

Abstract
APOL1 kidney risk variants (RVs) were identified in 2010 as major drivers of glomerular, tubulointerstitial and renal microvascular disease in individuals with sub-Saharan African ancestry. In December 2020, the “APOL1 at Ten” conference summarized the first decade of progress and discussed controversies and uncertainties that remain to be addressed. Topics included trypanosome infection and its role in the evolution of APOL1 kidney RVs, clinical phenotypes in APOL1-associated nephropathy, relationships between APOL1 RVs and background haplotypes on cell injury and molecular mechanisms initiating disease, the role of clinical APOL1 genotyping, and development of novel therapies for kidney disease. Future goals were defined, including improved characterization of various APOL1 RV phenotypes in patients and experimental pre-clinical models; further dissection of APOL1-mediated pathways to cellular injury and dysfunction in kidney (and other) cells; clarification of gene-gene and gene-environment interactions; and evaluation of the role for existing and novel therapies.
Keywords: APOL1, African Americans, chronic kidney disease, Trypanosomiasis, glomerulosclerosis, apolipoprotein L1
Trypanosomes, evolutionary history
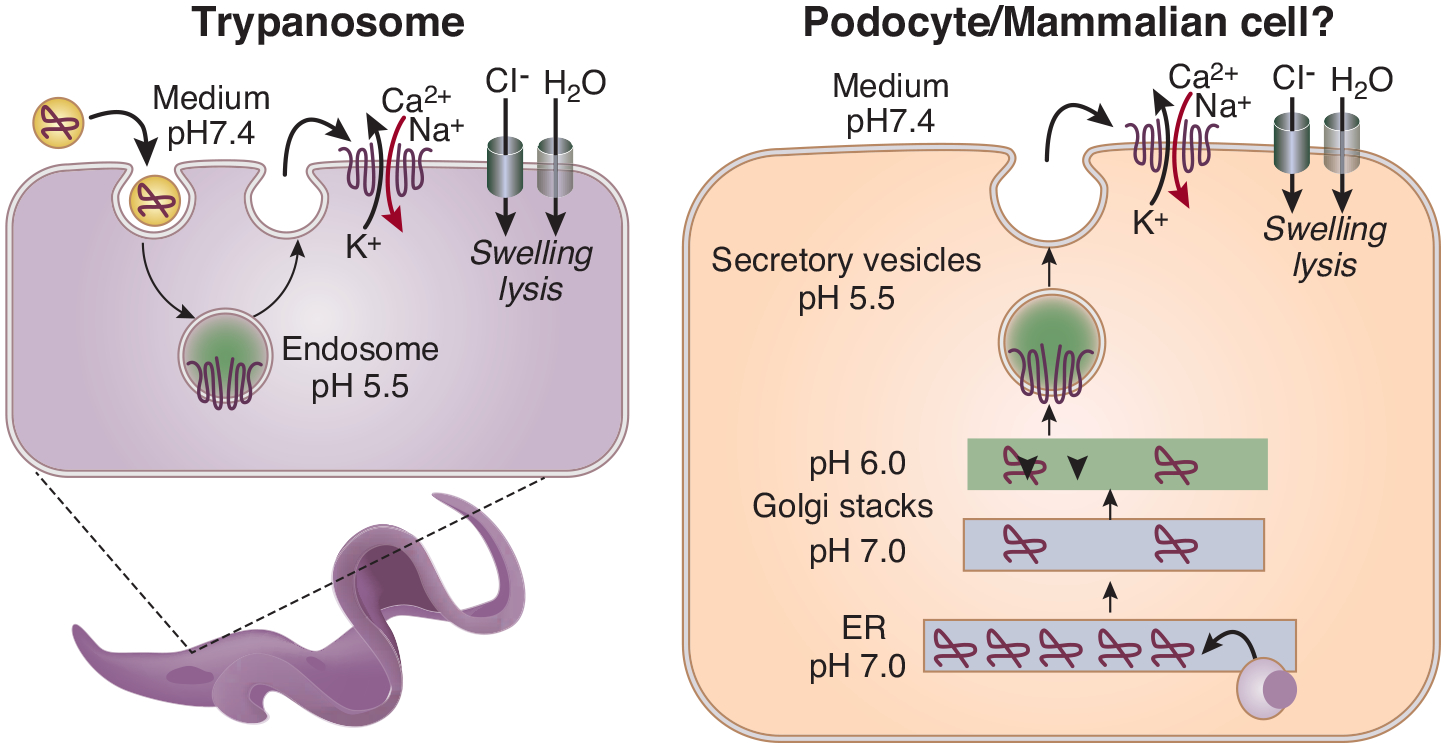
Trypanosoma are unicellular parasitic protozoa. Trypanosoma brucei ssp. causes potentially fatal human sleeping sickness as they move between the mammalian host and the tsetse fly. Human blood contains trypanolytic factors (TLF) comprised of haptoglobin related protein, apolipoprotein L1 (APOL1), APOA-1 and Immunoglobulin M1. APOL1 is the main lethal factor in the TLF. After endocytosis of the TLF by trypanosomes, APOL1 is activated by a two-step process: activation at acidic pH followed by channel opening at neutral pH2. APOL1 protein then creates cation selective channels in membranes leading to depolarization, a continuous influx of sodium and calcium, followed by chloride and subsequent osmotic swelling of the cell and internal organelles (Figure1).
