

Abstract

Ideal controlled pulmonary drug delivery systems provide sustained release by retarding lung clearance mechanisms and efficient lung deposition to maintain therapeutic concentrations over prolonged time. Here, we use atomic layer deposition (ALD) to simultaneously tailor the release and aerosolization properties of inhaled drug particles without the need for lactose carrier. In particular, we deposit uniform nanoscale oxide ceramic films, such as Al2O3, TiO2, and SiO2, on micronized budesonide particles, a common active pharmaceutical ingredient for the treatment of respiratory diseases. In vitro dissolution and ex vivo isolated perfused rat lung tests demonstrate dramatically slowed release with increasing nanofilm thickness, regardless of the nature of the material. Ex situ transmission electron microscopy at various stages during dissolution unravels mostly intact nanofilms, suggesting that the release mechanism mainly involves the transport of dissolution media through the ALD films. Furthermore, in vitro aerosolization testing by fast screening impactor shows a ∼2-fold increase in fine particle fraction (FPF) for each ALD-coated budesonide formulation after 10 ALD process cycles, also applying very low patient inspiratory pressures. The higher FPFs after the ALD process are attributed to the reduction in the interparticle force arising from the ceramic surfaces, as evidenced by atomic force microscopy measurements. Finally, cell viability, cytokine release, and tissue morphology analyses verify a safe and efficacious use of ALD-coated budesonide particles at the cellular level. Therefore, surface nanoengineering by ALD is highly promising in providing the next generation of inhaled formulations with tailored characteristics of drug release and lung deposition, thereby enhancing controlled pulmonary delivery opportunities.
Keywords: atomic layer deposition, inhalation, controlled release, isolated perfused rat lung, particle-to-cell deposition, dry powder inhaler, budesonide
Introduction
Drug delivery by inhalation offers a targeted therapy for lung diseases such as asthma and chronic obstructive pulmonary disease (COPD).1 Moreover, by providing higher bioavailability than oral and parenteral routes for several small-molecule therapeutics, it has emerged as a route of administration for the treatment of systemic diseases such as diabetes mellitus.2 The large absorptive surface area of the lungs arising from the large number of alveoli—ranging from 200 to 600 million—coupled with the thin alveolar–vascular permeable layer allows the efficient deliver of drugs to the bloodstream.3 Hence, besides enabling direct access to the site of action for lung diseases, pulmonary delivery has the potential to ensure a rapid onset of action for systemic diseases.4 This may allow for a lower dose than oral administration or injection. However, the rapid absorption of inhaled drugs from the lungs may limit their local therapeutic effect, as they can be quickly cleared from the lung tissue.5 Frequent drug doses may thus be needed to maintain their local concentration in the lung tissue within the therapeutic window, often leading to poor patient compliance with the therapeutic regime. Furthermore, the clearance of inhaled particles is determined by their initial deposition pattern in the lungs, which depends on the physical and chemical properties of the aerosol such as particle interactions and aerodynamic particle size distribution as well as on physiological factors such as airflow and breathing patterns.6 Therefore, controlled pulmonary drug delivery in terms of lung deposition, dissolution, and absorption is highly desirable for the treatment of chronic diseases, as a reduction in dosing frequency and optimization in powder load may be obtained.7
A number of devices to generate respirable drug-containing particles or droplets have been developed for inhaled delivery, such as nebulizers, pressurized metered dose inhalers (pMDIs), and dry powder inhalers (DPIs).8,9 Both nebulizers and pMDIs deliver drugs in solution or suspension aerosols. In particular, the drug is dissolved or suspended in water for nebulizers and in a nonpolar volatile propellant for pMDIs. Nebulizers are not often used for the therapy of chronic diseases, as they are less portable and noisy and necessitate continuous inhalations by the patient over an extended period of time. pMDIs make use of greenhouse gases, e.g., hydrofluoroalkanes, as propellants, leading to high-velocity dose emission arising from the pressurization, which requires patients to coordinate the “press and breath” maneuver; otherwise the drug may wrongly deposit in the oropharynx. While pMDIs are designed for low drug loads, DPIs can deliver much higher drug loads. However, as DPIs do not use any propellant, it is required by the patient to generate the inspiratory force needed to aerosolize the powder. Moreover, compared to nebulizers, DPIs often have superior chemical stability and are seen as much easier and faster to use. Yet, due to the strong cohesive forces of the small drug particles relevant for inhalation, the dry drug powder in DPIs is often blended with carrier excipient particles, usually lactose, to reduce drug cohesiveness and improve aerosolization.10 However, the addition of carrier particles increases the total inhaled powder mass with a drug-to-carrier ratio typically not exceeding 1:90, causing decreased drug delivery efficiency.11,12
Substantial efforts have been devoted to design carrier-free DPI formulations using various particle engineering techniques such as controlled agglomeration, referred to as spheronization, of micronized particles to form soft aggregates, i.e., spheroids, and physical vapor deposition or spray drying for encapsulation of inhalable particles by polymers, lipids, or amino acids.13 At the same time, such approaches can modify the drug release and enhance the aerodynamic properties of the powder.7 For instance, large porous particles made of poly(lactic-co-glycolic acid) (PLGA) embedding the drug have been prepared to ease particle dispersion and diminish drug clearance due to their large geometric diameter by escaping phagocytosis by alveolar macrophages.14−16 Moreover, conjugation of drugs to polyethylene glycol, i.e., PEGylation, can prolong the retention of small inhaled drugs in the lungs.17 Liposomes, which consist of artificial vesicles formed of lipid bilayers, encapsulating the drug may instead modulate the release by adjusting the lipid composition:18 for example, rigid phospholipids or cholesterol reduces the fluidity and thus the permeability of the liposome membrane.19 Still, such formulations can only offer limited drug loadings, up to at best 50%, which lowers the amount of drug delivered for the same fine particle fraction.7,16,20 In an attempt to increase the drug loading of traditional sustained-release formulations, thin coatings of PLGA on micronized budesonide particles have been fabricated by pulsed laser deposition (PLD).21,22 PLGA-coated budesonide showed a biexponential dissolution profile with an initial burst of free drug around 30–40%, likely arising from the lack of coating uniformity and conformality, followed by a slower release with increasing coating amounts.21,22 PLD as well as conventional physical and chemical vapor deposition can in fact fail to uniformly and conformally coat irregularly shaped structures such as inhaled drug particles and, moreover, operate at high vacuum conditions.23−25 Currently, there is still no established, scalable, and cost-effective technology to develop controlled inhaled delivery systems, which provide sustained release combined with enhanced aerosolization, while retaining high drug loadings.26,27
Atomic layer deposition (ALD) is a vapor-phase technique for depositing ultrathin conformal films on any substrate with thickness control at the sub-nanometer level.28,29 Alternating exposures of a precursor and a co-reactant in the vapor phase separated by purging steps enable self-terminating reactions with the substrate surface, thus resulting in an atomically controlled film growth. Widely used for the functionalization of wafers in the semiconductor industry with oxide ceramic films, ALD has also been applied to a large variety of powders, recently also including pharmaceutical particles, due to its ability to conformally coat complex three-dimensional substrates.30−37 Crucially, nanophase ceramics have already been used in a broad spectrum of biomedical and drug delivery applications, as they can modulate drug release kinetics, incorporate multifunctional molecules, and target action sites.38 ALD of oxide ceramics can therefore extend release and improve flow properties and solid-state stability of virtually any pharmaceutical substance.32−36,39 Nanoscale oxide ceramic ALD films have been shown to slow down the dissolution rate of acetaminophen particles, while preventing drug chemical degradation and cytotoxicity.34 Similarly, we have demonstrated that nanosized Al2O3-based films grown via ALD in a fluidized bed reactor at nearly ambient conditions can greatly sustain the release and improve the dispersibility of budesonide and lactose particles.32,36 In addition, Al2O3 ALD films with a thickness of 30–35 nm have been proven to maintain a stable plasma concentration for indomethacin, when administered subcutaneously in rats, up to over 12 weeks.35 However, neither in vitro nor ex vivo biorelevant studies of ALD-coated inhaled drugs have been reported yet. Moreover, the effect of the ALD films on drug dissolution and other functional properties such as flowability is not well understood.35,36,39,40
In this work, we use ALD to simultaneously prolong release and improve aerosolization of budesonide dry particles, a corticosteroid for the treatment of various respiratory diseases including asthma and COPD, while avoiding the addition of carriers in the final formulation. The surface of micronized budesonide was engineered by SiO2, TiO2, and Al2O3 nanoscale films. The ALD process is carried out at 40 °C in an atmospheric-pressure fluidized bed reactor for a range of cycles from 10 to 50 for TiO2 and Al2O3 ALD and from 10 to 100 for SiO2 ALD. The film morphology, thickness, and amount were analyzed by transmission electron microscopy (TEM) and inductively coupled plasma optical emission spectrometry (ICP-OES). The dissolution of biorelevant (<5 μm) aerosol fractions of uncoated and ALD-coated budesonide collected after dispersion via a modified Andersen cascade impactor (mACI) was monitored by high-performance liquid chromatography and ex situ TEM to understand the particle–medium interaction. The drug absorption was quantified using the ex vivo isolated perfused rat lung (IPRL) model. Furthermore, the aerosolization performance was evaluated by a fast screening impactor collecting particles with an aerodynamic diameter smaller than 5 μm, which correspond to the fine particle fraction (FPF). To explain the FPF results, contact force measurements by atomic force microscopy (AFM) were carried out. Finally, safety and efficacy parameters of ALD-coated budesonide were assessed by measuring cell viability of human epithelial alveolar A549 cells as well as cytokine release and histology of primary human bronchial epithelial cells cultured at 3D.
Results and Discussion
Deposition of SiO2, TiO2, and Al2O3 Nanoscale Films on Micronized Budesonide
Gram-scale batches of micronized budesonide particles were engineered with nanoscale films by ALD in fluidized bed reactors. The latter are advantageous technologies due to their scale-up prospects31 and long-standing use in the pharmaceutical industry. The deposition of SiO2 was carried out up to 100 cycles, whereas that of TiO2 and Al2O3 up to 50 cycles. The morphology of the bare and ALD-coated budesonide particles was first investigated by TEM analysis. Conformal films were obtained on each individual particle for each deposition process. In SiO2 ALD, average film thicknesses of 1.5, 3, 6, and 10 nm were found after 10, 25, 50, and 100 cycles, respectively (see Figure 1a). A nearly linear evolution of the film thickness with the number of cycles was obtained (see Figure 1b), as according to the typical ALD growth. The growth per cycle (GPC), which corresponds to the slope of the fitting line of film thickness vs number of cycles, was thus ∼0.1 nm, in agreement with the 0.09–0.11 nm GPC reported for SiCl4/H2O ALD.41,42 In TiO2 ALD, the average film thicknesses were 3, 8, and 15 nm after 10, 25, and 50 cycles, which translate into a GPC of ∼0.3 nm (see Figure 1d,e). This GPC is slightly higher than that typically reported for TiCl4/H2O ALD, i.e., ∼0.1 nm,43 due to the operation at nearly ambient conditions, which makes the removal of unreacted H2O during the purging steps extremely challenging, thus resulting in additional growth by chemical vapor deposition (CVD) reactions. Instead, in Al2O3 ALD, 20, 35, and 50 nm thick films on average were observed after 10, 25, and 50 cycles, respectively (see Figure 1g,h). Such high thicknesses arise from the penetration of trimethylaluminum (TMA), the Al precursor used in the Al2O3 ALD process, which is known to infiltrate biomaterials and polymers, where organometallics such as TMA can prolongedly reside due to attractive chemical interactions with functional groups of the biomaterial.44,45 In this instance, TMA does not readily react on the budesonide surface and mostly diffuses into the budesonide outer core, where it is kinetically trapped.46 This results in subsurface growth and the formation of films consisting of an Al2O3–budesonide mixture at the near surface region of budesonide. During the first exposure of TMA, the precursor might display both noncovalent and covalent interaction with budesonide, due to the presence of two hydroxyl and carbonyl groups in its molecular structure. Upon exposure to the oxygen source, here O3, aluminum oxyhydroxide nuclei exhibiting hydroxyl-terminated surfaces are formed, with which TMA is more likely to react in the next cycles. In this process, also referred to as sequential infiltration synthesis, the extent of infiltration and thus the final film thickness are determined by the cumulative duration of exposure and partial pressure of the precursor vapor as well as of the following purging step, rather than solely by the number of cycles. Therefore, a GPC cannot be defined for Al2O3 ALD on budesonide. For simplification, the film thicknesses are assigned later on only to Al2O3, but it is worth pointing out they refer to the mixture of Al2O3–budesonide.
