

Abstract
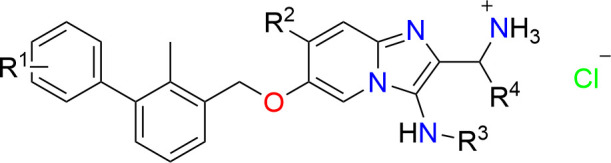
The PD-1/PD-L1 axis has proven to be a highly efficacious target for cancer immune checkpoint therapy with several approved antibodies. Also, small molecules based on a biphenyl core can antagonize PD-1/PD-L1, leading to the in vitro formation of PD-L1 dimers. However, their development remains challenging, as we do not yet fully understand their mode of action. In this work, we designed a new scaffold based on our previously solved high-resolution structures of low-molecular-weight inhibitors bound to PD-L1. A small compound library was synthesized using the Groebke–Blackburn–Bienaymé multicomponent reaction (GBB-3CR), resulting in the structure–activity relationship of imidazo[1,2-a]pyridine-based inhibitors. These inhibitors were tested for their biological activity using various biophysical assays giving potent candidates with low-micromolar PD-L1 affinities. An obtained PD-L1 cocrystal structure reveals the binding to PD-L1. Our results open the door to an interesting bioactive scaffold that could lead to a new class of PD-L1 antagonists.
Keywords: PD-L1 inhibitor; multicomponent reaction; Groebke−Blackburn−Bienaymé; imidazo[1,2-a]pyridine
In recent decades, oncology has been revolutionized by immunotherapy.1 In particular, immune checkpoint blockade (ICB) targeting the PD-1/PD-L1 axis has shown impressive clinical benefit, with durable regression and even cure in a subset of hard-to-treat cancers.2,3 In general, ICB-responsive cancers are characterized by high levels of mutations and corresponding neoantigens (“hot” tumors). These neoantigens can be recognized by immune effector T-cells, which under homeostatic conditions would result in cancer cell elimination.4 In cancer, this elimination is restrained by the immune checkpoint PD-1 expressed on T-cells. PD-1 binds to PD-L1 expressed on cancer cells, resulting in an inhibitory intracellular signaling cascade that prevents proper T-cell activation.5 Consequently, inhibition of the interaction of PD-L1 and PD-1 receptors promotes T-cell activation. Several PD-1/PD-L1-directed antibodies are in clinical use, and numerous experimental ones are under development. However, current PD-1/PD-L1-directed therapies are useful only for a small subset of patients, are expensive to produce, have a risk of adverse effects, and show development of resistance, which limits their utility.6,7 Therefore, novel therapeutic modalities such as small molecules or peptides exhibit a lot of promise.8,9 The only small-molecule inhibitor targeted against PD-L1 currently undergoing clinical trial is CA-170 (Figure 1B).10 However, by means of various functional cell assays it was recently proved not to be a direct binder to PD-1 or PD-L1, and its mode of action remains unclear.11 As part of our ongoing efforts to understand and develop small molecules that antagonize PD-1/PD-L1, we present here the design, synthesis, biological activity, and structural basis of imidazopyridines as PD-1/PD-L1 antagonists.8,11−14
Figure 1.
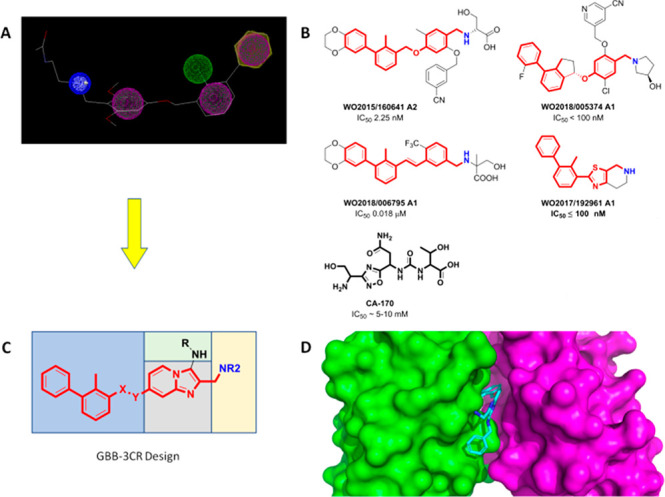
Design of PD-1/PD-L1 antagonists. (A) Generalized pharmacophore model of a PD-L1 antagonist. Aromatic (purple), hydrophobic (green), and basic (positive charged, blue) pharmacophores are included. (B) Examples of potent PD-L1 dimerizers taken from the patent literature. The pharmacophore is indicated by red and blue colors. (C) Design of imidazopyridines accessible by GBB-3CR. The four variable parts of the scaffold are indicated by the different colored boxes. (D) Modeling of an imidazopyridine into a PD-L1 dimer structure (PDB ID:5NIX).
