

Abstract
Idiopathic pulmonary fibrosis (IPF) is a debilitating and fatal condition that causes severe scarring of the lungs. While the pathogenesis of IPF continues to be extensively studied and several factors have been considered, an exact cause has yet to be established. With inadequate treatment options and no cure available, overall disease prognosis is still poor. Existing oral therapies, pirfenidone and nintedanib, may attempt to improve the patients’ quality of life by mitigating symptoms and slowing disease progression, however chronic doses and systemic deliveries of these drugs can lead to severe side effects. The lack of effective treatment options calls for further investigation of restorative as well as additional palliative therapies for IPF. Nanoparticle-based sustained drug delivery strategies can be utilized to ensure targeted delivery for site-specific treatment as well as long-acting therapy, improving overall patient compliance. This review provides an update on promising strategies for the delivery of anti-fibrotic agents, along with an overview of key therapeutic targets as well as relevant emerging therapies currently being evaluated for IPF treatment.
Keywords: Idiopathic Pulmonary Fibrosis, Nanoparticles, Pathophysiology, Formulations, Inflammatory Growth Factors, Anti-fibrotic Agents
Graphical Abstract
1. Introduction
Lung fibrosis is a chronic lung condition characterized by excessive extracellular matrix (ECM) deposition and alveolar tissue remodeling resulting in scar tissue formation, (Figure 1), leading to functional failures and complications [1]. Idiopathic pulmonary fibrosis (IPF), a progressive and chronic disorder, is the most common type of lung fibrosis with a survival rate of only 2–5 years [2]. The yearly incidence and prevalence rates in the United States have steadily increased within the last decade, reaching 494.5 cases per 100 000 persons in 2011 [2]. In the United Kingdom, there were a total of 343 million prevalent cases during 2011–2016 at a normalized mortality rate of 5.59 cases per 100 000 persons annually [3]. The trend has been progressing upward with estimated mortality rates of about 8.5 cases per 100 000 persons annually during 2017–2019 [3].
Figure 1.

Schematic of healthy vs fibrotic lung. Alveolar wall of healthy lung (left) shows thin layer of epithelial tissue, whereas in a fibrotic state, lung with IPF is characterized by thickening of alveolar epithelium and resulting scar tissue [10]. Figure created using BioRender (https://biorender.com/)
In 2011, pirfenidone received approval from the European Medicines Agency (EMA) as the first drug to be licensed for treatment of IPF in Europe [4]. In 2014, following extensive clinical trials, the FDA approved pirfenidone and nintedanib for IPF treatment [5]. Although these drugs work to suppress symptoms such as inflammation and scarring of the lungs, there remains a need for treatments that can potentially cure fibrosis, as pirfenidone and nintedanib do not halt or reverse the fibrosis established in IPF [6]. Clinical trials that have assessed the tolerability of pirfenidone and nintedanib have also shown that adverse reactions often occur during treatment [7]. According to the recommended dosing schedule when prescribing Esbriet® (pirfenidone), the full daily dose of pirfenidone is 9 pills (267 mg each, yellow tablets or white capsules) [8]. These systemic and chronic daily doses can lead to off-target toxicities and significant patient discomfort. For example, pooled data from the INPULSIS and TOMORROW studies showed that adverse events such as diarrhea was experienced in more than 60% of all patients in the nintedanib treatment group [9]. Similarly, studies have shown pirfenidone treatment leading to adverse events such as nausea, abdominal pain, and photosensitive rash [9].
Despite these reported off-target toxicities, pirfenidone and nintedanib remain the only two drugs approved for IPF treatment so far. Therefore, there is a need for effective approaches for site-specific and sustained delivery of these drugs to maximize their efficacy. An approach to increase the effectiveness and reduce the overall toxicity of drugs is to encapsulate them within nanoparticle (NP) or microparticle (MP) formulations, which can also be used to achieve sustained drug delivery. For instance, inhalable NP formulations are preferred in order to achieve localized therapy in a fast, easy and patient-compliant manner [11]. In addition, NP formulations offer the opportunity to encapsulate multiple therapeutic agents within a single formulation, for combination therapy. Evaluating combination therapies to treat IPF is an ongoing process given that there are no FDA-approved combination therapies for IPF thus far. With combination therapy, two drugs are together expected to have an additive or a synergistic effect in combating fibrosis, which cannot be achieved using a single drug. This review discusses potential targets and emerging therapies that can benefit from encapsulation within drug carriers and delivery by pulmonary route, and provides an exhaustive overview of current literature on the development of NP-based as well as MP-based formulations for anti-fibrotic drug delivery to treat IPF.
2. Pathogenesis and Therapeutic Targets
In order to identify effective therapeutic targets for the treatment of IPF, it is necessary to learn more about the various pathways that can play a key role in fibrosis. This section reviews important fibrosis mediators relevant to the IPF disease pathway as well as existing and emerging targets for treatment. These can potentially be encapsulated within advanced drug delivery formulations such as NPs, for site-specific therapy.
The alveoli within the lungs is lined by 2 major types of cells (pneumocytes), alveolar epithelial cells type 1 (AEC1, squamous cells) and type 2 (AEC2, cuboidal cells consisting of microvilli) [12]. AEC2 have the special ability to differentiate further into both AEC1 and AEC2. A layer of interstitial tissue between the AEC1/AEC2 and capillaries contain macrophages and fibroblasts [13]. Any injury to alveolar lining triggers AEC2-mediated proliferation of fibroblasts, which is further differentiated into myofibroblasts (mesenchymal cell type). Myofibroblasts are fibroblastic smooth muscle cells which secretes reticular collagen fibers that maintain the structural integrity and elastic fibers that provides elasticity to lungs. In normal conditions, following the repair process, myofibroblasts undergo apoptosis thereby restoring the normal functioning of the lungs [13]. However, in IPF, physiologic apoptosis of myofibroblasts is severely disturbed and recurrent microinjuries to alveoli leads to aberrant wound healing and progressive fibrosis (Figure 2). The imbalance between the synthesis and lysis of myofibroblasts ultimately leads to excessive collagen deposition. This results in issues with ventilation, and imbalance in the oxygen supply to the body. Furthermore, loss of elastic fibers and increase in collagen contribute to the stiffness of the lungs hampering the breathing process [14]. This has been demonstrated in progressive fibrosis, where patients demonstrate significant physiological impairment in the lungs which affects the amount of oxygenated blood in the body [15]. Although the complex etio-pathogenesis of IPF has not been completely explored, genetic influence has been found to a have role in the disease onset and progression [16]. Mutations are known to affect the genes expressed by AECs. Altered gene expression leads to generation of abnormal proteins and mediators responsible for increasing the fibrosis and inflammatory response. Polymorphism at the promoter region of MUC5B, a mucin gene, is the strongest and most replicated genetic risk factor for IPF [17][18][19]. In addition, mutations in surfactant protein genes such as SFTPC and SFTPA2, and telomerase-related genes such as TERC and TERT, are characterized in familial pulmonary fibrosis cases [20]. Another study showed that gene AKAP13 - a Rho guanine nucleotide exchange factor regulates activation of profibrotic pathways via RhoA. Identification of this specific gene can be a helpful tool for providing targeted treatment via inhibition of RhoA pathway in IPF patients [16].
Figure 2.

Schematic representation of IPF pathogenesis. Microvascular injury triggers release of inflammatory mediators, platelet activation and secretion of pro-fibrotic cytokines leading to the activation, recruitment, proliferation and differentiation of fibroblasts into myofibroblasts, resulting in the release of extracellular matrix (ECM) components and promotion of wound healing. Abnormal pulmonary wound repair and excessive formation of irreversible scar tissue leads to the development and progression of IPF. Sustained release of anti-fibrotic drugs from a NP drug platform can inhibit the fibrosis mechanism, thus slowing the progression of IPF [21][22]. Figure created using BioRender (https://biorender.com/)
Fibrotic changes seen in IPF are orchestrated by key growth factors such as transforming growth factor (TGF-β1) [23], platelet-derived growth factor (PDGF) [24], vascular endothelial growth factor (VEGF) [25] and fibroblast growth factor (FGF) [26][27][28]. These growth factors act in collaboration through multiple signaling pathways such as TGFβ1- SMAD [23], WNT-Catenin [29], sonic hedgehog [30], Notch [1] and FGF-FGFR [26]. All these pathways are known to have a key role in alveolar epithelial cells (AEC) – myofibroblast (mesenchymal cell) interaction (AMI) responsible for generation of fibrotic condition [31]. In the following sections, we have attempted to explain the role and mechanism of these signaling pathways in the pathophysiology of IPF and how they can be manipulated for drug discovery.
Transforming growth factor-β (TGF-β) is a key multifunctional biological mediator belonging to a large family of polypeptides and is presented in three isoforms TGF-β1, β2, and β3, with TGF-β1 being the most critical in the pathogenesis of IPF [32]. The main role of TGF-β1 is to modulate various cellular activities, such as proliferation, differentiation, and programmed cell death, thus maintaining the tissue homeostasis [33]. TGF-β1 is secreted in a latent form which gets activated in IPF condition. AECs in IPF express integrin that binds to TGF-β1-associated latent proteins and transforms TGF-β1 into active forms. Recently, there has been an increasing evidence supporting the association of TGF-β1 activation pathways in chronic lung diseases, including IPF. SMADs are a family of intracellular signaling proteins that play crucial role in activation of TGF-β1 [34]. TGF-β/SMAD signaling plays a significant role in differentiation of AEC2s into AECls during alveolar phase. Following tissue injury, activated TGF-β1 mediates fibroblasts differentiation to myofibroblasts stimulating formation of extracellular matrix (ECM) and inhibition of matrix metalloproteinases mediated degradation of ECM. Furthermore, these activities of TGF-β1 are closely orchestrated by fibronectin, VEGF, and PDGF [23]. In this line, a study evaluated the anti-fibrotic role of N-acetyl-l-cysteine (NAC) on mono and three-dimensional human fetal lung fibroblasts cell culture model. NAC downregulated the expression of TGF-β1 mediated pro-fibrotic players such as fibronectin, VEGF and reversed the TGF-β1-stimulated α-smooth muscle actin expression signifying its therapeutic role in IPF [35]. Fibronectin is a multifunctional glycoprotein present in ECM that serves as a potent chemoattractant for lung fibroblasts. It is released in increased levels by fibroblasts and epithelial cells on stimulation with TGF-β1 [36]. VEGF is a multifunctional growth factor which play a significant role in the survival of endothelial cells [37]. Understanding the role of these mediators in these signaling pathways have led to the development of the FDA approved drug Pirfenidone for the treatment of IPF, which is known to suppress TGF-β activation [38].
Fibroblast growth factors (FGFs) are another group of proteins known for their potential to regulate cell proliferation, survival, migration and differentiation. Fibroblast growth factor receptor (FGFR) present in the cell surface possesses tyrosine kinase activity. These receptors are important for FGF to produce signals during the repair process via various complicated pathways. A study assessing the anti-fibrotic effect of Nintedanib on clinical isolates of fibroblasts from IPF patients demonstrated that Nintedanib, a triple kinase inhibitor that block the activities of VEGF, FGF, and PDGF, enabled the decrease in the protein and mRNA levels of ECM protein, fibronectin, and collagen. Furthermore, the drug also inhibited TGF-β1 mediated myofibroblast differentiation [39]. Several studies have also demonstrated the role of FGF-2 in IPF [26]. For example, another study looking at the effect of Nintedanib observed higher levels of PDGF and FGF-2 receptors in clinically isolated fibroblasts from IPF patients than in cells from non-IPF patients (control group). Nintedanib inhibited the pro-proliferative effect caused by PDGF, FGF, and VEGF. The study also showed that Nintedanib increased the expression of pro-MMP-2 and inhibited TGF mediated collagen generation [40]. Nintedanib is now USFDA approved and is known to act by inhibition of FGFR signaling pathway to control fibrosis [38].
