

Abstract
A high incidence of acute megakaryoblastic leukemia (AMKL) in Down syndrome patients implies that chromosome 21 genes have a pivotal role in AMKL development, but the functional contribution of individual genes remains elusive. Here, we report that SON, a chromosome 21-encoded DNA- and RNA-binding protein, inhibits megakaryocytic differentiation by suppressing RUNX1 and the megakaryocytic gene expression program. As megakaryocytic progenitors differentiate, SON expression is drastically reduced, with mature megakaryocytes having the lowest levels. In contrast, AMKL cells express an aberrantly high level of SON, and knockdown of SON induced the onset of megakaryocytic differentiation in AMKL cell lines. Genome-wide transcriptome analyses revealed that SON knockdown turns on the expression of pro-megakaryocytic genes while reducing erythroid gene expression. Mechanistically, SON represses RUNX1 expression by directly binding to the proximal promoter and two enhancer regions, the known +23 kb enhancer and the novel +139 kb enhancer, at the RUNX1 locus to suppress H3K4 methylation. In addition, SON represses the expression of the AP-1 complex subunits JUN, JUNB and FOSB which are required for late megakaryocytic gene expression. Our findings define SON as a negative regulator of RUNX1 and megakaryocytic differentiation, implicating SON overexpression in impaired differentiation during AMKL development.
INTRODUCTION
Acute megakaryoblastic leukemia (AMKL) is a subtype of acute myeloid leukemia (AML), which involves excessive proliferation of immature megakaryoblasts with impaired differentiation along the megakaryocytic lineage [1] . Although AMKL is fairly rare, accounting for about 1% of total adults diagnosed with AML, its occurrence is greatly contrasted in children, with the incidence rate rising to 4-15% of patients diagnosed with AML [2–4]. This is likely due to the high occurrence rate of AMKL in children with Down syndrome (DS), who are at a 500-fold increased risk of developing this malignancy compared with the general population [2, 5].
To date, the knowledge about any mechanisms that contribute to AMKL development is scarce. In non-Down syndrome AMKL (non-DS-AMKL), only a few chromosomal translocation events that generate fusion genes (e.g. RBM15-MKL1 and CBFA2T3-GLIS2) have been identified as underlying molecular mechanisms [6]. Since incidence of AMKL is greatly increased in individuals with DS, chromosome 21-encoded genes have been studied for their roles in DS-AMKL development in cooperation with other hits, such as GATA1 mutations [7]. Although multiple studies have proved that the presence of trisomy 21 perturbed normal hematopoietic differentiation [8–11], the exact roles of individual chromosome 21 genes in this process remain elusive. Among chromosome 21 genes, RUNX1, ERG, ETS2, DYRK1 and microRNA 125-b2 are implicated to create pre-leukemic conditions [12–18]. Paradoxically, RUNX1 expression is decreased in DS-AMKL despite the increased gene dosage [19], suggesting that chromosome 21 genes may inter-regulate gene expression.
SON is a large nuclear speckle protein possessing both DNA- and RNA-binding abilities, and its expression is particularly high in pluripotent embryonic stem cells and hematopoietic organs/cells [20–24]. Our group and others have reported that SON functions as a splicing co-factor required for correct RNA processing of a group of genes involved in cell cycle, DNA repair, epigenetic regulation and stem cell pluripotency [21, 24, 25]. We also recently reported that heterozygous loss-of-function mutations in the SON gene in human patients cause a novel developmental disorder designated as ZTTK syndrome (Zhu-Tokita-Takenouchi-Kim syndrome) [26]. Interestingly, a partial SON gene was first cloned from a screening of DNA-binding factors and it was also identified as negative regulator of transcription from the human hepatitis B virus genome [22, 27]. Recently, our group reported that SON inhibits the assembly of MLL complex, a methyltransferase complex responsible for histone-3-lysine-4 tri-methylation (H3K4me3), at the promoters of multiple genes, resulting in transcriptional repression [28].
Importantly, the SON gene is located on human chromosome 21 and several studies have implied that SON may play a role in DS-associated leukemogenesis. A study with human induced pluripotent stem cell (iPSC)-derived hematopoietic progenitors demonstrated that SON is significantly increased in trisomy-21 iPSCs compared to euploid controls [10]. Nevertheless, how SON expression is associated with hematopoietic lineage differentiation and whether SON contributes to AMKL development still remain unexplored.
Here, we demonstrate that SON is upregulated in AMKL cells and SON depletion induces differentiation of both non-DS- and DS-AMKL cell lines. Furthermore, we identified that SON depletion leads to the onset of the megakaryocytic gene expression program through upregulation of the transcription factor RUNX1 as well as AP-1 complex components, JUN, JUNB and FOSB. Our results reveal SON as a critical repressor of megakaryocytic differentiation, implicating its increased dosage in AMKL pathogenesis.
MATERIALS AND METHODS
Cell culture and transfection
The cell lines, MEG-01 (non-DS AMKL), K562 (chronic myeloid leukemia; erythroblastic leukemia), Kasumi-1 (t(8;21)-positive acute myeloid leukemia) and HL60 (acute promyelocytic leukemia), were purchased from ATCC (Manassas, VA). The CMY and CMK cell lines (DS-AMKL) were kindly provided by Dr. Shai Izraeli (Tel Aviv University). The cell lines were grown in RPMI-1640 medium supplemented with 10% fetal bovine serum and 4mM L-glutamine. For differentiation experiments, AMKL cell lines (MEG-01, CMY and CMK) were treated either with DMSO (control) or with 5 nM phorbol 12-myristate 13-acetate (PMA; Sigma Aldrich, St. Louis, MO) and incubated for 4 and 8 days to evaluate SON expression during megakaryocytic differentiation. Day 0 samples were used as a control for each treatment group. For knockdown experiments, cells were nucleofected with Amaxa Nucleofector II for siRNA transfection and incubated for 2 to 5 days depending on the purpose of experiments. The SON siRNA sequence used for nucleofection is GCAUUUGGCCCAUCUGAGAtt (Silencer Select siRNA custom synthesis product by Life Technologies/ThermoFisher, Waltham, MA) which was verified for its effectiveness and specificity in previous studies [21, 28]. RUNX1 siRNA (siRNA ID s229352) and negative control siRNA (UAACGACGCGACGACGUAAtt; custom synthesis product) was purchased from ThermoFisher Scientific.
Wright-Giemsa Staining
The cells were cyto-spun on a slide and fixed in ice cold methanol. First, 300 μL of the Wright stain (Sigma Aldrich) was added to the slide for 3 min, then topped with 600 μL 1X PBS for 6 min. The slide was washed with deionized water and then dried. Next, 600 μL 1:10 diluted Giemsa stain (Sigma Aldrich) was added to the slide and incubated for 10-15 min at room temperature. Finally, the slide was washed and dried before adding the Permount solution with the microscope cover glass.
Western Blot
Protein lysates were subjected to 7–12% SDS-PAGE and electro-transferred to Polyvinylidene difluoride (PVDF) membranes. The membranes were then blocked in 5% nonfat dry milk for 1 h at room temperature, lightly washed, and incubated with the indicated primary antibodies overnight at 4 °C on a shaker. The membranes were further washed for 1 h, incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (ThermoFisher) for 1 h at room temperature, washed again, and finally visualized using Bio-Rad Clarity Western ECL. Primary antibodies used are: Anti-SON (SON-N Ab) generated against amino acids 74-88 of human SON; Anti-RUNX1/AML (D33G6) (Rabbit polyclonal; catalog #4336; Cell Signaling, Danvers, MA); Anti-JUNB (Rabbit polyclonal; catalog # A302-704A-M; Bethyl, Laboratories Montgomery, TX); Anti-c-JUN (Rabbit polyclonal; catalog # A302-959A-M; Bethyl Laboratories); Anti-Actin (Mouse monoclonal; catalog # A5441-.2ML; Sigma-Aldrich); Anti-GAPDH (Mouse monoclonal; catalog # MAB374; Millipore, Burlington, MA).