Our recently published cocrystal structures of several small molecules binding to PD-L1 have been used to propose a generalized pharmacophore model for small-molecule PD-L1 binders (Figure 1A).9,14−17 These structures triggered a wave of small-molecule designs and subsequent patent applications.18,19 Accordingly, a twisted biphenyl moiety is linked via a two-atom linker to a planar (hetero)aromatic ring fragment that has a methanamine para to the linker moiety. Symmetrical central biphenyl moieties with two times the linker (hetero)aromatic fragment have also been described as highly potent PD-L1 binders.20 The diversity of PD-L1 small-molecule scaffolds based on our proposed pharmacophore model and claimed in patents is great (Figure 1B).18,19 The biphenyl component allows for fewer variations, but the linker and (hetero)aromatic moieties can be executed in a variety of designs. Finally, the water-exposed part of the molecule allows for many variations useful to tune drug-like properties such as water solubility.
To circumvent the lengthy and linear sequential syntheses of many small-molecule PD-1/PD-L1 antagonists, we decided to explore multicomponent reactions for the one-pot assembly of the central (hetero)aromatic part of the pharmacophore model. For this, we chose the Groebke–Blackburn–Bienaymé reaction (GBB-3CR) which is a versatile three-component reaction of heterocyclic amidines, aldehydes, and isocyanides that gives access to drug-like molecules (Figure 1C).21−23 A key fragment of the scaffold is a bicyclic imidazo ring.
In our design, the bicyclic heteroaromatic moiety (Figure 1 C, gray) is the central element of the scaffold, to which three suitable substituents are attached, namely, the biphenyl (Figure 1 C, blue), methanamine (Figure 1 C, yellow), and amino (Figure 1 C, green) moieties. To decide on the position of the substituents, we used molecular modeling performed via Moloc and Scorpion software (Figure 1D).24,25 The best fit into the receptor was to introduce the methanamine moiety by the GBB-3CR aldehyde component and the biphenyl moiety through a C–O coupling to the aminopyridine linker.
Following the design and docking studies (Figure 1D), we opted for the imidazopyridine-containing scaffold (Figure 1C) and started on the development of a corresponding synthesis route that would yield the established scaffold.
We envisioned a synthetic route in which the step with the highest introduction of variation should be carried out last. Therefore, the GBB reaction was chosen as one of the last stages. Consequently, the preparation of the biphenyl-substituted amidines was approached first (Scheme 1). We started synthesizing the twisted biphenyl moiety via the Suzuki cross-coupling reaction between (3-bromo-2-methylphenyl)methanol (2) and unprotected boronic acids 1a–c. Using Pd(dppf)Cl2 as the catalyst in a solvent system consisting of toluene, ethanol, and saturated aqueous sodium hydrogen carbonate solution (5:1:5) and heating this for 12 h at 85 °C generated compounds 3a–c in excellent yields of 90–97%.
Scheme 1. Synthetic Route to Compounds 5a–f.

Reagents and conditions: (a) Pd(dppf)Cl2, toluene/ethanol/NaHCO3 (sat. aq.) (5:1:5) (0.3 M), 85 °C, 12 h; (b) 5-fluoro-2-nitropyridine (4a) or 5-fluoro-4-methyl-2-nitropyridine (4b), KOH, dry DMSO, 0 °C to rt, 5 min to 1 h; (c) Fe, HCl; EtOH/H2O (5:1) (0.1 M), 2 h, reflux. R1 = H, p-F, [3,4]-(OC2H4O); R2 = H, CH3.
Following the Suzuki reaction, we selected the nucleophilic aromatic substitution involving 3a–c and 5-fluoro-2-nitropyridine (4a) or 5-fluoro-4-methyl-2-nitropyridine (4b, see the Supporting Information) as the best option to prepare the precursors to amidines 6a–d.
