

SUMMARY
Parkinson’s disease-causing mutations in the LRRK2 gene hyperactivate LRRK2 kinase activity and cause increased phosphorylation of Rab GTPases, important regulators of intracellular trafficking. We found that the most common LRRK2 mutation, LRRK2-G2019S, dramatically reduces the processivity of autophagosome transport in neurons in a kinase-dependent manner. This effect was consistent across an overexpression model, neurons from a G2019S knock-in mouse, and human iPSC-derived neurons gene-edited to express the G2019S mutation, and was reversed by genetic or pharmacological inhibition of LRRK2. Furthermore, LRRK2 hyperactivation induced by overexpression of Rab29, a known activator of LRRK2 kinase, disrupted autophagosome transport to a similar extent. Mechanistically, we found that hyperactive LRRK2 recruits the motor adaptor JIP4 to the autophagosomal membrane, inducing abnormal activation of kinesin that we propose leads to an unproductive tug-of-war between anterograde and retrograde motors. Disruption of autophagosome transport correlated with a significant defect in autophagosome acidification, suggesting that the observed transport deficit impairs effective degradation of autophagosomal cargo in neurons. Our results robustly link increased LRRK2 kinase activity to defects in autophagosome transport and maturation, further implicating defective autophagy in the pathogenesis of Parkinson’s disease.
Graphical Abstract
In Brief / eTOC blurb:
Boecker et al. find that hyperactivation of LRRK2 by a Parkinson’s disease-associated mutation disrupts the axonal transport of autophagosomes through the dysregulation of associated motors. Defective transport leads to impaired autophagosome maturation. Thus, dysfunctional autophagy is predicted to contribute to Parkinson’s disease pathogenesis.
INTRODUCTION
The leucine-rich repeat kinase 2 (LRRK2) gene encodes a large multidomain protein linked to the pathogenesis of Parkinson’s disease (PD).1 LRRK2 mutations are the most common genetic cause of PD, accounting for ~1% of sporadic and ~5% of familial forms.2 The most frequent pathogenic mutation, LRRK2-G2019S, is an autosomal dominant missense mutation located in the kinase domain that enhances kinase activity.3-5 Increased LRRK2 kinase activity is also induced by a PD-causing mutation in the VPS35 gene,6 and has been observed in patients with idiopathic PD.7 Together, these observations indicate that elevated activation of LRRK2 kinase may be a common pathogenic driver of PD, although the mechanism by which increased LRRK2 activity leads to neurodegeneration remains unknown.
Phospho-proteomic analyses identified a subset of Rab GTPases, including Rab8, Rab10, and Rab29, as bona fide substrates of LRRK2 kinase.8,9 Rab proteins are master regulators of intracellular trafficking.10 Increased LRRK2 kinase activity enhances Rab phosphorylation, altering interactions with downstream effector proteins such as the motor adaptor protein JNKinteracting protein 4 (JIP4).11,12 In particular, Rab29 is genetically linked to PD13 and functions as both a substrate and activator of LRRK2 kinase.14-16
Effective intracellular transport is essential for neurons because of their unique morphology with an axonal arbor ranging up to hundreds of meters in total length within the human CNS.17 The extensive axonal arborization of dopaminergic nigrostriatal neurons may make these cells especially vulnerable to transport deficits.18,19 In particular, axonal transport of autophagic vesicles (AVs) in neurons is tightly regulated and vital to neuronal homeostasis.20,21 AVs are preferentially formed at the distal tip of the axon and then transported retrogradely towards the cell soma, maturing en route by fusion with late endosomes/lysosomes.21-24 Inhibition of AV transport impairs AV acidification, causing defective cargo degradation.24,25 Defects in autophagy have repeatedly been linked to PD in general, and LRRK2-mediated pathophysiology in particular.26-30
Here, we investigated the effects of the LRRK2-G2019S mutation on organelle transport and neuronal autophagy. We examined three model systems: LRRK2-G2019S overexpression, primary G2019S knock-in (KI) neurons,31,32 and gene-edited human iPSC-derived G2019S KI neurons. In all three models, LRRK2-G2019S decreased the processivity of AV transport through increased kinase activity. Impaired AV transport was accompanied by defects in axonal AV acidification, indicating defective AV maturation. LRRK2 hyperactivation induced by Rab29 overexpression decreased the processivity of AV transport to a similar extent. Downstream of hyperactive LRRK2, our data implicate the enhanced recruitment of the kinesin-activating motor adaptor protein JIP4 in the disruption of AV transport, resulting in an unregulated tug-of-war between anterograde and retrograde motors. Our data linking LRRK2 hyperactivation to the defective trafficking of neuronal autophagosomes further establish autophagy as a key pathway in PD pathogenesis.
RESULTS
Overexpression of LRRK2-G2019S disrupts the processivity of AV transport.
Given the data implicating LRRK2 in the regulation of autophagy, we asked whether overexpression of LRRK2-G2019S affects axonal AV transport.26-30 Rat hippocampal neurons were transfected with EGFP-LRRK2-WT or EGFP-LRRK2-G2019S. In parallel, we used the kinase-inactive variant EGFP-LRRK2-G2019S-D1994N to assess the dependency of the effect of LRRK2-G2019S on its increased kinase activity (Figure 1A). In all three groups, most mCherry-LC3 labeled AVs were transported retrogradely (~80%, Figures S1A-B) as previously described in neurons both in vitro and in vivo.21-24,33,34 The density of AVs in the mid-axon was similar in all groups (Figure S1C), suggesting that neither overexpression of hyperactive nor kinase-dead LRRK2 affects the overall rate of autophagosome production in the distal axon.
Figure 1. Overexpression of LRRK2-G2019S disrupts processivity of axonal AV transport.

(A) Kymographs of mCherry-LC3 vesicles in EGFP-LRRK2-WT, −G2019S, and −G2019S-D1994N expressing axons. Asterisks indicate pauses. Arrowheads point to reversals. See also Video S1. (B-C) Bar graph and frequency distribution of (B) pause number per vesicle and (C) pause duration during AV transport in WT, G2019S, and G2019S-D1994N expressing neurons. Frequency distribution does not show pauses > 42 sec (WT: 2.54%, G2019S: 2.87%, G2019S-D1994N: 3.49%). (D) Fraction of time paused (as measured by seconds paused per minute), (E) Number of reversals per AV in WT, G2019S, and G2019S-D1994N expressing axons (mean ± SEM; n = 87-139 AVs from 17-20 neurons from 3 independent cultures; ns, not significant, p=0.44; **p=0.0008; ***p<0.0001; Kruskal-Wallis with Dunn’s multiple comparisons test). See also Figures S1A-C for quantification of AV directionality and density. Figure S3 and Figure S4 show the effect of LRRK2-G2019S overexpression on microtubule and LAMP1-vesicle dynamics, respectively.
Strikingly, we observed that AVs in neurons expressing LRRK2-G2019S paused frequently during retrograde organelle motility (Figure 1A, Video S1). The number of pauses per vesicle in LRRK2-G2019S expressing neurons was more than 2-fold higher than in neurons expressing LRRK2-WT (Figure 1B). In contrast, AVs in LRRK2-G2019S-D1994N neurons paused at a similar frequency as in LRRK2-WT neurons (Figure 1B), suggesting that increased pausing is dependent on the hyperactive kinase activity of LRRK2-G2019S. The average pause duration in LRRK2-G2019S neurons was not significantly different from LRRK2-WT or LRRK2-G2019S-D1994N cells (Figure 1C), but due to the higher frequency of pauses, the fraction of time paused per vesicle was significantly increased (Figure 1D). AVs in LRRK2-G2019S neurons also changed direction more frequently (Figure 1A, Video S1). The number of reversals was significantly increased in LRRK2-G2019S neurons as compared to either LRRK2-WT or LRRK2-G2019S-D1994N (Figure 1E).
Endogenous LRRK2-G2019S disrupts AV transport in mouse cortical neurons and iPSC-derived neurons in a kinase-dependent manner.
Next, we investigated whether LRRK2-G2019S affects axonal AV transport when expressed at endogenous levels. Western Blots confirmed similar LRRK2 expression levels in cortical neurons from WT or G2019S KI mice (Figures S1D-E). In both WT and G2019S KI neurons, the majority of axonal EGFP-LC3 labeled AVs moved in the retrograde direction (Figure S1F). The number of pauses during AV transport was significantly increased in G2019S KI neurons, with AVs pausing ~2 times more frequently (Figures 2A-B, Video S2). The average pause duration was not affected (Figure 2C). Thus, the increased fraction of time paused of AVs in G2019S KI neurons (Figure 2D) is due to the higher pause number. The number of reversals during AV transport was also significantly increased in G2019S KI neurons (Figure 2E). To quantify how this higher number of reversals affects the overall processivity of AV transport, we calculated the difference between total and net run length of each vesicle (Δ run length; Figure S1G). To ensure robust run length quantification, we imaged for longer time periods and with a larger field of view. Notably, Δ run length was significantly higher in G2019S KI neurons, indicating a loss of directed processivity in AV transport due to an increase of non-processive motility (Figure 2F).
Figure 2. AV transport is disrupted in mouse LRRK2-G2019S knock-in neurons and rescued by LRRK2 kinase inhibition.

(A) Kymographs of axonal EGFP-LC3 vesicles in WT and G2019S KI mouse cortical neurons. See also Video S2. (B-C) Bar graph and frequency distribution of (B) pause number per vesicle and (C) pause duration during AV transport in WT and G2019S KI neurons. Frequency distribution does not show pauses > 48 sec (WT: 2.95%, G2019S: 6.89%). (D) Fraction of time paused, (E) Number of reversals, (F) Δ run length (difference between total run length and net run length) of AVs in WT and G2019S KI neurons (mean ± SEM; n = 112-126 AVs from 26 neurons from 3 independent cultures; ns = not significant, p=0.78; ***p<0.0001; Mann-Whitney test). Figures S1D-G show LRRK2 expression and AV directionality in WT and G2019S KI neurons. (G) Kymographs of axonal EGFP-LC3 vesicles in mouse G2019S KI neurons treated overnight with DMSO or 100 nM MLi-2. See also Video S3. (H) Pause number, (I) Pause duration, (J) Fraction of time paused, (K) Number of reversals, (L) Δ run length of AVs in G2019S KI neurons treated with DMSO or MLi-2 (mean ± SEM; n = 106-116 AVs from 24-28 neurons from 3 independent cultures; ns = not significant, p=0.02; ***p<0.0001; Mann-Whitney test with Bonferroni correction for multiple testing: p<0.0125 was considered statistically significant). Dotted lines indicate the respective average observed in untreated WT neurons. See also Figures S2A-B for phospho-Rab Western blots of WT or G2019S KI neurons +/− MLi-2.
Microtubule dynamics in G2019S KI neurons and the effect of MLi-2 on microtubule dynamics are shown in Figures S2 and S3. Figure S4 shows LAMP1-vesicle dynamics in G2019S KI neurons.
To investigate whether the loss of processivity was caused by increased LRRK2 activity, we tested whether the LRRK2 kinase inhibitor MLi-235 rescues AV transport in G2019S KI neurons. Treatment with 100 nM MLi-2 markedly decreased LRRK2 kinase activity in G2019S KI neurons, as measured by reduced levels of LRRK2-phosphorylated pT73 Rab10 and pS106 Rab12 (Figures S2A-B). LRRK2 inhibition by MLi-2 significantly reduced pause number and fraction of time paused for axonal AVs in G2019S KI neurons (Figures 2G-J, Video S3). Both parameters were reduced to levels lower than in untreated WT neurons (see dotted lines in Figures 2H-L). MLi-2 treatment also rescued non-processive motility in G2019S KI neurons, as measured by number of reversals and Δ run length (Figures 2K-L).