Figure1.
APOL1 forms cytotoxic cation channels in a pH-dependent manner
These effects of APOL1 are inhibited in human-infective T.b. rhodesiense3 and T.b. gambiense4, 5, each through a different resistance mechanism. This leads susceptible individuals to suffer from sleeping sickness, an acute infective form more common in East Africa caused by T.b. rhodesiense, or an indolent form more common in West Africa caused by T.b. gambiense. In turn, APOL1 G1 and G2 RVs modify susceptibility to African sleeping sickness. The G2 allele, even in the heterozygous state, confers protection from infection by T.b. rhodesiense, while G1 reduces severity of illness due to T.b. gambiense. However, homozygosity and compound heterozygosity (high risk genotypes: HR) increase susceptibility to nephropathy. The recent H3Africa study showed that the highest G1 frequencies were in West Africa, such as in the Côte d’Ivoire (43%) and the Fon population in Benin (34%)6. This is consistent with geographical correlation between the prevalence of APOL1 RVs and different forms of sleeping sickness. Interestingly, the frequency of G1 was markedly different between the Bantu vs Nilo-Sharan speaking Ugandans (14 vs 2%), in contrast to similar frequencies of G2 in these groups6?.
In summary, the only currently known biological function of APOL1 is conferring protection against trypanosomiasis. To increase protection from a potentially deadly disease, the allele frequency RV APOL1 rapidly increased in Africa7. However, the newly evolved APOL1 RVs in homozygous form is associated with an increased long-term risk of kidney disease. Future studies should examine the role of the full spectrum of observed coding variants in APOL1 and their role in trypanosomiasis and kidney disease.
Clinical phenotypes
Familial aggregation of ESKD in African American families suggested an inherited basis for progressive nephropathy nearly 30 years ago8–10. This concept was validated with the discovery of APOL1 association with a spectrum of non-diabetic forms of glomerulosclerosis in individuals with recent African ancestry11, 12, including focal segmental glomerulosclerosis or FSGS (all varieties), collapsing glomerulopathy (collapsing FSGS), and the syndrome of solidified or diffuse glomerulosclerosis with low level proteinuria (often mis-labelled arterionephrosclerosis or hypertensive nephropathy)13–15. As shown in Table 1, kidney disease associated with sickle cell disease, systemic lupus erythematosus, and allograft failure in kidneys transplanted from donors with APOL1 HR genotypes also reside in the APOL1-associated nephropathy spectrum16–19.
Table 1.
APOL1-associated disease spectrum
| Diagnosis | Manifestations, risk conditions, diagnosis | APOL1 association | Odds ratio |
|---|---|---|---|
| Primary FSGS | Nephrotic proteinuria, low serum albumin | Yes | 17 |
| Post-adaptive FSGS | Risk: Low birth weight, prematurity, increased body size Feature: normal serum albumin |
Yes | ND |
| APOL1 FSGS | May mimic other forms | Yes | See other |
| Virus-associated FSGS | HIV-1, cytomegalovirus, Epstein—Barr virus, Sars-CoV-2, others | Yes: HIV-1 | 29 (USA), 89 (South Africa) |
| Drug-associated FSGS | Many medications | Interferon | ND |
| Hypertension-attributed ESKD or presumed arterionephrosclerosis | Hypertension, sub-nephrotic proteinuria and progressive loss of eGFR | Yes | 7 |
| Sickle cell nephropathy | Proteinuria, progressive renal disease | Yes | 2.7–5.4 |
| Pre-eclampsia | Hypertension, proteinuria | Yes | 1.8, 1.9 |
One end of the disease spectrum, which shows the strongest association with APOL1 RV is collapsing glomerulopathy. It is seen with extremely high interferon levels, including interferon administration, HIV infection (HIV-associated nephropathy, HIVAN), systemic lupus, and SARS-CoV-2 infection (COVID-19 associated nephropathy, COVAN)20, 21. This fits the concept that interferon drives APOL1 transcription, resulting in abundant APOL1 RV protein in kidney cells. It is possible that massive immune system activation related to SARS-CoV-2 may occasionally cause nephropathy in individuals with G1G0 genotype, as reported with HIVAN on the African continent and a recipient of a G1G0 deceased donor kidney22.
While kidney disease is the major manifestation of RV APOL1, they may also be associated with other phenotypes, such as sepsis and pre-eclampsia23, 24. In three independent studies, the high-risk genotype was associated with increased odds of pre-eclampsia in African-Americans (odds ratio [OR] 1.4 to 3.3), two under a recessive model and one under a dominant model23, 25, 26. In each study, risk was conferred by the genotype of the fetus. The mother’s country of origin (USA vs Haiti) and discordance between maternal and fetal APOL1 genotype seemed to modify the association. In two independent studies of black patients with nephrotic syndrome, a high risk (HR) APOL1 genotype was associated with increased odds of having been born preterm (OR 2.7, 9.6)27, 28. Finally, ~50% of black children with nephrotic syndrome or proteinuric kidney disease in the Nephrotic Syndrome Study Network (NEPTUNE) and Chronic Kidney Disease in Children (CKiD) had an APOL1 HR genotype and these children had a significantly lower estimated glomerular filtration rate (eGFR) at presentation and more rapid decline of eGFR27.
