

Abstract
Rationale:
Thioredoxin-interacting protein (Txnip) is a novel molecular target with translational potential in diverse human diseases. Txnip has several established cellular actions including binding to thioredoxin, a scavenger of reactive oxygen species (ROS). It has been long recognized from in vitro evidence that Txnip forms a disulfide bridge through cysteine 247 (C247) with reduced thioredoxin to inhibit the anti-oxidative properties of thioredoxin. However, the physiological significance of the Txnip-thioredoxin interaction remains largely undefined in vivo.
Objective:
A single mutation of Txnip, C247S, abolishes the binding of Txnip with thioredoxin. Using a conditional and inducible approach with a mouse model of a mutant Txnip that does not bind thioredoxin, we tested whether the interaction of thioredoxin with Txnip is required for Txnip’s pro-oxidative or cytotoxic effects in the heart.
Methods and Results:
Overexpression of Txnip C247S in cells resulted in a reduction in ROS, due to an inability to inhibit thioredoxin. Hypoxia (1% O2, 24 hrs)-induced killing effects of Txnip were decreased by lower levels of cellular ROS in Txnip C247S-expressing cells compared with wild-type Txnip-expressing cells. Then, myocardial ischemic injuries were assessed in the animal model. Cardiomyocyte-specific Txnip C247S knock-in mice had better survival with smaller infarct size following MI compared to control animals. The absence of Txnip’s inhibition of thioredoxin promoted mitochondrial anti-oxidative capacities in cardiomyocytes, thereby protecting the heart from oxidative damage induced by myocardial infarction. Furthermore, an unbiased RNA sequencing screen identified that hypoxia-inducible factor 1 signaling pathway was involved in Txnip C247S-mediated cardioprotective mechanisms.
Conclusion:
Txnip is a cysteine-containing redox protein that robustly regulates the thioredoxin system via a disulfide bond-switching mechanism in adult cardiomyocytes. Our results provide the direct in vivo evidence that regulation of redox state by Txnip is a crucial component for myocardial homeostasis under ischemic stress.
Keywords: ROS, Metabolism, Mitochondria, Cell death
INTRODUCTION
Acute myocardial infarction (MI) is a common cardiac emergency with the potential for substantial morbidity and mortality. The high mortality rate among patients with cardiogenic shock after acute MI remains a challenge in need of solutions [1]. Thus, a better understanding of pathophysiology of MI is warranted to identify novel molecular targets for therapeutic intervention.
Excessive production of reactive oxygen species (ROS) has been implicated in MI. In the ischemic heart, oxygen delivery to the myocardium is not sufficient to complete the process of substrate oxidation in the mitochondria, resulting in electron leakage from the electron transport chain and increased superoxide generation [2]. Irreversible ischemic damage is characterized by oxidative modifications of essential proteins and lipids by ROS, which ultimately lead to tissue necrosis and apoptosis.
The thioredoxin system, along with the glutathione system, is a major mechanism by which the intracellular environment is maintained in the reduced state [3]. Thioredoxin activity is mediated by a pair of cysteine thiols at its active site (Cys32 and Cys35) [4]. The reduced form of thioredoxin facilitates the reduction of other proteins by cysteine thiol-disulfide exchange. Because of this function, thioredoxin acts as an active scavenger of ROS and plays a protective role in myocardial ischemia [5, 6] and cardiac hypertrophy [7].
Thioredoxin-Interacting Protein (Txnip), a member of arrestin-domain-containing protein superfamily, was originally identified as a negative regulator of thioredoxin [8]. We previously reported that a Txnip cysteine 247 (Cys247) forms an intrermolecular disulfide bond with reduced thioredoxin by disulfide exchange, yielding a stable mixed disulfide [9]. In this manner, Txnip inhibits thioredoxin’s reducing activity [10]. The protein crystal structure of the Txnip-thioredoxin complex confirms the formation of a stable interaction interface mediated by an intermolecular disulfide bond between Txnip Cys247 and thioredoxin Cys32 [11]. Biochemical assays have further established the critical role of Txnip Cys247 for regulating thioredoxin activity [9]. For instance, thioredoxin activity is reduced in Txnip-overexpressing cells, while it is unaltered in Txnip Cys247-to-Ser mutant (C247S)-overexpressing cells [9].
In contrast to this clear biochemical evidence, surprisingly little evidence supports a direct role for Txnip in regulation of thioredoxin activity in vivo. In particular, multiple in vivo studies have revealed no differences in available thioredoxin in the tissues of Txnip-deficient animals compared with wild-type animals [12, 13]. Txnip-deficient mice exhibit interesting phenotypes in metabolism [14] and cancer growth [15], and mouse models have specifically established a central physiological role for Txnip in glucose metabolism [16, 17]. However, these metabolic effects are mainly mediated by a thioredoxin-independent function of Txnip [18, 19]. Given the original discovery of Txnip as an intrinsic binding partner for thioredoxin, an obvious question remains unresolved: could Txnip function as an inhibitor of thioredoxin under pathological conditions associated with oxidative stress in vivo?
In the present work, we have generated a novel inducible, cell type-specific knock-in mouse model in which Txnip Cys247 is converted to a serine. This point mutation abolishes binding of Txnip to thioredoxin, thus eliminating all direct effects of Txnip on thioredoxin function specifically in adult cardiomyocytes. Using this mouse, we provide the first direct in vivo evidence that the Txnip-thioredoxin interaction plays a key role in redox homeostasis in the heart. The data also demonstrate that myocardial redox balance maintained by thioredoxin through a Txnip-dependent mechanism is crucial for prevention of cardiac death during the early post-MI period.
METHODS
Cell culture, adenoviral vectors and plasmid DNA transfection
HT1080 cells, SKBR3 cells, and H9c2 cardiomyocytes cells were obtained from the American Type Culture Collection. Mouse embryonic fibroblasts (MEFs) were from a systemic Txnip-null mouse [17]. Cells were maintained in Dulbecco’s modified Eagle’s medium containing penicillin/streptomycin and 10% fetal bovine serum. Recombinant adenoviral constructs expressing the mouse Txnip or Txnip C247S mutant in tandem with constitutive red fluorescent protein mCherry were generated using the pAV[Exp]-CMV vector system. For controls, the identical virus expressing empty vector (EV) was used. Cells were infected with the adenovirus at a multiplicity of infection of 20. For transient transfection, human Txnip or Txnip C247S mutant were subcloned into the pCDH-CMV-MCS-EF1-GreenPuro backbone and transfected using PureFection transfection reagent (System Biosciences). For hypoxic experiments, cells were incubated at 1% O2 balanced with 94% N2 and 5% CO2 at 37℃ in a hypoxic chamber (Coy Laboratory Products Inc.).
Gene and protein expression analyses
Gene expression was analyzed by real-time PCR with specific oligonucleotides. RNA was extracted by RNeasy Plus Mini Kit (Qiagen). Each sample was adjusted to 1.0 ng/μL using DeNovix DS-11 (DeNovix Inc.). Complementary DNA was produced by SuperScript IV VILO Master Mix, and quantitative real-time PCR was performed using QuantStudio 3 system and TaqMan primers (Thermo Fisher Scientific). Relative amounts of mRNA were normalized with glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Protein expression was analyzed by Western blotting. After adjusting protein concentrations, each sample was separated by electrophoresis on NuPAGE Bis-Tris gels (Thermo Fisher Scientific). Immunoblotting was performed using the antibodies against Txnip (clone JY2, MBL International), thioredoxin-1 (BD Biosciences), thioredoxin-2 (Abcam), ubiquitin C-terminal hydrolase L1 (UCHL1) (Sigma), actin, and GAPDH (Invitrogen).
Assays for thioredoxin activity, ROS, cell death, glucose uptake, and ATP content
Thioredoxin activity was measured with an insulin disulfide reduction assay in cytosolic or mitochondrial fractions of heart homogenates [20]. Glutathione level was quantified using EarlyTox Glutathione Assay Kit (Molecular Devices), and the GSH/GSSG ratio was estimated.[20] Cryosections were stained with the cell-permeant 2’,7’-dichlorodihydrofluorescein diacetate (H2DCFDA) (Thermo Fisher Scientific). A cellular response for oxidative stress was evaluated by an activation of nuclear factor erythroid 2–related factor 2 (Nrf2) using OKD Oxidative Stress Detector system (Cosmo Bio). Tissue levels of lipid peroxide (malondialdehyde) were estimated in heart homogenates with Lipid Peroxidation (MDA) Assay Kit (Abcam). These values were normalized by total protein content measured by Bradford assay.
Cellular lactate dehydrogenase (LDH) release was quantified by CytoTOX-One homogeneous membrane integrity assay (Promega). To distinguish types of cell death, Apoptotic/Necrotic/Healthy Cells Detection Kit (PromoCell) and Apoptosis/Necrosis Assay Kit (Abcam) were used. An apoptotic response was measured as caspase-3/7 activities using EarlyTox Caspase 3/7 R110 Assay Kit (Molecular Devices). For glucose uptake assay, cells were incubated with 100 μM 2-deoxyglucose and 125 μM 2-[3H]deoxyglucose (1 μCi/ml, PerkinElmer Life Science) for 30 min. The radiolabel in the cell lysates was measured by liquid scintillation counting as described [21]. Cellular amounts of ATP were measured using CellTiter-Glo Luminescent Cell Viability Assay (Promega).
Generation of Txnip C247S knock-in mice
We generated a tissue-specific Txnip C247S knock-in mouse line by replacing the wild-type Txnip gene exons 1–8 (NM_023719.2) with the conditional targeted frame of Txnip (Figure 3). Mouse genomic fragments containing homology arms were amplified from BAC clone by using high fidelity Taq and were assembled into a target vector with a neomycin cassette driven by the phosphoglycerate kinase (PGK) promoter. The linearized targeting construct was gel-purified, microinjected into the C57BL/C mouse blastocytes, and transferred into pseudo-pregnant foster mothers at Cyagen Biosciences Inc. (Santa Clara, CA). The neomycin selection cassette was flanked by self-deletion anchor (SDA) sites and removed in germ cells. Founder mice were generated on a C57BL/6 background. The genotypes were determined by PCR of tail-extracted DNA to verify proper targeting to the Txnip locus. PCR primers for the knock-in allele were sense 5’-CACTTTCTGAGGTACTGTTTCCTG-3’ and antisense 5’-GAAGCTCGAAGCCGAACTTGTACTC-3’.
Figure 3. The Cre-loxP recombination strategy for conditional cardiomyocyte-specific knock-in mutation of Txnip.