As a third independent model, we investigated the effect of G2019S mutation on AV transport in human iPSC-derived neurons (i3Neurons),34,36 which have been shown to express LRRK2.37 We used CRISPR/Cas9 gene editing to generate i3N iPSCs heterozygous for the G2019S mutation. EGFP-LC3 labeled AVs in G2019S KI i3Neurons paused significantly more frequently than in isogenic WT i3Neurons (Figures 3A-B, Video S4). While the average pause duration was not affected by the G2019S mutation (Figure 3C), the fraction of time paused, number of reversals, and Δ run length were significantly increased (Figures 3D-F). Treatment with 100 nM MLi-2 rescued the number of pauses and fraction of time paused in G2019S KI i3Neurons (Figures 3G-J). While MLi-2 did not have a significant effect on Δ run length, it reduced the number of reversals to a similar level as observed in WT neurons (Figures 3K-L). Thus, consistent with the data from overexpression model and mouse G2019S KI neurons, we observed a significant disruption of AV transport in human G2019S KI neurons that was rescued by pharmacological LRRK2 kinase inhibition.
Figure 3. AV transport is disrupted in human iPSC-derived LRRK2-G2019S knock-in neurons and rescued by LRRK2 kinase inhibition.

(A) Kymographs of axonal EGFP-LC3 vesicles in WT and G2019S KI i3Neurons. See also Video S4. (B) Pause number, (C) Pause duration, (D) Fraction of time paused, (E) Number of reversals, (F) Δ run length of AVs in WT and G2019S KI i3Neurons (mean ± SEM; n = 133-189 AVs from 32-33 neurons from 3 independent experiments; ns = not significant, p=0.0299; ***p<0.0001; Mann-Whitney test with Bonferroni correction for multiple testing: p<0.0125 was considered statistically significant). (G) Kymographs of axonal EGFP-LC3 vesicles in G2019S KI i3Neurons treated overnight with DMSO or 100 nM MLi-2. (H) Pause number, (I) Pause duration, (J) Fraction of time paused, (K) Number of reversals, (L) Δ run length of AVs in G2019S KI i3Neurons treated with DMSO or MLi-2 (mean ± SEM; n = 225-279 AVs from 41-43 neurons from 3 independent cultures; ns = not significant, p>0.1368; **p=0.0021; ***p=0.0001; Mann-Whitney test). Dotted lines indicate the respective average in untreated WT i3Neurons.
LRRK2-G2019S does not affect axonal microtubule dynamics.
Previous reports have proposed that LRRK2 mutants may alter microtubule dynamics by affecting microtubule stability, which could potentially contribute to the defects in AV transport we observed.38-40 We expressed fluorescently labeled end binding protein 3 (EB3) in mouse WT and G2019S KI neurons to visualize the plus-ends of growing microtubules and measure microtubule dynamics (Figure S2C). Quantification revealed no differences in run length, run time, velocity, or density of EB3 comets between WT and G2019S KI neurons (Figures S2D-G). Similarly, overexpression of EGFP-LRRK2-WT or EGFP-LRRK2-G2019S in rat hippocampal neurons did not affect run length, run time, or number of EB3 comets when compared to the expression of EGFP only (Figures S3A-H).
Since the LRRK2 inhibitor MLi-2 has been shown to increase the affinity of LRRK2 for microtubules,41 we investigated whether MLi-2 treatment affects microtubule dynamics at endogenous LRRK2 expression levels. We observed no differences in run time, run length, velocity, or density of EB3 comets between DMSO or MLi-2 treated mouse WT neurons (Figures S3I-P).
LRRK2-G2019S does not affect axonal transport of LAMP1 vesicles.
To investigate whether LRRK2-G2019S affects the axonal transport of other cargoes, we examined the motility of LAMP1-RFP positive late endosomes/lysosomes in WT and G2019S KI neurons. First, we photobleached a segment of the axon to deplete the signal of stationary vesicles,42 aiding visualization of mobile vesicles and allowing quantification of pause events (Figure S4A). In contrast to the robust retrograde motility of AVs, about two-thirds of mobile LAMP1 vesicles moved in the anterograde direction (Figure S4G). Measurements of pausing for motile LAMP1 vesicles revealed no difference between WT and G2019S KI neurons (Figures S4B-C). Analysis of retrograde LAMP1 vesicles only showed a small but nonsignificant trend towards a higher pause number and higher fraction of time paused in G2019S KI neurons (Figures S4D-E). Co-expression of EGFP-LC3 and LAMP1-RFP showed that only a small fraction of retrograde LAMP1-RFP vesicles was positive for EGFP-LC3 (~ 13%, Figure S4H). Similar to the observations in G2019S KI neurons, overexpression of LRRK2-G2019S did not significantly alter axonal trafficking of LAMP1-RFP vesicles in rat hippocampal neurons (Figures S4F-M).
Impairment of axonal transport by LRRK2-G2019S affects maturation of axonal AVs.
Retrograde transport facilitates AV maturation in the axon through en route fusion with lysosomal vesicles. Disrupted AV transport has been shown to impair AV acidification, leading to defective cargo degradation.25,43 To probe whether LRRK2-G2019S affects AV maturation, we transfected mouse WT or G2019S KI neurons with mCherry-EGFP-LC3 (Figure 4A). The EGFP signal of this tandem construct is quenched in an acidic environment, allowing for differentiation between non-acidified (mCherry- and EGFP-positive) and acidified (mCherry-only positive) LC3 vesicles.
Figure 4. LRRK2-G2019S disrupts acidification of axonal AVs.

(A) Kymographs of mCherry-EGFP-LC3 vesicles in the proximal axon of WT or G2019S KI mouse cortical neurons. Magenta arrowheads: mCherry-positive traces; green arrowheads: EGFP-positive traces; white arrowheads: mCherry- and EGFP-positive traces. (B-D) Percentage of acidified (= mCherry-only positive) AVs in (B) the distal (mean ± SEM; n = 17-20 neurons from 4 independent cultures; ns, not significant, p=0.55; Mann-Whitney test), (C) the proximal axon of WT and G2019S KI neurons (mean ± SEM; n = 29-30 neurons from 4 independent cultures; **p=0.0084; Mann-Whitney test), (D) the proximal axon of G2019S KI neurons treated with DMSO or 100 nM MLi-2 overnight (mean ± SEM; n = 24-26 neurons from 3 independent cultures; *p=0.0123; Mann-Whitney test).
For acidification of axonal LAMP1 vesicles see Figure S5.
In the distal axon of both WT and G2019S KI neurons, only ~10% of AVs were acidified (Figure 4B). This is not surprising, as de novo formation of immature AVs takes place at the distal tip.22,44 Consistent with AV maturation during retrograde transport, we observed a higher fraction of acidified AVs in the proximal axon. However, the fraction of acidified AVs in the proximal axon of G2019S KI neurons (~20%) was significantly lower compared to WT neurons (~40%) (Figures 4A and 4C). LRRK2 inhibition by MLi-2 fully rescued impaired AV acidification in the proximal axon of G2019S KI neurons (Figure 4D). Impairment of autophagosome transport and acidification correlated with a trend towards a higher number of LC3 vesicles in the proximal axon of G2019S KI neurons (Figure S5A). Treatment with MLi-2 significantly decreased the number of LC3 vesicles in the proximal axon of G2019S KI neurons (Figure S5B). Together, these findings demonstrate that G2019S mutation substantially impairs the maturation of axonal AVs in a kinase-dependent manner.
In contrast to AV transport, axonal trafficking of LAMP1-positive late endosomes/lysosomes was not affected by expression of the G2019S mutation (Figure S4). Furthermore, using SEP-LAMP1-RFP as a dual-color reporter for late endosome/lysosome acidification,45,46 we did not observe a difference in the acidification of axonal LAMP1 vesicles between WT and G2019S KI neurons (Figures S5C-E). These findings suggest that impaired AV acidification in G2019S KI neurons was not induced by lysosomal defects.
Rab29 is localized to axonal AVs.
Rab29 functions as both a substrate and an activator of LRRK2 kinase.8,14,16,32,47 Overexpression of Rab29 has been shown to increase LRRK2 kinase activity.14,16 To investigate whether Rab29 may be involved in the LRRK2 pathway controlling processivity of AV transport, we transiently expressed EGFP-Rab29 together with mCherry-LC3 in mouse cortical neurons. Intriguingly, we observed robust comigration of EGFP-Rab29 with axonal LC3 vesicles (Figure 5A, Video S5). We also noticed a distinct population of dimmer EGFP-Rab29 vesicles moving along the axon that did not colocalize with LC3-positive AVs (Figures 5A and 5G). Consistent with previous observations in cell lines,14,16 there was a Golgi-like distribution of EGFP-Rab29 in the neuronal soma (Figure S6A), spatially separated from the axonal, AV-associated population.
Figure 5. Overexpression of Rab29 disrupts AV transport.

(A) Time lapse images of axonal mCherry-LC3 and EGFP-Rab29 vesicles in a WT mouse cortical neuron. Scale bar, 5 μm. See also Video S5. (B) Fluorescence recovery after photobleaching Halo-Rab29 signal of an EGFP-LC3 labeled AV. Arrowheads point to the position of the AV. Scale bar, 5 μm. See also Figures S6A-C and Video S6. (C) Proteins associated with the outer membrane of isolated AVs are degraded after treatment with Proteinase K. AV cargo is only degraded by Proteinase K after membrane permeabilization. (D) Western Blot of pT71 Rab29 and total Rab29 from brain lysate, autophagosome fraction, and autophagosome fraction after treatment with Proteinase K. PK, Proteinase K. (E-F) Western Blot quantification of (E) pT71 Rab29 levels (mean ± SEM; n = 8-9 biological replicates; *p=0.022; Mann-Whitney test) and (F) total Rab29 levels (mean ± SEM; n = 7-9 biological replicates; ns, not significant, p=0.25; Mann-Whitney test) in autophagosome fraction of WT and G2019S KI mice. Data shown are normalized to total protein and relative to whole brain lysate. Western Blots of LRRK2, GM130, and LC3 are shown in Figures S6D-F. (G) Kymographs of axonal EGFP-Rab5 or EGFP-Rab29 vesicles, and mCherry-LC3 vesicles in mouse WT cortical neurons. Magenta arrowheads point to tracks of mCherry-LC3 vesicles; green arrowheads highlight EGFP-Rab29 tracks that colocalize with mCherry-LC3 tracks. (H) Pause number, (I) Fraction of time paused, (J) Δ run length of AVs in WT and G2019S KI neurons overexpressing Rab5 or Rab29, and in Rab29 overexpressing WT neurons treated with DMSO or MLi-2 (Overexpression of Rab5 or Rab29: mean ± SEM; n = 107-149 AVs from 27-38 neurons from 3-4 independent cultures; ns, not significant, p>0.06; **p<0.001; ***p<0.0001; Kruskal-Wallis with Dunn’s multiple comparisons test. Treatment of Rab29 overexpressing neurons with DMSO or MLi-2: mean ± SEM; n = 78-109 AVs from 26-27 neurons from 3 independent cultures; ns, not significant, p=0.19; ***p<0.0001; Mann-Whitney test). See also Figures S6G-H.
The observed colocalization of Rab29 and LC3 in the axon could be explained by either an association of Rab29 with the outer AV membrane, or by engulfment of Rab29 within the AV lumen as an autophagosomal cargo. To distinguish between these possibilities, we photobleached Halo-Rab29 on EGFP-LC3 positive axonal AVs and analyzed fluorescence recovery. We observed that Halo-Rab29 signal recovered over several minutes (Figures 5B, S6B, Video S6). This indicates that photobleached Halo-Rab29 is gradually replaced by fluorescent Halo-Rab29 from the cytosol, suggesting that Rab29 is bound to the outer AV membrane. If Rab29 was located in the AV lumen, fluorescence recovery would not be observed. Quantification of Halo-Rab29 intensity after photobleaching showed a similar recovery in WT and G2019S KI neurons (Figure S6C).