The phenotypic heterogeneity associated with APOL1 HR genotypes is poorly understood and future studies shall focus on deeper mechanistic understanding. Risk conferred by APOL1 in non-renal diseases appear much smaller than for collapsing FSGS, a role for APOL1 RVs in cardiovascular disease and hypertension independent of kidney disease has not yet been established24, 29, 30. APOL1-associated nephropathy is an autosomal recessive disorder with incomplete penetrance. While, one APOL1 RV is sufficient to protect from trypanosomiasis, increased disease risk is predominantly observed in people carrying 2 RVs.
APOL1-induced cell injury in mammalian cells
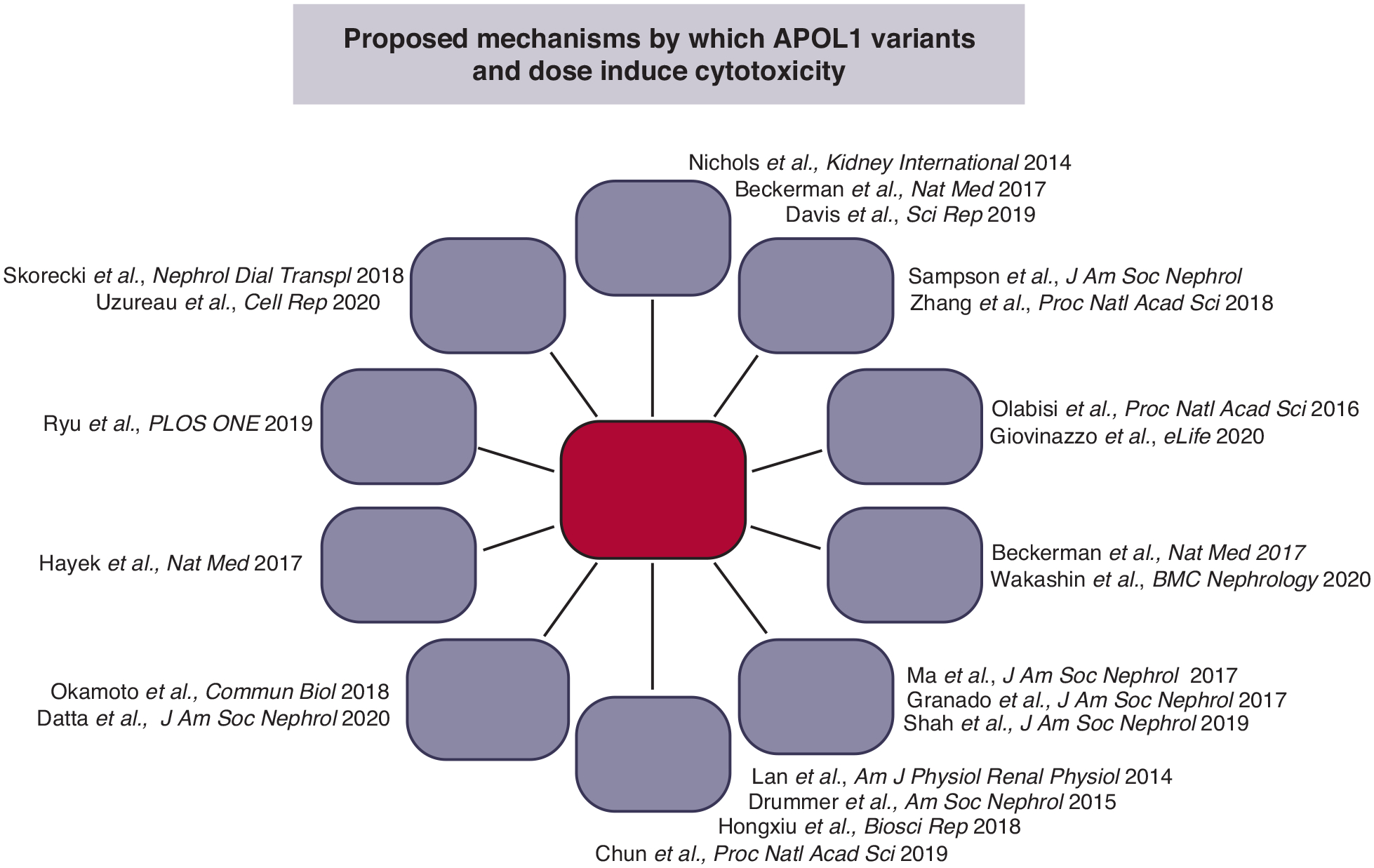
APOL1-induced trypanosome killing has been studied for decades. However, APOL1-induced toxicity in mammalian cells remains poorly understood. Drs. Pollak, Olabisi, Scales, Raper and Susztak addressed aspects of cell injury5, 28, 31 and molecular mechanisms32, 33 using cell culture and animal models34 (see Figure 2).
Figure 2.