RT-PCR analysis
Total RNA was isolated using the RNeasy Mini Kit (Qiagen, Germantown, MD). The cDNA was synthesized using SuperScript III Reverse Transcriptase (ThermoFisher). The mRNA expression was analyzed with quantitative PCR analysis with iTaq Universal SYBR Green SMX 500 master mix (Bio-Rad, Hercules, CA). Gene expression was quantified using the 2-ΔΔCt methods. GAPDH, ACTB or YWHAZ were used as endogenous reference genes. Primer sequences are in Supplementary Table (Table S1).
RNA sequencing and data analysis
MEG-01 AMKL cells were transfected with negative control siRNA or SON siRNA (100 pmol/106 cells) and harvested after 48 h (no significant morphological changes observed at this time point) for total RNA isolation using the RNeasy Mini Kit (Qiagen). The RNA was poly(A)-selected and the libraries were prepared using TruSeq stranded total RNA Library Prep kit (Illumina, San Diego, CA) and sequenced (150 bp paired-end) using DNA nanoball sequencing (DNBseq) technology (BGI, Cambridge, MA). All sequencing data have been deposited to the NCBI GEO database under accession number GSE157178.
For data analysis, Kallisto [29] was used for pseudoalignments to the transcriptome and transcript quantifications. The output from Kallisto was used for differential expression analyzed by Sleuth [30]. Differentially expressed genes were further analyzed for Gene Set Enrichment Analysis (GSEA; http://www.broad.mit.edu/gsea/ [31]) and the enrichment map was visualized using Cytoscape [32].
Flow cytometry analysis
MEG-01 and CMY cell lines were transfected with siRNA and incubated for 4 and 5 days, respectively. Once cells were harvested and washed with 1X PBS, cells were incubated with anti-CD61-PE (Clone: VI-PL2, BD Biosciences, San Jose, CA) for 30 min at RT, washed with 1X PBS, and resuspended in 1X PBS for analyzation. The analysis was performed on BD Bioscience FACS Canto II Flow Cytometer.
Chromatin immunoprecipitation (ChIP) and ChIP-qPCR
MEG-01, CMY, and CMK cells were incubated with 1% formaldehyde in 5 ml growth medium for 10 min at room temperature and cross-linking reaction was terminated by incubation with 125 mM glycine for 5 min. Subsequently, cells were incubated for 20 min at 4°C with lysis buffer (5 mM HEPES pH 8.0 / 85 mM KCl / 0.5% NP-40 / 1x Complete Protease Inhibitor Cocktail [Roche]), collected by centrifugation for 5 min at 3,000g and resuspended in RIPA buffer (150 mM NaCl / 50 mM Tris-HCl, pH 8.0/ 1 mM EDTA / 1% sodium deoxycholate / 0.1% SDS / 1% Triton X-100 / 1x Complete Protease Inhibitor Cocktail) for another 20 min. To shear chromatin to lengths ranging between 200–500 base pairs, crude nuclei were sonicated with the Ultrasonic disintegrator Sonicator S-4000 (Misonix). Sonicated DNA from each sample were incubated at 4°C overnight with 1–5 μg of specific antibodies or normal immunoglobulin G (IgG) as controls and magnetic bead protein A or G (Dynabeads Protein A or Protein G, ThermoFisher). The magnetic beads were washed 5 times for 3 min at 4°C on a rotating platform with 1 ml wash buffer (100 mM Tris pH 7.5 / 500 mM LiCl / 1% NP-40 / 1% Sodium deoxycholate) and washed once with TE (10 mM Tris pH 7.5 / 0.1 mM EDTA). After washing, the washed beads were eluted by heating for 2 hr at 65°C in elution buffer (1% SDS / 0.1 M NaHCO3) with proteinase K. ChIP DNA were purified and concentrated using the QIAquick PCR Purification Kit (Qiagen). For ChIP-qPCR, all ChIP signals were normalized to total input or H3 ChIP, and each experiment was performed at least three times independently. The primer sets for ChIP-qPCR were listed in the Supplementary Table (Table S2). Antibodies used for ChIP are: Anti-SON (SON-N Ab) generated against amino acids 74-88 of human SON; Anti-Histone H3 antibody - ChIP Grade (Rabbit polyclonal; catalog # ab1791; Abcam, Cambridge, United Kingdom); Anti-Histone H3 (tri methyl K4) antibody - ChIP Grade (Rabbit polyclonal; catalog # ab8580; Abcam); Anti-Histone H3 (mono methyl K4) antibody - ChIP Grade (Rabbit polyclonal; catalog # ab8895; Abcam).
Statistical analysis
All experiments are the results of at least three biological replicates. Data are presented as the mean ± SD with P-values calculated by two-tail student’s t-test or one-way ANOVA or Shapiro-Wilk normality test. All statistics were calculated with GraphPad Prism version 7.02.
RESULTS
SON is minimally expressed in differentiating megakaryocytic progenitors and mature megakaryocytes, but its expression is aberrantly increased in AMKL.
To address how SON expression is regulated during megakaryocytic differentiation, we used the BloodSpot database (www.bloodspot.eu, [33]), a repository of hematopoietic gene expression. The analysis of SON expression in normal human hematopoiesis showed that SON is highly expressed in hematopoietic stem cells (HSCs), especially, in CD133(+) CD34(dim) HSCs, and its level decreases when HSCs differentiate into common myeloid progenitors (CMPs) (Fig. 1A), which is consistent with our previous observation [23]. Interestingly, the SON expression pattern showed striking dynamics when megakaryocyte-erythroid progenitors (MEPs) differentiate into megakaryocytes and erythrocytes. SON remains highly expressed in erythroblasts along the erythroid maturation process with a transient increase to reach the highest level in the CD34(−) CD71(+) glycophorin A (GlyA)(−) erythroblast populations. In contrast, the SON expression is drastically decreased in megakaryocytic progenitors (colony-forming-unit [CFU]-megakaryocytic) and mature megakaryocytes (Fig. 1A). Furthermore, our analysis of data from another RNA-sequencing study (data deposited in the European Genome-phenome Archive, EGAD00001000745; [34]), revealed that mature megakaryocytes have a lower level of SON compared to erythroblasts and MEPs (Fig. 1B). These data demonstrate that SON expression is precisely regulated during megakaryocytic-erythroid lineage differentiation and mature megakaryocytes have the lowest level of SON within this lineage.
Figure 1. SON is minimally expressed in differentiated megakaryocytes but is aberrantly upregulated in AMKL.

A. Hierarchical differentiation tree of relative SON mRNA expression levels in the indicated human hematopoietic subpopulations. Normal human hematopoiesis-DMAP dataset (GSE24759) was analyzed and retrieved from BloodSpot). Note the high levels of SON in HSCs and differentiating erythroid cells, which is in sharp contrast to the low level of SON in differentiating megakaryocytes.
B. SON expression in human megakaryocyte-erythroid progenitors (MEPs) compared with erythroblasts and megakaryocytes. The data were retrieved from the European Genome-phenome Archive accession number EGAD00001000745. n = 3 - 5.
C. SON expression in bone marrow cells from AML, non-DS-AMKL, DS-AMKL, and TMD patients. The data were retrieved from the GSE83449 dataset. The results from three different probes for SON (Affymetrix Human U133A) were shown; 214988_s_at, detecting the 3’UTR region; 201086_x_at, detecting exon 12 and 3’UTR; 201085_s_at, detecting exon 3. *p<0.5, **p<0.01 compared to AML.
D. SON expression in flow-sorted leukemic blasts isolated from bone marrow cells of non-DS-AMKL and DS-AMKL patients. The data were retrieved from the GSE16677 dataset. *p<0.5 compared to non-DS-AMKL.