Next, we investigated whether endogenous Rab29 and LRRK2 are present on AVs. We isolated AVs through sequential ultracentrifugation of mouse brain lysates (Figure S6D),21,48 adding Gly-Phe-β-naphthylamide (GPN) to inactivate and deplete lysosomal vesicles. We detected total Rab29, LRRK2-phosphorylated phospho-T71 (pT71) Rab29, and LRRK2 in immunoblots of isolated AVs from WT or G2019S KI mice. Total Rab29, pT71 Rab29, and LRRK2 were all lost after treating isolated AVs with Proteinase K (Figures 5C-D and S6E). This indicates that Rab29 and LRRK2 are bound to the outer AV membrane, corroborating the results of our Halo-Rab29 photobleaching experiments as well as previous reports of the recruitment of LRRK2 to autophagosomes.47,49,50 Luminal proteins such as LC3-II were protected by the AV membrane from Proteinase K treatment and only degraded after membrane permeabilization by Triton X (Figure S6E). The Golgi marker GM130 was not detected in the isolated AV fraction, excluding the possibility that Rab29 signal was due to contamination of isolated AVs with Golgi membrane (Figure S6E). Notably, pT71 Rab29, but not total Rab29, was enriched in the AV fraction compared to the total brain lysate input (Figure 5D). Consistent with higher LRRK2 kinase activity, we detected increased levels of pT71 Rab29 on AVs from G2019S KI mice (Figure 5E). There was no difference in the amount of total Rab29 or LRRK2 between WT and G2019S KI AVs (Figures 5F and S6F).
Rab29 overexpression disrupts axonal AV transport similar to G2019S mutation.
In non-neuronal cells, overexpression of Rab29 has been shown to recruit LRRK2 to the membrane of the Golgi and increase LRRK2 kinase activity.14,16 Recent studies found that membrane association but not membrane identity is important for LRRK2 activation by Rab29.15,51
Since we detected Rab29 on axonal AVs, we investigated whether overexpression of Rab29 affects the processivity of AV transport in a similar manner to the LRRK2-G2019S mutation. We transiently expressed EGFP-Rab29 and mCherry-LC3 in WT or G2019S KI mouse cortical neurons. EGFP-Rab5 was used as a negative control, since Rab5 is not known to affect LRRK2 kinase activity.14 As expected, AVs in G2019S KI neurons overexpressing Rab5 paused more frequently than in WT neurons overexpressing Rab5 (Figures 5G-H). The pause frequency in WT neurons overexpressing Rab29 was significantly increased compared to WT neurons overexpressing Rab5 but similar to G2019S KI neurons overexpressing Rab5. Rab29 overexpression in G2019S KI neurons did not have an additive effect but resulted in a pause number similar to either WT neurons overexpressing Rab29 or G2019S KI neurons overexpressing Rab5 (Figure 5H). While pause number was significantly increased, overexpression of Rab29 did not affect average pause duration (Figure S6G). Fraction of time paused and Δ run length were significantly increased, but again, overexpression of Rab29 did not produce an additive effect in G2019S KI neurons (Figures 5I-J). These observations are consistent with reports that LRRK2 and Rab29 act within the same pathway.52,53
LRRK2 kinase inhibition rescues AV transport in Rab29 overexpressing neurons.
Next, we tested whether Rab29 overexpression affects AV transport through activation of LRRK2 kinase activity; if so, the LRRK2 kinase inhibitor MLi-2 should rescue defective transport in Rab29 overexpressing cells. While the co-migration of axonal LC3 and Rab29 vesicles was not affected by MLi-2 (Figure S6H), LRRK2 kinase inhibition significantly reduced pause number, fraction of time paused, and Δ run length of axonal AVs in WT neurons expressing EGFP-Rab29 (Figures 5H-J). All parameters were rescued to levels similar to WT neurons expressing Rab5 as a negative control. Consistent with earlier experiments, pause duration was not affected by MLi-2 (Figure S6G). Together, our data demonstrate that increased LRRK2 activity, caused by either the G2019S mutation or Rab29 overexpression, disrupts the processivity of axonal AV transport.
LRRK2-G2019S enhances recruitment of JIP4 to the AV membrane and increases kinesin activity.
Both the anterograde motor kinesin and the retrograde motor dynein are bound to AVs; the opposing activities of these motors are regulated by adaptor proteins that inhibit kinesin and promote dynein activity.21,43 The observed increases in reversals and non-processive motility suggest that LRRK2 hyperactivation may increase kinesin activity and thus disrupt AV transport by causing an unregulated tug-of-war between anterograde and retrograde motors.
JIP3 and JIP4 are motor adaptor proteins that bind LRRK2-phosphorylated Rab proteins: Rab8, Rab10, and Rab35.11,12,51 In astrocytes, expression of LRRK2-G2019S increased recruitment of JIP4 to damaged lysosomes by enhancing Rab phosphorylation.12 In line with these findings, we detected significantly higher levels of JIP4 on autophagosomes isolated from the brains of G2019S KI mice compared to WT mice (Figure 6A). JIP3 levels showed a similar trend but did not reach statistical significance (Figure S7A). Both Rab8 and Rab10 were present on the outer AV membrane (Figure S7B). Western Blots using a pan-specific phosphothreonine (pT) Rab antibody (detecting JIP4-interacting phospho Rab8, 10, and 35) showed enrichment of pT Rabs in the AV fraction and increased levels of LRRK2-phosphorylated Rab proteins on AVs from G2019S KI mice (Figure 6B). LRRK2-phosphorylated pT73 Rab10, but not total Rab10, was enriched in the autophagosome fraction (Figure S7B).
Figure 6. LRRK2-G2019S recruits JIP4 to the AV membrane and activates kinesin.

(A-D) Representative Western Blot and quantification of (A) JIP4 (mean ± SEM; n = 8 biological replicates; **p=0.0047; Mann-Whitney test), (B) phospho Threonine Rabs (mean ± SEM; n = 6-7 biological replicates; *p=0.0135; Welch’s t test), (C) p150Glued (mean ± SEM; n = 7-8 biological replicates; *p=0.61; Mann-Whitney test), (D) KHC (mean ± SEM; n = 6-7 biological replicates; *p=0.014; Mann-Whitney test) in the autophagosome fraction of WT and G2019S KI mouse brain lysates. Data shown are normalized to total protein and relative to whole brain lysate. (E-F) Microtubule pelleting assay (E) with lysates of WT or G2019S KI MEFs incubated with GMPCPP-stabilized microtubules (5 μM) in the presence of 10 mM AMP-PNP and quantification (F) of KHC bound to the microtubule pellet (mean ± SEM; n = 4 biological replicates; *p=0.0216; Welch’s t test). (G-H) Microtubule pelleting assay (G) with lysates of G2019S KI MEFs treated with 200 nM MLi-2 overnight and quantification (H) of KHC bound to the microtubule pellet (mean ± SEM; n = 4 biological replicates; *p=0.0286; Welch’s t test). S, supernatant; P, pellet. Figure S7 shows additional Western Blots and IF staining.
JIP4 activates kinesin by binding kinesin light chain and kinesin heavy chain (KHC).54,55 Binding of the GTPase Arf6 to the leucine zipper II domain of JIP4 interferes with JIP4’s association with kinesin but favors interaction with the dynein activator dynactin.56,57 While JIP4 levels were increased on G2019S KI AVs, we detected only low levels of Arf6 associated with AVs and found no difference between Arf6 levels on WT and G2019S KI AVs (Figure S7C). Further, levels of p150Glued, a subunit of dynactin, were similar on WT and G2019S KI AVs (Figure 6C). In contrast and consistent with recruitment of kinesin by JIP4, KHC levels were significantly higher on G2019S KI AVs than on WT AVs (Figure 6D). The presence of JIP4, KHC, and p150Glued on axonal AVs in G2019S KI neurons was confirmed by IF staining (Figures S7D-E). A microtubule pelleting assay in the presence of AMP-PNP (a non-hydrolysable ATP analog) showed increased microtubule binding of KHC from G2019S KI MEF lysates compared to WT, indicating that LRRK2 hyperactivation increases levels of activated kinesin (Figures 6E-F). Treatment with MLi-2 rescued this effect (Figures 6G-H). This suggests that by recruiting JIP4, hyperactive LRRK2 increases the amount of active kinesin on the autophagosomal membrane, resulting in a tug-of-war with the retrograde motor dynein that is predicted to induce more pausing and directional reversals, thus decreasing processive retrograde motility.
JIP4 overexpression disrupts axonal AV transport.
To test whether increased JIP4 association interferes with processive axonal AV motility, we analyzed the transport of EGFP-LC3 vesicles in Halo-JIP4 overexpressing mouse WT neurons. We observed robust comigration of EGFP-LC3 and Halo-JIP4 vesicles (Figures 7A-B, Video S7). Strikingly, Halo-JIP4 overexpression was sufficient to increase the percentage of AVs moving either bidirectionally or in the anterograde direction while decreasing the fraction of retrograde AVs (Figure 7C), consistent with higher kinesin activity on AVs in JIP4 overexpressing neurons. Similar to the effect of G2019S mutation and Rab29 overexpression, JIP4 overexpression increased the number of pauses and time paused per minute of axonal AVs (Figures 7D-E). JIP4 overexpression also elevated the number of reversals and Δ run length during AV transport to levels about twice as high as in G2019S KI or Rab29 overexpressing neurons (Figures 7F-G). Thus, JIP4 overexpression resulted in a similar but more pronounced impairment of AV transport as compared to the effects of LRRK2 hyperactivation. Overall, our findings support a model in which hyperactive LRRK2 recruits JIP4 to the AV membrane via its binding to LRRK2-phosphorylated Rab proteins. JIP4 then recruits and activates kinesin, resulting in a tug-of-war with the retrograde motor dynein and disruption of processive AV transport (Figures 7H-I).
Figure 7. Overexpression of JIP4 disrupts AV transport.

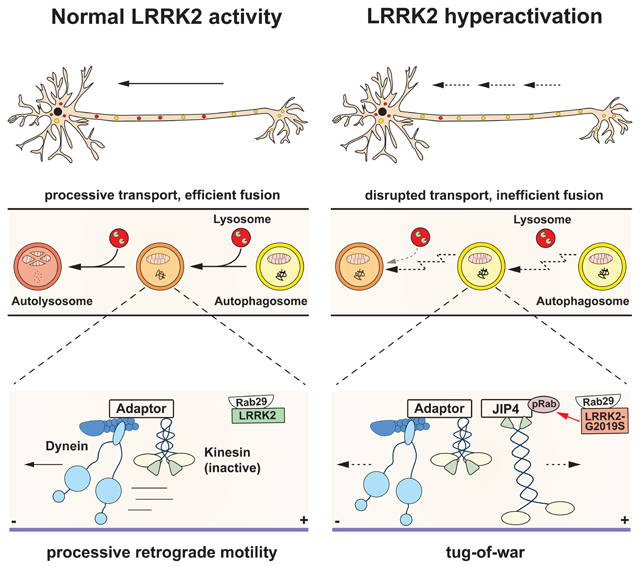
(A) Time lapse images of axonal EGFP-LC3 and Halo-JIP4 vesicles in a mouse WT cortical neuron. Scale bar, 5 μm. See also Video S7. (B) Kymographs of axonal EGFP-LC3 and Halo-JIP4 vesicles. Arrowheads highlight comigration. (C) Directionality of EGFP-LC3 vesicles in Halo-Tag only and Halo-JIP4 overexpressing WT neurons. Antero + bidir., anterograde + bidirectional; retro., retrograde; stat., stationary (mean ± SEM; n = 24-25 neurons from 3 independent cultures; ***p<0.0001; Two-way ANOVA). (D) Pause number, (E) Fraction of time paused, (F) Number of reversals, (G) Δ run length of AVs in Halo-Tag only and Halo-JIP4 overexpressing neurons (mean ± SEM; n = 68-87 AVs from 24-25 neurons from 3 independent cultures; ***p<0.0001; Mann-Whitney test). Dotted lines indicate the respective average observed in mouse G2019S KI neurons. (H-I) Model depicting the effect of increased LRRK2 kinase activity on axonal AV transport and maturation. (H) Normal LRRK2 activity allows for processive retrograde AV transport, facilitating efficient fusion en route with lysosomal vesicles. Motor adaptors inhibit kinesin and promote dynein activity, resulting in processive retrograde transport. (I) LRRK2 hyperactivation by LRRK2-G2019S mutation or Rab29 overexpression disrupts processive AV transport, leading to inefficient autophagosome-lysosome fusion and impaired AV acidification. Hyperactive LRRK2 enhances recruitment of JIP4 to the AV membrane via binding to phospho-Rabs, resulting in abnormal kinesin activation and a tug-of-war between anterograde and retrograde motors.
