

Abstract

Among heterogeneous electrocatalysts, gold comes closest to the ideal reversible electrocatalysis of CO2 electrochemical reduction (CO2RR) to CO and, vice versa, of CO electroxidation to CO2 (COOR). The nature of the electrolyte has proven to crucially affect the electrocatalytic behavior of gold. Herein, we expand the understanding of the effect of the widely employed bicarbonate electrolytes on CO2RR using gold monocrystalline electrodes, detecting the CO evolved during CO2RR by selective anodic oxidation. First, we show that CO2RR to CO is facet dependent and that Au(110) is the most active surface. Additionally, we detect by in situ FTIR measurements the presence of adsorbed COtop only on the Au(110) surface. Second, we highlight the importance of acid–base equilibria for both CO2RR and COOR by varying the electrolyte (partial pressure of CO2 and the concentration of the bicarbonate) and voltammetric parameters. In this way, we identify different regimes of surface pH and bicarbonate speciation, as a function of the current and electrolyte conditions. We reveal the importance of the acid–base bicarbonate/carbonate couple, not only as a buffering equilibrium but also as species involved in the electrochemical reactions under study.
Introduction
In the search for more efficient energy conversion technologies, the ultimate goal is to find a material that can catalyze an electrochemical process reversibly. For a given Ox/Red couple, such a material exhibits high rates for both the electrochemical reduction and oxidation with zero overpotential. Platinum is a good example of such a reversible catalyst, as it is able to catalyze reversibly H+/H2 conversion,1,2 i.e., the hydrogen evolution reaction (HER) and the hydrogen evolution oxidation (HOR). Reversible catalysts have been predicted to exist for two-electron transfer reactions3 since such reactions typically have only a single catalytic intermediate, whose binding energy needs to be optimized. In relation to the electrocatalytic CO2 reduction reaction (CO2RR), researchers would like to find an electrocatalyst interconverting reversibly CO2/CO, that is, the following two-electron transfer reaction:
| 1 |
where AH is a Brønsted acid and A– is its conjugated base. In neutral-alkaline media, the acid–base couple is commonly considered to be H2O/OH–. However, other acid–base couples may be taken into account, e.g., H2CO3/HCO3–, HCO3–/ CO32–, or H2PO4–/HPO42–.
In nature there is an enzyme, the Ni-containing carbon monoxide dehydrogenase (Ni-CODH), which can catalyze in a quasi-reversible fashion the interconversion between CO2/CO.4,5 Enzymes for the reversible conversion of CO2 and formic acid also exist.6 For this latter conversion, (nearly) reversible electrocatalysts have also been identified in heterogeneous electrocatalysis7 and molecular electrocatalysis.8 However, reversible synthetic heterogeneous electrocatalysts for the CO2/CO conversion have, to the best of our knowledge, not yet been identified. According to a computational study by Hansen et al., this is related to the existence of two intermediates (adsorbed CO and COOH) in the catalytic pathway, whose binding energies have a scaling relationship.9 The enzyme can break this scaling by the presence of a second coordination sphere which stabilizes COOH.9 Among the known metal electrocatalysts, gold has the highest activity for both CO2 electrochemical reduction to CO (CO2RR)9,10 and CO electrooxidation (COOR).11,12 Therefore, understanding the details of the mechanism of the CO2/CO conversion on gold, and especially the role of the electrolyte (the electrochemical “second coordination sphere”), is important for developing more efficient catalysts for CO2RR to CO.
Studies of the structure dependence of CO2RR on gold low-index monocrystalline surfaces revealed that (110) is the most active facet.13−15 The structure sensitivity trend for CO2RR to CO on the low-index facets points to the central role of low-coordinated surface sites in CO2RR.14,16,17 Analogously, the Au(110) surface was measured to be the most active facet for COOR, both in acidic and alkaline media.11,18,19
Besides the electrode surface structure, various properties of the electrolyte (e.g., anions, cations, and pH) have been shown to play a crucial role in the electrocatalysis of the CO2RR and COOR. In particular, bicarbonate solution has been proven to lead to higher CO2RR efficiency, compared to other buffered solutions.20 Understandably, bicarbonate is the most widely employed electrolyte in CO2RR electrocatalysis. Aside from its buffering ability, bicarbonate can also act as a supplier of CO2 through the solution equilibrium between CO2 and bicarbonate21,22 and as a a proton donor for HER.23 Nonetheless, attempts to fully understand the multifaceted role of acid–base bicarbonate equilibria and the role of the different species in solution are complicated by the interconnection between surface speciation and reaction rates.24 Recently, numerous studies have identified the importance of current-driven changes in the local environment close to the electrode surface compared to the bulk of the solution on the outcome of CO2RR.25−27
In this study, we assess the structure and electrolyte dependence of CO2RR on monocrystalline Au(hkl) electrodes. Based on the ability of the gold electrode to selectively oxidize CO, we detect the CO produced during CO2RR by applying anodic potentials.28 In this way, we gain insights into the selectivity-potential trend for CO2RR in bicarbonate electrolyte through a fast semiquantitative method. Subsequently, we extend the measurements of CO2RR on Au(110) in a variety of electrolyte conditions, by changing the partial pressure of CO2 and bicarbonate concentration, and voltammetric parameters, such as the negative vertex potential and the scan rate. These electrochemical measurements of CO2RR activity together with in situ FTIR studies help us to draw a more detailed picture of the effect of bicarbonate surface speciation and equilibria not only on CO2RR but also on COOR on well-defined gold surfaces.
Experimental Section
Chemicals and Materials
Electrolytes were prepared from H2SO4 (96%, Merck Suprapur), HClO4 (70%, Merck Suprapur), KHCO3 (EmsureACS Merck), and KClO4 (99.995%, Aldrich Ultrapure) using Milli-Q water (resistivity ≥18.2 MΩ cm). Prior to experiments, the electrolytes were purged for 20 min with Ar (6.0 purity, Linde), CO2 (4.5 purity, Linde), or CO (4.7 purity, Linde). To obtain the selected partial pressure of CO2, the flow of CO2 and Ar was set accordingly using two mass flow controllers (SLA5850, Brooks Instrument).
Experimental Procedure
The glassware was stored overnight in a 1 g L–1 KMnO4 solution. Prior to experiments, the residual KMnO4 was removed by addition of a diluted Piranha solution and the glassware was boiled in Milli-Q water for seven times. The electrochemical experiments were performed using two electrochemical cells in a three-electrode configuration at room temperature using a Bio-Logic VSP300 potentiostat. Both cells contained a coiled gold counter electrode (99.99% purity). As a reference electrode, we employed in cell 1 a homemade reversible hydrogen electrode (RHE) and in cell 2 a Ag/AgCl electrode (KCl-saturated, Pine Research Instrumentation). In cell 2, the pH of the electrolytes purged for 20 min with the selected gas atmosphere was determined with a pH meter (SI Analytics Lab 855 Benchtop Meter), and thus, the potential was calculated according to ERHE = EAgAgCl + 0.199 V + (0.059 × pH).