E. SON expression in bone marrow cells from pediatric AMKL patients with NUP98-KDM5A fusion. CD34+ cord blood hematopoietic stem/progenitor cells were used as normal controls. The data were retrieved from the GSE123485 dataset. *p<0.5 compared to CD34+ cord blood cells.
While our group and others previously observed that SON is highly expressed in AML [28, 35], whether SON expression is altered in other hematologic malignancies is largely unknown. In addition, the SON gene is located on human chromosome 21 and overexpressed in Down syndrome individuals who show a high risk of AMKL development. Knowing that SON expression is decreased during normal megakaryocytic differentiation (Figs. 1A and 1B), we sought to examine whether SON expression is altered in AMKL. We analyzed the data reporting differential gene expression in total bone marrow cells from patients with AML, non-DS-AMKL, DS-AMKL and transient myeloproliferative disorder (TMD), a pre-leukemic condition characterized by abnormal excessive proliferation of myeloblasts frequently found in newborn babies with Down syndrome (GSE83449; [19]). The data from three different microarray probe sets consistently showed that SON expression is increased in bone marrow cells of patients with non-DS-AMKL as well as DS-AMKL compared to AML (Fig. 1C). Our analyses of another data set comparing gene expression in flow-sorted leukemic blasts from patients (GSE16677; [36]) showed a higher SON level in DS-AMKL compared to Non-DS-AMKL leukemic blasts (Fig. 1D). Furthermore, SON is significantly upregulated in non-DS-AMKL (NUP98-KDM5A fusion-associated pediatric AMKL) when compared to normal CD34(+) cord blood cells which are enriched with HSCs (Fig. 1E; the data set was obtained from GSE123485; [37]). These findings suggest that while a high level of SON is always observed in DS-AMKL leukemic blasts due to trisomy 21, SON expression is also elevated in non-DS-AMKL, sometimes comparably as high as the DS-AMKL SON level.
In addition, the TCGA AML data set showed that AMKL, which is AML-M7 based on the French-American-British (FAB) classification of AML, showed the highest level of SON among different subtypes of AML (Supplementary Fig. S1A). We also found that the DS-AMKL cell lines CMY and CMK and a chronic myelogenous leukemia (CML) cell line K562 (which has characteristics of the megakaryocytic-erythroid lineage) have a higher level of SON expression compared to the monocyte lineage AML cell lines, Kasumi-1 and HL60 (Supplementary Fig. S1B). Taken together, our extensive data analyses revealed that a marked reduction of SON expression occurs during normal megakaryocytic differentiation, and aberrant upregulation of SON is a prominent feature of AMKL, which is associated with impaired megakaryocytic differentiation.
SON expression is decreased upon induction of megakaryocytic differentiation, and SON knockdown leads to the onset of megakaryocytic differentiation in AMKL cells.
Next, we questioned whether SON expression is altered during megakaryocytic differentiation of AMKL cells. To this end, we treated the MEG-01 (non-DS-AMKL) and CMY (DS-AMKL) cell lines with phorbol 12-myristate 13-acetate (PMA). It has been well known that PMA induces differentiation of human erythrocytic and megakaryocytic leukemia cell lines within the broad range of concentration between 0.1 nM and 100 nM with optimum effects seen at 2 nM −10 nM [38–41]. As shown in Figures 2A and 2B, we observed marked decreases of the SON mRNA and protein expression levels in 4 and 8 days after 5 nM PMA treatment, indicating that SON is indeed downregulated when AMKL cells are differentiated. Megakaryocytic differentiation of the cells was confirmed by upregulation of CD61 (also known as integrin beta 3; ITGB3) and von Willebrand factor (vWF) (Fig. 2A).
Figure 2. SON expression is decreased during PMA-induced megakaryocytic differentiation and SON knockdown leads to the onset of megakaryocytic differentiation in AMKL cells.

A. Quantitative RT-PCR detection of SON, CD61, and vWF after treating the AMKL cell lines, MEG-01 (non-DS-AMKL) and CMY (DS-AMKL), with 5 nM PMA for the indicated time to induce megakaryocytic differentiation. Data are expressed as mean ±SD; n=3, *p<0.5, **p<0.01.
B. Western blot demonstrating the protein levels of SON and Actin in DMSO-treated (control) or PMA-treated MEG-01 and CMY cells.
C. Representative images of Wright-Giemsa stained MEG-01, CMY, and CMK cells 72 h after control siRNA or SON siRNA. Scale bar: 100μm.
D. Quantitative RT-PCR detection of SON, CD61 and vWF 48 h after control siRNA or SON siRNA transfection. Data are expressed as mean±SD; n=2-6, *p<0.05, **p<0.01 compared to control.
While we confirmed the downregulation of SON during megakaryocytic differentiation of AMKL cells, it was still not clear whether reduction of SON is a driving force that induces megakaryocytic differentiation, or if it is merely a consequence of differentiation. Interestingly, when we reduced the SON level by siRNA transfection into MEG-01, CMY and CMK cells, these cells showed noticeable morphological changes; cells became much larger and multinucleated cells were observed (Fig. 2C), suggesting megakaryocytic differentiation. Furthermore, CD61 and vWF were significantly upregulated upon SON depletion (Fig. 2D). These results demonstrate that depletion of SON is indeed able to induce megakaryocytic differentiation in AMKL cell lines.
SON knockdown in AMKL cells reveals the genome-wide transcriptome changes and the affected cellular pathways.
Since SON is a nuclear protein that regulates both transcription and RNA splicing [21, 28], we next sought to examine the genome-wide transcriptome changes upon SON knockdown in AMKL cells. Our RNA-sequencing results identified the genes differentially expressed upon SON knockdown in MEG-01 cells (Fig. 3A). Using the Wald test with q-value ≤ 0.05 (Wald test beta, a biased estimate of the natural log transformation, >0.25 for upregulated genes and <−0.25 for downregulated genes), we identified 2,718 upregulated genes and 2,670 downregulated genes (Supplementary Tables S3 – S4). Gene Set Enrichment Analysis (GSEA) followed by visualization of the enrichment map using Cytoscape revealed statistically enriched pathways among differentially expressed genes (Fig. 3B). Consistent with previous reports [21, 25], we found that multiple pathways associated with cell cycle and DNA damage response were enriched, including the pathways regulating G1 phase, S phase (DNA synthesis), mitotic phase, spindle checkpoint, chromatid mitotic phase cohesion, response to irradiation and p53 downstream pathway. Our data also revealed that pathways associated with nonsense-mediated RNA decay and protein translation, as well as extracellular matrix-integrin pathways, are significantly enriched among differentially expressed genes (Fig. 3B). In addition, from individual GSEA enrichment plots, we found that, upon SON knockdown, genes involved in metabolism, such as NADH metabolism, citrate cycle and pyruvate metabolism, were significantly downregulated (Supplementary Fig. S2). Taken together, these analyses established the gene expression profiles and identified the affected pathways upon SON knockdown in AMKL cells.
Figure 3. SON knockdown in AMKL cells reveals the genome-wide transcriptome changes and the affected cellular pathways.