DISCUSSION
There is a growing body of evidence linking dysfunctional autophagy to PD pathogenesis.58,59 Previous reports support a role for LRRK2 as a regulator of autophagy and suggest that pathogenic LRRK2 mutations cause dysregulation of the autophagic pathway.28-30,60-63 However, the underlying mechanism is not yet clear. The identification of a subgroup of Rab proteins as LRRK2 substrates links LRRK2 kinase activity to dynamic processes of intracellular trafficking and transport. Here, we used live-imaging to study the effect of the most frequent pathogenic LRRK2 mutation, LRRK2-G2019S, on neuronal autophagy. We found that LRRK2-G2019S disrupts processive retrograde transport of axonal AVs in a kinase-dependent manner by increasing pause frequency and promoting non-processive motility. This effect was consistent across a LRRK2-G2019S overexpression model, primary G2019S KI neurons, and human G2019S KI iPSC-derived neurons.
Recent studies indicate that Type 1 LRRK2 kinase inhibitors such as MLi-2 can promote binding of LRRK2 to microtubules and have raised the possibility that microtubule-bound LRRK2 may act as a roadblock for molecular motors.41,64 However, at endogenous LRRK2 expression levels, MLi-2 does not appear to impair transport along microtubules. In fact, we found that MLi-2 improved the transport processivity of AVs in both G2019S KI and Rab29 overexpressing neurons, consistent with a beneficial effect of Type 1 LRRK2 kinase inhibitors across multiple models of PD-related neurodegeneration.65-67 We further found that neither treatment with MLi-2 nor expression of the G2019S mutation affected axonal microtubule dynamics.
In cell lines, overexpressed Rab29 recruits LRRK2 to the Golgi or to stressed lysosomes, leading to localized activation.14,16,47,68,69 While membrane association is essential for LRRK2 activation by Rab29, specific membrane identity is not required.15 We observed in neurons that EGFP-Rab29 robustly co-migrated with axonal AVs and found endogenous Rab29 to be present on the outer membrane of isolated autophagosomes. Overexpression of EGFP-Rab29 in mouse WT neurons disrupted AV transport similar to expression of the G2019S mutation. Treatment with MLi-2 rescued AV processivity in Rab29 overexpressing neurons, confirming that Rab29 overexpression disrupts AV transport by increasing LRRK2 kinase activity. Rab29 overexpression in G2019S KI neurons did not have an additive effect but resulted in a pause number similar to G2019S KI neurons without Rab29 overexpression and to WT neurons overexpressing Rab29. The non-additive effect of G2019S mutation and Rab29 overexpression suggests that both act within the same pathway, and that reaching a certain threshold of LRRK2 activity causes the observed impairment of AV transport. Our result is in line with genetic studies showing that variants of LRRK2 and the Rab29 containing PARK16 locus do not increase PD risk in an additive manner.70
How does increased LRRK2 activity mediate the observed disruption of AV transport by LRRK2-G2019S or Rab29 overexpression? Physiologically, the scaffold proteins huntingtin, HAP1, and JIP1 (structurally distinct from JIP4) enable processive retrograde AV transport by inhibiting kinesin and promoting dynein activity (Figure 7H).25,43 AVs from G2019S KI mice have increased levels of phospho-Rabs and JIP4, a motor adaptor protein that has been shown to bind to LRRK2-phosphorylated Rab proteins.11,12 JIP4 recruits and activates the anterograde motor kinesin, unless kinesin binding is competitively blocked by the GTPase Arf6.57 We observed that increased levels of JIP4 on G2019S AVs were accompanied by increased levels of kinesin; and increasing JIP4 levels by overexpression was sufficient to induce a significantly higher fraction of anterogradely and bidirectionally moving AVs in live-imaging experiments. These results support a model in which hyperactive LRRK2 recruits JIP4 to the AV membrane via its binding to LRRK2-phosphorylated Rab proteins. JIP4 then recruits and activates kinesin, resulting in a tug-of-war with the retrograde motor dynein that is activated by other AV-resident motor adaptors (Figure 7I). Further work will be required to determine which specific phospho-Rab(s) mediate recruitment of JIP4 to the AV membrane. The described model is supported by the phenotype observed in our live-imaging experiments, where LRRK2 activation by G2019S mutation or Rab29 overexpression caused an increase in reversals and non-processive AV motility.
In contrast, we did not detect an effect of LRRK2-G2019S on the axonal transport of LAMP1-vesicles. This is somewhat surprising, as previous reports have found LRRK2-mediated recruitment of JIP4 to the membrane of LAMP1 vesicles in non-neuronal cells.12 Notably, motility of axonal LAMP1 vesicles is overall less processive than transport of AVs (LAMP1 vesicles: ~50% bidirectional/stationary, 25% anterograde, 25% retrograde; AVs: ~80% retrograde vesicles).34,42,71,72 Thus, it is possible that JIP4 recruitment by hyperactive LRRK2 only results in a measurable effect for AVs, where increased JIP4 levels disrupt mechanisms that normally ensure robust retrograde AV motility. Alternatively, the related kinase LRRK1, rather than LRRK2, may play a role in controlling the retrograde motility of LAMP1-positive late endosomes/lysosomes. The late endosome marker Rab7 is phosphorylated by LRRK1 but not LRRK2 at Ser72.73 Rab7 phosphorylation by LRRK1 has been implicated in mediating the recruitment of Rab7-interacting lysosomal protein (RILP),74 a scaffold protein that promotes retrograde transport of late endosomes/lysosomes by enhancing dynein activity.75
Previous work has shown that efficient autophagosomal cargo degradation is dependent on functional retrograde transport.76 Disruption of retrograde axonal AV transport in a model of Huntington’s disease caused ineffective autophagosome-lysosome fusion and impaired autophagosomal cargo degradation.25 Knockdown of the motor scaffold protein JIP1 was also sufficient to disrupt retrograde AV transport and reduce acidification.43 Similarly, we found that disrupted transport in G2019S KI axons was accompanied by a defect in AV acidification. While we observed no change in density, transport, or acidification of axonal LAMP1 vesicles inG2019S neurons, we cannot fully exclude the potential contribution of lysosomal defects to impaired AV maturation. In line with our results, overexpression of LRRK2-G2019S in rat neurons decreased autophagic flux, but did not affect lysosomal pH.30 Disruption of transport and ineffective acidification may delay autophagosomal cargo degradation and over time result in buildup of protein aggregates and/or dysfunctional organelles in the axon. Indeed, accumulation of alpha-synuclein aggregates in the axon (‘Lewy Neurites’) has been shown to occur early during PD pathogenesis.77-79 Evidence from human histopathology and animal models suggests that axonal pathology precedes damage to the neuronal cell body in PD, supporting a disease model of ‘retrograde axonal degeneration’.80-82
In summary, our work robustly links dysregulation of the LRRK2 pathway to defects in neuronal AV transport. We show across different model systems that increased LRRK2 kinase activity induced by either G2019S mutation or Rab29 overexpression strikingly disrupts the processivity of axonal AV transport. Mechanistically, we found that LRRK2 hyperactivation recruits JIP4 to the AV membrane, activating kinesin and resulting in a tug-of-war between anterograde and retrograde motors. Our data connect dysfunction of LRRK2, a kinase that affects intracellular trafficking by phosphorylating Rab proteins, with defects in autophagy, a pathway that has long been implicated in the pathogenesis of Parkinson's disease.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Erika Holzbaur (holzbaur@pennmedicine.upenn.edu).
Materials Availability
Plasmids and iPSC lines generated in this study are available upon request to the lead contact.
Data and Code Availability
The custom MATLAB scripts used in this study to manually track kymographs (KymoSuite) are available at https://github.com/jnirschl/kinesin-3_guedes-dias_2018.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Primary neuron culture
Mouse cortex was dissected from homozygous LRRK2-G2019S or WT embryos of either sex at day 15.5. All experiments were performed following protocols approved by the Institutional Animal Care and Use Committee at the University of Pennsylvania. LRRK2-G2019S knock-in mice (model #13940) and B6NTac (model #B6) were obtained from Taconic, Cambridge City, Indiana production site. Cortical neurons were isolated by digestion with 0.25% Trypsin and trituration through a small-bore serological pipette. Rat hippocampal neurons from E18 Sprague Dawley rats were obtained from the Neuron Culture Service Center at the University of Pennsylvania. Neurons were plated in Attachment Media (MEM supplemented with 10% horse serum, 33 mM D-glucose and 1 mM sodium pyruvate) on poly-L-lysine coated 35 mm glassbottom imaging dishes (P35G-1.5-20-C; MatTek). After 5 hours, Attachment Media was replaced with Maintenance Media (Neurobasal [Gibco] supplemented with 2% B-27 [Gibco], 33 mM D-glucose [Sigma], 2 mM GlutaMAX [Gibco], 100 U/mL penicillin and 100 mg/mL streptomycin [Sigma]). AraC (1 μM) was added the day after plating to prevent glia cell proliferation. Every 3-4 days, 40% of the media was replaced with fresh Maintenance Media. Transfections of mouse cortical neurons (DIV 6-7) or rat hippocampal neurons (DIV 6) were performed 16-24 hours before imaging with Lipofectamine 2000 Transfection Reagent (ThermoFisher) and 0.4 – 1.0 μg of total plasmid DNA. For transduction with EGFP-LC3B, mouse cortical neurons (125,000 cells per imaging dish) were incubated with BacMam EGFP-LC3B reagent (ThermoFisher, P36235) 48 hours before imaging.
Human iPSC culture and neuronal differentiation
Human i3N iPSCs that harbor a doxycycline-inducible mNGN2 transgene in the AAVS1 safeharbor locus were a gift from M. Ward (National Institutes of Health, Maryland) and have been described previously.34,83 Cytogenetic analysis of G-banded metaphase cells demonstrated a normal male karyotype (Cell Line Genetics). Mycoplasma testing was negative. i3N iPSCs were cultured on Growth Factor Reduced Matrigel (Corning) coated plates and fed daily with Essential 8 medium (ThermoFisher). Differentiation into i3Neurons was performed following an established protocol.36 In brief, i3N iPSCs were split with Accutase (Sigma) and plated on Matrigel-coated dishes in Induction Medium (DMEM/F12 containing 2 μg/mL doxycycline, 1% N2-supplement [Gibco], 1% NEAA [Gibco] and 1% GlutaMAX [Gibco]). After 3 days, pre-differentiated i3Neurons were dissociated with Accutase and cryo-preserved. On day of use, pre-differentiated i3Neurons were thawed and plated on poly-L-ornithine coated live-imaging dishes (MatTek) at a density of 300,000 cells per dish. For each experimental condition, cells from at least two different batches of induction were used. i3Neurons were cultured in BrainPhys Neuronal Medium (StemCell) supplemented with 2% B27 (Gibco), 10 ng/mL BDNF (PeproTech), 10 ng/mL NT-3 (PeproTech) and 1 μg/mL Laminin (Corning). Every 3–4 days, 40% of the medium was replaced with fresh culture medium. Live-imaging experiments were performed 21 days after thawing pre-differentiated i3Neurons (DIV21). i3Neurons were transfected 72 hours before live-imaging with Lipofectamine Stem Transfection Reagent (ThermoFisher) and 1 μg of plasmid DNA.
METHOD DETAILS
Plasmids
Plasmids used include EGFP-LRRK2-WT (subcloned from FLAG-LRRK2-WT, gift from W. Smith, Johns Hopkins University, Maryland), EGFP-LRRK2-G2019S (subcloned from FLAG-LRRK2-G2019S, gift from W. Smith, Johns Hopkins University, Maryland), EGFP-LRRK2-G2019S-D1994N (subcloned from FLAG-LRRK2-G2019S-D1994N, gift from W. Smith, Johns Hopkins University, Maryland), mCherry-LC3B (subcloned from EGFP-LC3, a gift from T. Yoshimori, Osaka University, Japan), PGK mCherry-LC3B (gift from Michael Ward, National Institutes of Health, Maryland), PGK EGFP-LC3B (subcloned from PGK mCherry-LC3B), mCherry-EGFP-LC3B (gift from T. Johansen, University of Tromso, Norway), EGFP-Rab29 (gift from D. Alessi, University of Dundee, United Kingdom), Halo-Rab29 (subcloned from EGFP-Rab29), EGFP-Rab5 (gift from M. Zerial, Max Planck Institute Dresden, Germany), LAMP1-RFP (Addgene #1817), EB3-mCherry (gift from A. Akhmanova, Utrecht University, the Netherlands), Halo-JIP4 (subcloned from pGEXP1-JIP4 #DU27651, acquired from MRC PPU Reagents and services, University of Dundee), SEP-LAMP1-RFP (kindly provided by J. Bonifacino, NIH, Bethesda). Unless stated otherwise plasmids used CMV promoter to initiate transcription.
