lymphoid-like cells (ILCs) play critical roles in early responses to many pulmonary infections.
The distinctive lineages of effector CD4+ T cells are designated as T helper Th1, Th2, Th17, and Treg cells. These cells are classified by the effector cytokines they produce and the expression of different clusters of differentiation markers (CD) and additional transcription factors. The cytokines produced by Th1 cells are interferon-γ, IL-2, TNF-α producers.23 In contrast, Th2 produces the signature cytokines IL-4, IL-5, and IL-13. The Th1/Th2 can have negative regulatory feedback limiting each other’s ability to expand and execute an effort function.24 The third distinctive effector lineage of CD4+ T cells, termed Th17 cells, was first demonstrated in 2000 in mouse models of autoimmune encephalitis.24–25
Memory immunity contributing to the protection against M. tuberculosis
It has well been thought that CD4+ and CD8+ T cells can form immunological memory and benefit the host when the host encounters secondary challenges from the same or related pathogens. The purpose of vaccination is to establish a stable population of long-lived memory T cells.26 Existing information indicates that both effector and central memory cells can be generated, depending on the experimental conditions. Still, both in animal models and in clinical studies, it is evident that effector memory T cells are the predominant subset.26–27 There exists an urgent unmet medical need to make new Mtb vaccines targeting different immune mechanisms. Immune correlates of protection against Mtb are not yet fully understood.28
Currently, whether an Mtb vaccine would function better if it targeted one specific T cell subset rather than another remains completely unknown. Further complicating the question is new evidence that suggests other subsets such as IL-17 secreting CD4+ T cells and cells with stem cell-like qualities may also play important roles.26–29
The use of chemotherapy in early studies to remove a primary infection with Mtb and subsequently evaluate the immune response status led to the expansion of memory T cells in mice.6 This was shown by the passive cell transfer technique followed by the recipient challenge, showing CD4+ T cells were expanding before the challenge.6 It was subsequently demonstrated that such cells had a wide variety of antigen recognition15 and comprised of late expanding CD4+ T cells expressing a phenotype indicating that they were primarily activated effector or effector memory T cells.
The precise origin of memory T cells is not fully understood. Recent evidence indicates that most activated CD4+ effector T cells die after antigen clearance, but some remain memory T cells. This can happen rapidly in certain situations. A study using cells from transgenic mice demonstrated that just three days of resting in culture in vitro permitted effectors cells to become cells with features of “memory”.18 This supports the theory that an abrupt switch from effector cells to a resting memory phenotype is established in the periphery, leaving a population of cell phenotypes with a high sensitivity to the antigen. This is desirable for the host because if the Mtb returns, it will infect in reduced numbers before it attempts to replicate and will generate heightened and responsive memory T cells to become secondary effectors cells. These secondary effector cells have the ability upon secondary antigen recognition to rapidly mount an immune response to Mtb resulting in the pathogen’s death.
The majority of this information has been understood from transgenic mouse models engineered to recognize specific peptides or acute viral infection models. Most of our understanding of chronic diseases come from models of viral infection. However, memory immunity has been dominantly studied (i) using animal models is a sterile environment impact gut commensal bacilli,30 (ii) dominate use of laboratory-adapted strains24–31, (iii) bacterial growth with tween a detergent which causes loss of the capsule and immunomodulatory antigens.32 The consensus to date is that, most likely, CD4+ memory T cells are far less well understood than CD8+ memory T cells. Moreover, less is known about the required induction of an ideal generation of memory T cells or the kinetics of effector to memory transition. An additional restraint is that most memory T cell studies have utilized short-term in vitro assays or recall in vivo using small intervals before re-challenge.17
The urgent need for vaccine development requires information on vaccine design and validation of correlates of markers that specifically identify correlated memory immunity produced in response to the vaccination. However, this highly sought after memory T cell marker has not been elucidated. However, it is now possible to get information based on the analysis of specific T cell markers. Some associated memory markers are CD44+. However, this is less useful due to its changes in density on multiple cell subsets, but CD62L is better because it relates to migration patterns of effector and memory T cell subsets through or away from lymphoid tissues. Currently, since a phenotype by itself is not a complete picture, it most likely is better to try to define memory T cell based on not just the phenotype but also in terms of what cytokine/chemokines are produced and what these cells actually can accomplish and their kinetics of emergence and persistence.
In the context of memory immunity, we comprehend more about CD8+ memory than CD4+, and we think there is a change between naïve, effector, and memory T cell populations.19 Data exists (Table 1) that supports the idea that there are two populations of memory T cells for CD4+ and CD8+, effector memory cells [TEM] and central memory T cells [TCM].20, 33, 34 Their site can differentiate these cell phenotypes in lymphoid and non-lymphoid tissues, combined with their expression of an organization of cell surface markers [CD62L, CCR7], other markers of cytokine, and chemokine receptors, summarized in Table 1. In addition, the degree of their response contrasts. For example, TCM are quicker than TEM, and the cytokines they secrete are IFN-γ, TNF-α, and IL-2.33–35
Table 1.
Markers associated with effector and memory immunity.
| Cluster of differentiation marker | Function |
|---|---|
| CD44 | Cell traffic |
| CD62L | Cell traffic |
| CCR7 | Chemokine directed cell traffic |
| CD127 | Alpha component of the IL-7 receptor |
| CD122 | One of three subunits of the IL-15 receptor |
| KLRG1 | Possible marker of memory T cell terminal differentiation |
| PD-1 | Possible marker of memory T cell exhaustion |
| CD45 | Family of transmembrane glycoproteins |
| CXCR6 | Chemokine receptor implicated in cellular influx into the lung |
Evidence exists (Table 1) that supports the notion that there are two populations of memory T cells for CD4+ and CD8+, effector memory cells [TEM] and central memory T cells [TCM] (20, 33, 34). Their location can differentiate these cell phenotypes in lymphoid and non-lymphoid tissues, combined with their expression of an arrangement of cell surface markers [CD62L, CCR7], additional markers of cytokine, and chemokine receptors, summarized in Table 1. Also, the rate of their response differs. For example, TCM are faster than TEM, and the cytokines they produce are IFN-γ, TNF-α, and IL-2 (33–35).
The up-to-date concept supported by investigational data shows that peripheral tissues for instance the lungs contain CD62Llo TEM, which acts as the primary defense against Mtb.35 The pathogen’s ability to escape the first line of defense frequently leads to the pathogen reaching the lymphoid tissues. Then this is met by CD62Lhi TCM. It has been suggested36 that after activation by secondary exposure to antigen effector memory cells can only become short-lived effectors. However, in contrast, central memory T cells can either become effectors or turn into effector memory cells. Additionally suggested20 is that effector memory cells cooperate with dendritic cells by CD40L and central memory cells, which express far less CD40L. However, increased numbers of dendritic cells occur in the lungs as well as lymph nodes. Effector memory cells do not usually go into the lymph nodes, even though this apparently can happen if the effector memory cell acquires CXCR3, which can bind to CD62P on high endothelial venules.
Central memory T cells have an heightened proliferative ability and rapidly become effectors upon subsequent re-exposure to an antigen.37 Human central memory cells expressing CCR7hi CD62Lhi can begin to look like effector memory cells after TCR stimulation in the presence of cytokines or on the expression of the chemokine receptors CXCR3, CCR4, and CXCR5.38 One marker that may be very useful is CXCR6, which appears to be a specific marker on T cells traveling to the lungs in response to CXCL16 produced in great quantities by macrophages.38
Central memory T cells are usually CD62Lhi CD127hi, while effector memory T cells are CD62Llo CD127hi.39 Also, central memory can further be divided into populations based on the cell’s capacity to make effector cytokines, with cells expressing KLRG-1 more likely to do so; these KLRG-1+ cells are also CD127hi. However, there is increasing evidence that KLRG-1 could be used as a possible vaccine efficacy marker.40 Currently, it is considered as a marker of terminal differentiation, although it should be noted that high expression of this marker can, in some cases, be reversed.41
Lastly, the issue of whether or not IFN-γ levels translates into protection.42 It should perhaps be framed in terms of which cluster of differentiation antigens are associated with a subset of memory T cell making IFN-γ cytokine, where these cells are located, and at what stage in the kinetic response to Mtb. For example, in humans, this can only be measured in T cells in the blood and not at infection sites or within lymphoid tissues. So, this information has to come from animal models of infection and vaccination.
However, it remains to be seen that this is the case with all TB patients, who many times get reinfected with a different strain.26 The published mouse Mtb reinfection mouse model43 has shown that reinfection drives a substantial memory T cell response, quickly collapsing. Interestingly, the publication by Verver and her colleagues in South Africa44 showed that patients with TB disease who had earlier been successfully treated with chemotherapy were four times more likely to present with a new infection (reinfection). We decided to model this in mice and infected them with the optimal dose of virulent W-Beijing strain HN878 and then treated them with chemotherapy to sterilize them.43 Both the protection and the memory response was soon lost with a concomitant increase in expression of the exhaustion marker PD-1 on these cells, which might point to the underlying cause. Additional studies, with Dr. Nardell, Harvard Medical School, using a natural transmission of Mtb patient to guinea pig transmission model.36 This model sought to examine the ability of naturally transmitted multidrug-resistant Mtb to establish infection and progress to active TB disease in guinea pigs. Guinea pigs were continuously exposed for four months to the exhaust air of a 6-bed multidrug-resistant TB inpatient hospital ward in South Africa. Serial tuberculin skin test reactions were measured to determine infection. All animals were then evaluated for histologic disease progression at necropsy. Although 75% of the 362 exposed guinea pigs had positive skin test reactions [≥6 mm], only 12% had histopathologic evidence of active disease.