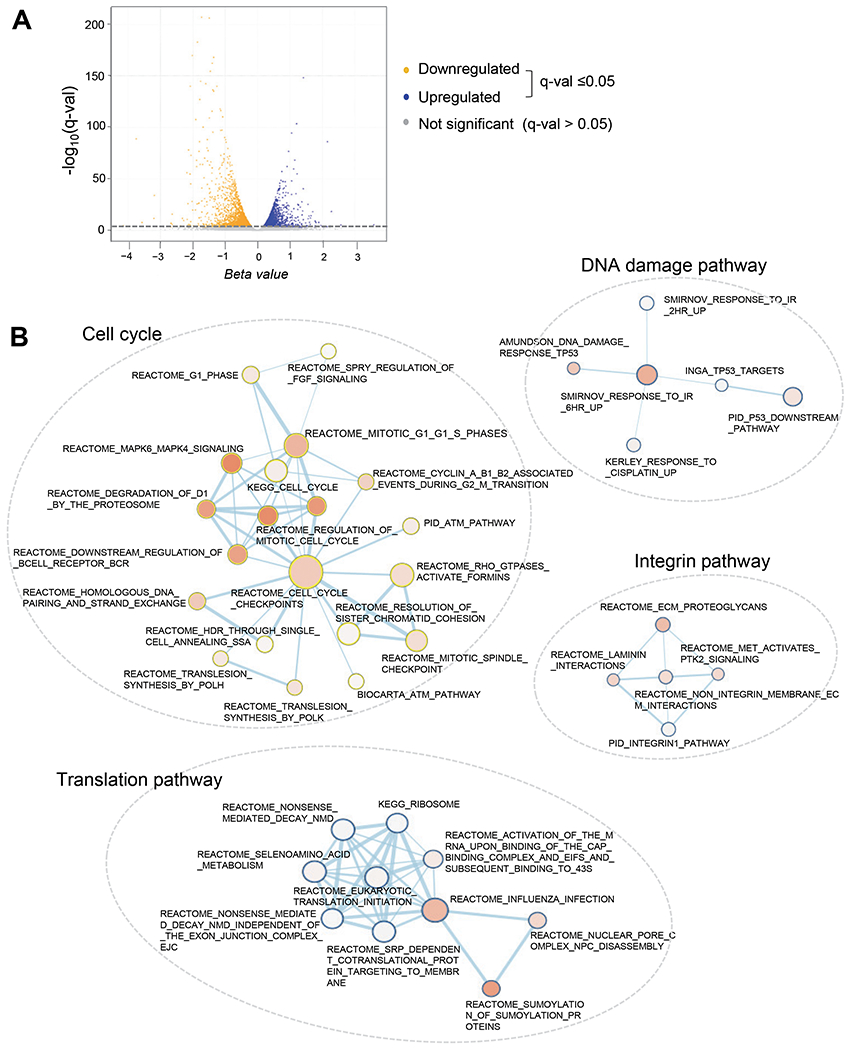
A. Volcano plot showing the profiles of differentially expressed genes between control and SON siRNA-transfected MEG-01 AMKL cells.
B. Enrichment map (Cytoscape) visualizing the representative results of Gene Set Enrichment Analysis (GSEA) of the genes differentially expressed upon SON knockdown in MEG-01 cells. The descriptions on next to each node are the gene set names found in the Molecular Signatures Database (MSigDB). Enrichment results as a network of gene-sets (nodes) related by their similarity (edges). Enrichment significance is encoded by the node color intensity and node size represents the gene-set size and edge thickness represents the degree of overlap between two gene-sets.
SON knockdown in AMKL cells leads to activation of genes promoting megakaryocytic differentiation.
Although our groups and others previously reported the list of SON-regulated genes identified by microarray or RNA-sequencing [21, 24, 25], none of those studies have been done in hematopoietic cells. Therefore, how SON regulates genes associated with hematopoietic differentiation remains completely unknown. Interestingly, through GSEA, we found that the genes previously known to be enriched in MEPs when compared to HSCs (i.e. genes downregulated in HSCs versus MEPs) are upregulated upon SON knockdown (Fig. 4A, top; enrichment plot), which indicates that SON knockdown suppresses HSC-like gene signature while inducing MEP-like gene signature. The list includes the genes that have been verified for their functional significance in megakaryocytic differentiation (Fig. 4A, bottom; heatmap), such as genes inducing differentiation of stem cells and leukemia cells (LZTFL1, BACH2, and CEP68) [42–45], genes promoting megakaryocytic differentiation (PLEKHO1, ADCY6, and PDK1) [46–48], and a gene conferring sensitivity to anti-leukemic reagents (SESN3) [49].
Figure 4. SON knockdown leads to upregulation of pro-megakaryocytic genes.

A. GSEA enrichment plots demonstrating genes previously known to be enriched in MEPs when compared to HSCs are upregulated upon SON knockdown (top panel). The corresponding Blue-Pink O’gram of core enrichment genes generated by GSEA (bottom panel).
B. GSEA enrichment plots demonstrating genes previously known to be enriched in MEPs when compared to GMPs are upregulated upon SON knockdown (top panel). The corresponding Blue-Pink O’gram of core enrichment genes generated by GSEA (bottom panel).
C. Heatmap showing the expression profiles of selected pro-megakaryocytic and pro-erythroid genes.
In addition, GSEA revealed that genes enriched in MEPs compared to GMPs (i.e. gene upregulated in MEPs versus GMPs) were indeed upregulated upon SON knockdown (Fig. 4B, top; enrichment plot). These genes include many of those required for megakaryocyte maturation/platelet activation (PTPRE, PLEK, F2RL2, and PTPRJ) [50–53] and cell adhesion/cytoskeletal organization (LAMC1, ZYX, and DOCK5) [54–56] (Fig. 4B, bottom; heatmap). These findings reveal that SON knockdown in AMKL cells strongly induces the gene signature of megakaryocytic-erythroid lineage differentiation.
Knowing that SON knockdown in AMKL cells leads to upregulation of genes associated with MEP lineage differentiation in the sake of HSC- and GMP-associated genes, we further examined whether megakaryocytic and erythroid genes are differentially regulated in response to SON knockdown. We generated a list of pro-megakaryocytic genes and pro-erythroid genes (based on the information from Harmonizome, TISSUES Experimental Tissue Protein Expression Evidence Scores dataset; http://amp.pharm.mssm.edu/Harmonizome/), and analyzed their expression levels from our RNA-seq data. Our analysis showed that pro-megakaryocytic genes, including the transcription factors that favor megakaryocytic differentiation (RUNX1, JUN, FOSB, PU.1, and GABPA) are upregulated upon SON depletion, while several transcription factors known to function in erythroid lineage differentiation (KLF1, GATA2, MYC, and MYB) are decreased upon SON depletion (Fig. 4C). Taken together, these data demonstrate that SON depletion in AMKL cells turns on the pro-megakaryocytic gene expression program and suppresses the HSC-associated and pro-erythroid gene expression programs. Therefore, it is highly likely that the marked difference in SON expression between differentiating megakaryocytes and erythroblasts (Fig. 1A) plays as a key driving force to induce lineage-specific gene expression programs required for terminal differentiation.
SON depletion leads to upregulation of RUNX1, promoting megakaryocytic differentiation.
At the megakaryocytic-erythroid lineage bifurcation, the cooperation and antagonisms among transcription factors play critical roles in determining the lineage fate. One of the essential regulators of megakaryocytic differentiation is RUNX1 [57]. RUNX1 is an upstream regulator that can shift the balance between kruppel-like factor 1 (KLF1) and friend leukemia integration 1 (FLI1), the two master transcription factors dictating erythroid- and megakaryocytic differentiation, respectively [58]. It has been shown that RUNX1 directly binds to the KLF1 promoter and downregulates KLF1, thereby suppressing erythroid gene expression while activating megakaryocytic gene expression [59]. Our RNA-sequencing data revealed the increase of RUNX1 and the decrease of KLF1 expression upon SON knockdown (Fig. 4C), and we confirmed those changes in 3 different AMKL cell lines, MEG-01, CMY and CMK, by qPCR and Western blot (Figs. 5A and 5B).
Figure 5. SON depletion leads to upregulation of RUNX1, resulting in activation of RUNX1-regulated megakaryocytic transcription programs.

A. Quantitative RT-PCR detection of RUNX1 and KLF1, in control and SON-siRNA transfected AMKL cell lines (48 h post- transfection). Data are expressed as mean±SD; n=3-6, *p<0.05, **p<0.01.