By adding finely ground potassium hydroxide in dry DMSO at 0 °C to generate a superbasic medium in the presence of 3a–c and 4a–b, we were able to synthesize the corresponding products 5a–d with quantitative conversion and with brief reaction times of 5 min for 5a–c and 1 h for 5d.26 An extraction step was performed, and the resulting crude oil was utilized in the subsequent step. Lastly, to generate the desired amidines 6a–d, reduction of 5a–d was performed by the use of hydrochloric acid and iron powder in ethanol/water (5:1) under reflux conditions to avoid possible benzyl ether cleavage of the biphenylic component, i.e., via a classical Pd/C H2 reduction. Following 2 h of reaction time and chromatographic purification, the required amidines were obtained in yields of 72–92%.
Having the desired amidines in hand, we were able to execute the GBB reaction (Scheme 2). On the basis of the docking studies, we selected phenyl-containing isocyanides for the possibility of generating π stacking with Tyr-123 in the pocket of the PD-L1 dimer. As the aldehyde component, we chose polar substrates to increase the polarity, solubility, and possibility of hydrogen bonding. Therefore, we used tert-butyl (2-oxoethyl)carbamate and both enantiomers of the Garner aldehyde, (S)- and (R)-(−)-3-Boc-2,2-dimethyloxazolidine-4-carboxaldehyde.
Scheme 2. GBB Reaction.

Reagents and conditions: (A) Sc(OTf)3 (10 mol %), DCM/MeOH (2:1) (0.3 M), 1 h, 120 °C, microwave-assisted heating; (B) 7 N HCl in 2-propanol, rt, 20 h.
We found the optimal reaction conditions for the used substrates to be scandium triflate (10 mol %) as the catalyst, 2:1 DCM/MeOH as the solvent system, a concentration of 0.3 M with regard to the amidine, and 1.7 equiv. of the isocyanide and aldehyde components. Microwave-assisted heating for 1 h generated the corresponding GBB products in good to excellent yields (48–86%). Subsequently, a chromatographic purification over silica was performed. This purification step yielded generally still slightly impure compounds. Therefore, we continued with the deprotection of the tert-butyl carbamate group and, in the case of compounds 9d and 9e, the additional aminal protecting group. Performing the deprotection with 7 N HCl in 2-propanol at room temperature for 20 h resulted in full conversion. Following an additional chromatographic purification over silica, the final compounds 9a–k (Chart 1) were obtained and were analyzed via high-resolution mass spectrometry and 1H and 13C NMR spectroscopy (see the Supporting Information).
Chart 1. Compound Library of Imidazopyridine PD-L1 Antagonists.

For compound 9f, an adjusted synthesis route was chosen in which instead of (3-bromo-2-methylphenyl)methanol we used 1-(benzyloxy)-3-bromo-2-methylbenzene (A) for the Suzuki reaction, yielding 3-(benzyloxy)-2-methyl-1,1′-biphenyl (B) (see the Supporting Information). Additionally, a hydrogenation was performed with Pd/C and H2 (1 bar) in methanol for 2 h at 40 °C, reaching full conversion to 2-methyl-[1,1′-biphenyl]-3-ol (C). The remainder of the synthesis route was performed analogously to the other compounds, leading to compound 9f.
Moreover, we also explored a postsynthetic modification of compound 9h wherein we formed a tetrazole from the cyanide function on the isocyanide substrate, resulting in compound 9i. Compound 9h was reacted with sodium azide and zinc chloride in n-propanol at 95 °C for 20 h. After chromatographic purification and subsequent deprotection, we achieved a 39% yield of compound 9i. Conceptually, we expected that this modification would increase the affinity via hydrogen bonding between the tetrazole (as a carboxylic acid isostere) and Arg125.
To support our binding hypothesis from protein–ligand docking, we performed binding studies, which included NMR studies of ligand–PD-L1 binding and a homogeneous time-resolved fluorescence (HTRF) assay with PD-1 and PD-L1 proteins (Figure 2). Their binding to PD-L1 was verified using NMR titrations and HTRF assays (see the Supporting Information). The results of the NMR titration experiments demonstrated that our synthesized compounds generally disrupt the PD-1/PD-L1 complex and bind to PD-L1 as well as induced imerization of PD-L1.
Figure 2.