One emerging target for IPF treatment is the WNT5A of the signaling lipid modified glycoproteins WNT family. WNT5A signals and induces proliferation of lung fibroblasts [41], and it has been observed to be elevated in IPF fibroblasts [41][42][43]. The WNT proteins are glycoproteins known for their role in regulating matrix metalloproteinases (MMPs), cell-cycle regulators, angiogenic growth factors, promote proliferation and survival of lung fibroblasts as well as increase in ECM production. WNT signaling has a crucial role in epithelial cell proliferation, AMI, myofibroblast differentiation, and collagen synthesis. WNT/β-catenin signaling along with TGF-β has shown to produce anti-apoptotic and pro-fibrotic phenotypes in lung fibroblasts enhancing fibroblast proliferation, and myofibroblast differentiation and survival. Furthermore, activation of WNT/β-catenin pathway in AEC2 increased secretion of pro-inflammatory cytokines such as IL-1β, which subsequently lead to pro-fibrotic response. MMP-7 is activated by upregulation of WNT/β-catenin pathway facilitating breakdown of ECM and directional fibroblast movement. Overall, TGF-β and WNT interrelation is an integral part of IPF pathogenesis [29].
Among the hedgehog family, sonic hedgehog (SHH) plays a significant role in organ morphogenesis of the adult lung and keep the progenitor cells in the dormant state. Overexpression of hedgehog pathways has been associated with IPF. SHH pathway has shown to initiate myofibroblast differentiation and enhanced collagen production. In the lung, SHH increased fibroblast proliferation, survival, migration, and ECM production [30].
The role of autotaxin and lysophosphatidic acid (LPA) receptors in the pathogenesis of diseases such as IPF and lung cancer has been extensively researched in recent years. Autotaxin (ATX) is an enzyme responsible for most extracellular LPA production in the body [44]. ATX and LPA concentrations are increased in lung tissue and bronchoalveolar lavage fluid, respectively, in IPF patients [45]. LPA mediated signaling has been linked with fibroproliferative responses from pulmonary cells such as activation of TGF-β signaling and stimulation of fibroblast accumulation [44][46]. Several recent studies revealed that increased ATX/LPA activity contribute to chronic fibrosing of the lung, and in turn, inhibition of ATX can attenuate IPF [47][48].
A recent study determined that the protein Galectin-3 is upregulated in the fibrosis pathway with high levels found to be present in human fibrotic lung tissue [49]. Galectin-3 is a type of β-galactoside- binding lectins secreted by monocytes, macrophages and epithelial cells and other cell types. It mediates various cell-cell and cell-ECM interactions while contributing to regulation of biological responses, such as the inflammatory response, and has recently been shown to play a role in liver cirrhosis and IPF [50]. Upregulation of Galectin-3 has been found to activate myofibroblasts, which impacts several fibrotic pathways leading to scar formation and fibrosis [51][51]. Targeting and inhibiting Galectin-3 is therefore an attractive approach for the treatment of IPF.
Overall, in IPF, it can be said that activation of the above pathways are interconnected, and the activation of these pathways can trigger the transition from AEC to mesenchymal cells such as fibroblasts and myofibroblasts. This in turn leads to enhanced ECM and collagen production characteristic of IPF.
3. Existing and Emerging Pharmacological Therapies
As mentioned previously, pirfenidone and nintedanib, are currently the only antifibrotic agents approved for the treatment of IPF given that both drugs reduce rate of disease progression and have shown to provide stabilization of lung function test parameters in IPF patients in recent studies [38][52]. In both pre-clinical animal model research and clinical studies, these drugs have been shown to reduce the expression of collagen, TGF-β1 and alpha smooth muscle actin (α-SMA), which are elevated in fibrotic states as mentioned previously [38]. Although the exact mechanism of action is uncertain, both drugs when taken orally are capable of attenuating these mediators along with their downstream targets. Pirfenidone is an anti-inflammatory agent that inhibits TGF-β1-mediated fibroblast activity and collagen production, whereas nintedanib is a tyrosine kinase inhibitor acting primarily on the receptors for PDGF, FGF, and VEGF, ultimately reducing fibroblast proliferation and matrix deposition [53][54]. Despite the different roles they play in achieving an antifibrotic effect, a study in which pirfenidone and nintedanib were delivered over a 24 month period to IPF patients revealed that both drugs were equally effective in reducing the decline of forced vital capacity (FVC) and diffusing capacity of the lungs for carbon monoxide (DLCO) [53].
As mentioned in the previous section, following injury to the lung epithelium various proteins are involved in signaling of profibrotic mediators which in turn induce fibroproliferative responses. Therefore, IPF research is increasingly focused on developing new therapies that can potentially inhibit these proteins. A recent study demonstrated that the inhibition of Galectin-3 with novel inhibitor, TD139, in turn attenuated fibrosis [55]. This was demonstrated in a bleomycin-induced fibrotic mouse model, where Galectin-3 expression was elevated in the presence of active fibrosis, and the expression was significantly reduced following treatment with TD139 along with the reduction of fibrosis, confirming its activity in the fibrosis pathway. Not only do these results point to the potential of Galectin-3 as a therapeutic target, but also indicate that TD139 could be a promising agent for development for IPF treatment, as it is currently undergoing phase 2 clinical trials [56]. As noted earlier, ATX activity has also been linked to the progression of IPF. In previous studies, inhibition of ATX and similar enzymes via genetic deletion or pharmacological inhibition has reduced lung fibrosis [47]. Ziritaxestat (GLPG1690), an orally active potent autotaxin inhibitor, is currently in phase 3 clinical trials for treatment of IPF [46]. The lead compound, GLPG1690, demonstrated effective in vivo reduction of LPA in a bleomycin (BLM) -induced pulmonary fibrosis model in rodents [57]. To enter the clinic, the compound was structurally modified to improve pharmacokinetic properties. The clinical candidate showed robust ATX inhibition and greater efficacy when compared to the reference compound, pirfenidone, in the BLM mouse model [57]. The FLORA study was conducted to explore the effects of GLPG1690 in patients with IPF over 12 weeks. The findings of this trial showed significant increase in forced vital capacity and consistent reduction in LPA concentrations, with mild-to-moderate treatment-emergent adverse events in patients and zero mortality [58]. A phase 1 trial completed in 2019 investigated the ability of BBT-877, a small molecule inhibitor of ATX, to treat IPF [59]. This phase 1 trial was carried out with 80 healthy subjects and a variety of single ascending doses and multiple ascending doses. Safety and efficacy were analyzed based on AEs, ECG, vital signs, laboratory biochemical/hematology, and urinalysis. Overall, the trial yielded very promising results. All doses of BBT-877 were very well tolerated. Any adverse events were very mild and there were no significant changes in clinical related findings. BBT-877 is an orally available drug and is known to target a significant enzyme involved in the progression of IPF. This trial yielded promising results and phase 1 and 2 trials will continue.
Metformin, a well-established antidiabetic drug, has been showing promise in attenuating IPF in recent studies. Metformin not only exhibits significant antifibrotic effects but has been shown to reverse lung fibrosis in vitro. A study carried out using lung fibroblasts derived from IPF patients demonstrated resolution of fibrosis in groups treated concurrently with TGF-β1 and metformin along with an accumulation of lipid-droplets, downregulation of collagen-1 expression, and upregulation of PPARγ expression. These results are consistent with the researchers’ proposed anti-fibrotic mechanism of metformin, in which myofibroblasts are targeted by lipogenic differentiation induced by PPARγ signaling activation [60]. Metformin’s ability to alter myofibroblasts, the key effector cells responsible for ECM deposition, lead to inhibition of TGF-β1 signaling and suppression of collagen levels, ultimately improving lung structure and function. Another study found that activation of adenosine monophosphate-activated protein kinase (AMPK) by metformin in turn deactivates myofibroblasts and makes them susceptible to apoptosis in a bleomycin model, ultimately reversing the established lung fibrosis in mice [61]. It is important to note that AMPK signaling activation and myofibroblast-to-lipofibroblast transdifferentiation by metformin are independent of each other [60]. Given that current FDA-approved antifibrotic agents slow fibrosis progression, metformin’s alluring ability to reverse lung fibrosis through more than one pathway encourages further research into its role in IPF treatment.
Slowing disease progression is imperative in a chronic condition like IPF in which patient state deteriorates with increasing age. Cellular senescence, an irreversible replicative arrest state in cells, is known to play a role in pulmonary fibrosis through its contribution to release of inflammatory cytokines and pro-fibrotic growth factors. Senescent cells accumulate in tissues during the aging process and preventing this accumulation could help to treat IPF. Senolytics are drugs which induce death of senescent cells [62]. Combination treatment of senolytic agents dasatinib plus quercetin (DQ) have been explored in bleomycin-induced lung fibrosis mouse models and have shown to reduce dysfunction caused by IPF. The orally active tyrosine kinase inhibitor, dasatinib, has demonstrated reduction of inflammatory responses in asthmatic lungs in recent studies [63]. While quercitin is a natural flavonoid found in vegetables, it has been effective in selectively inducing death of senescent cells in several cultured cell models when combined with dasatinib [62]. The effect of DQ in attenuating lung fibrosis was further evaluated in a preliminary human clinical trial in which fourteen IPF patients were recruited and treated with 100 mg/day dasatinib and 1250 mg/day quercitin for three days a week over the course of three weeks. While there were no significant improvements in pulmonary function, physical function in the patients was noticeably improved with few but mostly tolerable adverse events and correlations to pro-inflammatory cytokines were observed [62]. Not only do the treatment results of this trial corroborate the results from previous bleomycin mouse models, they also encourage investigation into the future role of DQ in IPF treatment through larger randomized controlled trials. Currently in phase 2 clinical trials, DQ is seen as a feasible option for senolytic intervention in further studies as well as points towards the importance of combination drug treatments in IPF.
Drug combination approaches may be desirable to enhance the antifibrotic effect of existing IPF therapies when paired with similar agents or drugs exhibiting a similar effect. Pirfenidone-prednisolone is a combination therapy that has yielded promising results in vivo [64]. Prednisolone is a glucocorticoid sometimes used to treat inflammation of the lungs, but not necessarily in IPF. During this study, rats with paraquat (PQ)-induced pulmonary fibrosis were treated with a vehicle, pirfenidone, prednisolone, or pirfenidone plus prednisolone. Results displayed that pirfenidone along with prednisolone reduced the interstitial thickening, inflammatory responses, and collagen accumulation in comparison to the group treated with only pirfenidone or only prednisolone. The hydroxyproline content, oxidative stress parameters, and TGF-β1 and tumor necrosis factor-α (TNF-α) were also significantly reduced after treatment with pirfenidone and prednisolone, compared to only pirfenidone or prednisolone. The appeal of combination therapy is that it can simultaneously target multiple complex pathways and mechanisms involved in IPF, rather than a single drug. A glucocorticoid like prednisolone has been used in combination with other drugs to enhance their therapeutic effects, leading to the belief it could enhance the effects of an FDA-approved treatment for IPF.
Another combination treatment previously studied is nintedanib in combination with sildenafil, a phosphodiesterase inhibitor typically used to help with blood flow to the lungs. This combination treatment was originally studied given the potential for sildenafil to improve gas exchange as analyzed by the diffusion capacity of the lungs and to improve the quality of life in IPF patients [65]. IPF can result in vasoconstriction of blood vessels in the lungs, and sildenafil acts as vasodilator, indicating its potential in treating IPF along with nintedanib. After 12 weeks of treatment of 274 patients with a nintedanib group and a nintedanib-plus-sildenafil group, there was no significant difference in the SGRQ total score and dyspnea in IPF patients. Although this trial did not yield promising results about nintedanib and sildenafil as a successful combination therapy for IPF, it was able to provide information about what class of drugs could or could not potentially be used in combination with one of the FDA-approved treatments for IPF.