Generation of LRRK2-G2019S knock-in iPSCs
For CRISPR/Cas9 gene editing, i3N iPSCs were cultured on Matrigel coated plates in mTeSR medium (StemCell). Cells were transfected using Lipofectamine Stem Transfection Reagent (TermoFisher) with plasmids encoding Cas9-GFP and gRNA (5’-3’ sequence: TGCTCAGTACTGCTGTAGAATGG), as well as with ssDNA template for homology-directed repair. To generate clones heterozygous for G2019S mutation, iPSCs were transfected with a mix of ssDNA template with and without mutation.84 After 48 hours, iPSCs were sorted through FACS and GFP+ cells were plated on a Matrigel-coated 10 cm dish. Cells were grown for 10 days, then individual colonies were picked and screened for successful gene editing through a restriction site that was introduced as a silent mutation in the ssDNA repair template. Successful editing was confirmed by Sanger sequencing. Karyotype analysis (Cell Line Genetics) demonstrated a normal karyotype for all clones used in this study.
Live-cell imaging
Primary rat hippocampal neurons and mouse cortical neurons were imaged on DIV7-8 in low fluorescence Hibernate E medium (Brain Bits) supplemented with 2% B27 and 2 mM GlutaMAX. i3Neurons were imaged on DIV21 in low fluorescence Hibernate A medium (BrainBits) supplemented with 2% B27, 10 ng/mL BDNF and 10 ng/mL NT-3. Neurons were imaged in an environmental chamber at 37°C on a PerkinElmer UltraView Vox Spinning Disk Confocal system with a Nikon Eclipse Ti inverted microscope. Images were acquired with a Hamamatsu EMCCD C9100-50 camera controlled by Volocity software. Axons were identified based on morphological parameters.34,85 LC3 vesicles in mouse cortical neurons and i3Neurons, Rab5 vesicles, Rab29 vesicles, and EB3 comets were imaged with a Plan Apochromat 60x 1.40 NA oil immersion objective. LC3 vesicles in rat hippocampal neurons and LAMP1 vesicles were imaged using an Apochromat 100x 1.49 NA oil immersion objective. Time lapse recordings were acquired at a frame rate of 3 frames/sec for 5 minutes (LAMP1-RFP vesicles), 1.5 frames/sec for 5 minutes (SEP-LAMP1-RFP vesicles), 1 frame/sec for 5 minutes (EGFP-LC3 vesicles in mouse cortical and i3Neurons), 1 frame/sec for 3 minutes (mCherry-LC3 vesicles in rat hippocampal neurons), 0.8 frames/sec for 5 minutes (EGFP-Rab5/Rab29 + mCherry-LC3 co-trafficking, EGFP-LC3 + Halo-JIP4 co-trafficking, EGFP-LC3 + LAMP1-RFP co-trafficking, mCherry-EGFP-LC3 vesicles), or 0.5 frames/sec for 10 minutes (EB3-mCherry comets). Representative Halo-JIP4 images shown in Figure 7A were denoised using the “Despeckle” function in ImageJ. Recordings were acquired in the mid-axon (> 300 μm from soma and > 100 μm from distal axon terminal) unless stated otherwise. To aid visualization and accurate quantification of LAMP1-RFP vesicles, the area of interest was photobleached prior to the start of image acquisition using UltraView Photokinesis Device (PerkinElmer). For the Rab29 FRAP assay, Halo-Rab29 signal was photobleached ~ 10 μm proximal to distal of an axonal EGFP-LC3 vesicle. Halo-Rab29 and EGFP-LC3 signal were recorded at a frame rate of 0.8 frames/sec for 30 seconds before and 10 minutes after photobleaching.
Autophagosome isolation assay
Enriched autophagosome fractions were isolated following a protocol modified from Strømhaug et al.48 Briefly, one mouse brain was collected in a buffered 250mM sucrose solution, homogenized using a tissue grinder, and subsequently subjected to three differential centrifugations through Nycodenz and Percoll discontinuous gradients to isolate vesicles of the appropriate size and density. Following collection, the autophagosome enriched fraction (AP) was divided into three, one third was treated with 10 μg Proteinase K for 45 min at 37°C to degrade non-membrane protected proteins and enrich for internal autophagosome cargo (AP+PK), one third was membrane permeabilized by the addition of 0.2% Triton X-100 prior to the same Proteinase K treatment as a negative control (AP+Tx+PK), and the other third was left untreated for identification of all internal and externally-associated proteins on autophagosomes. For subsequent protein analysis, the input and the autophagosome enriched fractions were lysed in a buffer with a final concentration of 0.5% NP-40 with 1x protease and phosphatase inhibitors, PMSF and Pepstatin A. Protein concentration was measured by Bradford assay and equal amounts of protein in denaturing buffer were run on SDS-PAGE gels for further analysis by Western Blot.
Microtubule pelleting assay
WT or G2019S KI MEFs were lysed with BRB80 (80 mM PIPES, 1 mM EGTA, and 1 mM MgCl2) lysis buffer containing 0.5% Triton X-100, 2x Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher), and 2 μg/mL microcystin-LR (Sigma). For LRRK2 kinase inhibition, G2019S KI MEFs were treated with DMSO or 200 nM MLi-2 overnight before lysis. Lysates were incubated with 5 μM GMPCPP-stabilized microtubules at 37°C for 20 min, then centrifuged at 38,400 x g for 20 min at 25°C. Subsequently, pellet and supernatant were separated, and analyzed per SDS-PAGE as described below.
Immunoblotting
Neurons were washed twice with ice cold PBS and lysed with RIPA buffer (50 mM Tris-HCl, 150 mM NaCl, 0.1% Triton X-100, 0.5% deoxycholate, 0.1% SDS, 2x Halt Protease and Phosphatase inhibitor, 2 μg/mL microcystin-LR). Samples were centrifuged for 10 min at 17,000 g, and protein concentration of the supernatant was determinied by BCA assay. Proteins were resolved on 8% (LRRK2) - 15% (Rab proteins) acrylamide gels. Proteins were transferred to Immobilon-FL PVDF membranes (Millipore) using a wet blot transfer system. Membranes were then stained for total protein using Li-Cor Revert Total Protein Stain. Following imaging of total protein stain, membranes were destained, blocked for 1 hr with TrueBlack WB Blocking Buffer (Biotium), and incubated with primary antibodies diluted in TrueBlack WB Antibody Diluent + 0.2% Tween-20 (Biotium) overnight at 4°C. After three washes with TBS-Tween, membranes were incubated with secondary antibodies diluted in TrueBlack WB Antibody Diluent + 0.2% Tween-20 and 0.01% SDS for 1 hr at RT. Following three more washes with TBS-Tween, membranes were imaged using Odyssey CLx Infrared Imaging System (LI-COR). Western Blots were analyzed with Image Studio Software (Li-Cor).
Immunostaining
Mouse cortical neurons were fixed and permeabilized for 8 minutes at −20°C using ice-cold methanol. Cells were washed three times with PBS and blocked for 1.5 h hours with 5% goat serum and 1% BSA in PBS. Neurons were then incubated in primary antibodies diluted in blocking solution overnight at 4°C, washed three times with PBS, and incubated in secondary antibodies diluted in blocking solution for 1 hour at RT. After three washes with PBS and nuclear counterstaining with Hoechst (ThermoFisher, H21492), coverslips were mounted in ProLong Glass Antifade mountant (ThermoFisher, P36980). Images were acquired as z-stacks at 200 nm step-size using an Apochromat 100x 1.49 NA oil immersion objective on the Perkin Elmer spinning disk confocal setup described above.
QUANTIFICATION AND STATISTICAL ANALYSIS
LC3 and LAMP1 vesicle motility
Kymographs of LC3, Rab29, JIP4, and LAMP1 vesicles were generated with the Multiple Kymograph plugin for FIJI using a line width of 5 pixels. Vesicle tracks were traced manually with a custom MatLab GUI (KymoSuite). Motile vesicles were scored as anterograde (net displacement > 10 μm in the anterograde direction within time lapse acquistion) or retrograde (net displacement > 10 μm in the retrograde direction). Non-motile vesicles were binned into bidirectional (net displacement < 10 μm, but total displacement > 10 μm) and stationary vesicles (net and total displacement < 10 μm). A pause was defined as a single or consecutive instantaneous velocity value of < 0.083 μm/sec.86 Bidirectional and stationary vesicles were excluded from the quantification of pause number, pause duration and fraction of time paused. A reversal was defined as a change of movement direction succeeded by a run > 1 μm in the opposing direction. For quantification of Δ run length, the net run length of each vesicle was subtracted from its total run length. Analysis of LC3 vesicles in rat hippocampal neurons was performed unblinded, all other analyses were performed by a blinded investigator.
EB3 dynamics
Kymographs were generated using the KymographClear 2.0 macro toolset as previously described.86,87 KymographClear passes a Fourier filter on the original kymograph and allows for automated discrimination between anterograde, retrograde, or static components. The transformation improves signal-to-noise ratio of EB3 comets without affecting quantitative analysis of the data. All kymographs of EB3 comets shown in this paper are forward Fourier-filtered kymographs. Tracks of individual EB3 comets were manually traced using KymoSuite MatLab GUI and used to quantify run-length, run-time and velocity of each comet. Analysis was performed by a blinded investigator.
Autophagosome acidification (mCherry-EGFP-LC3 tandem construct)
Time lapse series were recorded in the proximal (< 150 μm from the cell soma) and distal axon (region immediately proximal to the axon tip). Kymographs were generated using the Multiple Kymograph plugin for FIJI as described above. Traces of LC3 vesicles were counted first in the EGFP, then in the mCherry kymograph by a blinded investigator.
Late endosome/lysosome acidification (SEP-LAMP1-RFP construct)
Time lapse series were recorded in the mid-axon. Kymographs were generated using the Multiple Kymograph plugin for FIJI. Traces of LAMP1 vesicles were counted first in the SEP, then in the RFP kymograph by a blinded investigator.
Rab29 FRAP assay
Halo-Rab29 fluorescence intensity of axonal autophagosomes was measured within a circular area of interest with 0.65 μm diameter. The localization of autophagosomes before and after Halo-Rab29 photobleaching was determined by their (not photobleached) EGFP-LC3 signal. Fluorescence intensity of Halo-Rab29 was quantified in every 5th frame (= every 6.25 sec) of the acquired time lapse recording.
Statistical analysis
All statistical analyses were performed with GraphPad Prism 9. Data were tested for normality using Shapiro-Wilk test. Non-normally distributed data were analyzed using nonparametric tests. Figure legends contain the statistical test used and specific p-values for each quantification. For statistical analysis of the multiple parameters used to quantify trafficking and pausing, Bonferroni correction was used to adjust significance levels for multiple testing. Figure legends indicate where p values < 0.05 were not considered statistically significant due to Bonferroni correction. For all quantifications at least three independent experiments were analyzed. All figures were prepared with Adobe Illustrator 2021.
Supplementary Material
Video S1. Overexpression of LRRK2-G2019S disrupts processivity of axonal AV transport. Related to Figure 1. Time lapse recordings of axonal mCherry-LC3 vesicles in rat hippocampal neurons transfected with EGFP-LRRK2-WT, EGFP-LRRK2-G2019S, or EGFP-LRRK2-G2019S-D1994N. Arrowheads highlight AV position during retrograde (towards the left) axonal transport.