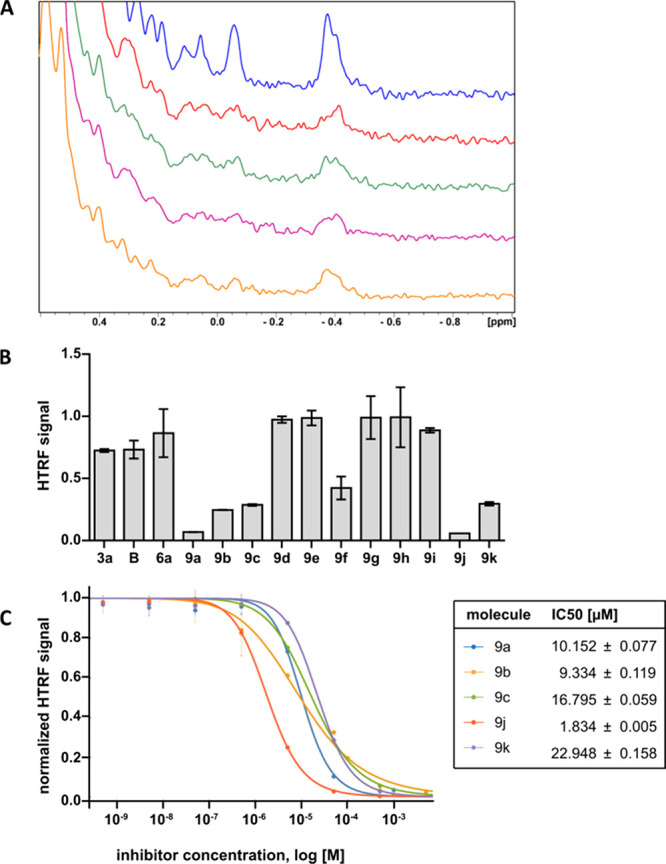
Binding studies. (A) Aliphatic part of the 1H NMR spectrum of PD-L1 (blue) and PD-L1 with compounds 9a (red), 9b (green), 9c (purple), and 9f (orange) in a 1:1 molar ratio. (B) HTRF scouting based on a concentration of 50 μM. (C) HTRF inhibition assay and calculated IC50 values for the best compounds from HTRF scouting.
HTRF experiments were conducted to further prove the ability of our compounds to disrupt the PD-1/PD-L1 complex and for IC50 value determination. The results of the HTRF experiments proved that all of the tested compounds were capable of disrupting the PD-1/PD-L1 complex, showing the potential of this scaffold. HTRF scouting revealed that parts of the GBB-3CR scaffold selected as binding parts in docking and cocrystallization studies (represented here as compounds 3a, 6, and B) have some disruption potential but that the full structure is needed for effective PD-L1 blocking, which we report in compounds 9a–e, 9j, and 9k. The phenyl ether (9f) and benzyl ether (9a) analogues show different affinity to the target protein: the presence of the linking methyl group increases the binding potential. Fluorine substitution on the biphenyl core is not tolerated (9g–i), whereas dioxane addition is accepted (9j and 9k). Additionally, the presence of a methyl group on the imidazopyridine ring (9j) improves the inhibitory activity of the GBB-3CR scaffold. IC50 values were determined for selected compounds and found to be in the range of 1.8–22.9 μM. The compounds with the lowest IC50 values were 9b (9.3 μM) and 9j (1.8 μM).
Next, the molecular basis of the interaction between 9c and the PD-L1 dimer was elucidated by X-ray structure analysis (Figure 3). The resolution was 2.45 Å, and the electron density of 9c was shown well, with the exception of the phenyl moiety of the phenylethyl fragment. 9c is mostly buried in the deep and elongated receptor site, which is composed of two PD-L1 monomers. In the absence of 9c the protein dimer complex has C2 symmetry. However, the binding of the small molecule breaks the symmetry. The biphenyl methylene moiety is embedded within two and three antiparallel β-sheets of monomers A and B, respectively. Monomer A (in green) contributes to the binding pocket with the 11 amino acids Lys124, Tyr56, Ala121, Ser93, Ser117, Ile54, Met115, Tyr123, Arg125, Lys124, and Asp122. Monomer B (in red) contributes to the binding pocket with the 12 amino acids Tyr56, Met115, Tyr123, Ala121, Ser117, Gln66, Ile54, Asp122, Ala121, Ser117, Ile54, and Gln66. The imidazopyridine moiety is embedded among Tyr56 and Gln66 of monomer B and Tyr123, Asp122, and Ala121 of monomer A. The phenylethylamine substituent is surrounded by Lys124, Arg125, and Tyr123 of monomer A and the hydroxy group of Tyr56 of monomer B. Finally, the aminomethyl group is largely water-exposed.