Proposed mechanisms of APOL1-mediated cytotoxicity
Scales emphasized the critical role of APOL1 haplotypes for G1 and G2-mediated cytotoxicity in cultured podocytes stably expressing APOL1 RVs35, 36. Specifically, the podocytotoxicity was dependent on the African APOL1 haplotype (E150/I228/K255), not simply the specific RV, even though there were no differences in localization or expression level between haplotypes37. She demonstrated that critical thresholds of expression at the plasma membrane must be surpassed to achieve cytotoxicity. African G1/G2 APOL1 exits the endoplasmic reticulum and reaches the cell surface to cause toxicity (in agreement with Giovinazzo et al.38), as indicated by the rescue effect of the ER-Golgi transport inhibitor Brefeldin A. Additionally, N-terminal splice variants of APOL1 lacking a fully functional signal sequence (isoforms B3 and C), which localize to the cytoplasmic face of the ER more than plasma membranes35, 36, 39, were non-toxic40. She emphasized that future publications should report the full sequence of the APOL1 variants used for in vitro and in vivo studies.
In a talk entitled “Re-envisioning the APOL1 cation channel,” Raper showed interesting results produced by studying the channel function of APOL1 reconstituted in an in vitro bilayer system4, 38. Her team showed a population of active channels at the plasma membrane, resulting in an influx of Na+ and Ca2+ ions in human cells expressing APOL1 RVs. Interestingly, wild type G0 and RV APOL1 did not differ in ion-conducting properties but manifested differences in pH gating and membrane insertion38. All variants of APOL1 traffic to the plasma membrane, en route they encounter acidification and neutralization along the secretory pathway, steps required for channel formation. However, while G1 and G2 are able to form cation channels when overexpressed, G0 does not. They hypothesize that G1 and G2 are activated at higher pH than G0, leading to channel formation (Figure 1). She suggested a model wherein APOL1-influenced channel activity is the upstream event causing cell death. Of particular importance, she also emphasized a potential public health issue due to the rapid evolution of Trypanosomal species in Africa in considering therapies which may only partially inhibit APOL1. These could lead to emergence of resistance mechanisms in parasites.
Susztak emphasized that conditional and inducible expression of G1 or G2 variants in mouse podocytes41 induced proteinuria, foot process effacement, global glomerulosclerosis and tubulointerstitial fibrosis; changes observed in patients with APOL1 HR genotypes42, 43. These studies firmly established that APOL1 RVs are disease-causative. Furthermore, gene expression analysis by RNA sequencing of tissue from mouse models with podocyte-specific RV expression showed striking similarities to those in patients with APOL1-associated FSGS. Disease severity in mice correlated with APOL1 genotype and total APOL1 expression. Her team observed differences in intracellular trafficking of APOL1, including a defect in acidification of endocytic vesicles causing a defect in autophagy and downstream activation of the inflammasome40, 44, 45.
Pollak noted that the G1 and G2 alleles are associated with different phenotypes in mice and humans, while G0 is generally less toxic in most but not all experimental systems46. Higher APOL1 RV expression levels associate with increasing toxicity in humans and experimental models. In most cases, two RVs are required for toxicity37, 47. The requirement for two RVs to cause disease and the recessive but apparent gain-of-function nature of the two RVs is poorly understood but may reflect a gene dosage effect, wherein by more APOL1 risk protein is made when two risk alleles are present.
Olabisi extended discussion of the two-RV requirement and the role of increased gene expression driving toxicity46, 48. He concluded that carriage of the RVs and their dose contribute to toxicity in vitro, with no evidence for a dominant protective effect of G0 on G1/G2-induced toxicity in HEK-293 cells. In addition, in vitro studies support that podocyte toxicity is associated with a mitochondrial defect49–51. This mitochondrial defect might mimic that observed in trypanosomes52.
While the exact molecular mechanism is not fully understood, several key themes emerged, such as the increased toxicity of RVs compared to reference G0, the dose-dependency of the toxicity, the critical role of background local haplotype and intracellular trafficking that together culminate into APOL1 channel activity and likely activation of multiple downstream pathways. In particular, we need to understand which injury mechanisms are important in which cells and under what conditions. There is the possibility that therapy directed against one or more injury pathways may slow, halt or reverse APOL1-associated nephropathy.
Biomarkers for predicting kidney disease
Although APOL1 RVs are major drivers of disease, only 15–20% of people with HR genotypes develop nephropathy that typically includes proteinuria. Of these, a small subset develop nephrotic syndrome, but most people with nephropathy lack heavy proteinuria. Thus, detection of sub-nephrotic proteinuria, possibly including microalbuminuria, and modest reductions in eGFR may be early biomarkers for intervention53.
Model system studies established the key role of inflammation, especially viral infection and interferon, in precipitating disease. To improve risk stratification among APOL1 HR individuals, relationships between APOL1 and biomarkers of immune system activation and kidney injury have been studied. The growing list of biomarkers now includes soluble urokinase-type plasminogen activator receptor (suPAR), tumor necrosis factor receptor 1 (TNFR1), TNFR2 and kidney injury molecule-1 (KIM1). Inflammation is associated with an increase in APOL1 expression triggering nephropathy in APOL1 HR genotype individuals54.