Figure 1.

Linear growth of SiO2, TiO2, and Al2O3 on micronized budesonide as a function of the ALD cycles. (a, d, g) TEM images of budesonide particles coated by (a) SiO2, (d) TiO2, and (g) Al2O3via ALD. The film thicknesses were measured by ImageJ. (b, e, h) Evolution of film thickness with the number of cycles for (b) SiO2, (e) TiO2, and (h) Al2O3 ALD. The error bars indicate 95% confidence intervals. (c, f, i) Evolution of the amount of deposited material, measured by ICP-OES and normalized with respect to the budesonide amount, with the number of cycles for (c) SiO2, (f) TiO2, and (i) Al2O3 ALD.
Moreover, the amount of deposited material after each ALD process was monitored as a function of the number of cycles by ICP-OES. This is crucial to estimate the drug loading in each ALD-processed budesonide formulation. Each deposited material loading essentially increases linearly with the number of cycles, which is consistent with the evolution of the film thicknesses (see Figure 1c,f,i). SiO2 loadings of 0.9, 2.8, 4.1, and 6.8 wt %, corresponding to budesonide loadings of 99.1, 97.2, 95.9, and 93.2 wt %, were obtained after 10, 25, 50, and 100 cycles, respectively (see Figure 1c). Instead, TiO2 loadings of 3.8, 10.7, and 21.1 wt %, equivalent to budesonide loadings of 96.2, 89.3, and 78.9 wt %, were found after 10, 25, and 50 cycles, respectively (see Figure 1f). The significantly lower loadings of SiO2 than TiO2 for comparable thickness are due to the lower density of SiO2, which is estimated to be 1.6 g/cm3,37 whereas the density of TiO2 after ALD at ∼40 °C is reported to be 3.3 g/cm3.47 The higher density of TiO2 is also evidenced by the darker films under TEM. Similarly to TiO2, Al2O3 loadings of 6.1, 14.4, and 18.7 wt %, corresponding to budesonide loadings of 93.9, 85.6, and 81.3 wt %, were measured after 10, 25, and 50 cycles, respectively (see Figure 1i). However, as shown previously, greatly higher thicknesses are observed after Al2O3 ALD for the same amount of deposited material, due to the infiltration of Al2O3 into the budesonide particles.
To further look into the morphology of the ALD films, elemental mappings were performed by energy dispersive X-ray spectroscopy (EDX) (see Figure 2). The maps of Si, Ti, Al, and related O confirm the uniform deposition of SiO2, TiO2, and Al2O3 on budesonide. In particular, the higher concentrations of Si, Ti, Al, and related O at the edges of the budesonide particles indicate the formation of core/shell structures. This is true even for Al2O3-coated budesonide, despite the penetration of Al2O3 into the outer core of the budesonide particles (see Figure 2c and Figure S1). To examine the crystallinity of the ALD-coated budesonide particles and the stability of their chemical structure upon the ALD process, X-ray powder diffraction (XRPD) analysis was carried out. The XRPD patterns before and after ALD do not display any difference, indicating the amorphous nature of the deposited films as well as the absence of changes in the crystal structure of budesonide (see Figure S2).
Figure 2.
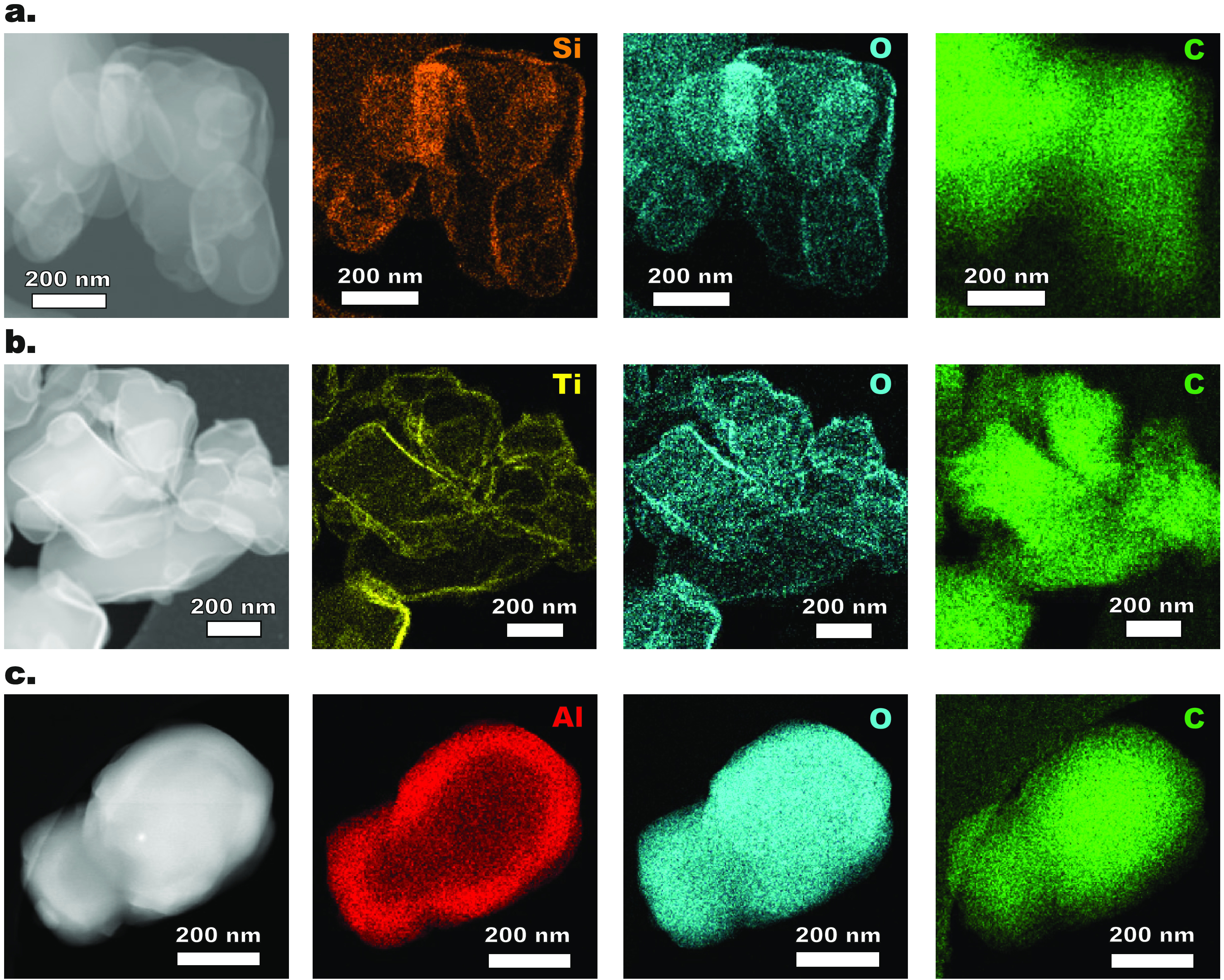
High-angle annular dark-field (HAADF) images and energy dispersive X-ray spectroscopy (EDX) mappings of (a) Si, O, and C in SiO2-coated budesonide after 100 cycles, (b) Ti, O, and C in TiO2-coated budesonide after 50 cycles, and (c) Al, O, and C in Al2O3-coated budesonide after 50 cycles.
Drug Release: In Vitro Dissolution and Ex Vivo Rat Lung Absorption
The first objective was to investigate the effect of the ALD films on the release of budesonide. Developing an in vitro dissolution test for inhaled formulations is challenging due to the complexity in reproducing the lung surfactants with the limited volume of fluid that lines the respiratory tract, approximately 10–20 mL/100 m2, and due to the competition between dissolution and mucociliary transport and uptake by macrophages. Moreover, the dissolution rate of inhaled formulations strongly depends on the physiological environment. Therefore, there are no regulatory requirements or established pharmacopeial techniques for the dissolution testing of inhaled drugs. However, as suggested by Velaga etal.,48 a dissolution method approaching biorelevance for orally inhaled products may still be obtained by using (i) a method for capturing a suitable fraction of aerosol, which needs to be nonagglomerated to mimic that delivered to the lungs by inhalation, (ii) a method for transferring the captured aerosol fraction to a dissolution system, (iii) a suitable dissolution media for dissolution under sink conditions, and (iv) a suitable dissolution apparatus, either large-volume systems with agitation or small-volume diffusion-controlled systems without agitation. In this work, in vitro dissolution of bare and ALD-coated budesonide dry powder was carried out in a stirred phosphate buffer solution at pH 6.8 with 0.5% sodium dodecyl sulfate (SDS) in a μDiss tube containing a 3D-printed holder for filters,49 which captured a biorelevant nonagglomerated aerosol fraction via an mACI.50 Scanning electron microscopy (SEM) and TEM images of the bare and ALD-coated budesonide particles after aerosolization with the mACI demonstrated their successful deagglomeration (see Figure S3). Interestingly, the agglomerate size of ALD-coated budesonide after mACI aerosolization was considerably smaller than bare budesonide, suggesting improved powder dispersibility after the ALD process, as we will show later. In any case, uniformly dispersed agglomerates with an open structure were found for both bare and ALD-coated budesonide. This is an essential requirement for ensuring an appropriate and immediate wetting of each individual particle at the initial stages of dissolution. To further verify the importance of the particle deagglomeration step, dissolution tests of different budesonide doses were performed with and without mACI aerosolization (see Figure S4). In case of no predispersion with the mACI, the dissolution profiles strongly depend on the budesonide dose. In particular, the dissolution rate significantly decreases with increasing budesonide doses. This arises from the presence of large closely packed agglomerates which prevent an adequate and prompt wetting, and thereby dissolution. The higher the budesonide dose, the higher the number of large agglomerates, the longer the wetting process, and the slower the dissolution process. Instead, no dose-dependent dissolution is observed for doses up to at least 120 μg when predispersing the powder with the mACI. Provided that sink conditions are fulfilled, the dissolution rate, having a first-order kinetics, must not depend on the drug dose. Therefore, using only the nonagglomerated aerosol fraction is crucial in dissolution testing of inhaled formulations.