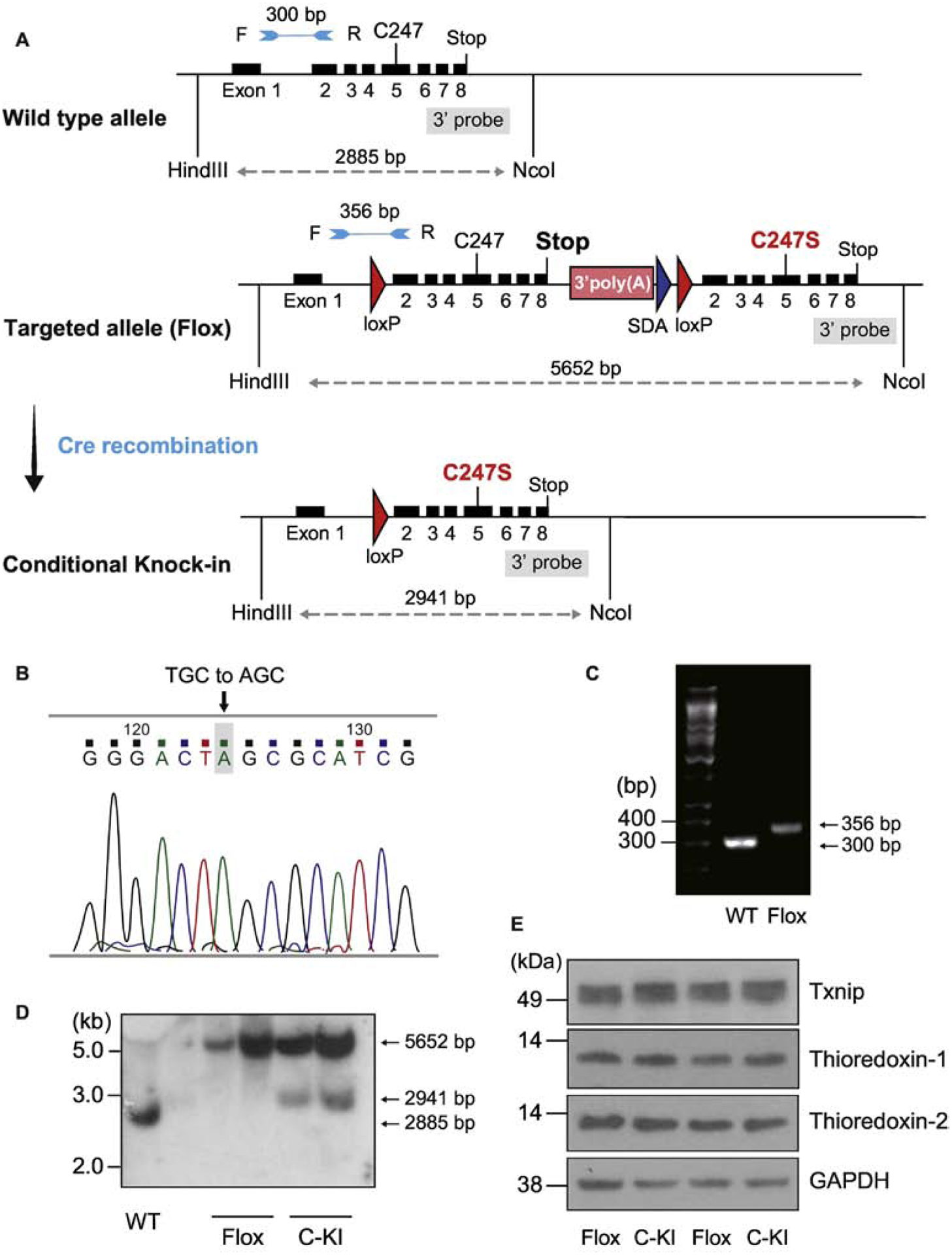
A. Two loxP sites were introduced in the targeting construct for Cre-mediated deletion of exons 2–8. Another set of exons 2–8 containing C247S mutation in exon5 was incorporated after the second loxP site. By breeding floxed mice with MerCreMer expressing mice, deletion of the floxed site was achieved by administration of tamoxifen, which resulted in the temporally regulated knock-in mutation of C247S under the control of wild-type (WT) gene regulatory element. B. Successful single mutation was confirmed by DNA sequencing in the mouse. C. WT and homozygous Txnip knock-in mice were characterized by PCR with forward (F) and reverse (R) primers, allowing the amplification of the loxP-containing deletion region; a 356-bp fragment for flox and a 300-bp fragment for WT allele. D. Successful recombination events were confirmed by Southern blot analyses. HindIII and NcoI-digested DNA was hybridized with the 3’ probe (5652-bp, 2911-bp, and 2885-bp bands represent the flox, conditional knock-in (C-KI), and WT allele, respectively). E. After the recombination, protein expression levels of Txnip, thioredoxin-1, and thioredoxin-2 were comparable in mouse hearts between flox mice and cardiomyocyte-specific knock-in mice.
To generate temporally regulated cardiomyocyte-specific Txnip knock-in mutation animals, a transgenic mouse overexpressing αMHC-MerCreMer in cardiomyocytes (The Jackson Laboratory) was mated with TxnipC247S/C247S mice. To induce Cre recombination, six-week-old animals were treated with tamoxifen or 4-hydroxytamoxifen (Sigma) as described [20, 22]. The successful homologous recombination and Cre-mediated deletion were confirmed by Southern analysis in genomic DNA from tails of αMHC-MerCreMer/TxnipC247S/C247S mice. The following oligonucleotide primers were used to generate the [γ32P]dCTP-labeled probe for Southern analysis: sense 5’-TCTCTTCAGCTCCTCCTTGC-3’ and antisense 5’-TCCGAGAAAGTGGTCAGGTC-3’.
Coronary artery ligation
All procedures involving animal use and surgeries were approved by the Institutional Animal Care and Use Committee. MI was produced in 12-week-old mice by a permanent ligation of the left anterior descending coronary artery [23]. To avoid the potential transient cardiotoxicity by tamoxifen-induced MerCreMer gene deletion [24], all procedures were performed at least four weeks after the completion of tamoxifen administration. Sham groups had the same procedure without the suture around the coronary artery. Echocardiography was performed without anesthesia to measure left ventricular parameters using GE Vivid 7 Dimension and GE i13L 10–14 MHz transducer. The heart was harvested and stained with 1% triphenyl tetrazolium to calculate infarct size. The harvested heart was paraffin embedded and stained with picrosirius red [23]. Apoptotic cell death was detected using In Situ Cell Detection Kit (Sigma) and quantified by fluorescence microscopy. Image analyses were performed by ImageJ (NIH Image).
Isolated perfused heart experiments
Isolated perfused heart experiments were performed in the Langendorff mode [20]. Briefly, the heart was excised and perfused with a constant pressure of 80 mmHg with modified Krebs-Henseleit buffer (in mM): 118 NaCl, 25 NaHCO3, 4.5 KCl, 2.5 CaCl2, 1.7 MgSO4, 1.2 KH2PO4, and 5.6 glucose, equilibrated with 95% O2-5% CO2 (pH 7.4). After an equilibration period, the heart was subjected to hypoxia with the buffer equilibrated with 95% N2-5% CO2. Isovolumic ventricular function was measured by a water-filled balloon inserted into the left ventricle and analyzed by a data-acquisition system (PowerLab, AD Instruments). At the end of the experiments, hearts were snap-frozen under liquid nitrogen. The nuclear fraction of the heart homogenates was extracted to measure HIF-1α levels using a HIF-1α Transcription Factor Assay Kit (Cayman chemical).
Next generation sequencing
Next generation sequencing was performed in the whole heart tissues (n=3 each) from wild-type or Txnip C247S male mice (12-week old age) at four weeks following the induction of C247S mutation. Complementary DNA libraries were constructed with NEBNext Ultra II RNA Library Prep kit (New England Biolabs). The libraries were sequenced by Illumina HiSeq 2×150bp configuration. Sequence reads were trimmed to remove possible adapter sequences and nucleotides with poor quality using Trimmomatic v.0.36. The trimmed reads were mapped to the mus musculus GRCm38 reference genome available on ENSEMBL using STAR ultrafast universal RNA-seq aligner. Standard RNA-seq was performed at GENEWIZ (South Plainfield, NJ). Sequencing depth (library size) was 20,000,000 paired-end reads per sample.
Statistical analysis
All data are presented as means±SEM. Statistical analysis was performed by GraphPad Prism. Comparisons between groups were tested by t-test, or by two-way ANOVA with a post hoc test of Tukey’s least significance difference. A chi-square test was used for comparisons between genders. For RNA-seq data, the differential expression analysis was performed by DESeq2 (Bioconductor). The Wald test was used to generate P-values and log2 fold changes. Genes with an adjusted P-value <0.05 and absolute log2 fold change >1 were considered as differentially expressed genes. Functional interactions of genes were visualized using STRING (ELIXIR).
RESULTS
Induction of oxidative stress by Txnip is mediated by the Cys247 that is required for interaction with thioredoxin.
We previously showed that the Txnip C247S mutant abolishes binding to thioredoxin, identifying Txnip Cys247 as a critical site for inhibition of thioredoxin activity in vitro [9]. In the present study, we took advantage of the Txnip C247S mutant to test whether the interaction of Txnip with thioredoxin is required for Txnip’s pro-oxidant function. To cleanly test the effect of the Txnip C247S mutant without a confounding effect from endogenous Txnip, we overexpressed wild-type Txnip or Txnip C247S mutant by adenoviral transduction in MEFs derived from a Txnip-knockout mouse [21]. As expected, adenoviral gene transfer of wild-type Txnip induced oxidative stress compared with that of an empty vector control. This was evident from two different analyses: 1) a decrease in the level of the anti-oxidant glutathione (Figure 1A); and 2) an increase in the cellular response to oxidative stress as shown by Nrf2 transcriptional activation (Figure 1B). In contrast, the Txnip C247s mutant did not induce cellular oxidative stress as measured by either glutathione levels or Nrf2 activation. These results indicate that Txnip’s pro-oxidant function is dependent on its ability to bind thioredoxin in vitro and that the Txnip Cys247 is required for induction of ROS.
Figure 1. Expression of Txnip C247S in cells results in a reduction in oxidative stress.