Moreover, Reversions (≥6 mm change) in skin test reactivity were seen in 22% of animals, exclusively among those with reactions of 6–13 mm. Notably, only two of 86 guinea pigs with reversion had histological evidence of disease compared to 47% (31/66) of guinea pigs with large, non-reversing reactions. In guinea pigs that reverted a skin test, a second positive reaction in 27 (33%) of them strongly suggested re-infection due to ongoing exposure by TB patients. These results show that a large majority of guinea pigs naturally exposed to human-sourced strains of multidrug-resistant tuberculosis became infected, denoted by a tuberculin skin reaction. Still, this reaction waned, and no Mtb disease developed while other guinea pigs would become infected multiple times (skin test positive and then negative multiple times).36 Ultimately, these animals went on to develop clinical Mtb disease. Thus, suboptimal doses of bacilli exposed numerous times may be required to create a primary infection or reinfection.
Another plausible explanation is that individuals with a predisposed immune susceptibility succumb to Mtb. Individuals with specific acquired or genetic/heritable disorders that compromise the lung architecture or lung immune system are more vulnerable to Mycobacteria lung disease. Some of the more well-accepted acquired disorders include smoking-related emphysema, bronchiectasis as a sequela of prior unrelated infections, pneumoconiosis such as silicosis, chronic aspiration, and use of corticosteroids, and other immunosuppressive agents such as TNF-α antagonists. Individuals with genetic/heritable disorders in which bronchiectasis, emphysema, or lung immune defects are important sequelae are also predisposed to M. tuberculosis primary disease and subsequent reinfection with Mtb.
Regulatory T-cell functions contributing to reduced tissue damage with concomitant suppression of effector immunity
Recently, we have been able to understand the process of immune homeostasis, leading to the insight that Tregs are fundamental to immune regulation and immune homeostasis formation.17 Treg can be defined as T cells with specific suppressive abilities, such that Tregs down-regulate proliferation and effector functions of different phenotypes of immune cells.
The discovery that natural Tregs were required to prevent autoimmune disease happened when neonatal thymectomized mice developed an autoimmune disease precluded by transferring T cells from adult mice.45–46 However, it was the finding that the subset of T cells accountable for suppression of autoimmunity expressed high levels of CD25 (the high-affinity IL-2 receptor α) and the transcription factor forkhead box P3 (Foxp3+) as the ‘master control gene’ that well-defined this subset as a distinct T cell lineage.47–50 The FoxP3 intracytoplasmic receptor act’s as a transcriptional regulator. It can bind to the promoters for genes involved in Treg cell function and subsequently suppress the transcription of crucial genes following T cell receptors’ stimulation.51 Also, Foxp3 suppressive activity’s role was identified as the genetic defect underlying lethal inflammatory autoimmunity in scurfy mice.52–53 More importantly, the scurfy mouse’s autoimmune phenotype was reversed by transfer of CD4+ T cells but not CD8+T cells,54–55 confirming a dominant suppressive role for CD4+Foxp3+ cells.56 The lack of Foxp3+ T cells due to mutations in their FoxP3-locus is also present in human disease. It causes IPEX syndrome, which results in a disorder characterized by immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome, and XLAAD (X-linked autoimmunity allergic dysregulation syndrome), highlighting the dominant role of Tregs in the preservation of normal immune homeostasis.57 Regardless, without bone marrow transplantation, the IPEX and XLAAD58 syndromes result in a lethal disease showing the essential role of Foxp3 for immune homeostasis in humans.56 Patients with a deficiency of CD25 develop a similar IPEX-like syndrome caused by defective production of the anti-inflammatory cytokine IL-10.59
Regulatory T-cell functions in immunity against M. tuberculosis
Treg cells can exert a suppressive function on immune responses for a short or long time interval. Treg populations are typically classified based on their site of development, phenotype, and function.60 Therefore, these cells are classified due to site of development into natural Tregs (nTregs) or induced Tregs (iTregs) and adaptive Tregs. The Natural Tregs develop in the thymus by process of MHC class II T cell receptor (TCR) dependent interactions with self-antigens leading to a high avidity selection.48–50 The naturally occurring CD4+CD25+FoxP3+ Tregs typically constitute 2–3% of peripheral blood,61 and this population can be expanded and recirculate in the human body with antigen-stimulation. Foxp3+ Tregs have been recognized that could be produced from mature conventional CD4+ T cells outside the thymus by many environments such as chronic antigen stimulation, limited co-stimulation, and persistence of the anti-inflammatory cytokine TGFβ62 or IL-10.63 Notably, Tregs are predominately identified by co-expression of FoxP3 and CD25. Both these clusters of differentiation markers are also expressed by newly activated conventional T cells and are thus not specific to Tregs.64 However, other surface markers expression has been adopted to expand the identification of distinct Treg populations. For instance, co-expression with or GITR or CTLA-4 with concomitant down-regulation of definite Tregs receptors such as CD49d or CD127 represent better signatures of true Tregs. Regardless, we currently lack the identification of a single marker specific to Tregs, which is not up-regulated upon activation of conventional CD4+ T cells.
Supporting information for a Role in Regulatory T cells Promoting Mycobacteria Disease
Treg Mechanisms of Suppression against M. tuberculosis
Since the pathways of development of nTregs and iTregs are different, these cells have non-overlapping specificities causing immune suppression in other tissue immune target cells and sites. Their versatility of suppression has characterized Tregs through multiple mechanisms encompassing both production of soluble factors and cell-contact dependent receptor-ligand interactions (reviewed in21 ). For that reason, the mechanisms that aid immune suppression involve (i) secretion of anti-inflammatory cytokines such as TGF-β and IL-10, which can directly prevent the function of responder T cells and myeloid cells65 or production of other factors that affect amino acid accessibility and energy metabolism, as well as (ii) For example, cell membrane-bound receptors, the high-affinity TCR, and other molecules with regulatory properties, together with CTLA-4 GITR, LAG3, CD39, PD-1, and Nrp1.62 Tregs’ common undesirable effects are suppression of cell growth, reduced production of inflammatory cytokines, and cytolytic activity. Compromised production of Th1 (IL-2, IFN-γ) and Th17 cytokines (IL-17) lead to reduced proliferation and defective stimulation of antimicrobial effector functions. Treg cells that express high levels of the IL-2Rα chain (CD25) can compete with effector T cells for IL-2, resulting in cytokine-mediated deprivation of the effector cells and Bim-mediated apoptosis.65
Moreover, the role of antigen-presenting cells (APC) can be inhibited by down-regulation of costimulatory molecules, such as CD80/CD86, that affect APCs’ inability to stimulate proinflammatory responses.66 Finally, (iii) Treg cells can kill APCs through a granzyme B-dependent mechanism.67 Thus, activated Treg cells may function to directly kill effector cells in a means similar to CD8+ cytotoxic cells.65 Also, (v) stimulated Treg cells may also express known receptors (e.g., galectin-1) on their cell surface that can interact with receptors on effector T cells, causing cell cycle arrest.65 Instances of suppressive mechanisms mediated by cell membrane-bound receptors on the Treg cells inhibit APCs’ function or other cells of the innate immune system.65
Treg antigen-presenting cell Suppression against M. tuberculosis
Additional regulatory immune cell phenotypes
Regulation of cellular homeostasis also includes other kinds of regulatory immune cell subsets, for example, regulatory B cells (Breg),22 tolerogenic dendritic cells (DCs),68 and modified macrophages. Although naturally-occurring CD4+CD25+FoxP3+ Treg cells are the best characterized regulatory subset, there is also a complex network of innate and adaptive regulatory cells and mechanisms that contribute to the overall control of immune homeostasis. These regulatory subsets may be tightly connected in that Treg cells can induce specific regulatory subsets, whereas other regulatory subsets can also support Treg cells’ activation and recruitment.
Regulatory B cells (Breg)
Similar to T cells, B cells can have functionally different purposes in the control of immune homeostasis. B cells are also a large cellular component of Mtb granulomas in humans, non-human primates, and mice.69–70 It has been described that a subset of regulatory B cells (Breg cells) controls inflammation and autoimmunity in both humans71 and mice.72 On the contrary to effector B cells, which can produce cytokines to maintain the differentiation of either Th1 or Th2 cells, Breg cells can suppress the potential inflammation of effector T cells and alter APCs’ activity.73 Consequently, CD19+CD24hiCD38hi Breg cells can suppress T cell functions, including the differentiation of TNF-α producing, IFN-γ Th1 cells and IL-17 producing Th17 cells.74–75 Strong CD19+CD1d+CD5+ Bregs cellular immunity have been found to be elevated in Patients with active Mtb and specifically suppress Th17 responses, which may cause a adverse impact on the clinical outcome of Mtb.76 Breg cells also suppress the production of TNF-α by macrophages.71 A functional cross-talk seems to exist between Breg and Treg cells because Bregs might help pulmonary infiltration of FoxP3+ Treg cells that prevent allergic inflammation in a murine infection model77 and consequently induce Treg cell development in an autoimmune disease model.78
Likewise, CD19+CD24hiCD38hi Breg cells stimulate FoxP3+ Treg cells’ propagation with potent suppressive functions in healthy individuals. In contrast, Breg cells from patients with dissimilar autoimmune conditions have lost their suppressive ability and the maintenance of FoxP3+ Treg cells.74–75 Remarkably Immune suppression mediated by Breg cells is largely dependent on IL-10.71, 72, 74, 75 For instance, In transplantation tolerance, it has been shown that nTregs can lead to the differentiation of tolerogenic DCs that can work together with Breg cells in the transplant to stimulate the induction of iTregs from naïve T cells via the production of IL-10.66 Moreover, B reg cells can inhibit the proliferation of CD4+ T cells and concurrently promote the expansion of iTregs as well as an improved expression of FoxP3 and CTLA-4 in Treg cells that seem to be dependent on TGF-β production.79–80
Effector T-cell functions against M. avium
Nontuberculosis Mycobacteria infections are increasing worldwide, resulting in a severe public health problem.81 NTM’s are Mycobacteria species other than Mycobacterium tuberculosis complex and Mycobacterium leprae that can cause pulmonary and extrapulmonary disease in vulnerable individuals and are reported worldwide.4 The most common NTM causing disease and outbreaks in the United States are species within the MAC—comprised of at least nine species including M. avium, M. intracellulare, and M. chimaera, followed by Mycobacterium abscessus complex (including M. abscessus sensu stricto, M. massiliense, and M. bolletii), Mycobacterium chelonae and Mycobacterium kansasii.82
Given the ubiquitous nature of NTM in the environment, it is likely that repeated exposures from multiple sources- such as showerheads, swimming pools, and Jacuzzi baths and soil- increases the likelihood of established disease in susceptible individuals.83 Other reports demonstrate the possibility that cystic fibrosis patients acquire NTM infection by contact with fomites or from patient to patient transmission.4
Recent population-based studies have shown this worldwide increase in NTM began in 2000, and currently, in the United States, the prevalence of NTM lung disease exceeds that of TB by approximately10-fold.84 The amounts of inhaled NTMs and the number of exposures required for infection, and the disease’s ultimate progression remain unknown. Understanding whether reinfection plays a potential role in the pathogenesis of NTM lung disease is highly relevant since reinfection most likely occurs given the ubiquitous nature of NTM in humans’ environment. The missing gaps of knowledge in understanding the process of reinfection, colonization, dissemination, and disease progression in these individuals exposed to NTM and develop a progressive NTM disease remains unknown due to the intractability of transient infection in human patients.83
However, unlike Mtb, the ubiquitous nature of Mycobacterium avium complex organisms in the environment, relatively few infected people develop disease.85 The fact that M. avium is killed by effector immunity in most healthy individuals highlights the requirement of some degree of susceptibility due to either underlying lung disease or immunosuppression to develop a progressive M. avium disease. The frequency of pulmonary MAC disease is increasing in many areas, and the exact reasons are unknown.83 However, some Mtb patients also become dually infected with NTM, and Mtb this mixed infection poses issues with how to treat the patient because most NTMs do not respond to routine TB drugs.37
Individuals with certain acquired or genetic/heritable disorders that compromise the lung architecture or lung immune system are more vulnerable to NTM lung disease. Some of the more well-accepted acquired disorders include smoking-related emphysema, bronchiectasis as a sequela of prior unrelated infections, pneumoconiosis such as silicosis, chronic aspiration, and use of corticosteroids and other immunosuppressives such as TNF-α antagonists shown in Figure 2.