Inflammation and immune system activation increase plasma suPAR levels. Reiser et al. reported that a tripartite complex containing APOL1 protein, suPAR and integrin avb3 forms on the cell surface of podocytes. This complex can lead to cellular activation with podocyte detachment from the glomerular basement membrane, podocytopenia, proteinuria and glomerulosclerosis55. Higher plasma suPAR levels are associated with development of CKD in African Americans from the Emory Cardiovascular Biobank and African American Study of Kidney Disease and Hypertension (AASK). The authors proposed an APOL1-environment interaction that could cause CKD in the setting of high suPAR levels related to inflammation or immune activation.
Nadkarni and colleagues demonstrated that plasma levels of kidney injury marker 1 (KIM1), tumor necrosis factor receptor (TNFR)1, and TNFR2 levels helped stratify individuals with APOL1 HR genotypes at risk for nephropathy56. Among 498 HR APOL1 genotype participants in the BioMe Biobank Program in the Mount Sinai Health System 80 developed ESKD or sustained 40% decline in eGFR after 5.9 years. Baseline TNFR1, TNFR2, and KIM1 concentrations were independently associated with these composite renal outcomes and inclusion of these biomarkers significantly improved prediction. When the concentration of all three biomarker were elevated, the event rate was 40%, compared to 17% in participants who had an increase in only one or two biomarkers, and 7% with zero biomarkers. Machine learning algorithms incorporating electronic health records and biomarkers identified individuals with APOL1 HR genotypes at risk for nephropathy progression.
The absence of JC viruria presumably reflects a heightened immune response that prevented or eradicated viral infection; this heightened immune response could also be associated with immunologically driven APOL1 expression. For example, colonization of the kidney and lower urinary tract with JC polyoma virus occurs in ~30% of the general population. Individuals with JC polyoma viruria and APOL1 HR genotypes appear less likely to develop nephropathy compared to those lacking JC viruria. The heightened immune response in virus-free individuals presumably prevented or eradicated viral infection57. This finding extends to non-APOL1-associated forms of CKD in African Americans, supporting a role for immune system activation in nephropathy historically attributed to hypertension or hyperglycemia58.
Select environmental exposures may induce a more fulminant form of nephropathy in those with an APOL1 HR genotype. These include interferon administration and HIV-1 and SARS-CoV-2 infections triggering intense immune responses including interferon release20, 59. Air pollution, fine particulate matter <2.5 μm (PM2.5), might also be an environmental trigger for APOL1-associated nephropathy60. PM2.5 and APOL1 were independently associated with nephropathy in individuals with recent African ancestry in New York. However, the effect of PM2.5 was stronger in individuals with an APOL1 HR genotype. If replicated, air pollution could prove to be an environmental factor that accentuates CKD risk. The mechanisms whereby exposure to air pollution increases risk are unclear. In contrast to these environmental exposures, major APOL1-second gene interactions have yet to be identified, although genetic modifiers with low allele frequencies likely exist61.
Role of APOL1 genotyping
The role of APOL1 genotyping in kidney transplantation and nephrology remains controversial as the majority of individuals with HR genotypes will not develop CKD. Genetic risk may have a negative psychological impact on some people specially because effective therapies are yet to come. However, novel therapies are on the horizon and instances exist where genotyping may prove helpful62, 63. For example, APOL1 genotyping in those with recent African ancestry may improve compliance in patients with HIV infection or lupus at risk for nephropathy, guide physicians prior to administering interferon, provide a more definitive diagnosis/etiology in patients with FSGS or bland chronic kidney injury, detect non-diabetic nephropathy in individuals with diabetes mellitus, and assist with family planning. These require formal study, although prospective controlled trials involving provision of genetic information to subsets of participants will be challenging. APOL1 genotyping is required to determine eligibility for treatment trials using small molecule inhibitors and anti-sense oligonucleotide therapy and fortunately more such trials are likely. Widespread genotyping will likely only occur once effective therapies are available. Many transplant programs have incorporated genotyping in the living donor work-up and the APOL1 Long-term Kidney Transplant Outcomes (APOLLO) study is assessing transplant outcomes based on deceased and living kidney donor genotypes63.
Current quandaries, therapeutics and salient questions for future research
Although there was strong consensus on many key issues, several important topics require further investigation. One such issue is whether or not there are differences between G1 and G2 variants. While initial studies indicated similar trypanolytic activities for the APOL1 G1 and G2 variants, recent studies indicate differences in SRA binding by the G1 and G2 variants that lead to differences in protection against T.b. rhodesiense and T.b. gambiense. This is in line with recently observed differences in allele frequencies in the H3Africa Study. Differences in development of kidney disease have also been observed. For example, individuals with heterozygous G1 allele showed increased incidence of ESKD, FSGS and HIVAN compared to G0/G0 individuals, and incidence was also higher in G1 risk allele subjects compared to G2. G1 was associated with younger ESKD age, a finding not present for G2 subjects. Future studies shall focus on understanding differences between G1 and G2 alleles, local background haplotype structure and a variety of other coding variants in APOL1.