After assessing the quality of our dissolution testing procedure with different doses of bare budesonide, we measured the dissolution rate of each ALD-coated budesonide formulation over 3 h (see Figures 3 and S5). Bare budesonide particles reach a cumulative release of 77% already after 30 min and 95% after 2 h (see Figure 3). Instead, the ALD-coated budesonide particles dissolve at a decreasing rate with increasing film thickness. In particular, SiO2-coated budesonide displays a cumulative release after 30 min of ∼70% with the 6 nm film and ∼55% with the 10 nm film (see Figure 3a and Figure 4a). TiO2-coated budesonide shows a cumulative release after 30 min of ∼72% with the 3 nm film, ∼64% with the 8 nm film, and ∼51% with the 15 nm film (see Figure 3b and Figure 4a). Furthermore, Al2O3-coated budesonide exhibits a cumulative release after 30 min of ∼45% with the 20 nm film, ∼33% with the 35 nm film, and ∼28% with the 50 nm film (see Figure 3c and Figure 4a). Interestingly, the dissolution rates are not significantly affected by the nature of the coating material or of the surface composition, as their profiles clearly scale with the film thickness. Moreover, it is worth noting that the dissolution curves are S-shaped, with an initial delay attributed to the time the solution takes to diffuse through the film. As suggested by Vogel etal.,40 we already hypothesized the mechanism of solution diffusion through the voids of the ALD film in the dissolution of Al2O3-coated lactose particles.36 To prove this release mechanism, the morphology of the ALD-coated budesonide particles was now assessed by ex situ TEM analysis after 5 and 30 min dissolution (see Figure 4d). In particular, TEM grids were positioned at the collection plate of the mACI for drug deposition, then placed in between two glass fiber filters and transferred to the dissolution setup. In agreement with the dissolution profile, the bare budesonide particles dissolve quickly, as shown by the presence of bubbles arising from the dissolution medium into the particles already after 5 min and by the absence of intact bare particles after 30 min. On the other hand, the ALD-coated budesonide particles remain substantially intact during the whole dissolution process. After 5 min, no bubble is observed inside the ALD-coated particles, indicating the absence of dissolution medium in the particles. This is consistent with the delay at the initial stages of dissolution due to the time the medium takes to diffuse. Instead, bubbles are found after 30 min of dissolution, demonstrating the diffusion of the medium through the ALD films, without any noticeable film erosion or degradation. This analysis suggests that the mechanism behind dissolution of the ALD-coated particles mainly involves the transport of dissolution medium through sub-nanoscale pores and defects, typically present in ALD films synthesized at ambient conditions. Following inward diffusion, the medium dissolves the budesonide core, which then diffuses out and is loaded into the micelles composed of SDS. Therefore, the dissolution mechanism is essentially diffusion-based for each ALD-coated budesonide formulation.
Figure 3.
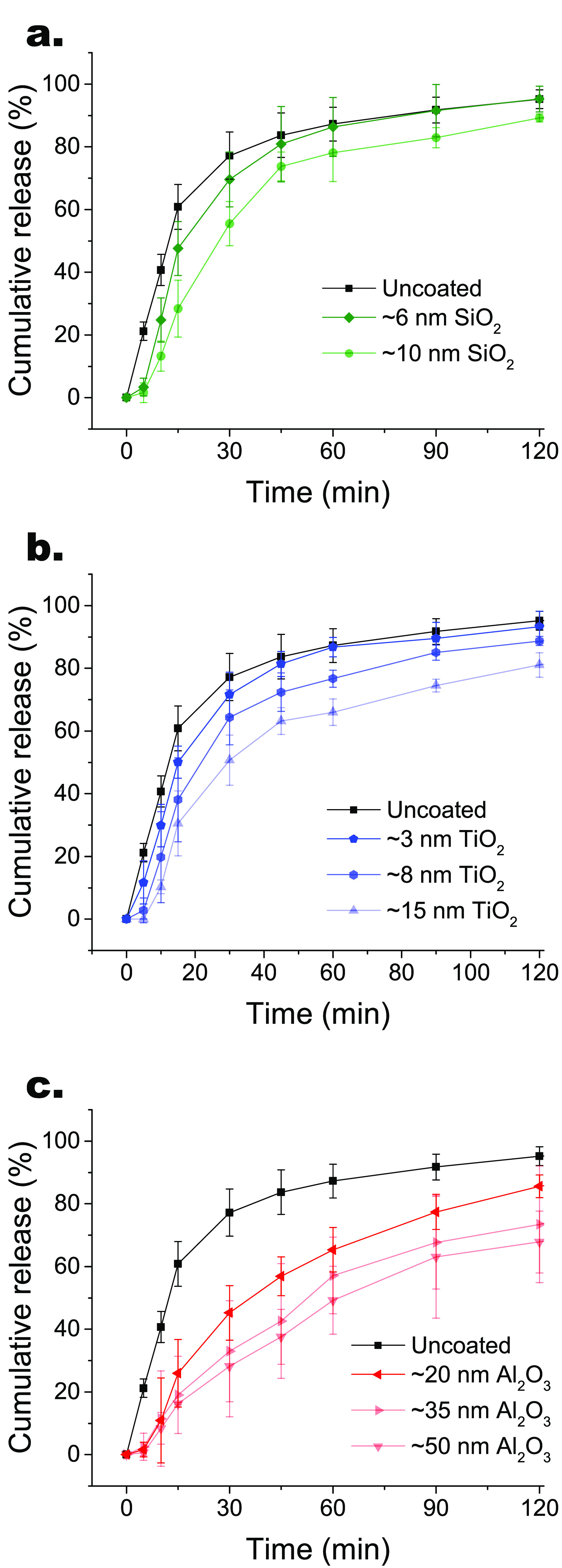
In vitro dissolution profiles of uncoated and ALD-coated budesonide after aerosolization using mACI, distinguished by ALD material: (a) SiO2, (b) TiO2, and (c) Al2O3. Increasing film thicknesses result in slower dissolution rates for each ALD material. The error bars indicate standard deviations.
Figure 4.

Drug release: in vitro dissolution and ex vivo IPRL absorption. (a) In vitro dissolution profiles of uncoated and ALD-coated budesonide after aerosolization using mACI. The error bars indicate standard deviations. (b) Ex vivo absorption profiles of uncoated and ALD-coated budesonide using isolated perfused rat lung (IPRL). The error bars indicate 95% confidence intervals. (c) Correlation of the time required for 50% drug release/absorption (t50) between in vitro and ex vivo. (d) TEM images of uncoated and ALD-coated budesonide after 5 and 30 min dissolution in sodium phosphate buffer. After 5 min bubbles from the medium are present only in the uncoated particles, and after 30 min they also appear in the ALD-coated particles. While after 30 min already a budesonide amount ranging from ∼30% to ∼55% is released from the ALD-coated particles, the films are still intact.
To study the absorption of the bare and ALD-coated budesonide formulations and thus obtain a more realistic correlation with in vivo performance, the ex vivo IPRL model was used.51 After 30 min, bare budesonide reaches a cumulative absorption of ∼60%, whereas 10-nm-SiO2-coated, 15-nm-TiO2-coated, and 20-nm-Al2O3-coated budesonide reach a cumulative absorption of ∼37% (see Figure 4b). Therefore, a significantly slowed absorption is observed for ALD-coated budesonide, in agreement with the slowed release in the in vitro dissolution tests. Moreover, the absorption profiles of ALD-coated budesonide were compared to that of the commercial budesonide suspension Pulmicort Respules, which is a very well-characterized reference (see Figure S6). A constant absorption rate over time is observed for both ALD-coated budesonide formulations compared to bare budesonide and Pulmicort Respules, whose profiles reach a maximum after 3 min and thereafter quickly drop. We then correlated the times required for 50% drug release in the in vitro dissolution test and drug absorption in the ex vivo IPRL test. As shown in Figure 4c, a linear correlation between t50 in vitro and ex vivo is found for both bare and ALD-coated budesonide. Even though more data points are required to increase the robustness of the correlation, this suggests that already the in vitro dissolution data offer the biorelevant capability to differentiate between rates of bare and ALD-coated budesonide formulations.
In Vitro Aerosolization
A high FPF is desired to increase the lung-deposited dose and thus reduce the total dose delivered to the patient, making more efficient use of the drug inhaled. In particular, a lower total dose may reduce at the same time drug cost and risks for side effects. The FPFs for both bare and ALD-coated budesonide formulations were therefore quantified by in vitro aerosolization measurements using the fast screening impactor, a two-stage abbreviated impactor collecting particles with an aerodynamic diameter smaller than 5 μm, at an air flow rate of 30 L/min, corresponding to a pressure drop of 0.8–1.0 kPa, i.e., a very low inspiratory pressure. The FPFs were measured both after long-term storage for 8–10 months at 20 °C and ∼0% RH and after storage under stressed conditions, i.e., open for 1 month at 40 °C and 75% RH (see Figure 5a). After long-term storage at 20 °C and ∼0% RH, an FPF of ∼30% is found for bare budesonide, whereas significantly higher FPF values ranging from ∼55% to ∼70% are obtained for 10-cycle-ALD-coated budesonide. In particular, a 2.3-fold increase in FPF is observed for ALD-coated budesonide after 10 SiO2 and Al2O3 cycles, whereas a 1.9-fold increase is observed for 10-cycle-TiO2-coated budesonide (see Figure S7a). The FPF values of each ALD-coated formulation tend to apparently decrease with increasing film thickness. However, this is attributed to the increase in powder agglomeration with increasing ALD cycles, rather than to a film thickness effect, as the surface morphology and composition are essentially unaltered. When increasing the number of ALD cycles from 10 to 50, the time the budesonide particles are subjected to mixing and collisions in the fluidized bed increases 5-fold, thus further promoting particle agglomeration, which in turn lowers FPF. Yet, even the FPFs of the most ALD-coated formulations remain higher than bare budesonide. It is worth noting that SiO2-coated budesonide seems to be the least affected by the increase in ALD cycles, as shown by the FPF evolution, suggesting that the combination of SiCl4/H2O precursors performs best in reducing powder agglomeration. After open storage for 1 month at 40 °C and 75% RH, bare budesonide exhibits an FPF of ∼45%, higher than the original value of ∼30%. This is attributed to surface relaxation promoted by the higher temperatures and relative humidities.52 The FPF of each SiO2-coated budesonide formulation stays essentially constant after stress testing (see also Figure S7c). A similar behavior is observed for Al2O3-coated budesonide, but only up to 25 cycles, as the 50-cycle-Al2O3-coated sample benefits from surface relaxation, which makes the FPF still higher than bare budesonide. Instead, TiO2-coated budesonide formulations show greatly higher FPFs after storage under stressed conditions. The FPF values increase by 40–50% after 10 and 25 TiO2 cycles.
Figure 5.

In vitro aerosolization and correlation with interparticle force. (a) Fine particle fraction (FPF), % of loaded mass below 5 μm, of uncoated and ALD-coated budesonide with different film thicknesses, resulting from different numbers of cycles, of SiO2, TiO2, and Al2O3 after long-term storage for 8–10 months at 20 °C and ∼0% RH and after storage for 1 month at 40 °C and 75% RH. The measurements were carried out at a flow rate of 30 L/min for 8 s using the monodose inhaler. The error bars indicate standard deviations. (b) Contact forces of uncoated and ALD-coated budesonide. The error bars indicate 95% confidence intervals. (c) Correlation between FPF and interparticle force after long-term storage for 8–10 months at 20 °C and ∼0% RH. The FPF decreases with increasing contact forces.
