

Abstract
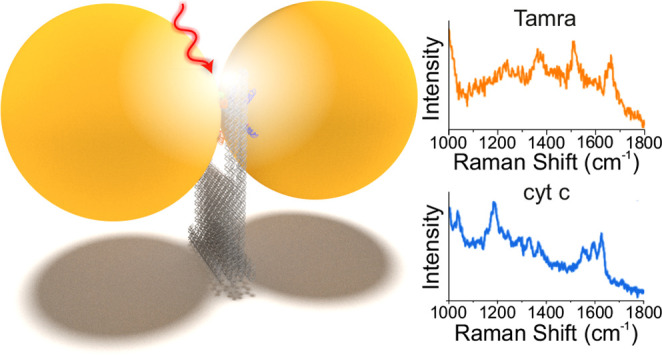
DNA origami technology allows for the precise nanoscale assembly of chemical entities that give rise to sophisticated functional materials. We have created a versatile DNA origami nanofork antenna (DONA) by assembling Au or Ag nanoparticle dimers with different gap sizes down to 1.17 nm, enabling signal enhancements in surface-enhanced Raman scattering (SERS) of up to 1011. This allows for single-molecule SERS measurements, which can even be performed with larger gap sizes to accommodate differently sized molecules, at various excitation wavelengths. A general scheme is presented to place single analyte molecules into the SERS hot spots using the DNA origami structure exploiting covalent and noncovalent coupling schemes. By using Au and Ag dimers, single-molecule SERS measurements of three dyes and cytochrome c and horseradish peroxidase proteins are demonstrated even under nonresonant excitation conditions, thus providing long photostability during time-series measurement and enabling optical monitoring of single molecules.
Keywords: DNA origami, surface-enhanced Raman scattering, nanoparticles, single molecules, proteins, plasmonics
The ultimate detection limit in analytical chemistry is the single molecule (SM), ideally without the necessity to label the target molecule. Furthermore, the ability to extract specific information about single molecular events allows for the screening of molecular property distributions, which are otherwise hidden within an ensemble averaged measurement.1 Up to now, several techniques have been reported that are capable of detecting molecules down to the SM regime, such as fluorescence spectroscopy,2 scanning probe microscopy,3 and surface-enhanced Raman scattering (SERS).4 Among them, SERS is a particularly promising technique because in addition to the simple detection of a target molecule, it can also provide detailed chemical and structural information through the Raman vibrational fingerprint of the molecule. However, SERS relies on the strong Raman signal enhancement upon excitation of localized surface plasmon resonances (LSPR), mainly in gold (Au) or silver (Ag) nanostructures.5,6 The Raman signal enhancement is the strongest in small, nanometer-sized volumes in-between coupled Au or Ag nanostructures, which are referred to as SERS hot spots. Therefore, the detection of SMs in these hot spots requires a precise control over the arrangement of nanoparticles as well as the placement of target molecules into these hot spots, which imposes severe challenges for nanofabrication. This is why most of the SM SERS measurements reported so far rely on the random adsorption of analyte molecules in the hot spots.4,7,8 In a complementary approach, the analyte molecules are precisely positioned into the hot spots via covalent or noncovalent interactions using DNA.9 DNA origami is a powerful nanoengineering tool,10 which is based on the programmed self-assembly into various two- (2D) and three-dimensional (3D) shapes.11,12 DNA origami nanostructures are formed from a long single-stranded scaffold strand and a suitable set of short artificial staple strands,10 and several computation tools have been developed to automate the design process of DNA origami.13−17 Due to the rich chemical functionalization schemes of DNA, molecules can be positioned along the DNA origami surface with nanometer precision.18 Furthermore, various kinds of nanoparticles can be arranged on the nanoscale to create sophisticated plasmonic nanostructures and to perform SERS19−28 and surface-enhanced fluorescence (SEF)29,30 measurements. Past examples of such structures include different platforms to form dimers of spherical particles,19,26,29,31−33 dimers of Au nanostars,27 bowtie antennas,28 bimetallic nanostructures,20,32 DNA origami nanocavities,34,35 and plasmonic nanostructures consisting of three or more spherical particles.21,22,24,36 While DNA origami-based nanoantennas with larger interparticle gaps in the range of 10–30 nm are required for SEF, SERS requires small gaps below about 5 nm to provide strong signal enhancement. Consequently, DNA origami-based SM SERS measurements have mainly focused on small molecules such as organic dyes that can be placed in small interparticle gaps either by random adsorption or by DNA conjugation. Here, we introduce a versatile 3D plasmonic DNA origami nanofork antenna (DONA) structure representing Au and Ag dimers suitable for SM SERS measurements of not only small molecules or DNA but also more complex biomolecules such as cytochrome c (cyt c) and horseradish peroxidase (HRP). Through oxDNA simulation and atomic force microscopy (AFM), we can demonstrate that the structure exhibits better overall rigidity compared to the previous SERS substrates and the ability to place biomolecules precisely into the dimeric hot spot. The addressable binding sites in the as-designed DONA structures allow for precise placement of basically arbitrary molecules of different dimension in the gap between two Au or Ag nanoparticles, where the electric field enhancement is strong enough to detect SMs by SERS. Here, the increase in the gap size to accommodate larger molecules comes at the expense of lowered field enhancement in the hot spot but with the increase of the overall hot spot volume. By varying the gap size, we can show that DONAs with larger gap sizes can still produce SM signals, although the DONAs with smaller gap sizes are flexible enough to accommodate proteins to produce a high yield of SM SERS spectra. Our findings suggest that the proteins, even though having the dimensions larger than the gap size, can at least partially occupy the hot spot and thus exhibit characteristic Raman spectra.
Results and Discussion
To create complex arrangements of Au and Ag nanoparticles on DNA origami nanostructures, the particles are coated with thiolated single-stranded DNA that is hybridized to complementary DNA extensions protruding from the DNA origami.21,37,38 A frequently used nanoparticle configuration for SERS is the nanoparticle dimer. Importantly, even a small offset from the hot spot can lead to a drastic drop in the field enhancement, which calls for precise positioning of molecules in-between the nanoparticles, which, however, has been challenging. To overcome this, we have designed a DNA origami nanofork structure forming the basis of the plasmonic DONA structures. It consists of a rectangular base with two arms, as illustrated in Figure 1A and Figure S1 (Supporting Information, SI). Two nanoparticles can be bound to the arms such that each particle is connected to both arms. The two arms are connected by a bridge consisting of two DNA double helices. The main purpose of the bridge is to serve as an anchor point for target analyte molecules, which can be precisely placed into the plasmonic gap of the two nanoparticles, at the location where the SERS signal enhancement is the strongest.
Figure 1.

Scheme and characterization of the DONA structures. (A) Schematic representation of the DNA origami nanofork having a DNA bridge that is 90 nt long. (B) AFM and TEM images of the DNA origami nanofork. The sizes of the close-up AFM and TEM images on the lower left are both approximately 90 nm × 100 nm. (C) Two differently coated nanoparticles can be attached selectively via DNA hybridization to the two different sequences of DNA capture strands on the arms and the bridge of the DNA origami to form DONA structures. A side-view and a top-view scheme of the assembled DONA structures is shown. The position of an analyte molecule attached to the DNA bridge is illustrated as a green glow. The DNA hybridization of nanoparticles and DNA origami in the “zipper” configuration is shown in the insets. (D) Representative TEM image of an assembled smaller gap size Au DONA (more examples shown in Figure S2), along with distributions of gap sizes determined from TEM images of smaller and larger gap size DONAs with an average gap size of 1.17 and 1.40 nm, respectively. The number (N) of the counted DONAs was 109 and 95 for the smaller and larger gap sizes, respectively.
The outer dimensions of the DNA origami nanofork are estimated to be 48 nm × 71 nm, based on an interhelical distance of 2.536 nm for a honeycomb lattice.39 The base of the nanofork is designed so that the distance between the two arms is about 31 nm when the arms are straight. Each arm is equipped with four ssDNA capture strands (capture sequences a = 5′-A24 and b = 3′-(AAC)8 in Figure 1 and Figure S1, to bind two differently coated nanoparticles), extending to different sides and designed to bind complementary sequences on Au or Ag nanoparticles. The bridge includes four staple strands, each crossing over once between the two helices to increase the stability and rigidity of the bridge. In addition, the free 3′ and 5′ ends of these staple strands allow for modifications to be added to the bridge. Further, to increase the precision of the nanoparticle placement, two of the bridge staple strands are equipped with capture strands on each end. Hence, each nanoparticle will be bound by a total of six capture strands. Although the base sets the overall distance between the arms, the arm-to-arm distance and hence the bridge placement or position can fluctuate due to the flexibility of the DNA origami. To assess the magnitude of fluctuation and the precision of the bridge position, we initially considered nanoforks with the bridge length of either 90 nucleotides (nt) (31.5 nm, short version) or 120 nt (42 nm, long version).