APOL1-conferred protection from trypanosomiasis is due to its ion channel function. Mechanistic studies indicate that APOL1 can also function as an ion channel in mammalian cells (Figure 1). Channel activation patterns and differences between risk and reference allele are poorly understood. APOL1 undergoes complex intracellular trafficking, where changes in intracellular pH appear to be important for membrane insertion and channel function. In vitro reprogrammed and CRISPR-edited podocytes appear to be a promising new model system to understand these processes. Cell-type specific conditional inducible RV APOL1 transgenic mice or BAC transgenic animals have proven to recapitulate aspects of human disease and should be important for preclinical analyses. Ongoing clinical studies are targeting either the APOL1 channel function or the APOL1 dose, consistent with molecular studies.
Other critical issues include who should be treated and when should treatment start. Detection of biomarkers prospectively identifying high risk individuals remain critical for development of therapeutics. Early studies target the highest risk group, such as those with FSGS and heavy proteinuria. Another key question is whether the disease is still driven by APOL1 dose or channel activity at this stage. Furthermore, while these therapeutic modalities might be practical in areas where there is little risk for trypanosomiasis, it will be critical to understand their effect on parasitic infection. Finally, it will be important to determine whether patients with APOL1-associated nephropathy respond to SGLT2 inhibitors, a drug class with protective effects on kidney and cardiovascular disease.
Conclusions
Remarkable progress has been made in the past 10 years, since identification of APOL1 as the gene responsible for much of the increased risk of CKD among African Americans. We have a better understanding of the global distribution of populations affected by APOL1-associated nephropathy, clinical manifestations of disease and molecular pathways contributing to dysfunction in cultured cells, animal models, human cells, and tissues. Most exciting, new therapies are being developed and one is in a phase II trial stage.
The first ten years of this emerging field were characterized by productive collaborations across an array of disciplines, including molecular biology, protein chemistry, cell biology, animal models, pharmaceutical development, clinical research, and sociology. This comes at a time of renewed hope for preventing or slowing CKD progression in the clinic. Above all, the perspectives of patients, families, and communities was sought, considered, and acted upon in manuscripts and multi-disciplinary discussions. These interactions engendered efforts to communicate research advances and therapeutic options to the African American community and empower community members to craft appropriate messages and serve as messengers. Together, we believe novel therapies will emerge that change the narrative for many individuals currently destined to require renal replacement therapy leading to lives with preserved kidney function.
Acknowledgement.
BIF is supported by NIH grants R01 DK070941, R01 DK084149, R01 MD009055, and U01 DK116041. MS is supported by NIH RO1 DK108805, DK119380, and RC2 DK122397. KS is supported by NIH R01 DK105821, DK087635 and DK076077.
Disclosures:
BIF and Wake Forest University Health Sciences have rights to a US patent involving APOL1 genotyping. BIF is a consultant for AstraZeneca and RenalytixAI and he receives research support from AstraZeneca. MS is a consultant for Janssen and Maze and is on the advisory board of Natera. KS is a consultant for Bayer, Astra Zeneca, Jnana, Maze biotech, on the advisory board of Jnana and received research support from Bayer, Novartis, Gilead, Merck, Boehringer Ingelheim, GSK and Regeneron.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Lugli EB, Pouliot M, Portela Mdel P, et al. Characterization of primate trypanosome lytic factors. Mol Biochem Parasitol 2004; 138: 9–20. [DOI] [PubMed] [Google Scholar]
- 2.Thomson R, Finkelstein A. Human trypanolytic factor APOL1 forms pH-gated cation-selective channels in planar lipid bilayers: relevance to trypanosome lysis. Proc Natl Acad Sci U S A 2015; 112: 2894–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perez-Morga D, Vanhollebeke B, Paturiaux-Hanocq F, et al. Apolipoprotein L-I promotes trypanosome lysis by forming pores in lysosomal membranes. Science 2005; 309: 469–472. [DOI] [PubMed] [Google Scholar]
- 4.Cooper A, Capewell P, Clucas C, et al. A Primate APOL1 Variant That Kills Trypanosoma brucei gambiense. PLoS Negl Trop Dis 2016; 10: e0004903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uzureau S, Lecordier L, Uzureau P, et al. APOL1 C-Terminal Variants May Trigger Kidney Disease through Interference with APOL3 Control of Actomyosin. Cell Rep 2020; 30: 3821–3836 e3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choudhury A, Aron S, Botigue LR, et al. High-depth African genomes inform human migration and health. Nature 2020; 586: 741–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ko WY, Rajan P, Gomez F, et al. Identifying Darwinian selection acting on different human APOL1 variants among diverse African populations. American journal of human genetics 2013; 93: 54–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Freedman BI. Renal microvascular susceptibility in African American pedigrees. Transplant Proc 1993; 25: 2423–2425. [PubMed] [Google Scholar]
- 9.Freedman BI, Spray BJ, Tuttle AB, et al. The familial risk of end-stage renal disease in African Americans. Am J Kidney Dis 1993; 21: 387–393. [DOI] [PubMed] [Google Scholar]