To explain that as well as the drastic increase in FPF of ALD-coated budesonide formulations, interparticle forces (F) measurements were performed (see Figure 5b). First, a decrease in the contact force of bare budesonide after stressed storage from ∼75 to ∼50 nN is found. Surface relaxation results in a lower surface energy, and thus in lower contact force, which translates into a higher FPF. Crucially, a substantial drop in the contact force for each ALD-coated budesonide formulation is observed. In particular, a descending-order force scale Funcoated > FAl2O3 > FTiO2 > FSiO2 can be extrapolated. Specifically, the average contact forces for Al2O3-coated, TiO2-coated, and SiO2-coated budesonide are ∼50, ∼42, and ∼30 nN. This force trend is consistent with the Hamaker constants, which are directly proportional to the van der Waals interactions, of the ceramic materials under consideration.53 In fact, the Hamaker constant of SiO2 is significantly lower than that of TiO2 and Al2O3. The higher levels of FAl2O3 over FTiO2 might be explained by the contribution of budesonide arising from Al2O3 infiltration or by the higher water adsorption of Al2O3, which increases the capillary force contribution. The contact forces remain constant with increasing ALD cycles. This is expected, as the surface morphology and composition do not change after 10 ALD cycles. Therefore, the reduced interparticle force in each ALD-coated budesonide formulation can be explained by the modified surface composition, which diminishes the van der Waals force. An additional decline in FTiO2 after stressed storage is noticed. At first sight, this might be surprising, as the capillary contribution to the interparticle force typically increases with thicker water adsorbed layers. However, for TiO2 nanosized particles, the water adsorbed layer thickness strongly depends on humidity, consequently affecting the humidity dependence of the interparticle force. In particular, molecular dynamics simulations by Laube etal.54 revealed a force decrease at increasing humidity for smooth, nearly spherical TiO2 particles, in agreement with our observed drop in FTiO2 after storage at 75% RH. Finally, the FPF and interparticle force of uncoated and ALD-coated budesonide were correlated. As shown in Figure 5c, a linear correlation between FPF and interparticle force is obtained. This suggests that the superior aerosolization performance, i.e., improved FPF, of ALD-coated budesonide results from the reduced interparticle force due to the ceramic surface. Importantly, the ALD-coated budesonide formulations retain their higher FPFs under stressed conditions, thus demonstrating excellent stability. This holds great promise for the development of carrier-free DPI formulations.
Safety and Efficacy Assessment
To investigate safety and efficacy parameters of ALD-coated budesonide, two different in vitro cell based systems, i.e., A549 and EpiAirway, were used. A549 is a cancer cell line representing the alveolar epithelium and is cultured as a monolayer completely submerged in culture medium. The 3D human bronchial model, EpiAirway,55 is made up of primary cells with the ability to differentiate into in vivo-like pseudostratified epithelium mimicking the epithelium of the central airways. Moreover, the 3D model reconstitutes a polarized tissue with a basolateral side that is in contact with the culture medium and an apical side that is exposed to air, allowing for inhalation studies by powder deposition. A first safety screening against A549 cells was carried out using a CCK-8 assay. After incubation of the cells with both uncoated and ALD-coated budesonide for 24 h, no cytotoxicity was observed (see Figure S8). The cell viability stays consistent across the tested concentration range 1–100 μM of budesonide formulation. This is in agreement with the findings from Ivask etal.,56 who demonstrated that SiO2, TiO2, and Al2O3 nanoparticles showed no or negligible toxic effects against human epithelial alveolar A549 cells, human epithelial colorectal Caco2 cells, and the murine fibroblast cell line Balb/c 3T3 at concentrations smaller than 100 μg mL–1 with the same incubation time of 24 h. To further investigate whether the deposited ceramic materials could induce toxicity upon longer incubation time, the cells were treated with uncoated and ALD-coated budesonide concentrations up to 1000 μM for 48 h. The viability measure of cells exposed to 1% SDS (v/v) or 50 μM menadione was used as positive controls.57,58 The mean cell viability for a concentration of budesonide formulation of 20 μM is above 90% for uncoated and Al2O3-coated budesonide and above 80% for SiO2-coated and TiO2-coated budesonide (see Figure 6a). The cell viability of both uncoated and ALD-coated budesonide drops from >80–90% at 20 μM to ∼20–30% at 500 and 1000 μM of budesonide formulation. This reduction is typically reported for long-term, e.g., 48 h, exposures to high concentrations of budesonide.59 Crucially, no significant difference in cell viability of uncoated and ALD-coated budesonide is found at each concentration of budesonide formulation, indicating that the cytotoxicity arises from budesonide itself rather than from the ceramic materials. Therefore, each ALD-coated budesonide formulation appears to be tolerated in terms of A549 cell viability for concentrations of budesonide formulation up to 100 μM for 24 h incubation and up to 20 μM for 48 h incubation.
Figure 6.
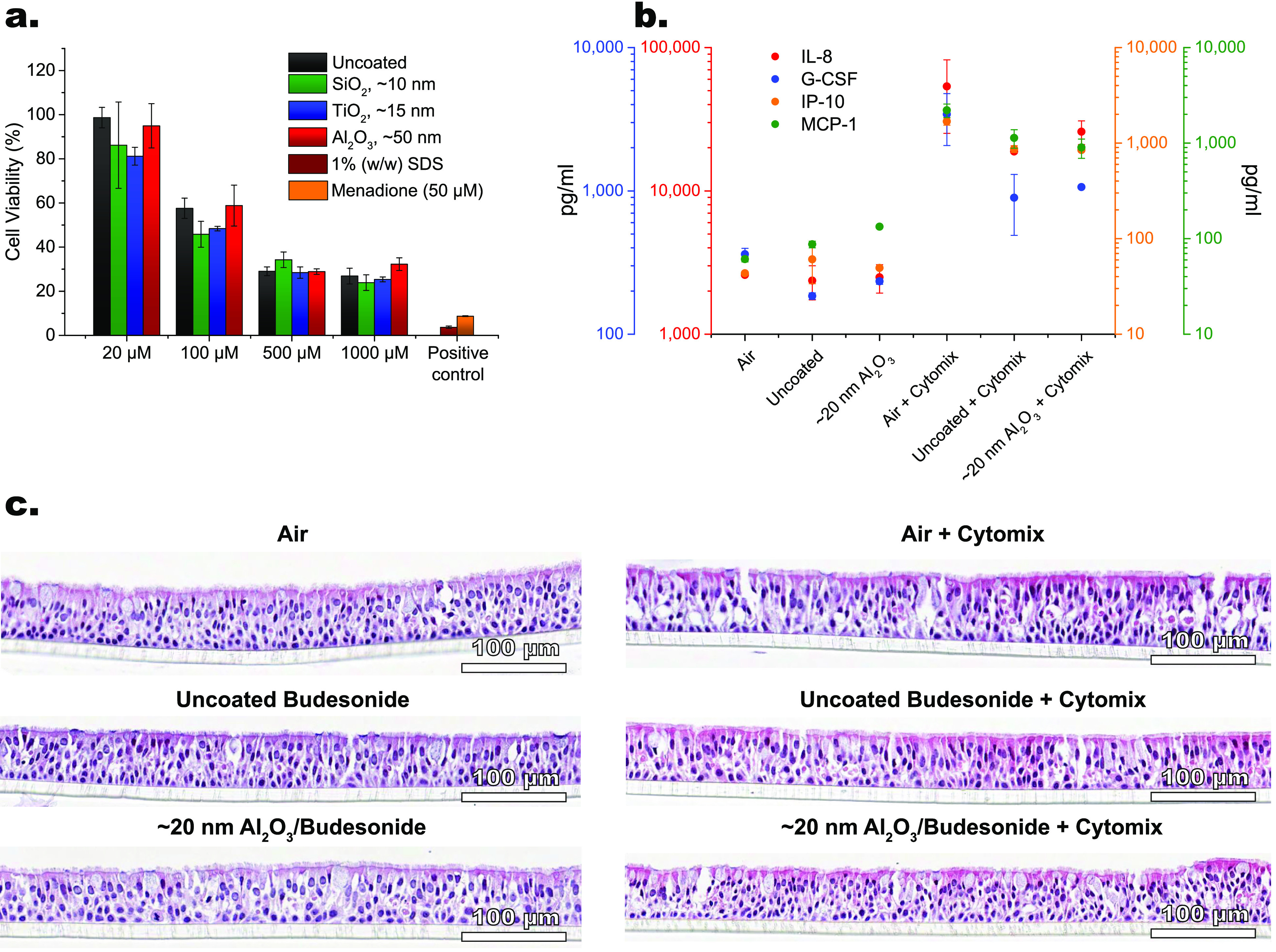
Safety and efficacy assessment. (a) Cell viability after treatment with uncoated and ALD-coated budesonide in the concentration range of 20–1000 μM for 48 h on A549 cells. The error bars indicate standard errors. (b) Cytokine analysis of IL-8, G-CSF, IP-10, and MCP-1 and (c) histology analysis of lung tissue morphology upon exposure of 3D human bronchial epithelial cells to air and uncoated and Al2O3-coated budesonide for 24 h with and without cytomix (0.2 mg/mL LPS and 500 ng/mL TNF-α).
The characteristics and performance of each formulation in terms of drug loading, release, aerosolization, and safety are categorized in Table 1. The highest drug loadings for the same thickness are obtained by Al2O3 ALD. The infiltration of Al2O3 in the budesonide core leads to thick films only partially consisting of Al2O3, thus resulting in higher drug loadings. SiO2 ALD offers higher drug loadings than TiO2 ALD due to the lower SiO2 density, which results in a lower amount of deposited material for the same thickness. The infiltration mechanism of Al2O3 is also beneficial in forming thick films far faster than SiO2 and TiO2 ALD, which have GPCs of 0.1 and 0.3 nm, respectively, as shown earlier. Given the highest thickness, Al2O3-budesonide films translate into the slowest dissolution rates, as indicated by the highest t50 values. The best aerosolization properties are exhibited by SiO2-coated budesonide, due to the lower van der Waals force of SiO2. However, even TiO2-coated and Al2O3-coated budesonide show very high FPFs, if processed up to 10–25 cycles. With respect to safety parameters, the cell viability of Al2O3-coated budesonide is the closest to that of bare budesonide. Taken together, Al2O3 is the most promising material in sustaining release and improving aerosolization while retaining a high cell viability, high drug loading, and relatively short ALD processing time, especially when deposited for a low number of cycles.
Table 1. Drug Loading, Dissolution (t50 in Vitro), Aerosolization (FPF), and Safety Properties (Cell Viability at 20 μM for 48 h) of Uncoated and ALD-Coated Budesonide as Well as Their ALD Processing Time.
| loading | ALD time | t50 in vitro | FPF | cell viability | ||
|---|---|---|---|---|---|---|
| sample | (wt %) | (h) | (min) | (%) | (%) | |
| uncoated budesonide | 100 | 12.3 | 29.4 | 98.7 ± 4.6 | ||
| SiO2/budesonide | 6 nm | 95.9 | 9.4 | 16.7 | 68.3 | |
| 10 nm | 93.2 | 18.8 | 27.0 | 58.4 | 86.2 ± 19.6 | |
| TiO2/budesonide | 3 nm | 96.2 | 1.9 | 15.0 | 56.8 | |
| 8 nm | 89.3 | 4.8 | 21.8 | 52.8 | ||
| 15 nm | 78.9 | 9.6 | 29.5 | 36.7 | 81.2 ± 4.0 | |
| Al2O3/budesonide | 20 nm | 93.9 | 2 | 36.1 | 68.1 | |
| 35 nm | 85.6 | 5 | 52.6 | 53.0 | ||
| 50 nm | 81.3 | 10 | 61.6 | 30.3 | 95.0 ± 10.0 |
In this view, additional safety and efficacy tests were carried out using the 3D bronchial epithelial cultures for 10-cycle-Al2O3-coated budesonide. The respiratory epithelium is known to secrete chemoattractants and proinflammatory cytokines in response to lung injury or infections. Therefore, a range of cytokines, including interleukin 8 (IL-8), granulocyte-colony stimulating factor (G-CSF), interferon gamma-induced protein 10 (IP-10), and monocyte chemoattractant protein 1 (MCP-1), were measured to study the response of epithelial cells to uncoated and Al2O3-coated budesonide formulation with and without a cytomix treatment (see Figure 6b).60 Specifically, without the addition of the cytomix cocktail, safety can be assessed, whereas with the cytomix efficacy can be evaluated. The cytomix cocktail comprises cytokines chosen to reflect the activation of the innate immune system, as their secretion can recruit further immune cells such as monocytes and neutrophils to the site of injury or insult. Without the cytomix treatment, no significant change in cytokine levels was detected for either uncoated or Al2O3-coated budesonide. This suggests that Al2O3 does not induce a proinflammatory innate immune response and appears tolerable by nature. On the other hand, the cytomix treatment activates the early innate inflammatory cytokine release by epithelial cells. However, both uncoated and Al2O3-coated budesonide reduce the cytokine levels induced by the cytomix. Crucially, Al2O3 does not affect the anti-inflammatory activity of budesonide, thus retaining its efficacy.