The fabrication process of Ag or Au DONAs is shown in Figures 1A–C. The nanoforks with 90 nt bridge are characterized by atomic force microscopy (AFM) and transmission electron microscopy (TEM), as shown in Figure 1B. The images clearly show the successful formation of the nanoforks including the DNA bridge. In the zoomed-in AFM image the lower part of the base of the DNA origami fork appears elevated, which corresponds to the additional rows of DNA double helices that are also shown in the scheme of the DNA origami structure. The nanoforks are then hybridized with Au or Ag nanoparticles of 60 nm diameter to form the DONAs (Figure 1C, AFM image of DONAs in Figure S3). To assess the fabrication yield, we calculated the single NP to DONA dimer ratio for both Ag and Au DONAs (see Figure S4). The average ratios of single NPs to DONA dimers are determined to be 3.5:1 and 2.3:1 for Ag and Au DONAs, respectively.
For optimal SERS measurements, a narrow gap between the nanoparticles and precise placement of target molecule therein is required to establish a strong SERS signal enhancement. Two different gap sizes are considered during the experiments, where the gap size is adjusted by adding an extra 3 nt spacer sequence between the thiol group and the segment of the sequence binding to the coating strand (see Methods). The larger gap size was used to better accommodate proteins in the gap. We have measured the gap distance of the Au DONA structures containing the short and long coating sequences by TEM, and a typical TEM image of a short coating strand DONA is shown in Figure 1D, along with a histogram of gap sizes. The most probable gap distance is found to be 1.17 and 1.4 nm for the short and the long coating strands, respectively. As the DNA origami nanofork itself is not properly visible in the TEM images (see SI), we confirmed the binding of two differently coated 40 nm AuNPs by separately hybridizing each AuNP to the DNA origami nanofork and characterizing the binding in AFM (Figure S5). Here, we chose the 40 nm particles to allow a direct visualization of the nanofork. The dimensions of the nanofork are set as such that 60 nm AuNPs cover the fork in a way that the bottom of particles and the nanofork are on the same level. The use of the 60 nm AuNPs throughout the study is partly due to the fact that the bottom of the particles and the nanofork are leveled so that the bottom of the fork is barely touching the substrate in a DONA assembly, which should minimize some of the bending or shear forces exerted by the nanoparticles. It should be noted that the diameter of the bridge DNA does not impose a lower limit of the gap size because the bridge DNA is flexible enough to allow positioning of the analyte just beside the hot spot (i.e., the point of smallest distance between the nanoparticles), while at the same time the analyte molecule can direct itself from the bridge into the hot spot.
In order to further analyze and optimize the nanofork structure, we have performed molecular dynamics simulations using the coarse-grained model oxDNA2.16,17 The oxDNA model is ideally suited to analyze the effect of bridge length on the distance of the two DNA arms and the general displacement and fluctuation of the DNA bridge, which is important for the precision of the placement of target molecules. We simulated the nanofork with bridge lengths of 120 nt and 90 nt as well as no bridge. Figure 2A shows sides and top views of an equilibrated conformation of the DNA origami fork model with a 90 nt bridge, which confirms the overall rigidity of the structure. Figure 2B shows the probability distributions of the distance between the tips of the two DNA arms (dotted lines). The probability distribution is very broad when no bridge is present, with a maximum around 35 nm, which is slightly shorter than the nominal distance of 40 nm shown in Figure 1A. With a long bridge of 120 nt, the maximum of the probability function is shifted to 45 nm. With a shorter bridge of 90 nt, the distribution gets narrower with a most probable distance of 40 nm, corresponding to the designed value. An analysis of AFM images of corresponding nanofork samples yields results that match the simulations (Figure 2B, solid lines, and Figures S6 and S7). Since the short bridge has more accurate positioning and smaller fluctuation compared to the long bridge, for further experiments we have chosen a bridge length of 90 nt. The simulations also allow to estimate the thermal variation of the center of the bridge along the plane of the nanofork (Figure 2C). The plot shows that a shorter DNA bridge increases the stiffness of the bridge and that for the 90 nt bridge, a full-width at half-maximum (fwhm) of 3.14 nm is found, indicating the precision with which target molecules placed in the bridge could be located at the plasmonic junction.
Figure 2.
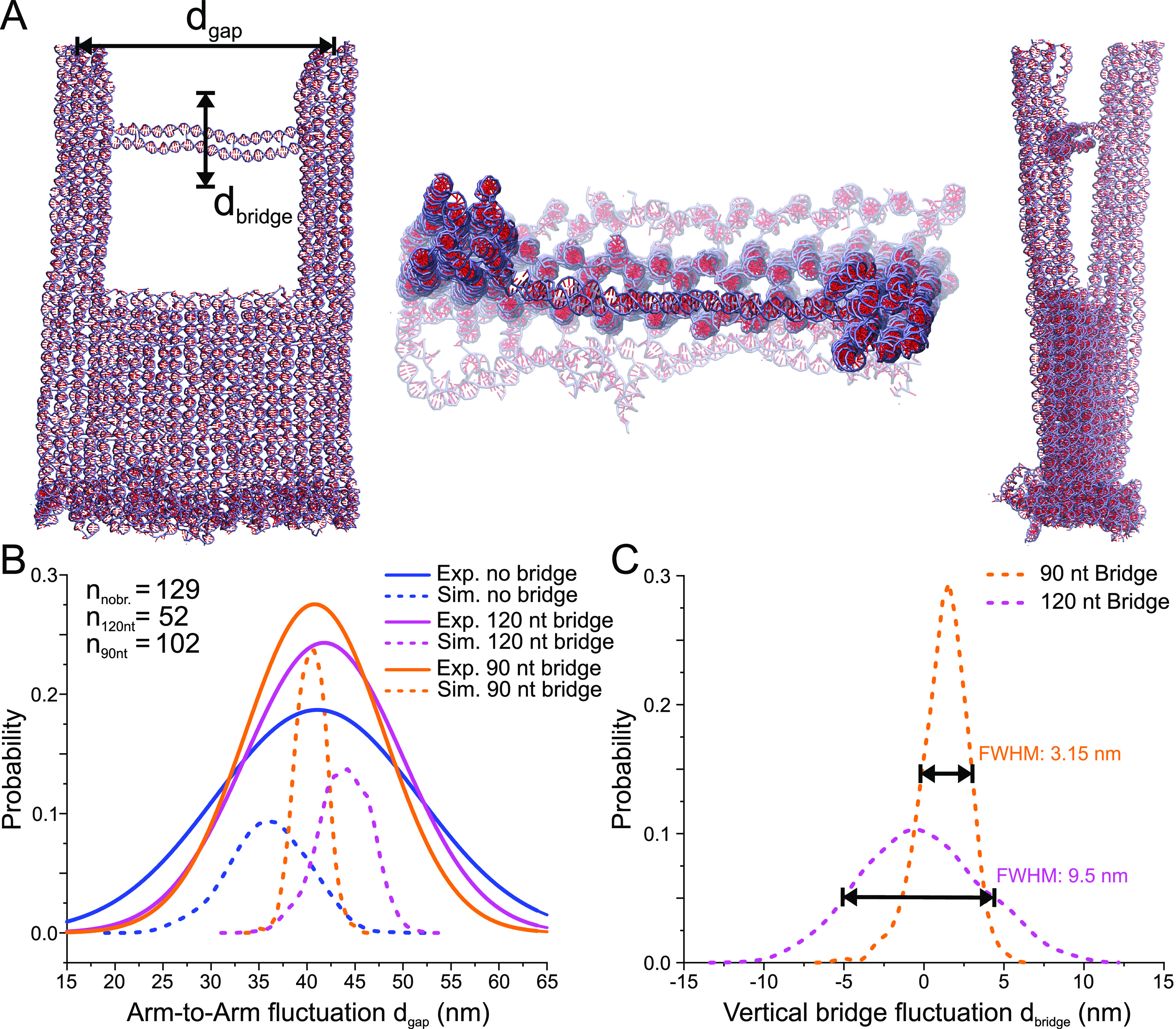
Coarse-grained oxDNA model of the DNA origami nanofork. (A) Snapshots of two side views and one top-view of the nanofork with 90 nt DNA bridge as simulated using the oxDNA model. (B) Plot of the simulated probability distribution of distance between the tips of the two arms and comparison with experimental values obtained from AFM images (for no bridge and 90 nt and 120 nt bridges, the maxima are 41.13, 40.82, and 41.84 nm, respectively). The number of nanoforks analyzed by AFM is given in the left legend. (C) Plot of the simulated probability distribution of the flexibility of the center of a 90 nt and a 120 nt DNA bridge with respect to the straight bridge position, with the maxima at 1.56 nm and −0.6 nm, respectively.