Video S2. LRRK2-G2019S knock-in disrupts processivity of axonal AV transport in mouse cortical neurons. Related to Figure 2A-F. Time lapse recordings of axonal EGFP-LC3 vesicles in WT and G2019S KI mouse cortical neurons. Arrowheads highlight AV position during retrograde (towards the left) axonal transport.
Video S3. LRRK2 kinase inhibitor MLi-2 rescues processivity of AV transport in mouse G2019S KI neurons. Related to Figure 2G-L. Time lapse recordings of axonal EGFP-LC3 vesicles in G2019S KI neurons treated with DMSO or MLi-2. Arrowheads highlight AV position during retrograde (towards the left) axonal transport.
Video S4. LRRK2-G2019S knock-in disrupts processivity of axonal AV transport in human iPSC-derived neurons. Related to Figure 3. Time lapse recordings of axonal EGFP-LC3 vesicles in WT and G2019S KI i3Neurons. Arrowheads highlight AV position during retrograde (towards the left) axonal transport.
Video S5. Comigration of axonal mCherry-LC3 and EGFP-Rab29 vesicles in a WT mouse cortical neuron. Related to Figure 5A. Arrowheads highlight position of an mCherry-LC3 and EGFP-Rab29 positive vesicle during retrograde (towards the left) axonal transport.
Video S6. Fluorescence recovery after photobleaching Halo-Rab29 signal of an axonal EGFP-LC3 labeled AV. Related to Figure 5B. Time lapse recording showing recovery of Halo-Rab29 signal over several minutes. Arrowheads point to the position of the AV.
Video S7. Comigration of axonal EGFP-LC3 and Halo-JIP4 vesicles in a WT mouse cortical neuron. Related to Figure 7. Arrowheads highlight position of an EGFP-LC3 and Halo-JIP4 positive vesicle during retrograde (towards the left) axonal transport.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-LRRK2, Rabbit Monoclonal | Abcam | Cat# ab133474, RRID:AB_2713963 |
| Anti-Rab29, Rabbit Monoclonal | Abcam | Cat# ab256526 RRID:AB_2884877 |
| Anti-Rab29 (phospho T71), Rabbit Monoclonal | Abcam | Cat# ab241062 RRID:AB_2884878 |
| Anti-Rab10, Rabbit Monoclonal | Abcam | Cat# ab237703 RRID:AB_2884879 |
| Anti-Rab10 (phospho T73), Rabbit Monoclonal | Abcam | Cat# ab230261, RRID:AB_2811274 |
| Anti-Rab10 (phospho T73), Rabbit Monoclonal (for IF staining) | Abcam | Cat# ab241060, RRID:AB_2884876 |
| Anti-Rab8A, Rabbit Monoclonal | Cell Signaling | Cat# 6975, RRID:AB_10827742 |
| Anti-Rab8A (phospho T72), Rabbit Monoclonal | Abcam | Cat# ab230260, RRID:AB_2814988 |
| Anti-Rab12, Rabbit Polyclonal | Proteintech | Cat# 8843-1-AP, RRID:AB_10603469 |
| Anti-Rab12 (phospho S106), Rabbit Monoclonal | Abcam | Cat# ab256487, RRID:AB_2884880 |
| Anti-LC3B, Rabbit Polyclonal | Abcam | Cat# ab168831 |
| Anti-GM130, Mouse Monoclonal | BD Biosciences | Cat# 610823, RRID:AB_398142 |
| Anti-JIP3, Mouse Monoclonal | Santa Cruz | Cat# sc-46663, RRID:AB_627840 |
| Anti-JIP4/SPAG9, Rabbit Monoclonal | Cell Signaling | Cat# 5519, RRID:AB_10828724 |
| Anti-JIP4, Mouse Monoclonal | Santa Cruz | Cat# sc-271492, RRID:AB_10659098 |
| Anti-Kinesin Heavy Chain, Mouse Monoclonal | EMD millipore | Cat# MAB1614, RRID:AB_94284 |
| Anti-p150 [Glued], Mouse Monoclonal | BD Biosciences | Cat# 610474, RRID:AB_397846 |
| Anti-Arf6, Rabbit Polyclonal | Abcam | Cat# ab77581, RRID:AB_2058475 |
| Anti-Rabbit IgG-IRDye 800CW, Donkey Polyclonal | Li-COR Biosciences | Cat# 926-32213, RRID:AB_621848 |
| Anti-Rabbit IgG-IRDye 680RD, Donkey Polyclonal | Li-COR Biosciences | Cat# 926-68073, RRID:AB_10954442 |
| Anti-Mouse IgG-IRDye 800CW, Donkey Polyclonal | Li-COR Biosciences | Cat# 926-32212, RRID:AB_621847 |
| Anti-Mouse IgG Light Chain-AlexaFluor680, Goat Polyclonal | Jackson Immuno Research Labs | Cat# 115-625-174, RRID:AB_2338937 |
| Bacterial and Virus Strains | ||
| BacMam mEGFP-LC3 | Thermo Fisher | Cat# P36235 |
| NEB Turbo Competent E. coli | New England Biolabs | Cat# C2984 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| MLi-2 | Tocris | Cat# 5756 |
| DMSO | Sigma-Aldrich | Cat# D2650 |
| Janelia Fluor 646 HaloTag | Promega | Cat# GA1120 |
| PLL (mol wt 70,000 – 150,000) | Sigma-Aldrich | Cat# P1274 |
| HBSS (10x) | Thermo Fisher | Cat# 14185-052 |
| 1M HEPES | Thermo Fisher | Cat# 15630-080 |
| 2.5% Trypsin | Thermo Fisher | Cat# 15090-046 |
| Minimum essential medium (MEM) | Thermo Fisher | Cat# 11095-072 |
| Horse serum (heat inactivated) | Thermo Fisher | Cat# 16050-122 |
| Sodium pyruvate | Corning | Cat# 36017004 |
| D-Glucose solution 45% | Sigma-Aldrich | Cat# G8769 |
| GlutaMAX | Thermo Fisher | Cat# 35050061 |
| B27 Supplement | Thermo Fisher | Cat# 17504-044 |
| Neurobasal Medium | Thermo Fisher | Cat# 21103-049 |
| Penicillin-Streptomycin | Thermo Fisher | Cat# 15140-122 |
| AraC | Sigma-Aldrich | Cat# C6645 |
| Lipofectamine 2000 Transfection Reagent | Thermo Fisher | Cat# 11668019 |
| Matrigel Growth Factor Reduced | Corning | Cat# 354230 |
| Essential 8 Medium | Thermo Fisher | Cat# A1517001 |
| mTeSR Medium | Stemcell Technologies | Cat# 85850 |
| ReLeSR | Stemcell Technologies | Cat# 05872 |
| Accutase | Stemcell Technologies | Cat# 07920 |
| ROCK Inhibitor Y-27632 | Selleckchem | Cat# S1049 |
| Tet System Approved FBS | Takara | Cat# 631107 |
| DMEM/F-12, HEPES | Thermo Fisher | Cat# 11330032 |
| N2 Supplement | Thermo Fisher | Cat# 17502048 |
| Non-essential Amino Acids (NEAA) | Thermo Fisher | Cat# 11140050 |
| Doxycycline | Sigma-Aldrich | Cat# D9891 |
| Poly-L-Ornithine | Sigma-Aldrich | Cat# P3655 |
| BrainPhys Neuronal Medium | Stemcell Technologies | Cat# 05790 |
| Laminin | Corning | Cat# 354232 |
| BDNF | PeproTech | Cat# 450-02 |
| NT-3 | PeproTech | Cat# 450-03 |
| Lipofectamine Stem Transfection Reagent | Thermo Fisher | Cat# STEM00003 |
| Guanosine-50-[(ab)-methyleno]triphosphate, sodium salt (GMPCPP) | Jena Bioscience | NU-405 |
| AMP-PNP | Sigma-Aldrich | A2647 |
| Microcystin-LR | Sigma-Aldrich | 475815 |
| Halt Protease and Phosphatase Inhibitor Cocktail | Thermo Fisher | 78442 |
| Critical Commercial Assays | ||
| BCA Protein Assay Kit | Thermo Fisher | Cat# 23225 |
| Plasmid Maxi Kit | QIAGEN | Cat# 12163 |
| Experimental Models: Cell Lines | ||
| Human: NGN2 iPSCs | M. Ward (National Institutes of Health) [76] | N/A |
| Human: LRRK2-G2019S NGN2 iPSCs (heterozygous) | This paper | N/A |
| Human: LRRK2-WT NGN2 iPSCs | This paper | N/A |
| Mouse: LRRK2-G2019S knock-in MEFs | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: LRRK2-G2019S knock-in | Taconic | Model #13940 |
| Mouse: C57BL/6NTac WT | Taconic | Model #B6 |
| Rat: hippocampal neuron cultures from embryonic day 18 rat brains (Sprague Dawley) | University of Pennsylvania Neuron Culture Service Center | N/A |
| Oligonucleotides | ||
| gRNA for LRRK2-G2019S knock-in: TGCTCAGTACTGCTGTAGAATGG |
This paper | N/A |
| LRRK2-G2019S ssDNA HDR template: TTTTCACACTGTATCCCAATGCTGCCATCATTGCAAAGATTGCTGACTACAGCATTGCGCAATATTGCTGCCGGATGGGGATAAAAACATCAGAGGGCACACCAGGTAGGTGATCAGGTCTGTC |
This paper | N/A |
| LRRK2-WT ssDNA HDR template: TTTTCACACTGTATCCCAATGCTGCCATCATTGCAAAGATTGCTGACTACGGCATTGCGCAATATTGCTGCCGGATGGGGATAAAAACATCAGAGGGCACACCAGGTAGGTGATCAGGTCTGTC |
This paper | N/A |
| Recombinant DNA | ||
| Plasmid: EGFP-LRRK2-WT | Modified from FLAG-LRRK2-WT (gift from W. Smith, Johns Hopkins) | N/A |
| Plasmid: EGFP-LRRK2-G2019S | Modified from FLAG-LRRK2-G2019S (gift from W. Smith, Johns Hopkins) | N/A |
| Plasmid: EGFP-LRRK2-G2019S-D1994N | Modified from FLAG-LRRK2-G2019S-D1994N (gift from W. Smith, Johns Hopkins) | N/A |
| Plasmid: mCherry-LC3B | Modified from EGFP-LC3 (gift from T. Yoshimori, Osaka University, Japan) | N/A |
| Plasmid: PGK EGFP-LC3B | Modified from PGK mCherry-LC3B (gift from Michael Ward, NIH) | N/A |
| Plasmid: mCherry-EGFP-LC3B | Gift from T. Johansen, University of Tromso, Norway | N/A |
| Plasmid: EGFP-Rab29 | Gift from D. Alessi (D. Alessi (University of Dundee, United Kingdom) | N/A |
| Plasmid: Halo-Rab29 | Modified from EGFP-Rab29 | N/A |
| Plasmid: EGFP-Rab5 | Gift from M. Zerial (Max Planck Institute, Dresden) | N/A |
| Plasmid: LAMP1-RFP | Gift from W. Mothes (Yale) | Addgene #1817 |
| Plasmid: SEP-LAMP1-RFP | Gift from J. Bonifacino (NIH) | N/A |
| Plasmid: EB3-mCherry | Gift from A. Akhmanova (Utrecht University) | N/A |
| Plasmid: Halo-JIP4 | This paper | Modified from MRC PPU #DU27651 |
| Software and Algorithms | ||
| FIJI | NIH, USA | https://imagej.net/Fiji |
| Kymograph Clear 2.0 (FIJI plugin) | [80] | https://sites.google.com/site/kymographanalysis/ |
| Prism 8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Matlab R2018b | MathWorks | https://www.mathworks.com/products/matlab.html |
| KymoSuite (custom Matlab script) | [79] | https://github.com/jnirschl/kinesin-3_guedes-dias_2018 |
| Volocity | PerkinElmer | https://www.perkinelmer.com |
| Adobe Illustrator 2021 | Adobe | https://www.adobe.com/products/illustrator.html |
| Other | ||
| 35 mm #1.5 glass bottom imaging dishes, 14 mm glass diameter | MatTeK | Cat# P35G-1.5-14-C |
| 35 mm #1.5 glass bottom imaging dishes, 20 mm glass diameter | MatTek | Cat# P35G-1.5-20-C |
Highlights:
Parkinson’s disease LRRK2-G2019S mutation disrupts axonal autophagosome transport
Rab29-induced LRRK2 hyperactivation also impairs autophagosome transport
Hyperactive LRRK2 recruits JIP4, causing a tug-of-war between autophagosome motors
Impaired axonal transport is accompanied by defective autophagosome acidification
ACKNOWLEDGEMENTS
We thank Andrea Stavoe and Cameron Thompson for sharing their initial observations on LRRK2 and autophagy. We thank Mariko Tokito for assistance in the cloning of plasmid constructs, Jean Ann Maguire and the staff of the Human Pluripotent Stem Cell Core at the Children's Hospital of Philadelphia for expertise in the generation of CRISPR knock-in iPSCs, and Shalini Padmanabhan (Michael J. Fox Foundation) for helpful discussions. This work was supported by the German Research Foundation (DFG; BO 5434/1-1 to C.A. Boecker), the National Institutes of Health (R37 NS060698 to E.L.F. Holzbaur) and the Michael J. Fox Foundation (Grant #15100 to E.L.F. Holzbaur).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES:
- 1.Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, et al. (2004). Mutations in LRRK2 cause autosomaldominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607. [DOI] [PubMed] [Google Scholar]
- 2.Healy DG, Falchi M, O’Sullivan SS, Bonifati V, Durr A, Bressman S, Brice A, Aasly J, Zabetian CP, Goldwurm S, et al. (2008). Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. 7, 583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, and Dawson TM (2005). Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl. Acad. Sci. U. S. A 102, 16842–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, and Ross CA (2006). Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat. Neurosci 9, 1231–1233. [DOI] [PubMed] [Google Scholar]
- 5.Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug MP, Beilina A, Blackinton J, Thomas KJ, et al. (2006). Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol. Dis 23, 329–341. [DOI] [PubMed] [Google Scholar]