Figure 2.
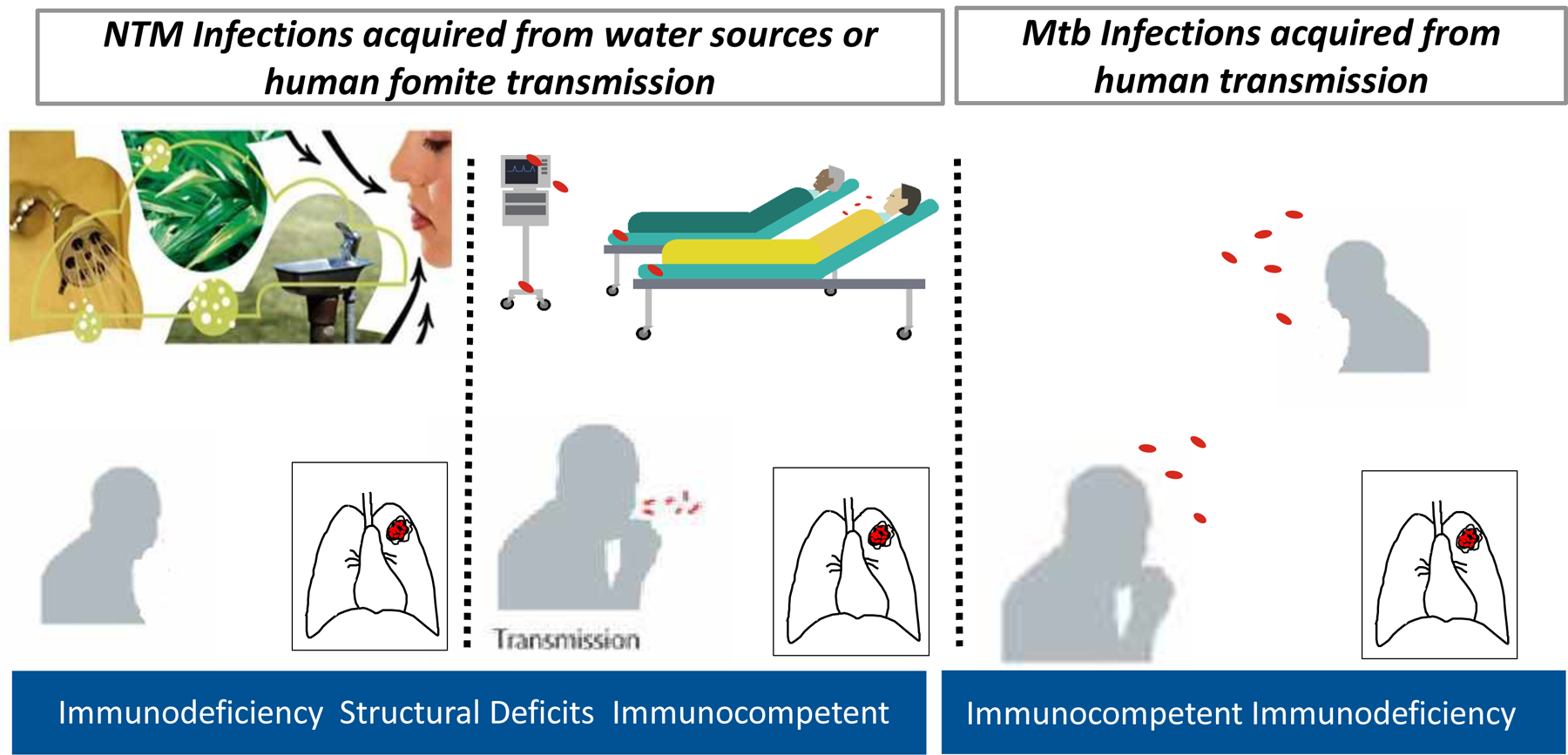
Host defense defects causing M. tuberculosis exist less frequently than in NTM while immunocompetent individuals readily succumb to M. tuberculosis infection. However, disseminated NTM disease and the determinants of susceptibility to NTM pulmonary disease are better understood. Host factors implicated in increased susceptibility include structural lung abnormalities, genetic disorders affecting mucociliary clearance, and use of steroids or other immunomodulatory drugs, such as TNF-α blockers.
Individuals with genetic/heritable disorders in which bronchiectasis, emphysema, or lung immune defects are important sequelae are also predisposed to NTM lung disease; such underlying disorders include cystic fibrosis (CF), primary ciliary dyskinesia, alpha-1-antitrypsin deficiency, congenital bronchial cartilage deficiency (Williams-Campbell syndrome), tracheobronchomegaly (Mounier-Kuhn syndrome), Sjogren’s syndrome, pulmonary alveolar proteinosis, and common variable immunodeficiency.86 It is important to emphasize that acquired and genetic/heritable predispositions are not mutually exclusive. Thus it is a plausible supposition that the more risk factors an individual has, the greater the likelihood of developing NTM lung disease.
The early methods for identifying NTM were developed in the 1950s by Ernest Runyon. They were grounded on growth rates and pigmentation, but the improvement of 16S rRNA gene sequencing substituted these classical methods, permitting the identification of over 150 species of NTM.83 Data was generated through numerical taxonomy studies that were completed from the late 1960s through the 1970s. With the advancement of DNA-DNA hybridization analyses, recognized relationships among NTM strains and delivered an essential link between the purely phenotypic Runyon classification method, and 16S rRNA gene sequencing.86 Additionally, taxonomic classifications based on DNA-DNA hybridizations were always confirmed by 16S rRNA gene sequencing and later by whole-genome sequencing. This allowed for these applications of molecular techniques permitting genetic analysis of NTM simplified classification and subsequent epidemiological studies. This may partially explain why increased rates of NTMs patients are being better diagnosed.
Isolation of MAC from a respiratory specimen does not necessarily mean that treatment is required. The treatment decision involves the synthesis of clinical, radiographic, and microbiologic information and weighing the risks and benefits for the individual patient. Successful treatment requires a multipronged approach that includes antibiotics, aggressive pulmonary hygiene, and sometimes sectioning the diseased lung.3, 87 A combination of azithromycin, rifampin, and ethambutol administered three times weekly is recommended for nodular bronchiectatic disease. In contrast, the same regimen may be used for cavitary disease but administered daily and often with the inclusion of a parenteral aminoglycoside.88 (for an outstanding review on this specific topic, see reference116). Disseminated MAC (DMAC) is almost exclusively seen in patients with late-stage AIDS. It can be treated with a macrolide in combination with ethambutol, with or without rifabutin: the most critical intervention in this setting is gaining HIV control with the use of potent antiretroviral therapy. Treatment outcomes for many patients with MAC disease remain suboptimal, so new drugs and treatment regimens are needed. Given the high rate of reinfection after cure, one of the greatest needs is a better understanding of where infection occurs and how this can be prevented.
Mycobacterium avium is an emerging pathogen, and less is understood about the effector and suppressor immune responses. While NTM are generally considered to be less virulent than Mtb, they can cause pulmonary and extrapulmonary disease in vulnerable individuals. The most common NTM causing disease and outbreaks in the United States are species within the MAC, comprising at least nine species, including M. avium, M. intracellulare, and M. chimaera. Other reports demonstrate the possibility that cystic fibrosis patients acquire NTM infection by contact with fomites or from patient to patient transmission.4 It has also been suggested that healthy immunocompetent individuals colonized with NTMs may transmit to NTMs to more vulnerable populations.89 Recent population-based studies have demonstrated this worldwide increase in NTM began in 2000. In the United States, the prevalence of NTM lung disease exceeds that of tuberculosis by approximately 10-fold.84
M. avium are capable of surviving in intracellular compartments in phagocytic cells.83 Immunity against Mycobacteria requires a balance between immune responses that kill or at least constrain Mycobacteria replication and immune regulation to dampen tissue-damaging inflammation.3 One example that strongly supports the importance of a protective cellular response against Mtb and, to a lesser degree M. avium90–91 is individuals with the acquired immunodeficiency syndrome (AIDS) increased susceptibility to Mtb due to a deficiency of CD4+ T cells. Before the development of the highly-active antiretroviral cocktail therapies that are currently used against HIV infection, disseminated infections due to the NTM, particularly with MAC, were relatively common, further revealing the importance of the CD4+ T cells in host defense against mycobacteria. However, with the development of an effective anti-HIV drug cocktail and restoration of the immune system, disseminated MAC infections are rarely seen in this population however Mtb infections in HIV seropositive individuals still frequently occurs.