To test the performance of the DONAs for SM SERS, we have placed single dye molecules (TAMRA, Cy3.5 and Cy5) at the center of the DNA bridge using dye-modified staple strands and then assembled Au and Ag DONAs (see Methods and Table S1). In general, to achieve single molecular sensitivity, a system should produce high enough electric (E) field enhancements to achieve a signal enhancement in the order of 109–1012, according to the forth power approximation.8 To investigate this, we calculated the field enhancement (FE) in the gap between the particles with 60 nm diameter as a function of wavelength using the finite difference time domain (FDTD) method (see Methods and SI for details). We have considered different gap sizes ranging from 1.2 nm (corresponding to the mean gap size found in TEM images) to 3.5 nm as shown in Figure 3A for Au and in Figure 3B for Ag dimers for polarization along the dimer axis. The data in Figure 3A and Figure 3B allow for an analysis of the SERS signal enhancement of nanoparticle dimers with different gap sizes at specific Raman excitation wavelengths and in respect to the most common excitation lines used in Raman spectroscopy (i.e., 785 nm, 633 nm, 532 nm and 488 nm). Although the highest FE is predicted for the narrowest gap size (1.2 nm) around 736 nm for the Au dimer, the FDTD simulations indicate that an excitation at 633 nm for Au dimers is still advantageous because dimers with different gap sizes give rise to a SERS signal enhancement of 109 or higher at this wavelength. For example, the FE at 633 nm excitation wavelength is 255 for a 1.2 nm gap and 312 for a 2.5 nm gap, corresponding to a SERS signal enhancement of around 4.2 × 109 and 9.5 × 109, respectively, using the E4 approximation.40
Figure 3.

FDTD simulations of DNA coated Au and Ag dimers and SM SERS spectra of different dyes. The dashed line in (A) and (B) indicates a field enhancement of 109 that is assumed to be the requirement for SM detection. (A) FDTD simulations of the electric field enhancement of a Au dimer consisting of DNA coated 60 nm diameter particles on a silicon substrate upon excitation along the dimer axis. (B) FDTD simulations of the electric field enhancement of a DNA coated Ag dimer consisting of 60 nm diameter particles on a silicon substrate upon excitation along the dimer axis. (C) SM SERS spectra of TAMRA, Cy3.5, and Cy5 obtained using Au and Ag DONAs (Cy5 was measured only using Au DONAs). The inset shows SEM images of the specific DONAs, from which the SM SERS spectra have been obtained. The size of the inset SEM images is 450 × 450 nm.
Besides the plasmonic enhancement, the SERS signal depends also on the optical properties of the analyte molecules, that is, their optical absorption. The SERS signal can either be further enhanced by the resonance Raman effect by matching the molecule’s optical absorption with the LSPR,41 or nonresonant molecule excitation can be used to avoid photobleaching.
Figure 3C shows examples of SM SERS spectra of TAMRA, Cy3.5, and Cy5, which are obtained from Au DONAs excited at 633 nm and Ag dimers excited at 532 nm (see Methods and Figures S8–S11). The black spectra (Figure 3C) are reference spectra of the respective dyes (see Methods, Figures S12 and S13 for SERS spectra, and Figure S14 for the absorption spectra). Excitation at 532 nm as used for Ag dimers results in a SM SERS signal with considerably higher signal-to-noise ration than in previous SM SERS studies.20 Interestingly, when using 633 nm excitation for the corresponding Au DONA, SM SERS of TAMRA can be measured, although TAMRA does not absorb at 633 nm, and therefore resonant Raman contributions can be excluded in this case. This indicates that the plasmonic field enhancement at 633 nm is indeed strong enough to detect a SM under nonresonant conditions, which is also supported by our FDTD simulations. The same behavior is observed for Cy3.5, which can be excited by the 532 nm laser, but not sufficiently by the 633 nm laser. Nevertheless, strong and characteristic SM SERS spectra are obtained for both Au and Ag DONAs. The strongest absorption of Cy5 is observed at around 650 nm (Figure S14), close to the 633 nm excitation used for the Au dimers. Consequently, a strong and characteristic SM SERS spectrum28 is obtained for Cy5 using the Au DONAs.
The presented dye spectra also include several peaks that cannot be attributed to dye spectra such as the peak at 1608 cm–1 in the case of the TAMRA-Ag-DONA in the Figure 3C. The most probable source of these peaks is the DNA from the DNA coating layer and the DNA bridge,42 which is inherently present in the system. To rule out that the observed spectra would originate from DNA and not from the dyes, we performed control measurements using nanoforks with pure DNA bridges (i.e., no dyes) and averaged several spectra for both the dyes (see Figure S15) and the pure DNA bridges (see Figures S15–S17). Although in some cases the pure DNA-bridge DONAs contained similar peaks as in the case of the dyes, the average spectra did not resemble any identifiable DNA fingerprint spectra, indicating that these peaks appear only randomly and we are not systematically measuring the surrounding DNA. In contrast, the average DONA-dye spectra clearly displayed the characteristic peaks of the set dyes, and thus the dye characteristic peaks observed in the Figure 3C can be attributed to the dyes. Therefore, the success of SM SERS measurements using different dyes, nanoparticle species, and excitation wavelengths demonstrates the strength and versatility of DONAs, that is, the ability to tune the plasmonic properties according to the needs of the molecular systems to be investigated.
Typically, SM SERS spectra show strong fluctuations of the signal intensity with time, which reveals information about the stability and dynamics of molecules in the hot spot. One possible way to characterize plasmonic properties of DONA samples is to employ dark-field (DF) microscopy. This then provides the basis to record time-dependent SM SERS spectra of them. Before we discuss time-dependent SM SERS spectra, we further characterize the gap sizes of the DONAs by measuring the polarization-dependent DF scattering spectra of several gold DONAs. We used an adjusted version of the FDTD model to correlate the LSPR peak of the gap mode from each dimer to the corresponding gap size (see SI and Methods for details).
Figure 4A shows the simulated DF scattering spectra for a 60 nm AuNP dimer. The main LSPR peak associated with the gap mode is red shifting, as the gap size decreases due to stronger coupling between the AuNPs. For the simulations in Figure 4A, the average nanoparticle diameter of 60 nm is assumed, but as can be seen in the TEM images, the dimensions of nanoparticles can differ from the nominal value. For the DONA shown in the inset of Figure 4B, polarization-dependent DF scattering spectra were measured, and the FDTD model was adjusted to match the dimensions of the slightly ellipsoidal particles (72 nm × 53 nm × 64.5 nm and 75 nm × 65 nm × 72 nm). The simulated gap size was varied so that the simulated and the experimental curves associated with the gap mode matched each other (the dashed lines and the solid blue curve in Figure 4B). The solid blue and orange lines in the inset indicate the analyzer angles, where the corresponding colored experimental curves were measured (labeled as the gap mode and the transverse mode). The simulated curve corresponding to the excitation along the gap axis (the dashed blue curve, normalized for visualization purposes) matched the measured peak around 678 nm, corresponding to a gap size of roughly 1.9 nm. Since the excitation light is unpolarized, we are considering electric field polarizations parallel, perpendicular, and in a 45° angle with respect to the gap axis in the simulations. The dashed black curve contains the contribution of all the polarization angles, and it fits quite well with the overall shape of the curve. This would suggest that first the peak around 580 nm corresponds to the contributions from the off-gap-axis electric field polarizations and second the analyzer angle is mostly probably not perfectly matched with the dimer axis, since one can observe the off-gap-axis excitations. The same peak is also observed when the scattered light is extracted perpendicular to the gap axis (the solid orange curve), and we attribute this to the single particle scattering that is shifted due to the silicon surface and proximity of the other particle.
Figure 4.

Experimental and simulated DF scattering spectra and SM SERS time series measurements of TAMRA using Au DONA. (A) Simulated DF scattering spectrum of AuNP dimer when the gap size of two 60 nm AuNPs is varied between 1.2 and 3.5 nm. The graph on the lower right shows the LSPR dependency on the gap size. (B) Experimental and simulated DF scattering spectra from single DONA. The orange and the blue line in the inset indicate the measured polarization axis. The simulated blue curve, where the excitation is along the dimer axis, is fitted to the peak around 678 nm by varying the gap size of the two ellipsoidal particles. The dashed black curve combines the contribution of all of the excitations. The size of the inset is 500 nm × 500 nm. (C) Time vs Raman shift contour plot for a single TAMRA molecule. The dotted lines indicate the main TAMRA peaks (1654, 1537, 1509). The dotted green line indicates a band at 1347, which can be either a shifted TAMRA band or a DNA peak. The white arrows indicate different times at which the SERS spectra shown in (D) are extracted. The size of the inset image is 250 nm × 250 nm. (D) Full single TAMRA spectra extracted from the contour plot at 18, 33, 36, 57, 75, and 78 s. The same TAMRA bands are indicated with lines as in (C).
The fine-tuned LSPR peak of the gap mode does not differ much from the corresponding nominal 60 nm DONA spectra (2 nm) in Figure 4A, and hence we use the simulated 60 nm spectra in Figure 4A to evaluate the gap size of DONAs. Applying these findings to similar DONA spectra (see Figures S18 and S19) yields gap sizes ranging between 1.9 and 3.5 nm, but due to instrumentational limitations of the excitation lamp, we are unable to cover the spectral region beyond 686 nm and to determine gap sizes well below 1.9 nm. However, some of the DONA spectra plateaued around the gap mode or had a slight upward slope (Figure S19C,D). This indicates that the gap mode peak has shifted beyond 700 nm, further proving that the gap sizes below 2 nm exist.