- 6.Mir R, Tonelli F, Lis P, Macartney T, Polinski NK, Martinez TN, Chou M-Y, Howden AJM, Konig T, Hotzy C, et al. (2018). The Parkinson’s disease VPS35[D620N] mutation enhances LRRK2-mediated Rab protein phosphorylation in mouse and human. Biochem. J 475, 1861–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di Maio R, Hoffman EK, Rocha EM, Keeney MT, Sanders LH, De Miranda BR, Zharikov A, Van Laar A, Stepan AF, Lanz TA, et al. (2018). LRRK2 activation in idiopathic Parkinson’s disease. Sci. Transl. Med 10, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steger M, Tonelli F, Ito G, Davies P, Trost M, Vetter M, Wachter S, Lorentzen E, Duddy G, Wilson S, et al. (2016). Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife 5, 1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steger M, Diez F, Dhekne HS, Lis P, Nirujogi RS, Karayel O, Tonelli F, Martinez TN, Lorentzen E, Pfeffer SR, et al. (2017). Systematic proteomic analysis of LRRK2-mediated rab GTPase phosphorylation establishes a connection to ciliogenesis. Elife 6, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kiral FR, Kohrs FE, Jin EJ, and Hiesinger PR (2018). Rab GTPases and Membrane Trafficking in Neurodegeneration. Curr. Biol 28, R471–R486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Waschbüsch D, Purlyte E, Pal P, McGrath E, Alessi DR, and Khan AR (2020). Structural Basis for Rab8a Recruitment of RILPL2 via LRRK2 Phosphorylation of Switch 2. Structure 28, 406–417.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bonet-Ponce L, Beilina A, Williamson CD, Lindberg E, Kluss JH, Saez-Atienzar S, Landeck N, Kumaran R, Mamais A, Bleck CKE, et al. (2020). LRRK2 mediates tubulation and vesicle sorting from lysosomes. Sci. Adv 6, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, et al. (2009). Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet 41, 1308–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Purlyte E, Dhekne HS, Sarhan AR, Gomez R, Lis P, Wightman M, Martinez TN, Tonelli F, Pfeffer SR, and Alessi DR (2017). Rab29 activation of the Parkinson’s disease-associated LRRK2 kinase. EMBO J. 37, e201798099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gomez RC, Wawro P, Lis P, Alessi DR, and Pfeffer SR (2019). Membrane association but not identity is required for LRRK2 activation and phosphorylation of Rab GTPases. J. Cell Biol 218, 4157–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Z, Bryant N, Kumaran R, Beilina A, Abeliovich A, Cookson MR, and West AB (2018). LRRK2 phosphorylates membrane-bound Rabs and is activated by GTPbound Rab7L1 to promote recruitment to the trans-Golgi network. Hum. Mol. Genet 27, 385–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guedes-Dias P, and Holzbaur ELF (2019). Axonal transport: Driving synaptic function. Science 366, eaaw9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsuda W, Furuta T, Nakamura KC, Hioki H, Fujiyama F, Arai R, and Kaneko T (2009). Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J. Neurosci 29, 444–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giguere N, Burke Nanni S, and Trudeau L-E (2018). On Cell Loss and Selective Vulnerability of Neuronal Populations in Parkinson’s Disease. Front. Neurol 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stavoe AKH, and Holzbaur ELF (2019). Autophagy in Neurons. Annu. Rev. Cell Dev. Biol 35, 477–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maday S, Wallace KE, and Holzbaur ELF (2012). Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J. Cell Biol 196, 407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maday S, and Holzbaur ELF (2014). Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev. Cell 30, 71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stavoe AKH, Hill SE, Hall DH, and Colon-Ramos DA (2016). KIF1A/UNC-104 Transports ATG-9 to Regulate Neurodevelopment and Autophagy at Synapses. Dev. Cell 38, 171–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neisch AL, Neufeld TP, and Hays TS (2017). A STRIPAK complex mediates axonal transport of autophagosomes and dense core vesicles through PP2A regulation. J. Cell Biol 216, 441–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wong YC, and Holzbaur ELF (2014). The Regulation of Autophagosome Dynamics by Huntingtin and HAP1 Is Disrupted by Expression of Mutant Huntingtin, Leading to Defective Cargo Degradation. J. Neurosci 34, 1293–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yue M, Hinkle KM, Davies P, Trushina E, Fiesel FC, Christenson TA, Schroeder AS, Zhang L, Bowles E, Behrouz B, et al. (2015). Progressive dopaminergic alterations and mitochondrial abnormalities in LRRK2 G2019S knock-in mice. Neurobiol. Dis 78, 172–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schapansky J, Khasnavis S, DeAndrade MP, Nardozzi JD, Falkson SR, Boyd JD, Sanderson JB, Bartels T, Melrose HL, and LaVoie MJ (2018). Familial knockin mutation of LRRK2 causes lysosomal dysfunction and accumulation of endogenous insoluble α-synuclein in neurons. Neurobiol. Dis 111, 26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramonet D, Daher JPL, Lin BM, Stafa K, Kim J, Banerjee R, Westerlund M, Pletnikova O, Glauser L, Yang L, et al. (2011). Dopaminergic Neuronal loss, Reduced Neurite Complexity and Autophagic Abnormalities in Transgenic Mice Expressing G2019S Mutant LRRK2. PLoS One 6, 13–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Albanese F, Novello S, and Morari M (2019). Autophagy and LRRK2 in the Aging Brain. Front. Neurosci 13, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wallings R, Connor-Robson N, and Wade-Martins R (2019). LRRK2 interacts with the vacuolar-type H+-ATPase pump a1 subunit to regulate lysosomal function. Hum. Mol. Genet 28, 2696–2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matikainen-Ankney BA, Kezunovic N, Mesias RE, Tian Y, Williams FM, Huntley GW, and Benson DL (2016). Altered Development of Synapse Structure and Function in Striatum Caused by Parkinson’s Disease-Linked LRRK2-G2019S Mutation. J. Neurosci 36, 7128–7141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lis P, Burel S, Steger M, Mann M, Brown F, Diez F, Tonelli F, Holton JL, Ho PW, Ho S-L, et al. (2018). Development of phospho-specific Rab protein antibodies to monitor in vivo activity of the LRRK2 Parkinson’s disease kinase. Biochem. J 475, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maday S, and Holzbaur ELF (2016). Compartment-Specific Regulation of Autophagy in Primary Neurons. J. Neurosci 36, 5933–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boecker CA, Olenick MA, Gallagher ER, Ward ME, and Holzbaur ELF (2020). ToolBox: Live Imaging of intracellular organelle transport in induced pluripotent stem cellderived neurons. Traffic 21, 138–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fell MJ, Mirescu C, Basu K, Cheewatrakoolpong B, DeMong DE, Ellis JM, Hyde LA, Lin Y, Markgraf CG, Mei H, et al. (2015). MLi-2, a Potent, Selective, and Centrally Active Compound for Exploring the Therapeutic Potential and Safety of LRRK2 Kinase Inhibition. J. Pharmacol. Exp. Ther 355, 397–409. [DOI] [PubMed] [Google Scholar]
- 36.Fernandopulle MS, Prestil R, Grunseich C, Wang C, Gan L, and Ward ME (2018). Transcription Factor-Mediated Differentiation of Human iPSCs into Neurons. Curr. Protoc. Cell Biol 79, e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bieri G, Brahic M, Bousset L, Couthouis J, Kramer NJ, Ma R, Nakayama L, Monbureau M, Defensor E, Schüle B, et al. (2019). LRRK2 modifies α-syn pathology and spread in mouse models and human neurons. Acta Neuropathol. 137, 961–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kett LR, Boassa D, Ho CCY, Rideout HJ, Hu J, Terada M, Ellisman M, and Dauer WT (2012). LRRK2 Parkinson disease mutations enhance its microtubule association. Hum. Mol. Genet 21, 890–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Law BMH, Spain VA, Leinster VHL, Chia R, Beilina A, Cho HJ, Taymans JM, Urban MK, Sancho RM, Ramirez MB, et al. (2014). A direct interaction between leucine-rich repeat kinase 2 and specific β-Tubulin isoforms regulates tubulin acetylation. J. Biol. Chem 289, 895–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gillardon F (2009). Leucine-rich repeat kinase 2 phosphorylates brain tubulin-beta isoforms and modulates microtubule stability - A point of convergence in Parkinsonian neurodegeneration? J. Neurochem 110, 1514–1522. [DOI] [PubMed] [Google Scholar]
- 41.Deniston CK, Salogiannis J, Mathea S, Snead DM, Lahiri I, Matyszewski M, Donosa O, Watanabe R, Bohning J, Shiau AK, et al. (2020). Structure of LRRK2 in Parkinson’s disease and model for microtubule interaction. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moughamian AJ, and Holzbaur ELF (2012). Dynactin Is Required for Transport Initiation from the Distal Axon. Neuron 74, 331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fu M, Nirschl JJ, and Holzbaur ELF (2014). LC3 Binding to the Scaffolding Protein JIP1 Regulates Processive Dynein-Driven Transport of Autophagosomes. Dev. Cell 29, 577–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stavoe AK, Gopal PP, Gubas A, Tooze SA, and Holzbaur EL (2019). Expression of WIPI2B counteracts age-related decline in autophagosome biogenesis in neurons. Elife 8, 1–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Evans CS, and Holzbaur EL (2020). Degradation of engulfed mitochondria is ratelimiting in Optineurin-mediated mitophagy in neurons. Elife 9, 1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Farías GG, Guardia CM, De Pace R, Britt DJ, and Bonifacino JS (2017). BORC/kinesin-1 ensemble drives polarized transport of lysosomes into the axon. Proc. Natl. Acad. Sci 114, E2955–E2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eguchi T, Kuwahara T, Sakurai M, Komori T, Fujimoto T, Ito G, Yoshimura S, Harada A, Fukuda M, Koike M, et al. (2018). LRRK2 and its substrate Rab GTPases are sequentially targeted onto stressed lysosomes and maintain their homeostasis. Proc. Natl. Acad. Sci 115, E9115–E9124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stromhaug PE, Berg TO, Fengsrud M, and Seglen PO (1998). Purification and characterization of autophagosomes from rat hepatocytes. Biochem. J 335, 217–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alegre-Abarrategui J, Christian H, Lufino MMP, Mutihac R, Venda LL, Ansorge O, and Wade-Martins R (2009). LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum. Mol. Genet 18, 4022–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schapansky J, Nardozzi JD, Felizia F, and LaVoie MJ (2014). Membrane recruitment of endogenous LRRK2 precedes its potent regulation of autophagy. Hum. Mol. Genet 23, 4201–4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kluss JH, Beilina A, Lewis PA, Cookson MR, and Bonet-Ponce L (2020). Membrane targeting activates Leucine-rich repeat kinase 2 with differential effects on downstream Rab activation. bioRxiv, 2020.12.01.406223. [Google Scholar]