Early in vitro studies showed M. avium were slowed but not killed when cultured in activated macrophages.92 At this time, the role of Th1 cytokines began to be investigated, and an early study suggested that TNF-α was more effective than IFN-γ in growth inhibition.92 This highlights an essential difference between Mtb bacterial control with IFN-γ and M. avium control with TNF-α.93
More recent studies have shown that mice can control M. avium infections independently of macrophage activation by IFN-γ, with TNF- α compensating for the lack of IFN-γ activation by IFN-γ.93 Moreover, the M. avium granuloma development forms without the aid of IFN-γ activation of macrophages.93
Early studies showed that M. avium strains could show different morphotypes, which could impact the strain’s virulence properties. It was shown that a virulent colony morphotype smooth transparent (SmT) isolate induced a delayed, relatively small TNF-α response. In contrast, the smooth domed (SmD) equivalent induced a rapid, large TNF-α response.92 It was then found that while macrophage activation generated reactive oxygen species in response to M. avium, isolates expressing the virulent SmT form were unaffected.94 Further studies showed that, in general, SmT isolates were virulent, and SmD isolates were much less so, with rough variants showing an intermediate phenotype. As predicted, there were large differences between the inflammatory and protective cytokine/chemokine profiles of SmT and Smd/SmO infections.95
Activated macrophages also produced nitric oxide (NO). Still, blocking of this event did not influence growth inhibition, and subsequent studies raised the possibility that acidification of the bacterial phagosome was, in fact, more critical in the control of the infection.96 In vivo, CD4 involvement was crucial for both protection and granuloma formation, and it was suggested that the TH1 cytokines IFN-γ and TNF-α produced by these cells act in synergy.96 Consistent with the Th1 pathway hypothesis, depletion of interleukin-12 (IL-12) also reduced resistance to M. avium infection.97 In addition to CD4 cells, IL-12 was also implicated in TNF-α production by NK cells in M. avium infections.98 An early role for IL-6 was suggested, as was a late response involving IL-10.99 More recently, a role for TNF-α has been illustrated by infections occurring in patients treated with anti-TNF-α biologic reagents for rheumatoid arthritis.99
Consistent with earlier studies, T cells harvested from mice infected with M. avium transferred protection only if live bacteria were used. This was, of course, consistent with observations at the time that only live M. tuberculosis was capable of generating protective T cells.100 While CD4+ T cells were the primary source of protection, some studies suggested a contribution by CD8+ cells, although the latter was eventually shown not to be critical.
A 1999 study101 provided the unexpected result that mice that lack the NOS2 gene and cannot make NO within their macrophages were more resistant than wild-type controls to M. avium (the opposite of the result obtained with M. tuberculosis), at least partially explaining why virulent M. avium strains grow in mice despite robust Th1 responses. Subsequent studies also demonstrated a superior fibrotic response in the lungs of gene knockout mice. They showed that the increased resistance was consistent when multiple virulent M. avium strains were used.102 A possible explanation was the observation that, along with TNF-α, NO controlled granuloma integrity rather than being directly antimicrobial.102
Regulatory T-cell functions against M. avium
Regulatory T cells (Tregs) express the transcription factor Foxp3+ and are essential to counterbalance any excessive pro-inflammatory Th1 responses.17 Clinical studies have shown that M. avium regulatory T cells tend to express PD-1 markers of immune exhaustion, a state where a cell persists; however, this cell cannot exert a function to kill the pathogen. These clinical studies have demonstrated the percentage of PD-1 on CD4+ (PD-1+CD4+) lymphocytes, and monocyte-derived suppressor cells were higher in the M. avium patients with pulmonary disease than healthy controls.103 There were no intergroup differences regarding CTLA-4+CD4+ lymphocytes. Higher PD-1+CD4+ lymphocytes were found in M. avium patients compared to healthy controls.103 Positive sputum acid-fast stains and fibrocavitary radiographic lesions correlated with elevated PD-1+CD4+ lymphocytes and regulatory T cells.103 Moreover, the percentage of PD-1+CD4+ lymphocytes at the initial and two months of follow-up significantly predicted subsequent radiographic progression.103
Regulatory T cells’ involvement in M. avium disease progression seems to be dominant during chronic lung disease.104 Studies using normal mice infected intravenously with8 viable M. avium develop persistent infections of the lung resulted in a progressive disease. The presence of suppressor T cells within the heavily infected lung was present. Additional research is necessary to provide correlates of protection with M. avium as its emergence currently is a global threat and as an emerging Mycobacteria pathogen able to the high ability to mutate and evolve into a pathogen able to infect immunocompetent individuals.
Effector T-cell functions against M. abscessus
Even less is known about the early stages of M. abscessus effector immunity than we know about Mtb and M. avium and invasion in human patients’ upper respiratory tract. However, our studies using M. abscessus strains expressing fluorescent markers using whole-body mouse xenogen imaging (which allows for tracking the specific site of infection) after pulmonary infection shows M. abscessus persists in the nasopharyngeal passageways before dissemination into the lower airways.105 M. abscessus species are intracellular pathogens that primarily infect macrophages5 The fact that autophagy processing, which is required for control of M. abscessus infection within human primary macrophages106– 107, is defective in cystic fibrosis (CF) combined with the presence of macrophages producing IL-10 macrophages suggests that M. abscessus may persist better in human CF cells. The less efficient internalization of M. abscessus strains by human neutrophils and the reduced pro-inflammatory response they trigger compared to Staphylococcus aureus may be further factors contributing to M. abscessus ability to survive to persist in neutrophil-rich settings such as the CF airway.25
Studies exposing a high-dose aerosol to different mouse strains, C57BL/6, and ob/ob mice, developed an established infection and a pulmonary immune response consisting of an early influx of IFN-γ+ CD4+ T cells.105 However, this immune response preceded the successful clearance of M. abscessus in both strains of mice, although Mycobacteria elimination was delayed in the ob/ob mice. Infected guinea pigs showed an increased influx of lymphocytes into the lungs with bacterial clearance by Day 60.105 A high-dose aerosol, C57BL/6, and ob/ob mice developed an established infection and a pulmonary immune response consisting of an early influx of IFN-γ+ CD4+ T cells. This immune response preceded the successful clearance of M. abscessus in both strains of mice. However, Mycobacteria elimination was delayed in the ob/ob mice. Infected guinea pigs showed an increased influx of lymphocytes into the lungs with bacterial clearance by day 60.
In contrast to the C57BL/6 and ob/ob mice and guinea pigs, IFN-γ knockout (GKO) mice challenged with a LDA or HDA of M. abscessus showed a progressive lung infection despite a robust influx of T cells, macrophages, and dendritic cells, culminating in extensive lung consolidation. Furthermore, with HDA challenge of the GKO mice, the emergence of IL-4- and IL-10-producing CD4+ and CD8+ T cells was seen in the lungs.105 In conclusion, IFN-γ may be important in the host defense against M. abscessus. However, TNF-α appears to be essential for controlling disease. IFN-γ knockout mice challenged with a LDA or HDA of M. abscessus showed a progressive lung infection despite a robust influx of T cells, macrophages, and dendritic cells, culminating in extensive lung consolidation. Furthermore, with the GKO mice’s challenge, the emergence of IL-4- and IL-10-producing CD4+ and CD8+ T cells was seen in the lungs. In conclusion, TNF-α is critically important in the host defense against M. abscessus in small mouse models and humans.
Suppressor T-cell functions against M. abscessus
Mycobacterium abscessus infections are ubiquitous in the environment and are responsible for colonization, infection, and pseudo-outbreaks in healthcare settings throughout the world.108–110 It is plausible that cystic fibrosis patients are exposed daily to one or more sources of M. abscessus in their routines, and studies support showing NTM patient’s disease was caused by identical strains obtained from their homes.110–113 A recent collaboration involving a consortium of institutions funded by the cystic fibrosis trust in the UK revealed exogenous reinfection by cystic fibrosis patients encountering residual M. abscessus fomites in clinical settings may be occurring globally.4, 114 More importantly, our studies demonstrated that these globally circulating M. abscessus clones showed increased ability to infect human macrophages and resulted in increased bacterial burden and pulmonary damage in mice.4
Mycobacterium abscessus is a rapidly growing Mycobacteria species, regarded as less virulent than M. avium and Mtb, however still an important pathogen responsible for a wide array of clinical manifestations in humans, ranging from cutaneous infections to severe chronic pulmonary infections, usually encountered in immunocompromised and in cystic fibrosis (CF) patients and are often refractory to antibiotic therapy.115 Mycobacterium abscessus represents a crucial respiratory pathogen among the rapidly-growing nontuberculous mycobacteria. The underlying immunopathological mechanisms of pathogenesis remain mostly unknown. A significant reason for the low advances in M. abscessus research has been a lack of adequate models to study the acute and chronic stages of the disease, leading to a delayed evaluation of potentially active antibiotics’ therapeutic efficacy. However, the recent development of more sophisticated immunocompromised mice has been exploited to dissect the immune and inflammatory responses to M. abscessus.5, 115
Autophagy processing has been shown to be required for control of M. abscessus infection within human primary macrophages106–107 It is defective in cystic fibrosis combined with macrophages producing IL-10 suggests that M. abscessus may persist better in human cystic fibrosis cells. The less efficient internalization of M. abscessus strains by human neutrophils and the reduced pro-inflammatory response they trigger is due to PD-1 expression compared to Staphylococcus aureus may be further factors contributing to M. abscessus to survive and persist in neutrophil-rich settings such as the cystic fibrosis airway.25 Upon infection, M. abscessus isolates may persist in upper airways reinfecting the lungs by the cough reflex or remain latent in patients’ lungs for years or even decades before causing a clinically apparent infection.116 It is plausible in the host that M. abscessus upper airway reinfection or exogenous reinfection can occur, leading to immune exhaustion and/or reduced regulatory T cell function since cystic fibrosis patients at risk for M. abscessus infection have been identified to express markers of immune exhaustion, and dysregulation in type 1 cytokine (IFNγ and TNFα) release.117 Moreover, CF patients have markers of immune exhaustion during bacterial infection,117–118 regulatory T cell (Tregs) impairment,119 and neutrophil arginase-1 T cell suppression.120
Future directions:
Despite years of intensive research, Bacille Calmette-Guerin (BCG) remains the only licensed vaccine and has variable efficacy. It protects against childhood TB but is not effective in adult pulmonary TB. As a result of intense research in understanding TB vaccinology, there are many new vaccine candidates in clinical development and many more in pre-clinical trials that aim to replace or boost BCG vaccine. NTM interference has been implicated in reducing BCG vaccine efficacy in healthy individuals. However, studies using NTMs and Mtb to demonstrate NTM interference with BCG has been derived from mouse models with the NTMs ability of bacilli persisting in the mouse stomach. New models in which NTMs don’t persist in the stomach should be developed, such as the guinea pig and nonhuman primate model of BCG vaccination and the NTM interference.