Figure 4C,D presents a continuous time series SERS measurement of a Au DONA with a TAMRA dye at 633 nm excitation over the course of 120 s, recorded using combined DF and Raman microscopy (see Methods and Figures S20 and S21). The characteristic signals of TAMRA at 1654, 1537, and 1509 cm–1 can be clearly identified, while additional signals attributed to DNA are also observed. The characteristic Raman bands can be observed over several minutes, indicating that no photobleaching of the dye occurs, which is attributed to the fact that TAMRA does not absorb the 633 nm light of the excitation laser. The signal intensity steadily decreases over a time course of several minutes, while the reference signal (Si at 520 cm–1) remains rather constant (Figure S22). This effect could be due to a soldering of the nanoparticles during the constant laser excitation43 or the DNA structure is damaged or disintegrated possibly due to plasmonic heating.44 On a shorter time scale (as displayed in Figure 4), clear fluctuations of the signals are observed, which correspond to SERS blinking.4,45−47 One example is the bands at 1537 and 1509 cm–1: For the first 70 s, the band at 1537 cm–1 is clearly stronger than the band at 1509 cm–1, while the band at 1537 cm–1 almost disappears between 70 and 100 s, and subsequently the original intensity ratio is restored. Both bands are assigned to C–C stretching vibrations,48 the intensity of which varies according to thermal movements of the molecule within the SERS hot spot, giving rise to SERS blinking. Furthermore, it should be noted that the band expected at 1360 cm–1 for TAMRA appears either very weak during the time series measurements or it is shifted to 1347 cm–1, for example, due to the vibrational Stark effect.49
After demonstrating the potential of DONAs to measure single dye molecules even under nonresonant conditions, we further extend our study to highlight the whole potential of DONAs to directly measure single protein molecules by SERS. This can be achieved with different coupling schemes exploiting either noncovalent or covalent binding of proteins. Here, we have used the heme proteins cyt c and HRP as model systems to demonstrate their direct, label-free, and nonresonant SM SERS detection using DONAs (Figure 5). Cyt c is found in the inner membranes of mitochondria and is involved in biological electron-transfer processes, while HRP is involved in oxidation processes.50,51 Heme proteins strongly absorb at around 420 nm (Soret or B band) and 550 nm (Q band),52 however, we use the 633 nm excitation laser for the SM measurements, which is nonresonant with the heme proteins. In this way, not only the heme prosthetic group is visible in the spectra but also the amino acid part of the proteins can be directly detected.
Figure 5.

Single-molecule SERS measurements of cyt c and HRP. (A) Field enhancement plots of the DONA Au dimers, when the gap size is 1.2 nm (smaller gap) and 3 nm (larger gap). (B) Distributions of the smaller and larger gap sizes determined from TEM images of cyt c with the average gap sizes of 1.45 and 1.83 nm, respectively. The number (N) of counted dimers was 101 and 109 for the smaller and larger gaps, respectively. (C) SM SERS spectra of cyt c using Au DONA structures and 633 nm excitation. (D) Distributions of the smaller and larger gap sizes determined from TEM images of HRP with the average gap sizes of 1.21 and 2.53 nm. The number (N) of counted dimers was 108 and 95 for the small and large gaps, respectively. (E) SM SERS spectra of HRP using Au DONA structures and 633 nm excitation. The insets in (C) and (E) show SEM images of the specific DONAs, from which the SM SERS spectra have been obtained. The sizes of the SEM images are from top to bottom 360 × 280 nm, 360 × 420 nm, and 300 × 360 nm and 240 × 240 nm, 360 × 420 nm, and 300 × 300 nm, respectively.
For the noncovalent, but specific binding of cyt c to the DONAs, a 4-pyridine moiety53 is coupled via an amide bond and a C5 linker to a staple strand at the center of the DNA bridge (the sequence shown in Table S1). HRP is covalently coupled to the NHS ester group of a Sulfo-SMCC cross-linker via a lysine residue. The maleimide group of the cross-linker is then bound to a thiolated staple strand (Table S1), which in turn is incorporated into the DNA bridge of the DNA origami nanofork (the binding schemes of cyt c and HRP are shown in Figure S23). Both, cyt c and HRP are bound to the DNA origami nanofork prior to the binding of the 60 nm Au nanoparticles. For synthesis of DONAs with smaller and larger gap sizes, we utilized the same particle coating sequences as in the case of dyes and similar sequences with added 3 nt spacers at the thiol-end, respectively. The reason for this is two-fold: the benefit for having a smaller gap size is the higher EF, as demonstrated in Figure 2, but the physical size of the protein is larger than the dye, and thus it would be more beneficial to increase the hot spot size to fully incorporate the larger molecules. Figure 5A shows simulations of the field enhancement distributions within the interparticle gaps for two different minimal gap sizes (1.2 and 3.0 nm). Based on the simulations, the effective hot spot, that is, the volume where a field enhancement of 109 is still achieved, is roughly 11 nm3 for the 1.2 nm gap size and approximately 27 nm3 for the 3 nm gap size. For the larger gap size, the volume encloses a signal enhancement of ≥109 along the dimer axis, which is available for a protein is approximately 133 nm3. This would suggest that an increase in the gap size should overall benefit the incorporation and detection of larger proteins. The volume of cyt c is approximately 14 nm3,54 while the volume of HRP is about 41 nm3,55 indicating that cyt c is small enough to be completely affected by a signal enhancement sufficient for SM detection in the larger gap hot spot, while only part of the HRP can be localized in the volume of highest signal enhancement.
Figure 5B,D shows the gap distances of DONAs with shorter and longer coating strands containing cyt c or HRP, respectively, as determined from TEM images. Accordingly, even for the shorter coating strands, the gap sizes increase when the proteins are present compared to the dye containing DONAs shown in the Figure 1D. Apparently, the presence of the proteins prevents the nanoparticles from coming closer, which is another indirect proof of successful positioning of proteins into the hot spot. With longer coating strands, the DONAs offer more flexibility, and the gap size increases on average from 1.45 nm (shorter coating strands) to 1.83 nm for cyt c and from 1.21 to 2.53 nm for HRP, while at the same time the gap distributions become broader. Examples of SM SERS spectra of cyt c and HRP for the shorter and longer coating strand DONAs (blue and red spectra, respectively) recorded at 633 nm excitation are shown in Figure 5C,E along with the corresponding SEM images of Au dimers and a reference spectrum (black spectra, see also Figures S24 and S25). The reference spectra were obtained from clusters of 60 nm Au nanoparticles with unspecifically bound cyt c or HRP, respectively (see Methods). The characteristic band appearing around 1370 cm–1 in both the cyt c and the HRP spectra is assigned to the stretching vibration of the porphyrin ring (ν4) of the heme group, indicating the oxidation state of the central iron ion (Fe3+).52,56 The strong signal around 1580 cm–1 appearing in the cyt c spectra is assigned to the ν19 band, the exact position of which is strongly dependent on the spin state of the central iron ion.52 The band at 1628–1630 cm–1 is due to the ν10 band, the spectral position of which is also indicative of the oxidized form of cyt c. In general, characteristic cyt c signals can be observed for both the smaller and larger gap sizes, which supports the general conclusion that also larger gap sizes can lead to signal enhancements sufficient for SM detection. But we find a better spectral reproducibility for the shorter gap size, suggesting a more pronounced conformational flexibility in the case of larger gap sizes. Apart from the signals arising from the heme unit of cyt c, the vibrational ring-breathing mode of the amino acid phenylalanine (Phe) can also be clearly distinguished at 1001 cm–1,57 but interestingly only for the smaller gap size DONAs. Cyt c possesses four Phe residues, and the possibility to detect this band in a single cyt c molecule by SERS using DONAs indicates the high sensitivity of the method. Also for HRP, characteristic SERS signals can be clearly detected in the case of DONAs with shorter coating strands. In addition to the ν4 band at 1370 cm–1 (which is broader in the reference spectrum with a shift to smaller wavenumber because both oxidation states appear in the reference), also the oxidation state marker at 1630 cm–1 (ν10) is visible, indicating the presence of Fe3+.52 With HRP we were not able to collect SM SERS spectra for the larger gap sizes, which might be an indication for a nonsufficient signal enhancement due to the larger gap sizes. Nevertheless, SM SERS spectra of HRP could be detected for the smaller gap size DONAs.
Lastly, although we purify the solution from an excess of free proteins after binding them to the forks, we have a minute amount of proteins left in the solution, which could bind unspecifically to the AuNPs or to the nanoforks. To test the possibility of contributions by unspecifically bound proteins to the measured SM-SERS protein spectra, we repeated the same sample fabrication process using nanoforks without any capture mechanism (i.e., no thiol or pyridine in the bridge). Interestingly, the DONA structures for each protein did not exhibit any spectra that could be identified as either cyt c or HRP (Figures S26 and S27), proving that the SERS spectra in the Figure 5 originate from a single protein in the bridge rather than from any unspecifically bound protein.