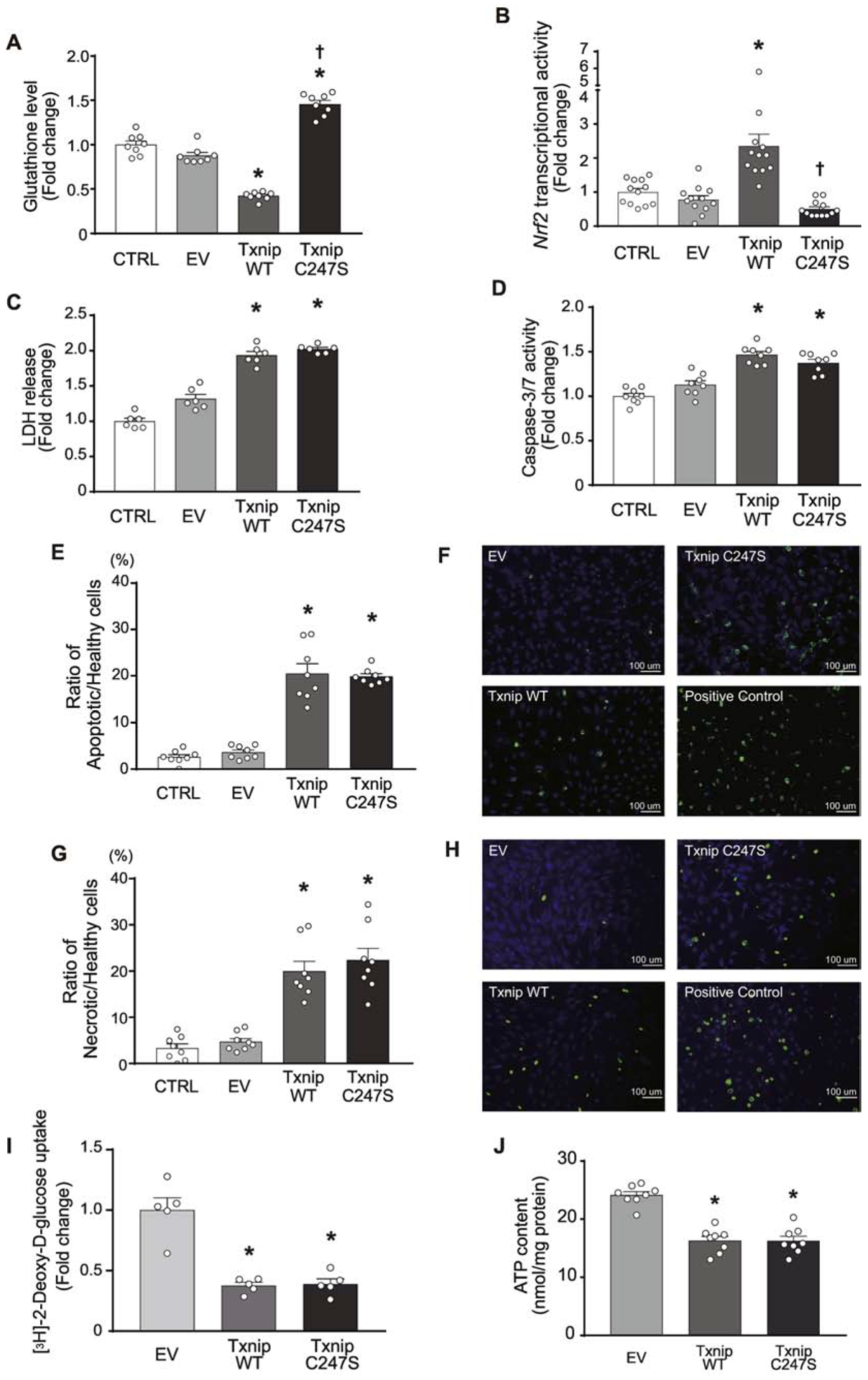
A. The amount of glutathione was quantified in live cells. MEFs derived from Txnip-knockout mice were reconstituted with adenoviral gene vector of Txnip WT, C247S, or empty vector (EV). CTRL are the cells without adenovirus. B. Nrf2 transcription factor activation, a sensitive cellular response for oxidative stressor, was quantified in HT1080 cells that were transfected with a plasmid containing a reporter gene in which luciferase was fused with the Nrf2 ubiquitination domain. In this system, oxidative stress stabilizes the luciferase-fusion protein and increases luciferase activities. Adenoviral overexpression of Txnip WT but not C247S induced this response. C and D. Lactate dehydrogenase (LDH) release into culture media and caspase-3/7 activity were measured in MEFs. E-H. The number of apoptotic (green stained with FITC-Annexin V in E and F) or necrotic (green stained with Nuclear Green DCS1 in G and H) cells were counted over that of healthy (blue stained with CytoCalcein Violet 450) HT1080 cells treated with adenoviral vectors. Staurosporine (2 μM for 2 hrs) was used for a positive control. I and J. 2-[3H]deoxyglucose uptake and cellular ATP levels were measured in Txnip-knockout MEFs after adenoviral overexpression of Txnip WT, C247S or EV. Values were normalized to total protein content. Glucose uptake was expressed as fold change to the mean value of uptake for EV. *P<0.01 vs. EV and CTRL, and †P<0.01 vs. Txnip WT.
Increased oxidative stress can induce cellular damage, and overexpression of Txnip is known to be cytotoxic [25]. To investigate whether Txnip Cys247 has a specific role in this effect, we overexpressed wild-type Txnip or Txnip C247S mutant in Txnip-knockout MEFs and analyzed several indicators of cellular damage. Overexpression of wild-type Txnip was cytotoxic as demonstrated by increased cellular LDH release and activation of caspase-3/7 (Figure 1C and 1D). This was accompanied by increased levels of Annexin V-positive apoptotic cell death (Figure 1E and 1F) and DNA Nuclear Green DCS1-positive necrotic cell death (Figure 1G and 1H). However, overexpression of the Txnip C247S mutation caused similar levels of cytotoxicity as wild-type Txnip.
One of the established functions of Txnip is an inhibition of glucose metabolism [21]. Thus, we determined the effect of Txnip C247S on cellular glucose uptake. Overexpression of wild-type Txnip decreased 2-[3H]deoxyglucose uptake in Txnip-knockout MEFs compared with that of an empty vector control (Figure 1I). Interestingly, overexpression of Txnip C247S also strongly decreased glucose uptake. Further analysis demonstrated that decreased glucose uptake by overexpression of Txnip WT or C247S was accompanied by reduced cellular ATP contents (Figure 1J). These data confirm that Txnip inhibits cellular glucose metabolism and decreases intracellular ATP levels through a thioredoxin-independent mechanism, which could mediate baseline cell death [26].
Txnip C247S induces less cellular ROS and cytotoxicity under hypoxia.
Since overexpression of Txnip under basal conditions appeared to cause cytotoxicity independent of thioredoxin, we hypothesized that effects due to inhibition of thioredoxin may be more apparent under challenge with ROS-induced stress. Overexpression of wild-type Txnip and Txnip C247S in Txnip-null MEFs induced similar levels of LDH release under baseline conditions (Figure 2A). Incubation of Txnip-null MEFs with 200–400 μM hydrogen peroxide for 3 hrs caused a modest further increase in LDH release from empty vector-transfected and wild-type Txnip-transfected cells. Note that cytotoxicity of high levels of hydrogen peroxide was relatively mild in empty vector-transfected cells, because Txnip-null cells were resistant to oxidative stress compared with wild-type MEFs (Supplemental figure 1A). Interestingly, overexpression of Txnip C247S in Txnip-null cells induced less LDH release than overexpression of wild-type Txnip under 400 μM hydrogen peroxide (Figure 2A). This beneficial effect was abolished by treatment with a potent inhibitor of thioredoxin (PX-12, 10 μM) or thioredoxin reductase (D9, 20 nM), supporting the notion that Txnip Cys247 mediates cytotoxicity through inhibition of thioredoxin (Supplemental figure 1B).
Figure 2. Txnip Cys247 regulates the induction of oxidative stress and cell death under hypoxic conditions.
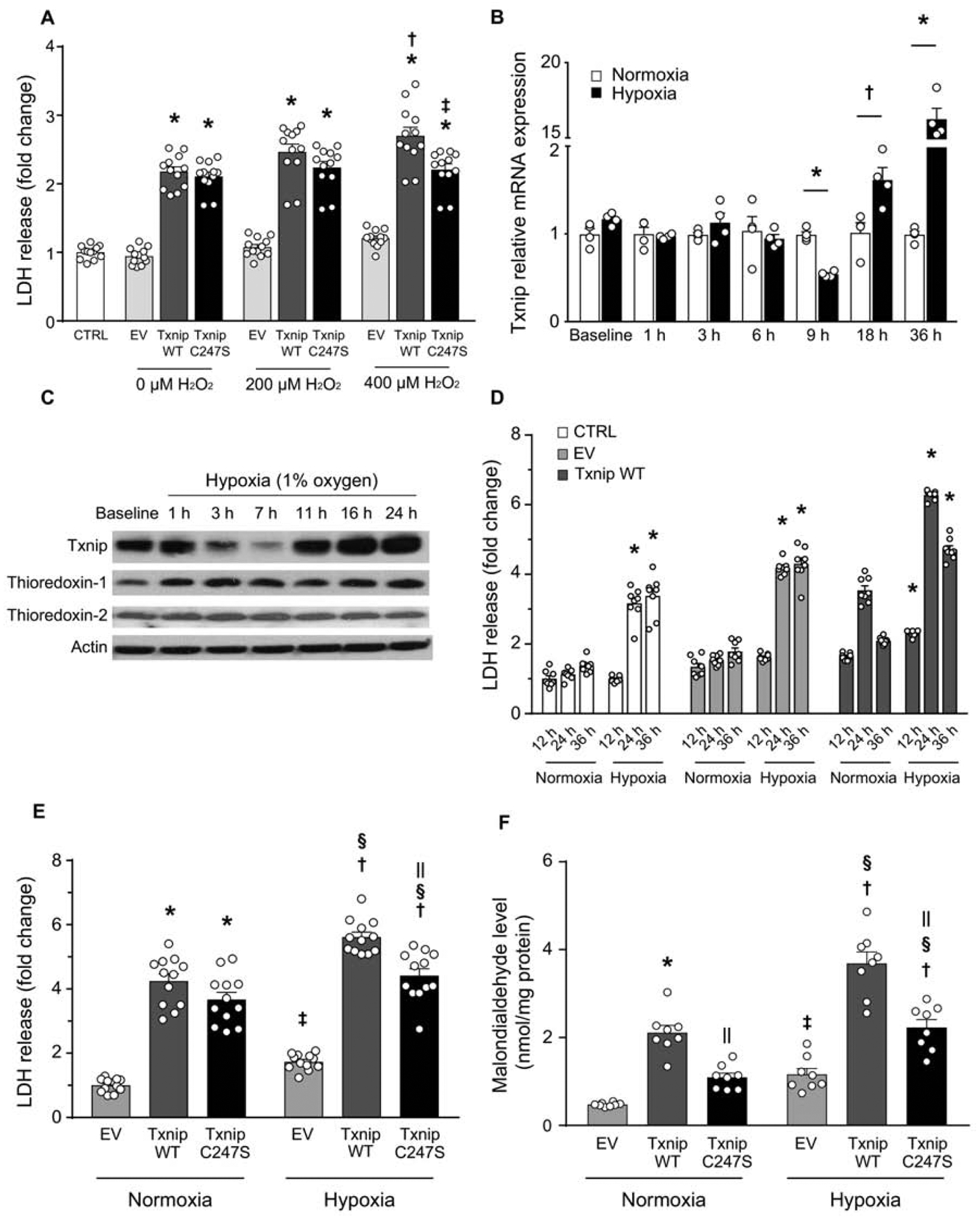
A. Txnip C247S blocked ROS-induced cell death. MEFs derived from Txnip-knockout mice were reconstituted with adenoviral gene transfer of Txnip wild-type (WT), C247S, or empty vector (EV). Cells were incubated with indicated concentrations of hydrogen peroxide (H2O2) for 3 hrs. *P<0.01 EV, †P<0.01 vs. 0 μM Txnip WT, ‡P<0.01 vs. 400 μM Txnip WT. B. Time-dependent changes of Txnip mRNA expression by hypoxia (1% O2) in SKBR3 cells. *P<0.01 and †P<0.05 vs. normoxia. C. Western blot analyses in H9c2 rat cardiomyocytes. Txnip expressions were sensitive to oxygen levels. D. Adenoviral overexpression of Txnip enhanced lactate dehydrogenase (LDH) release from SKBR3 cells under hypoxia. *P<0.01 vs. normoxia. E and F. Txnip-null MEFs were transfected with adenoviruses encoding Txnip WT, C247S, or EV. Hypoxia (1% O2, 24 hrs) increased LDH release and cellular production of malondialdehyde, a marker of oxidative stress. C247S reduced hypoxia-induced cellular damage and formation of ROS. *P< 0.01 vs. EV normoxia, †P<0.01 vs. EV hypoxia, ‡P<0.05 vs. EV normoxia, §P<0.01 vs. the same adenovirus under normoxia, and ||P<0.01 vs. Txnip WT under the same condition.