Moreover, studies have shown BCG induces cross-protective T cell immunity against M. avium and M. abscessus in immunocompetent small animal models. Human TB and NTM cross-protective T cells were quantified using flow cytometric assays.39
Additional studies need to be conducted to models reflective of the susceptible population of humans who are at risk of NTM infection. Cystic fibrosis human studies and cystic fibrosis animal models that best reproduce the target susceptible population. The primary model that can be developed and mimic humans is the guinea pig and NHP model, which has a low stomach pH, so NTMs don’t survive. Primary and secondary granulomas are similar to humans and the NHP model. In summary, the scientific community must use models that represent NTM infection and disease and how BCG vaccination, NTM persistence, and Mtb outcome impact the human host.
Summary Conclusions:
The Majority of the Mtb vaccine community considerer’s effector immune responses to be vital in the control of Mtb. There is a consensus that regulatory T cells are a double-edged sword because they help reduce tissue-damaging inflammation; however, they can also reduce the Th1 immunity required for control of M. tuberculosis. Although, in regards to the emerging pathogen M. avium, it is clear the effector functions required for killing are not reliant on IFN-γ as dominantly as with M. tuberculosis. Moreover, both M. avium and M. abscessus are opportunistic infections PD-1 expression and immune exhaustion are more dominant than regulatory T cell suppression. M. abscessus is less well understood and thrives on uncontrolled inflammation due to diseases such as in cystic fibrosis and chronic obstructive pulmonary diseases. The common thread in Mtb, M. avium, and M. abscessus are the ability to infect through aerosol exposure, increased capacity to develop drug resistance, and lack of a cure. These Mycobacteria pathogens Mtb, M. avium, and M. abscessus are evolving, and pose a potential threat to global human health.
Figure 1. Regulatory T cell suppression of antigen-presenting cells (APC).
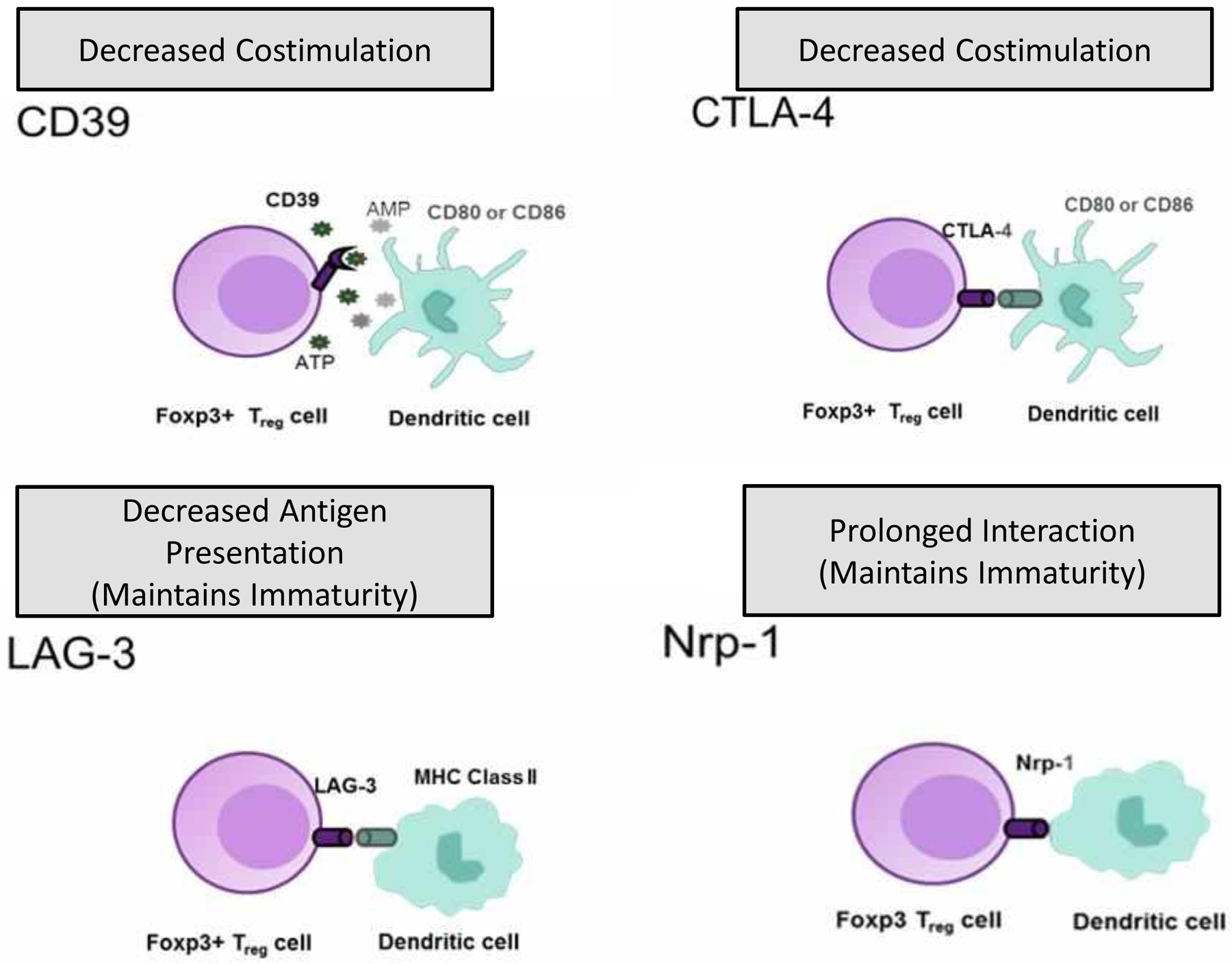
CTLA-4 on Treg cells’ surface can prevent or depress the up-regulation of CD80 and CD86, the major costimulatory molecules on APC. LAG-3 on Treg cells can interact with MHC class II on APC by binding LAG-3 to MHC class II molecules on immature DCs, causing an inhibitory signal that suppresses DC maturation and immaturity. Tissue destruction results in extracellular ATP that functions as an indicator and exerts inflammatory effects on DCs. Catalytic inactivation of extracellular ATP by CD39 represents an anti-inflammatory mechanism that may be used by Treg cells to prevent the harmful effects of ATP on antigen-presenting cell function. In contrast, Nrp-1 (neuropilin) promotes extended interactions between Treg cells and immature DCs and limits access of the effector cells to APC.
Acknowledgments:
This research received funding from We thank our colleagues in the Mycobacteria Research Laboratories for their contributions to the development of the methods described here. This article was funded with Cystic Fibrosis Foundation grant number ORDWAY19G0. Several of the more modern approaches were developed using funding from NIH programs Dr. Ordway is an NIH Innovation award recipient grant number 1DP2OD006450, and NIH/ NIAID task order HHSN272201000009I/HHSN27200001, subtask 6, principal investigator [PI], Anne J. Lenaerts, co-PI, Diane J. Ordway) (program officers, Christine Sizemore and Andre McBride, and Jim Boyce). Additional funding leading to the development of the guinea pig model was generously provided by the Bill & Melinda Gates Foundation grant number OPP1081476.
Footnotes
Conflicts of Interest: “The authors declare no conflict of interest.”