Along with investigating therapies that can be combined with FDA approved drugs for IPF, it is pertinent to consider strategies for more effective delivery and release of the therapeutic agents for effective therapy. For example, NP delivery of combinations of drugs such as prostaglandin E and siRNAs (targeting MMP3, CCL12 and HIF1A) in IPF treatment have increased survival rates of mice to 80% after 3 weeks [66]. The survival rates for single use of either prostaglandin E or siRNAs were 60% and 0%, respectively, after three weeks. Those rates after two weeks were 80%, 60% and 20%, respectively, compared to controls [66]. This further supports that the development of NPs designed together with combination of emerging drugs would open new avenues for therapeutic intervention in IPF patients.
4. Drug Delivery Approaches for IPF Treatment
Several drug delivery strategies and routes of administration have been explored especially in the past decade, for the site-specific and effective delivery of therapies to the deep lungs to treat IPF with minimal off-target effects. Pulmonary delivery of therapeutics is an attractive alternative over oral or parenteral drug delivery offering several advantages like direct delivery of drugs at the targeted site resulting in rapid onset of action, limiting adverse side-effects by minimizing the entry of toxic therapeutics into the systemic circulation, non-invasive administration, and improved patient compliance [67][68]. There is a significant difference in the distribution of inhaled therapeutics within the lungs when compared to the more conventional routes like oral administration, where the therapeutics would need to undergo first-pass metabolism in the liver before entering the blood circulation and eventually arriving at the lungs (Figure 3) [69][70]. However, free drugs that are directly inhaled may be subject to poor stability or inadequate absorption [71]. To overcome these limitations, the drugs may be encapsulated within engineered nano- or micro-particles thus improving their therapeutic efficacy and activity. Numerous particle carriers like liposomes, polymer/ lipid-based particles and mesoporous silica NPs when inhaled have shown to significantly accumulate and retain longer in the lungs thus improving the pharmacokinetics of the encapsulated drug [72][73][74].
Figure 3.
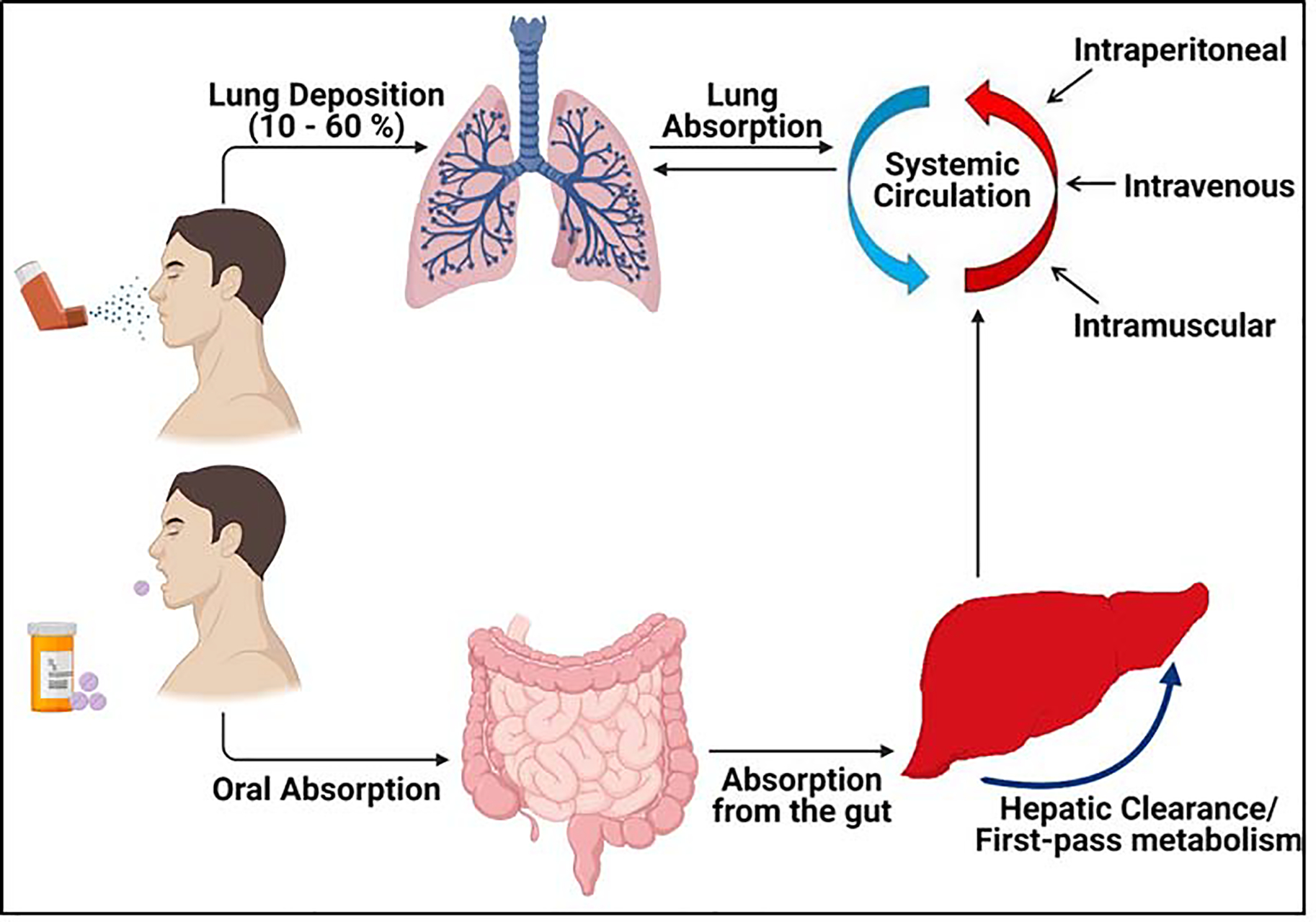
Comparison of delivery of NPs by inhalation and the more conventional methods such as oral and parenteral delivery. Significant increase in the local concentrations of the drug is observed in lungs after inhalation drug delivery when compared to oral drug delivery systems as well as other routes as shown above [67]. Figure created using BioRender (https://biorender.com/).
Among inhalable NP formulations, we have previously demonstrated that Poly(d,l-lactic-co-glycolic acid) (PLGA) - a polymer in use in several FDA-approved products, is promising for drug delivery to the deep lung tissue by nebulization [75][76]. Drug-containing MPs and NPs can be surface decorated with cell-specific targeting ligands for site-specific delivery of therapeutics [77][78]. When coupled with the biocompatibility and biodegradability characteristics of polymers like PLGA, MPs and NPs provide versatility in the delivery of drugs through more than one route of administration with capabilities for controlled and sustained delivery, reduced dosage, and overall increased effectiveness of encapsulated drugs [79]. Recent studies have shown that monodispersed drug particles with a mass median aerodynamic diameter (MMAD) of ~1.5 μm were deposited within the peripheral areas of the lungs of IPF patients following nebulization [80]. Jet nebulizers in particular have been shown to deposit therapeutic agents within the deep lung tissue of IPF patients with minimal airway deposition [81]. These clinical studies confirm that pulmonary drug delivery is a suitable approach for the treatment of IPF patients, and that despite the reduced lung function in this disease, inhaled therapeutics can be deposited in the deep lung tissue following inhalation. In this section, we review the numerous drug delivery approaches for IPF treatment while comparing different components and routes of administration explored, with emphasis on inhaled nano- and micro-formulations.
4.1. Nano-formulations
Pulmonary delivery allows at least three times greater local distribution of NPs than by oral administration and systemic injection [82]. Since the lungs have an open airway, delivery of therapeutic drugs to the lungs via inhalation is an attractive alternative to traditional methods of administration used in the treatment of pulmonary fibrosis. There are several factors that affect the deposition of inhaled drugs within the lung. For instance, inhaled aerosolized therapeutics are subject to distribution mechanisms in the lung based on the size of the particles [83][84]. Inhaled aerosol particles with diameters between 1–5 μm are generally considered ideal for efficient lung deposition as they are deposited in smaller airways by gravitation sedimentation [85], however they tend to be phagocytosed by alveolar macrophages. Particles in the 100–1000 nm diameter range tends to penetrate deeper as they are deposited in alveoli via Brownian diffusion and are more rapidly absorbed, whereas particles larger than 5 μm are subject to inertial impaction in larger airways such as bronchi and trachea [85]. Larger particles are also more likely to be cleared by mucociliary clearance mechanisms in the lung, therefore, achieving the optimal size is integral to successful absorption of the inhaled drug (Figure 4) [85]. In one study, albuterol was delivered in varying aerosol particle sizes to asthma patients with greater lung deposition and deeper airway penetration observed in smaller sizes of 1.5 μm (1500 nm) and 3 μm than sizes of 6 μm and greater [84]. Drug encapsulation within MPs or NPs not only allows for greater penetration, but also provides a vehicle whose size can be optimized accordingly for more effective inhalation delivery.
Figure 4.

Pulmonary delivery of NPs via inhalation. Aerosolized drug-loaded NPs when inhaled passes through the respiratory tract and gets deposited deep in the alveolar region [86]. The NP platform facilitates sustained release of the encapsulated drugs thus improving the localized distribution of drugs in the lungs. Figure created using BioRender (https://biorender.com/)
NP-based formulations in the 50–200 nm are suitable for alveolar deposition and cellular internalization post-nebulization, while evading uptake by alveolar macrophages [87]. An extensive list of drug delivery approaches developed and tested for IPF treatment has been provided in Table 1. Rawal et al. utilized chitosan NPs (diameter = 109.7 nm) for pulmonary delivery of bedaquiline (BDQ) by dry powder inhaler (DPI) to reduce side effects associated with oral BDQ administration. In vivo toxicity studies of inhaled BDQ NPs in male Wistar rats showed better safety profile than orally administered BDQ solution [88]. Several different types of NPs including liposomes, solid/lipid NPs, polymeric NPs, and dendrimers, have been prepared for delivery either as dry powder for aerosolization, or as suspensions for nebulization [82]. Since the lung surfactants secreted from AEC2 cells are composed of w/w 90% lipids, liposomes and lipid-coated solid NP formulations are most frequently used to evade major barriers of the lung tissues [82][89]. Specifically, dipalmitoylphosphatidylcholine (DPPC), which makes up 80% of the lung surfactant is used as a coating to reduce recognition and phagocytosis by alveolar macrophages, which account for 90% of airway immune cells [68][89]. A combination of DPPC and dipalmitoylphosphatidylglycerol (DPPG) was used as a carrier for anti-cancer agent paclitaxel and delivered as respirable dry powder. In vitro studies on A549 lung cancer cells showed enhanced chemotherapeutic activity of the formulation when compared to free paclitaxel. Furthermore, the formulation showed significant deposition in the lower stages (nm aerodynamic size) of a Next Generation Impactor (NGI), indicating that the DPPC/DPPG-based formulation would be an effective carrier for inhalational drug delivery [90].
Table 1.