The gold single-crystal disk electrodes (⌀ 7.0 mm, 99.999%, aligned with an accuracy ∼0.1°, Surface Preparation Laboratory) were prepared by carefully flame annealing them until red-hot, with cooling in air and rinsing with Milli-Q water.29 For all the electrochemical experiments, the single-crystal disks were brought in contact with the electrolyte in a hanging meniscus configuration under potential control at 0.08 V vs RHE. First, in cell 1 we measured the cyclic voltammetry (CV) of the Au(hkl) disk between 0.08 and 1.2 V vs RHE in Ar-purged 0.1 M H2SO4 at 50 mV s–1 (see Figure S2). The measured characterization CVs in 0.1 M H2SO4 are consistent with well-ordered flame-annealed gold single-crystal surfaces.29−31 As Au(hkl) surfaces present specific sulfate adsorption peaks in the double-layer region,29 we could characterize Au(hkl) crystals without reaching the oxidizing potential, thus avoiding surface dissolution and roughening. Transferring the crystal to cell 2, we evaluated the Ohmic resistance by electrochemical impedance spectroscopy (EIS) at 0.1 V vs RHE, and we applied 85% Ohmic drop compensation to all the following measurements. Next, we measured the CV of Au(hkl) crystals in the double-layer region in the tested bicarbonate electrolyte (see Figure S3). Finally, the catalytic activity of the Au(hkl) surfaces for CO2RR was measured by CV starting at 0.08 V vs RHE to a selected cathodic potential value and back to the upper limit of the double layer (1.2 V vs RHE) at 50 mVs–1, or by chronoamperometry (CA) measurements. The disk currents were normalized by the electrochemical active surface area (ECSA), which was determined by integrating the reduction peak from the CV in 0.1 M H2SO4 divided by the charge corresponding to a gold monolayer 390 (μC cm–2) (see Figure S1).32
The in situ Fourier-transform infrared spectroscopy (FTIR) measurements were performed in external reflection mode with an incident angle of 60° using a Bruker Vertex 80v IR spectrophotometer. A detailed description of the setup is outlined elsewhere.33 The gold single-crystal disk (prepared and characterized as described above) was located in a spectroelectrochemical glass cell mounted on a 60° bevelled CaF2 prism (MaTeck). The reference electrode was a Ag/AgCl electrode (KCl-saturated), and the counter electrode was a gold ring surrounding the working electrode to ensure homogeneous potential across the disk surface. The disk electrode was pressed against the prism in a thin layer configuration biased at the reference potential (E0 = +0.1 V vs RHE). After stabilization of the thin layer, the background spectrum was recorded at +0.1 V vs RHE. To minimize the disruption of the thin layer, the spectra were recorded while the potential was pulsed (0.02 s) in between the selected and the reference potential. Each spectrum was obtained in reflectance mode by averaging over 100 scans with a resolution of 4 cm–1. In this fashion, a positive (negative) band corresponds to an increase (decrease) in the transmittance at the selected potential compared to the reference. Hence, a positive (negative) band is associated with a decrease (increase) in the concentration of an IR-absorbing species on the surface and/or in the thin layer.
Results and Discussion
Structure Dependence of CO2RR
In this section, we will discuss the structure dependence of CO2RR to CO on the three low index planes of gold, i.e., Au(111), Au(100), and Au(110), as determined by cyclic voltammetry (CV) and chronoamperometry (CA) experiments in 0.1 M KHCO3. Our way of measuring the CO2RR electrocatalytic activity by subsequent cathodic and anodic CAs is a fast semiquantitative way to capture the structure dependence trend for CO2RR, as well as the selectivity to CO2RR vs HER as a function of the applied potential. Next, we investigated the presence of possible CO2RR reaction intermediates on Au(hkl) by means of in situ FTIR experiments.
Figure 1 A shows the cyclic voltammograms of Au(111), Au(100), and Au(110) in CO2-saturated 0.1 M KHCO3. First, the potential was scanned to negative values, where CO2 is reduced to CO, and then to positive values, where the CO formed during CO2RR is reoxidized. Under reduction conditions, the cathodic current measured for Au(110) is one order of magnitude larger than the current measured for Au(111) and Au(100) electrodes. However, the cathodic current is due to the contribution of two electrochemical processes, namely, CO2RR and HER. To unravel the contribution of CO2RR to the total cathodic current, the potential was scanned to values where the gold electrode is selective for CO electrooxidation over HOR28 with comparable catalytic activity for all Au(hkl).19 Still, this method is not quantitative, as part of the CO generated at the surface during CO2RR will diffuse away. The fraction of CO diffusing to the bulk depends on the time of the measurement and, hence, on the scan rate (see Figure S15). Integration of the current due to selective CO electrooxidation provides the oxidation charge (QOx), which will be used as a parameter to evaluate CO2RR activity througout this work. Clearly, the CO reoxidation current in Figure 1 follows the trend Au(110) > Au(111) ≈ Au(100). This result agrees with previous investigations of the structure sensitivity of CO2RR to CO,13,14 validating our method to estimate CO2RR activity by in situ CO electrooxidation.
Figure 1.

For the different Au(hkl) crystals in CO2-saturated 0.1 M KHCO3: (A) Cyclic voltammetry at 50 mVs–1. (B) Number of moles of CO formed (left axis, full square) and number of moles of CO divided the total number of moles reduced (right axis, empty square) as a function of the applied negative potential.
To gain more insight on the CO2RR activity as a function of the applied potential, we performed CA measurements at different cathodic potentials and, subsequently, stepped the potential to 0.8 V vs RHE to oxidize the CO formed (see Figure S4). Figure 1 B illustrates the number of moles of CO (nCO) evolved during CO2RR at different negative applied potentials for the different Au(hkl) in CO2-saturated 0.1 M KHCO3. The nCO were calculated by integrating the oxidative CAs profiles, considering that the number of electrons transferred is equal to 2 for each oxidized CO molecule. Similarly to the voltammetric response, the CA measurements exhibit the same structure dependence for CO2RR to CO, i.e., Au(110) > Au(111) ≈ Au(100). Notably, while for Au(111) and Au(100) nCO keeps increasing as a function of the more negative applied potential, for Au(110), after an initial steep increase, the nCO levels out at ca. −0.6 V vs RHE and decreases at −1.0 V vs RHE. A similar trend emerges in the CVs in Figure 1 for Au(110), where the increase in the cathodic current plateaus around −4 mA cm–2 at a potential of ca. −0.65 V vs RHE. This decrease for both nCO and the cathodic current may be ascribed to a mass transport limitation in CO2 and by its consumption by the homogeneous reactions, as a response to the increase in the local alkalinity (a more detailed discussion will be given in the next section). Figure 1 B shows on the right axis the number of moles of CO divided the total number of moles (nCO/ntot) converted during the CA at cathodic potential (assuming the formation of two-electron transfer products, CO and H2). Hence, nCO/ntot is an evaluation of the selectivity to CO2RR over HER. In fact, it is a lower estimate because some of the CO will diffuse away before it is oxidized. For Au(110), nCO/ntot gives a maximum between −0.4/–0.5 V vs RHE and then decreases for more negative applied potentials due to the increasing HER current.
Discussing the observed structure dependence of
CO2RR on gold electrodes,
numerous studies have argued that the low-coordination surface sites
(step- or edge-like) are the active sites for CO2RR.14,16,17,34 On the other hand, the potential of zero charge (pzc) of a surface also plays a central role in interfacial charge transfer
processes, as it relates directly to the interfacial electric field,35,36 and therefore in the stabilization of adsorbed charged species.
Namely, one would expect that for CO2RR a surface with a lower pzc leads to a better stabilization of the negatively charged
first reaction intermediate (*CO2•–).35,37,38 For the same applied potential (Eapp vs reference electrode), the absolute negative
interfacial electric field (Eapp vs Epzc) decreases for a surface
with less positive pzc, favoring the adsorption of
negatively charged CO2. Similarly, the effect of cation
identity on CO2RR has been rationalized in terms of reduction of the
electric field at the liquid–solid interface as a function
of the size of the hydrated cation in the outer Helmholtz plane.37 Concerning the surface structure of Au(hkl) crystals, we should consider that they undergo potential-induced
reconstruction at potentials more negative than ca. −0.4 V
vs SHE.31,39 Hence, during CO2RR conditions the basal
planes of gold reconstruct to Au(111)-(22x ), Au(100)-(hex),
and Au(110)-(1 ×
2) or -(1 × 3).15 We attribute the
observed structure dependence of CO2RR on Au(hkl)
to the parallel effect of the (Epzc) and of the geometry of the surface sites. Consistently, the
least active surfaces for CO2RR are the reconstructed Au(111) and
Au(100), which are both (111)-like terraces surfaces with high-coordination
surface atoms and with a similar pzc (+0.564 and
+0.544 V vs SHE).31,40 The reconstructed Au(110), being
a stepped surface with low-coordination surface atoms and with a lower pzc (+0.204 V vs SHE), is the most active surface for CO2RR.