References
- 1.Togun T, Kampmann B, Stoker NG, Lipman M. Anticipating the impact of the COVID-19 pandemic on TB patients and TB control programmes. Annals of clinical microbiology and antimicrobials. 2020. May 23;19(1):21. Epub 2020/05/25. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kato-Maeda M, Shanley CA, Ackart D, et al. Beijing sublineages of Mycobacterium tuberculosis differ in pathogenicity in the guinea pig. Clinical and vaccine immunology : CVI. 2012. August;19(8):1227–37. Epub 2012/06/22. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Verma D, Stapleton M, Gadwa J, Vongtongsalee K, et al. Mycobacterium avium Infection in a C3HeB/FeJ Mouse Model. Front Microbiol. 2019;10:693. Epub 2019/04/20. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bryant JM, Grogono DM, Rodriguez-Rincon D, Everall I, et al. Emergence and spread of a human-transmissible multidrug-resistant nontuberculous mycobacterium. Science. 2016. November 11;354(6313):751–7. Epub 2016/11/16. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernut A, Herrmann JL, Ordway D, et al. The Diverse Cellular and Animal Models to Decipher the Physiopathological Traits of Mycobacterium abscessus Infection. Frontiers in cellular and infection microbiology. 2017;7:100. Epub 2017/04/20. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Houben RM, Dodd PJ. The Global Burden of Latent Tuberculosis Infection: A Re-estimation Using Mathematical Modelling. PLoS medicine. 2016. October;13(10):e1002152. Epub 2016/10/26. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Conradie F, Diacon AH, Ngubane N, et al. Treatment of Highly Drug-Resistant Pulmonary Tuberculosis. The New England journal of medicine. 2020. March 5;382(10):893–902. Epub 2020/03/05. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaufmann SHE. New vaccines against tuberculosis. Bundesgesundheitsblatt, Gesundheitsforschung, Gesundheitsschutz. 2020;63(1):56–64. ger. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu B, Dockrell HM, Ottenhoff THM, et al. Tuberculosis vaccines: Opportunities and challenges. Respirology. 2018. April;23(4):359–68. Epub 2018/01/18. eng. [DOI] [PubMed] [Google Scholar]
- 10.Dockrell HM, Smith SG. What Have We Learnt about BCG Vaccination in the Last 20 Years? Frontiers in immunology. 2017;8:1134-. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brandt L, Feino Cunha J, Weinreich Olsen A, et al. Failure of the Mycobacterium bovis BCG vaccine: some species of environmental mycobacteria block multiplication of BCG and induction of protective immunity to tuberculosis. Infect Immun. 2002. February;70(2):672–8. Epub 2002/01/18. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Verma D, Chan ED, Ordway DJ. Non-Tuberculous Mycobacteria Interference with BCG-Current Controversies and Future Directions. Vaccines. 2020. November 16;8(4). Epub 2020/11/20. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pereira SM, Dantas OM, Ximenes R, [BCG vaccine against tuberculosis: its protective effect and vaccination policies]. Rev Saude Publica. 2007. September;41 Suppl 1:59–66. Epub 2007/12/06. Vacina BCG contra tuberculose: efeito protetor e políticas de vacinação. por. [DOI] [PubMed] [Google Scholar]
- 14.Mangtani P, Nguipdop-Djomo P, Keogh RHS, et al. The duration of protection of school-aged BCG vaccination in England: a population-based case-control study. Int J Epidemiol. 2018. February 1;47(1):193–201. Epub 2017/10/13. eng. [DOI] [PubMed] [Google Scholar]
- 15.Guyot-Revol V, Innes JA, Hackforth S, et al. Regulatory T cells are expanded in blood and disease sites in patients with tuberculosis. American journal of respiratory and critical care medicine. 2006. April 1;173(7):803–10. Epub 2005/12/13. eng. [DOI] [PubMed] [Google Scholar]
- 16.Savage ND, de Boer T, Walburg KV, et al. Human anti-inflammatory macrophages induce Foxp3+ GITR+ CD25+ regulatory T cells, which suppress via membrane-bound TGFbeta-1. Journal of immunology (Baltimore, Md : 1950). 2008. August 1;181(3):2220–6. Epub 2008/07/22. eng. [DOI] [PubMed] [Google Scholar]
- 17.Brighenti S, Ordway DJ. Regulation of Immunity to Tuberculosis. Microbiology spectrum. 2016. December;4(6). Epub 2017/01/15. eng. [DOI] [PubMed] [Google Scholar]
- 18.Larson RP, Shafiani S, Urdahl KB. Foxp3(+) regulatory T cells in tuberculosis. Advances in experimental medicine and biology. 2013;783:165–80. Epub 2013/03/08. eng. [DOI] [PubMed] [Google Scholar]
- 19.Sia JK, Rengarajan J. Immunology of Mycobacterium tuberculosis Infections. Microbiology spectrum. 2019. July;7(4). Epub 2019/07/13. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mangtani P, Abubakar I, Ariti C, et al. Protection by BCG vaccine against tuberculosis: a systematic review of randomized controlled trials. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2014. February;58(4):470–80. Epub 2013/12/18. eng. [DOI] [PubMed] [Google Scholar]
- 21.Chen K, Kolls JK. T cell-mediated host immune defenses in the lung. Annual review of immunology. 2013;31:605–33. Epub 2013/03/23. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ordway D, Palanisamy G, Henao-Tamayo M, et al. The cellular immune response to Mycobacterium tuberculosis infection in the guinea pig. Journal of immunology (Baltimore, Md : 1950). 2007. August 15;179(4):2532–41. Epub 2007/08/07. eng. [DOI] [PubMed] [Google Scholar]
- 23.Henao-Tamayo MI, Obregón-Henao A, Arnett K, et al. Effect of bacillus Calmette-Guérin vaccination on CD4+Foxp3+ T cells during acquired immune response to Mycobacterium tuberculosis infection. J Leukoc Biol. 2016. April;99(4):605–17. Epub 2015/11/22. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ordway D, Henao-Tamayo M, Harton M, et al. The hypervirulent Mycobacterium tuberculosis strain HN878 induces a potent TH1 response followed by rapid down-regulation. Journal of immunology (Baltimore, Md : 1950). 2007. July 1;179(1):522–31. Epub 2007/06/21. eng. [DOI] [PubMed] [Google Scholar]
- 25.Ordway D, Henao-Tamayo M, Orme IM, er al. Foamy macrophages within lung granulomas of mice infected with Mycobacterium tuberculosis express molecules characteristic of dendritic cells and antiapoptotic markers of the TNF receptor-associated factor family. Journal of immunology (Baltimore, Md : 1950). 2005;175:3873–81. [DOI] [PubMed] [Google Scholar]
- 26.Henao-Tamayo M, Ordway DJ, Orme IM. Memory T cell subsets in tuberculosis: what should we be targeting? Tuberculosis (Edinburgh, Scotland). 2014. September;94(5):455–61. Epub 2014/07/06. eng. [DOI] [PubMed] [Google Scholar]
- 27.Orme IM, Ordway DJ. Mouse and Guinea Pig Models of Tuberculosis. Microbiology spectrum. 2016. August;4(4). Epub 2016/10/12. eng. [DOI] [PubMed] [Google Scholar]
- 28.Cardona P, Cardona PJ. Regulatory T Cells in Mycobacterium tuberculosis Infection. Frontiers in immunology. 2019;10:2139. Epub 2019/10/02. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McShane H. Insights and challenges in tuberculosis vaccine development. The Lancet Respiratory medicine. 2019. September;7(9):810–9. Epub 2019/08/17. eng. [DOI] [PubMed] [Google Scholar]
- 30.Arias L, Goig GA, Cardona P, et al. Influence of Gut Microbiota on Progression to Tuberculosis Generated by High Fat Diet-Induced Obesity in C3HeB/FeJ Mice. Front Immunol. 2019;10:2464. Epub 2019/11/05. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ordway DJ, Shang S, Henao-Tamayo M, et al. Mycobacterium bovis BCG-mediated protection against W-Beijing strains of Mycobacterium tuberculosis is diminished concomitant with the emergence of regulatory T cells. Clinical and vaccine immunology : CVI. 2011. September;18(9):1527–35. Epub 2011/07/29. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prados-Rosales R, Carreño LJ, Weinrick B, et al. The Type of Growth Medium Affects the Presence of a Mycobacterial Capsule and Is Associated With Differences in Protective Efficacy of BCG Vaccination Against Mycobacterium tuberculosis. The Journal of infectious diseases. 2016. August 1;214(3):426–37. Epub 2016/05/29. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaveh DA, Bachy VS, Hewinson RG, et al. Systemic BCG immunization induces persistent lung mucosal multifunctional CD4 T(EM) cells which expand following virulent mycobacterial challenge. PloS one. 2011;6(6):e21566. Epub 2011/07/02. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fulton SA, Martin TD, Redline RW,et al. Pulmonary immune responses during primary mycobacterium bovis- Calmette-Guerin bacillus infection in C57Bl/6 mice. American journal of respiratory cell and molecular biology. 2000. March;22(3):333–43. Epub 2000/03/01. eng. [DOI] [PubMed] [Google Scholar]
- 35.Bhatt K, Verma S, Ellner JJ, Salgame P. Quest for correlates of protection against tuberculosis. Clinical and vaccine immunology : CVI. 2015. March;22(3):258–66. Epub 2015/01/16. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dharmadhikari AS, Basaraba RJ, Van Der Walt ML, et al. Natural infection of guinea pigs exposed to patients with highly drug-resistant tuberculosis. Tuberculosis (Edinburgh, Scotland). 2011. July;91(4):329–38. Epub 2011/04/12. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Otchere ID, Asante-Poku A, Osei-Wusu S, et al. Isolation and characterization of nontuberculous mycobacteria from patients with pulmonary tuberculosis in Ghana. International journal of mycobacteriology. 2017. Jan-Mar;6(1):70–5. Epub 2017/03/21. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fatima S, Kumari A, Das G, et al. Tuberculosis vaccine: A journey from BCG to present. Life sciences. 2020. July 1;252:117594. Epub 2020/04/20. eng. [DOI] [PubMed] [Google Scholar]