B. Western blot demonstrating RUNX1 protein expression 48 h after SON siRNA transfection into the indicated AMKL cell lines.
C. Representative histograms from flow cytometric analyses detecting surface expression of CD61 in MEG-01 cells 96 h after transfection of single- or combinational siRNA (SON siRNA or/and RUNX1 siRNA).
D. Average percentage of CD61-positive cells obtained from four biological replicates of flow cytometric analyses. Data are expressed as mean±SD; n=3, *p<0.05, **p<0.01; Black asterisk, compared to control; Red asterisk, between two indicated samples.
In consideration of RUNX1 being a master megakaryocytic lineage regulator, we decided to investigate whether RUNX1 is crucial in megakaryocytic differentiation induced by SON depletion. To address this question, we knocked down SON and RUNX1 by transfecting MEG-01 and CMY cells with single or combinational siRNA (SON siRNA and/or RUNX1 siRNA). We noticed that large, multinucleated cells were formed by SON siRNA alone as well as the combinational siRNA treatment, but not by RUNX1 siRNA (Supplementary Figs. S3A and S4A). Quantitative measurements of mRNA by qPCR showed that vWF level was indeed increased by SON siRNA, but this effect was reduced upon addition of RUNX1 siRNA (Supplementary Figs. S3B and S4B). These results suggest that RUNX1 plays a role in megakaryocytic differentiation induced by SON depletion. We further examined the expression of CD61 at the cell surface by flow cytometry. The results showed that SON knockdown upregulated CD61 at the cell surface and, in contrast, RUNX1 knockdown led to reduction of CD61. We found that CD61 upregulation upon SON knockdown was partially abrogated when RUNX1 was concurrently reduced in MEG-01 cells (Figs. 5C and 5D). However, RUNX1 knockdown could not significantly hinder SON siRNA-mediated CD61 upregulation in CMY cells (Supplementary Figs. S4C and S4D). These results suggest that SON’s action in blocking CD61 expression during megakaryocytic differentiation is only partially through RUNX1.
SON directly binds to the RUNX1 promoter and two enhancer regions and represses RUNX1 transcription.
RUNX1 transcription is driven by two different promoters, the distal promoter (P1) and the proximal promoter (P2) [60, 61]. The RUNX1 gene also contains a well-known enhancer element, located 23 kb downstream of the P1 promoter (+23 kb). This +23 enhancer is known to regulate the activity of both P1 and P2 promoters of RUNX1 and is established as a critical enhancer in normal hematopoiesis [62]. Since we previously discovered that SON binds to the gene promoters and suppresses transcriptional activation by reducing the H3K4me3 levels [28], we examined whether SON directly binds to the promoter sequences in the RUNX1 locus in AMKL cells by SON chromatin immunoprecipitation and qPCR (ChIP-qPCR). First, ChIP-seq data for several histone modifications indicating open and active chromatin status (H3K4me3, H3K4me1, and H3K27ac) as well as RNA polymerase II (Pol II) (available from from ENCODE) were analyzed to identify locations for primer design in the P1 and P2 promoters and the +23 enhancer. Interestingly, besides these promoters and enhancer, we noticed two additional regions showing the feature of potential cis-regulatory elements enriched with active promoter/enhancer marks and Pol II-binding; the region located 57 kb upstream of the P1 promoter (−57 kb) and 139 kb downstream of the P1 promoter (+139 kb) (Fig. 6A).
Figure 6. SON directly binds to the RUNX1 promoter and two enhancer regions and represses RUNX1 transcription.

A. Integrative Genomics Viewer (IGV) images showing histone modifications (H3K4me3, H3K4me1 and H3K27ac) and RNA polymerase II enrichment (total RNA pol II and pSer-5) at the RUNX1 genomic locus in K562 cells. ChIP-seq data were retrieved from ENCODE. The location of the distal promoter (P1), proximal promoter (P2), +23 kb enhancer as well as two putative enhancers we identified (the −57 kb and +139 kb regions) are indicated as labeled and highlighted in dotted boxes. Primers used in ChIP-qPCR (b and c) were designed to target the regions within the dotted boxes.
B. ChIP-qPCR analyses of SON-binding to the indicated promoters and enhancer regions at the RUNX1 locus in MEG-01, CMY, and CMK cells. Negative control region (Neg cont) were also included to verify that SON enrichment is observed only at specific sites. Data are expressed as mean ±SD; n=3, *p<0.05, **p<0.0.
C. ChIP-qPCR analyses of histone modification levels (H3K4me3 and H3K4me1) at the proximal promoter (P2), +23 kb enhancer and a putative +139 kb enhancer upon SON knockdown (SON siRNA). Data are expressed as mean±SD; n=3-10, *p<0.05, **p<0.01.
Our SON ChIP-qPCR using three different AMKL cell lines, MEG-01, CMY and CMK, demonstrated that SON is enriched at the P2 promoter and the +23 enhancer as well as the +139 kb region (Fig. 6B). We then examined whether histone modifications are a potential mechanism of SON-mediated RUNX1 repression, and if so, whether these modifications are altered in these regions upon SON knockdown. Our ChIP-qPCR revealed that H3K4me3 is increased upon SON knockdown, especially at the P2 promoter. In addition, H3K4me1 is consistently increased at the +23 kb enhancer as well as the P2 promoter (Fig. 6C), indicating that the proximal promoter and the +23 kb enhancer become more open and active when SON is reduced. More interestingly, we found that H3K4me1 is also increased in the +139 kb region upon SON knockdown (Fig. 6C), indicating that the +139 kb region potentially functions as an enhancer for RUNX1 expression. Taken together, these results showed that SON directly binds to the +23 kb enhancer and the putative +139 kb enhancer as well as the P2 promoter at the RUNX1 locus and suppresses RUNX1 transcription through lowering the H3K4me3 and H3K4me1 modifications.
SON represses transcription of JUN, JUNB, and FOSB through direct promoter binding.
To further delineate the molecular mechanism by which SON inhibits megakaryocytic differentiation, we examined SON regulation of other pro-megakaryocytic transcription factors identified from our RNA-sequencing results (Fig. 4C). Among them is the Activator Protein-1 (AP-1) complex, which plays a role as a RUNX1 partner in transcription of late megakaryocytic genes [63]. The AP-1 transcription factor is a complex formed by the FOS (c-FOS, FOSB, FOSB2, FRA-1, and FRA-2) and JUN (c-JUN, JUNB, and JUND) families. Our qPCR and Western blot confirmed that SON knockdown indeed upregulates JUN, JUNB, and FOSB in AMKL cell lines (Figs. 7A and 7B)
Figure 7. SON represses transcription of JUN, JUNB, and FOSB through direct promoter binding.

A. Quantitative RT-PCR detection of SON and the indicated AP-1 subunits in control and SON-siRNA transfected AMKL cell lines (48 h post-siRNA transfection; mean±SD; n=2-6, *p<0.05, **p<0.01).
B. Western blot analysis of c-JUN and JUNB upon SON knockdown (48 h post-siRNA transfection). GAPDH was blotted as a loading control.
C. ChIP-qPCR analyses of SON-binding to the JUN, JUNB, and FOSB promoters (mean±SD, n=3-4, *p<0.05, **p<0.01).
D. ChIP-qPCR analyses of the H3K4me3 levels at the JUN, JUNB, and FOSB promoters in control or SON siRNA-transfected CMY cells (48 h post-siRNA transfection; mean±SD; n=3-8, *p<0.05, **p<0.01).