Figure 3.
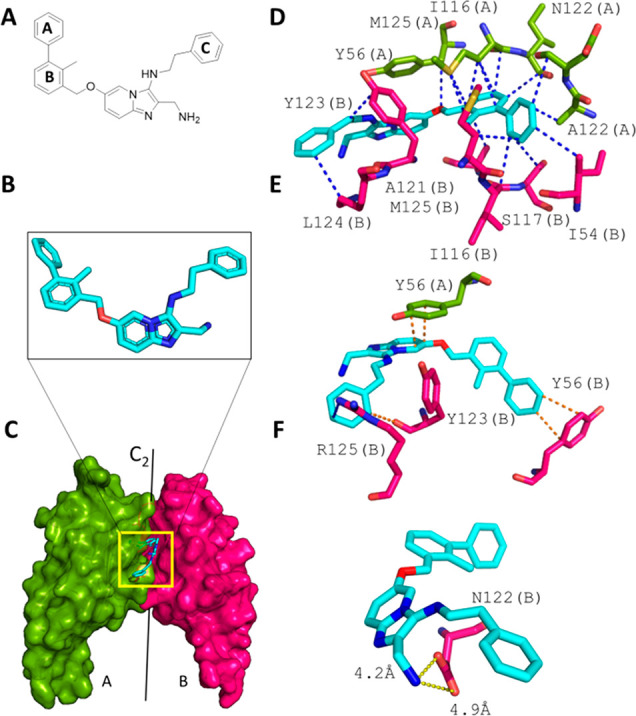
Binding of 9c to the PD-L1 dimer. (A) 2D structure of 9c. (B) Extracted 3D structure of 9c. (C) Overall view of 9c at the interface of two PD-L1 monomers (A and B, shown in green and red, respectively). (D–F) Scorpion software ligand–receptor interaction analysis. (D) vdW interactions between 9c and the PD-L1 dimer. (E) π stacking interactions. (F) Charge–charge interactions between Asp122 and the solvent-exposed aminomethyl substituent.
9c undergoes a diversity of hydrophobic, π-stacking, and charge–charge interactions (Figure 3D). Ring A of the biphenyl moiety is the deepest-buried part of 9c. The ring makes a short T-shaped π-stacking contact of 3.8 Å with Tyr56 of monomer A (Figure 3E). Plenty of hydrophobic contacts between ring A and Tyr123(B), Ile54(A), Ala121(B), Ser117(A), and Met115(A) can be observed. The twisted C–C bond connecting rings A and B is approximately at the position of the C2 axis of the imaginary PD-L1 dimer complex (A–B).
The dihedral angle defined by the two planes of the biphenyl is 124° (Figure 3B). This is comparable to other dihedral angles of similar molecules (e.g., BMS116617). The o-methyl group helps to keep the biphenyl moiety in a twisted shape close to the receptor conformation. In other compound designs, the o-methyl group was replaced by a bromide, chloride, or nitrile at the ortho position.12,27 The oxymethylene linker between the biphenyl and imidazopyridine fragments is rather flexible and in other designs can be an alkene, although structural data are missing.28
Why does imidazopyridine compound 9c show a rather low PD-L1 affinity? Detailed analysis of the cocrystal structure reveals that the π-stacking interaction of the pyridine and Tyr56(A) is suboptimal because of the low overlap of the two aromatic rings. Also, the electrostatics of this interaction is poor because the two dipole moments are parallel-aligned (Figure 3E). The distances between the Asp122(B) oxygens and the solvent-exposed aminomethyl substituent are rather long (4.2 and 4.8 Å), resulting in a poor charge–charge interaction (Figure 3F). Moreover, the largely hydrophobic pocket formed by Ile54(A), Val68(A), and Gln66(A) is not filled.
To understand the molecular basis accounting for the differences in affinity between 9c and 9j, we docked compound 9j to the dimer of PD-L1 (PDB entry 6R3K). The predicted binding mode of 9j matches perfectly the overall orientation of 9c and reveals the origins of the greater potency of 9j (Figure 4). First, the presence of the extended biphenyl is predicted to induce structural rearrangement of Tyr56 from monomer B. Thus, 9c occupies a larger surface area of the PD-L1 dimer and forms an open channel between monomers, as shown previously for compounds bearing an “extended” biphenyl ring system with the 2,3-dihydrobenzo[b][1,4]dioxine moiety.17 Second, the application of a shorter linker between the imidazopyridine core and ring C locates the latter one closer to the surface of monomer B and allows for additional π stacking with Tyr123, which is not observed in case of 9c (Figure 4).