- 39.Abate G, Hamzabegovic F, Eickhoff CS, et al. BCG Vaccination Induces M. avium and M. abscessus Cross-Protective Immunity. Frontiers in immunology. 2019;10:234. Epub 2019/03/07. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bénard A, Sakwa I, Schierloh P, et al. B Cells Producing Type I IFN Modulate Macrophage Polarization in Tuberculosis. American journal of respiratory and critical care medicine. 2018. March 15;197(6):801–13. Epub 2017/11/22. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Henao-Tamayo M, Irwin SM, Shang S, et al. T lymphocyte surface expression of exhaustion markers as biomarkers of the efficacy of chemotherapy for tuberculosis. Tuberculosis. 2011 2011/July/01/;91(4):308–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goldsack L, Kirman JR. Half-truths and selective memory: Interferon gamma, CD4+ T cells and protective memory against tuberculosis. Tuberculosis. 2007 2007/November/01/;87(6):465–73. [DOI] [PubMed] [Google Scholar]
- 43.Henao-Tamayo M, Obregón-Henao A, Ordway DJ, Shang S, et al. A mouse model of tuberculosis reinfection. Tuberculosis (Edinburgh, Scotland). 2012. May;92(3):211–7. Epub 2012/03/21. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Verver S, Warren RM, Beyers N, et al. Rate of reinfection tuberculosis after successful treatment is higher than rate of new tuberculosis. American journal of respiratory and critical care medicine. 2005. June 15;171(12):1430–5. Epub 2005/04/16. eng. [DOI] [PubMed] [Google Scholar]
- 45.Sakaguchi S, Takahashi T, Nishizuka Y. Study on cellular events in postthymectomy autoimmune oophoritis in mice. I. Requirement of Lyt-1 effector cells for oocytes damage after adoptive transfer. The Journal of experimental medicine. 1982. December 1;156(6):1565–76. Epub 1982/12/01. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Asano M, Toda M, Sakaguchi N, et al. Autoimmune disease as a consequence of developmental abnormality of a T cell subpopulation. The Journal of experimental medicine. 1996. August 1;184(2):387–96. Epub 1996/08/01. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sakaguchi S, Sakaguchi N, Asano M, et al. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995. August 1;155(3):1151–64. Epub 1995/08/01. eng. [PubMed] [Google Scholar]
- 48.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003. February 14;299(5609):1057–61. Epub 2003/01/11. eng. [DOI] [PubMed] [Google Scholar]
- 49.Khattri R, Cox T, Yasayko SA, et al. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nature immunology. 2003. April;4(4):337–42. Epub 2003/03/04. eng. [DOI] [PubMed] [Google Scholar]
- 50.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nature immunology. 2003. April;4(4):330–6. Epub 2003/03/04. eng. [DOI] [PubMed] [Google Scholar]
- 51.Marson A, Kretschmer K, Frampton GM, et al. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature. 2007. February 22;445(7130):931–5. Epub 2007/01/24. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sharma R, Jarjour WN, Zheng L, et al. Large functional repertoire of regulatory T-cell suppressible autoimmune T cells in scurfy mice. J Autoimmun. 2007. August;29(1):10–9. Epub 2007/05/25. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ochs HD, Gambineri E, Torgerson TR. IPEX, FOXP3 and regulatory T-cells: a model for autoimmunity. Immunol Res. 2007;38(1–3):112–21. Epub 2007/10/06. eng. [DOI] [PubMed] [Google Scholar]
- 54.Mayer CT, Tian L, Hesse C, et al. Anti-CD4 treatment inhibits autoimmunity in scurfy mice through the attenuation of co-stimulatory signals. J Autoimmun. 2014. May;50:23–32. Epub 2013/10/01. eng. [DOI] [PubMed] [Google Scholar]
- 55.Torgerson TR, Ochs HD. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome: a model of immune dysregulation. Curr Opin Allergy Clin Immunol. 2002. December;2(6):481–7. Epub 2004/01/31. eng. [DOI] [PubMed] [Google Scholar]
- 56.Brunkow ME, Jeffery EW, Hjerrild KA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001. January;27(1):68–73. Epub 2001/01/04. eng. [DOI] [PubMed] [Google Scholar]
- 57.Bennett CL, Ochs HD. IPEX is a unique X-linked syndrome characterized by immune dysfunction, polyendocrinopathy, enteropathy, and a variety of autoimmune phenomena. Curr Opin Pediatr. 2001. December;13(6):533–8. Epub 2001/12/26. eng. [DOI] [PubMed] [Google Scholar]
- 58.Li B, Samanta A, Song X, Iacono KT, et al. FOXP3 is a homo-oligomer and a component of a supramolecular regulatory complex disabled in the human XLAAD/IPEX autoimmune disease. Int Immunol. 2007. July;19(7):825–35. Epub 2007/06/26. eng. [DOI] [PubMed] [Google Scholar]
- 59.Caudy AA, Reddy ST, Chatila T, et al. CD25 deficiency causes an immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like syndrome, and defective IL-10 expression from CD4 lymphocytes. The Journal of allergy and clinical immunology. 2007. February;119(2):482–7. Epub 2007/01/02. eng. [DOI] [PubMed] [Google Scholar]
- 60.Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annual review of immunology. 2012;30:531–64. Epub 2012/01/10. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kang SM, Tang Q, Bluestone JA. CD4+CD25+ regulatory T cells in transplantation: progress, challenges and prospects. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2007. June;7(6):1457–63. Epub 2007/05/22. eng. [DOI] [PubMed] [Google Scholar]
- 62.Curotto de Lafaille MA, Lafaille JJ. Natural and adaptive foxp3+ regulatory T cells: more of the same or a division of labor? Immunity. 2009. May;30(5):626–35. Epub 2009/05/26. eng. [DOI] [PubMed] [Google Scholar]
- 63.Groux H, O’Garra A, Bigler M, et al. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 1997. October 16;389(6652):737–42. Epub 1997/10/24 21:29. eng. [DOI] [PubMed] [Google Scholar]
- 64.Sakaguchi S Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annual review of immunology. 2004;22:531–62. Epub 2004/03/23. eng. [DOI] [PubMed] [Google Scholar]
- 65.Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity. 2009. May;30(5):636–45. Epub 2009/05/26. eng. [DOI] [PubMed] [Google Scholar]
- 66.Wood KJ, Bushell A, Hester J. Regulatory immune cells in transplantation. Nature reviews Immunology. 2012. June;12(6):417–30. Epub 2012/05/26. eng. [DOI] [PubMed] [Google Scholar]
- 67.Magnani CF, Alberigo G, Bacchetta R, et al. Killing of myeloid APCs via HLA class I, CD2 and CD226 defines a novel mechanism of suppression by human Tr1 cells. European journal of immunology. 2011. June;41(6):1652–62. Epub 2011/04/07. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim H, Kwon KW, Kim WS, et al. Virulence-dependent induction of interleukin-10-producing-tolerogenic dendritic cells by Mycobacterium tuberculosis impedes optimal T helper type 1 proliferation. Immunology. 2017. June;151(2):177–90. Epub 2017/02/01. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tsai MC, Chakravarty S, Zhu G, et al. Characterization of the tuberculous granuloma in murine and human lungs: cellular composition and relative tissue oxygen tension. Cellular microbiology. 2006. February;8(2):218–32. Epub 2006/01/31. eng. [DOI] [PubMed] [Google Scholar]
- 70.Ulrichs T, Kosmiadi GA, Trusov V, et al. Human tuberculous granulomas induce peripheral lymphoid follicle-like structures to orchestrate local host defence in the lung. The Journal of pathology. 2004. October;204(2):217–28. Epub 2004/09/18. eng. [DOI] [PubMed] [Google Scholar]
- 71.Iwata Y, Matsushita T, Horikawa M, et al. Characterization of a rare IL-10-competent B-cell subset in humans that parallels mouse regulatory B10 cells. Blood. 2011. January 13;117(2):530–41. Epub 2010/10/22. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yanaba K, Bouaziz JD, Haas KM, et al. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity. 2008. May;28(5):639–50. Epub 2008/05/17. eng. [DOI] [PubMed] [Google Scholar]
- 73.Lund FE, Randall TD. Effector and regulatory B cells: modulators of CD4+ T cell immunity. Nature reviews Immunology. 2010. April;10(4):236–47. Epub 2010/03/13. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Blair PA, Norena LY, Flores-Borja F, et al. CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic Lupus Erythematosus patients. Immunity. 2010. January 29;32(1):129–40. Epub 2010/01/19. eng. [DOI] [PubMed] [Google Scholar]
- 75.Flores-Borja F, Bosma A, Ng D, Reddy V, Ehrenstein MR, et al. CD19+CD24hiCD38hi B cells maintain regulatory T cells while limiting TH1 and TH17 differentiation. Science translational medicine. 2013. February 20;5(173):173ra23. Epub 2013/02/22. eng. [DOI] [PubMed] [Google Scholar]
- 76.Zhang M, Zheng X, Zhang J, et al. CD19(+)CD1d(+)CD5(+) B cell frequencies are increased in patients with tuberculosis and suppress Th17 responses. Cellular immunology. 2012;274(1–2):89–97. Epub 2012/03/01. eng. [DOI] [PubMed] [Google Scholar]
- 77.Amu S, Saunders SP, Kronenberg M, et al. Regulatory B cells prevent and reverse allergic airway inflammation via FoxP3-positive T regulatory cells in a murine model. The Journal of allergy and clinical immunology. 2010. May;125(5):1114–24 e8. Epub 2010/03/23. eng. [DOI] [PubMed] [Google Scholar]
- 78.Watanabe R, Ishiura N, Nakashima H, et al. Regulatory B cells (B10 cells) have a suppressive role in murine lupus: CD19 and B10 cell deficiency exacerbates systemic autoimmunity. J Immunol. 2010. May 1;184(9):4801–9. Epub 2010/04/07. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee KM, Stott RT, Zhao G, et al. TGF-beta-producing regulatory B cells induce regulatory T cells and promote transplantation tolerance. European journal of immunology. 2014. June;44(6):1728–36. Epub 2014/04/05. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kessel A, Haj T, Peri R, et al. Human CD19(+)CD25(high) B regulatory cells suppress proliferation of CD4(+) T cells and enhance Foxp3 and CTLA-4 expression in T-regulatory cells. Autoimmunity reviews. 2012. July;11(9):670–7. Epub 2011/12/14. eng. [DOI] [PubMed] [Google Scholar]