Next, we sought to examine whether SON potentially binds to the promoters of JUN, JUNB and FOSB to directly regulate their transcription. We designed primers based on the RNA polymerase II peaks at the promoters of JUN, JUNB, and FOSB (Supplementary Fig. S5). Our ChIP-qPCR results indicated that SON indeed binds to the promoter regions of JUN, JUNB, and FOSB (Fig. 7C). Furthermore, by our ChIP-qPCR experiments, we determined that H3K4me3 level is increased at the promoter regions of JUN, JUNB and FOSB upon SON knockdown (Fig. 7D), indicating SON as a repressor of these specific AP-1 subunits. Taken together, our data demonstrated that SON suppresses the promoter activation of not only RUNX1 but also AP-1 complex components, thereby weakening the megakaryocytic gene expression program (Fig. 8).
Figure 8. Models of the SON function in regulating megakaryocytic differentiation and its proposed contribution to AMKL development.

A. To induce the onset of megakaryocytic differentiation, the SON expression level should be decreased, which weakens the repressive effect of SON on H3K4me at the promoters and/or enhancers of RUNX1 and the AP-1 complex subunit genes, JUN, JUNB and FOSB. Upregulated RUNX1 inhibits KLF1 expression while activating FLI1 expression, promoting megakaryocytic gene expression. AP-1 complex components may also boost megakaryocytic gene expression.
B. When progenitors from various stages in the megakaryocyte-erythroid lineage fail to downregulate SON due to various mechanisms (trisomy 21 or other mechanisms activating SON expression), RUNX1 is not sufficiently expressed, which leads to impaired megakaryocytic differentiation while still allowing KLF1-mediated erythroid gene expression. AP1 complex components are also downregulated, weakening megakaryocytic gene expression. Impaired megakaryocytic differentiation may cooperate with other oncogenic mutations, contributing to the increased risk of AMKL development.
DISCUSSION
AMKL is associated with poor prognosis, especially, in adult non-DS-AMKL patients, due to insufficient information of underlying molecular mechanisms and treatment targets. Non-DS-AMKLs are frequently found to have complex karyotype, oncogene fusions, and/or activating mutations in JAK2 or MPL. In DS-AMKL, it has been believed that increased gene dosage from chromosome 21 in cooperation with GATA1 mutations initiate the disease [7]. However, besides the GATA1 mutations, how other molecules/pathways specifically affect megakaryocytic differentiation remains elusive. Since the DS population is at a much higher risk of developing AMKL, chromosome 21 genes are a valid starting point to identify the factors that contribute to the initiation and progression of this type of leukemia. Our current study demonstrated that SON, one of the chromosome 21 genes, is aberrantly overexpressed in AMKL, and depletion of SON markedly increases megakaryocytic gene expression. We further demonstrated that SON functions as a transcriptional repressor of RUNX1 and AP-1 complex component genes, such as JUN, JUNB and FOSB, revealing underlying molecular mechanisms for AMKL development.
Bourquin et al. previously identified that SON is the second most upregulated chromosome 21-encoded potential transcription factor after BACH1 in DS-AMKL [19]. Our extensive analyses using various data sets and sources revealed that the SON expression level is indeed high in DS-AMKL leukemia blasts. Interestingly, from the patient bone marrow cell analyses, it has been shown that SON is also upregulated in non-DS-AMKL patients when compared to myelomonocytic AML patients, and the level is sometimes as high as the DS-AMKL SON level. Our groups and others previously demonstrated that SON expression is increased in myelomonocytic AML compared to normal hematopoietic progenitors [28, 35], therefore indicating that SON expression is exceptionally high in AMKL. The high level of SON expression in both DS-AMKL and non-DS-AMKL suggests that while trisomy 21 can assure SON upregulation in DS-AMKL without any additional regulatory abnormalities, which may naturally increase the risk of AMKL development in individuals with DS, there could be other mechanisms responsible for SON upregulation in non-DS-AMKL.
Interestingly, Bourquin et al. also report that the expression level of RUNX1, a master regulator of megakaryocytic differentiation, is decreased in DS-AMKL patients despite its increased gene dosage by being located on chromosome 21 [19]. Since RUNX1 is essential for normal megakaryopoiesis [57, 59, 64], reduction of RUNX1 in DS-AMKL could contribute to impaired megakaryocytic differentiation. However, the reason that RUNX1 expression is paradoxically low in DS-AMKL [19], despite its genic location on chromosome 21, has remained a puzzling question for a long time. We showed that SON directly binds to the proximal promoter (P2) and the +23 kb enhancer as well as the novel +139 kb enhancer of at the RUNX1 locus, revealing a previously unidentified regulatory mechanism of RUNX1 expression. We noticed the increased H3K4 methylation levels at the P2 promoter and the enhancer regions upon SON depletion, indicating SON exerts its function as a transcriptional repressor through binding to the promoter as well as two enhancer regions at the RUNX1 locus. A recent report identified chromatin-chromatin contacts within the RUNX1 locus and suggested that there are multiple promoter-enhancer interactions regulating RUNX1 expression [65]. Since we detected SON enrichment at the promoter and two enhancers, it will be interesting to investigate whether SON is involved in regulating long-range chromatin-chromatin interactions between promoters and enhancers. Both RUNX1 and SON genes are located on human chromosome 21, and our finding provides an example of complex inter-regulations among chromosome 21 genes.
Interestingly, while we indeed observed inverse correlations between SON expression and RUNX1 expression in some AML, AMKL and TMD patients that were included in the study by Bourquin et al, some of the patients did not show correlations between SON and RUNX1 expression (Supplementary Fig. S6). Particularly, some patients showed extremely low levels of RUNX1 regardless of SON expression status (Supplementary Fig. S6). These observations suggest that although SON is one of the factors repressing RUNX1, there should be additional factors responsible for the extremely low level of RUNX1 in some patients.
SON has dual abilities to interact with DNA and RNA and functions as a splicing co-factor as well as transcriptional repressor [21, 28]. Our group and others reported that many of the genes that are downregulated upon SON depletion are targets of SON-mediated RNA splicing [21, 24–26]. When SON is depleted, RNA splicing at selective splice sites is compromised, resulting in mis-splicing and subsequent nonsense-mediated RNA decay and gene downregulation. In diverse cell types, these groups of genes are associated with cell cycle, DNA repair and metabolism [21, 24–26]. In our current study, we also observed similar groups of genes downregulated in MEG-01 AMKL cells. In contrast, the target genes of SON-mediated transcriptional repression are generally upregulated upon SON depletion due to de-repression of the gene promoters [28]. Importantly, in this current study with AMKL cells, we found that genes associated with megakaryocytic differentiation are noticeably increased upon SON knockdown, unveiling SON’s function in repressing differentiation of a specific hematopoietic lineage.
Hematopoietic differentiation is largely controlled by lineage-specific transcription factors that activate highly specific gene expression programs that subsequently determine cell fates. Only a few transcription factors have been identified as regulators of megakaryocytic lineage differentiation, including GATA1, RUNX1 and FLI1. Our study revealed SON as a novel transcription factor that suppresses megakaryocytic differentiation. Based on our finding, we speculate that overexpression of SON caused by trisomy 21 (in DS-AMKL) or other unknown mechanisms (in non-DS-AMKL) may contribute to AMKL development when other hits (e.g. GATA1 mutations) are present (models in Fig. 8B). Testing this hypothesis would be an interesting direction for future research and will provide novel insights into molecular mechanisms of hematologic disorders associated with dysregulation of megakaryocytic lineage differentiation.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Shai Izraeli (Tel Aviv University) for kindly providing the CMY and CMK cell lines. This work was supported by the NIH grants (R01CA190688 and R01CA236911 to E.E.A. and R01HL136432 to S.T.L.) and institutional support from the University of Alabama at Birmingham School of Medicine, Department of Pathology, and the UAB O’Neal Comprehensive Cancer Center (to E.E.A.).