Conclusions
In conclusion, the designed 3D DNA origami nanofork has successfully been demonstrated for its suitability to assemble both nanoparticles and complex analyte molecules with nanoscale precision for SM SERS measurements. OxDNA simulations were employed to assess the structural dynamics of the nanofork and to choose the most suitable bridge length. The DNA bridge connecting the two arms of the nanofork allows for precise positioning of the target analyte in between the assembled nanoparticles of Au and Ag. This produces SERS hot spots on the scale of a few nanometers, creating a high enough field enhancement to enable label-free detection of single small molecules and even proteins (cyt c and HRP) via SERS, even at larger gap sizes around 2.5 nm. Importantly, the flexible integration of either Au and Ag nanoparticles supports SM SERS measurements both in resonant and nonresonant conditions. Time-dependent SM SERS of TAMRA under nonresonant conditions shows spectral blinking but no significant photobleaching, strikingly reflecting the single molecular SERS behavior. The experimental findings are supported by FDTD simulations that predict field enhancements high enough for SM measurements under nonresonant conditions. DNA origami is a sophisticated technique that offers options to achieve control over single molecular entities, thereby enabling different types of nanomachines.18,58 Here, we used this principle to build a platform for the direct, chemically specific, and label-free optical detection of single small molecules and single proteins. This presents a way to study biomolecular reactions at the SM level. One critical aspect of our design is the ability to generate arbitrary dimer compositions of any arbitrarily sized and shaped nanoparticles. The DONA structures could be used to monitor enzymatic action in different environments, perhaps even for multienzyme reaction cascades. At the same time, sensing of single small molecules represents one of the greatest challenges in analytical chemistry which could be achieved with the present DONA structure. This therefore presents a way to study single reaction intermediates which could prove invaluable for understanding chemical reaction pathways.
Methods
DNA Origami Nanofork Folding and Purifications
The staple strands used for the assembly of the DNA origami nanofork are shown in Table S2. Depending on the experiment, some strands were substituted with the modified versions of the same strand (see Table S1). The folding solution was made by mixing 10 μL of 10× buffer (10× TAE, 150 mM MgCl2), 67.5 μL of Millipore water, 20 μL of staple solution (the concentration of individual staple = 0.497 μM), and 2.5 μL of the scaffold (m13mp18, 0.1 μM). During folding, the ratio between staples and the scaffold is 40:1. The folding solution was heated to 80 °C and then cooled down to 20 °C over 15 h timespan. The solution was purified from excess oligos using 100 kDa centrifugal filters (Amicon, Ultracel, Centrifugal filter, 100 kDaA): The DNA origami solution (100 μL) was added to the filter, and 400 μL of Millipore water was mixed in. The filter was spun in 4700 rcf for 5 min, the filtrate was discarded, and 450 μL of Millipore water was added. The centrifugation was repeated 3 times, and the dead volume from the filter was finally recovered. The concentration of TAE and MgCl2 was adjusted to 1× and 5 mM, respectively. A UV–vis spectrometer was used to measure the DNA concentration of the final solution that was used to calculate the DNA origami concentration. Bruker Multimode 8 atomic force microscope (Billerica, Massachusetts, US) was used to acquire AFM images of the nanoforks.
Nanoparticle Coating Protocols
Gold (Au nanosphere NanoXact, citrate coated, 60 nm, 2.4 × 1010 particles/mL) and silver (Ag nanosphere NanoXact, citrate coated, 60 nm, 1.9 × 1010 particles/mL) nanoparticles were purchased from nanoComposix (San Diego, CA, USA). The DNA strands in the Table S2 and trisodium citrate dihydrate (TSC) were acquired from Sigma-Aldrich (Sigma-Aldrich Chemie GmbH, Munich, Germany). Tris(2-carboxyethyl)phosphine (TCEP) was purchased from RIZ Biochem.
The AuNPs were coated using a modified salt aging method, whereas the coating for silver nanoparticles was done using a pH altering method.59 For AuNPs, 400 μL of 60 nm AuNP stock solution was centrifuged once (2900 rcf, 5 min), the supernatant was removed, and the volume was adjusted to 26.5 μL by Milli-Q water. The disulfide bonds in the dithiol phosphoramidite (DTPA)-modified coating strands were precleaved using TCEP: 4 μL of the coating strand solution (100 μM) was mixed with 1 μL of TCEP (100 mM) and incubated at RT for 10 min. The 5 μL of cleaved DNA strand solution and 3.5 μL of 0.2% SDS were added to the AuNP solution and incubated 40 min to 1h and at 40 °C. After the incubation, the concentration of NaBr was gradually, in a stepwise manner, increased to 300 mM using initially 400 mM NaBr buffer and then 1 M buffer, where after each step the solution was incubated for 10 min at 40 °C: The initial 4 steps were 1.7 μL (400 mM), the next two 2.1 μL and 2.3 μL (400 mM), followed by 3× 2.3 μL and 1 M, and finally 6.5 μL and 1 M. After the NaBr addition, 1× TAE and 5 mM MgCl2 was added twice (6 μL and 6.6 μL) in a similar stepwise manner. Finally, the excess coating strands were purified by centrifugation: The sample was spun down using 2900 rcf for 5 min, supernatant was removed, and buffer added. During the first two round of centrifugation, 1× TAE, 5 mM MgCl2, and 0.02% SDS buffer was used, and during the final two rounds, the buffer was switched to 1× TAE and 5 mM MgCl2.
For silver particles, 400 μL of the 60 nm AgNP solution was centrifuged once (2900 rcf, 5 min), the supernatant was removed, and the volume was adjusted to 26.5 μL by Milli-Q water. The coating strands were cleaved similarly as in the case of AuNPs using TCEP. 3.5 μL of 0.2% SDS and 5 μL of DNA solution were added to the AgNP solution and the solution was incubated for 40 min at RT. Then the pH of the solution was adjusted close to 3 by adding 1.4 μL of 0.5 M TSC (pH 3, adjusted using HCl) to the mixture. After 3 min incubation at RT, NaCl (1 M) was added in stepwise manner until the final NaCl concentration of the AgNP solution was 0.3 M: 2 μL of NaCl was added 6 times followed by a final addition of 3.6 μL, where in-between additions the solution was rapidly vortexed and let stand still for few seconds. The particles were incubated for 5 min after the final addition of the NaCl and purified similarly as the AuNPs.
DONA Assembly and Gel Electrophoresis Purifications
DONAs were fabricated by mixing DNA origami nanoforks with two different 60 nm AuNP/AgNP batches, where for the shorter gap size DONAs one of the particles is coated with the T28 sequence and the other one with the (GTT)8T4 sequence. For the larger gap size DONAs, the sequences were T28GGT and (GTT)8T7, respectively. All materials were dissolved in 1× TAE and 5 mM MgCl2 buffer. The ratio of NP-T28: NP-(GTT)8T4:nanofork is 1.5:1.5:1, and the final concentration of nanoforks is between 0.1 and 0.2 nM. The ratio was the same for the longer sequences. The mixture was heated in a thermocycler to 40 °C and then cooled down to 20 °C in 3.5 h timespan. In some cases such as nanoforks with dyes, agarose gel electrophoresis was used to extract the dimers from the final product. Shortly, samples were run in 1% agarose gel made in 1× TAE and 5 mM MgCl2 for 60 min using 80 V. The dimer band was cut out of the gel, and the dimers were squeezed out using two microscope slides. Examples of Au and Ag DONA gels are shown in Figure S28.
TEM Imaging of DNA Origami Nanoforks and DONAs
All samples were imaged in JEOL JEM 1011 transmission electron microscope (JEOL, Akishima, Tokyo, Japan) equipped with Olympus MegaView G2 camera and using 80 kV acceleration voltage. In the TEM imaging, copper grids containing 1 nm carbon layer on top of 10 nm Formvar film (EFCF400-Cu-50, Science Services GmbH, Unterhachinger Straße 75, Munich, Germany) were used. To characterize the dimer gap sizes, the DONA solution was deposited on a TEM grid. For DONA deposition, no plasma treatment was required, whereas, for nanoforks, the TEM grids were plasma treated for 15 s using Diener Electronic Zepto plasma cleaner before deposition of the DNA origami and the staining. In the case of DONAs, 3 μL of DONA solution was pipetted to the grid and incubated for 2–3 min. The excess solution was blotted away, and 3 μL of staining solution was added. After 2 min incubation, the excess staining solution was blotted away, and the film was washed twice by adding 3 μL of Millipore water to the grid and blotting away the excess liquid. The grid was left to dry in ambient conditions.
In the case of DNA origami nanofork, after the plasma treatment, roughly 3 μL of DNA origami solution was pipetted on a grid and incubated for 5 min. The magnesium and fork concentrations were 20 mM and 0.6 nM, respectively. The excess solution was blotted away, and 3 μL of the same staining solution was added. After 2 min incubation, the excess solution was blotted away, and the sample was left to dry in ambient conditions without any washing. Figure S29 contains extra images of the DNA origami nanoforks.
Sample Preparation for SERS, SERS Measurements, SEM Imaging, and SERS-SEM Correlation
DONAs were deposited on a 7 mm × 7 mm silicon chip labeled with a scratch marker. A diamond cutter was used to draw a cross-shape on the chip, the surface was scrubbed clean from dust in acetone solution using a cotton stick, and cleaning was finalized by sonicating the chip in ethanol solution for 30 s. The clean chip was plasma treated for 10 min. For the deposition, the magnesium concentration of DONA solution was adjusted to 50 mM, the solution was pipetted to plasma-treated silicon surface, and incubated for roughly 1.5 h. The chip was washed with ethanol–water solution (1:1 ratio) and blown dry.