- 81.Prevots DR, Marras TK. Epidemiology of human pulmonary infection with nontuberculous mycobacteria: a review. Clin Chest Med. 2015. March;36(1):13–34. Epub 2015/02/14. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.De Groote MA, Huitt G. Infections Due to Rapidly Growing Mycobacteria. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2006. June 15, 2006;42(12):1756–63. [DOI] [PubMed] [Google Scholar]
- 83.Orme IM, Ordway DJ. Host response to nontuberculous mycobacterial infections of current clinical importance. Infect Immun. 2014. September;82(9):3516–22. Epub 2014/06/11. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Epson E, Cassidy M, Marshall-Olson A, et al. Patients with nontuberculous mycobacteria: comparison of updated and previous diagnostic criteria for lung disease. Diagnostic Microbiology and Infectious Disease. 2012 2012/September/01/;74(1):98–100. [DOI] [PubMed] [Google Scholar]
- 85.To K, Cao R, Yegiazaryan A, et al. General Overview of Nontuberculous Mycobacteria Opportunistic Pathogens: Mycobacterium avium and Mycobacterium abscessus. Journal of clinical medicine. 2020;9(8):2541. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chan ED, Bai X, Kartalija M, et al. Host immune response to rapidly growing mycobacteria, an emerging cause of chronic lung disease. American journal of respiratory cell and molecular biology. 2010. October;43(4):387–93. Epub 2010/01/19. eng. [DOI] [PubMed] [Google Scholar]
- 87.Philley JV, DeGroote MA, Honda JR, et al. Treatment of Non-Tuberculous Mycobacterial Lung Disease. Current treatment options in infectious diseases. 2016. December;8(4):275–96. Epub 2017/05/23. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Brown-Elliott BA, Nash KA, Wallace RJ. Antimicrobial Susceptibility Testing, Drug Resistance Mechanisms, and Therapy of Infections with Nontuberculous Mycobacteria. Clinical Microbiology Reviews. 2012;25(3):545–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hoffner S, Chan MM, Chan ED, et al. Chapter 12 - Drug discovery targeting drug-resistant nontuberculous mycobacteria. In: Kesharwani P, Chopra S, Dasgupta A, editors. Drug Discovery Targeting Drug-Resistant Bacteria: Academic Press; 2020. p. 361–76. [Google Scholar]
- 90.Chan ED, Verma D, Ordway DJ. Animal Models of Mycobacteria Infection. Curr Protoc Immunol. 2020. June;129(1):e98. Epub 2020/06/05. eng. [DOI] [PubMed] [Google Scholar]
- 91.Dharmadhikari AS, Nardell EA. What animal models teach humans about tuberculosis. American journal of respiratory cell and molecular biology. 2008. November;39(5):503–8. Epub 2008/06/17. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bermudez LE, Young LS. Tumor necrosis factor, alone or in combination with IL-2, but not IFN-gamma, is associated with macrophage killing of Mycobacterium avium complex. The Journal of Immunology. 1988;140(9):3006–13. [PubMed] [Google Scholar]
- 93.Resende M, Cardoso MS, Fróis-Martins R, et al. TNF-Mediated Compensatory Immunity to Mycobacterium avium in the Absence of Macrophage Activation by IFN-γ. The Journal of Immunology. 2019:ji1801594. [DOI] [PubMed] [Google Scholar]
- 94.Bermudez LEM, Young LS. Oxidative and non-oxidative intracellular killing of Mycobacterium avium complex. Microbial Pathogenesis. 1989 1989/October/01/;7(4):289–98. [DOI] [PubMed] [Google Scholar]
- 95.Appelberg R, Castro AG, Pedrosa J, et al. Role of gamma interferon and tumor necrosis factor alpha during T-cell-independent and -dependent phases of Mycobacterium avium infection. Infection and immunity. 1994. September;62(9):3962–71. Epub 1994/09/01. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Appelberg R Protective Role of Interferon Gamma, Tumor Necrosis Factor Alpha and Interleukin-6 in Mycobacterium tuberculosis and M. avium Infections. Immunobiology. 1994 1994/January/01/;191(4):520–5. [DOI] [PubMed] [Google Scholar]
- 97.Castro AG, Silva RA, Appelberg R. Endogenously produced IL-12 is required for the induction of protective T cells during Mycobacterium avium infections in mice. Journal of immunology (Baltimore, Md : 1950). 1995. August 15;155(4):2013–9. Epub 1995/08/15. eng. [PubMed] [Google Scholar]
- 98.Bermudez LE, Wu M, Young LS. Interleukin-12-stimulated natural killer cells can activate human macrophages to inhibit growth of Mycobacterium avium. Infection and immunity. 1995;63(10):4099–104. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Appelberg R, Castro AG, Pedrosa J, et al. Role of interleukin-6 in the induction of protective T cells during mycobacterial infections in mice. Immunology. 1994;82(3):361–4. eng. [PMC free article] [PubMed] [Google Scholar]
- 100.Orme IM. Induction of nonspecific acquired resistance and delayed-type hypersensitivity, but not specific acquired resistance in mice inoculated with killed mycobacterial vaccines. Infection and immunity. 1988;56(12):3310–2. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Orme IM, Ordway DJ. Host Response to Nontuberculous Mycobacterial Infections of Current Clinical Importance. Infection and immunity. 2014;82(9):3516–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lousada S, Flórido M, Appelberg R. Regulation of granuloma fibrosis by nitric oxide during Mycobacterium avium experimental infection. International Journal of Experimental Pathology. 2006;87(4):307–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shu CC, Pan SW, Feng JY, et al. The Clinical Significance of Programmed Death-1, Regulatory T Cells and Myeloid Derived Suppressor Cells in Patients with Nontuberculous Mycobacteria-Lung Disease. Journal of clinical medicine. 2019. May 23;8(5). Epub 2019/05/28. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Watson SR, Collins FM. The specificity of suppressor T cells induced by chronic Mycobacterium avium infection in mice. Clinical and experimental immunology. 1981. January;43(1):10–9. Epub 1981/01/01. eng. [PMC free article] [PubMed] [Google Scholar]
- 105.Ordway D, Henao-Tamayo M, Smith et al. Animal model of Mycobacterium abscessus lung infection. Journal of leukocyte biology. 2008. June;83(6):1502–11. Epub 2008/03/04. eng. [DOI] [PubMed] [Google Scholar]
- 106.Renna M, Schaffner C, Brown K, et al. Azithromycin blocks autophagy and may predispose cystic fibrosis patients to mycobacterial infection. The Journal of clinical investigation. 2011. September;121(9):3554–63. Epub 2011/08/02. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bruscia EM, Bonfield TL. Cystic Fibrosis Lung Immunity: The Role of the Macrophage. Journal of innate immunity. 2016;8(6):550–63. Epub 2016/11/02. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Shang SGS, Henao-Tamayo M, Shanley CA, et al. Increased Virulence of an Epidemic Strain of Mycobacterium massiliense in Mice. PloS one. 2011;6:e24726. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Falkinham J, Norton C, LeChevallier M. Factors influencing numbers of Mycobacterium avium, Mycobacterium intracellulare, and other mycobacteria in drinking water distribution systems. Appl Environ Microbiol. 2001;67(3):1225 – 31. 10.1128/AEM.67.3.1225-1231.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Falkinham JD. Epidemiology of infection by nontuberculous mycobacteria. Clin Microbiol Rev. 1996;9:177–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Falkinham JO 3rd. Current Epidemiologic Trends of the Nontuberculous Mycobacteria (NTM). Current environmental health reports. 2016. June;3(2):161–7. Epub 2016/03/30. eng. [DOI] [PubMed] [Google Scholar]
- 112.Falkinham JO 3rd. Environmental sources of nontuberculous mycobacteria. Clinics in chest medicine. 2015. March;36(1):35–41. Epub 2015/02/14. eng. [DOI] [PubMed] [Google Scholar]
- 113.Iakhiaeva E, Howard ST, Brown Elliott BA, et al. Variable-Number Tandem-Repeat Analysis of Respiratory and Household Water Biofilm Isolates of “Mycobacterium avium subsp. hominissuis” with Establishment of a PCR Database. Journal of clinical microbiology. 2016. April;54(4):891–901. Epub 2016/01/08. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Parkins MD, Floto RA. Emerging bacterial pathogens and changing concepts of bacterial pathogenesis in cystic fibrosis. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society. 2015. May;14(3):293–304. Epub 2015/04/18. eng. [DOI] [PubMed] [Google Scholar]
- 115.Obregon-Henao A, Arnett KA, Henao-Tamayo M, et al. Susceptibility of Mycobacterium abscessus to antimycobacterial drugs in preclinical models. Antimicrob Agents Chemother. 2015. November;59(11):6904–12. Epub 2015/08/26. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sniezek PJ, Graham BS, Busch HB, et al. Rapidly growing mycobacterial infections after pedicures. Archives of dermatology. 2003. May;139(5):629–34. Epub 2003/05/21. eng. [DOI] [PubMed] [Google Scholar]
- 117.Lutzky VP, Ratnatunga CN, Smith DJ, et al. Anomalies in T Cell Function Are Associated With Individuals at Risk of Mycobacterium abscessus Complex Infection. Frontiers in immunology. 2018;9:1319. Epub 2018/06/27. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Avendano-Ortiz J, Llanos-Gonzalez E, Toledano V, et al. Pseudomonas aeruginosa colonization causes PD-L1 overexpression on monocytes, impairing the adaptive immune response in patients with cystic fibrosis. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society. 2018. November 12. Epub 2018/11/18. eng. [DOI] [PubMed] [Google Scholar]
- 119.Hector A, Schafer H, Poschel S, et al. Regulatory T-cell impairment in cystic fibrosis patients with chronic pseudomonas infection. American journal of respiratory and critical care medicine. 2015. April 15;191(8):914–23. Epub 2015/01/31. eng. [DOI] [PubMed] [Google Scholar]
- 120.Ingersoll SA, Laval J, Forrest OA, et al. Mature cystic fibrosis airway neutrophils suppress T cell function: evidence for a role of arginase 1 but not programmed death-ligand 1. Journal of immunology (Baltimore, Md : 1950). 2015. June 1;194(11):5520–8. Epub 2015/05/01. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]