Cell survival is also threatened when cells are deprived of oxygen. During hypoxia, the mitochondrial electron transport chain generates ROS that act as signals to trigger diverse cellular responses [27]. Thus, we sought to determine the impact of Txnip C247S on hypoxia-induced cell death. SKBR3 cells or H9c2 cardiomyocytes were incubated under hypoxia (1% O2). Txnip mRNA (Figure 2B) and protein (Figure 2C) expressions were regulated in a biphasic manner by hypoxia, with an initial early decrease (at 7–9 hrs) followed by a significant increase with prolonged hypoxia (at 16–36 hrs). The long exposure to hypoxia (24 hrs) significantly increased the protein expression level of Txnip in cardiomyocytes (Supplemental figure 1C). However, hypoxia did not alter thioredoxin expression levels, suggesting that the level of Txnip may be a critical regulator of the anti-oxidant activity of thioredoxin under prolonged hypoxia. Hypoxic stimuli (24–36 hrs) increased cellular LDH release in Txnip-knockout MEFs (Figure 2D). Adenoviral reconstitution of wild-type Txnip further enhanced the hypoxia-induced cytotoxicity. This effect was partially but significantly reduced by Txnip C247S (Figure 2E). Furthermore, Txnip C247S-expressing MEFs had a lower level of oxidative stress measured by cellular production of malondialdehyde compared with wild-type Txnip-expressing MEFs (Figure 2F). These results demonstrate that Txnip expression is sensitive to cellular oxygen level, and suggest that cellular survival under hypoxia is dependent on redox balance regulated by Txnip through its inhibition of thioredoxin.
Generation of inducible cardiomyocyte-specific Txnip C247S knock-in mice.
To address the physiological role of Txnip’s interaction with thioredoxin, we used the Cre-loxP system to generate mice with a tissue-specific Txnip C247S knock-in mutation. Two loxP sites were introduced in the targeting construct for Cre-mediated deletion of wild-type Txnip gene exons 2–8 including a stop codon (Figure 3A). The second loxP site was followed by second construct of Txnip gene exons 2–8, containing a single mutation of C247S in exon 5. Upon Cre-mediated deletion of the original Txnip exons 2–8, the exons 2–8 containing the C247S mutation took their place in the Txnip gene. Thus, knock-in of the Txnip mutation can be achieved in a tissue of interest only.
Txnip(loxP- exons2–8-loxP)C247S/(loxP- exons2–8-loxP)C247S mice were crossed with mice expressing αMHC-MerCreMer. Treatment with 4-hydroxytamoxifen caused Cre recombination of the targeted allele, eliminating wild-type exons 2–8 and allowing expression of the replacement exons 2–8 containing C247S in exon 5 in cardiomyocytes only. DNA sequencing verified the single base pair substitution resulting in a serine codon in place of cysteine at the targeted allele (Figure 3B). Genomic PCR confirmed the homologous recombination in mouse tails. Only homologous integration events yielded PCR products of 356 bp, whereas the size of wild type PCR products was 300 bp (Figure 3C). Genomic DNA was digested with HindIII and NcoI and hybridized with the 3’ probe (Figure 3A). Southern analysis (Figure 3D) identified the existence of the conditional knock-in allele (C-KI, 2941 bp) in the heart after treatment with 4-hydroxytamoxifen (used as Txnip C247S knock-in mice), but not with vehicle lacking 4-hydroxytamoxifen (used as Txnip wild-type control animals). No aberrant Txnip protein was synthesized from the knock-in allele (Figure 3E). There are at least two types of thioredoxins in mammalian cardiomyocytes: cytosolic thioredoxin-1 and mitochondrial thioredoxin-2 [3]. Txnip binds both types of thioredoxins [28]. Western blot analysis demonstrated that expression levels of Txnip, thioredoxin-1 and thioredoxin-2 were all similar in heart tissues between Txnip C247S mice and their Txnip wild-type littermate controls (Figure 3E, Supplemental figure 2). Consistently, reducing activities of thioredoxin-1 and thioredoxin-2 were comparable in these heart tissues between genotypes (Figure 5A and B) at baseline. These results demonstrate that mutation of Txnip Cys247 was successfully engineered within the myocardium in vivo, but that the basal thioredoxin activities were not affected by the C247S mutation, consistent with the previous failure to identify effects of Txnip deletion on baseline thioredoxin activity in Txnip-deficient animals [12, 13]. To assess cardiac structure and function in these animals, echocardiographic parameters were measured. There were no differences between Txnip wild-type and C247S mice in left ventricular dimensions and wall thickness at baseline (Supplemental table 1).
Figure 5. Cardiomyocyte-specific Txnip C247S hearts have increased anti-oxidative activities following myocardium infarction.
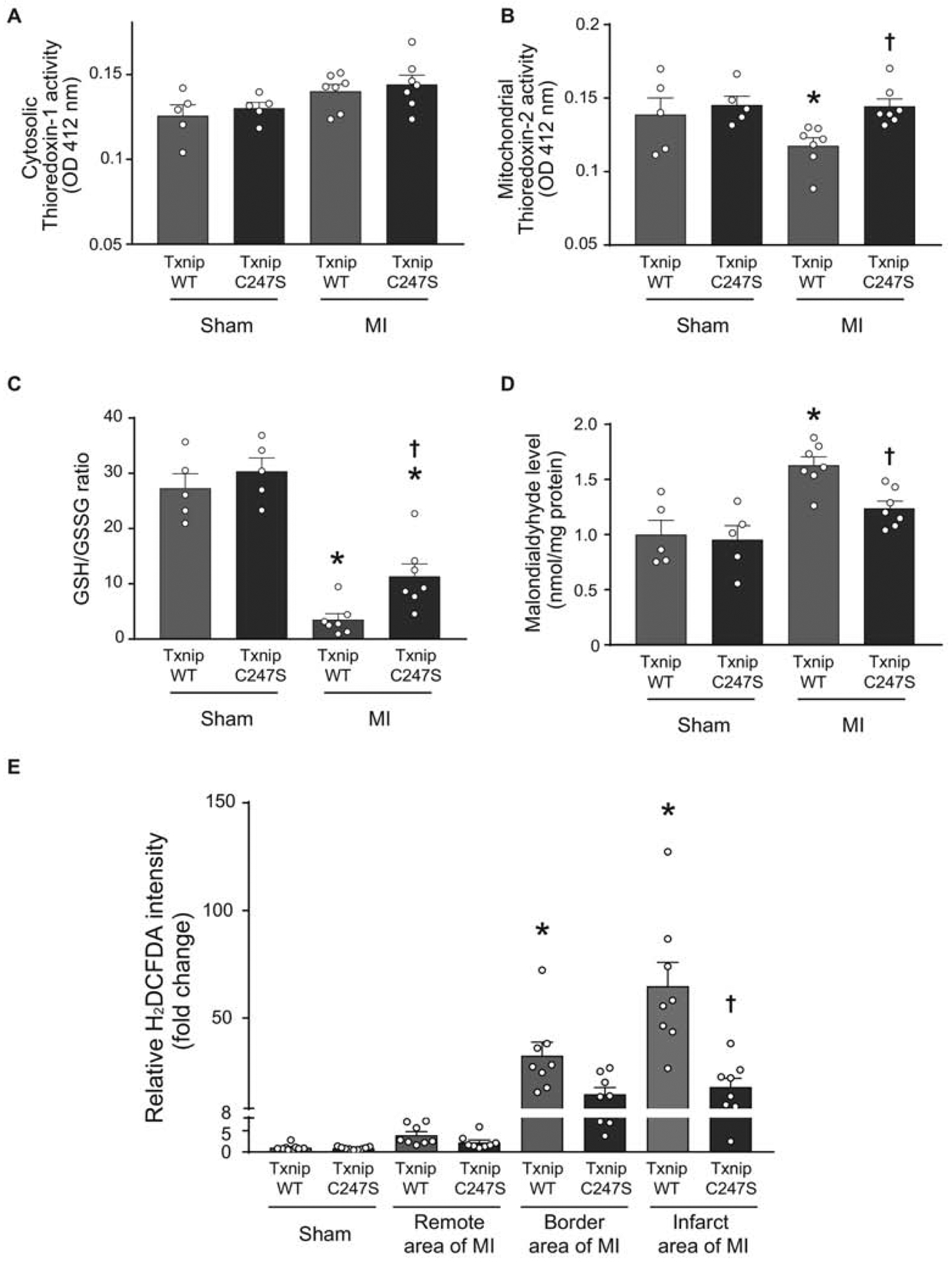
A and B. Thioredoxin’s reducing activities were measured in cytosolic (A) and mitochondria (B)-enriched fractions from the heart tissues of mice that underwent experimental MI or sham surgeries. *P<0.05 vs. Sham, †P<0.05 vs. WT MI. C and D. Tissue levels of GSH/GSSG ratio (C) and malondialdehyde (D) were analyzed in the mouse heart as markers of oxidative stress. *P<0.01 vs. Sham, †P<0.05 vs. WT MI. E. Frozen sections of the mouse heart were stained with a fluorogenic dye H2DCFDA that measures ROS activity within the myocardium. The fluorescence intensity was expressed in the remote, border, or infarct area as a relative change over the Sham group. *P<0.01 vs. Sham and remote area, †P<0.01 vs. WT infarct area.
Cardiomyocyte-specific Txnip C247S mutation is protective against ischemic injury in mice.
Txnip gene expression was induced by hypoxia in isolated cells. We therefore hypothesized that, even though thioredoxin activity was not changed in Txnip-C247S or Txnip-null [12, 13] tissues at baseline, Txnip might regulate thioredoxin activity under hypoxia. Furthermore, previous studies have reported that targeted deletion [20] or degradation of Txnip mRNA [29] protects the myocardium from ischemic injury. Given the detrimental effects associated with elevated Txnip, the question then arises as to what extent Txnip’s interaction with thioredoxin contributes to the cardioprotective response to ischemia. To address these issues, Txnip C247S mutant and their age-matched littermate control mice (both genders) were subjected to sham or experimental MI surgeries. Of the 12 Txnip wild-type (seven males and five females) and 12 C247S (five males and seven females) mice that underwent the sham surgeries, no mouse died by the sham procedure throughout the protocol. Of the 43 Txnip wild-type (20 males and 23 females) and 38 C247S (17 males and 21 females) mice that underwent MI surgeries, 12 Txnip wild-type (seven males and five females) and three C247S (one male and two females) mice died during the protocol. No mouse died during the perioperative phase (within 12 hrs after surgery). The survival rates were statistically different between genotypes determined by Kaplan-Meier analysis test (Figure 4A), but there was no gender difference in survival by the chi-square test.