Footnotes
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest
REFERENCES
- 1.Pagano L, Pulsoni A, Vignetti M, Mele L, Fianchi L, Petti MC et al. Acute megakaryoblastic leukemia: experience of GIMEMA trials. Leukemia 2002; 16: 1622–1626. [DOI] [PubMed] [Google Scholar]
- 2.Lange B The management of neoplastic disorders of haematopoiesis in children with Down’s syndrome. Br J Haematol 2000; 110: 512–524. [DOI] [PubMed] [Google Scholar]
- 3.Athale UH, Razzouk BI, Raimondi SC, Tong X, Behm FG, Head DR et al. Biology and outcome of childhood acute megakaryoblastic leukemia: a single institution’s experience. Blood 2001; 97: 3727–3732. [DOI] [PubMed] [Google Scholar]
- 4.Barnard DR, Alonzo TA, Gerbing RB, Lange B, Woods WG, Children’s Oncology G. Comparison of childhood myelodysplastic syndrome, AML FAB M6 or M7, CCG 2891: report from the Children’s Oncology Group. Pediatr Blood Cancer 2007; 49: 17–22. [DOI] [PubMed] [Google Scholar]
- 5.Hitzler JK, Zipursky A. Origins of leukaemia in children with Down syndrome. Nat Rev Cancer 2005; 5: 11–20. [DOI] [PubMed] [Google Scholar]
- 6.Gruber TA, Downing JR. The biology of pediatric acute megakaryoblastic leukemia. Blood 2015; 126: 943–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roberts I, Izraeli S. Haematopoietic development and leukaemia in Down syndrome. Br J Haematol 2014; 167: 587–599. [DOI] [PubMed] [Google Scholar]
- 8.Chou ST, Opalinska JB, Yao Y, Fernandes MA, Kalota A, Brooks JS et al. Trisomy 21 enhances human fetal erythro-megakaryocytic development. Blood 2008; 112: 4503–4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tunstall-Pedoe O, Roy A, Karadimitris A, de la Fuente J, Fisk NM, Bennett P et al. Abnormalities in the myeloid progenitor compartment in Down syndrome fetal liver precede acquisition of GATA1 mutations. Blood 2008; 112: 4507–4511. [DOI] [PubMed] [Google Scholar]
- 10.Chou ST, Byrska-Bishop M, Tober JM, Yao Y, Vandorn D, Opalinska JB et al. Trisomy 21-associated defects in human primitive hematopoiesis revealed through induced pluripotent stem cells. Proc Natl Acad Sci U S A 2012; 109: 17573–17578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roy A, Cowan G, Mead AJ, Filippi S, Bohn G, Chaidos A et al. Perturbation of fetal liver hematopoietic stem and progenitor cell development by trisomy 21. Proc Natl Acad Sci U S A 2012; 109: 17579–17584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Korbel JO, Tirosh-Wagner T, Urban AE, Chen XN, Kasowski M, Dai L et al. The genetic architecture of Down syndrome phenotypes revealed by high-resolution analysis of human segmental trisomies. Proc Natl Acad Sci U S A 2009; 106: 12031–12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Vita S, Canzonetta C, Mulligan C, Delom F, Groet J, Baldo C et al. Trisomic dose of several chromosome 21 genes perturbs haematopoietic stem and progenitor cell differentiation in Down’s syndrome. Oncogene 2010; 29: 6102–6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salek-Ardakani S, Smooha G, de Boer J, Sebire NJ, Morrow M, Rainis L et al. ERG is a megakaryocytic oncogene. Cancer Res 2009; 69: 4665–4673. [DOI] [PubMed] [Google Scholar]
- 15.Stankiewicz MJ, Crispino JD. ETS2 and ERG promote megakaryopoiesis and synergize with alterations in GATA-1 to immortalize hematopoietic progenitor cells. Blood 2009; 113: 3337–3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ng AP, Hyland CD, Metcalf D, Carmichael CL, Loughran SJ, Di Rago L et al. Trisomy of Erg is required for myeloproliferation in a mouse model of Down syndrome. Blood 2010; 115: 3966–3969. [DOI] [PubMed] [Google Scholar]
- 17.Malinge S, Bliss-Moreau M, Kirsammer G, Diebold L, Chlon T, Gurbuxani S et al. Increased dosage of the chromosome 21 ortholog Dyrk1a promotes megakaryoblastic leukemia in a murine model of Down syndrome. J Clin Invest 2012; 122: 948–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klusmann JH, Li Z, Bohmer K, Maroz A, Koch ML, Emmrich S et al. miR-125b-2 is a potential oncomiR on human chromosome 21 in megakaryoblastic leukemia. Genes Dev 2010; 24: 478–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bourquin JP, Subramanian A, Langebrake C, Reinhardt D, Bernard O, Ballerini P et al. Identification of distinct molecular phenotypes in acute megakaryoblastic leukemia by gene expression profiling. Proc Natl Acad Sci U S A 2006; 103: 3339–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hickey CJ, Kim JH, Ahn EY. New discoveries of old SON: a link between RNA splicing and cancer. J Cell Biochem 2014; 115: 224–231. [DOI] [PubMed] [Google Scholar]
- 21.Ahn EY, DeKelver RC, Lo MC, Nguyen TA, Matsuura S, Boyapati A et al. SON controls cell-cycle progression by coordinated regulation of RNA splicing. Mol Cell 2011; 42: 185–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun CT, Lo WY, Wang IH, Lo YH, Shiou SR, Lai CK et al. Transcription repression of human hepatitis B virus genes by negative regulatory element-binding protein/SON. J Biol Chem 2001; 276: 24059–24067. [DOI] [PubMed] [Google Scholar]
- 23.Ahn EE, Higashi T, Yan M, Matsuura S, Hickey CJ, Lo MC et al. SON protein regulates GATA-2 through transcriptional control of the microRNA 23a~27a~24-2 cluster. J Biol Chem 2013; 288: 5381–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu X, Goke J, Sachs F, Jacques PE, Liang H, Feng B et al. SON connects the splicing-regulatory network with pluripotency in human embryonic stem cells. Nat Cell Biol 2013; 15: 1141–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharma A, Markey M, Torres-Munoz K, Varia S, Kadakia M, Bubulya A et al. Son maintains accurate splicing for a subset of human pre-mRNAs. J Cell Sci 2011; 124: 4286–4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim JH, Shinde DN, Reijnders MRF, Hauser NS, Belmonte RL, Wilson GR et al. De Novo Mutations in SON Disrupt RNA Splicing of Genes Essential for Brain Development and Metabolism, Causing an Intellectual-Disability Syndrome. Am J Hum Genet 2016; 99: 711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mattioni T, Hume CR, Konigorski S, Hayes P, Osterweil Z, Lee JS. A cDNA clone for a novel nuclear protein with DNA binding activity. Chromosoma 1992; 101: 618–624. [DOI] [PubMed] [Google Scholar]
- 28.Kim JH, Baddoo MC, Park EY, Stone JK, Park H, Butler TW et al. SON and Its Alternatively Spliced Isoforms Control MLL Complex-Mediated H3K4me3 and Transcription of Leukemia-Associated Genes. Mol Cell 2016; 61: 859–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 2016; 34: 525–527. [DOI] [PubMed] [Google Scholar]
- 30.Pimentel H, Bray NL, Puente S, Melsted P, Pachter L. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat Methods 2017; 14: 687–690. [DOI] [PubMed] [Google Scholar]