- 10.Freedman BI, Iskandar SS, Appel RG. The link between hypertension and nephrosclerosis. Am J Kidney Dis 1995; 25: 207–221. [DOI] [PubMed] [Google Scholar]
- 11.Genovese G, Friedman DJ, Ross MD, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 2010; 329: 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tzur S, Rosset S, Shemer R, et al. Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Hum Genet 2010; 128: 345–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freedman BI, Kopp JB, Langefeld CD, et al. The apolipoprotein L1 (APOL1) gene and nondiabetic nephropathy in African Americans. Journal of the American Society of Nephrology : JASN 2010; 21: 1422–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nicholas Cossey L, Larsen CP, Liapis H. Collapsing glomerulopathy: a 30-year perspective and single, large center experience. Clin Kidney J 2017; 10: 443–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Freedman BI, Cohen AH. Hypertension-attributed nephropathy: what’s in a name? Nat Rev Nephrol 2016; 12: 27–36. [DOI] [PubMed] [Google Scholar]
- 16.Larsen CP, Beggs ML, Saeed M, et al. Apolipoprotein L1 risk variants associate with systemic lupus erythematosus-associated collapsing glomerulopathy. J Am Soc Nephrol 2013; 24: 722–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ashley-Koch AE, Okocha EC, Garrett ME, et al. MYH9 and APOL1 are both associated with sickle cell disease nephropathy. Br J Haematol 2011; 155: 386–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Freedman BI, Langefeld CD, Andringa KK, et al. End-stage renal disease in African Americans with lupus nephritis is associated with APOL1. Arthritis Rheumatol 2014; 66: 390–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Freedman BI, Locke JE, Reeves-Daniel AM, et al. Apolipoprotein L1 Gene Effects on Kidney Transplantation. Semin Nephrol 2017; 37: 530–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu H, Larsen CP, Hernandez-Arroyo CF, et al. AKI and Collapsing Glomerulopathy Associated with COVID-19 and APOL 1 High-Risk Genotype. J Am Soc Nephrol 2020; 31: 1688–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Velez JCQ, Caza T, Larsen CP. COVAN is the new HIVAN: the re-emergence of collapsing glomerulopathy with COVID-19. Nat Rev Nephrol 2020; 16: 565–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kasembeli AN, Duarte R, Ramsay M, et al. APOL1 Risk Variants Are Strongly Associated with HIV-Associated Nephropathy in Black South Africans. J Am Soc Nephrol 2015; 26: 2882–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reidy KJ, Hjorten RC, Simpson CL, et al. Fetal-Not Maternal-APOL1 Genotype Associated with Risk for Preeclampsia in Those with African Ancestry. Am J Hum Genet 2018; 103: 367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bajaj A, Ihegword A, Qiu C, et al. Phenome-wide association analysis suggests the APOL1 linked disease spectrum primarily drives kidney-specific pathways. Kidney Int 2020; 97: 1032–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hong X, Rosenberg AZ, Zhang B, et al. Joint Associations of Maternal-Fetal APOL1 Genotypes and Maternal Country of Origin With Preeclampsia Risk. Am J Kidney Dis 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller AK, Azhibekov T, O’Toole JF, et al. Association of preeclampsia with infant APOL1 genotype in African Americans. BMC Med Genet 2020; 21: 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ng DK, Robertson CC, Woroniecki RP, et al. APOL1-associated glomerular disease among African-American children: a collaboration of the Chronic Kidney Disease in Children (CKiD) and Nephrotic Syndrome Study Network (NEPTUNE) cohorts. Nephrol Dial Transplant 2017; 32: 983–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sampson MG, Robertson CC, Martini S, et al. Integrative Genomics Identifies Novel Associations with APOL1 Risk Genotypes in Black NEPTUNE Subjects. J Am Soc Nephrol 2016; 27: 814–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grams ME, Surapaneni A, Ballew SH, et al. APOL1 Kidney Risk Variants and Cardiovascular Disease: An Individual Participant Data Meta-Analysis. J Am Soc Nephrol 2019; 30: 2027–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bick AG, Akwo E, Robinson-Cohen C, et al. Association of APOL1 Risk Alleles With Cardiovascular Disease in Blacks in the Million Veteran Program. Circulation 2019; 140: 1031–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Skorecki KL, Lee JH, Langefeld CD, et al. A null variant in the apolipoprotein L3 gene is associated with non-diabetic nephropathy. Nephrol Dial Transplant 2018; 33: 323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ryu JH, Ge M, Merscher S, et al. APOL1 renal risk variants promote cholesterol accumulation in tissues and cultured macrophages from APOL1 transgenic mice. PLoS One 2019; 14: e0211559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Okamoto K, Rausch JW, Wakashin H, et al. APOL1 risk allele RNA contributes to renal toxicity by activating protein kinase R. Commun Biol 2018; 1: 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang JY, Wang M, Tian L, et al. UBD modifies APOL1-induced kidney disease risk. Proc Natl Acad Sci U S A 2018; 115: 3446–3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gupta N, Wang X, Wen X, et al. Domain-Specific Antibodies Reveal Differences in the Membrane Topologies of Apolipoprotein L1 in Serum and Podocytes. J Am Soc Nephrol 2020; 31: 2065–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scales SJ, Gupta N, De Maziere AM, et al. Apolipoprotein L1-Specific Antibodies Detect Endogenous APOL1 inside the Endoplasmic Reticulum and on the Plasma Membrane of Podocytes. J Am Soc Nephrol 2020; 31: 