Drug delivery approaches in IPF treatment models.
| Drug | Dosage form; route of administration | Excipients; method of preparation | Characterization; In vitro/in vivo outcomes | Ref |
| Interleukin-10 (IL-10) | Hydrogel; intranasal administration | Heprasil and the crosslinker Extralink was mixed to for an in-situ hydrogel | IL-10 decreased the expression of fibrotic markers on treatment with TGFβ-1 activated fibroblasts. | [92] |
| AMD3100; siRNA | Nano-emulsion for intratracheal administration | Combination of fluorinated polymeric CXCR4 antagonist AMD3100 and siRNA; sonication method | In vitro cell culture assays suggested that siRNA decreased expression of α-SMA in fibrotic cells. | [93] |
| Nintedanib and colchicine | MMP-2 responsive peptide (peptide E5) modified liposomes; intravenous injection | Cholesterol, Soybean phospholipids, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine polyethylene glycol maleimide, 2000 Da (DSPE-PEG2K-Mal) and DSPE-PEG2K, peptide E5; thin film hydration | Peptide E5 modification improved the cellular uptake of liposomes and demonstrated synergistic effect. | [96] |
| Tetrandrine (TET) | TET encapsulated HP-β-CD inclusion nanosuspension complex for endotracheal administration | Hydroxy-propyl-β-cyclodextrin (HP-βCD); Kneading method | Study demonstrated the improved efficacy of inhaled TET inclusion complexes compared to intravenous administration of free TET. | [99] |
| Caveolin scaffolding domain peptide-7 (CSP7) | Excipient free micronized CSP7 peptide powder for inhalation delivery | Excipient free formulation formulated by micronizing of peptide power using airjet milling technology | Lung fibrosis in mice was significant reduced after treatment with CSP7 inhalation powder though nose-only inhalation exposure system. | [104] |
| Thymoquinone (TQ) | PLGA-PVA NPs; intratracheal administration | PVA; double emulsion method using probe sonicator | Developed NPs showed significant reduction in fibrosis in rats when compared with disease control group | [94] |
| Combination of astaxanthin (AST) and trametinib (TRA) | Monocyte-derived multipotent cell (MOMC) surface-engineered NPs (PER NPs); intravenous injection | Peptide E5, poly (lactide-co-glycolide) block-poly (ethylene glycol) methyl ether maleimide (PLGA-PEG-Mal) and PLGA-PEG-c (RGDfc), Monocyte-derived multipotent cell (MOMC) isolated from rat blood; antisolvent precipitation method | Developed surface modified dual drug loaded PLGA NPs showed better antifibrotic activity then a current clinically USFDA approved drug – Pirfenidone. | [106] |
| Pirfenidone | CHI-SA NPs loaded with PFD as efficient transdermal drug delivery system | Pre-gelation method used to synthesize Chitosan and Sodium Alginate polymeric NPs | Release profile of the CHI-SA NPs showed sustained release of PFD from synthesized NPs. Fluorescent microscope images of PFD loaded CHI-SA NPs showed that NPs transfer through skin successfully. | [107] |
| Inhaled treprostinil prodrug hexadecyl-treprostinil (C16TR) formulated in a lipid NP (INS1009) | Lipid NP for delivery by nebulization; INS1009 was given by nose-only inhalation | INS1009 was formulated at a stock concentration of 0.3 mg/mL in PBS and appropriate dilutions were prepared to achieve the target doses | Evaluating anti-fibrotic effects through therapeutic dosing in a rat model of bleomycin-induced pulmonary fibrosis. Treatment of rats with inhaled INS1009 produced robust and dose-dependent reductions in the lung hydroxyproline content. | [108] |
| Fluorofenidone (AKF; 5-methyl-1-[3-fluorophenyl]-2-[1H]-pyridone) | Spermidine (Spd)-modified PEG-PLGA NPs as a lung-targeted delivery system for AKF; Both Spd-AKF-PLGA NPs and AKF-PLGA NPs were distributed to lung after intravenous injection. | Carboxylate-functionalized copolymer PLGA-PEG-COOH was synthesized by conjugating COOH-PEG-NH2 to PLGA-COOH. Emulsification-solvent evaporation was employed for the preparation of AKF NPs. | Biodistribution study suggested that Spd-AKF-PLGA NPs accumulated effectively in the lung. Fluorescence analysis showed that the Spd-AKF-PLGA NPs had high affinity for A549 cells and facilitated endocytosis. | [109] |
| Tacrolimus | Nebulized Tac-NPs or Tac by I.P. injection; Inhalation via a nose-only dosing chamber | Liquid dispersion of colloidal Tac and lactose aerosolized used vibrating mesh nebulizer | In vivo; Higher survival with inhaled Tac vs. injected Tac and reduced inflammation and fibrosis | [97] |
| Tacrolimus | 60 μ Tac; Intratracheal instillation via microspray aerosolizer | Tac (2.5mg) and cholesterol (5mg) dissolved in .1ml of 9:1 solution of chloroform and ethanol; BSA (50mg) dissolved in 5 ml deionized water; high-pressure homogenizer nine times at 20,000psi; rotary evaporated and centrifuged | In-vivo; Promising therapeutic efficacy | [110] |
| Tacrolimus | 180 μ Tac twice a week; Direct inhalation | O/w emulsification diffusion method to prepare tacrolimus-loaded chitosan-coated poly (lactic-co-glycolic acid) NPs | In-vivo; Better efficacy than oral administration daily | [98] |
| Montelukast (montelukast loaded nanostructured lipid carriers (MNLC)) | 5 mg montelukast for in vitro; equivalent of 1 mg aerosolized for in vivo; Intratracheal instillation | Melt-emulsification-ultrasonication method; prepared with 3% mannitol | In vivo and in vitro; MNLC-DPI improved drug deposition in lungs; high potential for pulmonary targeting | [111] |
Delivery of nano-formulations via intranasal and intratracheal routes in IPF treatment models in recent studies attests to the versatility and vast capability of these methods of administration. Trivedi et al., developed PLGA NPs containing pirfenidone, which were administered intratracheally to C57BL/6 mice. The NPs were able to maintain significantly higher levels of pirfenidone in the lungs compared to the free drug for up to 1 week, leading to significantly lower hydroxyproline levels indicating enhanced anti-fibrotic effect [91]. In addition to pirfenidone and nintedanib, new therapeutics in preclinical and clinical stages of development are also being encapsulated within NP formulations and explored for IPF therapy. Shamskhou et al., developed an intranasal hydrogel consisting of interleukin −10 (IL-10) entrapped in a mixture of Hyaluronan (to retain moisture) and heparin (to bind IL-10) [92], for IPF treatment. In vivo therapeutic and prophylactic treatment of hydrogels in bleomycin-induced fibrosis in mice significantly preserved lung architecture, as shown by minimal fibrosis with reduced collagen content and α-SMA. Although the study has demonstrated the effect of the hydrogel on 2D monoculture models, the effect of IL-10 loaded hydrogel on more complex 2D and 3D cell culture models consisting of alveolar cells, macrophages, lymphocytes will substantiate the claims. Wang et al., demonstrated the intratracheal administration of AMD3100 (CXCR4 chemokine receptor and plasminogen activator inhibitor-1 antagonist) and siRNA nano-emulsion for the treatment of IPF [93]. Formulated emulsion polyplexes demonstrated that the nanovesicles had a hydrodynamic diameter of 140 nm and zeta potential of +28 mV with spherical morphology and efficient siRNA binding to the polymer. In vitro cell culture assays suggested perfluorocarbon emulsion polyplexes containing a fluorinated polymeric CXCR4 antagonist showed enhanced uptake of siRNA in comparison to conventional siRNA polyplexes and decreased expression of α-SMA after treatment. Combination of siRNA and AMD3100 showed synergistic effect in both acute and chronic in vivo bleomycin induced fibrotic mice. Saghir et al., formulated thymoquinone (TQ) loaded PLGA NPs for intratracheal delivery [94]. TQ, a bioactive constituent of the Nigella sativa seeds, is well known for its use as a potential antioxidant and anti-inflammatory agent [95]. It is known to mitigate lung fibrosis by decreasing the production of pro-fibrotic proteins and oxidative stress. However, its use is limited due to its poor solubility and bioavailability. The intra-tracheal administration of TQ loaded PLGA NPs in rats with bleomycin-induced pulmonary fibrosis led to inhibition of lung inflammation and suppression of oxidative stress when compared to the disease control. These outcomes were also supported by the reduced expression of IL-10 and TGF-β1 in the treatment group.
As mentioned previously, since the two FDA-approved agents can only slow down the progression of IPF and not cure the condition, more effective treatments and treatment strategies are required. Dual drug-containing NP formulations have been explored to achieve synergistic effects of anti-fibrotic agents simultaneously. Chang et al., demonstrated the synergistic effect of liposomes containing Nintedanib (antifibrotic drug) and colchicine (for polarization of M1 macrophages into M2 macrophages) for IPF treatment [96]. Their matrix metalloproteinase 2 (MMP-2) responsive peptide (peptide E5) modified liposomes were roughly 100 nm in diameter with spherical morphology. Nintedanib was encapsulated in the lipid layer whereas colchicine in hydrophilic central core of liposomes. In vitro mono and co-culture studies demonstrated the stability of liposomes with better cellular uptake and in vivo experiments proved convincing synergistic activity of both drugs owing to improved cell targeting. Garbuzenko et al., developed an inhalable nanostructured lipid carrier encapsulating siRNA and prostaglandin E and effectively demonstrated a success in the treatment of IPF [66]. These results attest to the potential of NP formulations for delivering combination therapies for site-specific and effective treatment.
There are many reports in literature that on the implementation of inhalational delivery of drugs as aerosols for IPF treatment, to provide localized therapy. Shivshankar et al., explored tacrolimus (Tac) as a possible treatment for IPF. The drug has anti-fibrotic properties yet has many inflammatory side effects when taken orally [97]. By formulating a nebulized Tac loaded-NP, some of these side effects such as inflammation and kidney issues may be avoided, while still preventing fibrosis in the lungs. NPs were prepared via liquid dispersion of colloidal Tac and lactose and aerosolized using a vibrating mesh nebulizer in bleomycin-injured mice. Tac delivered via I.P. injection was used for comparison and the results were analyzed after 21 days of treatment. The results displayed that there was a higher survival rate with the inhaled Tac treatment vs. the injected Tac treatment. Histological analysis showed improvement in the fibrotic lesions in the lungs. The nebulized NPs containing Tac are a promising treatment for IPF but will require more extensive studies. Another study was done exploring Tac-loaded chitosan-coated PLGA NPs by Lee et al. [98], who administered these NPs on a bleomycin-induced pulmonary fibrosis mouse model twice a week via direct inhalation. This reduced frequency could aid in increased compliance with patients. H&E and Masson’s trichrome staining displayed reduction in inflammation and collagen deposition in the lung tissue after treatment with the NPs, indicating the promising treatment of tacrolimus. Mice treated with the NPs showed a two-fold reduction in hydroxyproline level compared to mice that were not treated. These findings indicate that inhalable NP formulations could potentially replace oral formulations for anti-fibrotic treatment.
The effectiveness of inhalation delivery has also been compared to other routes of administration. Su et al., demonstrated the use of active ingredient Tetrandrine (TET) isolated from the Chinese herb Stephania tetrandra for the treatment of IPF [99]. TET was encapsulated in hydroxy propyl-β-cydodextrin (HP-β-CD) by the kneading method to form dried inclusion complex. The formulated nanosuspension was suspended in saline and homogenized to form nanosuspension for endotracheal administration using a micro spray aerosolizer. In vivo pharmacokinetics and pharmacodynamics of the nanosuspension (630 nm) on bleomycin induced fibrotic rat model demonstrated improved efficacy of inhaled TET inclusion complexes compared to intravenous administration of free TET based on the rat lung studies, hydroxyproline content, and histopathology analysis. Hydroxyproline content was significantly decreased in TET inclusion complexes when compared to free TET and the disease control group. These studies further indicate that the inhalation delivery route may be superior when concerned with pulmonary fibrosis.