Figure 4. Cardiomyocyte-specific Txnip C247S knock-in mice exhibit better survival with smaller infarct size following coronary artery ligation compared to control animals.
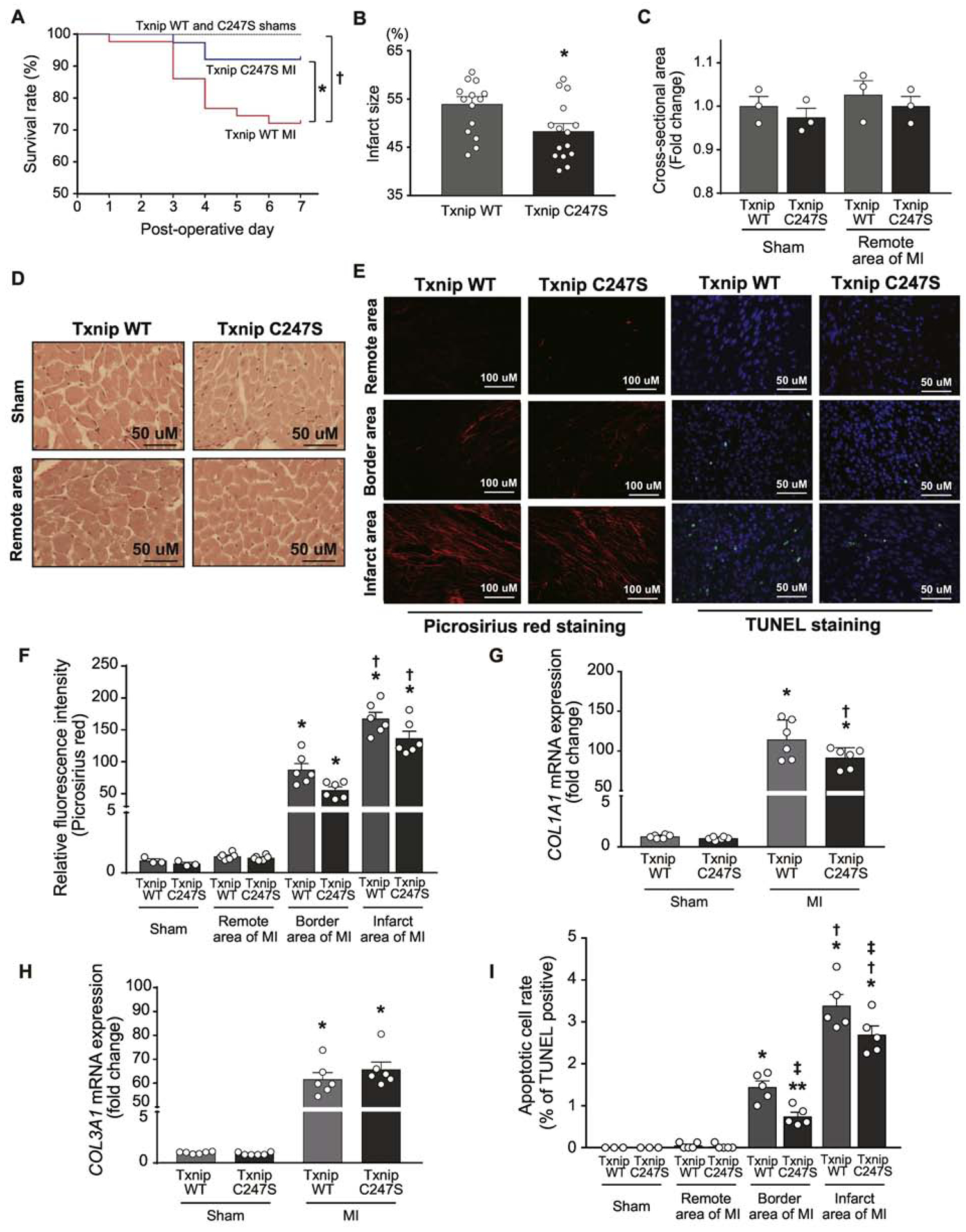
A. Kaplan-Meier curves show better survival rate at Day 7 following experimental myocardial infarction (MI) in Txnip C247S mice than in wild-type (WT) control mice. †P<0.05 between WT sham (n=12) and WT MI (n=43), *P<0.05 between WT MI and C247S MI (n=38), and P=N.S. between C247S sham (n=12) and C247S MI. B. Txnip C247S mice had smaller infarct size than that of Txnip WT mice. *P<0.05. C-F. Histological assessments with picrosirius red staining revealed no significant change in myocyte cross-sectional area (C and D) or myocardial collagen contents (E and F) between genotypes following MI. *P<0.01 vs. sham and remote area, and †P<0.01 vs. border area. G and H. The mRNA expression levels of COL1A1 and COL3A1 were measured by quantitative PCR in the apical areas of the heart tissue. *P<0.01 vs. sham, †P<0.05 vs. Txnip WT. E and I. Apoptotic cells were measured with TUNEL (green) and DAPI (blue). Txnip C247S mice had a decreased number of TUNEL-positive cells in the infarct area. *P<0.01 or **P<0.05 vs. sham and remote area, †P<0.01 vs. border area, and ‡P<0.05 vs. Txnip WT under the same area of condition.
Following coronary artery ligation, there were no differences between genotypes in the area at risk evaluated by phthalocyanine blue staining and the degree of ST-segment elevation on electrocardiograms (Supplemental figure 3). However, consistent with the difference in survival, infarct size was significantly smaller in Txnip C247S mice than in Txnip wild-type mice (Figure 4B). Left ventricular function was assessed by echocardiography before and after MI or sham surgeries. The result revealed that while left ventricular function was significantly reduced by MI, both groups subjected to MI had similar trends in fractional shortening and left ventricular mass (Table 1). We speculate that the discrepancy between infarct size and echocardiographic findings is attributed to the limitations of M-mode echocardiography, in which two-dimensional volume was reconstructed in hearts with anterior wall motion abnormalities. Acute MI did not induce cardiac hypertrophy in remote area, measured by myocyte cross-sectional area (Figure 4C and 4D) or heart weight normalized by tibial length in Txnip wild-type (Sham 56±2 vs. MI 57±2 mg/cm, P=N.S.) and C247S (Sham 53±2 vs. MI 57±1 mg/cm, P=N.S.) mice. MI led to fibrosis within the infarct and borderline myocardium. There was a trend towards less collagen depositions in Txnip C247S hearts than wild-type hearts (Figure 4E and F). The transcript level of collagen type I alpha 1 chain (COL1A1), but not type III alpha 1 chain (COL3A1), supported this trend, showing less induction of COL1A1 in MI hearts from Txnip C247S mice than wild-type animals (Figure 4G and H). Interestingly, Txnip-C247 mutant hearts exhibited a decreased number of TUNEL-positive apoptotic cells compared with the hearts from wild-type controls (Figure 4E and 4I). These results suggest that cardiomyocyte-specific single mutation of Cys247 in Txnip allowed animals to better survive with smaller infarct size and less cardiomyocyte death following MI.
Table 1.
Echocardiographic parameters in Txnip wild-type and C247S mice seven days after sham or MI surgeries.
| Sham | MI | |||
|---|---|---|---|---|
| WT | C247S | WT | C247S | |
| Echocardiography, n | 12 | 12 | 25 | 25 |
| Heart rate, beats/min | 688 ± 16 | 720 ± 11 | 700 ± 10 | 709 ± 7 |
| End-diastolic dimension, mm | 3.0 ± 0.1 | 3.0 ± 0.1 | 4.2 ± 0.1* | 3.9 ± 0.1† |
| End-systolic dimension, mm | 1.1 ± 0.1 | 1.1 ± 0.1 | 3.0 ± 0.1* | 2.7 ± 0.1† |
| Anterior wall thickness, mm | 0.6 ± 0.01 | 0.6 ± 0.02 | 0.4 ± 0.01* | 0.4 ± 0.01† |
| Posterior wall thickness, mm | 0.6 ± 0.01 | 0.6 ± 0.01 | 0.5 ± 0.02** | 0.6 ± 0.01 |
| Fractional shortening, % | 64 ± 2 | 64 ± 2 | 28 ± 1* | 29 ± 1† |
| Left ventricular mass per body weight | 2.3 ± 0.1 | 2.6 ± 0.2 | 3.2 ± 0.2* | 3.0 ± 0.2 |
P < 0.01 or
P < 0.05 vs. wild-type (WT) sham,
P < 0.01 vs. C247S sham. P=N.S. between WT and C247S for any parameters.
Cardiomyocyte-specific Txnip C247S mutation reduces oxidative stress following MI.
To determine whether functional benefits of Txnip C247S hearts under ischemia are attributable to decreased levels of ROS, we measured reducing activities of cytosolic thioredoxin-1 and mitochondrial thioredoxin-2 in MI hearts. No differences in myocardial activities of cytosolic thoredoxin-1 were seen at baseline or after MI between Txnip wild-type and C247S mice (Figure 5A). Mitochondrial thioredoxin-2 activities were also comparable between Txnip wild-type and C247S mice at baseline. However, mitochondrial thioredoxin-2 activities were significantly decreased by MI in Txnip wild-type hearts but not in Txnip C247S hearts (Figure 5B). This result indicates that the Txnip C247S mutation restored the thioredoxin2 activity in MI hearts to the non-MI baseline level.
Because thioredoxin-2 is a key mitochondrial protein that regulates cellular redox by suppressing mitochondrial ROS generation in cardiomyocytes [30], the ratio of GSH to GSSG was measured in these MI hearts. The anti-oxidative activity assessed by the GSH/GSSG ratio was not different between genotypes at baseline, but it was significantly decreased by MI in both groups (Figure 5C). However, Txnip C247S hearts showed a higher level of GSH/GSSG ratio after MI than wild-type hearts, consistent with the higher activity of thioredoxin-2 in the same hearts. The levels of cellular lipid peroxide, an indicator of oxidative stress estimated as malondialdehyde, were increased by MI in Txnip wild-type hearts but not in C247S hearts (Figure 5D). Myocardial ROS generation, as assessed by staining of the green fluorescence dye H2DCFDA, was also increased by ischemic damage but maintained lower levels in Txnip C247S hearts than in wild-type hearts (Figure 5E). These experiments revealed that Txnip C247S mice lacked the ability to inhibit mitochondrial thioredoxin-2 after MI and that the beneficial effect was correlated with decreased levels of ROS production in Txnip C247S hearts.
UCHL1-HIF-1 axis is a part of Txnip C247S-mediated cardioprotection against hypoxia.