- 31.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005; 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003; 13: 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bagger FO, Sasivarevic D, Sohi SH, Laursen LG, Pundhir S, Sonderby CK et al. BloodSpot: a database of gene expression profiles and transcriptional programs for healthy and malignant haematopoiesis. Nucleic Acids Res 2016; 44: D917–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen L, Kostadima M, Martens JHA, Canu G, Garcia SP, Turro E et al. Transcriptional diversity during lineage commitment of human blood progenitors. Science 2014; 345: 1251033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang E, Lu SX, Pastore A, Chen X, Imig J, Chun-Wei Lee S et al. Targeting an RNA-Binding Protein Network in Acute Myeloid Leukemia. Cancer Cell 2019; 35: 369–384 e367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klusmann JH, Godinho FJ, Heitmann K, Maroz A, Koch ML, Reinhardt D et al. Developmental stage-specific interplay of GATA1 and IGF signaling in fetal megakaryopoiesis and leukemogenesis. Genes Dev 2010; 24: 1659–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cardin S, Bilodeau M, Roussy M, Aubert L, Milan T, Jouan L et al. Human models of NUP98-KDM5A megakaryocytic leukemia in mice contribute to uncovering new biomarkers and therapeutic vulnerabilities. Blood Adv 2019; 3: 3307–3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Long MW, Heffner CH, Williams JL, Peters C, Prochownik EV. Regulation of megakaryocyte phenotype in human erythroleukemia cells. J Clin Invest 1990; 85: 1072–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hocevar BA, Morrow DM, Tykocinski ML, Fields AP. Protein kinase C isotypes in human erythroleukemia cell proliferation and differentiation. J Cell Sci 1992; 101 ( Pt 3): 671–679. [DOI] [PubMed] [Google Scholar]
- 40.Shelly C, Petruzzelli L, Herrera R. K562 cells resistant to phorbol 12-myristate 13-acetate-induced growth arrest: dissociation of mitogen-activated protein kinase activation and Egr-1 expression from megakaryocyte differentiation. Cell Growth Differ 2000; 11: 501–506. [PubMed] [Google Scholar]
- 41.Racke FK, Lewandowska K, Goueli S, Goldfarb AN. Sustained activation of the extracellular signal-regulated kinase/mitogen-activated protein kinase pathway is required for megakaryocytic differentiation of K562 cells. J Biol Chem 1997; 272: 23366–23370. [DOI] [PubMed] [Google Scholar]
- 42.Jiang H, Promchan K, Lin BR, Lockett S, Chen D, Marshall H et al. LZTFL1 Upregulated by All-Trans Retinoic Acid during CD4+ T Cell Activation Enhances IL-5 Production. J Immunol 2016; 196: 1081–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kato H, Igarashi K. To be red or white: lineage commitment and maintenance of the hematopoietic system by the “inner myeloid”. Haematologica 2019; 104: 1919–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Itoh-Nakadai A, Matsumoto M, Kato H, Sasaki J, Uehara Y, Sato Y et al. A Bach2-Cebp Gene Regulatory Network for the Commitment of Multipotent Hematopoietic Progenitors. Cell Rep 2017; 18: 2401–2414. [DOI] [PubMed] [Google Scholar]
- 45.Hamada T, Murasawa S, Yokoyama A, Hayashi S, Kobayashi Y, Asahara T. Changing modified regions in the genome in hematopoietic stem cell differentiation. Biochem Biophys Res Commun 2009; 381: 135–138. [DOI] [PubMed] [Google Scholar]
- 46.Fan J, Wang Y, Shen Y, Liu Q, Gao R, Qiu Y et al. A novel role of CKIP-1 in promoting megakaryocytic differentiation. Oncotarget 2017; 8: 30138–30150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu XL, Yuan JY, Zhang JW, Zhang XH, Wang RX. Differential gene expression in human hematopoietic stem cells specified toward erythroid, megakaryocytic, and granulocytic lineage. J Leukoc Biol 2007; 82: 986–1002. [DOI] [PubMed] [Google Scholar]
- 48.Geue S, Aurbach K, Manke MC, Manukjan G, Munzer P, Stegner D et al. Pivotal role of PDK1 in megakaryocyte cytoskeletal dynamics and polarization during platelet biogenesis. Blood 2019; 134: 1847–1858. [DOI] [PubMed] [Google Scholar]
- 49.Vakana E, Arslan AD, Szilard A, Altman JK, Platanias LC. Regulatory effects of sestrin 3 (SESN3) in BCR-ABL expressing cells. PLoS One 2013; 8: e78780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Senis YA. Protein-tyrosine phosphatases: a new frontier in platelet signal transduction. J Thromb Haemost 2013; 11: 1800–1813. [DOI] [PubMed] [Google Scholar]
- 51.Raslova H, Kauffmann A, Sekkai D, Ripoche H, Larbret F, Robert T et al. Interrelation between polyploidization and megakaryocyte differentiation: a gene profiling approach. Blood 2007; 109: 3225–3234. [DOI] [PubMed] [Google Scholar]
- 52.Kahn ML, Zheng YW, Huang W, Bigornia V, Zeng D, Moff S et al. A dual thrombin receptor system for platelet activation. Nature 1998; 394: 690–694. [DOI] [PubMed] [Google Scholar]
- 53.Marconi C, Di Buduo CA, LeVine K, Barozzi S, Faleschini M, Bozzi V et al. Loss-of-function mutations in PTPRJ cause a new form of inherited thrombocytopenia. Blood 2019; 133: 1346–1357. [DOI] [PubMed] [Google Scholar]
- 54.Sanders MA, Ampasala D, Basson MD. DOCK5 and DOCK1 regulate Caco-2 intestinal epithelial cell spreading and migration on collagen IV. J Biol Chem 2009; 284: 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Timpl R Macromolecular organization of basement membranes. Curr Opin Cell Biol 1996; 8: 618–624. [DOI] [PubMed] [Google Scholar]
- 56.Yoshigi M, Hoffman LM, Jensen CC, Yost HJ, Beckerle MC. Mechanical force mobilizes zyxin from focal adhesions to actin filaments and regulates cytoskeletal reinforcement. J Cell Biol 2005; 171: 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goldfarb AN. Transcriptional control of megakaryocyte development. Oncogene 2007; 26: 6795–6802. [DOI] [PubMed] [Google Scholar]
- 58.Starck J, Cohet N, Gonnet C, Sarrazin S, Doubeikovskaia Z, Doubeikovski A et al. Functional cross-antagonism between transcription factors FLI-1 and EKLF. Mol Cell Biol 2003; 23: 1390–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kuvardina ON, Herglotz J, Kolodziej S, Kohrs N, Herkt S, Wojcik B et al. RUNX1 represses the erythroid gene expression program during megakaryocytic differentiation. Blood 2015; 125: 3570–3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ghozi MC, Bernstein Y, Negreanu V, Levanon D, Groner Y. Expression of the human acute myeloid leukemia gene AML1 is regulated by two promoter regions. Proc Natl Acad Sci U S A 1996; 93: 1935–1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Levanon D, Groner Y. Structure and regulated expression of mammalian RUNX genes. Oncogene 2004; 23: 4211–4219. [DOI] [PubMed] [Google Scholar]
- 62.Bee T, Ashley EL, Bickley SR, Jarratt A, Li PS, Sloane-Stanley J et al. The mouse Runx1 +23 hematopoietic stem cell enhancer confers hematopoietic specificity to both Runx1 promoters. Blood 2009; 113: 5121–5124. [DOI] [PubMed] [Google Scholar]
- 63.Pencovich N, Jaschek R, Tanay A, Groner Y. Dynamic combinatorial interactions of RUNX1 and cooperating partners regulates megakaryocytic differentiation in cell line models. Blood 2011; 117: e1–14. [DOI] [PubMed] [Google Scholar]
- 64.Ichikawa M, Asai T, Saito T, Seo S, Yamazaki I, Yamagata T et al. AML-1 is required for megakaryocytic maturation and lymphocytic differentiation, but not for maintenance of hematopoietic stem cells in adult hematopoiesis. Nat Med 2004; 10: 299–304. [DOI] [PubMed] [Google Scholar]
- 65.Marsman J, Thomas A, Osato M, O’Sullivan JM, Horsfield JA. A DNA Contact Map for the Mouse Runx1 Gene Identifies Novel Haematopoietic Enhancers. Sci Rep 2017; 7: 13347. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.