), Au(100)-(hex),
and Au(110)-(1 ×
2) or -(1 × 3).15 We attribute the
observed structure dependence of CO2RR on Au(hkl)
to the parallel effect of the (Epzc) and of the geometry of the surface sites. Consistently, the
least active surfaces for CO2RR are the reconstructed Au(111) and
Au(100), which are both (111)-like terraces surfaces with high-coordination
surface atoms and with a similar pzc (+0.564 and
+0.544 V vs SHE).31,40 The reconstructed Au(110), being
a stepped surface with low-coordination surface atoms and with a lower pzc (+0.204 V vs SHE), is the most active surface for CO2RR.
Next, we investigated by in situ FTIR the presence of surface intermediates under CO2RR conditions. Figure 2 shows the p-polarized FTIR spectra recorded in CO2-saturated 0.1 M KHCO3 at increasing negative potentials for Au(110) (Figure S5 shows the full FTIR spectra for Au(110) and Au(111)). Although p-polarized light is more sensitive to the species adsorbed or close to the electrode surface, we can still observe vibration modes of species present in solution. Namely, for both Au(111) and Au(110) spectra, we detected several bands related to species in solution at 1310, 1361, 1400, 1620, and 2343 cm–1. Besides the band at 1620 cm–1 due to the OH bending of water, all the other modes are related to the bicarbonate species in the thin layer as a function of the applied negative potential. The positive band at 2343 cm–1 is due to CO2,aq, which is consumed for increasingly negative potentials. The negative-going bands at 1361 cm–1 and at 1310 cm–1 are attributed to the stretching modes of bicarbonate in solution.22,41 Initially, the concentration of bicarbonate in the thin layer increases (negative bands), but for Au(110) at a potential of −0.5 V vs RHE, bicarbonate starts being consumed (positive band). Simultaneously, carbonate is formed as shown by the positive going band at 1400 cm–1 due to the asymmetric stretching of dissolved carbonate.41 Furthermore, at −0.5 V vs RHE the very pronounced positive water band at 1620 cm–1 indicates the onset of water reduction.42 Overall, the bicarbonate surface speciation is changing as a function of the applied potential (leading to a cathodic current), being CO2 and HCO3– at low overpotential and HCO3– and CO32– at more negative potential.
Figure 2.

FTIR spectra of Au(110) measured with p-polarized light in CO2-saturated 0.1 M KHCO3. The background was collected at +0.1 V vs RHE, and then, the potential was gradually increased to more negative values through pulsed (0.02 s) chronoamperometry (0.0, −0.1, −0.2, −0.25, −0.3, −0.35, −0.4, −0.45, and −0.5 V vs RHE).
Only on Au(110) at potentials more negative than −0.2 V vs RHE did we observe a band at 2100 cm–1 related to adsorbed CO. In the literature, this frequency has been attributed to CO adsorbed on top, COtop.43,44 In our study, the COtop band is a bipolar band with a Stark tuning slope of ca. 20–26 cm–1 V–1 (see Figure S6). The Stark tuning slope obtained in our study for COtop falls in the range (20–40 cm–1 V–1) measured for Au(hkl) during COOR.43,45 Compared to COtop previously detected on polycrystalline gold as measured in the attenuated total reflection mode (ATR),21,44,46 our results show a slightly different potential dependence of the COtop band. Namely, in the ATR spectra the COtop band disappears at potentials more negative than −0.1/–0.3 V vs RHE,21,46 while in our study we observed the COtop mode to potentials as negative as −0.5 V vs RHE. This different potential dependence of COtop may originate from the variations in the experimental conditions of this work compared to previous ones,21,44 i.e., the infrared technique (external vs internal reflection), reference potential (0.1 VRHE compared to ca. 1.0–1.2 VRHE), electrolyte nature (K+ compared to Na+), and electrode surface (monocrystalline compared to a high roughness Au polycrystalline surface). Finally, the FTIR measurements support the activity trend of CO2RR for Au(hkl) planes, as we detected the presence of the reaction product only on the most active surface, i.e., Au(110).
Role of Acid–Base Equilibria in Bicarbonate Electrolyte for CO2RR and COOR
In this section, we will investigate the role of the various acid–base equilibria present in bicarbonate electrolyte on the electrocatalysis of CO2RR and CO electrooxidation on the most active surface, Au(110). Importantly, a bicarbonate solution is a buffering system through two different acid/base equilibria. The first acid–base couple CO2/HCO3– has a pKa,1 = 6.3 being a good buffering agent for pH 5.3–7.3 according to
| 2 |
For higher pH 9.3–11.3, the buffering will take place through the HCO3–/CO32– equilibrium with a pKa,2 = 10.3 according to
| 3 |
In the bulk of the solution,
the bicarbonate speciation is dictated
by the bulk pH. In turn, the bulk pH of a bicarbonate electrolyte
depends on the partial pressure of CO2 (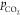 ) and on the initial bicarbonate
concentration.
However, in aqueous electrolytes the occurrence of electrochemical
processes at the electrode surface will lead to significant changes
in the pH near the electrode surface. Thus, the surface pH, and as
a result the bicarbonate surface speciation, will be determined by
the current at the electrode. In the absence of forced convection,
especially, large concentration gradients will build up between the
surface and the bulk. In this case, also the kinetics of these solution
reactions are important, and reaction 3 is several
orders of magnitude faster than reaction 2.47 To investigate the effect of reaction 2 and 3 on CO2RR and CO re-electrooxidation,
we will systematically change the following parameters: the partial
pressure of CO2, the bulk bicarbonate concentration, and
the applied cathodic potential.
) and on the initial bicarbonate
concentration.
However, in aqueous electrolytes the occurrence of electrochemical
processes at the electrode surface will lead to significant changes
in the pH near the electrode surface. Thus, the surface pH, and as
a result the bicarbonate surface speciation, will be determined by
the current at the electrode. In the absence of forced convection,
especially, large concentration gradients will build up between the
surface and the bulk. In this case, also the kinetics of these solution
reactions are important, and reaction 3 is several
orders of magnitude faster than reaction 2.47 To investigate the effect of reaction 2 and 3 on CO2RR and CO re-electrooxidation,
we will systematically change the following parameters: the partial
pressure of CO2, the bulk bicarbonate concentration, and
the applied cathodic potential.
Keeping the bulk concentration
of bicarbonate fixed, purging the
solution with different partial pressures of CO2 (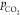 ) leads to different bulk
pH, as the equilibrium
of reaction 2 shifts to the right. Specifically,
by increasing the
) leads to different bulk
pH, as the equilibrium
of reaction 2 shifts to the right. Specifically,
by increasing the 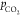 , the
pH drops, being ca. 9.0 in Ar-saturated
and 6.8 in CO2-saturated 0.1 M KHCO3 (see Figure 3 B).
, the
pH drops, being ca. 9.0 in Ar-saturated
and 6.8 in CO2-saturated 0.1 M KHCO3 (see Figure 3 B).
Figure 3.

(A) Cyclic voltammetry
of Au(110) in 0.1 M KHCO3 at
50 mVs–1 for different  . (B) Calculated oxidation
charges obtained
by integration of the CV up to 1.2 V vs RHE, after polarization to
−1.2 V vs SHE and experimentally measured bulk pH from (A).
. (B) Calculated oxidation
charges obtained
by integration of the CV up to 1.2 V vs RHE, after polarization to
−1.2 V vs SHE and experimentally measured bulk pH from (A).