- 52.Kuwahara T, Inoue K, D’Agati VD, Fujimoto T, Eguchi T, Saha S, Wolozin B, Iwatsubo T, and Abeliovich A (2016). LRRK2 and RAB7L1 coordinately regulate axonal morphology and lysosome integrity in diverse cellular contexts. Sci. Rep 6, 29945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mazza MC, Nguyen V, Beilina A, Ding J, and Cookson MR (2020). Combined knockout of Lrrk2 and Rab29 does not result in behavioral abnormalities in vivo. bioRxiv, 2020.05.13.093708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Watt D, Dixit R, and Cavalli V (2015). JIP3 activates kinesin-1 motility to promote axon elongation. J. Biol. Chem 290, 15512–15525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vilela F, Velours C, Chenon M, Aumont-Nicaise M, Campanacci V, Thureau A, Pylypenko O, Andreani J, Llinas P, and Ménétrey J (2019). Structural characterization of the RH1-LZI tandem of JIP3/4 highlights RH1 domains as a cytoskeletal motor-binding motif. Sci. Rep 9, 16036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cockburn JJB, Hesketh SJ, Mulhair P, Thomsen M, O’Connell MJ, and Way M (2018). Insights into Kinesin-1 Activation from the Crystal Structure of KLC2 Bound to JIP3. Structure 26, 1486–1498.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Montagnac G, Sibarita JB, Loubéry S, Daviet L, Romao M, Raposo G, and Chavrier P (2009). ARF6 Interacts with JIP4 to Control a Motor Switch Mechanism Regulating Endosome Traffic in Cytokinesis. Curr. Biol 19, 184–195. [DOI] [PubMed] [Google Scholar]
- 58.Lynch-Day MA, Mao K, Wang K, Zhao M, and Klionsky DJ (2012). The Role of Autophagy in Parkinson’s Disease. Cold Spring Harb. Perspect. Med 2, a009357–a009357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, and Agid Y (1997). Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol. Histopathol 12, 25–31. [PubMed] [Google Scholar]
- 60.Tong Y, Yamaguchi H, Giaime E, Boyle S, Kopan R, Kelleher RJ, and Shen J (2010). Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of -synuclein, and apoptotic cell death in aged mice. Proc. Natl. Acad. Sci 107, 9879–9884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Giaime E, Tong Y, Wagner LK, Yuan Y, Huang G, and Shen J (2017). Age-Dependent Dopaminergic Neurodegeneration and Impairment of the Autophagy-Lysosomal Pathway in LRRK-Deficient Mice. Neuron 96, 796–807.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sánchez-Danés A, Richaud-Patin Y, Carballo-Carbajal I, Jiménez-Delgado S, Caig C, Mora S, Di Guglielmo C, Ezquerra M, Patel B, Giralt A, et al. (2012). Diseasespecific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol. Med 4, 380–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Plowey ED, Cherra SJ, Liu YJ, and Chu CT (2008). Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J. Neurochem 105, 1048–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Watanabe R, Buschauer R, Bohning J, Audagnotto M, Lasker K, Lu T, Boassa D, Taylor S, and Villa E (2020). The In Situ Structure of Parkinson’s Disease-Linked LRRK2. Cell 182, 1508–1518.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Daher JPL, Abdelmotilib HA, Hu X, Volpicelli-Daley LA, Moehle MS, Fraser KB, Needle E, Chen Y, Steyn SJ, Galatsis P, et al. (2015). Leucine-rich repeat kinase 2 (LRRK2) pharmacological inhibition abates α-synuclein gene-induced neurodegeneration. J. Biol. Chem 290, 19433–19444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Volpicelli-Daley LA, Abdelmotilib H, Liu Z, Stoyka L, Daher JPL, Milnerwood AJ, Unni VK, Hirst WD, Yue Z, Zhao HT, et al. (2016). G2019S-LRRK2 expression augments α-synuclein sequestration into inclusions in neurons. J. Neurosci 36, 7415–7427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yao C, Johnson WM, Gao Y, Wang W, Zhang J, Deak M, Alessi DR, Zhu X, Mieyal JJ, Roder H, et al. (2013). Kinase inhibitors arrest neurodegeneration in cell and C. elegans models of LRRK2 toxicity. Hum. Mol. Genet 22, 328–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Beilina A, Rudenko IN, Kaganovich A, Civiero L, Chau H, Kalia SK, Kalia LV, Lobbestael E, Chia R, Ndukwe K, et al. (2014). Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc. Natl. Acad. Sci. U. S. A 111, 2626–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Madero-Pérez J, Fernández B, Lara Ordóñez AJ, Fdez E, Lobbestael E, Baekelandt V, and Hilfiker S (2018). RAB7L1-mediated relocalization of LRRK2 to the golgi complex causes centrosomal deficits via RAB8A. Front. Mol. Neurosci 11, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pihlstrøm L, Rengmark A, Bjørnarå KA, Dizdar N, Fardell C, Forsgren L, Holmberg B, Larsen JP, Linder J, Nissbrandt H, et al. (2015). Fine mapping and resequencing of the PARK16 locus in Parkinson’s disease. J. Hum. Genet 60, 357–362. [DOI] [PubMed] [Google Scholar]
- 71.Maday S, Twelvetrees AE, Moughamian AJ, and Holzbaur ELF (2014). Axonal Transport: Cargo-Specific Mechanisms of Motility and Regulation. Neuron 84, 292–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hendricks AG, Perlson E, Ross JL, Schroeder HW, Tokito M, and Holzbaur ELF (2010). Motor Coordination via a Tug-of-War Mechanism Drives Bidirectional Vesicle Transport. Curr. Biol 20, 697–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Malik AU, Karapetsas A, Nirujogi RS, Mathea S, Pal P, Lis P, Taylor M, Purlyte E, Gourlay R, Dorward M, et al. (2020). Deciphering the LRRK code: LRRK1 and LRRK2 phosphorylate distinct Rab proteins and are regulated by diverse mechanisms. bioRxiv, 2020.11.25.397836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hanafusa H, Yagi T, Ikeda H, Hisamoto N, Nishioka T, Kaibuchi K, Shirakabe K, and Matsumoto K (2019). LRRK1 phosphorylation of Rab7 at S72 links trafficking of EGFR-containing endosomes to its effector RILP. J. Cell Sci 132. [DOI] [PubMed] [Google Scholar]
- 75.Jordens I, Fernandez-Borja M, Marsman M, Dusseljee S, Janssen L, Calafat J, Janssen H, Wubbolts R, and Neefjes J (2001). The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Curr. Biol 11, 1680–1685. [DOI] [PubMed] [Google Scholar]
- 76.Ravikumar B, Acevedo-Arozena A, Imarisio S, Berger Z, Vacher C, O’Kane CJ, Brown SDM, and Rubinsztein DC (2005). Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat. Genet 37, 771–776. [DOI] [PubMed] [Google Scholar]
- 77.Braak H, Del Tredici K, Rüb U, De Vos RAI, Jansen Steur ENH, and Braak E (2003). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211. [DOI] [PubMed] [Google Scholar]
- 78.Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, and Lee VMY (2011). Exogenous α-Synuclein Fibrils Induce Lewy Body Pathology Leading to Synaptic Dysfunction and Neuron Death. Neuron 72, 57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lingor P, Koch JC, Tönges L, and Bähr M (2012). Axonal degeneration as a therapeutic target in the CNS. Cell Tissue Res. 349, 289–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chung CY, Koprich JB, Siddiqi H, and Isacson O (2009). Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV α-synucleinopathy. J. Neurosci 29, 3365–3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tagliaferro P, and Burke RE (2016). Retrograde Axonal Degeneration in Parkinson Disease. J. Parkinsons. Dis 6, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Burke RE, and O’Malley K (2013). Axon degeneration in Parkinson’s disease. Exp. Neurol 246, 72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang C, Ward ME, Chen R, Liu K, Tracy TE, Chen X, Xie M, Sohn PD, Ludwig C, Meyer-Franke A, et al. (2017). Scalable Production of iPSC-Derived Human Neurons to Identify Tau-Lowering Compounds by High-Content Screening. Stem Cell Reports 9, 1221–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Maguire JA, Cardenas-Diaz FL, Gadue P, and French DL (2019). Highly Efficient CRISPR-Cas9-Mediated Genome Editing in Human Pluripotent Stem Cells. Curr. Protoc. Stem Cell Biol 48, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kaech S, and Banker G (2006). Culturing hippocampal neurons. Nat. Protoc 1, 2406–2415. [DOI] [PubMed] [Google Scholar]
- 86.Guedes-Dias P, Nirschl JJ, Abreu N, Tokito MK, Janke C, Magiera MM, and Holzbaur ELF (2019). Kinesin-3 Responds to Local Microtubule Dynamics to Target Synaptic Cargo Delivery to the Presynapse. Curr. Biol 29, 268–282.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mangeol P, Prevo B, and Peterman EJG (2016). KymographClear and KymographDirect : two tools for the automated quantitative analysis of molecular and cellular dynamics using kymographs. Mol. Biol. Cell 27, 1948–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Overexpression of LRRK2-G2019S disrupts processivity of axonal AV transport. Related to Figure 1. Time lapse recordings of axonal mCherry-LC3 vesicles in rat hippocampal neurons transfected with EGFP-LRRK2-WT, EGFP-LRRK2-G2019S, or EGFP-LRRK2-G2019S-D1994N. Arrowheads highlight AV position during retrograde (towards the left) axonal transport.
Video S2. LRRK2-G2019S knock-in disrupts processivity of axonal AV transport in mouse cortical neurons. Related to Figure 2A-F. Time lapse recordings of axonal EGFP-LC3 vesicles in WT and G2019S KI mouse cortical neurons. Arrowheads highlight AV position during retrograde (towards the left) axonal transport.
Video S3. LRRK2 kinase inhibitor MLi-2 rescues processivity of AV transport in mouse G2019S KI neurons. Related to Figure 2G-L. Time lapse recordings of axonal EGFP-LC3 vesicles in G2019S KI neurons treated with DMSO or MLi-2. Arrowheads highlight AV position during retrograde (towards the left) axonal transport.
Video S4. LRRK2-G2019S knock-in disrupts processivity of axonal AV transport in human iPSC-derived neurons. Related to Figure 3. Time lapse recordings of axonal EGFP-LC3 vesicles in WT and G2019S KI i3Neurons. Arrowheads highlight AV position during retrograde (towards the left) axonal transport.
Video S5. Comigration of axonal mCherry-LC3 and EGFP-Rab29 vesicles in a WT mouse cortical neuron. Related to Figure 5A. Arrowheads highlight position of an mCherry-LC3 and EGFP-Rab29 positive vesicle during retrograde (towards the left) axonal transport.
Video S6. Fluorescence recovery after photobleaching Halo-Rab29 signal of an axonal EGFP-LC3 labeled AV. Related to Figure 5B. Time lapse recording showing recovery of Halo-Rab29 signal over several minutes. Arrowheads point to the position of the AV.
Video S7. Comigration of axonal EGFP-LC3 and Halo-JIP4 vesicles in a WT mouse cortical neuron. Related to Figure 7. Arrowheads highlight position of an EGFP-LC3 and Halo-JIP4 positive vesicle during retrograde (towards the left) axonal transport.
Data Availability Statement
The custom MATLAB scripts used in this study to manually track kymographs (KymoSuite) are available at https://github.com/jnirschl/kinesin-3_guedes-dias_2018.