To probe the signaling network mediated by Txnip Cys247 further, we performed an unbiased high-throughput RNA-seq to profile potential transcriptional responses in Txnip C247S animals (the Gene Expression Omnibus accession number GSE166252). Of the 60 genes that were identified as differentially expressed between Txnip C247S and wild-type mice, 24 and 36 genes were up- and down-regulated, respectively, in Txnip C247S hearts compared to wild-type hearts (Figure 6A, Supplemental table 2). Functional enrichment analysis predicted associations of these molecules (23 nodes and 27 edges) as an interactive network (Figure 6B). Particularly interesting was the upregulation of UCHL1, an upstream activator of hypoxia-inducible factor 1 (HIF-1). The increased expression of UCHL1 was confirmed at both mRNA and protein levels by quantitative real-time PCR and Western blot analyses, respectively (Figure 6C–E). HIF-1 is a master regulator of the cellular adaptive response to hypoxia. UCHL1 deubiquitinates HIF-1α protein [31], upregulates HIF-1 activity, and mediates cellular protection under hypoxia. To confirm that UCHL1-HIF-1 axis is mechanistically involved in the cardioprotection against hypoxia in Txnip C247S hearts, isolated mouse hearts were perfused with a hypoxic (95% N2, 5% CO2) or normoxic (95% O2, 5% CO2) solution in the Langendorff preparation. Left ventricular functional response, as measured by rate-pressure product (Figure 6F) or end-diastolic pressure (Figure 6G), was significantly impaired by continuous hypoxic perfusion compared with normoxic perfusion. However, Txnip C247S hearts preserved better mechanical function during 25 min of hypoxic perfusion than wild-type hearts. Thus, while two-dimensional echocardiography failed to detect LV functional differences in MI hearts between genotypes, the LV pressure analyses with the ex vivo approach revealed that Txnip C247S hearts maintained greater LV function during hypoxia than wild-type hearts. The mRNA level of UCHL1 remained increased in Txnip C247S hearts following hypoxia (1.55±0.22 fold increase vs. wild-type hearts, P<0.05, n=8 each). As expected, DNA binding activities of specific HIF-1α transcription factor in nuclear extracts from these hearts were significantly increased by hypoxic perfusion. Interestingly, Txnip C247S hearts showed an even higher level of HIF-1α transcription factor than wild-type hearts under hypoxia (Figure 6H). Thus, the increase in UCHL1 expression in Txnip C247S hearts led to a rapid accumulation of nuclear HIF-1α protein in response to acute hypoxic stimuli. These results indicate that Txnip C247S cardiomyocytes acquired a hypoxia-resistant phenotype mediated through a UCHL1-HIF-1 axis-associative mechanism.
Figure 6. An unbiased RNA sequencing screen identifies UCHL1-HIF-1 axis as a part of Txnip C247S-mediated molecular mechanisms.
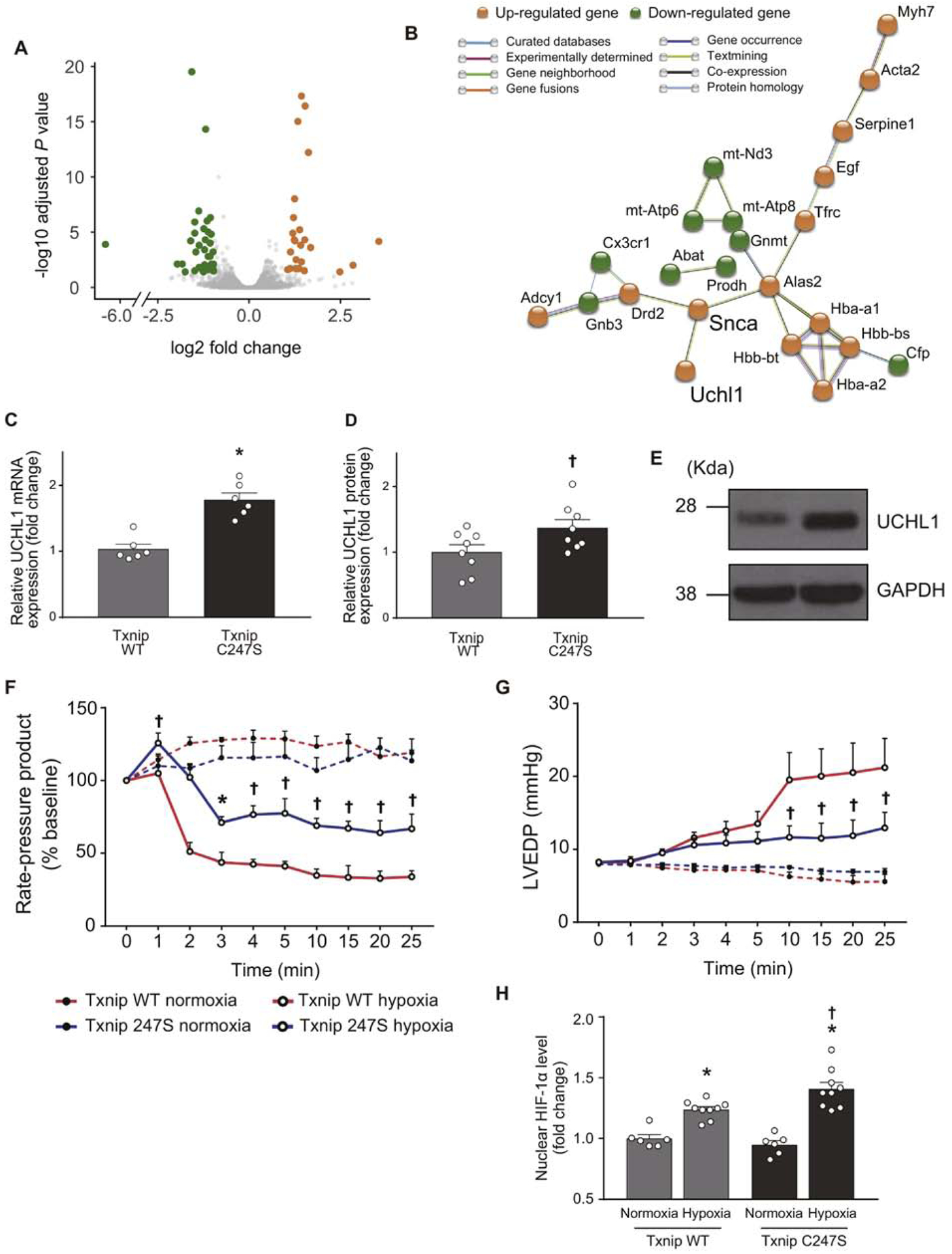
A. RNA-seq was performed in the heart tissues from Txnip wild-type (WT) and C247S mice (n=3 each). A volcano plot shows that 24 genes (orange) and 36 genes (green) were up- and down-regulated, respectively, in Txnip C247S hearts compared to control hearts. B. Functional enrichment analysis using STRING database identified a molecular interaction network mediated by Txnip C247S. UCHL1 is an activator of HIF1. C-E. Increased expression of UCHL1 were confirmed in Txnip C247S hearts by quantitative PCR (C) and Western blot analysis (D and E). *P<0.01 or †P<0.05 vs. WT. F-H. To test whether UCHL1-HIF-1 axis is functional, the Txnip C247S heart was subjected to global hypoxia (95% N2, 5% CO2; n=6) or normoxia (95% N2, 5% CO2; n=4) in Langendorff perfusion system. Txnip C247S hearts maintained better cardiac mechanical function during hypoxic perfusion, as shown by the higher rate-pressure product (F) and lower left ventricular end-diastolic pressure (LVEDP; G) than those in WT hearts. *P<0.05 or †P<0.01 vs. WT hypoxia. At the end of perfusion, HIF-1α levels were measured in nuclear extracts with ELISA. Txnip C247S hearts had higher HIF-1α levels to bind the HIF-1α response element under hypoxia (H). *P<0.01 vs. normoxia, and †P<0.05 vs. WT hypoxia.
DISCUSSION
Txnip inhibits thioredoxin activity in vitro by forming a mixed disulfide bond with thioredoxin at its catalytically active cysteines in a disulfide exchange reaction [9]. The molecular interaction between these two key molecules has for two decades generated speculation that Txnip is a key regulator of cellular redox balance [10]. However, until now it had been unclear whether the Txnip-thioredoxin interaction had any functional importance on in vivo physiology. In the present study, we generated a novel mouse with cardiomyocyte-specific “knock-in” of a mutant Txnip that does not interact with thioredoxin. Our results provide the first direct evidence that inhibition of mitochondrial thioredoxin-2 is required for the induction of oxidative stress by Txnip in the heart, which contributes to post-MI cardiotoxicity.
There are several strengths and limitations to note in this study. Firstly, our approach to generate conditional and inducible mutagenesis was useful to overcome limitations in interpreting results had Txnip been modified in all cells of the body. Txnip has wide-ranging functions including the inhibition of cellular growth, which impacts multiple organs including skeletal muscle [32] and adipose tissues [17]. Thus, we introduced cardiomyocyte-specific mutagenesis prior to inducing MI in adult animals. A disadvantage of the technique is that the efficiency of Cre excision does not normally exceed 75% [33]. Despite this issue, we believe that our approach was sufficient to demonstrate a cardiomyocyte-specific role for regulation of thioredoxin-2 by Txnip.
Secondly, our results reveal explanations for why previous investigations had yielded surprisingly little evidence for an in vivo role for regulation of thioredoxin by Txnip. While the Txnip C247S mutation abolishes the inhibition of thioredoxin by Txnip, we found no difference in thioredoxin activity between Txnip wild-type and C247S mutant hearts at baseline (Figure 5A). This is consistent with previous in vivo studies from independent groups that found that Txnip-deficient mice had no change in thioredoxin activity [12, 13]. We hypothesize that unlike forced overexpression in cells in vitro, physiological levels of Txnip do not regulate thioredoxin activity so drastically. On the other hand, we found that cellular Txnip expression was robustly increased by prolonged hypoxia (Figure 2B and 2C). Endogenous Txnip primarily exists in the nucleus in mammalian cells [34], but oxidative or ischemic stress leads to Txnip shuttling into the mitochondria, where Txnip binds to and inhibits mitochondrial thioredoxin-2 [20, 28]. Correspondingly, we observed a higher activity of mitochondrial thioredoxin-2 in MI hearts from Txnip C247S mice compared to those from wild-type mice (Figure 5B). Taken together, our results suggest that Txnip does function in vivo to inhibit thioredoxin, but it operates primarily in the mitochondria under specific conditions of pathological stress.
Thirdly, in vitro results raise the question as to why induction of cell death was equivalent between cells with overexpression of Txnip wild-type and those with C247S mutant under normoxia (Figure 1). Recent studies uncovered a thioredoxin-independent function of Txnip that may be attributable to its alpha-arrestin domain.[35] Txnip is a multifunctional protein that can interact with several binding partners involved in redox-independent cell signaling. For example, Txnip binds to glucose transporters (GLUTs), promoting internalization of GLUTs and inhibiting cellular glucose uptake [18, 19, 21]. These interactions are not mediated by Txnip Cys247, because Txnip C247S mutant still binds to GLUT1 and inhibits cellular glucose uptake [21, 35]. Therefore, the toxicity of both overexpressed Txnip and Txnip C247S under normoxia is consistent with a ROS-independent mechanism for cell death, such as metabolic inhibition.