A Witec Alpha 300 Raman microscope was used in the measurements (Witec, Ulm, Germany). For the 488 and 532 nm measurements, a spectrograph (Blaze 500, grating 600 gr/mm) equipped with Andor DV401-BV CCD-camera was used, and for the 633 nm measurements, a spectrograph (Blaze 750, grating 600 gr/mm) equipped with Andor DU401A-BR-DD-352 CCD camera was used. The estimated spot sizes were 1.32, 1.44, and 1.72 μm for 488, 532, and 633 nm, respectively. Raman maps were acquired using varying laser power densities and integration times per point (see SI). After a Raman measurement, the Raman map area was imaged using SEM (Thermo Fisher Phenom ProX Desktop SEM or FEI Quanta 250) where the scratch markers acted as reference points to identify the area scanned with the Raman microscope. For the dye references, particles coated with dye-T28 oligos were deposited on a silicon surface, and point spectra were measured using a 532 or 633 nm laser (see SI for more details). For protein samples, the proteins were incubated with the nanoparticles to adsorb them to the particle surface and cause aggregation. Then the aggregates were deposited on a silicon surface, and point measurements were performed using the 633 nm laser and 300 μW (200 μW) laser power and 8 s (12 s) integration time for cyt c (HRP).
The same Witec Alpha 300 Raman microscope was used in the DF scattering experiments but with several changes: Zeiss Hal-100 Halogen lamp (100 W), Zeiss EX EPIplan-NEOFLUAR 100× BD objective (NA 0.9), and an analyzer were employed to produce the DF excitation and to measure the polarization-dependent scattering from the DONAs. A fresh Au DONA sample was prepared and deposited following the procedures outlined in previous sections. An initial DF scattering map was captured, and the sample was transferred to AFM to map the same area and to find the DONAs. Then the sample was switched back to the Raman microscope, and the position of the fiber in the field of view was solved by illuminating light from the spectrometer end and centering the internal marker on the fiber spot. The orientation of the analyzer (from −90° to +90°) in respect to the field of view was figured out before the experiment by using a linear polarizer placed in between the silicon reference sample and the objective: The silicon peak at 520 cm–1 was monitored, where the minimum and maximum in the peak signal indicated the perpendicular and parallel orientations of the analyzer with respect to the polarizer angle that was fixed to either a horizontal or vertical direction with respect to the field of view. Polarization-dependent spectra were then recorded by placing the internal marker on top of an individual DONA, setting the analyzer angle to ±90°, recording the spectra and the background next to the DONA, and rotating to the analyzer to the next angle. The analyzer was turned from one extreme to the other initially in 30° steps. Then extra spectra were recorded close to the DONA gap mode orientation, which was identified from the AFM images and from the spectral behavior (red shifting of the spectrum). After measuring several DONA in such fashion, the sample was removed, a mirror surface was placed to the microscope, and the lamp spectrum and the dark current were recorded. The final DF scattering spectra were calculated by subtracting the background (IBG) from the DONA spectra (IDONA) and dividing it by the dark current (IDC) subtracted lamp spectrum (ILAMP): I = (IDONA– IBG)/(ILamp– IDC).
FDTD Simulations
A detailed overview of the FDTD simulations can be found in the SI. In brief, we used the Lumerical FDTD Solutions software (v 8.19.1584). The model included two gold or silver spheres (refractive index from Johnson and Christy)60 surrounded by 1.12 nm DNA layer (refractive index 1.7)25 and on top of a silicon substrate (refractive index from61) with 3 nm silicon dioxide (refractive index 1.44) on top of the Si. The DNA layer thickness was measured using AFM (Figures S30 and S31). The model is presented in Figure S32. Here, we are omitting the bridge and the DNA origami nanofork from the framework. The medium was defined as air. The distance between gold spheres was varied according to the measured distance from Figure 1 (from 1.2 to 3.5 nm), where a water layer was added between DNA layers in the case of 2.5 and 3 nm particle-to-particle distances. The electric field distributions were calculated, and the field maximum around the gap region was recorded. To solve the E0 case, we removed the DNA layers and the metal spheres. Electric field polarizations along the gap (the gap mode) and perpendicular to gap (the off-gap mode) were considered. The results show that the electric field is highly localized within the gap region in the case of the gap mode and the off-gap mode excitation results mainly in plasmonic mode that resembled single particle dipole excitation (Figures S33–S37). The volumes of the 1.2 and 3 nm gap size hot spots were calculated from Figure S38.
DF Scattering Simulations
The DF scattering simulations are based on the same model, but the excitation is fixed to 64° in respect to surface normal and the azimuthal angle is fixed to 90° (incident light is perpendicular to the gap axis, i.e., the gap mode). Since the excitation light is unpolarized, the electric field polarization angles of 0° (perpendicular to the gap axis), 45°, and 90° (parallel to the gap axis) angle were considered. For the 60 nm AuNP dimer, the gap distance was varied between 1.2 and 3.5 nm, and the corresponding scattering spectra were recorded between 450 and 750 nm. For the fine-tuned model, we measured the dimensions of an individual DONA from the SEM and AFM images assuming a roughly ellipsoidal shape (72 nm × 53 nm × 64.5 nm and 75 nm × 65 nm × 72 nm) and calculated the scattering spectra similarly as before. In the case of the height, the particle + the DNA layer size was set out to be the same as the height in the AFM image, while keeping still the DNA layer thickness as 1.12 nm.
OxDNA Simulations
We performed Langevin molecular dynamics simulations for the three nanofork structures considered (90 nt, 120 nt, and no bridge) using the coarse-grained DNA model oxDNA2,16,17 parametrized to reproduce mechanical and thermodynamic experimental properties of DNA. The initial oxDNA configurations were obtained converting the caDNAno files with the tacoxDNA package.62 The caDNAno model of the nanofork is shown in Figure S39. Simulations were carried out with the LAMMPS software implementation.63 The temperature was set to T = 300 K and the monovalent salt concentration to 1 M. Each system was simulated for about 2 × 106 simulation time units, for a total of 2000 configurations sampled.
Acknowledgments
We thank Sibylle Rüstig for assisting in TEM imaging. This research was supported by the European Research Council (ERC; consolidator grant no. 772752).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.1c00188.
The caDNAno design and the schematic view of the DNA nanofork origami (Figures S1 and S39), extra TEM images of Au DONAs and nanoforks (Figures S2 and S29), AFM and SEM images of Au DONA monomer and DNA nanoforks (Figures S3–S7), correlated RAMAN-SEM maps of Au/Ag DONAs containing the studied dyes and proteins (Figures S8–S11 and S24–S25), reference Raman and UV–vis spectra for the dyes (Figures S12–S14), average spectra of dyes and control DONAs (Figure S15), correlated RAMAN-SEM maps of the different Ag/Au DONA control samples (Figures S16–17 and S26–S27), and the list of the used DNA sequences (Tables S1 and S2). Detailed description for the combined DF and Raman time series measurement (Figures S18–S22), protein coupling schemes (Figure S23), examples of Au and Ag DONA gels (Figure S28), evaluation of the DNA coating layer of used nanoparticles (Figures S30 and S31), as well as the simulations of the plasmonic dimer structures (Figures S32–S38) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Claridge S. A.; Schwartz J. J.; Weiss P. S. Electrons, Photons, and Force: Quantitative Single-Molecule Measurements from Physics to Biology. ACS Nano 2011, 5 (2), 693–729. 10.1021/nn103298x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moerner W. E.; Fromm D. P. Methods of Single-Molecule Fluorescence Spectroscopy and Microscopy. Rev. Sci. Instrum. 2003, 74 (8), 3597. 10.1063/1.1589587. [DOI] [Google Scholar]