Figure 3 A shows
the cyclic voltammograms of Au(110) in 0.1 M KHCO3 solutions
purged with different  . In
general the cathodic current and, more
precisely, the quasi-plateau in the current (jlim) observed during the cathodic scan increases with the
. In
general the cathodic current and, more
precisely, the quasi-plateau in the current (jlim) observed during the cathodic scan increases with the  (see Figure S8). A more detailed discussion of the nature of jlim will be given later. Curiously, the anodic
part of
the CVs exhibits two oxidation waves for
(see Figure S8). A more detailed discussion of the nature of jlim will be given later. Curiously, the anodic
part of
the CVs exhibits two oxidation waves for 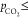 0.6
atm and a single oxidation peak for
0.6
atm and a single oxidation peak for  0.6
atm. According to the oxidation peak
potential, we name the peak at a potential of ca. +0.4 V vs RHE the
1st peak and the one at ca. +0.7 V vs RHE the 2nd peak. By integrating the total oxidation current (sum of the 1st and 2nd peak), we obtain the total oxidation
charge (QOx). Figure 3 B shows QOx calculated
from the CVs in 0.1 M KHCO3 solutions purged with different
0.6
atm. According to the oxidation peak
potential, we name the peak at a potential of ca. +0.4 V vs RHE the
1st peak and the one at ca. +0.7 V vs RHE the 2nd peak. By integrating the total oxidation current (sum of the 1st and 2nd peak), we obtain the total oxidation
charge (QOx). Figure 3 B shows QOx calculated
from the CVs in 0.1 M KHCO3 solutions purged with different  . The QOx scales
linearly with the
. The QOx scales
linearly with the 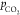 , suggesting
that both oxidation peaks (1st and 2nd) can
be attributed to a product of CO2RR.
For this reason, we investigated whether the origin of the early oxidation
wave may relate to the formation of a CO2RR products beyond CO, as
proposed by Narayanaru et al.48 During
CO2RR on a gold electrode, formic acid (HCOOH) was generally detected
with low Faraday efficiency (<5%).49−52 Additionally, a few reports have
claimed that at more negative potential CO2 could even
be reduced to methanol (CH3OH)48,52 through a path involving the formation of a formaldehyde intermediate
(H2CO), as calculated by DFT.52 We probed whether the 1st oxidation wave in the CV of
Au(110) may originate from CO2RR to any of these 1-carbon-containing
products, i.e., HCOOH, CH3OH, and H2CO, by adding
10 mM of each organic molecule to a solution of 0.2 atm of CO2 in 0.1 M KHCO3 (see Figure S9). The addition of these organic molecules did not result
in a net increase of any of the oxidation waves. Consequently, the
duality of the oxidation wave cannot be explained in terms of CO2RR
to any 1-carbon-containing molecule other than CO.
, suggesting
that both oxidation peaks (1st and 2nd) can
be attributed to a product of CO2RR.
For this reason, we investigated whether the origin of the early oxidation
wave may relate to the formation of a CO2RR products beyond CO, as
proposed by Narayanaru et al.48 During
CO2RR on a gold electrode, formic acid (HCOOH) was generally detected
with low Faraday efficiency (<5%).49−52 Additionally, a few reports have
claimed that at more negative potential CO2 could even
be reduced to methanol (CH3OH)48,52 through a path involving the formation of a formaldehyde intermediate
(H2CO), as calculated by DFT.52 We probed whether the 1st oxidation wave in the CV of
Au(110) may originate from CO2RR to any of these 1-carbon-containing
products, i.e., HCOOH, CH3OH, and H2CO, by adding
10 mM of each organic molecule to a solution of 0.2 atm of CO2 in 0.1 M KHCO3 (see Figure S9). The addition of these organic molecules did not result
in a net increase of any of the oxidation waves. Consequently, the
duality of the oxidation wave cannot be explained in terms of CO2RR
to any 1-carbon-containing molecule other than CO.
To further probe the effect of acid–base equilibria and the nature of the two electrooxidation peaks, we performed measurements in electrolytes of different bulk bicarbonate concentrations (i.e., different buffer strength). Figure 4 displays the cyclic voltammograms of Au(110) in bicarbonate electrolytes of different concentration (0.02, 0.1, and 0.5 M) purged with Ar. In Ar-saturated electrolytes, the cathodic current is mainly due to HER and increases with the bicarbonate concentration, in agreement with bicarbonate being a viable proton donor for HER.23 Still, as shown by the appearance of an oxidation wave after the cathodic polarization, we measured a small COOR current. Distinctly, in Ar-purged solutions the oxidation peak potential corresponds to the 1st peak and its charge is proportional to the bulk bicarbonate concentration. During in situ FTIR experiments in Ar-saturated 0.5 M KHCO3 on Au(110), we detect the presence of COtop at 2100 cm–1 (see Figure S7). However, no CO2 band at 2343 cm–1 was detected, suggesting that CO2 comes from acid/base equilibria in solution. Our results corroborates the hypothesis that bicarbonate is a source of CO2,aq through the following solution equilibrium:21,22,53
| 4 |
Figure 4.

Cyclic voltammetry of Au(110) at 50 mV s–1 in Ar-saturated KHCO3 solution of different concentrations (0.02, 0.1, and 0.5 M). Dotted lines show cyclic voltammetry prior to cathodic polarization in the given electrolyte.
Recent first-principles molecular dynamics simulations suggest a mechanism in which HCO3– is converted into CO2 by exchange of a proton from a neighboring water molecule to bicarbonate.54
Next, we investigated the effect of the negative vertex potential (and corresponding cathodic current) on the 1st and 2nd oxidation peaks. Figure 5 A shows potential opening cyclic voltammograms of Au(110) in 0.1 M KHCO3 purged with 0.2 atm of CO2. Figure 5 B displays the charge calculated from the integration of the anodic wave in (A), as the total oxidation charge, and for the 1st and 2nd peaks, separately. The peaks were fitted using an asymmetric double sigmoidal function in OriginLab. A specific potential-charge trend was observed for the two oxidation peaks. While the 2nd peak gradually increases with more negative potential until −0.8 V vs RHE, the 1st peak only appears at a potential more negative than −0.6 V vs RHE and keeps increasing. Interestingly, the growth of the 1st oxidation peak initiates concomitantly the appearance of the quasi-plateau in the cathodic current at −0.6 V vs RHE. Similarly, the decrease in the 2nd oxidation peak occurs simultaneously with the increase in the cathodic current after the semi-plateau at −0.8 V vs RHE, which is attributed to an increase in HER.
Figure 5.

(A) Cyclic voltammetry of Au(110) in 0.2 atm of CO2–0.1 M KHCO3 at 50 mV s–1 for increasing cathodic potentials. (B) Calculated oxidation charges for the different limiting applied cathodic potentials.
The trend observed for the oxidation peaks (1st and
2nd), by varying the  , the
concentration of KHCO3,
and the applied cathodic potential, indicates a strong pH effect.
Specifically, the growth of the 1st oxidation peak appears
to emerge after the manifestation of the cathodic quasi-plateau. To
have a deeper insight into the nature of the species leading to the
two well-separated anodic peaks and to the quasi-plateau in the cathodic
current, we performed voltammetry at different scan rates. According
to the Randles–Sevcik equation,55 for a diffusion limiting process the peak current varies linearly
with the square root of the scan rate (ν). From the slope of
the linear fitting it is then possible to derive the diffusion coefficient D (cm2 s–1) of the limiting
species by rearranging the Randles–Sevcik equation (see the
derivation in the Supporting Information):
, the
concentration of KHCO3,
and the applied cathodic potential, indicates a strong pH effect.
Specifically, the growth of the 1st oxidation peak appears
to emerge after the manifestation of the cathodic quasi-plateau. To
have a deeper insight into the nature of the species leading to the
two well-separated anodic peaks and to the quasi-plateau in the cathodic
current, we performed voltammetry at different scan rates. According
to the Randles–Sevcik equation,55 for a diffusion limiting process the peak current varies linearly
with the square root of the scan rate (ν). From the slope of
the linear fitting it is then possible to derive the diffusion coefficient D (cm2 s–1) of the limiting
species by rearranging the Randles–Sevcik equation (see the
derivation in the Supporting Information):
| 5 |
where s (A cm–2 V–0.5 s0.5) is the slope, n is the number of electron transferred, and c (mol cm–3) is the concentration.