Fourthly, because Txnip controls redox balance inside cardiomyocytes through a C247-mediated mechanism, a similar phenotype of the mutant mouse of Txnip C247S to Txnip knockout mouse might be expected under ischemic injuries. Indeed, we have discovered that Txnip C247S mutant mice exhibit a preserved cardiac mechanical function during hypoxia (Figure 6C and 6D) similar to mice with deletion of Txnip [20]. However, this does not necessarily suggest that Txnip C247S mutant animals share the same cardioprotective mechanism with Txnip-knockout animals. Inhibition of glucose metabolism by upregulation of Txnip may be a part of the pathological processes that were seen in hypoxic stress and resulted in extensive cell death, a significant portion of which was attributed to necrosis and apoptosis. In the case of the knockout model, deletion of Txnip in mice can enhance anaerobic glycolysis with a robust increase in myocardial glucose uptake, along with a high level of thioredoxin-2 activities during ischemia/reperfusion [20]. Txnip C247S mutant lacks the ability to affect glucose utilization [21, 35]. Therefore, while knockout experiments test pleiotropic cellular functions of Txnip, our knock-in model specifically identifies a thioredoxin-dependent function of Txnip.
Fifthly, we previously found that Txnip binds to protein disulfide isomerase (PDI) through Txnip Cys247 and shifts the PDI redox equilibrium. PDI contains domains closely similar in their amino acid sequences to that of thioredoxins, and facilitate protein folding inside the ER [36]. We analyzed myocardial expression of X-box binding protein-1 (XBP1) mRNA, a marker of ER stress, and found no difference in ER stress between Txnip wild-type and C247 hearts both at baseline and after hypoxic insults (Supplemental figure 4). Thus, the significance of the Txnip Cys247-PDI interaction is limited in ischemic hearts in vivo.
Finally, the next-generation RNA sequencing identified an interesting signaling network of UCHL1-HIF-1 axis mediated by Txnip Cys247. An increase in UCHL1 expression in Txnip C247S hearts was accompanied by a rapid accumulation of HIF-1α in response to acute hypoxic stimuli. HIF1 has been implicated in regulating a number of genes that are responsible for the metabolic adaptation to hypoxia. Acute HIF-1α stabilization protects the heart against acute ischemic injuries by promoting aerobic glycolysis and decreasing mitochondrial oxidative stress [37]. A relevant question is how Txnip Cys247 regulates UCHL1-HIF-1 expressions. Thioredoxin-1 has been described to increase HIF-1α DNA binding and its transcriptional activity [38], yet another report suggests that thioredoxin-1 and thioredoxin-2 have opposed regulatory functions on HIF-1α [39]. It is unclear whether the activation of HIF-1 that we observed in the Txnip C247S hearts was due to activation of thioredoxins. Another point is that Txnip C247S mutation might be able to confer anti-oxidant properties on the heart through alternative mechanisms. For example, HIF-1-mediated reprogramming may lead to decreased ROS, as activation of the UCHL1-HIF-1 axis induces the production of anti-oxidant GSH by stimulating the pentose phosphate pathway [40]. Furthermore, glutathione peroxidase activities can be enhanced by increasing expression of α-synuclein (Snca) [41] (Figure 6B and Supplemental table 2). Hence, an unbiased high-throughput RNA-seq revealed several putative regulatory pathways mediated by Txnip Cys247.
To summarize, we identified the Txnip-thioredoxin interaction as a crucial mechanism to regulate redox balance in the heart. While this concept has often been demonstrated in vitro, our results begin to resolve long-standing questions by providing the first direct evidence for the functional inhibition of thioredoxin by Txnip in vivo. Hypoxia-induced Txnip interferes with mitochondrial thioredoxin-2, after it translocates into the mitochondria by MI-induced ROS formation (Figure 7). However, Txnip C247S mutant hearts exhibit unperturbed thioredoxin-2 activity, thereby protecting the heart from ROS. These data provide a new mechanistic basis for understanding the pathophysiology of acute MI, and insights into the role of a single cysteine in Txnip that may be a molecular target for therapeutic intervention.
Figure 7. Mechanisms by which Txnip C247S achieves cardioprotection.
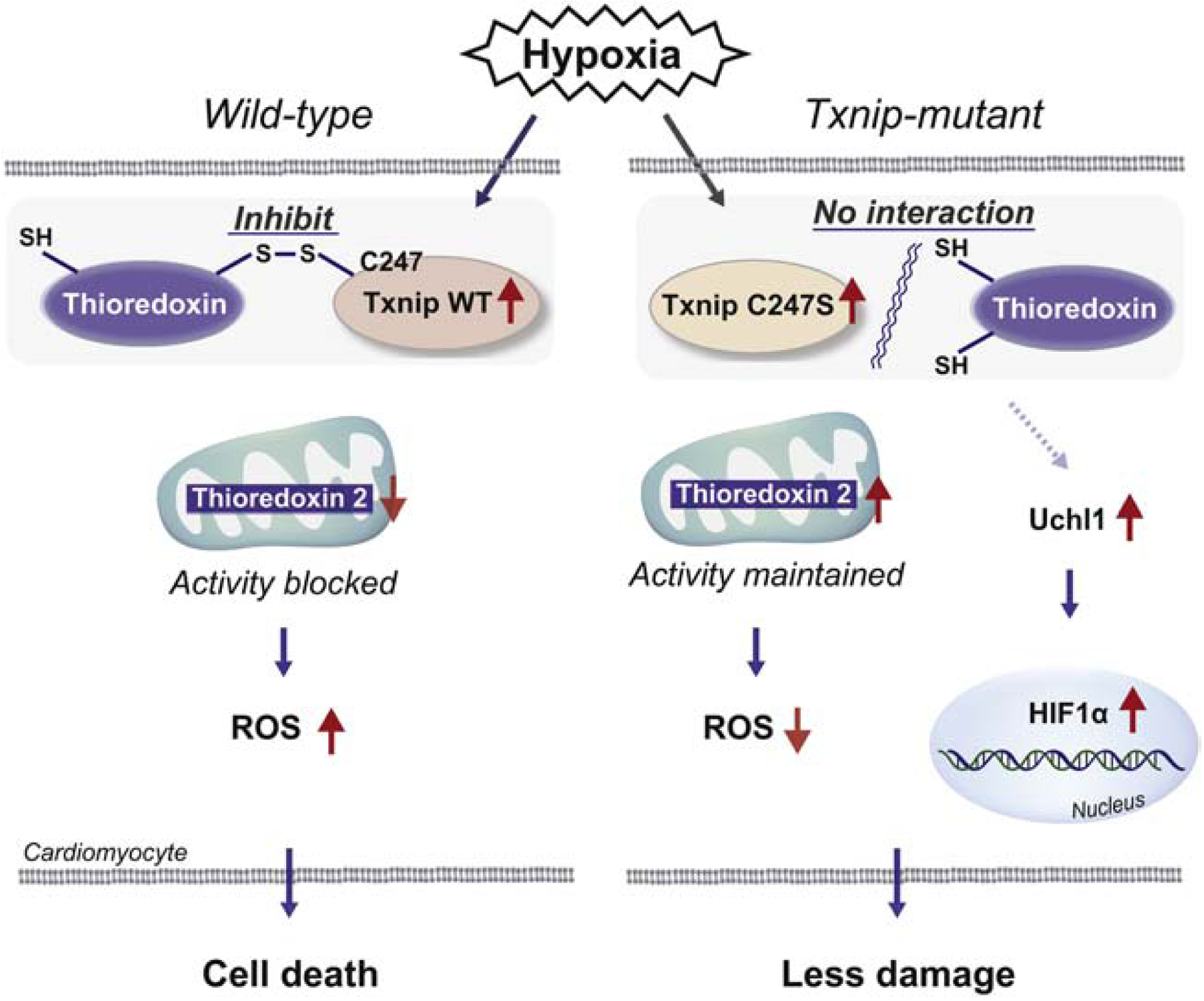
In response to ischemia, Txnip is shuttled into the mitochondria, where Txnip binds to thioredoxin-2 through a disulfide bond formation, resulting in inhibition of anti-oxidant activities and promotion of cardiomyocyte damage in wild type. Txnip C247S fails to form the complex with thioredoxins, thereby increases anti-oxidant activities and protects cardiomyocytes from ischemic insults. Another mechanism involved is activation of the UCHL1-HIF-1 axis, which confers hypoxic tolerance on the Txnip C247S heart.
Supplementary Material
Highlights.
Txnip is a cysteine-containing redox protein that inhibits antioxidant thioredoxin.
Txnip forms a disulfide bridge through Cys247 with thioredoxin.
Txnip C247S results in a reduction in ROS due to an inability to bind thioredoxin.
Txnip C247S knock-in mice have better outcome after myocardial infarction.
Regulation of redox balance by Txnip is crucial for myocardial homeostasis.
Sources of Funding
This work was supported by awards from the Philip V. & Anna S. Brown Foundation, the LaRue S. Fisher and Walter F. Fisher Memorial Trust, and by a NIH grant 1R01HL130861.
Non-standard Abbreviations and Acronyms
- αMHC
α-myosin heavy chain
- BAC
bacterial artificial chromosome
- C247S
cysteine 247-to-serine substitution
- COL1A1
collagen type I alpha 1 chain
- COL3A1
collagen type III alpha 1 chain
- dCTP
deoxycytidine triphosphate
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GLUT
glucose transporter
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- H2DCFDA
2’,7’-dichlorodihydrofluorescein diacetate
- HIF-1
hypoxia-inducible factor 1
- LDH
lactate dehydrogenase
- MerCreMer
tamoxifen inducible Cre recombinase
- MEF
mouse embryonic fibroblast
- MI
myocardial infarction
- Nrf2
nuclear factor erythroid 2–related factor 2
- ROS
reactive oxygen species
- Snca
α-synuclein
- TCA
Trichloroacetic acid
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
- Txnip
thioredoxin-interacting protein
- UCHL1
ubiquitin C-terminal hydrolase L1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures None.