- Pavliček N.; Gross L. Generation, Manipulation and Characterization of Molecules by Atomic Force Microscopy. Nat. Rev. Chem. 2017, 1 (1), 0005. 10.1038/s41570-016-0005. [DOI] [Google Scholar]
- Zrimsek A. B.; Chiang N.; Mattei M.; Zaleski S.; McAnally M. O.; Chapman C. T.; Henry A.-I.; Schatz G. C.; van Duyne R. P. Single-Molecule Chemistry with Surface- and Tip-Enhanced Raman Spectroscopy. Chem. Rev. 2017, 117 (11), 7583–7613. 10.1021/acs.chemrev.6b00552. [DOI] [PubMed] [Google Scholar]
- Pilot R.; Signorini R.; Durante C.; Orian L.; Bhamidipati M.; Fabris L. A Review on Surface-Enhanced Raman Scattering. Biosensors 2019, 9 (2), 57. 10.3390/bios9020057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer J.; Jimenez de Aberasturi D.; Aizpurua J.; Alvarez-Puebla R. A.; Auguié B.; Baumberg J. J.; Bazan G. C.; Bell S. E. J.; Boisen A.; Brolo A. G.; Choo J.; Cialla-May D.; Deckert V.; Fabris L.; Faulds K.; García de Abajo F. J.; Goodacre R.; Graham D.; Haes A. J.; Haynes C. L.; et al. Present and Future of Surface-Enhanced Raman Scattering. ACS Nano 2020, 14 (1), 28–117. 10.1021/acsnano.9b04224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. M.; Jin S. M.; Kim H. M.; Suh Y. D. Single-Molecule Surface-Enhanced Raman Spectroscopy: A Perspective on the Current Status. Phys. Chem. Chem. Phys. 2013, 15 (15), 5276–5287. 10.1039/c3cp44463e. [DOI] [PubMed] [Google Scholar]
- Le Ru E. C.; Etchegoin P. G. Single-Molecule Surface-Enhanced Raman Spectroscopy. Annu. Rev. Phys. Chem. 2012, 63, 65–87. 10.1146/annurev-physchem-032511-143757. [DOI] [PubMed] [Google Scholar]
- Lee J.-H.; Nam J.-M.; Jeon K.-S.; Lim D.-K.; Kim H.; Kwon S.; Lee H.; Suh Y. D. Tuning and Maximizing the Single-Molecule Surface-Enhanced Raman Scattering from DNA-Tethered Nanodumbbells. ACS Nano 2012, 6 (11), 9574–9584. 10.1021/nn3028216. [DOI] [PubMed] [Google Scholar]
- Rothemund P. W. K. Folding DNA to Create Nanoscale Shapes and Patterns. Nature 2006, 440 (7082), 297–302. 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]
- Dietz H.; Douglas S. M.; Shih W. M. Folding DNA into Twisted and Curved Nanoscale Shapes. Science 2009, 325 (5941), 725–730. 10.1126/science.1174251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenbauer K. F.; Sigl C.; Dietz H. Gigadalton-Scale Shape-Programmable DNA Assemblies. Nature 2017, 552 (7683), 78–83. 10.1038/nature24651. [DOI] [PubMed] [Google Scholar]
- Douglas S. M.; Marblestone A. H.; Teerapittayanon S.; Vazquez A.; Church G. M.; Shih W. M. Rapid Prototyping of 3D DNA-Origami Shapes with caDNAno. Nucleic Acids Res. 2009, 37 (15), 5001–5006. 10.1093/nar/gkp436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro C. E.; Kilchherr F.; Kim D.-N.; Shiao E. L.; Wauer T.; Wortmann P.; Bathe M.; Dietz H. A Primer to Scaffolded DNA Origami. Nat. Methods 2011, 8 (3), 221–229. 10.1038/nmeth.1570. [DOI] [PubMed] [Google Scholar]
- Kim D.-N.; Kilchherr F.; Dietz H.; Bathe M. Quantitative Prediction of 3D Solution Shape and Flexibility of Nucleic Acid Nanostructures. Nucleic Acids Res. 2012, 40 (7), 2862–2868. 10.1093/nar/gkr1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouldridge T. E.; Louis A. A.; Doye J. P. K. Structural, Mechanical, and Thermodynamic Properties of a Coarse-Grained DNA Model. J. Chem. Phys. 2011, 134 (8), 85101. 10.1063/1.3552946. [DOI] [PubMed] [Google Scholar]
- Snodin B. E. K.; Randisi F.; Mosayebi M.; Šulc P.; Schreck J. S.; Romano F.; Ouldridge T. E.; Tsukanov R.; Nir E.; Louis A. A.; Doye J. P. K. Introducing Improved Structural Properties and Salt Dependence into a Coarse-Grained Model of Dna. J. Chem. Phys. 2015, 142 (23), 234901. 10.1063/1.4921957. [DOI] [PubMed] [Google Scholar]
- Tapio K.; Bald I. The Potential of DNA Origami to Build Multifunctional Materials. Multifunct. Mater. 2020, 3 (3), 032001. 10.1088/2399-7532/ab80d5. [DOI] [Google Scholar]
- Prinz J.; Schreiber B.; Olejko L.; Oertel J.; Rackwitz J.; Keller A.; Bald I. DNA Origami Substrates for Highly Sensitive Surface-Enhanced Raman Scattering. J. Phys. Chem. Lett. 2013, 4 (23), 4140–4145. 10.1021/jz402076b. [DOI] [Google Scholar]
- Prinz J.; Heck C.; Ellerik L.; Merk V.; Bald I. DNA Origami Based Au-Ag-Core-Shell Nanoparticle Dimers with Single-Molecule SERS Sensitivity. Nanoscale 2016, 8 (10), 5612–5620. 10.1039/C5NR08674D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck C.; Prinz J.; Dathe A.; Merk V.; Stranik O.; Fritzsche W.; Kneipp J.; Bald I. Gold Nanolenses Self-Assembled by DNA Origami. ACS Photonics 2017, 4 (5), 1123–1130. 10.1021/acsphotonics.6b00946. [DOI] [Google Scholar]
- Heck C.; Kanehira Y.; Kneipp J.; Bald I. Placement of Single Proteins within the SERS Hot Spots of Self-Assembled Silver Nanolenses. Angew. Chem., Int. Ed. 2018, 57 (25), 7444–7447. 10.1002/anie.201801748. [DOI] [PubMed] [Google Scholar]
- Liu N.; Liedl T. Dna-Assembled Advanced Plasmonic Architectures. Chem. Rev. 2018, 118 (6), 3032–3053. 10.1021/acs.chemrev.7b00225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilo-Pais M.; Watson A.; Demers S.; LaBean T. H.; Finkelstein G. Surface-Enhanced Raman Scattering Plasmonic Enhancement Using DNA Origami-Based Complex Metallic Nanostructures. Nano Lett. 2014, 14 (4), 2099–2104. 10.1021/nl5003069. [DOI] [PubMed] [Google Scholar]
- Thacker V. V.; Herrmann L. O.; Sigle D. O.; Zhang T.; Liedl T.; Baumberg J. J.; Keyser U. F. DNA Origami Based Assembly of Gold Nanoparticle Dimers for Surface-Enhanced Raman Scattering. Nat. Commun. 2014, 5, 3448. 10.1038/ncomms4448. [DOI] [PubMed] [Google Scholar]
- Simoncelli S.; Roller E.-M.; Urban P.; Schreiber R.; Turberfield A. J.; Liedl T.; Lohmüller T. Quantitative Single-Molecule Surface-Enhanced Raman Scattering by Optothermal Tuning of DNA Origami-Assembled Plasmonic Nanoantennas. ACS Nano 2016, 10 (11), 9809–9815. 10.1021/acsnano.6b05276. [DOI] [PubMed] [Google Scholar]
- Tanwar S.; Haldar K. K.; Sen T. DNA Origami Directed Au Nanostar Dimers for Single-Molecule Surface-Enhanced Raman Scattering. J. Am. Chem. Soc. 2017, 139 (48), 17639–17648. 10.1021/jacs.7b10410. [DOI] [PubMed] [Google Scholar]
- Zhan P.; Wen T.; Wang Z.-G.; He Y.; Shi J.; Wang T.; Liu X.; Lu G.; Ding B. DNA Origami Directed Assembly of Gold Bowtie Nanoantennas for Single-Molecule Surface-Enhanced Raman Scattering. Angew. Chem., Int. Ed. 2018, 57 (11), 2846–2850. 10.1002/anie.201712749. [DOI] [PubMed] [Google Scholar]
- Acuna G. P.; Möller F. M.; Holzmeister P.; Beater S.; Lalkens B.; Tinnefeld P. Fluorescence Enhancement at Docking Sites of DNA-Directed Self-Assembled Nanoantennas. Science 2012, 338 (6106), 506–510. 10.1126/science.1228638. [DOI] [PubMed] [Google Scholar]
- Puchkova A.; Vietz C.; Pibiri E.; Wünsch B.; Sanz Paz M.; Acuna G. P.; Tinnefeld P. DNA Origami Nanoantennas with over 5000-Fold Fluorescence Enhancement and Single-Molecule Detection at 25 Mm. Nano Lett. 2015, 15 (12), 8354–8359. 10.1021/acs.nanolett.5b04045. [DOI] [PubMed] [Google Scholar]
- Prinz J.; Matković A.; Pešić J.; Gajić R.; Bald I. Hybrid Structures for Surface-Enhanced Raman Scattering: DNA Origami/Gold Nanoparticle Dimer/Graphene. Small 2016, 12 (39), 5458–5467. 10.1002/smll.201601908. [DOI] [PubMed] [Google Scholar]