Figure 6 A shows chronoamperometry measurements at −0.8 V vs RHE for 60 s on Au(110) in 0.1 M KHCO3 purged with 0.2 atm of CO2, followed by a linear-sweep voltammogram in the double layer (0.08 to 1.2 V vs RHE). The same measurement was repeated for different scan rates of the CVs. Figure 6 B displays the derived Randles–Sevcik plot for the dependence of the two electrooxidation peaks on the scan rate. Clearly, the current of both the 1st and 2nd peak scales linearly with the square root of the scan rate, indicating that both processes are diffusion limited. Hence, both the 1st and 2nd peak can be ascribed to CO bulk electrooxidation and not to the oxidation of irreversibly adsorbed CO, which was not detected during in situ FTIR experiments (see Figure 2). Interestingly, the slope of the linear fitting for the two peaks is different, suggesting a diffusion process limited by a different species.
Figure 6.

(A) Chronoamperometry of Au(110) at −0.8 V vs RHE for 60 s in 0.1 M KHCO3 purged with 0.2 atm of CO2, followed by linear-sweep voltammetry in the double layer region at different scan rates. (B) Randles–Sevcik plot for the 1st and 2nd CO electrooxidation peaks as measured in (A). (C) Voltammetry of polycrystalline Au rotating disk electrode in CO-saturated 0.1 M KClO4 with increasing concentration of equimolar HCO3–/CO32– at 50 mV s–1 and 1600 rpm.
To simplify the system, we controlled the effect of surface concentration gradient developing during the cathodic scan on the COOR by performing bulk COOR measurements ([CO] = 1 mM) on a Au rotating disk electrode (RDE). Figure 6 C shows voltammetry measurements of bulk COOR on Au RDE in CO-saturated 0.1 M KClO4 with increasing concentration of equimolar HCO3–/CO32– at 50 mV s–1 and 1600 rpm. We can clearly observe two different diffusion-limiting plateaus for COOR. For increasing concentration of HCO3–/CO32–, the 1st plateau increases while the 2nd plateau remains constant. The increase in the 1st plateau current mirrors the increase in HCO3–/CO32– concentration, but not the concentration of OH– (i.e., pH). It is worth noticing that we used equimolar solutions of HCO3–/CO32– to minimize the increase in the bulk pH. Nonetheless, the mere addition of HCO3– does not lead to the appearance of the 1st COOR plateau (see Figure S13). Hence, the observed rise in the 1st COOR plateau in Figure 6 C has to be ascribed to the increasing CO32– concentration.
Based on this information, we propose that the appearance of two
separated bulk CO electrooxidation peaks can be attributed to reaction
mechanisms mediated by two different oxygen donors. The marked pH
and electrolyte dependence of the two anodic peaks leads us to ascribe
the 2nd peak to COOR by H2O, while the 1st peak is due to COOR by CO32–. We base this on the observations
that the RDE experiments of bulk COOR reveal that the 1st plateau current is proportional to the CO32– concentration (and
not to OH– or HCO3– concentration), while the 2nd plateau is independent of the electrolyte nature. Therefore, in
the CO reoxidation experiments, the appearance of the 1st peak cannot be attributed to HCO3– being the oxygen donor, but to a
species generated in response to a buildup of a pH gradient, i.e.,
CO32–. Indeed, for the same bulk concentration of HCO3– (0.1 M), we measured
the 1st peak only for electrolytes with a lower buffer
capacity, caused by a lower 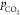 (Figure 3), and after the
development of a certain cathodic
current (Figure 5).
Though preliminary, these results indicate that the early onset peak
for COOR (1st) cannot be explained as free OH– being the oxygen donor, as we showed recently for the CO oxidation
peaks on a platinum electrode in an electrolyte not containing (bi)carbonate.56 Specifically, we suggest that CO32– does
not act as a direct oxygen donor in COOR, rather it is involved in
the generation of OH–/OHads– through its acid–base
equilibrium with H2O/HCO3– at the electrified interface. The
onset potential for COOR in the presence of CO32– is ca. 0.3 V vs RHE
and is comparable to the one observed for COOR in alkaline media,
where OH– is the oxygen donor.57 A study of OH– adsorption on Au(111)
shows that, at a potential as low as 0.3 V vs RHE, OH– is already adsorbed on the electrode with a low surface concentration.58 It is plausible to propose that the OH– generated from CO32– acid–base reaction results in adsorbed OH–, leading to a lower COOR overpotential. Performing
analogous COOR experiments with a RDE in a variety of electrolytes,
we observe that the role of anions as an oxygen donor “shuttle”
in COOR is a more generic feature of buffering anions, not limited
to CO32–. These experiments, together with infrared spectroscopic measurements,
will be discussed in a separate paper.
(Figure 3), and after the
development of a certain cathodic
current (Figure 5).
Though preliminary, these results indicate that the early onset peak
for COOR (1st) cannot be explained as free OH– being the oxygen donor, as we showed recently for the CO oxidation
peaks on a platinum electrode in an electrolyte not containing (bi)carbonate.56 Specifically, we suggest that CO32– does
not act as a direct oxygen donor in COOR, rather it is involved in
the generation of OH–/OHads– through its acid–base
equilibrium with H2O/HCO3– at the electrified interface. The
onset potential for COOR in the presence of CO32– is ca. 0.3 V vs RHE
and is comparable to the one observed for COOR in alkaline media,
where OH– is the oxygen donor.57 A study of OH– adsorption on Au(111)
shows that, at a potential as low as 0.3 V vs RHE, OH– is already adsorbed on the electrode with a low surface concentration.58 It is plausible to propose that the OH– generated from CO32– acid–base reaction results in adsorbed OH–, leading to a lower COOR overpotential. Performing
analogous COOR experiments with a RDE in a variety of electrolytes,
we observe that the role of anions as an oxygen donor “shuttle”
in COOR is a more generic feature of buffering anions, not limited
to CO32–. These experiments, together with infrared spectroscopic measurements,
will be discussed in a separate paper.
Finally, we analyze the
scan rate dependence of jlim during cathodic
polarization. Figure 7 A shows the voltammetry for Au(110) in CO2-saturated
0.1 M KHCO3 at different scan rates.
In Figure 7 B, we constructed
a Randles–Sevcik plot with the limiting cathodic current measured
at E = −1.0 V vs SHE. In a limited scan rate
range (2–25 mV s–1), there is a linear relationship
between jlim and the square root of the
scan rate, suggesting that the current is diffusion limited. Considering
that Au(110) is highly selective for CO2RR vs HER, that jlim depends linearly on the  (see Figure S8 B), and that it is comparable in electrolytes containing
different
bicarbonate concentration (see Figure 7 C), we propose that jlim is due to CO2RR becoming mass transport limited in CO2. However, the derivation of the diffusion coefficient from the slope
of the linear fitting in Figure 7 B gives a value that is two orders of magnitude lower
than the diffusion coefficient of CO2 (see Table S1). Most likely, the disagreement between
the calculated and the theoretical diffusion coefficient of CO2 is due to the interplay of homogeneous equilibria leading
to consumption of CO2 according to eq 2. Indeed, for each molecule of CO2 being reduced, two molecules of A– (i.e., OH– or CO32–) are produced according to reaction 1. Thus, CO2RR can be considered as a self-inhibiting
reaction; as CO2RR proceeds, CO2 is more and more consumed
by the product of its own reduction through solution reactions. A
similar plateau in the CO2RR current was observed for both gold24 and silver59 electrodes
and was ascribed to mass transport limitation in CO2. Still,
the CO2RR current exhibits an unusual scan rate dependence; it is
diffusion limited at low scan rates, but for higher scan rates, it
deviates from Randles–Sevcik behavior (see Figure 7 B). Under the latter conditions,
the plateau in the cathodic current disappears and the current becomes
almost scan rate independent, suggesting that CO2RR is not controlled
any more by diffusion. Consistently, improving mass transport by forced
convection has a marked effect on jlim at low scan rate, while its effect is more subtle for higher scan
rate (see Figure S11). Even if at low scan
rate under stationary conditions, the current at −1.1 V vs
SHE appears to be diffusion limited, and the Koutecky–Levich
analysis displays that the current is still partially kinetically
controlled (see Figure S12). This latter
result is in agreement with a mass transport limitation in CO2RR ascribed
to an “apparent” CO2 flux, due to a convolution
of mass transport with the kinetics of homogeneous reactions. Disentanglement
of the scan rate dependence requires many more detailed experiments
and presumably also kinetic simulations, which is outside of the scope
of this work.