4.2. Micro-formulations
MP formulations in the 1–1000 μm range are often explored for pulmonary drug delivery as they can preferentially deposit in the deep lung tissue; however these formulations, as mentioned previously, tend to be easily phagocytosed by alveolar macrophages which can reduce their retention time in the lungs [100][101][102]. Pirfenidone was loaded into sustained release MPs prepared using an Ethyl cellulose-Eudragit RS 100 combination. An aerodynamic diameter of <5 μm was observed, which can potentially maximize drug deposition in the central and peripheral regions of the lungs [103]. Zhang et al. developed an excipient-free micronized caveolin scaffolding domain peptide (CSP7) for the treatment of lung fibrosis, which was tested in vitro and in vivo [104]. After preparation by jet milling technology and achieving particle diameter size of 1.58 μm, stability studies were conducted and showed that the micronized peptide powder was stable over 6 months. In vivo studies in bleomycin induced fibrotic mice demonstrated the significant reduction of collagen in mice treated with CSP7 powder for inhalation. Hyaluronidase (HYAL) was also investigated as a potential treatment by da Silva Bitencourt et al. for IPF due to its ability to reduce bleomycin-induced fibrosis [105]. Assembling PLGA MPs and loading with HYAL aids in preserving the biological activity and making it a more effective treatment. The HYAL-MPs were formulated using the emulsion and solvent evaporation methods. In this experiment, 2mg of the HYAL-MPs were intranasally inoculated for treatment. Results displayed that the MPs were able to reduce neutrophil recruitment and collagen deposition more than soluble HYAL was able to. This indicates the potential for HYAL-MPs as an effective delivery system for treatment of IPF. Recently, Hu et al. developed inhalable large porous PLGA MPs containing curcumin for the treatment of IPF. Due to the porous structure, the MPs had low densities and smaller aerodynamic diameters which helped them evade alveolar macrophage uptake in vitro. Compared to curcumin powders, the MPs had a significant anti-fibrotic effect and reduced hydroxyproline content and collagen I synthesis following tracheal insufflation into bleomycin-treated rat models [102]. In this manner, new methods of maximizing deep lung deposition and longer retention of MPs following inhalation are being developed and optimized in order to ensure greater anti-fibrotic efficacy.
5. Conclusions and Future Outlook
IPF is a life-threatening condition with the worst prognosis among the interstitial lung diseases. It is being increasingly recognized as a complication in patients post COVID-19 infection, which further increases the urgency of identifying effective and site-specific therapies to reverse and potentially cure the disease [112][113]. The sole FDA-approved anti-fibrotic agents pirfenidone and nintedanib are prescribed to try to alleviate symptoms in IPF patients in an attempt to improve their prognosis. However, since neither of these drugs can reverse or cure the fibrosis occurring within the lungs, the patients are still left susceptible to disease- and drug-related complications. Alternative therapies are sought to overcome the side effects of chronic administration of these FDA-approved oral drugs. Given the increasing mortality rates and overwhelming complications associated with IPF, patients are in need of more restorative therapies as well as a variety of treatment options. An ideal and effective treatment approach would entail: (1) therapeutic agents that reverse the fibrosis to restore the lung architecture while having minimal or negligible off-target toxicities, and (2) formulations for non-invasive, sustained, and localized delivery of anti-fibrotic agents to improve treatment efficacy and patient compliance.
As the exact cause of IPF remains unknown, more research must be conducted in determining pathogenesis and identifying therapeutic targets. In addition to providing a descriptive explanation of the current conception of how the overproduction of myofibroblasts leads to distortion and fibrotic scarring of the lung tissue, we have included a detailed review of the following key fibrotic pathway mediators and their role in myofibroblast differentiation: TGF-β1, PDGF, VEGF and FGF. Profibrotic responses induced by these mediators have been linked with signaling of emerging therapeutic targets such as WNT5A, SHH, ATX, and LPA receptors in recent studies. Further investigation of these mediators could contribute to the understanding of fibroproliferative pathways in IPF and in turn help to explain the exact mechanism for antifibrotic action, ultimately advancing the drug development pipeline for IPF.
Given that there are only two antifibrotic treatments available on the market, there is a urgent need for new drug development as well as improvement of existing therapies. We have discussed several drugs currently being studied for IPF treatment and where they are in the drug development pipeline. In addition to the promising lead compounds GLPG1690, BBT-877, and TD139 all currently undergoing clinical trials for safety and efficacy in attenuating fibrosis, existing drugs used for other conditions such as metformin, DQ, and prednisolone have shown favorable results in attenuating fibrosis with the potential for combination with FDA-approved antifibrotic drugs. Combination therapies with existing antifibrotic agents pirfenidone and nintedanib should be explored to either add to or compliment the overall antifibrotic effect achieved. Metformin alone has yielded promising results in not only attenuating but also reversing the established fibrosis in vivo [60][61].
Long-acting drug delivery strategies such as NP and MP encapsulation may be a more effective way to deliver antifibrotic agents to the lung for sustained and localized action, avoiding systemic toxicity and reducing dosage frequency. We provided an extensive review of nano- and micro-formulation approaches in IPF treatment models in vitro and in vivo as can be found in Table 1. NPs assembled using biocompatible polymers such as PLGA or the DPPC lipid present in significant quantities in the lung surfactant coating, work to maintain a sustained release of the encapsulated drug while shielding particles from systemic clearance. Given their size, these particles have the capability of being delivered as aerosol directly to the lung while complementing its large surface area for increased absorption. Drugs encapsulated in NPs delivered directly to the lung via inhalation showed greater overall efficacy than drugs injected in vivo, as was in the example of tacrolimus loaded NPs. NP drug delivery systems can also be utilized to deliver a combination therapy as was in the case of nintedanib and colchicine liposomes, which displayed a synergistic effect in vitro.
To conclude, we have presented an ample number of examples where NP-based and MP-based formulations have been utilized to deliver drugs in IPF treatment models to achieve greater absorption, less frequent dosing, dual-drug therapy, and localized delivery to the lung where the fibrosis is occurring. Paired with the information provided on potential therapeutic targets and promising drug therapies, NP-based drug delivery strategies may be one of the major components necessary to make headway in the development of future IPF treatment methods.
Acknowledgements
We would like to acknowledge the Rhode Island Institutional Development Award (IDeA) Network of Biomedical Research Excellence from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM103430 (JUM) and P20GM103652 (YZ) for financial support. YZ is also supported by R01HL146498 from the National Heart, Lung, and Blood Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Martinez FJ, Collard HR, Pardo A, Raghu G, Richeldi L, Selman M, Swigris JJ, Taniguchi H, Wells AU, Idiopathic pulmonary fibrosis, Nat. Rev. Dis. Prim. 3 (2017). 10.1038/nrdp.2017.74. [DOI] [PubMed] [Google Scholar]
- [2].Raghu G, Chen SY, Hou Q, Yeh WS, Collard HR, Incidence and prevalence of idiopathic pulmonary fibrosis in US adults 18–64 years old, Eur. Respir. J. 48 (2016) 179–186. 10.1183/13993003.01653-2015. [DOI] [PubMed] [Google Scholar]
- [3].Navaratnam V, Hubbard RB, The Mortality Burden of Idiopathic Pulmonary Fibrosis in the United Kingdom, Am. J. Respir. Crit. Care Med. 200 (2019) 256–258. 10.1164/rccm.201902-0467LE. [DOI] [PubMed] [Google Scholar]
- [4].Cottin V, Maher T, Long-term clinical and real-world experience with pirfenidone in the treatment of idiopathic pulmonary fibrosis, Eur. Respir. Rev. 24 (2015) 58–64. 10.1183/09059180.00011514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Meyer KC, Decker CA, Role of pirfenidone in the management of pulmonary fibrosis, Ther. Clin. Risk Manag. 13 (2017) 427–437. 10.2147/TCRM.S81141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Dinh PUC, Paudel D, Brochu H, Popowski KD, Gracieux MC, Cores J, Huang K, Hensley MT, Harrell E, Vandergriff AC, George AK, Barrio RT, Hu S, Allen TA, Blackburn K, Caranasos TG, Peng X, Schnabel LV, Adler KB, Lobo LJ, Goshe MB, Cheng K, Inhalation of lung spheroid cell secretome and exosomes promotes lung repair in pulmonary fibrosis, Nat. Commun. 11 (2020). 10.1038/s41467-020-14344-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Galli JA, Pandya A, Vega-Olivo M, Dass C, Zhao H, Criner GJ, Pirfenidone and nintedanib for pulmonary fibrosis in clinical practice: Tolerability and adverse drug reactions, Respirology. 22 (2017) 1171–1178. 10.1111/resp.13024. [DOI] [PubMed] [Google Scholar]
- [8].Genentech USA Inc., Esbriet® (Pirfenidone) Dosage - How to Take Esbriet, Esbriet.Com. (2020). https://www.esbriet.com/taking-esbriet/how-to-take-esbriet.html. [Google Scholar]
- [9].Jo HE, Randhawa S, Corte TJ, Moodley Y, Idiopathic Pulmonary Fibrosis and the Elderly: Diagnosis and Management Considerations, Drugs and Aging. 33 (2016) 321–334. 10.1007/s40266-016-0366-1. [DOI] [PubMed] [Google Scholar]
- [10].Crouch E, Pathobiology of pulmonary fibrosis, Am. J. Physiol. - Lung Cell. Mol. Physiol. 259 (1990). 10.1152/ajplung.1990.259.41159. [DOI] [PubMed] [Google Scholar]