Figure 4.
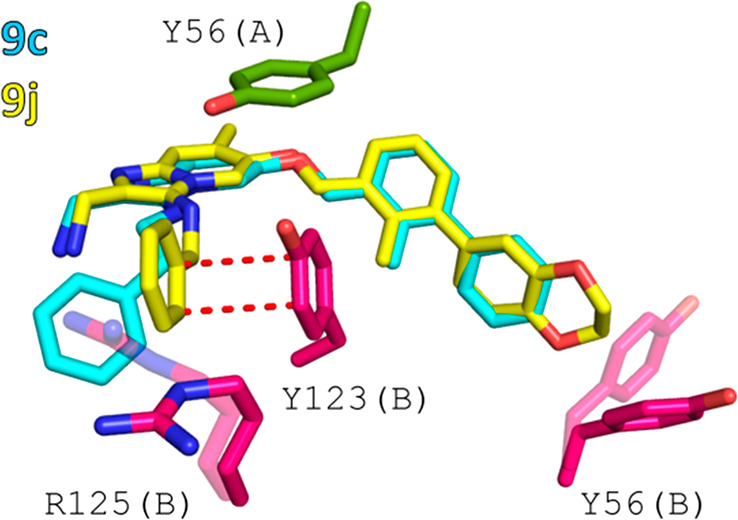
Comparison of the binding modes of 9c and 9j. (A) Superimposition of 9c from the cocrystal structure with the anticipated orientation of 9j obtained from molecular docking. (B) Structural basis of the better potency of 9j. Additional π–π stacking interactions are highlighted as red dashes, and transparent amino acids (R125 and Y56) denote residues anticipated to change their conformation upon binding of 9j to the PD-L1 dimer.
Inspired by our previously published cocrystal structures of PD-L1, we designed a novel scaffold, imidazopyridine, as a PD-L1 dimerizer. By exploitation of the multicomponent reaction (MCR) principle, imidazopyridines can be conveniently synthesized by the GBB-3CR with high variations of aldehydes, aminopyridines, and isocyanides. A small library consisting of 11 compounds was synthesized and tested for their efficacy through various biological assays, exhibiting IC50 values of 16.8–1.8 μM. We were further able to cocrystallize compound 9c with PD-L1, providing an explanation for the rather low affinities of the compound series. Overall, 9c can be regarded as a model compound that indicates the need of further improvements to achieve practical affinities, which are ongoing in our laboratory.
Acknowledgments
This research was funded by Project POIR.04.04.00-00-420F/17-00 (to T.A.H.), which is carried out within the TEAM Program of the Foundation for Polish Science cofinanced by the European Union under the European Regional Development Fund, COFUNDs ALERT (Grant Agreement 665250), and a KWF Kankerbestrijding grant (Grant Agreement 10504). Furthermore, we acknowledge the MCB Structural Biology Core Facility (supported by the TEAM TECH CORE FACILITY/2017-4/6 Grant from the Foundation for Polish Science) for valuable support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00033.