(see Figure S8 B), and that it is comparable in electrolytes containing
different
bicarbonate concentration (see Figure 7 C), we propose that jlim is due to CO2RR becoming mass transport limited in CO2. However, the derivation of the diffusion coefficient from the slope
of the linear fitting in Figure 7 B gives a value that is two orders of magnitude lower
than the diffusion coefficient of CO2 (see Table S1). Most likely, the disagreement between
the calculated and the theoretical diffusion coefficient of CO2 is due to the interplay of homogeneous equilibria leading
to consumption of CO2 according to eq 2. Indeed, for each molecule of CO2 being reduced, two molecules of A– (i.e., OH– or CO32–) are produced according to reaction 1. Thus, CO2RR can be considered as a self-inhibiting
reaction; as CO2RR proceeds, CO2 is more and more consumed
by the product of its own reduction through solution reactions. A
similar plateau in the CO2RR current was observed for both gold24 and silver59 electrodes
and was ascribed to mass transport limitation in CO2. Still,
the CO2RR current exhibits an unusual scan rate dependence; it is
diffusion limited at low scan rates, but for higher scan rates, it
deviates from Randles–Sevcik behavior (see Figure 7 B). Under the latter conditions,
the plateau in the cathodic current disappears and the current becomes
almost scan rate independent, suggesting that CO2RR is not controlled
any more by diffusion. Consistently, improving mass transport by forced
convection has a marked effect on jlim at low scan rate, while its effect is more subtle for higher scan
rate (see Figure S11). Even if at low scan
rate under stationary conditions, the current at −1.1 V vs
SHE appears to be diffusion limited, and the Koutecky–Levich
analysis displays that the current is still partially kinetically
controlled (see Figure S12). This latter
result is in agreement with a mass transport limitation in CO2RR ascribed
to an “apparent” CO2 flux, due to a convolution
of mass transport with the kinetics of homogeneous reactions. Disentanglement
of the scan rate dependence requires many more detailed experiments
and presumably also kinetic simulations, which is outside of the scope
of this work.
Figure 7.

(A) Voltammetry of Au(110) crystal in CO2-saturated 0.1 M KHCO3 at different scan rates. (B) Randles–Sevcik plot of the plateau current (jlim) as measured in (A). (C) Voltammetry of the Au(110) crystal in CO2-saturated 0.1, 0.05, 0.005, and 0.0 M KHCO3 and a constant concentration of K+ (0.1 M, by addition of KClO4) at 25 mV s–1.
Conclusions
In summary, we investigated the structure dependence of Au(hkl) for CO2RR by cyclic voltammetry, where the cathodic scan was directly followed by anodic polarization to detect by electrooxidation the CO evolved. The Au(110) surface exhibits the highest activity for CO2RR, and COtop is the only adsorbed species detected.
By performing cyclic voltammetry on Au(110) in different bicarbonate electrolyte conditions, we revealed the importance of the current-induced changes in the surface species on CO2RR and COOR, as sketched in Figure 8. As the cathodic reactions proceed, the surface pH increases, and CO2RR becomes mass-transport limited, resulting in a current plateau. The mass transport limitation in CO2RR exhibits an anomalous scan rate dependence, as it is the result of the interplay of the diffusion rate of different bicarbonate-related species and the kinetics of the homogeneous equilibria leading to CO2 consumption. Our results suggest that close to the surface, where the concentrations of bicarbonate-related species are constantly changing, the kinetics of the homogeneous equilibria (2 and 3) may be more important than its thermodynamics (pKa). In the anodic scan, the change in bicarbonate surface speciation leads to the appearance of an additional peak (1st) for COOR. We propose that the origin of this 1st peak can be explained in terms of COOR being mediated by CO32– species, while the 2nd peak is due to COOR by H2O.
Figure 8.

Schematics of the current-driven changes in the surface bicarbonate speciation during cyclic voltammetry of Au(110) in CO2-containing bicarbonate electrolytes and its effect on COOR.
In conclusion, we highlighted the importance of the electrolyte species as primary actors in the electrochemical reactions. Bicarbonate (HCO3–) participates both in CO2RR, as a supply of CO2, and in HER, as an available proton donor,23 while carbonate (CO32–) acts as an oxygen donor in COOR. As the change of species at the electrode surface compared to the bulk may lead to the presence of new actors in the catalytic pathways, the necessity of probing in situ the local composition of the electrified interface becomes crucially important.
Acknowledgments
This project is part of the Solar-to-Products program and of the Advanced Research Center for Chemical Building Blocks (ARC–CBBC) consortium, cofinanced by The Netherlands Organization for Scientific Research (NWO) and by Shell Global Solutions International B.V.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.langmuir.1c00703.
Electrochemical characterization, additional voltammograms and FTIR data, and derivation of the diffusion coefficient (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Nørskov J. K.; Bligaard T.; Logadottir A.; Kitchin J. R.; Chen J. G.; Pandelov S.; Stimming U. Trends in the Exchange Current for Hydrogen Evolution. J. Electrochem. Soc. 2005, 152, J23. 10.1149/1.1856988. [DOI] [Google Scholar]
- Sheng W.; Zhuang Z.; Gao M.; Zheng J.; Chen J. G.; Yan Y. Correlating hydrogen oxidation and evolution activity on platinum at different pH with measured hydrogen binding energy. Nat. Commun. 2015, 6, 5848. 10.1038/ncomms6848. [DOI] [PubMed] [Google Scholar]
- Koper M. T. Thermodynamic theory of multi-electron transfer reactions: Implications for electrocatalysis. J. Electroanal. Chem. 2011, 660, 254–260. 10.1016/j.jelechem.2010.10.004. [DOI] [Google Scholar]
- Ragsdale S. W. Life with Carbon Monoxide. Crit. Rev. Biochem. Mol. Biol. 2004, 39, 165–195. 10.1080/10409230490496577. [DOI] [PubMed] [Google Scholar]
- Parkin A.; Seravalli J.; Vincent K. A.; Ragsdale S. W.; Armstrong F. A. Rapid and Efficient Electrocatalytic CO2/CO Interconversions by Carboxydothermus hydrogenoformans CO Dehydrogenase I on an Electrode. J. Am. Chem. Soc. 2007, 129, 10328–10329. 10.1021/ja073643o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reda T.; Plugge C. M.; Abram N. J.; Hirst J. Reversible interconversion of carbon dioxide and formate by an electroactive enzyme. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 10654–10658. 10.1073/pnas.0801290105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortlever R.; Shen J.; Schouten K. J. P.; Calle-Vallejo F.; Koper M. T. M. Catalysts and Reaction Pathways for the Electrochemical Reduction of Carbon Dioxide. J. Phys. Chem. Lett. 2015, 6, 4073–4082. 10.1021/acs.jpclett.5b01559. [DOI] [PubMed] [Google Scholar]