2044–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lannon H, Shah SS, Dias L, et al. Apolipoprotein L1 (APOL1) risk variant toxicity depends on the haplotype background. Kidney Int 2019; 96: 1303–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Giovinazzo JA, Thomson RP, Khalizova N, et al. Apolipoprotein L-1 renal risk variants form active channels at the plasma membrane driving cytotoxicity. Elife 2020; 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dummer PD, Limou S, Rosenberg AZ, et al. APOL1 Kidney Disease Risk Variants: An Evolving Landscape. Semin Nephrol 2015; 35: 222–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lan X, Jhaveri A, Cheng K, et al. APOL1 risk variants enhance podocyte necrosis through compromising lysosomal membrane permeability. Am J Physiol Renal Physiol 2014; 307: F326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kopp JB, Anders HJ, Susztak K, et al. Podocytopathies. Nat Rev Dis Primers 2020; 6: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beckerman P, Bi-Karchin J, Park AS, et al. Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat Med 2017; 23: 429–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beckerman P, Susztak K. APOL1: The Balance Imposed by Infection, Selection, and Kidney Disease. Trends Mol Med 2018; 24: 682–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Davis SE, Khatua AK, Popik W. Nucleosomal dsDNA Stimulates APOL1 Expression in Human Cultured Podocytes by Activating the cGAS/IFI16-STING Signaling Pathway. Sci Rep 2019; 9: 15485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wakashin H, Heymann J, Roshanravan H, et al. APOL1 renal risk variants exacerbate podocyte injury by increasing inflammatory stress. BMC Nephrol 2020; 21: 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Datta S, Kataria R, Zhang JY, et al. Kidney Disease-Associated APOL1 Variants Have Dose-Dependent, Dominant Toxic Gain-of-Function. J Am Soc Nephrol 2020; 31: 2083–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chun J, Zhang JY, Wilkins MS, et al. Recruitment of APOL1 kidney disease risk variants to lipid droplets attenuates cell toxicity. Proc Natl Acad Sci U S A 2019; 116: 3712–3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Olabisi OA, Heneghan JF. APOL1 Nephrotoxicity: What Does Ion Transport Have to Do With It? Semin Nephrol 2017; 37: 546–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Granado D, Muller D, Krausel V, et al. Intracellular APOL1 Risk Variants Cause Cytotoxicity Accompanied by Energy Depletion. J Am Soc Nephrol 2017; 28: 3227–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shah SS, Lannon H, Dias L, et al. APOL1 Kidney Risk Variants Induce Cell Death via Mitochondrial Translocation and Opening of the Mitochondrial Permeability Transition Pore. J Am Soc Nephrol 2019; 30: 2355–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ma L, Chou JW, Snipes JA, et al. APOL1 Renal-Risk Variants Induce Mitochondrial Dysfunction. J Am Soc Nephrol 2017; 28: 1093–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Olabisi OA, Zhang JY, VerPlank L, et al. APOL1 kidney disease risk variants cause cytotoxicity by depleting cellular potassium and inducing stress-activated protein kinases. Proc Natl Acad Sci U S A 2016; 113: 830–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Freedman BI, Limou S, Ma L, et al. APOL1-Associated Nephropathy: A Key Contributor to Racial Disparities in CKD. Am J Kidney Dis 2018; 72: S8–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nichols B, Jog P, Lee JH, et al. Innate immunity pathways regulate the nephropathy gene Apolipoprotein L1. Kidney Int 2015; 87: 332–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hayek SS, Koh KH, Grams ME, et al. A tripartite complex of suPAR, APOL1 risk variants and alphavbeta3 integrin on podocytes mediates chronic kidney disease. Nat Med 2017; 23: 945–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nadkarni GN, Chauhan K, Verghese DA, et al. Plasma biomarkers are associated with renal outcomes in individuals with APOL1 risk variants. Kidney Int 2018; 93: 1409–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Divers J, Nunez M, High KP, et al. JC polyoma virus interacts with APOL1 in African Americans with nondiabetic nephropathy. Kidney international 2013; 84: 1207–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kruzel-Davila E, Divers J, Russell GB, et al. JC Viruria Is Associated With Reduced Risk of Diabetic Kidney Disease. J Clin Endocrinol Metab 2019; 104: 2286–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kopp JB, Nelson GW, Sampath K, et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. Journal of the American Society of Nephrology : JASN 2011; 22: 2129–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Paranjpe I, Chaudhary K, Paranjpe M, et al. Association of APOL1 Risk Genotype and Air Pollution for Kidney Disease. Clin J Am Soc Nephrol 2020; 15: 401–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Langefeld CD, Comeau ME, Ng MCY, et al. Genome-wide association studies suggest that APOL1-environment interactions more likely trigger kidney disease in African Americans with nondiabetic nephropathy than strong APOL1-second gene interactions. Kidney Int 2018; 94: 599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kopp JB, Winkler CA. Genetic Testing for APOL1 Genetic Variants in Clinical Practice: Finally Starting to Arrive. Clin J Am Soc Nephrol 2020; 15: 126–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Freedman BI, Moxey-Mims MM, Alexander AA, et al. APOL1 Long-term Kidney Transplantation Outcomes Network (APOLLO): Design and Rationale. Kidney Int Rep 2020; 5: 278–288. [DOI] [PMC free article] [PubMed] [Google Scholar]