The 3D bronchial epithelial cultures demonstrate sufficient differentiation with adequately distributed ciliated and goblet cells (see Figure 6c). Based on hematoxylin and eosin staining, no difference can be observed between air-treated, budesonide-treated, and Al2O3–budesonide-treated cells. Similarly, even the corresponding cytomix-treated cultures do not display significant alterations in the composition of ciliated and goblet cells as well as in their overall histological appearance. Basal cells are in close contact with the basement membrane, as expected. The only minor difference in the cytomix-treated cultures is given by the presence of condensed nuclei in each cell type and increased eosinophilic stain in the cytoplasm of the ciliated cells. This is attributed to the increased release in cytokines shown in Figure 6b, as a higher cell activity due to, for example, protein secretion is often coupled with a darker staining pattern. Overall, Al2O3-coated budesonide appears to be safe and efficacious with respect to the bronchial epithelial cells, in agreement with the results in alveolar A549 cells. Yet, to fully assess the safety profile of the ALD films, more comprehensive regulatory repeat-dose in vivo toxicity studies would be required. In particular, while the ALD films may be degraded and removed by clearance mechanisms in the lung, i.e., mucociliary clearance in the central airways and phagocytosis in the alveolar region by alveolar macrophages,61 long-term studies on pulmonary clearance and potential impact of ALD-coated budesonide would be needed to assess eventual bioaccumulation, biodegradation, or expulsion of the ALD materials.
Conclusion
In conclusion, we have developed inhaled budesonide powder formulations with controlled release and aerosolization properties depositing a range of ceramic nanoscale films by ALD, namely, SiO2, TiO2, and Al2O3. TEM and EDX reveal the deposition of uniform SiO2 and TiO2 surface nanofilms and the infiltration of Al2O3 into the outer core of the budesonide particles. Nonetheless, a core/shell structure is retained for each ALD-coated budesonide formulation. Nanoscale films of ∼10 nm and above are able to significantly sustain the release of budesonide both in vitro and ex vivo. The dissolution rate seems to be unaffected by the composition of the film, but mainly depends on its thickness, which dictates the diffusion of the medium. Importantly, the ceramic ALD films also improve the aerosolization performance of budesonide after both standard and stressed storage due to their lower van der Waals force. In particular, ALD-coated budesonide processed up to 10–25 cycles delivers FPFs up to 2.3 times higher than bare budesonide, also at a very low inhalation flow rate. Increasing the number of ALD cycles and thus the powder processing time in the fluidized bed promotes particle agglomeration, which in turn results in lower FPF, yet still higher than bare budesonide. Moreover, the deposited ceramic materials preserve the safe and efficacious attributes of budesonide, as no toxicity is detected in human epithelial alveolar A549 cells and 3D human bronchial epithelial cells. By providing sustained release and improved aerosolization while retaining high drug loadings up to ∼95% and cell viability, ALD proves to be a revolutionary route to fabricate controlled pulmonary drug delivery systems.
Experimental Section
Materials
Micronized budesonide particles with a particle size distribution ranging from 0.1 to 10 μm and a specific surface area of ∼5.6 m2/g were provided by AstraZeneca and used as received.32 The ALD precursors, trimethylaluminum (TMA), titanium tetrachloride (TiCl4), and silicon tetrachloride (SiCl4), were purchased from Nouryon, Strem Chemicals, and Alfa Aesar, respectively, and used as received. Ozone was employed as a co-reactant with TMA, whereas demineralized water as a co-reactant with TiCl4 and SiCl4. Each precursor was stored in a stainless steel bubbler under an inert atmosphere. Sodium phosphate monobasic monohydrate, disodium hydrogen phosphate anhydrous, sodium dodecyl sulfate, orthophosphoric acid (85%), and potassium dihydrogen phosphate were purchased from Sigma-Aldrich and used to prepare sodium phosphate buffer solutions at pH = 6.8. European Pharmacopoeia reference standard budesonide, supplied by Sigma-Aldrich, was used for calibration in the in vitro dissolution test. Acetonitrile and ethanol of HPLC grade were provided by Carl Roth GmbH.
ALD Experiments
The ALD experiments were carried out in a vibrated fluidized bed reactor operating at atmospheric pressure, as described elsewhere.32,36 The ALD precursors, i.e., TMA, TiCl4, and SiCl4, and co-reactants, i.e., O3 and H2O, were kept at around room temperature (see Table S1). N2 (99.999 v/v%) was used as both carrier and purging gas. The lines were kept at 30 °C above the bubblers’ temperature to avoid undesired condensation and underdelivery of precursors. The reactor was operated at a temperature of 40 °C for each ALD process (see Table S1). In each experiment, budesonide powder batches of 5 g were loaded into the reactor. Optimized gas flows of 1 NL min–1, corresponding to 3.4 cm/s at room temperature, were employed to deliver the precursors to the reactor and sufficiently mix the powder with the assistance of mechanical vibration. ALD of Al2O3 and TiO2 were run up to 50 cycles, whereas ALD of SiO2 was run up to 100 cycles. The precursors and their exposure times in each deposition process are reported in Table S1.
Surface Characterization
The morphology of the ALD films on the budesonide particles was investigated by TEM and scanning transmission electron microscopy (STEM). The samples were prepared by directly dispersing the powders on copper TEM grids of 3.05 mm in diameter. TEM and STEM images of several particles on the grid were taken using a JEOL JEM1400 electron microscope operating at 120 kV and a FEI Cs corrected cubed Titan operating at 300 kV, respectively. In STEM, the images were obtained in high-angle annular dark-field (HAADF) mode. In parallel with HAADF imaging, EDX measurements were acquired using an Oxford Instruments XMaxN100TLE detector. Elemental maps of Si, Ti, Al, C, and O were collected at several locations on the grids. The thickness of the Al2O3, TiO2, and SiO2 films was then evaluated with the ImageJ software. For each sample, the film thickness of 10–50 particle agglomerates was measured at various locations and averaged.
Elemental analysis of Si, Ti, and Al was carried out by ICP-OES. Approximately 30 mg of powder was destructed in 4.5 mL of 30% HCl, 1.5 mL of 65% HNO3 and 1 mL of 40% HF using the microwave Multiwave PRO. The destruction time in the microwave was 60 min at maximum power. After destruction, the samples were diluted to 50 mL with Milli-Q water and analyzed with a PerkinElmer Optima 5300 DV optical emission spectrometer. From the measured mass fractions of Si, Ti, and Al, the corresponding weight percentages of SiO2, TiO2, and Al2O3 were calculated.
The crystal structure of uncoated and ALD-coated budesonide was examined by XRPD. The diffractograms were obtained by a Bruker AXS D8 Discover diffractometer with Co Kα radiation. The angle 2θ was scanned from 5° to 50° with steps of 0.02°.
In Vitro Dissolution Test
Prior to the in vitro dissolution test, the uncoated and ALD-coated budesonide powders were dispersed using an mACI, as reported by Franek etal.50 The modified ACI consists of a throat, preseparator, stage 0, stage 1, five custom-made hollow stages and a collection plate with glass fiber filters (Whatman grade GF/C, binder free, 21 mm in diameter) where the powders were deposited. The cutoff diameter of stage 1 is 4.4 μm. To achieve particle deposition on the filter instead of impaction while ensuring full emptying of the capsule, the hollow stages have the same height as a standard stage, i.e., 2.6 cm. Four milligram amounts of uncoated and ALD-coated budesonide were filled into hydroxypropyl methylcellulose capsules (HPMC Vcaps, size 3 capsules Capsugel), which were inserted into a monodose inhaler. The inhaler device was attached to the throat of the mACI. The powders were aerosolized at the standard USP airflow conditions operated at a flow rate of 60 L/min and a flow time of 0.5 s and left to sediment for 40 min onto the glass fiber filters, which were then used in the dissolution tests.
The morphology of the budesonide powders after aerosolization with mACI was analyzed by SEM and TEM, respectively. A SEM sample holder with carbon tape and some TEM grids were placed at the collection plate of the mACI. SEM images were taken at several magnifications using a JEOL JSM-6010 scanning electron microscope, whereas TEM images were recorded at various locations and magnifications using a JEOL JEM-1400 electron microscope.
The dissolution of the budesonide powders deposited through gravity onto the filters was performed as described by Franek etal.50 One filter with deposited budesonide was joined with an empty filter, enclosing the deposited budesonide in between the filters. The assembled filters were then inserted into a 3D-printed holder, which was placed into a μDiss tube (Pion).49 The dissolution tests were carried out with a stirring speed of 500 rpm at 37 °C in 25 mL of 0.1 M degassed phosphate buffer solution with 0.5% SDS (pH 6.8). Dissolution occurred under sink conditions (budesonide dose/solution volume < 1/5 solubility). Samples of 1.4 mL were collected at different time points (5, 10, 15, 30, 45, 60, 90, 120, and 180 min), and subsequently 1.4 mL of fresh dissolution medium at 37 °C was refilled back to maintain a constant volume of dissolution medium. Remaining amounts of budesonide in/on the filter were further dissolved in pure ethanol with a sonication of 20 min. The total amount of budesonide loaded on the filter was determined by summing the released amount during the dissolution test and the remaining amount in/on the filter dissolved in ethanol after the dissolution test. The fraction of released budesonide was calculated by dividing the amount of budesonide released at each time point by the initial budesonide mass loaded on the filter.
The budesonide concentration was quantified using high-performance liquid chromatography with a UV–vis detector (HPLC-UV) operating at 254 nm using a Waters Acquity system H-Class equipped with a quaternary solvent delivery pump. The chromatographic separation was performed using an HPLC Nucleodur C18 column, 150 mm × 4.6 mm, 5 μm (Macherey-Nagel). The column temperature was maintained at 40 °C. The injected volume of each sample was 10 μL. The mobile phase consisted of phosphate buffer (pH = 3) and acetonitrile (32/68, v/v). The eluent was sonicated for 15 min before use. The flow rate was 1.0 mL/min. Data acquisition and processing were realized by using EMPOWER-3 software (Waters Corporation). Quantitation was achieved by measuring the peak area at the wavelength with the maximum absorption, i.e., 254 nm.