Experimental procedures, analytical data, NMR spectra, NMR binding assay, PD-L1 cocrystallizations, HRTF assay, and molecular docking (PDF)
Author Contributions
⊥ R.B. and M.W. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Dömling A.; Holak T. A. Programmed Death-1: Therapeutic Success after More than 100 Years of Cancer Immunotherapy. Angew. Chem., Int. Ed. 2014, 53 (9), 2286–2288. 10.1002/anie.201307906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P.; Allison J. P. Dissecting the Mechanisms of Immune Checkpoint Therapy. Nat. Rev. Immunol. 2020, 20 (2), 75–76. 10.1038/s41577-020-0275-8. [DOI] [PubMed] [Google Scholar]
- Okazaki T.; Honjo T. The PD-1-PD-L Pathway in Immunological Tolerance. Trends Immunol. 2006, 27 (4), 195–201. 10.1016/j.it.2006.02.001. [DOI] [PubMed] [Google Scholar]
- He X.; Xu C. Immune Checkpoint Signaling and Cancer Immunotherapy. Cell Res. 2020, 30, 660. 10.1038/s41422-020-0343-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai Y.; Ishida M.; Tanaka Y.; Okazaki T.; Honjo T.; Minato N. Involvement of PD-L1 on Tumor Cells in the Escape from Host Immune System and Tumor Immunotherapy by PD-L1 Blockade. Proc. Natl. Acad. Sci. U. S. A. 2002, 99 (19), 12293–12297. 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins F.; Sofiya L.; Sykiotis G. P.; Lamine F.; Maillard M.; Fraga M.; Shabafrouz K.; Ribi C.; Cairoli A.; Guex-Crosier Y.; Kuntzer T.; Michielin O.; Peters S.; Coukos G.; Spertini F.; Thompson J. A.; Obeid M. Adverse Effects of Immune-Checkpoint Inhibitors: Epidemiology, Management and Surveillance. Nat. Rev. Clin. Oncol. 2019, 16 (9), 563–580. 10.1038/s41571-019-0218-0. [DOI] [PubMed] [Google Scholar]
- Kalbasi A.; Ribas A. Tumour-Intrinsic Resistance to Immune Checkpoint Blockade. Nat. Rev. Immunol. 2020, 20 (1), 25–39. 10.1038/s41577-019-0218-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinidou M.; Zarganes-Tzitzikas T.; Magiera-Mularz K.; Holak T. A.; Dömling A. Immune Checkpoint PD-1/PD-L1: Is There Life Beyond Antibodies?. Angew. Chem., Int. Ed. 2018, 57, 4840–4848. 10.1002/anie.201710407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magiera-Mularz K.; Skalniak L.; Zak K. M.; Musielak B.; Rudzinska-Szostak E.; Berlicki Ł.; Kocik J.; Grudnik P.; Sala D.; Zarganes-Tzitzikas T.; Shaabani S.; Dömling A.; Dubin G.; Holak T. A. Bioactive Macrocyclic Inhibitors of the PD-1/PD-L1 Immune Checkpoint. Angew. Chem., Int. Ed. 2017, 56 (44), 13732–13735. 10.1002/anie.201707707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curis, Inc. A Study of CA-170 (Oral PD-L1, PD-L2 and VISTA Checkpoint Antagonist) in Patients with Advanced Tumors and Lymphomas (NCT02812875). https://clinicaltrials.gov/ct2/show/NCT02812875?term=PD-L1++small+molecule&draw=2&rank=3 (accessed 2020-11-07).
- Musielak B.; Kocik J.; Skalniak L.; Magiera-Mularz K.; Sala D.; Czub M.; Stec M.; Siedlar M.; Holak T. A.; Plewka J. CA-170 – A Potent Small-Molecule PD-L1 Inhibitor or Not?. Molecules 2019, 24, 2804. 10.3390/molecules24152804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konieczny M.; Musielak B.; Kocik J.; Skalniak L.; Sala D.; Czub M.; Magiera-Mularz K.; Rodriguez I.; Myrcha M.; Stec M.; Siedlar M.; Holak T. A.; Plewka J. Di-Bromo-Based Small-Molecule Inhibitors of the PD-1/PD-L1 Immune Checkpoint. J. Med. Chem. 2020, 63 (19), 11271–11285. 10.1021/acs.jmedchem.0c01260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musielak B.; Janczyk W.; Rodriguez I.; Plewka J.; Sala D.; Magiera-Mularz K.; Holak T. Competition NMR for Detection of Hit/Lead Inhibitors of Protein–Protein Interactions. Molecules 2020, 25, 3017. 10.3390/molecules25133017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzik K.; Zak K. M.; Grudnik P.; Magiera K.; Musielak B.; Törner R.; Skalniak L.; Dömling A.; Dubin G.; Holak T. A. Small-Molecule Inhibitors of the Programmed Cell Death-1/Programmed Death-Ligand 1 (PD-1/PD-L1) Interaction via Transiently Induced Protein States and Dimerization of PD-L1. J. Med. Chem. 2017, 60 (13), 5857–5867. 10.1021/acs.jmedchem.7b00293. [DOI] [PubMed] [Google Scholar]