- [11].Mangal S, Gao W, Li T, Zhou QT, Pulmonary delivery of nanoparticle chemotherapy for the treatment of lung cancers: Challenges and opportunities, Acta Pharmacol. Sin. 38 (2017) 782–797. 10.1038/aps.2017.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tesfaigzi Y, Roles of apoptosis in airway epithelia, Am. J. Respir. Cell Mol. Biol. 34 (2006) 537–547. 10.1165/rcmb.2006-00140C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Selman M, Pardo A, Role of epithelial cells in idiopathic pulmonary fibrosis: From innocent targets to serial killers, Proc. Am. Thorac. Soc. 3 (2006) 364–372. 10.1513/pats.200601-003TK. [DOI] [PubMed] [Google Scholar]
- [14].Sgalla G, Iovene B, Calvello M, Ori M, Varone F, Richeldi L, Idiopathic pulmonary fibrosis: Pathogenesis and management, Respir. Res. 19 (2018). 10.1186/s12931-018-0730-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lindell KO, Olshansky E, Song MK, Zullo TG, Gibson KF, Kaminski N, Hoffman LA, Impact of a disease-management program on symptom burden and health-related quality of life in patients with idiopathic pulmonary fibrosis and their care partners, Hear. Lung J. Acute Crit. Care. 39 (2010) 304–313. 10.1016/j.hrtlng.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Allen RJ, Porte J, Braybrooke R, Flores C, Fingerlin TE, Oldham JM, Guillen-Guio B, Ma SF, Okamoto T, John AE, Obeidat M, Yang IV, Henry A, Hubbard RB, Navaratnam V, Saini G, Thompson N, Booth HL, Hart SP, Hill MR, Hirani N, Maher TM, McAnulty RJ, Millar AB, Molyneaux PL, Parfrey H, Rassl DM, Whyte MKB, Fahy WA, Marshall RP, Oballa E, Bossé Y, Nickle DC, Sin DD, Timens W, Shrine N, Sayers I, Hall IP, Noth I, Schwartz DA, Tobin MD, Wain LV, Jenkins RG, Genetic variants associated with susceptibility to idiopathic pulmonary fibrosis in people of European ancestry: a genome-wide association study, Lancet Respir. Med. 5 (2017) 869–880. 10.1016/S2213-2600(17)30387-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhang Q, Wang Y, Qu D, Yu J, Yang J, The Possible Pathogenesis of Idiopathic Pulmonary Fibrosis considering MUC5B, Biomed Res. Int. 2019 (2019). 10.1155/2019/9712464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Seibold MA, Wise AL, Speer MC, Steele MP, Brown KK, Loyd JE, Fingerlin TE, Zhang W, Gudmundsson G, Groshong SD, Evans CM, Garantziotis S, Adler KB, Dickey BF, du Bois RM, Yang IV, Herron A, Kervitsky D, Talbert JL, Markin C, Park J, Crews AL, Slifer SH, Auerbach S, Roy MG, Lin J, Hennessy CE, Schwarz MI, Schwartz DA, A Common MUC5B Promoter Polymorphism and Pulmonary Fibrosis, N. Engl. J. Med. 364 (2011) 1503–1512. 10.1056/NEJMoa1013660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hancock LA, Hennessy CE, Solomon GM, Dobrinskikh E, Estrella A, Hara N, Hill DB, Kissner WJ, Markovetz MR, Grove Villalon DE, Voss ME, Tearney GJ, Carroll KS, Shi Y, Schwarz MI, Thelin WR, Rowe SM, Yang IV, Evans CM, Schwartz DA, Muc5b overexpression causes mucociliary dysfunction and enhances lung fibrosis in mice, Nat. Commun. 9 (2018) 1–10. 10.1038/s41467-018-07768-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Garcia CK, Idiopathic pulmonary fibrosis: Update on genetic discoveries, in: Proc. Am. Thorac. Soc, 2011: pp. 158–162. 10.1513/pats.201008-056MS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Castelino FV, Varga J, Interstitial lung disease in connective tissue diseases: Evolving concepts of pathogenesis and management, Arthritis Res. Ther. 12 (2010). 10.1186/ar3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hadjicharalambous MR, Lindsay MA, Idiopathic pulmonary fibrosis: Pathogenesis and the emerging role of long non-coding RNAs, Int. J. Mol. Sci. 21 (2020). 10.3390/ijms21020524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Han YY, Shen P, Chang WX, Involvement of epithelial-to-mesenchymal transition and associated transforming growth factor-/Smad signaling in paraquat-induced pulmonary fibrosis, Mol. Med. Rep. 12 (2015) 7979–7984. 10.3892/mmr.2015.4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Deng X, Jin K, Li Y, Gu W, Liu M, Zhou L, Platelet-Derived Growth Factor and Transforming Growth Factor β1 Regulate ARDS-Associated Lung Fibrosis Through Distinct Signaling Pathways, Cell. Physiol. Biochem. 36 (2015) 937–946. 10.1159/000430268. [DOI] [PubMed] [Google Scholar]
- [25].Murray LA, Habiel DM, Hohmann M, Camelo A, Shang H, Zhou Y, Coelho AL, Peng X, Gulati M, Crestani B, Sleeman MA, Mustelin T, Moore MW, Ryu C, Osafo-Addo AD, Elias JA, Lee CG, Hu B, Herazo-Maya JD, Knight DA, Hogaboam CM, Herzog EL, Antifibrotic role of vascular endothelial growth factor in pulmonary fibrosis, JCI Insight. 2 (2017). 10.1172/jci.insight.92192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].MacKenzie BA, Korfei M, Henneke I, Sibinska Z, Tian X, Hezel S, Dilai S, Wasnick R, Schneider B, Wilhelm J, El Agha E, Klepetko W, Seeger W, Schermuly R, Günther A, Bellusci S, Increased FGF1-FGFRc expression in idiopathic pulmonary fibrosis, Respir. Res. 16 (2015). 10.1186/s12931-015-0242-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wollin L, Wex E, Pautsch A, Schnapp G, Hostettler KE, Stowasser S, Kolb M, Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis, Eur. Respir. J. 45 (2015) 1434–1445. 10.1183/09031936.00174914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Amano H, Mastui Y, Ito Y, Shibata Y, Betto T, Eshima K, Ogawa F, Satoh Y, Shibuya M, Majima M, The role of vascular endothelial growth factor receptor 1 tyrosine kinase signaling in bleomycin-induced pulmonary fibrosis, Biomed. Pharmacother. 117 (2019). 10.1016/j.biopha.2019.109067. [DOI] [PubMed] [Google Scholar]
- [29].Shi J, Li F, Luo M, Wei J, Liu X, Distinct Roles of Wnt/ β-Catenin Signaling in the Pathogenesis of Chronic Obstructive Pulmonary Disease and Idiopathic Pulmonary Fibrosis, Mediators Inflamm. 2017 (2017). 10.1155/2017/3520581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hu B, Liu J, Wu Z, Liu T, Ullenbruch MR, Ding L, Henke CA, Bitterman PB, Phan SH, Reemergence of hedgehog mediates epithelial-mesenchymal crosstalk in pulmonary fibrosis, Am. J. Respir. Cell Mol. Biol. 52 (2015) 418–428. 10.1165/rcmb.2014-0108OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chanda D, Otoupalova E, Smith SR, Volckaert T, De Langhe SP, Thannickal VJ, Developmental pathways in the pathogenesis of lung fibrosis, Mol. Aspects Med. 65 (2019) 56–69. 10.1016/j.mam.2018.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Saito A, Horie M, Micke P, Nagase T, The role of TGF-β signaling in lung cancer associated with idiopathic pulmonary fibrosis, Int. J. Mol. Sci. 19 (2018). 10.3390/ijms19113611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sureshbabu A, Tonner E, Allan GJ, Flint DJ, Relative roles of TGF-β and IGFBP-5 in idiopathic pulmonary fibrosis, Pulm. Med. 2011 (2011). 10.1155/2011/517687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gauldie J, Kolb M, Ask K, Martin G, Bonniaud P, Warburton D, Smad3 signaling involved in pulmonary fibrosis and emphysema, in: Proc. Am. Thorac. Soc., Proc Am Thorac Soc, 2006: pp. 696–702. 10.1513/pats.200605-125SF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Sugiura H, Ichikawa T, Liu X, Kobayashi T, Wang XQ, Kawasaki S, Togo S, Kamio K, Mao L, Ann Y, Ichinose M, Rennard SI, N-acetyl-l-cysteine inhibits TGF-β1-induced profibrotic responses in fibroblasts, Pulm. Pharmacol. Ther. 22 (2009) 487–491. 10.1016/j.pupt.2009.04.002. [DOI] [PubMed] [Google Scholar]
- [36].Mitrović V, Mitrović S, & Todorović D , [Fibronectin--a multifunctional glycoprotein] - PubMed, Srp. Arh. Celok. Lek. 123 (1995) 198–201. https://pubmed-ncbi-nlm-nih-gov.uri.idm.oclc.org/17974429/ (accessed September 26, 2020). [PubMed] [Google Scholar]
- [37].Lee CG, Link H, Baluk P, Homer RJ, Chapoval S, Bhandari V, Kang MJ, Cohn L, Kim YK, McDonald DM, Elias JA, Vascular endothelial growth factor (VEGF) induces remodeling and enhances TH2-mediated sensitization and inflammation in the lung, Nat. Med. 10 (2004) 1095–1103. 10.1038/nm1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Myllärniemi M, Kaarteenaho R, Pharmacological treatment of idiopathic pulmonary fibrosis – preclinical and clinical studies of pirfenidone, nintedanib, and N-acetylcysteine, Eur. Clin. Respir. J. 2 (2015) 26385. 10.3402/ecrj.v2.26385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Rangarajan S, Kurundkar A, Kurundkar D, Bernard K, Sanders YY, Ding Q, Antony VB, Zhang J, Zmijewski J, Thannickal VJ, Novel mechanisms for the antifibrotic action of nintedanib, Am. J. Respir. Cell Mol. Biol. 54 (2016) 51–59. 10.1165/rcmb.2014-0445OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hostettler KE, Zhong J, Papakonstantinou E, Karakiulakis G, Tamm M, Seidel P, Sun Q, Mandal J, Lardinois D, Lambers C, Roth M, Anti-fibrotic effects of nintedanib in lung fibroblasts derived from patients with idiopathic pulmonary fibrosis, Respir. Res. 15 (2014). 10.1186/s12931-014-0157-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Huang C, Xiao X, Yang Y, Mishra A, Liang Y, Zeng X, Yang X, Xu D, Blackburn MR, Henke CA, Liu L, MicroRNA-101 attenuates pulmonary fibrosis by inhibiting fibroblast proliferation and activation, J. Biol. Chem. 292 (2017) 16420–16439. 10.1074/jbc.M117.805747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Martin-Medina A, Lehmann M, Burgy O, Hermann S, Baarsma HA, Wagner DE, De Santis MM, Ciolek F, Hofer TP, Frankenberger M, Aichler M, Lindner M, Gesierich W, Guenther A, Walch A, Coughlan C, Wolters P, Lee JS, Behr J, Königshoff M, Increased extracellular vesicles mediate WNT5A signaling in idiopathic pulmonary fibrosis, Am. J. Respir. Crit. Care Med. 198 (2018) 1527–1538. 10.1164/rccm.201708-1580OC. [DOI] [PubMed] [Google Scholar]
- [43].Ahangari F, Kaminski N, WNT5A in extracellular vesicles a new frontier for pulmonary fibrosis, Am. J. Respir. Crit. Care Med. 198 (2018) 1468–1470. 10.1164/rccm.201807-1321ED. [DOI] [PubMed] [Google Scholar]