Ex Vivo Isolated Perfused Rat Lung Absorption Studies
The IPRL method was used as previously described by Ewing etal.51 In brief, rats were euthanized using an overdose of pentobarbital. Lung and heart were dissected en bloc, and the pulmonary artery was catheterized. A plastic tube was fitted in the trachea, and the lung was placed in an artificial thorax. Hydrostatic pressure was used to perfuse pulmonary circulation with an albumin Krebs-Ringer buffer at neutral pH. Isolated lung preparation was ventilated using a rodent ventilator (No. 7025, Ugo Basile) at 75 breaths per minute and a tidal volume on average of 1.5 mL. IPRLs were perfused in a single-pass mode, prohibiting recirculation of perfusate in the lung. Short inhalation exposures of aerosolized uncoated and ALD-coated budesonide were delivered to IPRLs. A respiratory circuit was devised to pass aerosols and to allow dry powder pharmaceuticals to deposit in isolated rat lungs, using a downstream vacuum source and the inspiratory airflow produced by IPRLs. The targeted lung deposited dose was 20 nmol per lung, corresponding approximately to a lung deposited dose of 20 μg/kg bodyweight. Following inhalation, perfusate exiting the lung was collected using a sample collector. Perfusate was collected at several time points up to 1 h following drug administration, and aliquots (50 μL) were retained for bioanalysis. At the end of the experiment, the lungs were weighed and frozen prior to sample preparation and analysis.
The lungs were homogenized in a Ringer solution using Precellys Dual bead-beating technology (Bertin Technologies SAS). For the analysis, isolated perfused rat lung homogenate (50 μL) and lung perfusate (50 μL) were protein precipitated by the addition of methanol (180 μL), containing acetic acid (0.2%, v/v) and an analytical internal standard. After centrifugation (3440g, 20 min, 4 °C) the supernatants were diluted (1:1, v/v) with deionized water, containing acetic acid (0.2%, v/v) to match the initial mobile phase. If necessary, the samples were diluted further in 30% methanol and 0.2% acetic acid (v/v). Automated liquid handling during the sample preparation was done using the Bravo platform (Agilent Technologies Inc.). A budesonide stock solution (2 mM) was prepared in methanol and used for the calibration standard samples. Calibration curves were prepared from a dilution series in methanol, followed by a 1:10 dilution in the blank perfusate buffer and blank lung homogenate matrix, respectively. The calibration samples were precipitated and analyzed exactly as the samples. Three calibration standard curves, with at least six concentration levels, were interspersed with study samples within each analytical run. The samples were loaded and separated on a PAL HTC-xt autosampler (CTC Analytics AG)/Agilent 1200 LC pump system (Agilent Technologies Inc.) coupled to an API 5500 triple quadrupole mass spectrometer (Sciex) or a PAL HTC-xt autosampler/Shimazdu LC system (Shimazdu Corporation) coupled to an API 5000 triple quadrupole mass spectrometer in negative mode with the mass transition 489.4 > 357.5 at declustering potential and collision energy of −75 V and −20 V, respectively. Separation was achieved by applying a gradient elution on a Kinetex C18 column (50 mm × 2.1 mm, particle size 2.6 μm) (Phenomenex). The mobile phases consisted of 10 mM ammonium formate acetate and 0.2% acetic acid in deionized water (A) and 0.2% acetic acid in methanol (B). See Table S2 for the applied gradient. The software Analyst Version 1.6.2 (Sciex) was used for instrument control, data acquisition, and processing of all LCMS/MS-data concentrations and to determine valid concentrations. Budesonide concentrations were determined by fitting individual response values to the calibration curve made from the calibration standard samples. All calibration curves were best fitted with linear regression with a weighting factor of 1/x or 1/x2. Criteria for qualification of calibration levels were %CV < 25 at concentrations near the lower limit of quantification (LLOQ) and %CV < 15 at all other levels. LLOQ for lung perfusate and lung homogenate was 0.19 nM and 15 nM, respectively.
In Vitro Aerosolization and Interparticle Force Measurements
A fast screening impactor (FSI) (Copley Scientific Ltd.) was used as an abbreviated impactor to assess the aerodynamic performance of uncoated and ALD-coated budesonide samples. FSI consists of a coarse fraction collector that collects particles with an aerodynamic diameter larger than 5 μm and a fine fraction collector that collects particles with an aerodynamic diameter smaller than 5 μm. A 5 μm size cutoff plate is inserted at the bottom of the preseparator. The entire system was connected to a vacuum pump (Gast, model 1423) with a flow controller TrB III trigger box (AB FIA) in order to produce a pressure drop of ∼1 kPa over the inhaler. An accurately weighed amount of powder (uncoated or ALD-coated budesonide), equal to 4.5 mg, was manually introduced into a size 3 hard HPMC capsule and aerosolized with a flow rate of 30 L/min for 8 s (total volume of 4 L) and a critical flow with a P3/P2 ratio < 0.5, as required by the European Pharmacopoeia using a monodose inhaler (RS01, Plastiape). The fine particle fraction is defined as the mass fraction of particles < 5 μm of aerodynamic diameter with respect to the loaded mass. The fine particles passing through a 5 μm size cutoff plate were dissolved in ethanol–water (40/60, v/v) and sonicated for 40 min. After being filtered through a 0.2 μm filter, the concentration of budesonide was determined at a wavelength of 248 nm using a Hach Lange DR5000TM UV–vis spectrometer or determined by HPLC as described earlier. The fine particle fractions of all the samples were investigated both after long-term storage for 8–10 months at 20 °C and 0% RH and after storage in open glass vials for 1 month at 40 °C and 75% RH. The chamber at 40 °C and 75% RH consists of a closed box containing a saturated NaCl solution and placed inside an oven held at 40 °C.
AFM measurements were taken in an NT-MDT (NTEGRA) microscope with OLTESPA silicon nitride cantilevers (Bruker AFM probes) with a stiffness of 2 N/m. The AFM was mounted onto a vibration-isolated table. Small amounts of particles were heaped on a microscope glass slide, which was then knocked to remove loosely bound agglomerates. The measurements were performed at ambient conditions, i.e., 20 °C and ∼40% RH. Particle agglomerates for analysis were selected by a top view camera mounted on the AFM. Deflection curves (50–200 per sample) were recorded over a 100 × 100 μm area on several agglomerates to average out local inhomogeneities. Force curves were then obtained by converting the cantilever deflection using Hooke’s law and the thermal noise method on a cleaned microscope glass slide. The jumps in the force curves correspond to contacts between two or more particles. The contact force between two individual particles was determined by measuring the last jump in the force curves, as described by Salameh etal.62,63
Cell Viability Analysis
The cell viability analysis of uncoated and ALD-coated budesonide was carried out with the human epithelial alveolar A549 cell line. A549 cells were purchased from the American Type Culture Collection. The cells were cultured in 75 cm2 culture flasks (Corning Inc. Life Sciences) using 15 mL of Dulbecco’s modified Eagle’s medium in an incubator at 37 °C in an atmosphere of 5% CO2 and 95% relative humidity. The medium was supplemented with 10% heat inactivated fetal bovine serum (Sigma-Aldrich) and antimycotic solution (1% v/v). The growth medium was changed every other day until the time of use. A549 cells were seeded on 96-well plates at a density of 1 × 104 cells per well and cultured overnight. The media was aspirated from all the wells, and the cells were then treated with the desired concentration ranging from 20 to 1000 μM of budesonide formulation, which was dispersed and diluted in 100 μL of cell culture media for each well. Cells incubated with 1% SDS (v/v) or 50 μM menadione diluted in the cell culture media were used as positive controls. After incubation for 48 h, cell viability was evaluated by adding 10 μL of CCK-8 reagent (Sigma-Aldrich) to each well and incubated for an additional 2 h at 37 °C under the condition of 5% CO2 and 95% relative humidity. The optical density was measured by using a microplate reader (Multiskan FC, Thermo Scientific) at 450 μnm with a reference absorbance at 620 nm, according to the manufacturer’s protocol.
3D Human Bronchial in Vitro Model
The EpiAirway bronchial tissue models and culture media were procured from MatTek Corporation.55 Nondiseased primary human bronchial cells differentiated at MatTek facilities were obtained via accredited institutions, where experimental procedures were explained in full, and subjects provided informed consent. Bronchial cells were seeded on 12 mm Snapwell inserts in a six-well Transwell plate format (Corning, product number: 3801) and maintained in AIR-100-MM culture media supplemented with antibiotics/antimycotics according to the manufacturer’s description (cat. no. AIR-200-SNPPE). Upon arrival at AstraZeneca Gothenburg laboratories, the cells were brought to cell culture conditions at 37 °C and left to acclimatize for 7 days prior to experiments. During the adaptation period, the cultures were fed basolaterally with 2.5 mL/well maintenance medium every second day, and trans-epithelial electrical resistance (TEER) was recorded as quality control by overlaying cells on the apical side with 200 μL of culture media. On the day of the experiment, the cells were placed in fresh six-well plates with 2.5 mL of warm culture media on the basolateral side. Next, the mACI was used to dose the apical side of the cells with uncoated and Al2O3-coated budesonide particles, respectively.64,65 One milligram of powder was weighed into a screenhaler, which connected to the mACI into which the cells were placed. A suction of 60 L/min was applied for 0.3 s to disperse the powder into an aerosol, which was subsequently allowed to sediment onto the cells in the mACI during 5 min. To quantify the powder dose on the cells, additional cells were dosed alongside the cells used for safety and efficacy assessments. The budesonide dose was quantified from these cells by mass-balance determination using LC-MS/MS. The powder dose on the cells was within the range 0.5–1 μg for both uncoated and Al2O3-coated budesonide particles. In addition to the treated samples, a subset of cells were exposed to air only without any powder (i.e., “air” control). After dosing, the cells were placed back into plates containing culture media and divided into a toxicology (n = 3 per treatment) and an efficacy (n = 3 per treatment) cohort, respectively. Cells in the toxicology cohort were allowed to incubate for 24 h prior to end point measurements. Cells in the efficacy cohort were left to preincubate with compounds for 2 h in the incubator followed by cytomix treatment up to 24 h to trigger an inflammatory response. The cytomix consists of 0.2 mg/mL lipopolysaccharide (LPS) from Escherichia coli O26: B6 (Sigma-Aldrich, L8274) and 500 ng/mL recombinant human tumor necrosis factor alpha (rhTNF-α) (R&D Systems, 210-TA). After the final 24 h incubation step, supernatants from the basolateral side were collected and stored at −80 °C for subsequent cytokine analysis. Then, cells were fixed for histopathology analysis.
Collected supernatants were thawed on ice and centrifuged for 5 min at 1000 rpm at 4 °C. Neat supernatants were tested for IL-8, interferon gamma (IFNγ), IL-6, MCP-1, G-CSF, and IP-10 using MILLIPLEX MAP human cytokine/chemokine magnetic bead panel (HCYTOMAG-60K, Millipore Corporation) following the manufacturer’s instructions. The levels of IFNγ and IL-6 were below or close to the limit of quantification, and thus not reported. In brief, 25 μL samples, standards, and controls were incubated with antibody-immobilized beads and assay buffer for 17 h at 4 °C on an orbital shaker at 800 rpm. Plates were washed and incubated with 25 μL of detection antibodies for 1 h prior to incubation with 25 μL of streptavidin–phycoerythrin for 30 min. Plates were then washed, and 150 μL of Drive Fluid was added. Fluorescence was measured on Luminex MAGPIX using Luminex xMAP Technology (Luminex BV). All incubations took place on an orbital shaker at 800 rpm at room temperature, unless stated otherwise. Standard curves were generated using 5PL nonlinear regression and used to extrapolate cytokine concentrations. Acceptance criteria for controls and standard curve included 20% CV and percent recovery between 80% and 120%.