- Cunningham D. W.; Barlow J. M.; Velazquez R. S.; Yang J. Y. Reversible and Selective CO2 to HCO2- Electrocatalysis near the Thermodynamic Potential. Angew. Chem., Int. Ed. 2020, 59, 4443–4447. 10.1002/anie.201913198. [DOI] [PubMed] [Google Scholar]
- Hansen H. A.; Varley J. B.; Peterson A. A.; Nørskov J. K. Understanding Trends in the Electrocatalytic Activity of Metals and Enzymes for CO2 Reduction to CO. J. Phys. Chem. Lett. 2013, 4, 388–392. 10.1021/jz3021155. [DOI] [PubMed] [Google Scholar]
- Hori Y.; Kikuchi K.; Suzuki S. Production Of CO and CH4 in Electrochemical Reduction of CO2 at metal elctrodes in aqueous hydrogencarbonate solution. Chem. Lett. 1985, 14, 1695–1698. 10.1246/cl.1985.1695. [DOI] [Google Scholar]
- Edens G. J.; Hamelin A.; Weaver M. J. Mechanism of Carbon Monoxide Electrooxidation on Monocrystalline Gold Surfaces: Identification of a Hydroxycarbonyl Intermediate. J. Phys. Chem. 1996, 100, 2322–2329. 10.1021/jp9525604. [DOI] [Google Scholar]
- Rodriguez P.; Koper M. T. M. Electrocatalysis on gold. Phys. Chem. Chem. Phys. 2014, 16, 13583–13594. 10.1039/C4CP00394B. [DOI] [PubMed] [Google Scholar]
- Todoroki N.; Tei H.; Tsurumaki H.; Miyakawa T.; Inoue T.; Wadayama T. Surface Atomic Arrangement Dependence of Electrochemical CO2 Reduction on Gold: Online Electrochemical Mass Spectrometric Study on Low-Index Au(hkl) Surfaces. ACS Catal. 2019, 9, 1383–1388. 10.1021/acscatal.8b04852. [DOI] [Google Scholar]
- Mezzavilla S.; Horch S.; Stephens I. E. L.; Seger B.; Chorkendorff I. Structure Sensitivity in the Electrocatalytic Reduction of CO2 with Gold Catalysts. Angew. Chem., Int. Ed. 2019, 58, 3774–3778. 10.1002/anie.201811422. [DOI] [PubMed] [Google Scholar]
- Fu Y.; Ehrenburg M. R.; Broekmann P.; Rudnev A. V. Surface Structure Sensitivity of CO2 Electroreduction on Low-Index Gold Single Crystal Electrodes in Ionic Liquids. ChemElectroChem 2018, 5, 748–752. 10.1002/celc.201701209. [DOI] [Google Scholar]
- Feng X.; Jiang K.; Fan S.; Kanan M. W. Grain-Boundary-Dependent CO2 Electroreduction Activity. J. Am. Chem. Soc. 2015, 137, 4606–4609. 10.1021/ja5130513. [DOI] [PubMed] [Google Scholar]
- Mariano R. G.; McKelvey K.; White H. S.; Kanan M. W. Selective increase in CO2 electroreduction activity at grain-boundary surface terminations. Science 2017, 358, 1187–1192. 10.1126/science.aao3691. [DOI] [PubMed] [Google Scholar]
- Gallagher M.; Blizanac B.; Lucas C.; Ross P.; Marković N. Structure sensitivity of CO oxidation on gold single crystal surfaces in alkaline solution: Surface X-ray scattering and rotating disk measurements. Surf. Sci. 2005, 582, 215–226. 10.1016/j.susc.2005.03.018. [DOI] [Google Scholar]
- Rodriguez P.; Garcia-Araez N.; Koper M. T. M. Self-promotion mechanism for CO electrooxidation on gold. Phys. Chem. Chem. Phys. 2010, 12, 9373–9380. 10.1039/b926365a. [DOI] [PubMed] [Google Scholar]
- Wuttig A.; Yoon Y.; Ryu J.; Surendranath Y. Bicarbonate Is Not a General Acid in Au-Catalyzed CO2 Electroreduction. J. Am. Chem. Soc. 2017, 139, 17109–17113. 10.1021/jacs.7b08345. [DOI] [PubMed] [Google Scholar]
- Dunwell M.; Lu Q.; Heyes J. M.; Rosen J.; Chen J. G.; Yan Y.; Jiao F.; Xu B. The Central Role of Bicarbonate in the Electrochemical Reduction of Carbon Dioxide on Gold. J. Am. Chem. Soc. 2017, 139, 3774–3783. 10.1021/jacs.6b13287. [DOI] [PubMed] [Google Scholar]
- Zhu S.; Jiang B.; Cai W.-B.; Shao M. Direct Observation on Reaction Intermediates and the Role of Bicarbonate Anions in CO2 Electrochemical Reduction Reaction on Cu Surfaces. J. Am. Chem. Soc. 2017, 139, 15664–15667. 10.1021/jacs.7b10462. [DOI] [PubMed] [Google Scholar]
- Resasco J.; Lum Y.; Clark E.; Zeledon J. Z.; Bell A. T. Effects of Anion Identity and Concentration on Electrochemical Reduction of CO2. ChemElectroChem 2018, 5, 1064–1072. 10.1002/celc.201701316. [DOI] [Google Scholar]
- Zhang B. A.; Ozel T.; Elias J. S.; Costentin C.; Nocera D. G. Interplay of Homogeneous Reactions, Mass Transport, and Kinetics in Determining Selectivity of the Reduction of CO2 on Gold Electrodes. ACS Cent. Sci. 2019, 5, 1097–1105. 10.1021/acscentsci.9b00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwell M.; Yang X.; Setzler B. P.; Anibal J.; Yan Y.; Xu B. Examination of Near-Electrode Concentration Gradients and Kinetic Impacts on the Electrochemical Reduction of CO2 using Surface-Enhanced Infrared Spectroscopy. ACS Catal. 2018, 8, 3999–4008. 10.1021/acscatal.8b01032. [DOI] [Google Scholar]
- Yang K.; Kas R.; Smith W. A. In Situ Infrared Spectroscopy Reveals Persistent Alkalinity near Electrode Surfaces during CO2 Electroreduction. J. Am. Chem. Soc. 2019, 141, 15891–15900. 10.1021/jacs.9b07000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kas R.; Yang K.; Bohra D.; Kortlever R.; Burdyny T.; Smith W. A. Electrochemical CO2 reduction on nanostructured metal electrodes: fact or defect?. Chem. Sci. 2020, 11, 1738–1749. 10.1039/C9SC05375A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal A.; Marcandalli G.; Mints V. A.; Koper M. T. M. Competition between CO2 Reduction and Hydrogen Evolution on a Gold Electrode under Well-Defined Mass Transport Conditions. J. Am. Chem. Soc. 2020, 142, 4154–4161. 10.1021/jacs.9b10061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibler L. A. Preparation and Characterization of Noble Metal Single Crystal Electrode Surfaces. International Society of Electrochemistry 2003, 14, 20. [Google Scholar]
- Yoshida K.; Kuzume A.; Broekmann P.; Pobelov I. V.; Wandlowski T. Reconstruction and electrochemical oxidation of Au(110) surface in 0.1 M H2SO4. Electrochim. Acta 2014, 139, 281–288. 10.1016/j.electacta.2014.06.162. [DOI] [Google Scholar]
- Kolb D. M.; Dakkouri A. S.; Batina N. In Nanoscale Probes of the Solid/Liquid Interface; Gewirth A. A., Siegenthaler H., Eds.; Springer: The Netherlands, 1995; pp 263–284. [Google Scholar]
- Lukaszewski M.; Soszko M.; Czerwiński A. Electrochemical Methods of Real Surface Area Determination of Noble Metal Electrodes - an Overview. Int. J. Electrochem. Sci. 2016, 11, 4442–4469. 10.20964/2016.06.71. [DOI] [Google Scholar]
- García G.; Rodríguez P.; Rosca V.; Koper M. T. M. Fourier Transform Infrared Spectroscopy Study of CO Electro-oxidation on Pt(111) in Alkaline Media. Langmuir 2009, 25, 13661–13666. 10.1021/la902251z. [DOI] [PubMed] [Google Scholar]