- [44].Ninou I, Magkrioti C, Aidinis V, Autotaxin in pathophysiology and pulmonary fibrosis, Front. Med. 5 (2018). 10.3389/fmed.2018.00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Maher TM, van der Aar EM, Van de Steen O, Allamassey L, Desrivot J, Dupont S, Fagard L, Ford P, Fieuw A, Wuyts W, Safety, tolerability, pharmacokinetics, and pharmacodynamics of GLPG1690, a novel autotaxin inhibitor, to treat idiopathic pulmonary fibrosis (FLORA): a phase 2a randomised placebo-controlled trial, Lancet Respir. Med. 6 (2018) 627–635. 10.1016/S2213-2600(18)30181-4. [DOI] [PubMed] [Google Scholar]
- [46].Kraljić K, Jelić D, Žiher D, Cvrtila A, Dragojević S, Sinković V, Mesić M, Benzoxaboroles-novel autotaxin inhibitors, Molecules. 24 (2019). 10.3390/molecules24193419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ninou I, Kaffe E, Müller S, Budd DC, Stevenson CS, Ullmer C, Aidinis V, Pharmacologic targeting of the ATX/LPA axis attenuates bleomycin-induced pulmonary fibrosis, Pulm. Pharmacol. Ther. 52 (2018) 32–40. 10.1016/j.pupt.2018.08.003. [DOI] [PubMed] [Google Scholar]
- [48].Cao P, Aoki Y, Badri L, Walker NM, Manning CM, Lagstein A, Fearon ER, Lama VN, Autocrine lysophosphatidic acid signaling activates?-catenin and promotes lung allograft fibrosis, J. Clin. Invest. 127 (2017) 1517–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].MacKinnon AC, Gibbons MA, Farnworth SL, Leffler H, Nilsson UJ, Delaine T, Simpson AJ, Forbes SJ, Hirani N, Gauldie J, Sethi T, Regulation of transforming growth factor-β1-driven lung fibrosis by galectin-3, Am. J. Respir. Crit. Care Med. 185 (2012) 537–546. 10.1164/rccm.201106-0965OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Nishi Y, Sano H, Kawashima T, Okada T, Kuroda T, Kikkawa K, Kawashima S, Tanabe M, Goto T, Matsuzawa Y, Matsumura R, Tomioka H, Liu FT, Shirai K, Role of galectin-3 in human pulmonary fibrosis, Allergol. Int. 56 (2007) 57–65. 10.2332/allergolint.O-06-449. [DOI] [PubMed] [Google Scholar]
- [51].Slack RJ, Mills R, Mackinnon AC, The therapeutic potential of galectin-3 inhibition in fibrotic disease, Int. J. Biochem. Cell Biol. 130 (2021) 105881. 10.1016/j.biocel.2020.105881. [DOI] [PubMed] [Google Scholar]
- [52].Bargagli E, Piccioli C, Rosi E, Torricelli E, Turi L, Piccioli E, Pistolesi M, Ferrari K, Voltolini L, Pirfenidone and Nintedanib in idiopathic pulmonary fibrosis: Real-life experience in an Italian referral centre, Pulmonology. 25 (2019) 149–153. 10.1016/j.pulmoe.2018.06.003. [DOI] [PubMed] [Google Scholar]
- [53].Cerri S, Monari M, Guerrieri A, Donatelli P, Bassi I, Garuti M, Luppi F, Betti S, Bandelli G, Carpano M, Bacchi Reggiani ML, Tonelli R, Clini E, Nava S, Real-life comparison of pirfenidone and nintedanib in patients with idiopathic pulmonary fibrosis: A 24-month assessment, Respir. Med. 159 (2019). 10.1016/j.rmed.2019.105803. [DOI] [PubMed] [Google Scholar]
- [54].Hoffmann-Vold AM, Weigt SS, Saggar R, Palchevskiy V, Volkmann ER, Liang LL, Ross D, Ardehali A, Lynch JP, Belperio JA, Endotype-phenotyping may predict a treatment response in progressive fibrosing interstitial lung disease, EBioMedicine. 50 (2019) 379–386. 10.1016/j.ebiom.2019.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Hirani N, MacKinnon AC, Nicol L, Ford P, Schambye H, Pedersen A, Nilsson UJ, Leffler H, Sethi T, Tantawi S, Gavelle L, Slack RJ, Mills R, Karmakar U, Humphries D, Zetterberg F, Keeling L, Paul L, Molyneaux PL, Li F, Funston W, Forrest IA, Simpson AJ, Gibbons MA, Maher TM, Target-inhibition of Galectin-3 by Inhaled TD139 in Patients with Idiopathic Pulmonary Fibrosis, Eur. Respir. J. (2020) 2002559. 10.1183/13993003.02559-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Galecto Biotech AB, A Study to Test the Efficacy and Safety of Inhaled TD139 in Subjects With Idiopathic Pulmonary Fibrosis (IPF) - Full Text View - ClinicalTrials.gov, U.S. Natl. Libr. Med. Clin. (2019). NCT03832946 (accessed September 27, 2020). [Google Scholar]
- [57].Desroy N, Housseman C, Bock X, Joncour A, Bienvenu N, Cherel L, Labeguere V, Rondet E, Peixoto C, Grassot JM, Picolet O, Annoot D, Triballeau N, Monjardet A, Wakselman E, Roncoroni V, Le Tallec S, Blanque R, Cottereaux C, Vandervoort N, Christophe T, Mollat P, Lamers M, Auberval M, Hrvacic B, Ralic J, Oste L, Van der Aar E, Brys R, Heckmann B, Discovery of 2-[[2-Ethyl-6-[4-[2-(3-hydroxyazetidin-1-yl)-2-oxoethyl]piperazin-1-yl]-8-methylimidazo[1,2-a]pyridin-3-yl]methylamino]-4-(4-fluorophenyl)thiazole-5-carbonitrile (GLPG1690), a First-in-Class Autotaxin Inhibitor Undergoing Clinical Evaluation, J. Med. Chem. 60 (2017) 3580–3590. [DOI] [PubMed] [Google Scholar]
- [58].Li S, Hu X, Wang Z, Wu M, Zhang J, Different profiles of notch signaling in cigarette smoke-induced pulmonary emphysema and bleomycin-induced pulmonary fibrosis, Inflamm. Res. 64 (2015) 363–371. 10.1007/s00011-015-0816-y. [DOI] [PubMed] [Google Scholar]
- [59].Lee G, Kang S-U, Ryou J-H, Lim J-J, Lee D-Y, Kwon H-J, Ha G-H, Lee Y-H, BBT-877, a Potent Autotaxin Inhibitor in Clinical Development to Treat Idiopathic Pulmonary Fibrosis, in: Am. Thorac. Soc. Int. Conf. Meet. Abstr. Am. Thorac. Soc. Int. Conf. Meet. Abstr., American Thoracic Society, 2019: pp. A2577–A2577. 10.1164/ajrccm-conference.2019.199.1_meetingabstracts.a2577. [DOI] [Google Scholar]
- [60].Kheirollahi V, Wasnick RM, Biasin V, Vazquez-Armendariz AI, Chu X, Moiseenko A, Weiss A, Wilhelm J, Zhang JS, Kwapiszewska G, Herold S, Schermuly RT, Mari B, Li X, Seeger W, Günther A, Bellusci S, El Agha E, Metformin induces lipogenic differentiation in myofibroblasts to reverse lung fibrosis, Nat. Commun. 10 (2019). 10.1038/s41467-019-10839-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Rangarajan S, Bone NB, Zmijewska AA, Jiang S, Park DW, Bernard K, Locy ML, Ravi S, Deshane J, Mannon RB, Abraham E, Darley-Usmar V, Thannickal VJ, Zmijewski JW, Metformin reverses established lung fibrosis in a bleomycin model, Nat. Med. 24 (2018) 1121–1131. 10.1038/s41591-018-0087-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, Hashmi SK, Prata L, Masternak MM, Kritchevsky SB, Musi N, Kirkland JL, Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study, EBioMedicine. 40 (2019) 554–563. 10.1016/j.ebiom.2018.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Ryu KY, Lee HJ, Woo H, Kang RJ, Han KM, Park HH, Lee SM, Lee JY, Jeong YJ, Nam HW, Nam Y, Hoe HS , Dasatinib regulates LPS-induced microglial and astrocytic neuroinflammatory responses by inhibiting AKT/STAT3 signaling, J. Neuroinflammation. 16 (2019). 10.1186/s12974-019-1561-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Rasooli R, Pourgholamhosein F, Kamali Y, Nabipour F, Mandegary A, Combination Therapy with Pirfenidone plus Prednisolone Ameliorates Paraquat-Induced Pulmonary Fibrosis, Inflammation. 41 (2018) 134–142. 10.1007/s10753-017-0671-9. [DOI] [PubMed] [Google Scholar]
- [65].Kolb M, Raghu G, Wells AU, Behr J, Richeldi L, Schinzel B, Quaresma M, Stowasser S, Martinez FJ, Nintedanib plus sildenafil in patients with idiopathic pulmonary fibrosis, N. Engl. J. Med. 379 (2018) 1722–1731. 10.1056/NEJMoa1811737. [DOI] [PubMed] [Google Scholar]
- [66].Garbuzenko OB, Ivanova V, Kholodovych V, Reimer DC, Reuhl KR, Yurkow E, Adler D, Minko T, Combinatorial treatment of idiopathic pulmonary fibrosis using nanoparticles with prostaglandin E and siRNA(s), Nanomedicine Nanotechnology, Biol. Med. 13 (2017) 1983–1992. 10.1016/jmano.2017.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Chenthamara D, Subramaniam S, Ramakrishnan SG, Krishnaswamy S, Essa MM, Lin FH, Qoronfleh MW, Therapeutic efficacy of nanoparticles and routes of administration, Biomater. Res. 23 (2019) 1–29. 10.1186/s40824-019-0166-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Kuzmov A, Minko T, Nanotechnology approaches for inhalation treatment of lung diseases, J. Control. Release. 219 (2015) 500–518. 10.1016/j.jconrel.2015.07.024. [DOI] [PubMed] [Google Scholar]
- [69].Miller MR, Raftis JB, Langrish JP, McLean SG, Samutrtai P, Connell SP, Wilson S, Vesey AT, Fokkens PHB, Boere AJF, Krystek P, Campbell CJ, Hadoke PWF, Donaldson K, Cassee FR, Newby DE, Duffin R, Mills NL, Inhaled Nanoparticles Accumulate at Sites of Vascular Disease, ACS Nano. 11 (2017) 4542–4552. 10.1021/acsnano.6b08551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Savla R, Minko T, Nanotechnology approaches for inhalation treatment of fibrosis, J. Drug Target. 21 (2013) 914–925. 10.3109/1061186X.2013.829078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Beck-Broichsitter M, Merkel OM, Kissel T, Controlled pulmonary drug and gene delivery using polymeric nano-carriers, J. Control. Release. 161 (2012) 214–224. 10.1016/jjconreL201L12.004. [DOI] [PubMed] [Google Scholar]
- [72].Van Rijt SH, Bein T, Meiners S, Medical nanoparticles for next generation drug delivery to the lungs, Eur. Respir. J. 44 (2014) 765–774. 10.1183/09031936.00212813. [DOI] [PubMed] [Google Scholar]
- [73].Anderson CF, Grimmett ME, Domalewski CJ, Cui H, Inhalable nanotherapeutics to improve treatment efficacy for common lung diseases, Wiley Interdiscip. Rev. Nanomedicine Nanobiotechnology. 12 (2020) e1586. 10.1002/wnan.1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Chishti N, Jagwani S, Dhamecha D, Jalalpure S, Dehghan MH, Preparation, optimization, and in vivo evaluation of nanoparticle-based formulation for pulmonary delivery of anticancer drug, Med. 55 (2019). 10.3390/medicina55060294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Menon JU, Ravikumar P, Pise A, Gyawali D, Hsia CCW, Nguyen KT, Polymeric nanoparticles for pulmonary protein and DNA delivery, Acta Biomater. 10 (2014) 2643–2652. 10.1016/j.actbio.2014.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Ravikumar P, Menon JU, Punnakitikashem P, Gyawali D, Togao O, Takahashi M, Zhang J, Ye J, Moe OW, Nguyen KT, Hsia CCW, Nanoparticle facilitated inhalational delivery of erythropoietin receptor cDNA protects against hyperoxic lung injury, Nanomedicine Nanotechnology, Biol. Med. 12 (2016) 811–821. https://doi.org/10.1016Zj.nano.2015.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Jagwani S, Jalalpure S, Dhamecha D, Jadhav K, Bohara R, Pharmacokinetic and Pharmacodynamic Evaluation of Resveratrol Loaded Cationic Liposomes for Targeting Hepatocellular Carcinoma, ACS Biomater. Sci. Eng. 6 (2020) 4969–4984. 10.1021/acsbiomaterials.0c00429. [DOI] [PubMed] [Google Scholar]