- Weller L.; Thacker V. V.; Herrmann L. O.; Hemmig E. A.; Lombardi A.; Keyser U. F.; Baumberg J. J. Gap-Dependent Coupling of Ag–Au Nanoparticle Heterodimers Using DNA Origami-Based Self-Assembly. ACS Photonics 2016, 3 (9), 1589–1595. 10.1021/acsphotonics.6b00062. [DOI] [Google Scholar]
- Kühler P.; Roller E.-M.; Schreiber R.; Liedl T.; Lohmüller T.; Feldmann J. Plasmonic DNA-Origami Nanoantennas for Surface-Enhanced Raman Spectroscopy. Nano Lett. 2014, 14 (5), 2914–2919. 10.1021/nl5009635. [DOI] [PubMed] [Google Scholar]
- Chikkaraddy R.; Turek V. A.; Lin Q.; Griffiths J.; Nijs B.; Keyser U. F.; Baumberg J. J. Dynamics of Deterministically Positioned Single-Bond Surface-Enhanced Raman Scattering from DNA Origami Assembled in Plasmonic Nanogaps. J. Raman Spectrosc. 2020, 205, 291. 10.1002/jrs.5997. [DOI] [Google Scholar]
- Chikkaraddy R.; Turek V. A.; Kongsuwan N.; Benz F.; Carnegie C.; van de Goor T.; de Nijs B.; Demetriadou A.; Hess O.; Keyser U. F.; Baumberg J. J. Mapping Nanoscale Hotspots with Single-Molecule Emitters Assembled into Plasmonic Nanocavities Using DNA Origami. Nano Lett. 2018, 18 (1), 405–411. 10.1021/acs.nanolett.7b04283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang W.; Jia S.; Chao J.; Wang L.; Duan X.; Liu H.; Li Q.; Zuo X.; Wang L.; Wang L.; Liu N.; Fan C. Quantizing Single-Molecule Surface-Enhanced Raman Scattering with DNA Origami Metamolecules. Sci. Adv. 2019, 5 (9), eaau4506. 10.1126/sciadv.aau4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding B.; Deng Z.; Yan H.; Cabrini S.; Zuckermann R. N.; Bokor J. Gold Nanoparticle Self-Similar Chain Structure Organized by DNA Origami. J. Am. Chem. Soc. 2010, 132 (10), 3248–3249. 10.1021/ja9101198. [DOI] [PubMed] [Google Scholar]
- Shen X.; Asenjo-Garcia A.; Liu Q.; Jiang Q.; García de Abajo F. J.; Liu N.; Ding B. Three-Dimensional Plasmonic Chiral Tetramers Assembled by DNA Origami. Nano Lett. 2013, 13 (5), 2128–2133. 10.1021/nl400538y. [DOI] [PubMed] [Google Scholar]
- Fischer S.; Hartl C.; Frank K.; Rädler J. O.; Liedl T.; Nickel B. Shape and Interhelical Spacing of DNA Origami Nanostructures Studied by Small-Angle X-Ray Scattering. Nano Lett. 2016, 16 (7), 4282–4287. 10.1021/acs.nanolett.6b01335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S.-Y.; You E.-M.; Tian Z.-Q.; Moskovits M. Electromagnetic Theories of Surface-Enhanced Raman Spectroscopy. Chem. Soc. Rev. 2017, 46 (13), 4042–4076. 10.1039/C7CS00238F. [DOI] [PubMed] [Google Scholar]
- McNay G.; Eustace D.; Smith W. E.; Faulds K.; Graham D. Surface-Enhanced Raman Scattering (SERS) and Surface-Enhanced Resonance Raman Scattering (SERRS): A Review of Applications. Appl. Spectrosc. 2011, 65 (8), 825–837. 10.1366/11-06365. [DOI] [PubMed] [Google Scholar]
- Guerrini L.; Krpetić Ž.; van Lierop D.; Alvarez-Puebla R. A.; Graham D. Direct Surface-Enhanced Raman Scattering Analysis of DNA Duplexes. Angew. Chem., Int. Ed. 2015, 54 (4), 1144–1148. 10.1002/anie.201408558. [DOI] [PubMed] [Google Scholar]
- Salmon A. R.; Kleemann M.-E.; Huang J.; Deacon W. M; Carnegie C.; Kamp M.; de Nijs B.; Demetriadou A.; Baumberg J. J. Light-Induced Coalescence of Plasmonic Dimers and Clusters. ACS Nano 2020, 14 (4), 4982–4987. 10.1021/acsnano.0c01213. [DOI] [PubMed] [Google Scholar]
- Huschka R.; Zuloaga J.; Knight M. W.; Brown L. V.; Nordlander P.; Halas N. J. Light-Induced Release of DNA from Gold Nanoparticles: Nanoshells and Nanorods. J. Am. Chem. Soc. 2011, 133 (31), 12247–12255. 10.1021/ja204578e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh T.; Yamamoto Y. S. Recent Topics on Single-Molecule Fluctuation Analysis Using Blinking in Surface-Enhanced Resonance Raman Scattering: Clarification by the Electromagnetic Mechanism. Analyst 2016, 141 (17), 5000–5009. 10.1039/C6AN00936K. [DOI] [PubMed] [Google Scholar]
- Kitahama Y.; Nagahiro T.; Tanaka Y.; Itoh T.; Ozaki Y. Analysis of Blinking from Multicoloured SERS-Active Ag Colloidal Nanoaggregates with Poly-L-Lysine via Truncated Power Law. J. Raman Spectrosc. 2017, 48 (4), 570–577. 10.1002/jrs.5079. [DOI] [Google Scholar]
- Lombardi J. R.; Birke R. L.; Haran G. Single Molecule SERS Spectral Blinking and Vibronic Coupling. J. Phys. Chem. C 2011, 115 (11), 4540–4545. 10.1021/jp111345u. [DOI] [Google Scholar]
- Jaworska A.; Pyrak E.; Kudelski A. Comparison of the Efficiency of Generation of Raman Radiation by Various Raman Reporters Connected via DNA Linkers to Different Plasmonic Nano-Structures. Vib. Spectrosc. 2019, 101, 34–39. 10.1016/j.vibspec.2019.01.002. [DOI] [Google Scholar]
- Marr J. M.; Schultz Z. D. Imaging Electric Fields in SERS and TERS Using the Vibrational Stark Effect. J. Phys. Chem. Lett. 2013, 4 (19), 3268–3272. 10.1021/jz401551u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertini I.; Cavallaro G.; Rosato A. Cytochrome C: Occurrence and Functions. Chem. Rev. 2006, 106 (1), 90–115. 10.1021/cr050241v. [DOI] [PubMed] [Google Scholar]
- Veitch N. C. Horseradish Peroxidase: A Modern View of a Classic Enzyme. Phytochemistry 2004, 65 (3), 249–259. 10.1016/j.phytochem.2003.10.022. [DOI] [PubMed] [Google Scholar]
- Rygula A.; Majzner K.; Marzec K. M.; Kaczor A.; Pilarczyk M.; Baranska M. Raman Spectroscopy of Proteins: A Review. J. Raman Spectrosc. 2013, 44 (8), 1061–1076. 10.1002/jrs.4335. [DOI] [Google Scholar]
- Yamamoto H.; Liu H.; Waldeck D. H. Immobilization of Cytochrome C at Au Electrodes by Association of a Pyridine Terminated SAM and the Heme of Cytochrome. Chem. Commun. 2001, 11, 1032–1033. 10.1039/b101767p. [DOI] [Google Scholar]
- Mirkin N.; Jakoncic J.; Stojanoff V.; Moreno A.. Crystal Structure of Cytochrome C from Bovine Heart at 1.5 A Resolution. 10.2210/pdb2B4Z/pdb (accessed 12.2.2020). [DOI]
- Berglund G. I.; Carlsson G. H.; Hajdu J.; Smith A. T.; Szoke H.; Henriksen A.. Structure of Horseradish Peroxidase C1A Compound I. 10.2210/pdb1HCH/pdb (accessed 18.9.2019). [DOI]
- Kitahama Y.; Ozaki Y. Surface-Enhanced Resonance Raman Scattering of Hemoproteins and Those in Complicated Biological Systems. Analyst 2016, 141 (17), 5020–5036. 10.1039/C6AN01009A. [DOI] [PubMed] [Google Scholar]
- Yeo B.-S.; Mädler S.; Schmid T.; Zhang W.; Zenobi R. Tip-Enhanced Raman Spectroscopy Can See More: The Case of Cytochrome c. J. Phys. Chem. C 2008, 112 (13), 4867–4873. 10.1021/jp709799m. [DOI] [Google Scholar]
- Ramezani H.; Dietz H. Building Machines with DNA Molecules. Nat. Rev. Genet. 2020, 21 (1), 5–26. 10.1038/s41576-019-0175-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Servos M. R.; Liu J. Instantaneous and Quantitative Functionalization of Gold Nanoparticles with Thiolated DNA Using a Ph-Assisted and Surfactant-Free Route. J. Am. Chem. Soc. 2012, 134 (17), 7266–7269. 10.1021/ja3014055. [DOI] [PubMed] [Google Scholar]
- Johnson P. B.; Christy R. W. Optical Constants of the Noble Metals. Phys. Rev. B 1972, 6 (12), 4370–4379. 10.1103/PhysRevB.6.4370. [DOI] [Google Scholar]
- Pierce D. T.; Spicer W. E. Electronic Structure of Amorphous Si from Photoemission and Optical Studies. Phys. Rev. B 1972, 5 (8), 3017–3029. 10.1103/PhysRevB.5.3017. [DOI] [Google Scholar]
- Suma A.; Poppleton E.; Matthies M.; Šulc P.; Romano F.; Louis A. A.; Doye J. P. K.; Micheletti C.; Rovigatti L. TacoxDNA: A User-Friendly Web Server for Simulations of Complex DNA Structures, from Single Strands to Origami. J. Comput. Chem. 2019, 40 (29), 2586–2595. 10.1002/jcc.26029. [DOI] [PubMed] [Google Scholar]
- Henrich O.; Gutiérrez Fosado Y. A.; Curk T.; Ouldridge T. E. Coarse-Grained Simulation of DNA Using LAMMPS: An Implementation of the OxDNA Model and Its Applications. Eur. Phys. J. E: Soft Matter Biol. Phys. 2018, 41 (5), 57. 10.1140/epje/i2018-11669-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.