REFERENCES
- [1].Anderson JL, Morrow DA, Acute Myocardial Infarction, N Engl J Med 376(21) (2017) 2053–2064. [DOI] [PubMed] [Google Scholar]
- [2].Chen YR, Zweier JL, Cardiac mitochondria and reactive oxygen species generation, Circ Res 114(3) (2014) 524–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Yoshioka J, Thioredoxin superfamily and its effects on cardiac physiology and pathology, Compr Physiol 5(2) (2015) 513–30. [DOI] [PubMed] [Google Scholar]
- [4].Kallis GB, Holmgren A, Differential reactivity of the functional sulfhydryl groups of cysteine-32 and cysteine-35 present in the reduced form of thioredoxin from Escherichia coli, J Biol Chem 255(21) (1980) 10261–5. [PubMed] [Google Scholar]
- [5].Nicholson CK, Lambert JP, Molkentin JD, Sadoshima J, Calvert JW, Thioredoxin 1 is essential for sodium sulfide-mediated cardioprotection in the setting of heart failure, Arterioscler Thromb Vasc Biol 33(4) (2013) 744–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Adluri RS, Thirunavukkarasu M, Zhan L, Akita Y, Samuel SM, Otani H, Ho YS, Maulik G, Maulik N, Thioredoxin 1 enhances neovascularization and reduces ventricular remodeling during chronic myocardial infarction: a study using thioredoxin 1 transgenic mice, J Mol Cell Cardiol 50(1) (2011) 239–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Yamamoto M, Yang G, Hong C, Liu J, Holle E, Yu X, Wagner T, Vatner SF, Sadoshima J, Inhibition of endogenous thioredoxin in the heart increases oxidative stress and cardiac hypertrophy, J Clin Invest 112(9) (2003) 1395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kaimul AM, Nakamura H, Masutani H, Yodoi J, Thioredoxin and thioredoxin-binding protein-2 in cancer and metabolic syndrome, Free Radic Biol Med 43(6) (2007) 861–8. [DOI] [PubMed] [Google Scholar]
- [9].Patwari P, Higgins LJ, Chutkow WA, Yoshioka J, Lee RT, The interaction of thioredoxin with Txnip. Evidence for formation of a mixed disulfide by disulfide exchange, J Biol Chem 281(31) (2006) 21884–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Nishiyama A, Matsui M, Iwata S, Hirota K, Masutani H, Nakamura H, Takagi Y, Sono H, Gon Y, Yodoi J, Identification of thioredoxin-binding protein-2/vitamin D(3) up-regulated protein 1 as a negative regulator of thioredoxin function and expression, J Biol Chem 274(31) (1999) 21645–50. [DOI] [PubMed] [Google Scholar]
- [11].Hwang J, Suh HW, Jeon YH, Hwang E, Nguyen LT, Yeom J, Lee SG, Lee C, Kim KJ, Kang BS, Jeong JO, Oh TK, Choi I, Lee JO, Kim MH, The structural basis for the negative regulation of thioredoxin by thioredoxin-interacting protein, Nat Commun 5 (2014) 2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chutkow WA, Patwari P, Yoshioka J, Lee RT, Thioredoxin-interacting protein (Txnip) is a critical regulator of hepatic glucose production, J Biol Chem 283(4) (2008) 2397–406. [DOI] [PubMed] [Google Scholar]
- [13].Sheth SS, Castellani LW, Chari S, Wagg C, Thipphavong CK, Bodnar JS, Tontonoz P, Attie AD, Lopaschuk GD, Lusis AJ, Thioredoxin-interacting protein deficiency disrupts the fasting-feeding metabolic transition, J Lipid Res 46(1) (2005) 123–34. [DOI] [PubMed] [Google Scholar]
- [14].Bodnar JS, Chatterjee A, Castellani LW, Ross DA, Ohmen J, Cavalcoli J, Wu C, Dains KM, Catanese J, Chu M, Sheth SS, Charugundla K, Demant P, West DB, de Jong P, Lusis AJ, Positional cloning of the combined hyperlipidemia gene Hyplip1, Nat Genet 30(1) (2002) 110–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sheth SS, Bodnar JS, Ghazalpour A, Thipphavong CK, Tsutsumi S, Tward AD, Demant P, Kodama T, Aburatani H, Lusis AJ, Hepatocellular carcinoma in Txnip-deficient mice, Oncogene 25(25) (2006) 3528–36. [DOI] [PubMed] [Google Scholar]
- [16].Yoshihara E, Fujimoto S, Inagaki N, Okawa K, Masaki S, Yodoi J, Masutani H, Disruption of TBP-2 ameliorates insulin sensitivity and secretion without affecting obesity, Nat Commun 1 (2010) 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chutkow WA, Birkenfeld AL, Brown JD, Lee HY, Frederick DW, Yoshioka J, Patwari P, Kursawe R, Cushman SW, Plutzky J, Shulman GI, Samuel VT, Lee RT, Deletion of the alpha-arrestin protein Txnip in mice promotes adiposity and adipogenesis while preserving insulin sensitivity, Diabetes 59(6) (2010) 1424–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sullivan WJ, Mullen PJ, Schmid EW, Flores A, Momcilovic M, Sharpley MS, Jelinek D, Whiteley AE, Maxwell MB, Wilde BR, Banerjee U, Coller HA, Shackelford DB, Braas D, Ayer DE, de Aguiar Vallim TQ, Lowry WE, Christofk HR, Extracellular Matrix Remodeling Regulates Glucose Metabolism through TXNIP Destabilization, Cell 175(1) (2018) 117–132 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wu N, Zheng B, Shaywitz A, Dagon Y, Tower C, Bellinger G, Shen CH, Wen J, Asara J, McGraw TE, Kahn BB, Cantley LC, AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1, Mol Cell 49(6) (2013) 1167–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yoshioka J, Chutkow WA, Lee S, Kim JB, Yan J, Tian R, Lindsey ML, Feener EP, Seidman CE, Seidman JG, Lee RT, Deletion of thioredoxin-interacting protein in mice impairs mitochondrial function but protects the myocardium from ischemia-reperfusion injury, J Clin Invest 122(1) (2012) 267–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Myers RB, Fomovsky GM, Lee S, Tan M, Wang BF, Patwari P, Yoshioka J, Deletion of thioredoxin-interacting protein improves cardiac inotropic reserve in the streptozotocin-induced diabetic heart, Am J Physiol Heart Circ Physiol 310(11) (2016) H1748–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, Penninger JM, Molkentin JD, Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein, Circ Res 89(1) (2001) 20–5. [DOI] [PubMed] [Google Scholar]
- [23].Lindsey ML, Yoshioka J, MacGillivray C, Muangman S, Gannon J, Verghese A, Aikawa M, Libby P, Krane SM, Lee RT, Effect of a cleavage-resistant collagen mutation on left ventricular remodeling, Circ Res 93(3) (2003) 238–45. [DOI] [PubMed] [Google Scholar]
- [24].Koitabashi N, Bedja D, Zaiman AL, Pinto YM, Zhang M, Gabrielson KL, Takimoto E, Kass DA, Avoidance of transient cardiomyopathy in cardiomyocyte-targeted tamoxifen-induced MerCreMer gene deletion models, Circ Res 105(1) (2009) 12–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Chen J, Saxena G, Mungrue IN, Lusis AJ, Shalev A, Thioredoxin-interacting protein: a critical link between glucose toxicity and beta-cell apoptosis, Diabetes 57(4) (2008) 938–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tsujimoto Y, Apoptosis and necrosis: intracellular ATP level as a determinant for cell death modes, Cell Death Differ 4(6) (1997) 429–34. [DOI] [PubMed] [Google Scholar]
- [27].Guzy RD, Schumacker PT, Oxygen sensing by mitochondria at complex III: the paradox of increased reactive oxygen species during hypoxia, Exp Physiol 91(5) (2006) 807–19. [DOI] [PubMed] [Google Scholar]
- [28].Saxena G, Chen J, Shalev A, Intracellular shuttling and mitochondrial function of thioredoxin-interacting protein, J Biol Chem 285(6) (2010) 3997–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Xiang G, Seki T, Schuster MD, Witkowski P, Boyle AJ, See F, Martens TP, Kocher A, Sondermeijer H, Krum H, Itescu S, Catalytic degradation of vitamin D up-regulated protein 1 mRNA enhances cardiomyocyte survival and prevents left ventricular remodeling after myocardial ischemia, J Biol Chem 280(47) (2005) 39394–402. [DOI] [PubMed] [Google Scholar]
- [30].Huang Q, Zhou HJ, Zhang H, Huang Y, Hinojosa-Kirschenbaum F, Fan P, Yao L, Belardinelli L, Tellides G, Giordano FJ, Budas GR, Min W, Thioredoxin-2 inhibits mitochondrial reactive oxygen species generation and apoptosis stress kinase-1 activity to maintain cardiac function, Circulation 131(12) (2015) 1082–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Fang Y, Fu D, Shen XZ, The potential role of ubiquitin c-terminal hydrolases in oncogenesis, Biochim Biophys Acta 1806(1) (2010) 1–6. [DOI] [PubMed] [Google Scholar]
- [32].Parikh H, Carlsson E, Chutkow WA, Johansson LE, Storgaard H, Poulsen P, Saxena R, Ladd C, Schulze PC, Mazzini MJ, Jensen CB, Krook A, Bjornholm M, Tornqvist H, Zierath JR, Ridderstrale M, Altshuler D, Lee RT, Vaag A, Groop LC, Mootha VK, TXNIP regulates peripheral glucose metabolism in humans, PLoS Med 4(5) (2007) e158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sektas M, Specht M, Limited use of the Cre/loxP recombination system in efficient production of loxP-containing minicircles in vivo, Plasmid 53(2) (2005) 148–63. [DOI] [PubMed] [Google Scholar]
- [34].Dutta KK, Nishinaka Y, Masutani H, Akatsuka S, Aung TT, Shirase T, Lee WH, Yamada Y, Hiai H, Yodoi J, Toyokuni S, Two distinct mechanisms for loss of thioredoxin-binding protein-2 in oxidative stress-induced renal carcinogenesis, Lab Invest 85(6) (2005) 798–807. [DOI] [PubMed] [Google Scholar]
- [35].Patwari P, Chutkow WA, Cummings K, Verstraeten VL, Lammerding J, Schreiter ER, Lee RT, Thioredoxin-independent regulation of metabolism by the alpha-arrestin proteins, J Biol Chem 284(37) (2009) 24996–5003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lee S, Min Kim S, Dotimas J, Li L, Feener EP, Baldus S, Myers RB, Chutkow WA, Patwari P, Yoshioka J, Lee RT, Thioredoxin-interacting protein regulates protein disulfide isomerases and endoplasmic reticulum stress, EMBO Mol Med 6(6) (2014) 732–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ong SG, Lee WH, Theodorou L, Kodo K, Lim SY, Shukla DH, Briston T, Kiriakidis S, Ashcroft M, Davidson SM, Maxwell PH, Yellon DM, Hausenloy DJ, HIF-1 reduces ischaemia-reperfusion injury in the heart by targeting the mitochondrial permeability transition pore, Cardiovasc Res 104(1) (2014) 24–36. [DOI] [PubMed] [Google Scholar]
- [38].Naranjo-Suarez S, Carlson BA, Tobe R, Yoo MH, Tsuji PA, Gladyshev VN, Hatfield DL, Regulation of HIF-1alpha activity by overexpression of thioredoxin is independent of thioredoxin reductase status, Mol Cells 36(2) (2013) 151–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zhou J, Damdimopoulos AE, Spyrou G, Brune B, Thioredoxin 1 and thioredoxin 2 have opposed regulatory functions on hypoxia-inducible factor-1alpha, J Biol Chem 282(10) (2007) 7482–90. [DOI] [PubMed] [Google Scholar]
- [40].Nakashima R, Goto Y, Koyasu S, Kobayashi M, Morinibu A, Yoshimura M, Hiraoka M, Hammond EM, Harada H, UCHL1-HIF-1 axis-mediated antioxidant property of cancer cells as a therapeutic target for radiosensitization, Sci Rep 7(1) (2017) 6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Koo HJ, Yang JE, Park JH, Lee D, Paik SR, alpha-Synuclein-mediated defense against oxidative stress via modulation of glutathione peroxidase, Biochim Biophys Acta 1834(6) (2013) 972–6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.