- [78].Liu Y, Peng J, Wang S, Xu M, Gao M, Xia T, Weng J, Xu A, Liu S, Molybdenum disulfide/graphene oxide nanocomposites show favorable lung targeting and enhanced drug loading/tumor-killing efficacy with improved biocompatibility, NPG Asia Mater. 10 (2018) e458–e458. 10.1038/am.2017.225. [DOI] [Google Scholar]
- [79].Ding D, Zhu Q, Recent advances of PLGA micro/nanoparticles for the delivery of biomacromolecular therapeutics, Mater. Sci. Eng. C. 92 (2018) 1041–1060. 10.1016/j.msec.2017.12.036. [DOI] [PubMed] [Google Scholar]
- [80].Usmani OS, Biddiscombe MF, Yang S, Meah S, Oballa E, Simpson JK, Fahy WA, Marshall RP, Lukey PT, Maher TM, The topical study of inhaled drug (salbutamol) delivery in idiopathic pulmonary fibrosis, Respir. Res. 19 (2018) 25. 10.n86/s12931-018-0732-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Samuel J, Smaldone GC, Maximizing Deep Lung Deposition in Healthy and Fibrotic Subjects during Jet Nebulization, J. Aerosol Med. Pulm. Drug Deliv 33 (2020) 108–115. 10.1089/jamp.2019.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Yhee J, Im J, Nho R, Advanced Therapeutic Strategies for Chronic Lung Disease Using Nanoparticle-Based Drug Delivery, J. Clin. Med. 5 (2016) 82. 10.3390/jcm5090082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Williams RO, Carvalho TC, Peters JI, Influence of particle size on regional lung deposition - What evidence is there?, Int. J. Pharm. 406 (2011) 1–10. 10.1016/j.ijpharm.2010.12.040. [DOI] [PubMed] [Google Scholar]
- [84].Usmani OS, Biddiscombe MF, Barnes PJ, Regional lung deposition and bronchodilator response as a function of β2-agonist particle size, Am. J. Respir. Crit. Care Med. 172 (2005) 1497–1504. 10.1164/rccm.200410-1414OC. [DOI] [PubMed] [Google Scholar]
- [85].Dhand C, Prabhakaran MP, Beuerman RW, Lakshminarayanan R, Dwivedi N, Ramakrishna S, Role of size of drug delivery carriers for pulmonary and intravenous administration with emphasis on cancer therapeutics and lung-targeted drug delivery, RSC Adv. 4 (2014) 32673–32689. 10.1039/c4ra02861a. [DOI] [Google Scholar]
- [86].Klinger-Strobel M, Lautenschläger C, Fischer D, Mainz JG, Bruns T, Tuchscherr L, Pletz MW, Makarewicz O, Aspects of pulmonary drug delivery strategies for infections in cystic fibrosis-where do we stand?, Expert Opin. Drug Deliv. 12 (2015) 1351–1374. 10.1517/17425247.2015.1007949. [DOI] [PubMed] [Google Scholar]
- [87].Dandekar P, Venkataraman C, Mehra A, Pulmonary Targeting of Nanoparticle Drug Matrices, J. Aerosol Med. Pulm. Drug Deliv. 23 (2010) 343–353. 10.1089/jamp.2009.0784. [DOI] [PubMed] [Google Scholar]
- [88].Rawal T, Patel S, Butani S, Chitosan nanoparticles as a promising approach for pulmonary delivery of bedaquiline, Eur. J. Pharm. Sci. 124 (2018) 273–287. 10.1016/j.ejps.2018.08.038. [DOI] [PubMed] [Google Scholar]
- [89].Liu Q, Guan J, Qin L, Zhang X, Mao S, Physicochemical properties affecting the fate of nanoparticles in pulmonary drug delivery, Drug Discov. Today. 25 (2020) 150–159. 10.1016/j.drudis.2019.09.023. [DOI] [PubMed] [Google Scholar]
- [90].Meenach SA, Anderson KW, Hilt JZ, McGarry RC, Mansour HM, High-Performing Dry Powder Inhalers of Paclitaxel DPPC/DPPG Lung Surfactant-Mimic Multifunctional Particles in Lung Cancer: Physicochemical Characterization, In Vitro Aerosol Dispersion, and Cellular Studies, Ageing Int. 15 (2014) 1574–1587. 10.1208/s12249-014-0182-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Trivedi R, Redente EF, Thakur A, Riches DWH, Kompella UB, Local delivery of biodegradable pirfenidone nanoparticles ameliorates bleomycin-induced pulmonary fibrosis in mice, Nanotechnology. 23 (2012) 505101. 10.1088/0957-4484/23/50/505101. [DOI] [PubMed] [Google Scholar]
- [92].Shamskhou EA, Kratochvil MJ, Orcholski ME, Nagy N, Kaber G, Steen E, Balaji S, Yuan K, Keswani S, Danielson B, Gao M, Medina C, Nathan A, Chakraborty A, Bollyky PL, De Jesus Perez VA, Hydrogel-based delivery of I1–10 improves treatment of bleomycin-induced lung fibrosis in mice, Biomaterials. 203 (2019) 52–62. 10.1016/j.biomaterials.2019.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Wang Y, Ding L, Li Z, Chen G, Sun M, Oupicky D, Treatment of acute lung injury and early- and late-stage pulmonary fibrosis with combination emulsion siRNA polyplexes, J. Control. Release. 314 (2019) 12–24. 10.1016/jjconreL2019.10.030. [DOI] [PubMed] [Google Scholar]
- [94].Saghir SAM, Al-Gabri NA, Khafaga AF, El-Shaer NH, Alhumaidh KA, Elsadek MF, Ahmed BM, Alkhawtani DM, Abd El-Hack ME, Thymoquinone-PLGA-PVA nanoparticles ameliorate bleomycin-induced pulmonary fibrosis in rats via regulation of inflammatory cytokines and iNOS signaling, Animals. 9 (2019). 10.3390/ani9110951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Gholamnezhad Z, Havakhah S, Boskabady MH, Preclinical and clinical effects of Nigella sativa and its constituent, thymoquinone: A review, J. Ethnopharmacol. 190 (2016) 372–386. 10.1016/jjep.2016.06.061. [DOI] [PubMed] [Google Scholar]
- [96].Chang X, Xing L, Wang Y, Zhou TJ, Shen LJ, Jiang HL, Nanoengineered immunosuppressive therapeutics modulating M1/M2 macrophages into the balanced status for enhanced idiopathic pulmonary fibrosis therapy, Nanoscale. 12 (2020) 8664–8678. 10.1039/d0nr00750a. [DOI] [PubMed] [Google Scholar]
- [97].Shivshankar P, Payan H, Calhoun C, Levine S, Williams RO, Jagirdar J, Peters JI, Jourdan Le Saux C, Inhaled tacrolimus modulates pulmonary fibrosis without promoting inflammation in bleomycin-injured mice, J. Drug Deliv. Sci. Technol. 24 (2014) 469–477. 10.1016/S1773-2247(14)50090-1. [DOI] [Google Scholar]
- [98].Lee C, Seo J, Hwang HS, Thao LQ, Lee S, Lee ES, Lee EH, Choi HG, Youn YS, Treatment of bleomycin-induced pulmonary fibrosis by inhaled tacrolimus-loaded chitosan-coated poly(lactic-co-glycolic acid) nanoparticles, Biomed. Pharmacother. 78 (2016) 226–233. https://doi.org/10.10167j.biopha.2016.01.027. [DOI] [PubMed] [Google Scholar]
- [99].Su W, Liang Y, Meng Z, Chen X, Lu M, Han X, Deng X, Zhang Q, Zhu H, Fu T, Inhalation of Tetrandrine-hydroxypropyl-β-cydodextrin Inclusion Complexes for Pulmonary Fibrosis Treatment, Mol. Pharm. 17 (2020) 1596–1607. 10.1021/acs.molpharmaceut.0c00026. [DOI] [PubMed] [Google Scholar]
- [100].El-Sherbiny IM, El-Baz NM, Yacoub MH, Inhaled nano- and microparticles for drug delivery, Glob. Cardiol. Sci. Pract. 2015 (2015) 2. 10.5339/gcsp.2015.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Mohning MP, Thomas SM, Barthel L, Mould KJ, McCubbrey AL, Frasch SC, Bratton DL, Henson PM, Janssen WJ, Phagocytosis of microparticles by alveolar macrophages during acute lung injury requires merTK, Am. J. Physiol. - Lung Cell. Mol. Physiol. 314 (2018) L69–L82. 10.1152/ajplung.00058.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Hu Y, Li M, Zhang M, Jin Y, Inhalation treatment of idiopathic pulmonary fibrosis with curcumin large porous microparticles, Int. J. Pharm. 551 (2018) 212–222. 10.1016/j.ijpharm.2018.09.031. [DOI] [PubMed] [Google Scholar]
- [103].Pardeshi S, Patil P, Rajput R, Mujumdar A, Naik J, Preparation and characterization of sustained release pirfenidone loaded microparticles for pulmonary drug delivery: Spray drying approach, Dry. Technol. (2020) 1–11. 10.1080/07373937.2020.1833213. [DOI] [Google Scholar]
- [104].Zhang Y, Mackenzie B, Koleng JJ, Maier E, Warnken ZN, Williams RO, Development of an Excipient-Free Peptide Dry Powder Inhalation for the Treatment of Pulmonary Fibrosis, Mol. Pharm. 17 (2020) 632–644. 10.1021/acs.molpharmaceut.9b01085. [DOI] [PubMed] [Google Scholar]
- [105].Da Silva Bitencourt C, Gelfuso GM, Pereira PAT, De Assis PA, Tefé-Silva C, Ramos SG, Arantes EC, Faccioli LH, Hyaluronidase-loaded PLGA microparticles as a new strategy for the treatment of pulmonary fibrosis, Tissue Eng. - Part A. 21 (2015) 246–256. 10.1089/ten.tea.2013.0403. [DOI] [PubMed] [Google Scholar]
- [106].Chang X, Xing L, Xing L, Xing L, Xing L, Wang Y, Yang CX, He YJ, Zhou TJ, Gao XD, Li L, Hao HP, Jiang HL, Jiang HL, Jiang HL, Jiang HL, Monocyte-derived multipotent cell delivered programmed therapeutics to reverse idiopathic pulmonary fibrosis, Sci. Adv. 6 (2020). 10.1126/sciadv.aba3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Abnoos M, Mohseni M, Mousavi SAJ, Ashtari K, Ilka R, Mehravi B, Chitosan-alginate nano-carrier for transdermal delivery of pirfenidone in idiopathic pulmonary fibrosis, Int. J. Biol. Macromol. 118 (2018) 1319–1325. 10.1016/j.ijbiomac.2018.04.147. [DOI] [PubMed] [Google Scholar]
- [108].Corboz MR, Zhang J, LaSala D, DiPetrillo K, Li Z, Malinin V, Brower J, Kuehl PJ, Barrett TE, Perkins WR, Chapman RW, Therapeutic administration of inhaled INS1009, a treprostinil prodrug formulation, inhibits bleomycin-induced pulmonary fibrosis in rats, Pulm. Pharmacol. Ther. 49 (2018) 95–103. 10.1016/j.pupt.2018.01.012. [DOI] [PubMed] [Google Scholar]
- [109].Tang J, Li JM, Li G, Zhang HT, Wang L, Li D, Ding JS, Spermidine-mediated poly(Lactic-co-glycolic acid) nanoparticles containing fluorofenidone for the treatment of idiopathic pulmonary fibrosis, Int. J. Nanomedicine. 12 (2017) 6687–6704. 10.2147/IJN.S140569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Seo J, Lee C, Hwang HS, Kim B, Thao LQ, Lee ES, Oh KT, Lim JL, Choi HG, Youn YS, Therapeutic advantage of inhaled tacrolimus-bound albumin nanoparticles in a bleomycin-induced pulmonary fibrosis mouse model, Pulm. Pharmacol. Ther. 36 (2016) 53–61. 10.1016/j.pupt.2016.01.001. [DOI] [PubMed] [Google Scholar]
- [111].Patil-Gadhe A, Kyadarkunte A, Patole M, Pokharkar V, Montelukast-loaded nanostructured lipid carriers: Part II Pulmonary drug delivery and in vitro-in vivo aerosol performance, Eur. J. Pharm. Biopharm. 88 (2014) 169–177. 10.1016/j.ejpb.2014.07.007. [DOI] [PubMed] [Google Scholar]
- [112].Ojo AS, Balogun SA, Williams OT, Ojo OS, Pulmonary Fibrosis in COVID-19 Survivors: Predictive Factors and Risk Reduction Strategies, Pulm. Med. 2020 (2020). 10.1155/2020/6175964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Ahmad Alhiyari M, Ata F, Islam Alghizzawi M, Bint I Bilal A, Salih Abdulhadi A, Yousaf Z, Post COVID-19 fibrosis, an emerging complicationof SARS-CoV-2 infection, IDCases. 23 (2021) e01041. https://doi.org/10.1016yj.idcr.2020.e01041. [DOI] [PMC free article] [PubMed] [Google Scholar]