- Zak K. M.; Kitel R.; Przetocka S.; Golik P.; Guzik K.; Musielak B.; Dömling A.; Dubin G.; Holak T. A. Structure of the Complex of Human Programmed Death 1, PD-1, and Its Ligand PD-L1. Structure 2015, 23 (12), 2341–2348. 10.1016/j.str.2015.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zak K. M.; Grudnik P.; Guzik K.; Zieba B. J.; Musielak B.; Dömling A.; Dubin G.; Holak T. A. Structural Basis for Small Molecule Targeting of the Programmed Death Ligand 1 (PD-L1). Oncotarget 2016, 7 (21), 30323–30335. 10.18632/oncotarget.8730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skalniak L.; Zak K. M.; Guzik K.; Magiera K.; Musielak B.; Pachota M.; Szelazek B.; Kocik J.; Grudnik P.; Tomala M.; Krzanik S.; Pyrc K.; Dömling A.; Dubin G.; Holak T. A. Small-Molecule Inhibitors of PD-1/PD-L1 Immune Checkpoint Alleviate the PD-L1-Induced Exhaustion of T-Cells. Oncotarget 2017, 8 (42), 72167–72181. 10.18632/oncotarget.20050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarganes-Tzitzikas T.; Konstantinidou M.; Gao Y.; Krzemien D.; Zak K.; Dubin G.; Holak T. A.; Dömling A. Inhibitors of Programmed Cell Death 1 (PD-1): A Patent Review (2010–2015). Expert Opin. Ther. Pat. 2016, 26 (9), 973–977. 10.1080/13543776.2016.1206527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaabani S.; Huizinga H. P. S.; Butera R.; Kouchi A.; Guzik K.; Magiera-Mularz K.; Holak T. A.; Dömling A. A Patent Review on PD-1/PD-L1 Antagonists: Small Molecules, Peptides, and Macrocycles (2015–2018). Expert Opin. Ther. Pat. 2018, 28 (9), 665–678. 10.1080/13543776.2018.1512706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S.; Yang J.; Xu B.; Magiera-Mularz K.; Skalniak L.; Musielak B.; Kholodovych V.; Holak T. A.; Hu L. Design, Synthesis, Evaluation, and Structural Studies of C2-Symmetric Small Molecule Inhibitors of Programmed Cell Death-1/Programmed Death-Ligand 1 Protein–Protein Interaction. J. Med. Chem. 2019, 62 (15), 7250–7263. 10.1021/acs.jmedchem.9b00795. [DOI] [PubMed] [Google Scholar]
- Shaaban S.; Abdel-Wahab B. F. Groebke–Blackburn–Bienaymé Multicomponent Reaction: Emerging Chemistry for Drug Discovery. Mol. Diversity 2016, 20 (1), 233–254. 10.1007/s11030-015-9602-6. [DOI] [PubMed] [Google Scholar]
- Baenziger M.; Durantie E.; Mathes C. Development of an Industrial Process Based on the Groebke–Blackburn–Bienaymé Multicomponent Reaction: Efficient Preparation of 3-Aminoimidazo[1,2-a]Pyrazines. Synthesis 2017, 49 (10), 2266–2274. 10.1055/s-0036-1588130. [DOI] [Google Scholar]
- Boltjes A.; Dömling A. The Groebke-Blackburn-Bienaymé Reaction. Eur. J. Org. Chem. 2019, 2019 (42), 7007–7049. 10.1002/ejoc.201901124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber P. R.; Müller K. MAB, a Generally Applicable Molecular Force Field for Structure Modelling in Medicinal Chemistry. J. Comput.-Aided Mol. Des. 1995, 9 (3), 251–268. 10.1007/BF00124456. [DOI] [PubMed] [Google Scholar]
- Kuhn B.; Fuchs J. E.; Reutlinger M.; Stahl M.; Taylor N. R. Rationalizing Tight Ligand Binding through Cooperative Interaction Networks. J. Chem. Inf. Model. 2011, 51 (12), 3180–3198. 10.1021/ci200319e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y.; Thomé I.; Kim S. H.; Chen D.; Beyer A.; Bonnamour J.; Zuidema E.; Chang S.; Bolm C. Dimethyl Sulfoxide/Potassium Hydroxide: A Superbase for the Transition Metal-Free Preparation of Cross-Coupling Products. Adv. Synth. Catal. 2010, 352 (17), 2892–2898. 10.1002/adsc.201000575. [DOI] [Google Scholar]
- Li Z.; Wu L.; Yao W.. Heterocyclic Compounds as Immunomodulators. WO 2017205464 A1, 2017.
- Wang Y.; Xu Z.; Wu T.; He M.; Zhang N.. Aromatic Acetylene or Aromatic Ethylene Compound, Intermediate, Preparation Method, Pharmaceutical Composition and Use Thereof. WO 2018/006795 A1, 2018.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.