Cells were fixed in 4% paraformaldehyde for 15 min and washed three times in phosphate buffer solution. Dehydration in ethanol and xylene followed by paraffin (Merck) infiltration was done with a short program for biopsies (1 h and 55 min) on a Microm STP 120 spin tissue processor (Thermo Scientific). Cell layers were embedded in paraffin, and 4 μm thick sections were made using a microtome (Leica RM2165). Hematoxylin and eosin staining was performed using the standard protocol on a Leica ST5020. After staining, the sections were dehydrated in ethanol and xylene, mounted with Pertex mounting medium (Histolab), and scanned with an Aperio Scanscope slide scanner.
Acknowledgments
The authors thank Vera Wissel for part of the ALD experiments and acknowledge the financial support from AstraZeneca and Health–Holland, Top Sector Life Sciences & Health, to stimulate public–private partnerships.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.0c10040.
Experimental conditions; HAADF and EDX of Al2O3-coated budesonide; XRPD of uncoated and ALD-coated budesonide; SEM and TEM after mACI predispersion; dissolution for different budesonide doses with and without mACI predispersion; dissolution of ALD-coated budesonide over 3 h; comparison of ex vivo IPRL absorption with commercial Pulmicort Respules; FPF ratios; A549 cell viability for 24 h (PDF)
Author Contributions
△ D.L.Z. and F.S. contributed equally to this work.
The authors declare the following competing financial interest(s): J.R. van Ommen has a financial interest in Delft IMP.
Supplementary Material
References
- Chauhan B. F.; Ducharme F. M. Addition to Inhaled Corticosteroids of Long-Acting Beta2-Agonists versus Anti-Leukotrienes for Chronic Asthma. Cochrane Database of Systematic Reviews 2014, 1, CD003137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton J. S.; Byron P. R. Inhaling Medicines: Delivering Drugs to the Body through the Lungs. Nat. Rev. Drug Discovery 2007, 6, 67–74. 10.1038/nrd2153. [DOI] [PubMed] [Google Scholar]
- Labiris N. R.; Dolovich M. B. Pulmonary Drug Delivery. Part I: Physiological Factors Affecting Therapeutic Effectiveness of Aerosolized Medications. Br. J. Clin. Pharmacol. 2003, 56, 588–599. 10.1046/j.1365-2125.2003.01892.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton J. S.; Fishburn C. S.; Weers J. G. The Lungs as a Portal of Entry for Systemic Drug Delivery. Proc. Am. Thorac. Soc. 2004, 1, 338–344. 10.1513/pats.200409-049TA. [DOI] [PubMed] [Google Scholar]
- Todoroff J.; Vanbever R. Fate of Nanomedicines in the Lungs. Curr. Opin. Colloid Interface Sci. 2011, 16, 246–254. 10.1016/j.cocis.2011.03.001. [DOI] [Google Scholar]
- Hickey A. J. Controlled Delivery of Inhaled Therapeutic Agents. J. Controlled Release 2014, 190, 182–188. 10.1016/j.jconrel.2014.05.058. [DOI] [PubMed] [Google Scholar]
- Loira-Pastoriza C.; Todoroff J.; Vanbever R. Delivery Strategies for Sustained Drug Release in the Lungs. Adv. Drug Delivery Rev. 2014, 75, 81–91. 10.1016/j.addr.2014.05.017. [DOI] [PubMed] [Google Scholar]
- Dolovich M. B.; Dhand R. Aerosol Drug Delivery: Developments in Device Design and Clinical Use. Lancet 2011, 377, 1032–1045. 10.1016/S0140-6736(10)60926-9. [DOI] [PubMed] [Google Scholar]
- Garcia-Contreras L.; Ibrahim M.; Verma R. Inhalation Drug Delivery Devices: Technology Update. Med. Devices: Evidence Res. 2015, 8, 131–139. 10.2147/MDER.S48888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telko M. J.; Hickey A. J. Dry Powder Inhaler Formulation. Respiratory Care 2005, 50, 1209–1227. [PubMed] [Google Scholar]
- Sebti T.; Vanderbist F.; Amighi K. Evaluation of the Content Homogeneity and Dispersion Properties of Fluticasone DPI Compositions. J. Drug Delivery Sci. Technol. 2007, 17, 223–229. 10.1016/S1773-2247(07)50040-7. [DOI] [Google Scholar]
- Faulhammer E.; Wahl V.; Zellnitz S.; Khinast J. G.; Paudel A. Carrier-Based Dry Powder Inhalation: Impact of Carrier Modification on Capsule Filling Processability and in Vitro Aerodynamic Performance. Int. J. Pharm. 2015, 491, 231–242. 10.1016/j.ijpharm.2015.06.044. [DOI] [PubMed] [Google Scholar]
- Healy A. M.; Amaro M. I.; Paluch K. J.; Tajber L. Dry Powders for Oral Inhalation Free of Lactose Carrier Particles. Adv. Drug Delivery Rev. 2014, 75, 32–52. 10.1016/j.addr.2014.04.005. [DOI] [PubMed] [Google Scholar]
- Rawat A.; Majumder Q. H.; Ahsan F. Inhalable Large Porous Microspheres of Low Molecular Weight Heparin: in Vitro and in Vivo Evaluation. J. Controlled Release 2008, 128, 224–232. 10.1016/j.jconrel.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh Y. J.; Lee J.; Seo J. Y.; Rhim T.; Kim S.-H.; Yoon H. J.; Lee K. Y. Preparation of Budesonide-Loaded Porous PLGA Microparticles and Their Therapeutic Efficacy in a Murine Asthma Model. J. Controlled Release 2011, 150, 56–62. 10.1016/j.jconrel.2010.11.001. [DOI] [PubMed] [Google Scholar]
- Dhanda D. S.; Tyagi P.; Mirvish S. S.; Kompella U. B. Supercritical Fluid Technology Based Large Porous Celecoxib–PLGA Microparticles Do Not Induce Pulmonary Fibrosis and Sustain Drug Delivery and Efficacy for Several Weeks Following a Single Dose. J. Controlled Release 2013, 168, 239–250. 10.1016/j.jconrel.2013.03.027. [DOI] [PubMed] [Google Scholar]
- Bayard F.; Thielemans W.; Pritchard D.; Paine S.; Young S.; Bäckman P.; Ewing P.; Bosquillon C. Polyethylene Glycol-Drug Ester Conjugates for Prolonged Retention of Small Inhaled Drugs in the Lung. J. Controlled Release 2013, 171, 234–240. 10.1016/j.jconrel.2013.07.023. [DOI] [PubMed] [Google Scholar]
- Willis L.; Hayes D.; Mansour H. M. Therapeutic Liposomal Dry Powder Inhalation Aerosols for Targeted Lung Delivery. Lung 2012, 190, 251–262. 10.1007/s00408-011-9360-x. [DOI] [PubMed] [Google Scholar]
- Beck-Broichsitter M.; Rieger M.; Reul R.; Gessler T.; Seeger W.; Schmehl T. Correlation of Drug Release with Pulmonary Drug Absorption Profiles for Nebulizable Liposomal Formulations. Eur. J. Pharm. Biopharm. 2013, 84, 106–114. 10.1016/j.ejpb.2012.12.003. [DOI] [PubMed] [Google Scholar]
- Li Z.; Zhang Y.; Wurtz W.; Lee J. K.; Malinin V. S.; Durwas-Krishnan S.; Meers P.; Perkins W. R. Characterization of Nebulized Liposomal Amikacin (ArikaceTM) as a Function of Droplet Size. J. Aerosol Med. Pulm. Drug Delivery 2008, 21, 245–254. 10.1089/jamp.2008.0686. [DOI] [PubMed] [Google Scholar]
- Talton J.; Fitz-Gerald J.; Singh R.; Hochhaus G. Nano-Thin Coatings for Improved Lung Targeting of Glucocorticoid Dry Powders: in-Vitro and in-Vivo Characteristics.. Respiratory Drug Delivery VII 2000, 67–74. [Google Scholar]
- Singh R.; Kim W.-S.; Ollinger M.; Craciun V.; Coowantwong I.; Hochhaus G.; Koshizaki N. Laser Based Synthesis of Nanofunctionalized Particulates for Pulmonary Based Controlled Drug Delivery Applications. Appl. Surf. Sci. 2002, 197–198, 610–614. 10.1016/S0169-4332(02)00408-7. [DOI] [Google Scholar]
- Fitz-Gerald J.; Singh R. K.; Gao H.; Pennycook S. Nanometric Dry Powder Coatings Using a Novel Process. KONA 1999, 17, 173–182. 10.14356/kona.1999024. [DOI] [Google Scholar]
- Raula J.; Lähde A.; Kauppinen E. I. A Novel Gas Phase Method for the Combined Synthesis and Coating of Pharmaceutical Particles. Pharm. Res. 2008, 25, 242–245. 10.1007/s11095-007-9464-4. [DOI] [PubMed] [Google Scholar]
- Unger K.; Coclite A. M. Conformal Coating of Powder by Initiated Chemical Vapor Deposition on Vibrating Substrate. Pharmaceutics 2020, 12, 904. 10.3390/pharmaceutics12090904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weers J. G.; Bell J.; Chan H.-K.; Cipolla D.; Dunbar C.; Hickey A. J.; Smith I. Pulmonary Formulations: What Remains to Be Done?. J. Aerosol Med. Pulm. Drug Delivery 2010, 23, S-5–S-23. 10.1089/jamp.2010.0838. [DOI] [PubMed] [Google Scholar]
- Bäckman P.; Adelmann H.; Petersson G.; Jones C. B. Advances in Inhaled Technologies: Understanding the Therapeutic Challenge, Predicting Clinical Performance, and Designing the Optimal Inhaled Product. Clin. Pharmacol. Ther. 2014, 95, 509–520. 10.1038/clpt.2014.27. [DOI] [PubMed] [Google Scholar]
- Puurunen R. L. Surface Chemistry of Atomic Layer Deposition: a Case Study for the Trimethylaluminum/Water Process. J. Appl. Phys. 2005, 97, 121301. 10.1063/1.1940727. [DOI] [Google Scholar]
- Bui H. V.; Grillo F.; van Ommen J. R. Atomic and Molecular Layer Deposition: off the Beaten Track. Chem. Commun. 2017, 53, 45–71. 10.1039/C6CC05568K. [DOI] [PubMed] [Google Scholar]
- Weimer A. W. Particle Atomic Layer Deposition. J. Nanopart. Res. 2019, 21, 9. 10.1007/s11051-018-4442-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ommen J. R.; Goulas A. Atomic Layer Deposition on Particulate Materials. Materials Today Chemistry 2019, 14, 100183. 10.1016/j.mtchem.2019.08.002. [DOI] [Google Scholar]
- Zhang D.; Quayle M. J.; Petersson G.; van Ommen J. R.; Folestad S. Atomic Scale Surface Engineering of Micro- to Nano-Sized Pharmaceutical Particles for Drug Delivery Applications. Nanoscale 2017, 9, 11410–11417. 10.1039/C7NR03261G. [DOI] [PubMed] [Google Scholar]