- Dong C.; Fu J.; Liu H.; Ling T.; Yang J.; Qiao S. Z.; Du X.-W. Tuning the selectivity and activity of Au catalysts for carbon dioxide electroreduction via grain boundary engineering: a DFT study. J. Mater. Chem. A 2017, 5, 7184–7190. 10.1039/C6TA10733H. [DOI] [Google Scholar]
- Chen L. D.; Urushihara M.; Chan K.; Nørskov J. K. Electric Field Effects in Electrochemical CO2 Reduction. ACS Catal. 2016, 6, 7133–7139. 10.1021/acscatal.6b02299. [DOI] [Google Scholar]
- Liu T.; Xi C.; Dong C.; Cheng C.; Qin J.; Hu S.; Liu H.; Du X.-W. Improving Interfacial Electron Transfer via Tuning Work Function of Electrodes for Electrocatalysis: From Theory to Experiment. J. Phys. Chem. C 2019, 123, 28319–28326. 10.1021/acs.jpcc.9b09875. [DOI] [Google Scholar]
- Ringe S.; Clark E. L.; Resasco J.; Walton A.; Seger B.; Bell A. T.; Chan K. Understanding cation effects in electrochemical CO2 reduction. Energy Environ. Sci. 2019, 12, 3001–3014. 10.1039/C9EE01341E. [DOI] [Google Scholar]
- Ringe S.; Morales-Guio C. G.; Chen L. D.; Fields M.; Jaramillo T. F.; Hahn C.; Chan K. Double layer charging driven carbon dioxide adsorption limits the rate of electrochemical carbon dioxide reduction on Gold. Nat. Commun. 2020, 11, 33. 10.1038/s41467-019-13777-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnussen O.; Wiechers J.; Behm R. In situ scanning tunneling microscopy observations of the potential-dependent (1 × 2) reconstruction on Au(110) in acidic electrolytes. Surf. Sci. 1993, 289, 139–151. 10.1016/0039-6028(93)90893-O. [DOI] [Google Scholar]
- Lecoeur J.; Andro J.; Parsons R. The behaviour of water at stepped surfaces of single crystal gold electrodes. Surf. Sci. 1982, 114, 320–330. 10.1016/0039-6028(82)90474-5. [DOI] [Google Scholar]
- Arihara K.; Kitamura F.; Ohsaka T.; Tokuda K. Characterization of the adsorption state of carbonate ions at the Au(111) electrode surface using in situ IRAS. J. Electroanal. Chem. 2001, 510, 128–135. 10.1016/S0022-0728(01)00498-3. [DOI] [Google Scholar]
- Figueiredo M. C.; Ledezma-Yanez I.; Koper M. T. M. In Situ Spectroscopic Study of CO2 Electroreduction at Copper Electrodes in Acetonitrile. ACS Catal. 2016, 6, 2382–2392. 10.1021/acscatal.5b02543. [DOI] [Google Scholar]
- Rodriguez P.; Garcia-Araez N.; Koverga A.; Frank S.; Koper M. T. M. CO Electroxidation on Gold in Alkaline Media: A Combined Electrochemical, Spectroscopic, and DFT Study. Langmuir 2010, 26, 12425–12432. 10.1021/la1014048. [DOI] [PubMed] [Google Scholar]
- Wuttig A.; Yaguchi M.; Motobayashi K.; Osawa M.; Surendranath Y. Inhibited proton transfer enhances Au-catalyzed CO2-to-fuels selectivity. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E4585–E4593. 10.1073/pnas.1602984113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blizanac B. B.; Arenz M.; Ross P. N.; Marković N. M. Surface Electrochemistry of CO on Reconstructed Gold Single Crystal Surfaces Studied by Infrared Reflection Absorption Spectroscopy and Rotating Disk Electrode. J. Am. Chem. Soc. 2004, 126, 10130–10141. 10.1021/ja049038s. [DOI] [PubMed] [Google Scholar]
- Katayama Y.; Nattino F.; Giordano L.; Hwang J.; Rao R. R.; Andreussi O.; Marzari N.; Shao-Horn Y. An In Situ Surface-Enhanced Infrared Absorption Spectroscopy Study of Electrochemical CO2 Reduction: Selectivity Dependence on Surface C-Bound and O-Bound Reaction Intermediates. J. Phys. Chem. C 2019, 123, 5951–5963. 10.1021/acs.jpcc.8b09598. [DOI] [Google Scholar]
- Schulz K.; Riebesell U.; Rost B.; Thoms S.; Zeebe R. Determination of the rate constants for the carbon dioxide to bicarbonate inter-conversion in pH-buffered seawater systems. Mar. Chem. 2006, 100, 53–65. 10.1016/j.marchem.2005.11.001. [DOI] [Google Scholar]
- Narayanaru S.; Chinnaiah J.; Phani K. L.; Scholz F. pH dependent CO adsorption and roughness-induced selectivity of CO2 electroreduction on gold surfaces. Electrochim. Acta 2018, 264, 269–274. 10.1016/j.electacta.2018.01.106. [DOI] [Google Scholar]
- Hori Y.; Murata A.; Kikuchi K.; Suzuki S. Electrochemical reduction of carbon dioxides to carbon monoxide at a gold electrode in aqueous potassium hydrogen carbonate. J. Chem. Soc., Chem. Commun. 1987, 728–729. 10.1039/c39870000728. [DOI] [Google Scholar]
- Noda H.; Ikeda S.; Oda Y.; Imai K.; Maeda M.; Ito K. Electrochemical Reduction of Carbon Dioxide at Various Metal Electrodes in Aqueous Potassium Hydrogen Carbonate Solution. Bull. Chem. Soc. Jpn. 1990, 63, 2459–2462. 10.1246/bcsj.63.2459. [DOI] [Google Scholar]
- Hori Y.; Wakebe H.; Tsukamoto T.; Koga O. Electrocatalytic process of CO selectivity in electrochemical reduction of CO2 at metal electrodes in aqueous media. Electrochim. Acta 1994, 39, 1833–1839. 10.1016/0013-4686(94)85172-7. [DOI] [Google Scholar]
- Cave E. R.; Montoya J. H.; Kuhl K. P.; Abram D. N.; Hatsukade T.; Shi C.; Hahn C.; Nørskov J. K.; Jaramillo T. F. Electrochemical CO2 reduction on Au surfaces: mechanistic aspects regarding the formation of major and minor products. Phys. Chem. Chem. Phys. 2017, 19, 15856–15863. 10.1039/C7CP02855E. [DOI] [PubMed] [Google Scholar]
- Zhong H.; Fujii K.; Nakano Y.; Jin F. Effect of CO2 Bubbling into Aqueous Solutions Used for Electrochemical Reduction of CO2 for Energy Conversion and Storage. J. Phys. Chem. C 2015, 119, 55–61. 10.1021/jp509043h. [DOI] [Google Scholar]
- Dettori R.; Donadio D. Carbon dioxide, bicarbonate and carbonate ions in aqueous solutions under deep Earth conditions. Phys. Chem. Chem. Phys. 2020, 22, 10717–10725. 10.1039/C9CP06904F. [DOI] [PubMed] [Google Scholar]
- Bard A. J.; Faulkner L. R.. Electrochemical Methods: Fundamentals and Applications, 2nd ed.; John Wiley & Sons, 2000; p 163. [Google Scholar]
- Monteiro M. C. O.; Jacobse L.; Koper M. T. M. Understanding the Voltammetry of Bulk CO Electrooxidation in Neutral Media through Combined SECM Measurements. J. Phys. Chem. Lett. 2020, 11, 9708–9713. 10.1021/acs.jpclett.0c02779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez P.; Feliu J. M.; Koper M. T. Unusual adsorption state of carbon monoxide on single-crystalline gold electrodes in alkaline media. Electrochem. Commun. 2009, 11, 1105–1108. 10.1016/j.elecom.2009.03.018. [DOI] [Google Scholar]
- Chen A.; Lipkowski J. Electrochemical and Spectroscopic Studies of Hydroxide Adsorption at the Au(111) Electrode. J. Phys. Chem. B 1999, 103, 682–691. 10.1021/jp9836372. [DOI] [Google Scholar]
- Clark E. L.; Resasco J.; Landers A.; Lin J.; Chung L.-T.; Walton A.; Hahn C.; Jaramillo T. F.; Bell A. T. Standards and Protocols for Data Acquisition and Reporting for Studies of the Electrochemical Reduction of Carbon Dioxide. ACS Catal. 2018, 8, 6560–6570. 10.1021/acscatal.8b01340. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.