- Hellrup J.; Rooth M.; Johansson A.; Mahlin D. Production and Characterization of Aluminium Oxide Nanoshells on Spray Dried Lactose. Int. J. Pharm. 2017, 529, 116–122. 10.1016/j.ijpharm.2017.06.046. [DOI] [PubMed] [Google Scholar]
- Kääriäinen T. O.; Kemell M.; Vehkamäki M.; Kääriäinen M.-L.; Correia A.; Santos H. A.; Bimbo L. M.; Hirvonen J.; Hoppu P.; George S. M.; Cameron D. C.; Ritala M.; Leskelä M. Surface Modification of Acetaminophen Particles by Atomic Layer Deposition. Int. J. Pharm. 2017, 525, 160–174. 10.1016/j.ijpharm.2017.04.031. [DOI] [PubMed] [Google Scholar]
- Hellrup J.; Rooth M.; Mårtensson E.; Sigfridsson K.; Johansson A. Nanoshells Prepared by Atomic Layer Deposition – Long Acting Depots of Indomethacin. Eur. J. Pharm. Biopharm. 2019, 140, 60–66. 10.1016/j.ejpb.2019.04.019. [DOI] [PubMed] [Google Scholar]
- Zhang D.; La Zara D.; Quayle M. J.; Petersson G.; van Ommen J. R.; Folestad S. Nanoengineering of Crystal and Amorphous Surfaces of Pharmaceutical Particles for Biomedical Applications. ACS Applied Bio Materials 2019, 2, 1518–1530. 10.1021/acsabm.8b00805. [DOI] [PubMed] [Google Scholar]
- La Zara D.; Zhang F.; Sun F.; Bailey M. R.; Quayle M. J.; Petersson G.; Folestad S.; van Ommen J. R. Drug Powders with Tunable Wettability by Atomic and Molecular Layer Deposition: from Highly Hydrophilic to Superhydrophobic. Appl. Mater. Today 2021, 22, 100945. 10.1016/j.apmt.2021.100945. [DOI] [Google Scholar]
- Yang L.; Sheldon B.; Webster T. Nanophase Ceramics for Improved Drug Delivery: Current Opportunities and Challenges. Am. Ceram. Soc. Bull. 2010, 89, 24–32. [Google Scholar]
- Hirschberg C.; Jensen N. S.; Boetker J.; Madsen A. Ø.; Kääriäinen T. O.; Kääriäinen M.-L.; Hoppu P.; George S. M.; Murtomaa M.; Sun C. C.; Risbo J.; Rantanen J. Improving Powder Characteristics by Surface Modification Using Atomic Layer Deposition. Org. Process Res. Dev. 2019, 23, 2362–2368. 10.1021/acs.oprd.9b00247. [DOI] [Google Scholar]
- Vogel N. A.; Williams P. S.; Brozena A. H.; Sen D.; Atanasov S.; Parsons G. N.; Khan S. A. Delayed Dissolution and Small Molecule Release from Atomic Layer Deposition Coated Electrospun Nanofibers. Adv. Mater. Interfaces 2015, 2, 1500229. 10.1002/admi.201500229. [DOI] [Google Scholar]
- Ferguson J. D.; Weimer A. W.; George S. M. Atomic Layer Deposition of SiO2 Films on BN Particles Using Sequential Surface Reactions. Chem. Mater. 2000, 12, 3472–3480. 10.1021/cm000313t. [DOI] [Google Scholar]
- Sneh O.; Wise M.; Ott A.; Okada L.; George S. Atomic Layer Growth of SiO2 on Si(100) Using SiCl4 and H2O in a Binary Reaction Sequence. Surf. Sci. 1995, 334, 135–152. 10.1016/0039-6028(95)00471-8. [DOI] [Google Scholar]
- Niemelä J.-P.; Marin G.; Karppinen M. Titanium Dioxide Thin Films by Atomic Layer Deposition: a Review. Semicond. Sci. Technol. 2017, 32, 093005. 10.1088/1361-6641/aa78ce. [DOI] [Google Scholar]
- Lee S.-M.; Pippel E.; Gosele U.; Dresbach C.; Qin Y.; Chandran C. V.; Brauniger T.; Hause G.; Knez M. Greatly Increased Toughness of Infiltrated Spider Silk. Science 2009, 324, 488–492. 10.1126/science.1168162. [DOI] [PubMed] [Google Scholar]
- Waldman R. Z.; Mandia D. J.; Yanguas-Gil A.; Martinson A. B. F.; Elam J. W.; Darling S. B. The Chemical Physics of Sequential Infiltration Synthesis—A Thermodynamic and Kinetic Perspective. J. Chem. Phys. 2019, 151, 190901. 10.1063/1.5128108. [DOI] [PubMed] [Google Scholar]
- Parsons G. N.; Atanasov S. E.; Dandley E. C.; Devine C. K.; Gong B.; Jur J. S.; Lee K.; Oldham C. J.; Peng Q.; Spagnola J. C.; Williams P. S. Mechanisms and Reactions during Atomic Layer Deposition on Polymers. Coord. Chem. Rev. 2013, 257, 3323–3331. 10.1016/j.ccr.2013.07.001. [DOI] [Google Scholar]
- Piercy B. D.; Leng C. Z.; Losego M. D. Variation in the Density, Optical Polarizabilities, and Crystallinity of TiO2 Thin Films Deposited via Atomic Layer Deposition from 38 to 150 °C Using the Titanium Tetrachloride-Water Reaction. J. Vac. Sci. Technol., A 2017, 35, 03E107. 10.1116/1.4979047. [DOI] [Google Scholar]
- Velaga S. P.; Djuris J.; Cvijic S.; Rozou S.; Russo P.; Colombo G.; Rossi A. Dry Powder Inhalers: an Overview of the in Vitro Dissolution Methodologies and their Correlation with the Biopharmaceutical Aspects of the Drug Products. Eur. J. Pharm. Sci. 2018, 113, 18–28. 10.1016/j.ejps.2017.09.002. [DOI] [PubMed] [Google Scholar]
- Franek F.; Nilsson L.; Thörn H.; Fransson R.; Tehler U. Abstracts from The Aerosol Society Drug Delivery to the Lungs 29 Edinburgh International Conference Centre Edinburgh, Scotland, UK December 10–12, 2018. J. Aerosol Med. Pulmonary Drug Delivery 2019, 32, A17. [Google Scholar]
- Franek F.; Fransson R.; Thörn H.; Bäckman P.; Andersson P. U.; Tehler U. Ranking in Vitro Dissolution of Inhaled Micronized Drug Powders including a Candidate Drug with Two Different Particle Sizes. Mol. Pharmaceutics 2018, 15, 5319–5326. 10.1021/acs.molpharmaceut.8b00796. [DOI] [PubMed] [Google Scholar]
- Ewing P.; Eirefelt S. J.; Andersson P.; Blomgren A.; Ryrfeldt Å.; Gerde P. Short Inhalation Exposures of the Isolated and Perfused Rat Lung to Respirable Dry Particle Aerosols; the Detailed Pharmacokinetics of Budesonide, Formoterol, and Terbutaline. J. Aerosol Med. Pulm. Drug Delivery 2008, 21, 169–180. 10.1089/jamp.2007.0654. [DOI] [PubMed] [Google Scholar]
- Shur J.; Pitchayajittipong C.; Rogueda P.; Price R. Effect of Processing History on the Surface Interfacial Properties of Budesonide in Carrier-Based Dry-Powder Inhalers. Ther. Delivery 2013, 4, 925–937. 10.4155/tde.13.69. [DOI] [PubMed] [Google Scholar]
- Bergström L. Hamaker Constants of Inorganic Materials. Adv. Colloid Interface Sci. 1997, 70, 125–169. 10.1016/S0001-8686(97)00003-1. [DOI] [Google Scholar]
- Laube J.; Salameh S.; Kappl M.; Mädler L.; Ciacchi L. C. Contact Forces between TiO2 Nanoparticles Governed by an Interplay of Adsorbed Water Layers and Roughness. Langmuir 2015, 31, 11288–11295. 10.1021/acs.langmuir.5b02989. [DOI] [PubMed] [Google Scholar]
- Hayden P.; Jackson G. Long Term Culture of Pseudostratified Tracheal Bronchial EpiAirwayTM Tissues. MatTek Technical Reference 394, 2006. .
- Ivask A.; Titma T.; Visnapuu M.; Vija H.; Kakinen A.; Sihtmae M.; Pokhrel S.; Madler L.; Heinlaan M.; Kisand V.; Shimmo R.; Kahru A. Toxicity of 11 Metal Oxide Nanoparticles to Three Mammalian Cell Types in Vitro. Curr. Top. Med. Chem. 2015, 15, 1914–1929. 10.2174/1568026615666150506150109. [DOI] [PubMed] [Google Scholar]
- Inácio Â. S.; Mesquita K. A.; Baptista M.; Ramalho-Santos J.; Vaz W. L. C.; Vieira O. V. In Vitro Surfactant Structure-Toxicity Relationships: Implications for Surfactant Use in Sexually Transmitted Infection Prophylaxis and Contraception. PLoS One 2011, 6, e19850. 10.1371/journal.pone.0019850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeois B.; Owens J. W. The Influence of Hurricanes Katrina and Rita on the Inflammatory Cytokine Response and Protein Expression in A549 Cells Exposed to PM2.5 Collected in the Baton Rouge–Port Allen Industrial Corridor of Southeastern Louisiana in 2005. Toxicol. Mech. Methods 2014, 24, 220–242. 10.3109/15376516.2014.881945. [DOI] [PubMed] [Google Scholar]
- Fu H.; Zhang J.; Pan J.; Zhang Q.; Lu Y.; Wen W.; Lubet R. A.; Szabo E.; Chen R.; Wang Y.; Chen D.-R.; You M. Chemoprevention of Lung Carcinogenesis by the Combination of Aerosolized Budesonide and Oral Pioglitazone in A/J Mice. Mol. Carcinog. 2011, 50, 913–921. 10.1002/mc.20751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Hooper W. C.; Phillips D. J.; Tondella M. L.; Talkington D. F. Induction of Proinflammatory Cytokines in Human Lung Epithelial Cells during Chlamydia Pneumoniae Infection. Infect. Immun. 2003, 71, 614–620. 10.1128/IAI.71.2.614-620.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberdörster G. Lung Dosimetry: Pulmonary Clearance of Inhaled Particles. Aerosol Sci. Technol. 1993, 18, 279–289. 10.1080/02786829308959605. [DOI] [Google Scholar]
- Salameh S.; Schneider J.; Laube J.; Alessandrini A.; Facci P.; Seo J. W.; Ciacchi L. C.; Mädler L. Adhesion Mechanisms of the Contact Interface of TiO2 Nanoparticles in Films and Aggregates. Langmuir 2012, 28, 11457–11464. 10.1021/la302242s. [DOI] [PubMed] [Google Scholar]
- Salameh S.; Scholz R.; Seo J. W.; Mädler L. Contact Behavior of Size Fractionated TiO2 Nanoparticle Agglomerates and Aggregates. Powder Technol. 2014, 256, 345–351. 10.1016/j.powtec.2014.02.042. [DOI] [Google Scholar]
- Franek F.; Yousef G.; Balogh Sivars K.; Thorn H.; Fransson R.; Tehler U. Abstracts from The Aerosol Society Drug Delivery to the Lungs 28 Edinburgh International Conference Centre Edinburgh, Scotland, UK December 6–8, 2017. J. Aerosol Med. Pulmonary Drug Delivery 2018, 31, A17–A18. [DOI] [PubMed] [Google Scholar]
- Balogh Sivars K.; Sivars U.; Hornberg E.; Zhang H.; Brändén L.; Bonfante R.; Huang S.; Constant S.; Robinson I.; Betts C. J.; Åberg P. M. A 3D Human Airway Model Enables Prediction of Respiratory Toxicity of Inhaled Drugs in Vitro. Toxicol. Sci. 2018, 162, 301–308. 10.1093/toxsci/kfx255. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.